

Abstract
Enzyme-like catalysts by design have been a long sought-after goal of chemists but difficult to realize due to challenges in the construction of multifunctionalized active sites with accurately positioned catalytic groups for complex substrates. Hydrolysis of cellulose is a key step in biomass utilization and requires multiple enzymes to work in concert to overcome the difficulty associated with hydrolyzing the recalcitrant substrate. We here report methods to construct synthetic versions of these enzymes, through covalent molecular imprinting and strategic postmodification of the imprinted sites. The synthetic catalysts cleave a cellulose chain endolytically at multiple positions or exolytically from the nonreducing end by one or three glucose units at a time, all using the dicarboxylic acid motif found in natural cellulases. By mimicking endocellulase, exocellulase, and β-glucosidase, respectively, the synthetic catalysts hydrolyze cellulose in a synergistic manner, with an activity at 90 °C in pH 6.5 buffer more than doubled that of Aspergillus niger cellulase at pH 5 and 37 °C and 44% of that of a commercial cellulase blend (Novozyme). As robust cross-linked polymeric nanoparticles, the synthetic catalysts showed little change in activity after preheated at 90 °C for 3 days and could be reused, maintaining 76% of activity after 10 reaction cycles.
Graphical Abstract

INTRODUCTION
Fossil fuels continue to be the dominant source of energy and chemical feedstock to humanity but concerns for their sustainability and negative environmental/ecological impact have prompted many to look for alternatives. Lignocellulosic biomass is an abundant, renewable, and carbon-neutral raw material produced at a scale of 170–200 billion tons per year, with 35–50% being cellulose. These materials contain an enormous amount of energy and can be converted into many useful chemicals.1–6
Whether for chemical or fuel production, depolymerization of cellulose into soluble monomeric and oligomeric sugars is a prerequisite, usually through catalytic hydrolysis of the glycosidic linkages between the glucose repeat units. The glycosidic bonds in cellulose are stable under neutral and basic conditions, with a half-life of 4.5 million years at 25 °C over pH 7–14.7 Although acids accelerate the hydrolysis readily, sugar products formed from the initial hydrolysis undergo decomposition under strongly acidic conditions4,8 and some of side products (e.g., 5-hydroxymethylfurfural) inhibit microbial fermentations in downstream processing.9
Nature uses glycosidases to hydrolyze glycosidic bonds under mildly acidic conditions. The biocatalysts utilize a remarkably simple motif for the catalysis, i.e., a pair of carboxylic acids.10,11 One acid, in the protonated form, acts as a general acid to protonate the exocyclic glycosidic oxygen and the other, in the deprotonated form, works as either a general base or a nucleophile to attack the anomeric carbon of the glycosidic bond. Cooperative action of the two acids is key to the catalysis, as removal of one acid from the active site can lower the catalytic efficiency of the enzyme by >100,000 times.12,13
In addition to the stability of glycosidic bonds, another difficult challenge in cellulose hydrolysis is derived from its high crystallinity. To deal with the recalcitrant substrate, natural cellulase often operates through a processive mechanism in which the enzyme detaches a polymer chain from cellulose crystals while performing multiple rounds of hydrolysis before dissociating from the chain.14,15 An extraordinary binding energy (often > 20 kcal/mol)16 is required for the mechanism but meanwhile causes product inhibition, as cello-oligomers are also strongly bound by the tunnel- or cleft-shaped active site. β-Glucosidase can be added to alleviate product inhibition, by hydrolyzing the oligomers into weakly bound glucose. Hydrolytic efficiency can be further improved by employing both endocellulase and exocellulase in an enzyme blend, with the former targeting the amorphous region of cellulose fibers and the latter cleaving oligomers from the end of a cellulose chain.15
Although the above blend is capable of hydrolyzing cellulose, enzymes are fragile biomolecules generally difficult to recycle. Most enzymes have narrow operating windows with regard to reaction temperature and choice of solvents. Since cellulases represent a considerable cost in cellulose-based bioethanol production,17 great efforts have been made to enhance their stability and performance under challenging conditions.18,19
Chemists have long been interested in building synthetic catalysts with enzyme-like properties,20–22 including catalysts to hydrolyze glycosidic bonds.23–27 However, of the few reported artificial glycosidases, most only work for activated aryl glycosides. If synthetic enzymes with biological selectivity and efficiency but greater stability can be produced for glycan hydrolysis, multiple areas of research, development, and industries can benefit because of the prevalence and importance of carbohydrates.28
In this work, we report rational designs of synthetic catalysts that mimic endo- and exocellulase, with a biomimetic dicarboxylic motif in the active sites. By cleaving cellulose polymers both at internal sites and from chain ends, the synthetic catalysts hydrolyzed cellulose synergistically as natural or commercial cellulase cocktails. As robust cross-linked polymeric nanoparticles, they tolerated high temperatures including boiling water, with an activity at 90 °C in pH 6.5 buffer more than doubled that of cellulase from Aspergillus niger and 44% of that of a commercial cellulase blend at pH 5 and 37 °C. Unlike the easily denatured natural enzymes, the synthetic catalysts could be reused many times, maintaining 76% of activity after 10 cycles at 60 °C.
RESULTS AND DISCUSSION
Design and Synthesis of Synthetic Endocellulase.
Natural cellulases generally consist of one or more carbohydrate-binding modules (CBMs) linked to a catalytic domain (CD).15 CBM uses a number of polar and nonpolar amino acids in an extended cleft or groove to bind multiple sugar resides on a polysaccharide chain. In addition to bringing the CD to the target substrate, some CBMs can disrupt the crystalline cellulose chain to make the chain more accessible to the CD.29 Likewise, to construct a synthetic endocellulase, one has to first create binding sites for a segment of a cellulase chain—a cello-oligomer essentially—and then install acidic groups near the appropriate glycosidic bond(s) for hydrolytic cleavage. Although chemists have a long history of binding sugar derivatives using organic boronic acids,30,31 only the 4,6-diol of the glucose on the nonreducing end and the 1,2-diol on the reducing end can interact with boronic acids. Since the 2,3-trans-diol of an internal glucose does not form boronate bonds, a different strategy for binding is necessary.
Scheme 1 shows our method to create an endocellulase mimic, via covalent molecular imprinting and postmodification of the imprinted sites. Molecular imprinting is a powerful technique to create tailor-made binding sites in a cross-linked polymer network, using template molecules (T) as space holders and polymerizable functional monomers (FMs) to bind the templates.32–34 Molecularly imprinted materials have found wide applications35–37 including in catalysis.38–46 In our case, the imprinting was performed in cross-linked surfactant micelles,47 to afford polymeric nanoparticles (NPs) similar to proteins in size and water-solubility.
Scheme 1.

Preparation of endocellulase-mimicking NP1-CO2H (a) and NP1-CH2CH2CO2H (b). The surface ligands are omitted for clarity.
