

Abstract
Nitric oxide (NO) donors were recently shown to produce biphasic contractile effects in cardiac tissue, with augmentation at low NO levels and depression at high NO levels. We examined the subcellular mechanisms involved in the opposing effects of NO on cardiac contraction and investigated whether NO modulates contraction exclusively via guanylyl cyclase (GC) activation or whether some contribution occurs via cGMP/PKG-independent mechanisms, in indo 1–loaded adult cardiac myocytes. Whereas a high concentration of the NO donor S-nitroso-N-acetylpenicillamine (SNAP, 100 μmol/L) significantly attenuated contraction amplitude by 24.4±4.5% (without changing the Ca2+ transient or total cAMP), a low concentration of SNAP (1 μmol/L) significantly increased contraction amplitude (38±10%), Ca2+ transient (26±10%), and cAMP levels (from 6.2 to 8.5 pmol/mg of protein). The negative contractile response of 100 μmol/L SNAP was completely abolished in the presence of the specific blocker of PKG KT 5823 (1 μmol/L); the positive contractile response of 1 μmol/L SNAP persisted, despite the presence of the selective inhibitor of GC 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ, 10 μmol/L) alone, but was completely abolished in the presence of ODQ plus the specific inhibitory cAMP analog Rp-8-CPT-cAMPS (100 μmol/L), as well as by the NO scavenger oxyhemoglobin. Parallel experiments in cell suspensions showed significant increases in adenylyl cyclase (AC) activity at low concentrations (0.1 to 1 μ-mol/L) of SNAP (AC, 18% to 20% above basal activity). We conclude that NO can regulate both AC and GC in cardiac myocytes. High levels of NO induce large increases in cGMP and a negative inotropic effect mediated by a PKG-dependent reduction in myofilament responsiveness to Ca2+. Low levels of NO increase cAMP, at least in part, by a novel cGMP-independent activation of AC and induce a positive contractile response.
Keywords: nitric oxide signaling, cGMP, cAMP, contractility, cardiac myocyte
Nitric oxide (NO) has been implicated as a mediator of many cellular processes, including endothelium-dependent relaxation of blood vessels, chemical communication between peripheral nerves and smooth muscle, inhibition of platelet aggregation, immune responses, and neurotransmission.1 These effects of NO have been ascribed to the activation of soluble guanylyl cyclase (GC), leading to the production of cGMP and activation of cGMP-dependent protein kinase (PKG).1,2 Furthermore, NO and NO donors have been shown to elicit a wide range of effects on myocardial contractility,3-9 but the specific nature of the subcellular mechanisms underlying the diverse effects in heart is largely unknown.
Although the coronary endothelium is responsible for the bulk of the endogenous, physiological production of NO in the heart,10,11 NO can also be produced within the cardiac myocytes themselves (ie, autocrine production) by the constitutive NO synthase NOS-3.12,13 Dynamic regulation of NO production in the heart apparently yields beat-to-beat oscillations in response to changes in coronary flow and myocardial loading and achieves micromolar levels in close vicinity to the cardiac myocyte.11 Until recently, it was generally accepted that NO induced negative inotropic effects in cardiac preparations, mediated principally through cGMP-related mechanisms, specifically via the reduction of Ca2+ influx through L-type Ca2+ channels, either through activation of cGMP-dependent phosphodiesterase14-16 or PKG and/or phosphatases.17-20 cGMP has also been shown to decrease relative myofilament response to Ca2+ and therefore enhances myocardial relaxation and reduces diastolic tone.21 However, the hypothesis that cGMP-mediated processes are the only mechanisms responsible for the NO-mediated contractile effects has subsequently been challenged. Several studies have demonstrated a dissociation between cGMP concentrations and contractile state, because low concentrations of acetylcholine induced a negative inotropic effect even in the absence of any change in cGMP concentration.22-25 Recent reports show that under certain conditions, NO donors are able to enhance myocardial contractility,8,9 and that the basal intracardiac production and release of NO significantly augments the Frank-Starling response in the isolated heart.26 It has been demonstrated that the augmentation of contractility by exogenous NO could be the result of an elevation of the intracellular levels of cAMP due to the cGMP-dependent inhibition of cAMP hydrolysis by cGMP-inhibited phosphodiesterase (cGI-PDE or PDE type III).9 These observations, however, do not rule out the possibility of cAMP being increased by a direct (ie, GC/cGMP independent) modulation of the adenylyl cyclase (AC)/cAMP/cAMP-dependent protein kinase (PKA) pathway by NO.
Most reports on NO-mediated regulation of cardiac excitation and contraction have focused on the cGMP-dependent actions, whereas GC/cGMP-independent mechanisms have been largely unexplored. Relatively recently, a few publications have addressed “unconventional” pathways for NO signaling in the heart whereby NO may directly modulate protein function in a GC/cGMP-independent fashion, eg, via trans-nitrosylation of critical or regulatory thiols.27-29 Although NO certainly modulates cardiac contraction in part via cGMP/PKG-related mechanisms, considering the multiplicity of potential NO targets in the heart and the multifaceted effects of NO and NO-related compounds on cardiac contraction, it seems unlikely that NO exerts all of its physiologically relevant effects exclusively through activation of GC and PKG activation.
To examine whether NO potentially modulates the cAMP/PKA signaling pathway via GC-independent mechanisms, we studied the effects of the NO donor S-nitroso-N-acetylpenicillamine (SNAP), in the presence and absence of specific inhibitors of GC, PKG, and PKA, on contraction and the Ca2+ transient (Cai) in isolated rat cardiac myocytes, in parallel with measurements of intracellular cGMP, cAMP, and AC activity. Parallel functional experiments were performed with 3 - [2 - hydroxy - 1 - (1 - methylethyl) - 2 - nitrosohydrazino] - 1 - propanamine (NOC5), an NO amine complex that serves as an NO donor chemically distinct from SNAP, and with NO scavengers, to establish that the biological effects were the results of the action of NO. The results from the present study suggest that NO is capable of evoking either positive or negative inotropic responses in cardiac myocytes depending on the concentration of exposure. Whereas the decrease in contractile response seen principally at higher levels of NO can be attributed mainly to a cGMP-dependent reduction in myofilament responsiveness to Ca2+, the enhanced contractile response at lower NO levels is due to increased intracellular cAMP levels that are mediated, at least in part, through a novel NO-dependent, GC/cGMP-independent activation of AC. Preliminary findings of the present study have been previously published in abstract form.30,31
Materials and Methods
Cardiac Myocyte Isolation
Single cardiac myocytes were isolated via a previously described technique with minor modifications.32 Briefly, 2- to 4-month-old adult Sprague-Dawley rats (Charles River) were anesthetized with intraperitoneal sodium pentobarbital, and hearts were rapidly excised and perfused retrogradely with 25 mL nominally Ca2+-free bicarbonate buffer of the following composition (in mmol/L): NaCl 116.4, KCl 5.4, MgSO4 1.2, NaH2PO4 1.2, glucose 5.6, and NaHCO3 26.2, kept at 36±1°C and continuously gassed with 95% O2 and 5% CO2 to keep pH at 7.4. Perfusion was continued with a similar bicarbonate buffer containing 50 μmol/L Ca2+, 0.1% collagenase type B, 0.04 mg/mL protease XVI, and 0.1% BSA type V for ≈20 minutes. The left ventricle was then mechanically dissociated, and myocytes were resuspended in a series of HEPES buffers with gradually increasing Ca2+ concentration. Cells were finally suspended in HEPES buffer containing (in mmol/L) NaCl 137, KCl 4.9, MgSO4 1.2, NaH2PO4 1.2, glucose 15, HEPES 20, and CaCl2 1.0, (pH 7.4) and stored at room temperature until use. Noncardiac myocytes comprised ≪1% of cells prepared in this manner. Cardiac myocyte viability was typically 70% to 80%. All experiments were replicated at least 3 times (or as specifically stated) and represent cells from at least 2 (or more) different myocyte preparations. Experiments were performed under a protocol approved by the National Institute on Aging Animal Care Committee.
