


Abstract
Transcription of the Rhodobacter sphaeroides recA promoter (PrecA) is induced upon DNA damage in a lexA-dependent manner. In vivo experiments demonstrate that LexA protein represses and might also activate transcription of PrecA. Purified R.sphaeroides LexA protein specifically binds the SOS boxes located within the PrecA region. In vitro transcription analysis, using Escherichia coli RNA polymerase (RNAP), indicated that the presence of LexA may stimulate and repress transcription of PrecA. EMSA and DNase I footprinting experiments show that LexA and RNAP can bind simultaneously to PrecA. At low LexA concentrations it enhances RNAP binding to PrecA, stimulates open complex formation and strand separation beyond the transcription start site. At high LexA concentrations, however, RNAP-promoted strand separation is not observed beyond the +5 region. LexA might repress transcription by interfering with the clearance process instead of blocking the access of RNAP to the promoter region. Based on these findings we propose that the R.sphaeroides LexA protein performs fine tuning of the SOS response, which might provide a physiological advantage by enhancing transcription of SOS genes and delaying full activation of the response.
INTRODUCTION
DNA damage induces a set of cellular functions directly or indirectly involved in the repair of lesions and damage tolerance in bacteria (1,2). This network of damage-induced (din) genes is known as the SOS system, which is under the control of the lexA and recA gene products. LexA (also termed DinR in Bacillus subtilis) is a repressor which binds to an imperfect palindrome [the SOS box is 16 bp in Escherichia coli, CTGT(TA)4ACAG, and 13 bp in B.subtilis, CGAAC(AT)2GTTC] located near the promoters of the SOS genes (3–5). The E.coli and B.subtilis LexA repressors inhibit transcription by precluding RNA polymerase (RNAP) binding to the promoter region (6–8). Recently, Hamoen et al. (5) suggested that B.subtilis LexA blocks the interaction of the C-terminal domain of the α subunit (α-CTD) of RNAP with a putative UP element predicted as being between positions –46 to –58 upstream of the B.subtilis recA promoter (PrecA), and therefore precluding active RNAP binding to the promoter region.
RecA protein binds to single-stranded (ss)DNA regions, produced following DNA damage-mediated inhibition of replication or by enzymatic processing of broken DNA ends, and filaments on ssDNA (9,10). The RecA–ssDNA nucleofilament facilitates autocatalytic cleavage of LexA (11,12). The decrease in the cellular pool of LexA results in transcriptional derepression of the SOS genes (2,13). LexA repressor inactivation also leads to the synthesis of the rapidly hydrolysed SfiA (also termed SulA) checkpoint protein, which inhibits cell division (14). Once the DNA has been repaired, in E.coli cells shut-down of the SOS machinery occurs in a DinI-dependent manner (15,16). The level of LexA increases, with a subsequent repression of the SOS network, the synthesised SfiA protein is degraded by the Lon protease and cell division proceeds (17).
Recently, the SOS box in the Alpha group of Proteobacteria (e.g. Rhizobium etli and Rhodobacter sphaeroides, among others) has been established (18–21). LexA recognises two copies of the GAAC or GTTC motif which are separated from each other by 7 bp [SOS box, GAAC(N)7GAAC or GTTC(N)7GTTC], and a different number of copies of the box make up the operator of the SOS system (Fig. 1). The recognition of two direct repeats by a specific transcriptional regulator, instead of a dimer or a tetramer binding to the two halves of a palindromic sequence, is rare in bacteria.
Figure 1.

Nucleotide sequence of the non-transcribed strand of the PrecA region. The PrecA promoter is indicated by empty lines and the transcription start site of PrecA is denoted by a bent arrow. The translation start site, the SOS boxes (SOS1 and SOS2, framed) and the putative SOS-like boxes (underlined) are in bold. The position of the primer used in the primer extension assay is also indicated.
A R.sphaeroides lexA null allele [lexA(Def)], which presents constitutive expression of several din genes (e.g. recA, uvrA and lexA), does not show any decrease in cell viability (22). In contrast, E.coli lexA(Def) mutants are only viable when a mutation in the sfiA gene is also present (2). Likewise, B.subtilis lexA(Def) mutants seem to require an unidentified secondary compensatory mutation (23). Unlike E.coli and B.subtilis, constitutive expression of the SOS system in R.sphaeroides lexA(Def) cells does not increase cellular resistance against DNA-damaging agents (22). Furthermore, the DNA damage-mediated expression of SOS genes in both E.coli and B.subtilis wild-type cells is dramatically lower than under conditions of constitutive expression in lexA(Def) cells (23,24). However, in R.sphaeroides the expression of SOS genes in DNA-damaged wild-type cells is similar to or even higher than that shown in the lexA(Def) strain (22). It is likely that the organisation and control of the SOS network in the Alpha Proteobacteria might be quite distinct from the E.coli and B.subtilis SOS networks.
In this work we report that R.sphaeroides LexA protein, both in vivo and in vitro, represses transcription of the recA promoter (PrecA) and also works as an activator. In vitro the LexA protein binds to sites which overlap promoter sequences. LexA bound to DNA enhances binding of a heterologous RNAP to DNA without affecting open complex formation. In the presence of an excess of LexA, RNAP is stalled around the +5 region. The results presented provide the first evidence that a LexA repressor can play a dual regulatory function, derepressing SOS genes and delaying maximal activation before the SOS response is fully functional.