To bind an internal segment of a cellulose chain, we designed T–FM complex 1, prepared in one step from (blue-colored) 4-vinylbenzaldehyde (4-VB) as the FM and (red-colored) oligochitosan 2 as the template. The commercially available chitosan oligomer has 3–7 monomer units and closely resembles a portion of a cellulose chain (red colored in Scheme 1a, m = 2–6). The amino groups, meanwhile, serve as functional group handles for the desired manipulation.
The amphiphilic T–FM complex 1 is readily incorporated into the mixed micelle of 3a and 3b, which is cross-linked on the surface by the highly efficient click reaction, facilitated additionally by the proximity of the reactive groups on the surface of the micelle (Scheme 1a, step a). Surface decoration by monoazide 4 (step b) and free radical polymerization in the micellar core (step c) yield NP1. The core-cross-linking occurs among the methacrylates of the surfactants, micelle-solubilized 1 and divinylbenzene (DVB), and water-soluble N,N′-methylenebis(acrylamide) (MBAm), with 2,2-dimethoxy-2-phenylacetophenone (DMPA) as the photoinitiator. Hydrolysis in 6 M HCl at 95 °C removes the template by cleaving the imine bonds48 to afford NP1-CHO, with aldehyde groups in the imprinted sites (step d).
MBAm can be viewed as a hydrogen-bonding FM, useful for binding the glycan template. During the core-cross-linking (step c), the DMPA photoinitiator being hydrophobic prefers to reside in the core of the surface-cross-linked micelle and is covalently bound to the micelle once the polymerization starts. Water-soluble MBAm thus can only polymerize after they diffuse to the micelle, near the surfactant/water interface. These MBAm molecules are expected to interact with the glycan template, also located at the interface due to its hydrophilicity. Free radical polymerization/cross-linking then creates molecularly imprinted hydrogen-bonding sites for the oligochitosan template. Although hydrogen bonds are ineffective in water for molecular recognition due to solvent competition, they are stabilized inside a micelle49 and at the surfactant interface.50 A previous study shows that these MBAm-derived hydrogen bonds enhance the binding of 4-nitrophenyl-α-D-glucopyranoside by 180-fold.51
Our initial idea was to oxidize the (blue-colored) aldehydes into carboxylic acids (Scheme 1a, step e); in this way, 4VB not only serves as the functional monomer to recruit to the micelle the otherwise water-soluble oligochitosan template, but also represents a latent catalytic group to be activated by post-imprinting oxidation. Because the imine bonds of the template are formed with the amino groups of oligochitosan at the C2 positions, the carboxylic acids in NP1-CO2H should be close to the C2 hydroxyl of a bound cellulose chain. Ideally, one wants to position the acidic groups right next to the glycosidic linkages in a cellulose chain. However, doing so is challenging from a synthetic point of view and direct oxidation of the polymerized 4-VB is much more achievable. We reasoned that, as long as a bound cellulose chain could slide/wobble in the active site, some of the acidic groups of NP1-CO2H should be able to get close enough to a glycosidic oxygen(s) to catalyze the hydrolysis. Since the substrate is bound noncovalently (mainly by hydrogen bonds), it is expected to have considerable mobility.
To test the above hypothesis, we employed a well-established spectrophotometric assay to measure cellulose hydrolysis,19 with a commercial cellulase from Aspergillus niger for comparison. The commercial endocellulase is active toward both cellulose and cello-oligomers.52
The experiments confirmed our hypothesis. Whereas NP1-CHO and nonimprinted NP (NINP) was inactive in cellulose hydrolysis (Table 1, entries 2 and 3), NP1-CO2H at 90 °C displayed an activity within a factor of 5 to the natural enzyme (entry 4). LCMS indicated glucose was the dominant product formed in the hydrolysis (Figure S39).
Table 1.
Hydrolysis of cellulose by cellulases and NP-based artificial endocellulases.a
| entry | catalysts | pH | [catalyst] (mg/mL) | temp (°C) | [reducing sugar] (mg/mL) | enzyme activity (μmol mg−1 h−1) | relative activity (NP/enzyme) |
|---|---|---|---|---|---|---|---|
| 1 | endocellulase | 5.0 | 1 | 37 | 0.38 ± 0.03 | 0.177 ± 0.031 | 1 |
| 2 | NP1-CHO | 5.0 | 5 | 90 | 0 | 0 | 0 |
| 3 | NINP | 5.0 | 5 | 90 | 0 | 0 | 0 |
| 4 | NP1-CO2H | 5.0 | 5 | 90 | 0.41 ± 0.03 | 0.038 ± 0.004 | 0.21 |
| 5 | NP1-CH=CHCO2H | 5.0 | 5 | 90 | 0.70 ± 0.04 | 0.065 ± 0.002 | 0.37 |
| 6 | NP1-CH2CH2CO2H | 5.0 | 5 | 90 | 0.95 ± 0.03 | 0.088 ± 0.002 | 0.49 |
| 7 | NP1-CH2CH2CO2H | 5.0 | 4 | 90 | 0.81 ± 0.04 | 0.094 ± 0.002 | 0.53 |
| 8 | NP1-CH2CH2CO2H | 5.0 | 3 | 90 | 0.80 ± 0.02 | 0.125 ± 0.003 | 0.70 |
| 9 | NP1-CH2CH2CO2H | 5.0 | 2 | 90 | 0.63 ± 0.01 | 0.147 ± 0.002 | 0.83 |
| 10 | NP1-CH2CH2CO2H | 5.0 | 1 | 90 | 0.24 ± 0.01 | 0.114 ± 0.001 | 0.64 |
| 11 | NP1-CH2CH2CO2H | 5.0 | 2 | 60 | 0.15 ± 0.02 | 0.034 ± 0.001 | 0.19 |
| 12 | NP1-CH2CH2CO2H | 5.0 | 2 | 37 | 0.02 ± 0.01 | 0.004 ± 0.002 | 0.05 |
| 13 | NP6-CO2H | 6.5 | 5 | 90 | 1.38 ± 0.03 | 0.128 ± 0.002 | 0.72 |
| 14 | NP6-CO2H | 6.5 | 4 | 90 | 1.13 ± 0.04 | 0.131 ± 0.003 | 0.74 |
| 15 | NP6-CO2H | 6.5 | 3 | 90 | 1.01 ± 0.02 | 0.157 ± 0.003 | 0.88 |
| 16 | NP6-CO2H | 6.5 | 2 | 90 | 0.78 ± 0.01 | 0.181 ± 0.005 | 1.01 |
| 17 | NP6-CO2H | 6.5 | 1 | 90 | 0.29 ± 0.01 | 0.135 ± 0.002 | 0.76 |
| 18 | NP6-CO2H | 6.5 | 2 | 60 | 0.23 ± 0.01 | 0.053 ± 0.001 | 0.30 |
| 19 | NP6-CO2H | 6.5 | 2 | 37 | 0.04 ± 0.01 | 0.009 ± 0.001 | 0.05 |
Reactions were performed in duplicates with [Sigmacell cellulose] = 5.0 mg/mL in 1.0 mL 10 mM NaOAc buffer for 12 h.