Simultaneous Measurements of Contraction and Indo 1 Fluorescence
Changes in the Cai, assessed by indo 1 fluorescence, and cell length were measured in isolated myocytes as previously described.33 In brief, for measurements of [Ca2+]i, myocytes were loaded with the membrane-permeable acetoxymethyl ester of indo 1 (indo 1-AM), using a 10-minute exposure to a 25 μmol/L solution in HEPES buffer at room temperature. After loading, cells were transferred to a Lucite chamber with a glass coverslip on the stage of an inverted microscope and were continuously superfused with HEPES buffer. Myocytes were chosen for study according to previously established criteria,32 ie, a rod-shaped appearance with clear striations and no membrane blebs, a negative staircase of twitch performance on stimulation from rest, and the absence of spontaneous contractions. Cardiac myocyte contraction was produced via electrical field stimulation from 2 platinum electrodes (2- to 4-ms duration at 0.5 Hz) connected to a stimulator (model SD9, Grass Instrument Co).
Myocyte length at rest and during contraction was monitored from a red light (650 to 750 nm) bright field image of the cell projected onto a photodiode array (Reticon) with a 5-ms temporal resolution. For simultaneous measurement of the Cai in these cells, indo 1 fluorescence was excited by epi-illumination with 10-μs flashes of 350±5 nm of light focused onto the myocyte by a 100×/1.3NA Nikon UV fluor glycerine immersion objective. Epifluorescence collected every 5 ms was split into 391- to 434-nm (410 channel) and 457- to 507-nm (490 channel) wavelength bands by bandpass interference filters. The ratio of the indo 1 emission at the 2 wavelengths was calculated using a pair of fast integrator sample-and-hold circuits under the control of a specially modified PC, and it was taken as an index of the Cai. When isolated cardiac myocytes are loaded with indo 1-AM, there is a variable compartmentalization of the indicator into the mitochondria33 that prevents the use of a standard calibration curve. Thus, the present results in indo 1-AM–loaded myocytes will be expressed as fluorescence ratio rather than as absolute [Ca2+]i values. Both loading of the Ca2+ probe into the myocytes and the experiments were performed at 25°C to minimize the loss of the fluorescent Ca2+ indicator from the cell.33 Additionally, some experiments were implemented with cells that had not been loaded with indo 1, and only cell length was measured.
Assessment of Myofilament Response to Ca2+
Changes in myofilament responsiveness to Ca2+ were assessed using the steady-state relation between cell length and [Ca2+]i in intact single cardiac myocytes tetanized by high-frequency (10 Hz) stimulation after exposure to thapsigargin (0.2 μmol/L for 15 minutes), as described previously.34 Thapsigargin disables the sarcoplasmic reticulum35 and thus enables tetanization of otherwise intact myocytes. High-frequency electrical stimulation after thapsigargin treatment results in the effective summation of the repetitive transmembrane flux of Ca2+ current (because of the absence of sarcoplasmic reticulum Ca2+ sequestration and periodic release) to achieve a steady-state level of myoplasmic Ca2+ substantially elevated above resting levels, at the point that Ca2+ influx is balanced by the rate of Ca2+ extrusion (via Na/Ca exchange). With this approach, the Cai can be reversibly clamped near peak systolic levels during the tetanic contracture for 10 to 60 seconds (or longer) and then rapidly returned to resting levels on cessation of electrical stimulation via normal Na/Ca exchange mechanisms. The steady-state levels of Ca2+ achieved during tetanization, moreover, can be systematically regulated by adjustments in the concentration of bathing Ca2+ and can be matched between protocols to effectively gauge changes in myofilament Ca2+ sensitivity. Thus, changes in the degree of cell shortening between tetani clamped at the same Cai level can be attributed to changes in the relative myofilament responsiveness to Ca2+.
Determination of cGMP and cAMP
Suspensions of freshly isolated adult rat cardiac myocytes were pretreated for 30 minutes at 23°C with either control or 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) 10 μmol/L containing buffers and subsequently challenged with the indicated concentration of SNAP for the next 20 minutes. The cells were then lysed with 1 mL ice-cold 0.6 mmol/L perchloric acid. The cell lysates (950 μL) were transferred to microcentrifuge tubes, and the pH was adjusted to 7.0 using K2CO3. After centrifugation for 5 minutes at 8000g, the supernatant was vacuum-dried and then recovered in 200 μL Tris/EDTA buffer. After addition of 0.15 mmol/L Na2CO3 (20 μL) and 0.15 mmol/L ZnSO4 (20 μL) and incubation for 15 minutes on ice, the salt precipitate was removed by centrifugation for 5 minutes at 1200g, and 50 μL of supernatant was assayed using either a cAMP [3H] or cGMP [3H] assay kit (Amersham). Cellular protein was measured using the Bradford method (Bio-Rad) with bovine γ-globulin as standard.
Determination of AC Activity
Preparation of Purified Cardiac Sarcolemmal Membranes
After mincing and washing with saline EGTA (154 mmol/L NaCl, 0.1 mmol/L EGTA), freshly excised cardiac ventricular tissue was suspended into 5 volumes of homogenizing buffer (HB) (in mmol/L: HEPES 10, MgCl2 2, EGTA 0.1, and dithiothreitol 5; prepared the day of use). The suspension was homogenized 4 times for 10 seconds at 22 000 rpm with a polytron. Five more volumes of HB were added to the homogenate, and after centrifugation at 12 000g for 15 minutes at 4°C, the supernatants were poured into polypropylene ultracentrifuge tubes and centrifuged at 50 000g for 30 minutes at 4°C in an ultracentrifuge. The resulting pellets were then resuspended in a motorized Potter-Elvehjem glass/Teflon homogenizer with 0.75 HB per gram of original sample. Membrane protein was assayed by the BCA protein assay method (Pierce), and the homogenate was stored at −80°C until use.
AC Enzyme Activity
Two micrograms of membrane protein was added into a final volume of 200 μL containing (in mmol/L) HEPES 25, MgCl2 0.4, GTP 5, dithiothreitol 0.15, NaCl 50, creatine phosphate 2.5, creatine phosphokinase 5 U, 3-isobutyl-1-methylxanthine (IBMX) 10, and alamethicin (1 μg/μg of membrane protein). The tubes were transferred to a 30°C water bath, and the reaction with a given dose of SNAP was started by adding 5 mmol/L ATP. After 15 minutes, the reaction was stopped by addition of 1.8 mL of 80°C to 90°C H2O. Subsequently, cAMP was determined using the scintillation proximity assay according to the Amersham protocol furnished with the RPA 538 kit (Amersham).
Determination of Free NO Concentration Produced by NO Donors SNAP and NOC5
Free NO concentration was determined in the physiological buffer solutions containing either SNAP or NOC5 at the concentrations used in these experiments (90 minutes after preparation, 25°C) using a Sievers model 280 nitric oxide analyzer following the method recommended by the manufacturer (NO concentrations were also measured in the bathing solutions of some of the cardiac myocyte contraction experiments to ensure consistency of NO application). Calibration of the NO analyzer was obtained using reagent-grade NaNO3 prepared in nitrate-free, deionized water (standard solutions containing 10 nmol/L, 50 nmol/L, and 100 nmol/L and 1 μmol/L, 5 μmol/L, 10 μmol/L, and 100 μmol/L nitrate were used for calibration), in a nitrate-reducing reaction system of freshly prepared VCl3/HCl at 90°C as prescribed by Sievers, which results in the quantitative conversion of nitrate (and nitrite) to NO. The resulting NO was measured by the detection of chemiluminescence from its reaction with ozone. Within-run and between-run reproducibility was <±5%.