MATERIALS AND METHODS
Bacterial strains, plasmids and growth conditions
Escherichia coli strains DH5α, BL21(DE3), JM109 and S17 λpir (25–27) and R.sphaeroides strains UA8000 (wild-type) and UA8165 [lexA(Def)] were grown as reported previously (22). Plasmids pBSK, pET3b, pGEM-T, pHP45Km and pHRP309 were used (26,28,29). Plasmid-borne PrecA (pUA840) (19) and plasmid-borne PlacUV5 (pUC18) (25) were employed for promoter analysis. pET3b-borne lexA was used for overexpression. The pHRP309-borne PrecA:lacZ fusion was introduced into R.sphaeroides cells by biparental mating as described (22).
β-Galactosidase (β-Gal) assays
Mitomycin C (MMC) or UV radiation (at 30 J m–2) was applied at time zero and β-Gal experiments were carried out as previously described (19). All data are the averages of three independent assays. β-Gal levels were determined by the standard procedure (30).
DNA, enzymes and reagents
Nucleotides, DNA modification enzymes, RNase A and poly(dG·dC) were purchased from Boehringer-Mannheim and Promega. PCR amplifications were performed using Taq Expand DNA polymerase. Oligonucleotides were purchased from Gen Set Oligos and Boehringer-Mannheim. Gel-purified DNA fragments were end-labelled as described (25). The concentration of DNA was determined using molar extinction coefficients of 6500 M–1 cm–1 at 260 nm and expressed as mol DNA.
Escherichia coli RNAP was purchased from US Biochemical. The soluble 25 kDa LexA protein was purified in three steps by conventional column chromatography (phosphocellulose, S-Sepharose and Superose 12). The N-terminal amino acid sequence of the LexA protein was determined by automated Edman degradation. LexA protein concentration was determined using a molar extinction coefficient of 6400 M–1 cm–1 at 280 nm, as described previously (31). LexA is expressed in mol protein dimers.
Molecular mass determination
Gel filtration chromatography was carried out in buffer A (50 mM Tris–HCl, pH 7, 5% glycerol) containing 500 mM NaCl at 4°C at a flow rate of 0.5 ml min–1 and the A280 was measured. About 50–60 µg LexA protein was applied to the Superose 12 column. A standard curve of Kav versus the log of molecular mass was determined as recommended by Pharmacia.
Primer extension assays
Transcription assays were performed in a 25 µl reaction in buffer B [25 mM Tris–HCl pH 7.5, 2 mM MgCl2, 50 mM KCl, 5 mM (NH4)2SO4, 2% glycerol, 25 mM NaCl and 0.04 mM DTT] containing ATP, CTP, GTP and UTP (100 µM each), 2 nM linear plasmid (PrecA and PlacUV5 control) DNA and 40 nM RNAP. The reactions were incubated for 15 min at 30°C with and without increasing amounts of LexA protein, and the reactions were stopped by the addition of 50 µl of a solution containing 2% (w/v) SDS, 10 µg tRNA and 100 mM EDTA. The annealing and primer extension reactions were performed as described (32). The primers used hybridised downstream of PlacUV5 (5′-GTTTTCCCAGTCACGAC-3′ at coordinates 87–104, where 1 is the in vivo transcription start site) and PrecA (5′-GCCTTGTCCATCTCGCG-3′, Fig. 1). The reactions were stopped and precipitated. The cDNA was analysed by 6% denaturing urea–PAGE (dPAGE) and detected by autoradiography. The relative amounts of transcripts were estimated by laser scanning densitometry of the autoradiographs and the data presented are averages of three independent experiments.
Electrophoretic mobility shift assay (EMSA) experiments
For EMSA experiments, the 210 bp α-32P-labelled EcoRI–HindIII PrecA (2 nM) DNA fragment was incubated, in the presence of 1 µg poly(dG·dC) as a non-specific competitor DNA, in buffer C (25 mM Tris–HCl pH 7, 2 mM MgCl2, 50 mM KCl, 5% glycerol) with different amounts of LexA and/or RNAP for 10 min at 30°C in a 20 µl final volume. The reaction mixture was then immediately loaded onto the gel (25). The samples were separated by 5% non-denaturing (nd)PAGE (80:1 acrylamide/bis-acrylamide) gel. Gels were run with 1× Tris–glycine or 0.5× TAE at 150 V at room temperature and dried prior to autoradiography (32).
DNase I and KMnO4 footprinting
The 228 bp α-32P-labelled EcoRI–PstI PrecA DNA fragment (2 nM) was incubated, in the presence of 1 µg poly(dG·dC) as a non-specific competitor DNA, with different amounts of LexA and/or RNAP in buffer C for 10 min at 30°C in a 20 µl final volume. When indicated, the samples were preincubated with the appropriate NTPs (ATP, CTP and GTP) at 100 µM at 30°C for 10 min. After DNase I treatment (50 ng for 5 min at 37°C) the DNA was precipitated in the presence of 1 µg tRNA. KMnO4 footprinting was performed as indicated with DNase I. The samples were treated with 1 mM KMnO4 for 30 s at 37°C as previously described (33). The DNA was cleaved with piperidine and the samples were separated by 6% dPAGE, dried and analysed by autoradiography.
RESULTS
The R.sphaeroides recA gene is under negative and positive control
It is assumed that the maximal expression level of a LexA-regulated gene in DNA-damaged cells should be similar to that shown under conditions in which the SOS genes are being constitutively transcribed [e.g. lex null mutant cells, lexA(Def)]. The pHRP309-borne PrecA:lacZ fusion was introduced into both wild-type and lexA(Def) R.sphaeroides cells to address their kinetics of expression upon DNA damage. The recA gene is constitutively expressed in lexA(Def) mutant cells (22). Surprisingly, upon MMC treatment PrecA utilisation, measured as β-Gal accumulation, is up to three times greater in wild-type than in lexA(Def) cells (Fig. 2). The same results were found when UV radiation (at 30 J m–2) was used as the inducing agent (data not shown).