Aldehyde is a versatile functional group. Hypothesizing that flexible tethers to the acidic groups would increase the probability for the acids to find glycosidic bonds, we performed Knoevenagel condensation between NP1-CHO and malonic acid, using piperidine as the catalyst. The Doebner-modified reaction generally occurs with decarboxylation so that NP1-CH=CHCO2H could be produced (Scheme 1b, step a). The extension indeed was helpful, increasing the relative activity of the resulting catalyst to 37% of that of the natural endocellulase (Table 1, entry 5). For the reaction inside the imprinted sites, at least 10 equiv of malonic acids were needed for good results and higher amounts made little difference (Table S1).
Another possible postmodification of the active site is the reduction of the cinnamic double bond of NP1-CH=CHCO2H (Scheme 1b, step b), which could further increase the mobility of the acidic groups. To our delight, treatment with 50 equiv of hydrazine to reduce the C=C bonds further increased the relative activity (Table S1), up to 49% relative activity for NP1-CH2CH2CO2H (Table 1, entry 6).
Catalysts in cellulose hydrolysis can only react with the polymer chains on the surface of the crystals. The measured enzyme activity is thus a function of catalyst loading: too little catalyst cannot cover the surface and too high a loading results in idle catalysts in the solution. In our case, the best result was obtained at 2 mg/mL of NP1-CH2CH2CO2H (Table 1, entries 6–10). At this catalyst loading, the synthetic enzyme at 90 °C achieved 83% of the natural endocellulase’s activity at 37 °C (entry 9).
Natural cellulases generally use a pair of carboxylic acids in the active site for glycosidic cleavage.10 We did not attempt to introduce such a feature through the active-site aldehyde directly, reasoning that two acids would take up too much space in the active sites and interfere with the substrate binding (vide infra). Instead, we designed and synthesized template 6, prepared from oligochitoan 2 and aldehyde 5 in one step (Scheme 2). This template already has an extended carboxylic acid in the structure; the corresponding NP6-CO2H prepared following Scheme 1a thus should have two carboxylic acids per glucose-binding site, one from the oxidation of the benzaldehyde moiety and the other from the template itself. Since the pre-existing acid is already on the template, it is not expected to take up the space imprinted from the glycan but will be imprinted together with the rest of the template structure.
Scheme 2.

Preparation of NP6-CO2H from template 6.
The double acidic NP6-CO2H further improved the relative activity, up to 101% at 90 °C (entry 16). We also performed the Knoevenagel modification on the corresponding NP6-CHO, as well as the reduction of the C=C bonds but the resulting catalysts showed lower activities (data not shown).
Natural enzymes generally lose activity at high temperatures. Our synthetic endocellulases displayed an opposite trend. Both the monoacid catalyst (NP1-CH2CH2CO2H) and the diacid one (NP6-CO2H) exhibited low relative activity at 37 °C (5%, entries 12 and 19), but became much more active at higher temperatures, especially at 90 °C (entries 9 and 16).
Because glycosidase employs a protonated carboxylic acid and a deprotonated carboxylate for catalysis, their optimal pH (pHopt) generally falls between pH 4 to 6. Interestingly, both NP1-CH2CH2CO2H and NP6-CO2H gave a similar profile, with pHopt = 5.5 and 6.5, respectively (Figure 1). Loss of activity in natural glycosidases at low pH is normally caused by the protonation of the general base/nucleophile and loss at high pH by the deprotonation of the general acid.53,54 The similar profiles for NP1-CH2CH2CO2H and NP6-CO2H suggest cooperativity should also be present in our catalysts, with some acidic groups as general acids and others in the deprotonated forms as general bases or nucleophiles.
Figure 1.

Effects of solution pH on the hydrolysis of cellulose by synthetic endocellulases. [Sigmacell cellulose] = 5.0 mg/mL. [catalyst] = 2.0 mg/mL.
Both NP1-CH2CH2CO2H and NP6-CO2H followed Michaelis-Menten kinetics in their catalysis (Figure 2). These endocellulase mimics are designed to hydrolyze cello-oligomers. However, because a hydrolyzed oligomer can undergo additional hydrolysis in a secondary and even tertiary reactions by the same catalysts, we used cellobiose for the Michaelis-Menten studies, even though the substrate is anticipated to bind the catalyst less strongly than a longer cello-oligomer (vide infra). For a longer cell-oligomer, cleavage at any internal sites represents successful hydrolysis whereas for cellobiose only one particular cleavage is counted. For these reactions, the catalytic efficiency estimated from cellobiose (kcat/Km = 141 M−1 min−1 for NP1-CH2CH2CO2H and 255 M−1 min−1for NP6-CO2H) represent the lower limits of our catalysts.
Figure 2.

Michaelis-Menten plot for cellobiose hydrolysis by (a) NP1-CH2CH2CO2H and (b) in NP6-CO2H 10 mM MES buffer (pH 6.5) at 90 °C. [NP] = 10.0 μM.
We also studied the binding of various cello-oligomers by our catalysts using isothermal titration calorimetry (ITC). Figure 3 shows that the binding free energy (−ΔG) for all these NP hosts (including the non-catalytic NP1-CHO) increases with the chain length of the cello-oligomer. The result is reasonable because the nanoparticle hosts have been prepared from chitosan oligomers with 3–7 monomer units and are thus designed to bind a cello-oligomer with similar numbers of repeat units if the entire chain is imprinted successfully. Figure 3 also shows that NP1-CHO binds the cello-oligomer guests more strongly than the extended NP1-CH2CH2CO2H. The results is most likely caused by steric hindrance in the binding by the latter, as the CH2CH2CO2H groups introduced via postmodification would occupy part of the glycan-binding space in the imprinted sites. The effect could be offset partially by any potential (hydrogen-bonding) interactions between these acidic groups and the bound cello-oligomer.
Figure 3.

Binding free energies of NP hosts for cello-oligomers in 10 mM MES buffer at pH 6.5 at 298 K determined by ITC.
Design and Synthesis of Synthetic Exocellulase.