Materials
Collagenase type B was purchased from Boehringer Mannheim Corp; isoproterenol from Winthrop Pharmaceuticals; protease XVI, N-acetylpenicillamine (NAP), and IBMX from Sigma Chemical Co; indo 1-AM from Molecular Probes Inc; BSA type V, bovine hemoglobin, SNAP, and NOC5 from Calbiochem Corp; Rp-8-CPT-cAMPS from Biolog; and ODQ from Biomol Research Laboratories Inc. All other chemicals were of the purest reagent grade available.
Statistics
All data are presented as mean±SEM. Comparisons within groups were made by paired and unpaired Student t test, and values of P<0.05 were taken to indicate statistical significance.
Results
Reversible Effects of SNAP on Cardiac Myocyte Contraction
We assessed the effect of different concentrations of SNAP on contraction in electrically stimulated cardiac myocytes. Figure 1A shows representative examples of the opposing effects of high (100 μmol/L) and low (1 μmol/L) concentrations of SNAP on the unloaded contraction from 2 separate cardiac myocytes. At the high concentration (top tracing), SNAP induced a slowly evolving negative inotropic effect, which was maximal 15 to 20 minutes after administration of the drug, and this effect was completely reversible on washout. In contrast, low concentrations of SNAP induced a pronounced positive inotropic effect, which was also entirely reversible on washout. Figure 1B shows the average data from these experiments, indicating the time course of the opposing contractile responses induced by high and low concentrations of SNAP. After 20 minutes of SNAP exposure, twitch amplitude was reduced by 24±5% (n=5) at the high concentration but was increased by 38±10% (n=5) at the low concentration. Thus, the dynamic range of contractile regulation induced by the different concentrations of NO used in these experiments is about two thirds of the basal contraction amplitude.
Figure 1.
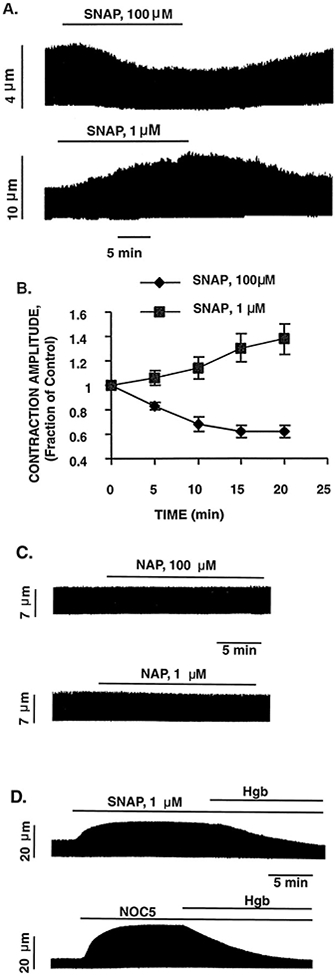
Effect of high (100 μmol/L) and low (1 μmol/L) concentrations of SNAP on the electrically stimulated myocyte contraction. A, Representative examples of the opposing contractile effects induced by high and low concentrations of SNAP from 2 different cardiac myocytes. The continuous chart recording of cell length (top) shows that a high concentration of SNAP induced a progressive decrease in twitch amplitude, whereas the bottom tracing shows that a low concentration of SNAP induced a progressive increase in contraction amplitude. Both negative and positive contractile effects of SNAP were entirely reversible on washout. B, Overall data of the time course of the contractile effects induced by high and low concentrations of SNAP. Data are mean±SE; n=5 cells per group. C, Representative examples of the lack of contractile effects of exposure to NAP 100 μmol/L and 1 μmol/L (the byproduct of SNAP after release of NO). D, Representative examples demonstrating that the positive contractile effects of both SNAP (1 μmol/L) and the unrelated NO donor NOC5 (1 μmol/L) were completely abolished by coincubation with the NO scavenger oxyhemoglobin (Hgb, 0.1 mmol/L).
As control experiments, cells were exposed to the byproduct of SNAP after release of NO, NAP, at the same concentrations as used in the SNAP experiments. Figure 1C shows that neither NAP at 100 μmol/L nor NAP at 1 μmol/L had any effect on myocyte contraction, indicating that the various contractile effects seen with SNAP do not result from the thiol byproduct after the production of NO. Figure 1D demonstrates that the positive contractile effect seen in these experiments was indeed the result of NO, per se, insofar as that seen both with SNAP (1 μmol/L) and an unrelated NO donor, NOC5 (1 μmol/L), was completely abolished by coincubation with the NO scavengers, oxyhemoglobin (10 μmol/L), and carboxy-PTIO (0.1 mmol/L; data not shown). The negative contractile effects seen at higher concentrations of both SNAP (100 μmol/L) and NOC5 (20 μmol/L) are similarly abolished by both oxyhemoglobin and carboxy-PTIO (data not shown). Similar results were obtained in at least triplicate observations in different cells under each of the conditions described above.
Effect of SNAP on [Ca2+]i, cGMP, and cAMP
Using indo 1–loaded cardiac myocytes, we investigated the effect of different concentrations of SNAP on contraction and Cai measured simultaneously. A representative example of the negative contractile effect of a high concentration of SNAP (100 μmol/L) and the associated Cai is depicted in Figure 2A. The progressive reduction in the twitch amplitude seen here was not associated with a decrease of the Cai, suggesting that SNAP (100 μmol/L) reduced the relative myofilament responsiveness to Ca2+. Also shown in the figure are the results from parallel experiments comparing intracellular levels of cGMP and cAMP in the presence and absence of SNAP. SNAP (100 μmol/L) induced a large increase in cGMP (31±6% versus basal, P<0.05) but had no significant effect on cAMP levels. In contrast, a low concentration of SNAP (1 μmol/L) elicited a positive contractile response that was associated with a significant increase in the Cai amplitude (26±10%, n=5 cells, P<0.05; Figure 2B). This low concentration of SNAP induced only a modest increase in cGMP (12±3% versus basal, P<0.05) but produced a large increment in cAMP levels (35±9% versus basal, P<0.05). The average effects of high and low concentrations of SNAP on contraction and Cai amplitude and kinetics are provided in the Table.
Figure 2.