Figure 2.

Expression of the PrecA:lacZ fusion in R.sphaeroides wild-type and lexA(Def) cells under SOS-induced conditions. Rhodobacter sphaeroides wild-type and lexA(Def) cells (closed and open symbols, respectively) were treated with MMC at 0.4 µg ml–1 and the level of expression of the PrecA:lacZ fusion measured. All determinations are the means of at least three experiments (each in triplicate) and a single standard error of any value was never >10%.
The data obtained cannot be explained by assuming solely a negative role for the LexA protein because the expression ratio of PrecA between the wild-type and lexA(Def) would increase until it reaches a plateau which would represent maximal promoter utilisation (derepression). Hence, we hypothesised that the kinetics of recA gene induction might be due to a combination of two overlapping processes: (i) transcriptional derepression of PrecA, as a consequence of LexA cleavage, which will enable cells to achieve the maximal level of expression obtained in lexA(Def) cells; (ii) activation of PrecA (direct effect) upon de novo synthesis of LexA. Alternatively, an uncharacterised activator, which only works upon LexA cleavage, could regulate both promoters. We have to assume that this hypothetical and uncharacterised activator is non-functional or not synthesised in the absence of LexA protein [e.g. in the lexA(Def) background] and the presence of cleaved LexA is required for its activity (indirect effect).
Purification and properties of the R.sphaeroides LexA protein
The 25 kDa LexA protein (with a predicted mass of 24 928 Da) was purified to >99% as assayed by SDS–PAGE and quantitative analysis of the N-terminal amino acid. The sequence of the first 10 N-terminal amino acids of the purified protein is in agreement with the predicted amino acid sequence derived from the lexA gene (data not shown).
The native molecular mass of purified LexA was estimated by size fractionation through a Superose 12 FPLC gel filtration column in a buffer containing 500 mM NaCl. From the elution profile of LexA and of a number of protein standards we estimated that the Mr of LexA is ∼50 000, about twice that of a LexA protomer. If we assume that LexA is spherical in shape, it is likely that LexA is a dimer in solution. This is consistent with the observation that E.coli LexA exists within the cells exclusively as a dimer (34). We cannot rule out, however, that R.sphaeroides LexA is an elongated monomer with a large Stokes radius.
Rhodobacter sphaeroides LexA protein represses transcription
Previously, the in vivo transcription start of PrecA has been mapped in R.sphaeroides upon SOS induction (19) (Fig. 1). A computer-based analysis of PrecA has pointed out the existence of two hexameric promoter elements located ∼10 and 35 bp upstream of the in vivo mapped transcription start site (+1) (19), however, the presence of the third promoter element (UP element; 35,36) in the –40 to –60 position is not obvious (Fig. 1). To test whether a heterologous RNAP (E.coli RNAP) recognises PrecA in vitro and to determine whether the LexA protein regulates promoter utilisation, transcription experiments were performed. The run-off transcripts produced in vitro by RNAP (40 nM) using a linear DNA substrate (2 nM), containing the R.sphaeroides PrecA and the E.coli PlacUV5 control, were analysed by primer extension using the primer denoted in Figure 1 for PrecA. A mRNA species of 117 nt in length was detected for PrecA (Fig. 3). From the position of the primer we could infer that the 117 nt long transcript started at the position mapped in vivo for PrecA (19). Furthermore, a mRNA species of 114 nt in length was detected for the PlacUV5 control (data not shown). It is likely, therefore, that E.coli RNAP recognises the PrecA promoter and initiates transcription, as does R.sphaeroides RNAP in vivo.
Figure 3.
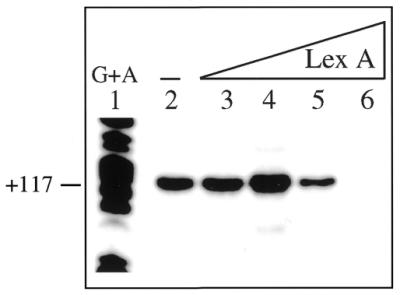
Primer extension assay of PrecA in the absence and presence of LexA. Linear plasmid DNA (2 nM) was incubated with 40 nM RNAP in the absence and presence of increasing amounts of LexA (1.7, 3.5, 7 and 14 nM) and subjected to in vitro transcription followed by primer extension. The length of the cDNA obtained is indicated. –, absence of LexA. The G + A sequencing reaction is presented. A standard error of any value was never >10%.
As revealed in Figure 3, the addition of 1.7 LexA dimers per DNA molecule increases PrecA utilisation, but the presence of 3 LexA dimers reduces promoter utilisation by ∼3-fold. Furthermore, 7 LexA dimers repress transcription initiation by >15-fold. Hence, at low LexA concentrations it controls PrecA positively, whereas at higher LexA concentrations promoter utilisation by RNAP is repressed.
Concentrations of LexA protein equal to or higher than those required to repress the PrecA promoter did not affect utilisation of the unrelated E.coli PlacUV5 control promoter (data not shown). From this result we can rule out that a contaminant RNase or any other non-specific effect could be responsible for the lack of RNA synthesis at higher LexA concentrations.
Rhodobacter sphaeroides LexA and RNAP interact with PrecA
The affinity of the LexA protein for the 210 bp α-32P-labelled EcoRI–HindIII PrecA DNA fragments (2 nM) was determined by EMSA following complex formation as a function of LexA concentration. PrecA has two SOS boxes spaced 13 bp apart and two half copies (Fig. 1). No binding was detected at LexA concentrations <0.3 nM, whereas at higher protein concentrations the amount of LexA–DNA complex formed is greatly increased until a plateau was observed (Fig. 4A). The exponential increase in complex formation suggests that LexA dimers bind to their cognate sequence in a cooperative manner. Three protein–DNA complexes (L1–L3) are observed (Fig. 4A). The apparent equilibrium constant (Kapp) of LexA–DNA complex formation is ∼3.5 nM at pH 7 and 30°C. Since the apparent binding stoichiometry to saturate the DNA substrate is ∼7 PrecA per LexA dimers, it is likely that the purified LexA protein is active. Furthermore, the stoichiometry of LexA observed by EMSA is in clear agreement with the amount of LexA required for PrecA repression.