Having created catalysts to cleave cellulose at internal sites, we set our next goal to build a synthetic exocellulase. It needs to bind the polymer at the chain end while having acidic groups near a glycosidic bond to be cleaved. For cellulose binding, we used a boroxole FM (8) to bind the 4,6-diol of the terminal glucose on a cellulase chain through reversible boronate bonds (Scheme 3). The installation of the catalytic group was made possible by template 7. The template molecule has a (red-colored) cellotriose conjugated to a (blue-colored) specially designed aryl hydrazine 9. The (magenta-colored) boroxole FM forms an anionic boronate with the glycan of the template in situ, stabilized by the cationic mixed micelle.55,56 Micellar imprinting affords NP7 with the template still bound in the imprinted site. Removal of the template by solvent washing vacates the imprinted site, which would be partially filled when 10 is added to the solution. Shape-similarity between 10 and hydrazine 9 (i.e., the aglycon of template 7) helps 10 occupy the imprinted space of the latter. Phenyl azides with fluoro groups at the 2,6-positions are well-known photoaffinity labels. Under photolysis, they form nitrenes that undergo C–H insertion readily,57 especially efficiently in a hydrophobic environment.58,59 The micellar core of NP7 has many C–H bonds from polymerized surfactants and DVB. Because the two carboxylic acids from the isophthalic acid moiety of 10 is supposed to occupy the space imprinted from the two meta nitro groups of the template, they should sandwich the exocyclic glycosidic oxygen of the third glucose unit in a bound cellulose and promote its hydrolysis.
Scheme 3.

Preparation of exocellulase-mimicking NP7-CO2H. The surface ligands are omitted for clarity.
Success of molecular imprinting is supported by the strong binding of template 7 by NP7, with a binding constant of Ka = 728 × 103 M−1 or a binding free energy of −ΔG = 8.03 kcal/mol in pH 6.5 buffer at 298 K (Table 2, entry 1). The photoaffinity label 10 was also bound strongly, with a binding constant nearly half of that of the full template (entry 2). Formation of the trisaccharide-binding active site in the photoaffinity-labelled catalyst was confirmed by the peaking of the binding at cellotriose by the final catalyst—NP7-(CO2H)2 (entries 3–8). This trend is very different from that exhibited by the endocellulase-mimicking catalysts in Figure 3.
Table 2.
ITC binding data for sugar guests by NP7-(CO2H)2.a
| entry | host | guest | pH | Ka (×103 M−1) | ΔG (kcal/mol) | N |
|---|---|---|---|---|---|---|
| 1 | NP7 | 7 | 6.5 | 728.2 ± 21.4 | −8.03 | 0.99 ± 0.05 |
| 2 | NP7 | 10 | 6.5 | 344.1 ± 11.2 | −7.55 | 1.04 ± 0.11 |
| 3 | NP7-(CO2H)2 | glucose | 6.5 | 7.34 ± 0.19 | −5.27 | 0.97 ± 0.03 |
| 4 | NP7-(CO2H)2 | cellobiose | 6.5 | 21.1 ± 0.23 | −5.89 | 1.11 ± 0.08 |
| 5 | NP7-(CO2H)2 | cellotriose | 6.5 | 75.2 ± 0.14 | −6.65 | 1.06 ± 0.03 |
| 6 | NP7-(CO2H)2 | cellotetraose | 6.5 | 39.3 ± 0.31 | −6.26 | 0.89 ± 0.04 |
| 7 | NP7-(CO2H)2 | cellopentaose | 6.5 | 11.4 ± 0.22 | −5.53 | 0.99 ± 0.05 |
| 8 | NP7-(CO2H)2 | cellohexaose | 6.5 | 4.22 ± 0.18 | −4.94 | 1.21 ± 0.08 |
The cross-linkable surfactants were a 3:2 mixture of 3a and 3b. The titrations were performed in 10 mM MES buffer at pH 5 at 298 K. The ITC titration curves are reported in the Figures S28–35, including the binding enthalpy and entropy. N is the average number of binding site per nanoparticle measured by ITC.
It is worth mentioning that bindings of the cello-oligomers by the endocellulase mimics (NP1-CH2CH2CO2H and NP6-CO2H) are noncovalent in nature and those by the exocellulase mimic (NP7-(CO2H)2) a combination of noncovalent and covalent (boronate) bonds. It is impressive that the noncovalent interactions in the endocellulase mimics (Figure 3) afford comparable binding strengths as the combined interactions in the exocellulase mimic, at least for the longer oligomers (Table 2).
Catalytic efficiency of NP7-(CO2H)2 was determined by its Michaelis-Menten plot, using cellotetraose as the substrate (Figure 4). The Michaelis constant (Km) was 373 μM and the catalytic turnover (kcat = 0.037 min−1). The catalytic efficiency (kcat/Km) was 99 M−1 min−1 in pH 6.5 buffer at 60 °C. Consistent with our design, cellotriose was the dominant product in the hydrolysis of both cellulose (Figure S40) and cellotetraose (Figure S41).
Figure 4.

Michaelis-Menten plot for the hydrolysis of cellotetraose by NP7-(CO2H)2 in pH 6.5 buffer at 60 °C. [NP] = 10.0 μM.
Hydrolysis of Cellulose by the Synthetic Enzyme Blend.
We thus have synthetic mimics of both endocellulase and exocellulase. Together with a β-glucosidase mimic, NP11-(CO2H)2, made from template 11 followed by the hydrolysis of the imine bonds and reductive amination to install the catalytic groups (Scheme 4, see Supporting Information for details),60 the synthetic enzyme blend should be able to cleave a cellulose endolytically in the amorphous regions (to increase the number of chain ends), exolytically from the nonreducing end, either three sugar residues or one residue at a time. All the catalysts—i.e., NP6-CO2H, NP7-(CO2H)2, and NP11-(CO2H)2—have the biomimetic dicarboxylic acid motifs for the catalytic hydrolysis.
Scheme 4.
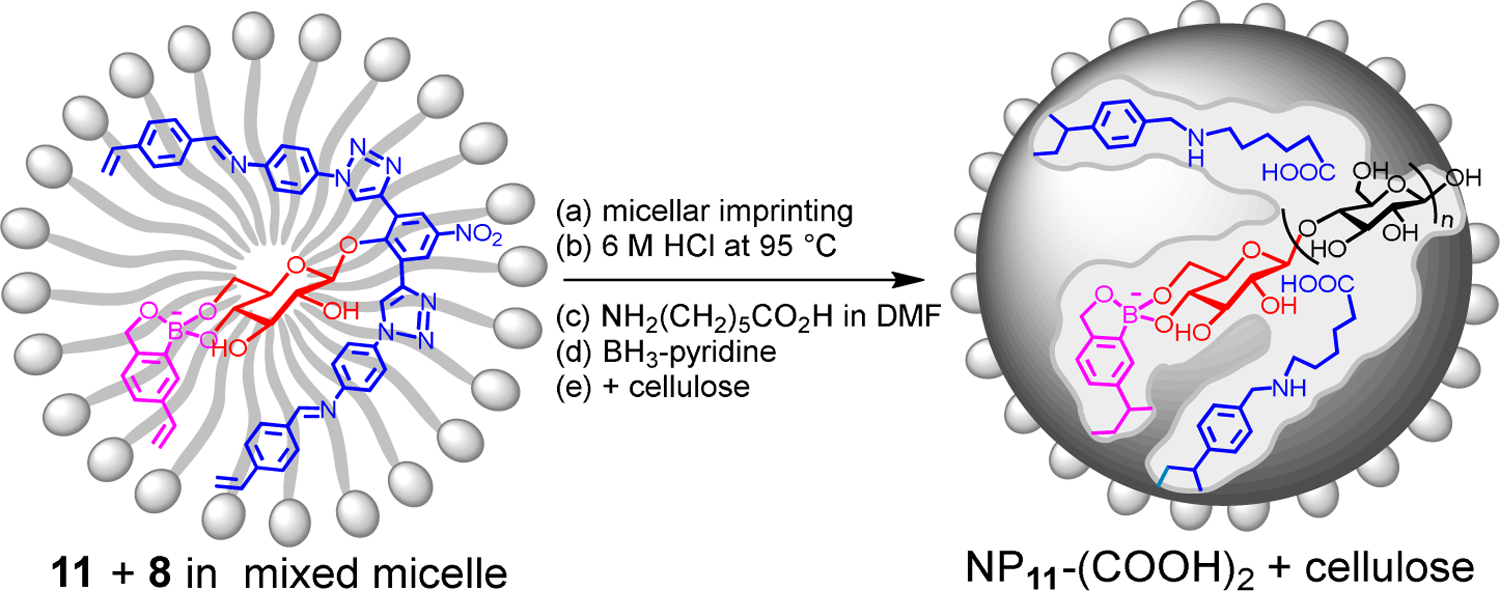
Preparation of β-glucosidase-mimicking NP11-(CO2H)2. The surface ligands are omitted for clarity.