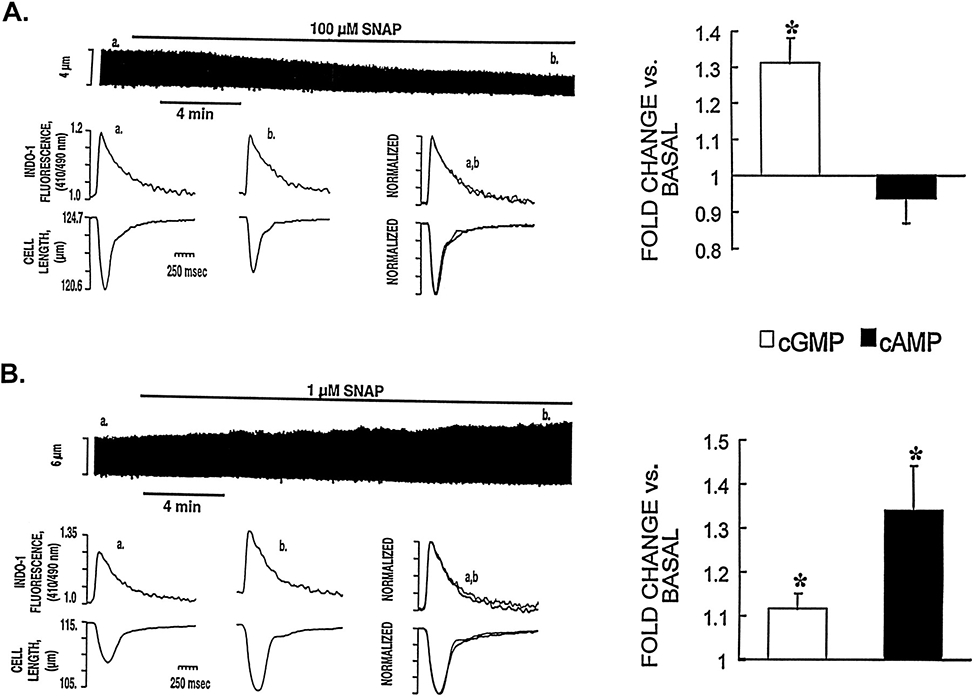
Effect of high (100 μmol/L) and low (1 μmol/L) concentrations of SNAP on [Ca2+]i, cGMP and cAMP levels, and contraction. A, Representative example of simultaneously recorded Cai and contraction traces showing the effects induced by a high concentration of SNAP in an indo 1-AM–loaded myocyte. The chart recording (top) shows that SNAP (100 μmol/L) induced a progressive decrease in twitch amplitude. The bottom tracings show, on an expanded time scale, the [Ca2+]i transients and cell length at control (a) and 15 to 20 minutes after addition of SNAP (b). The superimposed tracing (overlay of data from a and b; scales normalized to peak heights) shows that SNAP (100 μmol/L) reduced twitch duration and twitch amplitude without affecting the Cai. The right panel shows the effect of SNAP 100 μmol/L on cGMP and cAMP levels. The results are presented as fractions of the basal values of cGMP and cAMP (4.2±0.2 and 6.2±0.4 pmol/mg of protein, respectively). Each bar represents the mean±SEM of 3 independent experiments performed in triplicate. Using the same presentation scheme, Figure 2B shows the effect of low concentrations of SNAP (1 μmol/L) on [Ca2+]i, cGMP and cAMP levels, and contraction. Note that this concentration of SNAP was associated with a large increase in the Cai, cAMP levels, and contraction. *P<0.05 vs basal level.
Effect of SNAP on Myofilament Responsiveness to Ca2+
The results shown in Figure 2 suggest that high concentrations of SNAP are associated with a diminished sensitivity of the myofilaments to Ca2+. The effects of low concentrations of SNAP on myofilament sensitivity to Ca2+ are, however, more difficult to interpret, because both the amplitude of the Cai and the contraction change during the experimental protocol. To further characterize the effects of SNAP on myofilament responsiveness to Ca2+, steady-state tetanic contractures (which enable the Cai to be reversibly maintained at reproducible levels near that of peak systole for periods of 10 to 60 seconds) were performed in the presence and absence of SNAP. Figure 3A shows a representative example of the effect of SNAP (100 μmol/L) on the steady-state myofilament response to Ca2+. Despite the achievement of similar peak [Ca2+]i levels, in the presence of high concentrations of SNAP, steady-state cell contraction amplitude during the tetanus was significantly decreased, confirming that the phenomenon seen in Figure 2A was the result of a reduction in myofilament response to Ca2+. In contrast, perfusion with a low concentration of SNAP (1 μmol/L) did not significantly affect either peak steady-state Ca2+ or shortening during the tetanus (Figure 3B), thus indicating that low concentrations of SNAP do not exert an appreciable effect on myofilament responsiveness to Ca2+. Similar results were obtained in at least triplicate observations in different cells at each concentration of SNAP.
Figure 3.
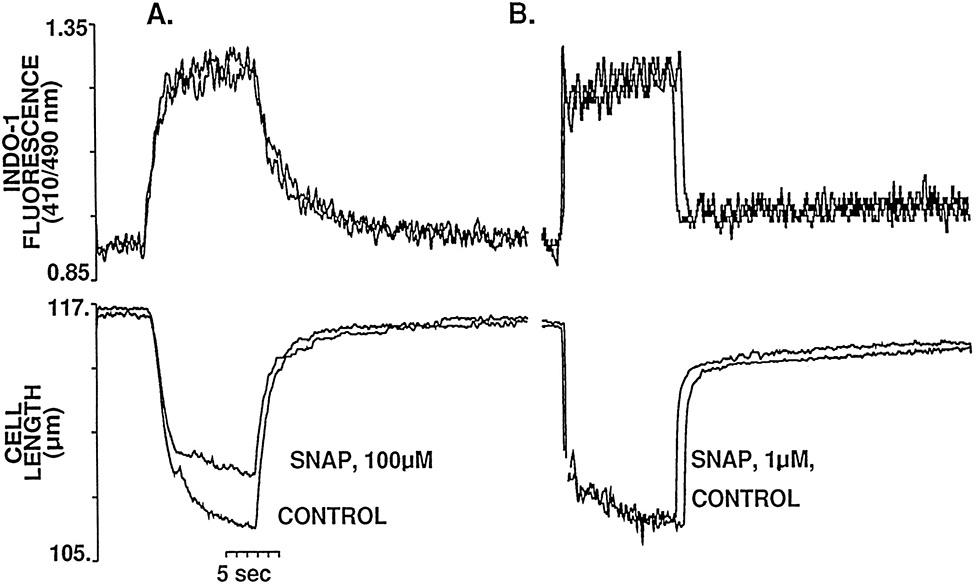
Representative example of the effects of SNAP 100 μmol/L (A) and 1 μmol/L (B) on the steady-state myofilament Ca2+ response during tetanic contraction (10 Hz) of myocytes pretreated with thapsigargin (0.2 μmol/L). The top tracings correspond to the superimposed Ca2+ at control and after 20 minutes of exposure to the indicated concentration of SNAP; immediately below are the superimposed tetanic contractions. Although SNAP had no effect on peak tetanic Ca2+ at either of the concentrations studied, the amplitude of the tetanic contraction is significantly decreased after exposure to high concentrations of SNAP but unchanged at the low concentration. It should be noted that the [Ca2+]i achieved during tetanization protocols is <1 μmol/L (ie, on the order of that achieved during peak systole), and, moreover, that Ca2+–indo 1 binding is far from saturation, even at the plateau of the tetanus.
To assess whether the increase in cGMP induced by a high concentration of SNAP (ie, Figure 2A, right panel) activates PKG and is, in turn, responsible for the observed reduction in myofilament responsiveness to Ca2+, steady-state tetanic contractures were also performed in cardiac myocytes pretreated with the specific blocker of PKG KT 5823 1 μmol/L. Figure 4 shows that inhibiting PKG activation with KT 5823 completely abolished the reduction in steady-state cell shortening during the tetany in the presence of high concentrations of SNAP (100 μmol/L). This protocol with KT 5823 has been shown to completely inhibit the negative contractile effect (of comparable magnitude to that seen here with SNAP 100 μmol/L) induced by 8-bromo-cGMP (50 μmol/L) in cardiac myocytes.21
Figure 4.
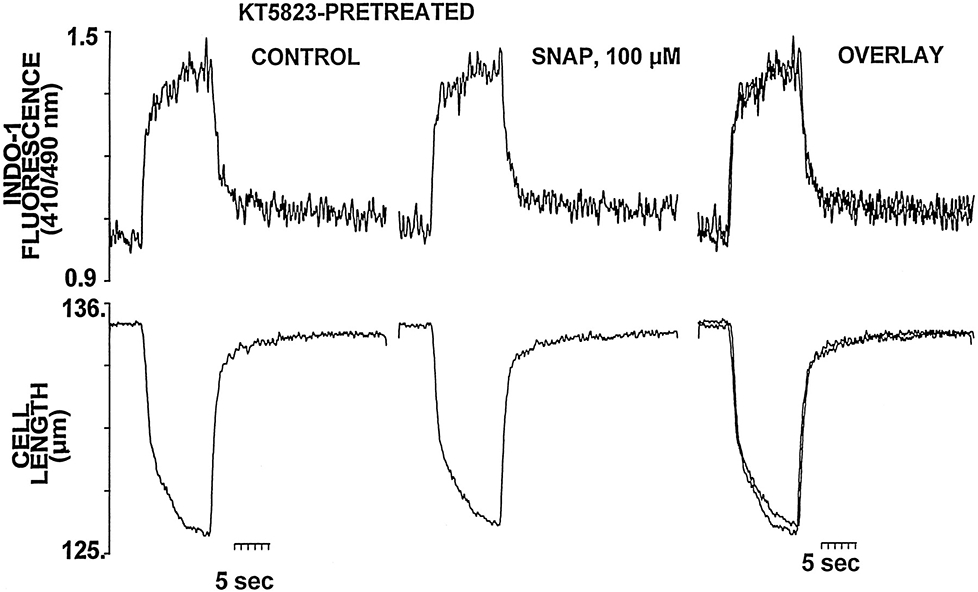
PKG inhibition with KT 5823 (1 μmol/L) prevents the negative myofilament effect of SNAP 100 μmol/L. Representative example of the effects of SNAP 100 μmol/L on the steady-state Cai and contraction during tetanic contractions of a myocyte pretreated and continuously perfused with KT 5823 1 μmol/L. The superimposed tracing shows the failure of SNAP to decrease the amplitude of the tetanic contraction in the presence of KT 5823.