Figure 4.

LexA protein does not displace RNAP from PrecA DNA. The 210 bp α-32P-labelled EcoRI–HindIII PrecA DNA fragment (2 nM) was incubated with increasing concentrations of LexA (1.7–28 nM) (A) or RNAP (2.5–40 nM) (B) in buffer C for 10 min at 30°C. Complexes were analysed by ndPAGE with a suboptimal amount of LexA (1.7 nM) and then with varying amounts of RNAP (1.2–5 nM) (C) or with a suboptimal amount of RNAP (1.2 nM) and then with varying amounts of LexA (1.7–7 nM) (D) in buffer C for 10 min at 30°C. The free DNA (FD), LexA complexes (L1, L2 and L3), RNAP complexes (R1 and R2) and RNAP–LexA complexes (RL1 and RL2) are indicated. –, absence of the denoted protein.
Few diffuse complexes are observed at low amounts of RNAP (10 nM). In the presence of 20 nM RNAP one major protein–PrecA DNA complex is formed (R1 complex), whereas in the presence of 40 nM a discrete complex (R2) is the major observed form (Fig. 4B). In quantitative terms, the Kapp of the RNAP–PrecA DNA complexes is ∼25 nM at pH 7 and 30°C.
The amount of heterologous RNAP required to form an RNAP–DNA complex was reduced at least 15-fold when LexA (1.7 nM) was preincubated with PrecA DNA (Fig. 4C, lanes 2–4). Enhanced binding of LexA was also observed when it was added to preformed RNAP–PrecA DNA complex (Fig. 4D, lanes 2–4). It is likely, therefore, that LexA and RNAP are present in the RL1 and RL2 ternary complexes, and this suggest that steric hindrance is not the mechanism of LexA repression (see below).
Since LexA helps loading of heterologous RNAP to the PrecA substrate DNA, we consider the above mentioned hypothesis of transcription activation by an uncharacterised LexA-dependent effector (indirect effector) unlikely.
RNAP and LexA protein form a ternary complex on PrecA
To localise the sequences recognised by both proteins and to address whether both proteins form a ternary complex with PrecA, the protein–DNA complexes were analysed by DNase I footprinting. As shown in Figure 5, RNAP (20–50 nM) protected discrete regions on the 228 bp PrecA DNA (2 nM) from nuclease attack. In the presence of 20 nM RNAP, sites protected from DNase I attack were not observed (Fig. 5B, lane 2). At higher RNAP concentrations (30–50 nM) protection from nuclease attack matches the location of the RNAP consensus sequences (–10 and –35; see Fig. 1) and it extended from position –48 to +23 (Fig. 5A, lanes 2 and 4, and B, lane 7). Hypersensitive sites spanning positions +75 to +94 were not observed at low RNAP (20 nM), but rather were detected at high concentrations (40–50 nM) (Fig. 5A, lanes 3 and 4, and B, lanes 2 and 7). In the presence of ATP, CTP and GTP (NTPs) or the absence of NTPs, hypersensitive sites spanning positions +75 to +94 were observed (Fig. 5A, lanes 2 and 4, and B, lanes 7 and 8). Only in the presence of 100 µM ATP, CTP and GTP, however, does RNAP protect broader regions from nuclease attack (positions –60 to +25) (Fig. 5B, lanes 7 and 8). It is likely, therefore, that under the experimental conditions used some RNAP molecules might leave the PrecA region.
Figure 5.

Interaction of RNAP and LexA with PrecA DNA. (A) The 228 bp α-32P-labelled EcoRI–PstI PrecA DNA (3 nM) fragment was incubated with RNAP (30–50 nM) or LexA (3.5–28 nM) in buffer C for 10 min at 30°C. (B) The 228 bp α-32P-labelled EcoRI–PstI PrecA DNA (2 nM) fragment was incubated with RNAP (20 and 40 nM) and then with varying amounts of LexA (7–28 nM) in the presence or absence of 100 µm NTPs (ATP, CTP and GTP) in buffer C for 10 min at 30°C. +, presence of the denoted factor; –, absence of the denoted factor. The location of the promoter, SOS boxes and the hypersensitive sites are indicated at the side of the gel. The coordinates, relative to the +1 (transcription initiation) site, are indicated.
The Kapp of the LexA–PrecA DNA complexes is ∼12 nM as determined by DNase I footprinting, slightly lower than that determined by EMSA. In the presence of low LexA (3.5–7 nM) concentrations, sites protected from DNase I attack were not observed, but a phosphodiester bond strongly hypersensitive to DNase I at position –10 was detected (Fig. 5A, lanes 5 and 6). At higher concentrations (14–28 nM), LexA protected discrete regions in the PrecA DNA from nuclease attack. The protected sites match the location of the SOS boxes (positions –47 to –20 and –18 to –11, SOS 1, and +1 to +9 and +11 to +19, SOS 2) and upstream sequences which coincide with the half-SOS box (positions +34 to +37, +40 to + 47, +56 to +59 and +76 to 79; see Fig. 1) were observed (Fig. 5A, lanes 7 and 8). A phosphodiester bond hypersensitive to DNase I at position –10 and protected bonds at positions –19, –1 and +10 were also observed. The hypersensitive site (position –10) and protected bonds at the same relative positions within SOS 1 (position –19) and SOS 2 (position +10) and at positions –1 and +20 are separated by 9 ± 1 nt, i.e. ∼1 helical turn (assuming 10.5 bp per turn) in double-stranded (ds)DNA. These periodic anomalies in the DNase I cleavage pattern suggest that the protein interacts with one face of the DNA helix.