The synthetic blend displayed a gradual increase of activity in cellulose hydrolysis over pH 4 to 6.5 and then a precipitous drop from pH 6.5 to 7 (Figure 5). The pH profile was similar to that of the endocellulase (Figure 1) and exocellulase mimic (Figure S43), which both peaked in performance at pH 6.5. The optimal pH for the β-glucosidase mimic, NP11-(CO2H)2, is 6.0.60
Figure 5.

Effects of solution pH on cellulose hydrolysis by the NP catalyst blend. [Sigmacell cellulose] = 5.0 mg/mL. [Total catalysts] = 2.0 mg/mL with a 1:1:2 ratio of NP6-CO2H, NP7-(CO2H)2, and NP11-(CO2H)2.
To evaluate the performance of the synthetic catalyst blend, we benchmarked it against a commercial cellulase blend and two cellulases from Aspergillus niger and Trichoderma reesei, respectively. The commercial blend was a product from Novozyme Corp. available from Sigma. It contains a range of enzymes including cellulases, β-glucosidase and hemicellulase and is often used for biofuels research. Our cellulose hydrolysis assay shows that the cellulase blend was more efficient than the Trichoderma reesei cellulase and the Aspergillus niger cellulase was the slowest (Table 3, entries 1–3).
Table 3.
Hydrolysis of cellulose by cellulases and NP catalysts.a
| entry | catalyst | [catalyst] (mg/mL) | temp (°C) | pH | [reducing sugar] (mg/mL) | enzyme activity (μmol mg−1 h−1) | relative activity |
|---|---|---|---|---|---|---|---|
| 1 | cellulase blend | 1 | 37 | 5.0 | 2.31 ± 0.03 | 1.075 ± 0.10 | 1 |
| 2 | cellulase from Aspergillus niger | 1 | 37 | 5.0 | 0.38 ± 0.03 | 0.177 ± 0.03 | 0.17 |
| 3 | cellulase from Trichoderma reesei | 1 | 37 | 5.0 | 1.91 ± 0.04 | 0.889 ± 0.01 | 0.83 |
| 4 | 1:0:0 endo/exo/beta | 2 | 90 | 6.5 | 0.78 ± 0.05 | 0.182 ± 0.01 | 0.17 |
| 5 | 0:1:0 endo/exo/beta | 2 | 90 | 6.5 | 0.26 ± 0.02 | 0.061 ± 0.01 | 0.06 |
| 6 | 0:0:1 endo/exo/beta | 2 | 90 | 6.5 | 0.31 ± 0.04 | 0.072 ± 0.01 | 0.07 |
| 7 | 1:3:0 endo/exo/beta | 2 | 90 | 6.5 | 0.95 ± 0.03 | 0.221 ± 0.004 | 0.21 |
| 8 | 1:2:0 endo/exo/beta | 2 | 90 | 6.5 | 1.04 ± 0.04 | 0.242 ± 0.010 | 0.22 |
| 9 | 1:1:0 endo/exo/beta | 2 | 90 | 6.5 | 1.23 ± 0.12 | 0.286 ± 0.009 | 0.27 |
| 10 | 2:1:0 endo/exo/beta | 2 | 90 | 6.5 | 0.98 ± 0.01 | 0.228 ± 0.002 | 0.21 |
| 11 | 3:1:0 endo/exo/beta | 2 | 90 | 6.5 | 0.92 ± 0.02 | 0.214 ± 0.006 | 0.20 |
| 12 | 3:0:1 endo/exo/beta | 2 | 90 | 6.5 | 0.82 ± 0.01 | 0.191 ± 0.002 | 0.18 |
| 13 | 2:0:1 endo/exo/beta | 2 | 90 | 6.5 | 0.96 ± 0.03 | 0.223 ± 0.002 | 0.21 |
| 14 | 1:0:1 endo/exo/beta | 2 | 90 | 6.5 | 1.05 ± 0.12 | 0.244 ± 0.009 | 0.23 |
| 15 | 1:0:2 endo/exo/beta | 2 | 90 | 6.5 | 1.41 ± 0.04 | 0.328 ± 0.010 | 0.30 |
| 16 | 1:0:3 endo/exo/beta | 2 | 90 | 6.5 | 1.12 ± 0.03 | 0.261 ± 0.004 | 0.24 |
| 17 | 0:3:1 endo/exo/beta | 2 | 90 | 6.5 | 0.42 ± 0.02 | 0.098 ± 0.003 | 0.09 |
| 18 | 0:2:1 endo/exo/beta | 2 | 90 | 6.5 | 0.48 ± 0.03 | 0.112 ± 0.004 | 0.10 |
| 19 | 0:1:1 endo/exo/beta | 2 | 90 | 6.5 | 0.59 ± 0.04 | 0.137 ± 0.002 | 0.13 |
| 20 | 0:1:2 endo/exo/beta | 2 | 90 | 6.5 | 0.67 ± 0.05 | 0.156 ± 0.006 | 0.15 |
| 21 | 0:1:3 endo/exo/beta | 2 | 90 | 6.5 | 0.69 ± 0.04 | 0.161 ± 0.005 | 0.15 |
| 22 | 1:1:2 endo/exo/beta | 2 | 90 | 6.5 | 2.02 ± 0.03 | 0.470 ± 0.004 | 0.44 |
| 23 | 1:1:2 endo/exo/betab | 2 | 90 | 6.5 | 1.98 ± 0.02 | 0.461 ± 0.002 | 0.43 |
| 24 | 1:1:2 endo/exo/beta | 1 | 90 | 6.5 | 0.45 ± 0.03 | 0.211 ± 0.003 | 0.20 |
| 25 | 1:1:2 endo/exo/beta | 3 | 90 | 6.5 | 3.13 ± 0.03 | 0.486 ± 0.011 | 0.45 |
| 26 | 1:1:2 endo/exo/beta | 4 | 90 | 6.5 | 3.74 ± 0.03 | 0.433 ± 0.008 | 0.40 |
| 27 | 1:1:2 endo/exo/beta | 2 | 100 | 6.5 | 2.24 ± 0.03 | 0.512 ± 0.001 | 0.48 |
| 28 | 1:1:2 endo/exo/beta | 2 | 60 | 6.5 | 0.42 ± 0.04 | 0.098 ± 0.01 | 0.09 |
| 29 | 1:1:2 endo/exo/beta | 2 | 37 | 6.5 | 0.09 ± 0.01 | 0.021 ± 0.001 | 0.02 |
The reactions were performed in duplicates with [cellulose] = 5.0 mg/mL [cellulase] = 1 mg/mL [NP] = 2 mg/mL in 1 mL of 10 mM NaOAc buffer for 12 h.