Effect of SNAP in the Presence of Inhibitors of GC
To investigate whether cGMP-independent mechanisms participate in the positive inotropic effect elicited by a low concentration of SNAP, experiments were performed in the presence of the selective inhibitor of NO-sensitive GC36 ODQ (10 μmol/L). Figure 5A shows the effect of SNAP (1 μmol/L) on the contraction in a representative cardiac myocyte pretreated and continuously perfused with ODQ. In spite of the presence of ODQ, the positive inotropic response to SNAP (1 μmol/L) persisted (32±7% increase in twitch amplitude) together with an increased Cai (20±6%) (n=6 cells). Perfusion with ODQ (10 μmol/L) alone did not significantly affect the baseline contraction or the Cai. Figure 5C shows a representative example of the lack of effect of SNAP (1 μmol/L) in the presence of ODQ (10 μmol/L) on the steady-state myofilament response to Ca2+ (similar to the result in Figure 3B), which indicates that the positive contractile effect seen in Figure 5A is purely the result of the increase in magnitude of the Cai (rather than a change in myofilament Ca2+ sensitivity).
Figure 5.
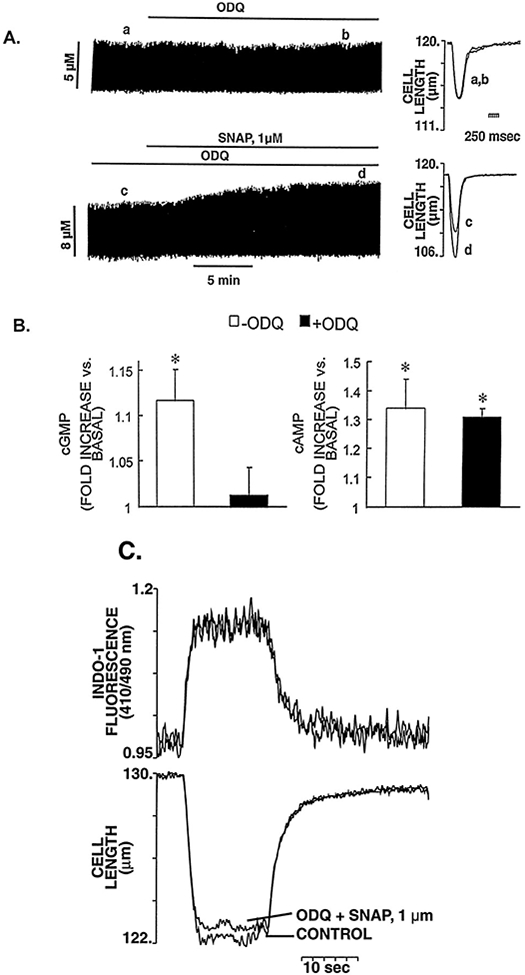
Failure of ODQ 10 μmol/L to block the positive contractile response of SNAP 1 μmol/L. A, Typical continuous chart recording of contraction amplitude in response to ODQ (10 μmol/L) alone (top tracing) and SNAP (1 μmol/L) in the continued presence of ODQ (10 μmol/L) (bottom tracing). The superimposed tracings of cell length on the right show the lack of effect of ODQ (10 μmol/L) on baseline contraction (top) and the increase of contraction amplitude in response to a low concentration of SNAP (1 μmol/L) in the continued presence of ODQ (10 μmol/L) (bottom). B, Parallel measurements of cGMP and cAMP in response to SNAP (1 μmol/L) in the presence and absence of ODQ. The results are presented as fractions of the basal values of cGMP and cAMP, 4.2±0.2 and 6.2±0.4 pmol/mg of protein, respectively. ODQ (10 μmol/L) alone did not affect basal cGMP or cAMP levels (4.1±0.2 and 5.2±0.7 pmol/mg of protein, respectively). ODQ completely abolished the SNAP-induced increase in cGMP but had no effect on increased cAMP levels. *P<0.05 vs basal level. C, Representative example of the lack of effect of SNAP (1 μmol/L) in the presence of ODQ (10 μmol/L) on the steady-state myofilament response to Ca2+ of a myocyte during tetanic contractions (10 Hz). This indicates that the positive contractile effect seen in panel A is purely the result of the increase in magnitude of the Cai.
The ability of ODQ to effectively inhibit cGMP production or to affect cAMP levels in response to SNAP was assessed in a parallel group of experiments in which cGMP and cAMP levels were measured in intact cardiac myocytes in the presence and absence of ODQ (10 μmol/L). That treatment with ODQ (10 μmol/L) was fully effective to block GC in these cardiac myocyte experiments is demonstrated by the fact it completely abolished the cGMP increase seen with 100 μmol/L SNAP: specifically, SNAP (100 μmol/L) alone increased basal cGMP (4.2±0.2 pmol/mg of protein) by 131±6%, whereas cGMP remained 98±4% of basal in the presence of SNAP (100 μmol/L)+ODQ (P<0.05). Figure 5B shows that ODQ completely abolished the increase in cGMP induced by SNAP 1 μmol/L, as expected, but did not affect the increased cAMP levels. Similar effects of ODQ on cGMP and cAMP levels were observed when higher concentrations of SNAP were used (data not shown). Moreover, preincubation of cardiac myocytes with ODQ was capable of transforming an otherwise typical negative inotropic response of a high dose of SNAP (10 μmol/L) to a sustained positive inotropic response (Figure 6). These results indicate that low concentrations of NO are able to increase the Cai and contraction in intact cardiac myocytes in a cGMP-independent fashion. Although previous reports have suggested that the positive contractile response induced by NO donors could be the result of an elevation of the intracellular levels of cAMP (and PKA activation) due to cGMP-dependent inhibition of PDE type III (cGMP-inhibited phosphodiesterase or cGI-PDE),8,9 our results indicate the presence of an important, additional pathway whereby NO donors may increase cAMP and exert a substantial positive inotropic action, ie, through a GC/cGMP-independent mechanism. Indeed, Figure 7 shows that even in the presence of IBMX (0.1 mmol/L), resulting in a background of tonically inhibited PDEs (including PDE type III), SNAP (1 μmol/L) can still exert a positive inotropic effect through an increase of the Cai, supporting the concept that the NO-mediated positive inotropy seen in these experiments can occur, at least in part, via PDE-independent mechanisms (representative of 4 cells). Because SNAP was able to elevate basal cAMP even when GC was completely inhibited by ODQ (10 μmol/L) in these cardiac myocytes, we hypothesized that the increase in cAMP and the positive inotropic effect observed with low concentrations of SNAP in the presence of ODQ could be attributed to a cGMP-independent activation of the AC/cAMP/PKA pathway by NO.
Figure 6.