Mutual binding of RNAP and LexA to PrecA, which matches both protein-binding sites, and the formation of ternary complexes which are cooperatively stimulated by the presence of both proteins were observed at low protein concentrations (Fig. 5B, lanes 3 and 9). In the presence of low RNAP (20 nM) and high LexA (14 nM), however, protection from nuclease attack at position +40 to +47 and hypersensitive sites at position +48 to +66 were also observed (Fig. 5B, lanes 4 and 6). The same general conclusions are reached when 100 µM ATP, CTP and GTP were added. The protection from nuclease attack at positions +40 to +47 and hypersensitive sites at positions +48 to +66 in the presence of both LexA and RNAP could correlate with the presence of imperfect SOS-like boxes or an SOS box having an unusual distance of the repeat spacer [GAAC(N)16GAAC(N)11GAAC] at positions +56 to +94 (see Fig. 1). The same features were detected in the presence of saturating amounts of RNAP (40 nM), but here the hypersensitive sites within positions +48 to +66 are more evident. It is likely, therefore, that (i) suboptimal LexA concentrations, in the absence of NTPs, enhance binding of a heterologous RNAP to PrecA DNA; (ii) the LexA repressor does not prevent RNAP from binding to PrecA, hence ruling out repression by steric hindrance; (iii) in the presence of 100 µM ATP, CTP and GTP, saturating amounts of RNAP present an extended footprint; (iv) the repression mechanism used by LexA is at a post-RNAP binding step. It is likely that LexA might bind to the promoter region together with RNAP in a way that hinders formation of either the open complex or promoter clearance by RNAP (37,38).
LexA protein enhances RNAP open complex formation
To determine how LexA is able to activate/repress transcription, its interaction with RNAP–PrecA DNA was investigated by monitoring KMnO4 modification of stable initiation complexes formed in the presence and absence of LexA. KMnO4 preferentially modifies single-stranded thymines (T) and to a lesser degree cytosines (C), and this will allow the characterisation of the strand-separated DNA in open RNAP transcription complexes.
The 228 bp α-32P-labelled EcoRI–PstI PrecA DNA fragment (2 nM) was incubated with LexA, RNAP or both proteins in the presence or absence of 100 µM ATP, CTP and GTP. The complexes were treated with KMnO4 and subjected to piperidine cleavage and the reaction mixture visualised by dPAGE. In the presence or absence of ATP, CTP and GTP, RNAP (40 nM) binds to and melts the dsDNA helix over a region which includes two T residues and a potential C in the –10 region, hence a nucleated complex and an incomplete open complex are suggested (Fig. 6, lanes 7 and 9). In the presence of 80 nM RNAP (∼3-fold Kapp) an incomplete open complex, defined as a melting of the dsDNA helix over the –10 region, was observed in the absence of NTPs (Fig. 6, lane 11), whereas RNAP-promoted melting of the dsDNA helix at T residues at positions +2, +3, +5 and beyond +5 was observed only in the presence of 100 µM ATP, CTP and GTP (Fig. 6, lane 13). The strand separation observed beyond +5 (+7 position) in the presence of 100 µM ATP, CTP and GTP may be due to slight contamination of the nucleotides with UTP or deamination of CTP and read-through at position +6 (first U in the mRNA). The activation of a second transcription start site, however, cannot be ruled out (see Fig. 3, lane 4). In the presence of 40 nM RNAP, nevertheless, the formation of a ternary RNA-producing complex was not observed (Fig. 6, lanes 7 and 9).
Figure 6.

LexA protein enhances RNAP open complex formation. The 228 bp α-32P-labelled EcoRI–PstI PrecA DNA fragment (2 nM) was incubated with suboptimal amounts of RNAP (5 nM) or saturating amounts of RNAP (40 and 80 nM) and with a saturating concentration of LexA (14 nM) in the presence or absence of 100 µM NTPs (ATP, CTP and GTP). The reaction was then treated with KMnO4 and piperidine and analysed by dPAGE.
The LexA protein (14 nM) increases RNAP-induced melting of the dsDNA helix over a region which includes three T residues in the +1 region at positions +2, +3 and +5 (Fig. 6, lanes 3 and 4). RNAP (5 nM) binding and promotion of strand separation at or near the start site is markedly increased (>16-fold) by LexA protein (14 nM) when compared to similar conditions in the presence of 80 nM RNAP and the absence of LexA (Fig. 6, lanes 4 and 11). The addition of 100 µM ATP, CTP and GTP does not seem to further increase open complex formation beyond the start site at a low ratio of RNAP over LexA (0.3 RNAP/LexA) (Fig. 6, lanes 5 and 6). It is likely that under this condition RNAP cannot leave the +1 to +5 region. It is also likely that under this condition formation of a ternary RNA-producing complex does not take place.