The catalyst mixture was preheated for 3 d at 90 °C in 1 mL of 10 mM NaOAc buffer (pH 6.5) before the hydrolysis experiment.
When used alone for cellulose hydrolysis, the endocellulase mimic NP6-CO2H was the most active among the synthetic catalysts, affording 17% of activity at 90 °C in pH 6.5 buffer relative to the commercial cellulase blend at 37 °C and pH 5 (entries 4–6).
Synergism clearly exists among these NP catalysts, as mixing the endo and the exo mimics (entries 7–11), the endo with the β-glucosidase mimic (entries 12–16), or the exo with the β-glucosidase mimic (entries 17–21) all improved the hydrolytic activity under the same overall catalyst loading. Among the three catalysts, the endocellulase mimic is the dominant contributor to the catalysis: not only was it the most active when used by itself, its mixture with any other catalysts also displayed higher activities than those without it.
The most active synthetic catalyst blend was the 1:1:2 mixture of NP6-CO2H, NP7-(CO2H)2, and NP11-(CO2H)2. The synthetic blend at 90 °C reached 44–45% of the activity of the commercial cellulase blend at 37 °C (Table 3, entries 22 and 25) and nearly 50% if the hydrolysis occurred in a boiling aqueous solution (entry 27). High temperature is necessary for the synthetic catalysts, as much lower activities were obtained at lower temperatures (entries 28–29).
As cross-linked polymeric nanoparticles, the synthetic catalysts could be reused multiple times. Figure 6 shows ten rounds of cellulose hydrolysis. In each catalytic cycle, a 5.0 mg of cellulose was added to a dialysis tubing with a MW-cutoff (MWCO) of 500 Da, which should let small sugar products escape but keep the catalysts and unreacted cellulose inside. After 12 h at 60 °C, another batch of cellulose was added, while the solution outside was analyzed. The recycling experiment shows that our synthetic glycosidase could maintain its activity very well, retaining 76% of the activity at the 10th reaction cycles. The reduced activity should not be caused by thermal instability of the catalyst because preheating at 90 °C for 3 days in pH 6.5 NaOAc buffer had a negligible effect on the catalysts (Table 3, compare entries 22 and 23). The most likely reason for the gradual decrease in the yield of hydrolysis over multiple reaction cycles was probably nonoptimal level of catalysts loading. Because cellulose hydrolysis was not carried to completion in each cycle, the catalyst loading would decrease over the reaction cycles. As shown in Table 3, the highest activity was obtained with 2–3 mg/mL of catalysts in the reaction mixture under our reaction conditions.
Figure 6.

Recyclability of NP catalyst blend for cellulose hydrolysis in 60 °C NaOAc buffer (pH 6.5).
CONCLUSIONS
Synthetic catalysts with enzyme-like performance have been a long-standing goal of chemists but their rational design has been hampered by the difficulty in constructing substrate-tailored active sites with accurately positioned catalytic groups.20–22 This work shows that molecular imprinting has a unique advantage in helping achieve this goal, by forming custom-made, functionalized imprinted sites conveniently through templated polymerization. When performed within the nanospace of a cross-linked micelle, the technique affords water-soluble nanoparticles with enzyme-like dimensions and a hydrophobic/hydrophilic core–shell morphology. As shown in this work, both noncovalent and covalent functionalities may be introduced into the imprinted sites to bind complex oligo- and polysaccharides. Solubility of the imprinted nanoparticles in water and selected organic solvents also allows facile postmodification by standard chemistry such as reductive amination and photoaffinity labelling. These features together enable the three main ingredients in cellulase blend to be mimicked through rational template design and strategic postfunctionalization, and, most importantly, the resulting synthetic blend is competitive in cellulose hydrolysis against natural enzymes. Meanwhile, as robust cross-linked polymeric nanoparticles, they tolerate high temperatures and organic solvents. The design principles and postmodification strategies should not be limited to a particular reaction, as long as suitable templates can be designed to allow the binding of the substrate and installation of catalytic groups at desired locations in the active site.
Supplementary Material
ACKNOWLEDGMENT
We thank NIGMS (R01GM138427) for supporting the research.