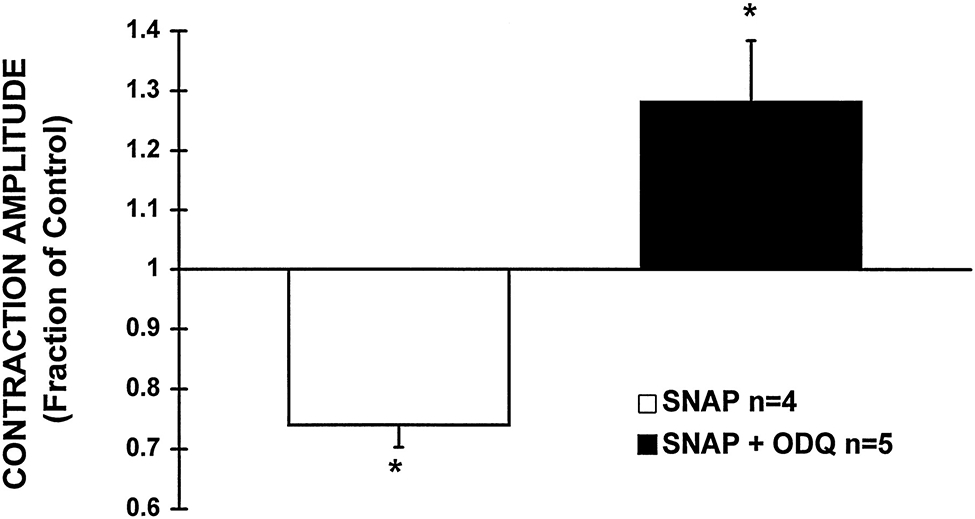
Inhibition of GC with ODQ (10 μmol/L) converts a negative contractile response of SNAP (10 μmol/L) to a sustained positive response. The bar graph depicts the average change in contraction amplitude, expressed as a fraction of the basal contraction, after 20 minutes of exposure of cells to SNAP (10 μmol/L) either in the absence or presence of ODQ (10 μmol/L). SNAP alone significantly attenuated contraction amplitude by 26±4%, whereas in the presence of ODQ, SNAP induced a pronounced increase in contraction, 28±10% above baseline. *P<0.05 vs basal level.
Figure 7.
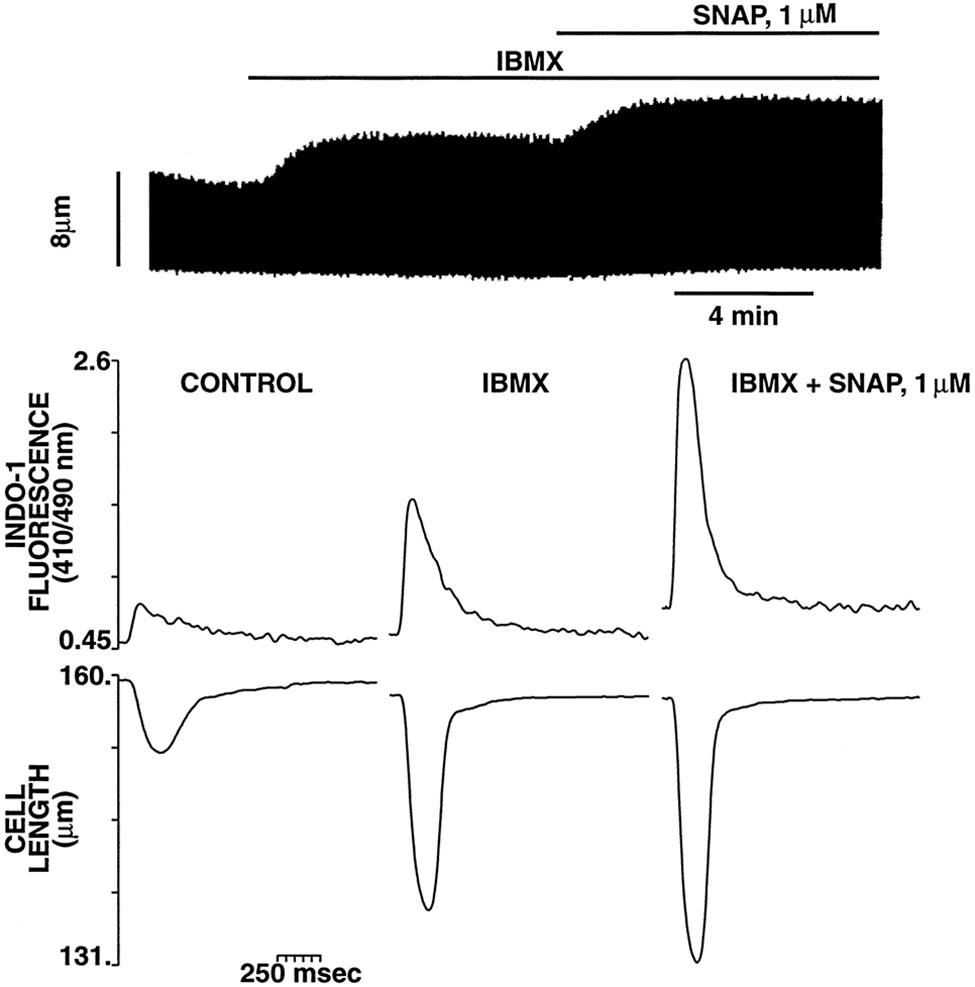
Tonic PDE inhibition with IBMX does not prevent the positive contractile response of SNAP. Top, Typical continuous chart recording of contraction amplitude in response to IBMX (0.1 mmol/L) followed by SNAP (1 μmol/L) in the continued presence of IBMX. Bottom, Representative example of the effect of IBMX and SNAP (same protocol as in the top panel) on contraction and [Ca2+]i from a different myocyte. Each tracing is taken at steady state under the condition listed. Note that even in the presence of IBMX, SNAP can still exert a positive inotropic effect via an increase of the Cai.
Effect of SNAP on AC Activity
Figure 8 illustrates the dose-response curve for the effect of SNAP on AC activity measured on cardiac sarcolemmal membranes. Basal AC activity was 92±6 pmol/mg of protein per minute. Low concentrations of SNAP (0.1 and 1 μmol/L, which produced 150 nmol/L and 500 nmol/L NO, respectively) showed significant increases of AC activity (19±6% and 16±4% above basal activity, respectively; P<0.05). At higher SNAP concentrations, AC activity was unchanged (at 10 μmol/L SNAP, which produced 1.4 μmol/L NO) or reduced (by 14±6% at 100 μmol/L SNAP, which produced 14.7 μmol/L NO). The increases of AC activity at low concentrations of SNAP (0.1 and 1 μmol/L) were 34% to 40% of the maximal Mn2+-stimulated AC activity.
Figure 8.
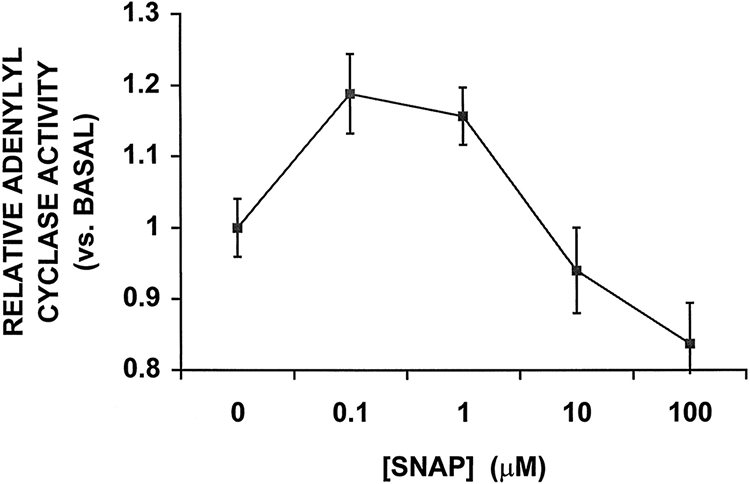
Effects of SNAP on AC activity in rat cardiac sarcolemmal membranes. AC activity of membranes prepared from rat heart was determined under basal conditions and in the presence of different concentrations of SNAP. SNAP produced a biphasic response in AC activity in a concentration-dependent fashion. Low concentrations of SNAP (0.1 to 1 μmol/L) were associated with significant increases in AC activity (P<0.05 vs control), whereas higher concentrations of SNAP resulted in either no change (10 μmol/L) or a small decrease (100 μmol/L) in AC activity. Average AC activity is expressed as a fraction of the basal activity (92±6 pmol/mg of protein per minute). Each point represents the mean±SEM of 9 independent experiments performed in triplicate. The increases in AC activity at 0.1 and 1 μmol/L SNAP were 40% and 34% of maximal Mn2+-stimulated AC activity, respectively.