In the presence of increasing amounts of RNAP (40 nM) over LexA (14 nM) the LexA enhances RNAP-promoted melting of the dsDNA helix at T residues at positions +2, +3 and +5, with a subsequent reduction in RNAP-induced melting at the –10 region even in the absence of NTPs when compared to the absence of LexA (Fig. 6, lanes 7 and 8). In the presence of both LexA and 100 µM ATP, CTP and GTP strand separation is near and beyond the start site (positions +2, +3 and +5 and +13 and +14) (Fig. 6, lanes 7–10). Read-through at position +6 was observed, but not at position +16 nor beyond (Fig. 6, lanes 10 and 14). The same general conclusions, strand separation promoted by RNAP at or near the start site in the presence of LexA, were reached when KMnO4 footprinting was performed with the non-template strand and in the presence or absence of heparin (150 µg ml–1). We have shown that formation of open complexes after the initial binding step is enhanced in the presence of LexA. At a high LexA over RNAP ratio (2.8 LexA/RNAP) RNAP cannot leave the +1 to +5 region, whereas at a low LexA/RNAP ratio (0.17) it leaves the +1 to +5 region. It is likely, therefore, that in the presence of an excess of LexA RNAP transcription is repressed by trapping RNAP at PrecA, whereas in the presence of low LexA it activates RNAP transcription of PrecA.
DISCUSSION
In vivo experiments carried out with the PrecA promoter in wild-type and lexA(Def) strains suggested a dual role of LexA protein in the control of R.sphaeroides SOS gene expression. Upon SOS induction PrecA utilisation increases 3-fold in wild-type cells when compared to the constitutive level (RNAP full promoter occupancy) of lexA(Def) cells. This result was confirmed by an analysis of promoter mutants presenting a reduction in transcription efficiency (data not shown).
We have shown in this study that R.sphaeroides LexA is a bona fide transcriptional repressor. Unlike the E.coli and B.subtilis LexA repressors, which block initial binding of the RNAP (5–8), we have documented that R.sphaeroides LexA can bind at the same time as RNAP and that the contacts with DNA in such a ternary complex are different from those made by either protein alone. The mutual binding of both proteins to PrecA and formation of the ternary complex is cooperatively stimulated by the presence of LexA and RNAP. The inhibitory effect of LexA on PrecA utilisation correlates with the amount of LexA–DNA complex formed, indicating the specificity of the repression system even in the presence of heterologous RNAP.
The contacts with DNA in such a ternary complex are slightly different from those made by either protein alone. The mechanism of LexA-dependent repression at PrecA can be explained as a trap of RNAP beyond template position +1 in the initial transcription complex, whereas in the presence of a low LexA/RNAP ratio LexA enhances promoter clearance. In both cases LexA increases the initial binding of RNAP to the promoter region and isomerisation from a closed to an open complex. In the absence of R.sphaeroides RNAP to test whether any of these hypotheses are correct, it is herein proposed that either LexA interacts with RNAP using a domain conserved between both E.coli and R.sphaeroides RNAP or LexA modifies the DNA topology, with subsequent generation of a discrete DNA structure which enhances or inhibits PrecA utilisation.
Few repressors have been shown to repress transcription by inhibiting promoter clearance: φ29-P4 (39,40), HNS at the rrnB P1 promoter (41), FIS at the gyrB promoter (42) and P22-Arc under certain conditions (43). Furthermore, sometimes the φ29-P4 and P22-Arc transcription factors, among others, show dual regulation of open complex formation and promoter clearance (38,43).
Replication forks are often inactivated under normal growth conditions. The accumulation of ssDNA regions resulting from stalled replication seems to be the induction signal for the SOS system (2). The pathways for fork reactivation involve the homologous recombination system (44). After the DNA lesions are healed, the induction signal disappears and the LexA pool accumulates in a passive manner (1,11). The biological significance of the dual function of R.sphaeroides LexA must be understood as an efficient way to adjust the range of SOS response to cell requirements. Rhodobacter sphaeroides is habitually present in the soil. Soil represents an ecological niche in which temperature fluctuations, osmotic stress, solar light and biological secondary metabolites excreted by different microbiota components constantly give rise to DNA-damaging stress situations. Rhodobacter sphaeroides has developed a finely tuned SOS response which is accomplished by having promoters of different strengths (19,22), by different LexA-binding affinities and by modulating the SOS response at the transcriptional level. Dual LexA regulation might have a buffering effect. On the one hand, ‘activated’ RecA protein accumulates at a stalled replication fork, RecA-dependent cleavage of LexA occurs and LexA repression of RNAP is relieved, allowing the system to respond rapidly to the release of repression. However, in the sub-induced SOS state (minor damage leading to low levels of ‘activated’ RecA protein) LexA would activate expression of SOS genes and quickly reach a repressive state, rather than producing the sub-induced SOS state as described for E.coli (45). On the other hand, when the damage signal accumulates, SOS cleavage of LexA occurs rapidly, leading to derepression of the SOS regulon. A low level of LexA might activate synthesis of the SOS genes. Concomitant with a reduction in the damage signal, LexA increases the speed of recovery to the repressed state. This DNA damage feedback response strategy is also utilised by mammalian cells (46). Indeed, CRT1 protein, which is the autoregulated key transducer of the transcriptional response to replicational stress and is induced by DNA-damaging agents, acts as a repressor of the transcription of repair genes and as a weak inducer (46–48).
Rhodobacter sphaeroides LexA binds to SOS boxes and represses expression of the SOS genes. LexA bound to its target sequences increases the initial binding of unbound RNAP to PrecA and, therefore, traps the RNAP and keeps it at PrecA, holding it ready for transcription. We assume that R.sphaeroides LexA inhibition of expression through trapping a functional transcription initiation complex at the promoter is of physiological advantage because the arrested RNAP can be relieved without decomposition of the initiation complex. Furthermore, the R.sphaeroides LexA feedback loop may bolster SOS induction in the presence of low levels of damage, and when the damage signal disappears few copies of LexA might permit rapid transcriptional repression. It is unlikely, therefore, that at partial occupancy (LexA bound to only one SOS box) LexA is acting as an activator and under full occupancy (LexA bound to both SOS boxes) as a repressor.