Footnotes
Supporting Information
Synthetic procedures, characterization of compounds and materials, ITC binding curves, additional tables and figures, and NMR data. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Himmel ME; Ding S-Y; Johnson DK; Adney WS; Nimlos MR; Brady JW; Foust TD Biomass Recalcitrance: Engineering Plants and Enzymes for Biofuels Production. Science 2007, 315, 804–807. [DOI] [PubMed] [Google Scholar]
- (2).Huber GW; Iborra S; Corma A Synthesis of Transportation Fuels from Biomass: Chemistry, Catalysts, and Engineering. Chem. Rev 2006, 106, 4044–4098. [DOI] [PubMed] [Google Scholar]
- (3).Luterbacher JS; Rand JM; Alonso DM; Han J; Youngquist JT; Maravelias CT; Pfleger BF; Dumesic JA Nonenzymatic Sugar Production from Biomass Using Biomass-Derived Γ-Valerolactone. Science 2014, 343, 277–280. [DOI] [PubMed] [Google Scholar]
- (4).Luterbacher JS; Martin Alonso D; Dumesic JA Targeted Chemical Upgrading of Lignocellulosic Biomass to Platform Molecules. Green Chem. 2014, 16, 4816–4838. [Google Scholar]
- (5).Robertson GP; Hamilton SK; Barham BL; Dale BE; Izaurralde RC; Jackson RD; Landis DA; Swinton SM; Thelen KD; Tiedje JM Cellulosic Biofuel Contributions to a Sustainable Energy Future: Choices and Outcomes. Science 2017, 356, eaal2324. [DOI] [PubMed] [Google Scholar]
- (6).Jing Y; Guo Y; Xia Q; Liu X; Wang Y Catalytic Production of Value-Added Chemicals and Liquid Fuels from Lignocellulosic Biomass. Chem 2019, 5, 2520–2546. [Google Scholar]
- (7).Wolfenden R; Lu X; Young G Spontaneous Hydrolysis of Glycosides. J. Am. Chem. Soc 1998, 120, 6814–6815. [Google Scholar]
- (8).Binder JB; Raines RT Fermentable Sugars by Chemical Hydrolysis of Biomass. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 4516–4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Almeida JRM; Bertilsson M; Gorwa-Grauslund MF; Gorsich S; Liden G Metabolic Effects of Furaldehydes and Impacts on Biotechnological Processes. Appl. Microbiol. Biotechnol 2009, 82, 625–638. [DOI] [PubMed] [Google Scholar]
- (10).Lairson LL; Withers SG Mechanistic Analogies Amongst Carbohydrate Modifying Enzymes. Chem. Commun 2004, 2243–2248. [DOI] [PubMed] [Google Scholar]
- (11).Zechel DL; Withers SG Glycosidase Mechanisms: Anatomy of a Finely Tuned Catalyst. Acc. Chem. Res 2000, 33, 11–18. [DOI] [PubMed] [Google Scholar]
- (12).MacLeod AM; Lindhorst T; Withers SG; Warren RAJ The Acid/Base Catalyst in the Exoglucanase/Xylanase from Cellulomonas Fimi Is Glutamic Acid 127: Evidence from Detailed Kinetic Studies of Mutants. Biochemistry 1994, 33, 6371–6376. [DOI] [PubMed] [Google Scholar]
- (13).MacLeod AM; Tull D; Rupitz K; Warren RAJ; Withers SG Mechanistic Consequences of Mutation of Active Site Carboxylates in a Retaining B-1,4-Glycanase from Cellulomonas Fimi. Biochemistry 1996, 35, 13165–13172. [DOI] [PubMed] [Google Scholar]
- (14).Igarashi K; Uchihashi T; Koivula A; Wada M; Kimura S; Okamoto T; Penttilä M; Ando T; Samejima M Traffic Jams Reduce Hydrolytic Efficiency of Cellulase on Cellulose Surface. Science 2011, 333, 1279–1282. [DOI] [PubMed] [Google Scholar]
- (15).Payne CM; Knott BC; Mayes HB; Hansson H; Himmel ME; Sandgren M; Ståhlberg J; Beckham GT Fungal Cellulases. Chem. Rev 2015, 115, 1308–1448. [DOI] [PubMed] [Google Scholar]
- (16).Payne CM; Jiang W; Shirts MR; Himmel ME; Crowley MF; Beckham GT Glycoside Hydrolase Processivity Is Directly Related to Oligosaccharide Binding Free Energy. J. Am. Chem. Soc 2013, 135, 18831–18839. [DOI] [PubMed] [Google Scholar]
- (17).Aden A; Ruth M; Ibsen K; Jechura J; Neeves K; Sheehan J; Wallace B; Montague L; Slayton A; Lukas J “Lignocellulosic Biomass to Ethanol Process Design and Economics Utilizing Co-Current Dilute Acid Prehydrolysis and Enzymatic Hydrolysis for Corn Stover,” NREL, 2002. [Google Scholar]
- (18).Wahlström RM; Suurnäkki A Enzymatic Hydrolysis of Lignocellulosic Polysaccharides in the Presence of Ionic Liquids. Green Chem. 2015, 17, 694–714. [Google Scholar]
- (19).Brogan APS; Bui-Le L; Hallett JP Non-Aqueous Homogenous Biocatalytic Conversion of Polysaccharides in Ionic Liquids Using Chemically Modified Glucosidase. Nat. Chem 2018, 10, 859–865. [DOI] [PubMed] [Google Scholar]
- (20).Breslow R Artificial Enzymes; Wiley-VCH: Weinheim, 2005. [Google Scholar]
- (21).Kirby AJ; Hollfelder F From Enzyme Models to Model Enzymes; Royal Society of Chemistry: Cambridge, UK, 2009. [Google Scholar]
- (22).Raynal M; Ballester P; Vidal-Ferran A; van Leeuwen PWNM Supramolecular Catalysis. Part 2: Artificial Enzyme Mimics. Chem. Soc. Rev 2014, 43, 1734–1787. [DOI] [PubMed] [Google Scholar]
- (23).Rousseau C; Nielsen N; Bols M An Artificial Enzyme That Catalyzes Hydrolysis of Aryl Glycosides. Tetrahedron Lett. 2004, 45, 8709–8711. [Google Scholar]
- (24).Ortega-Caballero F; Rousseau C; Christensen B; Petersen TE; Bols M Remarkable Supramolecular Catalysis of Glycoside Hydrolysis by a Cyclodextrin Cyanohydrin. J. Am. Chem. Soc 2005, 127, 3238–3239. [DOI] [PubMed] [Google Scholar]
- (25).Striegler S; Barnett JD; Dunaway NA Glycoside Hydrolysis with Sugar-Templated Microgel Catalysts. ACS Catal. 2012, 2, 50–55. [Google Scholar]
- (26).Sharma B; Striegler S Crosslinked Microgels as Platform for Hydrolytic Catalysts. Biomacromolecules 2018, 19, 1164–1174. [DOI] [PubMed] [Google Scholar]
- (27).Samanta M; Krishna VSR; Bandyopadhyay S A Photoresponsive Glycosidase Mimic. Chem. Commun 2014, 50, 10577–10579. [DOI] [PubMed] [Google Scholar]
- (28).National Research Council (NRC). Transforming Glycoscience: A Roadmap for the Future; National Academies Press: Washington, D.C., 2012. [PubMed] [Google Scholar]
- (29).Boraston AB; Bolam DN; Gilbert HJ; Davies GJ Carbohydrate-Binding Modules: Fine-Tuning Polysaccharide Recognition. Biochem. J 2004, 382, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wulff G; Vesper W Enzyme-Analogue Built Polymers .8. Preparation of Chromatographic Sorbents with Chiral Cavities for Racemic-Resolution. J. Chromatogr 1978, 167, 171–186. [Google Scholar]