Effect of SNAP in the Presence of Inhibitors of GC and PKA
To confirm that the persistent positive contractile response induced by low concentrations of SNAP in the absence of GC activation was mediated through cAMP-dependent PKA activation, experiments were performed using the cAMP analog Rp-8-CPT-cAMPS previously shown to effectively and specifically inhibit PKA-dependent processes.37 All cells were preincubated with Rp-8-CPT-cAMPS 100 μmol/L (at 37°C) for at least 1 hour before the experiment. In the continuous presence of the inhibitory cAMP analog Rp-8-CPT-cAMPS (100 μmol/L) and ODQ (10 μmol/L), low concentrations of SNAP were unable to elicit the positive inotropic response previously observed with SNAP alone. Figures 9A and 9B show representative examples of the failure of SNAP (1 μmol/L) to increase the contraction (or the associated Cai, as in Figure 9A) of intact rat cardiac myocytes continuously perfused with Rp-8-CPT-cAMPS with and without ODQ, respectively (similar results were observed in 4 other preparations). Similarly, NOC5 (1 μmol/L) also fails to elicit an increase in contraction in the presence of Rp-8-CPT-cAMPS (±ODQ, Figure 9C and 9D, respectively). These results demonstrate that the positive inotropic effect of low concentrations of NO is, at least in part, independent of cGMP and is mediated by increased cAMP and PKA activation. In these same cells, as well as in parallel experiments, the ability of Rp-8-CPT-cAMPS to successfully inhibit PKA activation was routinely confirmed by its efficacy to inhibit the positive inotropic effect induced by isoproterenol. The mean increase in contraction amplitude induced by 1 nmol/L isoproterenol was 171±10% of the control value (n=4 cells), which was completely abolished by Rp-8-CPT-cAMPS (94±5% of control value; n=4 cells).
Figure 9.
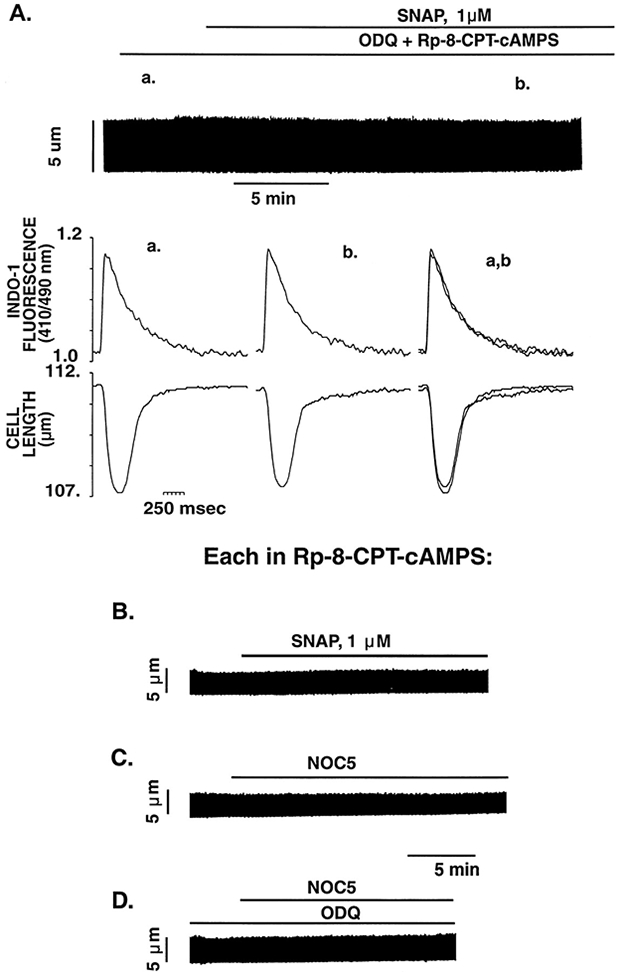
PKA inhibition with Rp-8-CPT-cAMPS (100 μmol/L) completely abolishes the positive contractile responses of SNAP and NOC5. A, Representative example of the effect of SNAP (1 μmol/L) in the continued presence of inhibitors of GC (ODQ 10 μmol/L) and PKA (Rp-8-CPT-cAMPS 100 μmol/L) on myocyte contraction and [Ca2+]i. The continuous chart recording shows the failure of SNAP (1 μmol/L) to increase the isotonic twitch contraction in a cardiac myocyte pretreated and continuously perfused with ODQ and Rp-8-CPT-cAMPS. The bottom tracings show, on an expanded time scale, [Ca2+]i and cell length at the times indicated (a, baseline; b, 10 minutes after addition of SNAP; and c, 15 to 20 minutes after SNAP). Under these conditions, SNAP had no significant effect on either contraction amplitude or Cai amplitude. Representative examples of the lack of effect on myocyte contraction of SNAP (1 μmol/L) (B), NOC5 (1 μmol/L) (C), and NOC5 (1 μmol/L) pretreated with the GC inhibitor ODQ (10 μmol/L) (D), each in the continued presence of the inhibitor of PKA (Rp-8-CPT-cAMPS 100 μmol/L).
Discussion
The findings in the present study indicate that the exogenous administration of the NO donor SNAP can induce opposing effects on myocyte contraction, in agreement with recent reports.8,9 Additional experiments using NOC5, an NO donor chemically distinct from SNAP, confirm these observations. Although the direction of the myocardial response clearly depended on the concentration of the NO donor used and on the level of intracellular cGMP reached, the principal novel finding of the present study is that a previously unrecognized GC/cGMP-independent mechanism involving the activation of AC is also critical in mediating this response.
In rat cardiac myocytes, high concentrations of SNAP (producing ≈14.7 μmol/L NO) induced large increases in cGMP and reduced myocyte contraction, whereas low concentrations of SNAP (producing in the range of ≈0.1 to 0.5 μmol/L NO) induced only moderate increases in cGMP and enhanced myocyte contraction. Furthermore, these contractile effects were not induced by the thiol byproduct of SNAP, NAP, after NO release. Similarly, concentrations of NOC5 producing ≈0.5 μmol/L NO also enhanced myocyte contraction. One can deduce that these contractile effects with SNAP and NOC5 are authentic effects of NO, because they are completely reversible in the presence of the NO scavengers oxyhemoglobin and carboxy-PTIO. More importantly, our results show that whereas a cGMP/PKG-mediated diminished sensitivity of the contractile proteins to Ca2+ is largely responsible for the negative inotropic effect at higher SNAP concentrations, the positive contractile response observed with low concentrations of SNAP is determined primarily by a cAMP/PKA-dependent, but GC/cGMP-independent, increase in the Cai. Although recent reports have addressed the wide range of actions of NO (and of NO donors) on myocardial contractility,3-9 the subcellular mechanisms involved in the opposing contractile responses have not been completely elucidated.