Acknowledgments
ACKNOWLEDGEMENTS
We thank S. Ayora, P. Mesa and F. Rojo for valuable comments, M. López and J. Ruiz for technical assistance and M. Llagostera for her interest in this project. This work was supported by grants BMC2001-2065 and 2001SGR-206 from the DGES and DURSI to J.B. and BMC2000-0548 from the DGES to J.C.A.
REFERENCES
- 1.Little J.W. and Mount,D.W. (1982) The SOS regulatory system of Escherichia coli. Cell, 29, 11–22. [DOI] [PubMed] [Google Scholar]
- 2.Friedberg E.C., Walker,G.C. and Siede,W. (1995) SOS responses and DNA damage tolerance in prokaryotes. In DNA Repair and Mutagenesis. American Society for Microbiology, Washington, DC, pp. 407–464.
- 3.Wertman K.F. and Mount,D.W. (1985) Nucleotide sequence binding specificity of the LexA repressor of Escherichia coli K-12. J. Bacteriol., 163, 376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Winterling K.W., Chafin,D., Hayes,J.J., Sun,J., Levine,A.S., Yasbin,R.E. and Woodgate,R. (1998) The Bacillus subtilis DinR binding site: redefinition of the consensus sequence. J. Bacteriol., 180, 2201–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hamoen L.W., Haijema,B., Bijlma,J.J., Venema,G. and Lovett,C.M. (2001) The Bacillus subtilis competence transcription factor, comK, overrides LexA-imposed transcriptional inhibition without physically displacing LexA. J. Biol. Chem., 276, 42901–42907. [DOI] [PubMed] [Google Scholar]
- 6.Brent R. and Ptashne,M. (1981) Mechanism of action of the lexA gene product. Proc. Natl Acad. Sci. USA, 78, 4204–4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Little J.W., Mount,D.W. and Yanisch-Perron,C.R. (1981) Purified LexA protein is a repressor of the recA and lexA genes. Proc. Natl Acad. Sci. USA, 78, 4199–4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bertrand-Burggraf E., Hurstel,S., Daune,M. and Schanarr,M. (1987) Promoter properties and negative regulation of the uvrA gene by the LexA repressor and its amino-terminal binding domain. J. Mol. Biol., 193, 293–302. [DOI] [PubMed] [Google Scholar]
- 9.Sassanfar M. and Roberts,J.W. (1990) Nature of the SOS-inducing signal in Escherichia coli. The involvement of DNA replication. J. Mol. Biol., 212, 79–96. [DOI] [PubMed] [Google Scholar]
- 10.Anderson D.G. and Kowalczykowski,S.C. (1998) Reconstitution of an SOS response pathway: derepression of transcription in response to DNA breaks. Cell, 95, 975–979. [DOI] [PubMed] [Google Scholar]
- 11.Little J.W. (1991) Mechanism of specific LexA cleavage: autodigestion and the role of RecA coprotease. Biochimie, 73, 411–422. [DOI] [PubMed] [Google Scholar]
- 12.Luo Y., Pfuettzner,R.A., Mosimann,S., Paetzel,M., Frey,E.A., Cherney,M., Kim,B., Little,J.W. and Strynadka,N.C.J. (2001) Crystal structure of LexA: a conformational switch for regulation of self-cleavage. Cell, 106, 585–594. [DOI] [PubMed] [Google Scholar]
- 13.Courcelle J., Khodursky,A., Peter,B., Brown,P.O. and Hanawalt,P.C. (2001) Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics, 158, 41–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huisman O. and d’Ari,R. (1981) An inducible DNA replication-cell division coupling mechanism in E.coli. Nature, 290, 797–799. [DOI] [PubMed] [Google Scholar]
- 15.Yasuda T., Morimatsu,K., Kato,R., Usukura,J., Takahashi,M. and Ohmori,H. (2001) Physical interactions between DinI and RecA nucleoprotein filament for the regulation of SOS mutagenesis. EMBO J., 20, 1192–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Voloshin O.N., Ramirez,B.E., Bax,A. and Camerini-Otero,R.D. (2001) A model for the abrogation of the SOS response by an SOS protein: a negatively charged helix in DinI mimics DNA in its interaction with RecA. Genes Dev., 15, 415–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gottesman S., Halpern,E. and Trisler,P. (1981) Role of sulA and sulB in filamentation by lon mutant of Escherichia coli K-12. J. Bacteriol., 148, 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tapias A. and Barbe,J. (1998) Mutational analysis of the Rhizobium etli recA operator. J. Bacteriol., 180, 6325–6331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernández de Henestrosa A.R., Rivera,E., Tapias,A. and Barbé,J. (1998) Identification of the Rhodobacter sphaeroides SOS box. Mol. Microbiol., 28, 991–1003. [DOI] [PubMed] [Google Scholar]
- 20.del Rey A., Diestra,J., Fernández de Henestrosa,A.R. and Barbé,J. (1999) Determination of the Paracoccus denitrificans SOS box. Microbiology, 145, 577–584. [DOI] [PubMed] [Google Scholar]
- 21.Tapias A. and Barbé,J. (1999) Regulation of divergent transcription from the uvrA-ssb promoters in Sinorhizobium meliloti. Mol. Gen. Genet., 262, 121–130. [DOI] [PubMed] [Google Scholar]
- 22.Tapias A., Campoy,S. and Barbé,J. (2000) Analysis of the expression of the Rhodobacter sphaeroides lexA gene. Mol. Gen. Genet., 263, 957–965. [DOI] [PubMed] [Google Scholar]
- 23.Haijema B.J., Sinderen,D., Winterling,K., Kooistra,J., Venema,G. and Hamoen,L.W. (1996) Regulated expression of the dinR and recA genes during competence development and SOS induction in Bacillus subtilis. Mol. Microbiol., 22, 75–85. [DOI] [PubMed] [Google Scholar]