- (31).James TD; Phillips MD; Shinkai S Boronic Acids in Saccharide Recognition; RSC Publishing: Cambridge, 2006. [Google Scholar]
- (32).Wulff G Enzyme-Like Catalysis by Molecularly Imprinted Polymers. Chem. Rev 2002, 102, 1–28. [DOI] [PubMed] [Google Scholar]
- (33).Haupt K; Mosbach K Molecularly Imprinted Polymers and Their Use in Biomimetic Sensors. Chem. Rev 2000, 100, 2495–2504. [DOI] [PubMed] [Google Scholar]
- (34).Ye L; Mosbach K Molecular Imprinting: Synthetic Materials as Substitutes for Biological Antibodies and Receptors. Chem. Mater 2008, 20, 859–868. [Google Scholar]
- (35).Pan J; Chen W; Ma Y; Pan G Molecularly Imprinted Polymers as Receptor Mimics for Selective Cell Recognition. Chem. Soc. Rev 2018, 47, 5574–5587. [DOI] [PubMed] [Google Scholar]
- (36).Zhang H Molecularly Imprinted Nanoparticles for Biomedical Applications. Adv. Mater 2020, 32, 1806328. [DOI] [PubMed] [Google Scholar]
- (37).Haupt K; Medina Rangel PX; Bui BTS Molecularly Imprinted Polymers: Antibody Mimics for Bioimaging and Therapy. Chem. Rev 2020, 120, 9554–9582. [DOI] [PubMed] [Google Scholar]
- (38).Wulff G; Liu J Design of Biomimetic Catalysts by Molecular Imprinting in Synthetic Polymers: The Role of Transition State Stabilization. Acc. Chem. Res 2012, 45, 239–247. [DOI] [PubMed] [Google Scholar]
- (39).Muratsugu S; Shirai S; Tada M Recent Progress in Molecularly Imprinted Approach for Catalysis. Tetrahedron Lett. 2020, 61, 151603. [Google Scholar]
- (40).Kirsch N; Hedin-Dahlström J; Henschel H; Whitcombe MJ; Wikman S; Nicholls IA Molecularly Imprinted Polymer Catalysis of a Diels-Alder Reaction. J. Mol. Catal. B: Enzym 2009, 58, 110–117. [Google Scholar]
- (41).Chen ZY; Xu L; Liang Y; Zhao MP Ph-Sensitive Water-Soluble Nanospheric Imprinted Hydrogels Prepared as Horseradish Peroxidase Mimetic Enzymes. Adv. Mater 2010, 22, 1488–1492. [DOI] [PubMed] [Google Scholar]
- (42).Servant A; Haupt K; Resmini M Tuning Molecular Recognition in Water-Soluble Nanogels with Enzyme-Like Activity for the Kemp Elimination. Chem.-Eur. J 2011, 17, 11052–11059. [DOI] [PubMed] [Google Scholar]
- (43).Shen X; Huang C; Shinde S; Jagadeesan KK; Ekström S; Fritz E; Sellergren B Catalytic Formation of Disulfide Bonds in Peptides by Molecularly Imprinted Microgels at Oil/Water Interfaces. ACS Appl. Mater. Interfaces 2016, 8, 30484–30491. [DOI] [PubMed] [Google Scholar]
- (44).Yuan Y; Yang Y; Faheem M; Zou X; Ma X; Wang Z; Meng Q; Wang L; Zhao S; Zhu G Molecularly Imprinted Porous Aromatic Frameworks Serving as Porous Artificial Enzymes. Adv. Mater 2018, 30, 1800069. [DOI] [PubMed] [Google Scholar]
- (45).Li S; Lieberzeit PA; Piletsky S; Turner APF Smart Polymer Catalysts and Tunable Catalysis; Elsevier: Amsterdam, Netherlands; Cambridge, MA, 2019. [Google Scholar]
- (46).Striegler S; Sharma B; Orizu I Microgel-Catalyzed Hydrolysis of Nonactivated Disaccharides. ACS Catal. 2020, 10, 14451–14456. [Google Scholar]
- (47).Awino JK; Zhao Y Protein-Mimetic, Molecularly Imprinted Nanoparticles for Selective Binding of Bile Salt Derivatives in Water. J. Am. Chem. Soc 2013, 135, 12552–12555. [DOI] [PubMed] [Google Scholar]
- (48).Xing X; Zhao Y Fluorescent Nanoparticle Sensors with Tailor-Made Recognition Units and Proximate Fluorescent Reporter Groups. New J. Chem 2018, 42, 9377–9380. [Google Scholar]
- (49).Nowick JS; Chen JS; Noronha G Molecular Recognition in Micelles - the Roles of Hydrogen-Bonding and Hydrophobicity in Adenine Thymine Base-Pairing in Sds Micelles. J. Am. Chem. Soc 1993, 115, 7636–7644. [Google Scholar]
- (50).Ariga K; Kunitake T Molecular Recognition at Air-Water and Related Interfaces: Complementary Hydrogen Bonding and Multisite Interaction. Acc. Chem. Res 1998, 31, 371–378. [Google Scholar]
- (51).Zangiabadi M; Zhao Y Selective Binding of Complex Glycans and Glycoproteins in Water by Molecularly Imprinted Nanoparticles. Nano Lett. 2020, 20, 5106–5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Okada G Cellulase of Aspergillus Niger. Meth. Enzymol 1988, 160, 259–264. [Google Scholar]
- (53).Li S-F; Cheng F; Wang Y-J; Zheng Y-G Strategies for Tailoring Ph Performances of Glycoside Hydrolases. Critical Reviews in Biotechnology 2021, 1–21. [DOI] [PubMed] [Google Scholar]
- (54).Becker D; Braet C; Brumer H 3rd; Claeyssens M; Divne C; Fagerström BR; Harris M; Jones TA; Kleywegt GJ; Koivula A; Mahdi S; Piens K; Sinnott ML; Ståhlberg J; Teeri TT; Underwood M; Wohlfahrt G Engineering of a Glycosidase Family 7 Cellobiohydrolase to More Alkaline Ph Optimum: The Ph Behaviour of Trichoderma Reesei Cel7a and Its E223s/A224h/L225v/T226a/D262g Mutant. Biochem. J 2001, 356, 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Gunasekara RW; Zhao Y A General Method for Selective Recognition of Monosaccharides and Oligosaccharides in Water. J. Am. Chem. Soc 2017, 139, 829–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Li X; Zhao Y Synthetic Glycosidases for the Precise Hydrolysis of Oligosaccharides and Polysaccharides. Chem. Sci 2021, 12, 374–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Leyva E; Young MJT; Platz MS High Yields of Formal Ch Insertion Products in the Reactions of Polyfluorinated Aromatic Nitrenes. J. Am. Chem. Soc 1986, 108, 8307–8309. [Google Scholar]
- (58).Bose I; Zhao Y Tandem Aldol Reaction from Acetal Mixtures by an Artificial Enzyme with Site-Isolated Acid and Base Functionalities. ACS Appl. Polym. Mater 2021, 3, 2776–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Bose I; Zhao Y Site-Selective Catalytic Epoxidation of Alkenes with Tunable Atomic Precision by Molecularly Imprinted Artificial Epoxidases. ACS Catal. 2022, 12, 3444–3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Li X; Zangiabadi M; Zhao Y Molecularly Imprinted Synthetic Glucosidase for the Hydrolysis of Cellulose in Aqueous and Nonaqueous Solutions. J. Am. Chem. Soc 2021, 143, 5172–5181. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.