- 24.Krueger J.H., Elledge,S.J. and Walker,G.C. (1983) Isolation and characterization of Tn5 insertion mutations in the lexA gene of Escherichia coli. J. Bacteriol., 153, 1368–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sambrook J., Maniatis,T. and Fritsch,E.F. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 26.Studier F.W. (1991) Use of bacteriophage T7 lysozyme to improve an inducible T7 expression system. J. Mol. Biol., 219, 37–44. [DOI] [PubMed] [Google Scholar]
- 27.de Lorenzo V., Herrero,M., Jakubzik,U. and Timmis,K.N. (1990) Mini-Tn5 transposon derivatives for insertion mutagenesis, promoter probing and chromosomal insertion of cloned DNA in Gram negative eubacteria. J. Bacteriol., 172, 6568–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simon R., Priefer,U. and Pühler,A. (1983) A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram-negative bacteria. Biotechnology, 1, 37–45. [Google Scholar]
- 29.Parales R.E. and Harwood,C.S. (1993) Construction and use of a new broad-host-range lacZ transcriptional fusion vector, pHRP309, for gram negative bacteria. Gene, 133, 23–30. [DOI] [PubMed] [Google Scholar]
- 30.Miller J.H. (1972) Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 31.Gill S.C. and von Hippel,P.H. (1989) Calculation of protein extinction coeficient from amino acid sequence data. Anal. Biochem., 182, 319–326. [DOI] [PubMed] [Google Scholar]
- 32.Rojo F. and Alonso,J.C. (1994) The beta recombinase from the streptococcal plasmid pSM19035 represses its own transcription by holding the RNA polymerase at the promoter region. Nucleic Acids Res., 20, 1855–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rojo F. and Salas,M. (1995) Transcriptional regulators: protein–DNA complexes and regulatory mechanisms. In Adolph,K.W. (ed.), Methods in Molecular Genetics. Academic Press, New York, NY, Vol. 6B, pp. 421–438.
- 34.Mohama-Borges R., Pacheco,A.B., Sousa,F.J., Foguel,D., Almeida,F.D. and Silva,L.J. (2000) LexA repressor forms stable dimers in solution. The role of specific DNA in tightening protein–protein interactions. J. Biol. Chem., 275, 4708–4712. [DOI] [PubMed] [Google Scholar]
- 35.Estream S.T., Ross,W., Gaal,T., Chen,Z.W.S., Niu,W., Ebright,R.H. and Gourse,R.L. (1999) Bacterial promoter architecture: subsite structure of UP elements and interactions with the carboxy-terminal domain of the RNA polymerase α subunit. Genes Dev., 13, 2134–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gourse R.L., Ross,W. and Gaal,T. (2000) Ups and downs in bacterial transcription initiation: role of the alpha subunit of RNA polymerase in promoter recognition. Mol. Microbiol., 37, 687–695. [DOI] [PubMed] [Google Scholar]
- 37.Roy S., Garges,S. and Adhya,S. (1998) Activation and repression of transcription by differential contact: two sides of a coin. J. Biol. Chem., 273, 14059–14062. [DOI] [PubMed] [Google Scholar]
- 38.Rojo F. (1999) Repression of transcription initiation in bacteria. J. Bacteriol., 181, 2987–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Monsalve M., Calles,B., Mencía,M., Salas,M. and Rojo,F. (1997) Transcription activation or repression by phage φ29 protein p4 depends on the strength of the RNA polymerase-promoter interactions. Mol. Cell, 1, 99–107. [DOI] [PubMed] [Google Scholar]
- 40.Mencia M., Monsalve,M., Rojo,F. and Salas,M. (1996). Transcription activation by phage phi29 protein p4 is mediated by interaction with the alpha subunit of Bacillus subtilis RNA polymerase. Proc. Natl Acad. Sci. USA, 93, 6616–6620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schröder O. and Wagner,R. (2000) The bacterial DNA-binding protein H-NS represses ribosomal RNA transcription by trapping RNA polymerase in the initiation complex. J. Mol. Biol., 298, 737–748. [DOI] [PubMed] [Google Scholar]
- 42.Schneider R., Travers,A., Kutateladze,T. and Muskhelishvili,G. (1999) A DNA architectural protein couples cellular physiology and DNA topology in Escherichia coli. Mol. Microbiol., 34, 953–964. [DOI] [PubMed] [Google Scholar]
- 43.Smith T.L. and Sauer,R.T. (1996) Dual regulation of open-complex formation and promoter clearance by Arc explains a novel repressor to activator switch. Proc. Natl Acad. Sci. USA, 93, 8868–8872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cox M.M., Goodman,M.F., Kreuzer,K.N., Sherratt,D.J., Sandler,S.J. and Marians,K.J. (2000) The importance of repairing stalled replication forks. Nature, 404, 37–41. [DOI] [PubMed] [Google Scholar]
- 45.Little J.W. (1983) The SOS regulatory system: control of its state by the level of RecA protease. J. Mol. Biol., 167, 791–808. [DOI] [PubMed] [Google Scholar]
- 46.Huang M., Zhou,Z. and Elledge,S.J. (1998) The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell, 94, 595–605. [DOI] [PubMed] [Google Scholar]
- 47.Li B. and Reese,J.C. (2000) Derepression of DNA damage-regulated genes requires yeast TAF(II)s. EMBO J., 19, 4091–4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou B.B. and Elledge,S.J. (2000) The DNA damage response: putting checkpoints in perspective. Nature, 408, 433–439. [DOI] [PubMed] [Google Scholar]