In frog cardiac myocytes and in human atrial cells, cGMP has been shown to decrease Ca2+ influx through L-type channels by the stimulation of PDE type II (cAMP phosphodiesterase).38,39 In rat cardiac myocytes, however, cGMP has been shown to modulate L-type channel activity through a PKG-dependent pathway that does not involve changes in cAMP levels.40,41 Because Ca2+ entry through L-type Ca2+ channels is the principal trigger for Ca2+ release from the sarcoplasmic reticulum, the inactivation of L-type channels by NO/cGMP should have a significant effect on the amount of Ca2+ released to the myofilaments and therefore contribute to the negative inotropic response. This seems not to be an important mechanism in our model system, in which the administration of SNAP (100 μmol/L) produced a slowly evolving negative inotropic effect that was not associated with a reduction in the Ci. Furthermore, in steady-state tetanic contraction experiments, SNAP (100 μmol/L) significantly decreased cell shortening during the tetanus, despite the achievement of similar peak [Ca2+]i levels compared with controls. Taken together, these results indicate that the negative inotropic response to high concentrations of SNAP, in isolated rat cardiac myocytes, is mediated primarily by a reduction in the myofilament sensitivity to Ca2+ rather than by a reduced Ca2+ availability at the myofibrils.
In rat cardiac myocytes, administration of relatively high concentrations of the cGMP analog 8-bromo-cGMP (50 μmol/L) produced strikingly similar effects to those observed with high concentrations of SNAP (ie, a comparable negative inotropic response without any change of the Cai).21 Evidence for a cGMP-mediated reduction in myofilament responsiveness to Ca2+ has also been demonstrated in studies performed on skinned cardiac fibers.42 Previous reports also show that PKG activation induces a rightward shift of the tension/pCa relation in skinned single rat ventricular myocytes, compatible with a reduction in Ca2+ sensitivity of troponin C,43 and that PKG mediates phosphorylation of troponin I at the same site as that phosphorylated by protein kinase A, a mechanism known to reduce the affinity of troponin C for Ca2+.44,45 The precise underlying mechanism for the reduced myofilament responsiveness to Ca2+ induced by high concentrations of SNAP and increased cGMP was not directly addressed in our experiments. However, the ability of the PKG inhibitor KT 5823 to completely abolish the decrease in intact myocyte shortening observed during the tetanus in the presence of SNAP (100 μmol/L) (as it was also shown to do in this model in the presence of 8-bromo-cGMP21) provides strong support for the concept that SNAP-induced PKG activation results in reduced myofilament responsiveness to Ca2+.
Although other reports have indicated that positive contractile responses are induced by low concentrations of NO donors and cGMP,8,9,22,46,47 the subcellular mechanisms involved have received relatively little attention. Some evidence suggests that the positive contractile response induced by NO donors could be the result of an elevation of the intracellular levels of cAMP (and PKA activation) due to a cGMP-dependent inhibition of the PDE type III (ie, cGMP-inhibited PDE).8,9 Furthermore, low concentrations of cGMP (0.1 to 10 μmol/L) as well as of the NO donor SIN-1 (0.1 to 10 nmol/L) were found to have a stimulatory effect on L-type Ca2+ current.15,48 It seems likely, therefore, that the inhibition of cAMP degradation, mediated by the inactivation of the PDE type III after low cGMP levels accumulate, could result in the cAMP/PKA-dependent stimulation of transmembrane Ca2+ influx, increasing the trigger for the Ca2+-induced Ca2+ release process, and producing a larger Cai and myocyte contraction. Nevertheless, in the current experiments, SNAP (1 μmol/L) was still capable of eliciting a pronounced positive contractile response (via increasing the Cai) in the presence of a background of tonically inhibited PDEs (via IBMX pretreatment; Figure 7). In this setting, it seems unlikely that cGMP accumulation, per se, could mediate additional PDE (type III) inhibition (and cAMP accumulation) beyond that tonically present, owing to the presence of IBMX, and this leads to the notion that the NO-mediated positive inotropy can occur, at least in part, via PDE-independent mechanisms.
However, a decrease in PDE activity is only one of the two possible major mechanisms capable of increasing cAMP in intact cells, the other being the “upstream” activation of AC. Our results show that the positive contractile response of SNAP (1 μmol/L) is indeed associated with a significant elevation of both basal cAMP and cGMP. Nevertheless, even when this basal cGMP increase was completely abolished by the presence of the selective inhibitor of soluble GC (ODQ) in the bathing solution (see Figure 5B), SNAP (1 μmol/L) was still able to induce a positive inotropic effect that was mediated by cAMP (Figures 5A and 9). In these settings, cAMP must be increased by a mechanism other than cGMP-mediated PDE inhibition, in all likelihood by a cGMP-independent activation of AC.
To evaluate the possible activation of AC by the NO donor SNAP, we determined AC activity in isolated sarcolemmal membranes from rat heart. SNAP induced a concentration-dependent biphasic response of AC activity, significantly increasing its activity at low doses (0.1 to 1 μmol/L) and either not changing (10 μmol/L) or marginally decreasing the activity at high doses (100 μmol/L). Consistent with our findings, recent studies provide evidence suggesting that NO released from endothelial cells can directly or indirectly activate AC in a cGMP-independent manner in isolated perfused rat kidneys.49 From the present experiments, we cannot ascertain whether NO activates AC directly or through a G protein (Gs/Gi)–dependent pathway. Indeed, recent reports have demonstrated that NO can modulate G protein function. Lander et al50 have demonstrated that treatment of intact human peripheral blood mononuclear cells with NO yielded membranes with enhanced GTPase activity, and that NO similarly enhanced the GTPase activity of pure, recombinant Gsα, Giαi, and p21ras. Miyamoto et al51 found in endothelial cells that NO can selectively inhibit G proteins of the Gi and Gq family but not those of the Gs family, and that this modulation of G proteins could have a permissive action on the Gs-AC pathway. Thus, investigation of this question of the potential modulation of G protein signaling by NO in cardiac myocytes should prove directly applicable to the issue of how NO mediates the activation of AC and the positive inotropic effect.
The final remaining possibility to explain changes in AC activation in these experiments is that NO may modulate the activity of the β-adrenergic receptor (βAR) itself. It is noteworthy that the cAMP/PKA-mediated positive contractile effect of low concentrations of NO was associated neither with changes in the Cai duration nor a lusitropic effect. Although this lack of effect on contraction and Cai kinetics does not resemble the typical abbreviation of these parameters observed on β1-adrenergic receptor (β1AR) stimulation (which is also mediated by cAMP/PKA activation), on the other hand, it closely resembles the effects of another cAMP/PKA signaling cascade activated by the specific stimulation of β2-adrenergic receptors β2ARs).52 Like the NO-induced positive inotropic effect, β2AR stimulation produces a slowly evolving increase in contraction amplitude (as compared with β1AR stimulation) without affecting the kinetics of contraction or the Cai.52 The differential regulation of cardiac excitation-contraction coupling by β1AR and β2AR stimulation has been attributed to a unique functional compartmentalization of cAMP.37 Thus, it is tempting to speculate that a simple and unifying explanation for all the experimental results at low NO concentrations involves the specific activation of β2ARs, or perhaps of G proteins specifically coupled to them, but this will require experimental verification.
The nature of the regulation of protein function by NO is an area of active investigation. Recent evidence suggests that thiols in proteins can recognize both nitrosative and oxidative events, which in turn may elicit distinct functional changes.27-29 For example, the extent of reversible poly-N-nitrosylation of thiol groups (R-S-NO) by NO was shown to correlate with the degree of activation of the cardiac Ca2+ release channel (ryanodine receptor), and this mode of posttranslational sulfhydryl modification has been proposed as a general model of NO-mediated regulation of protein function.27,29 These mechanisms could plausibly operate at each of the effector proteins potentially involved in the NO-stimulated production of cAMP in these experiments, including the β-ARs, G proteins, and AC, each of which has multiple potential sites available for S-nitrosylation (ie, thiols of the many cysteine residues). Indeed, posttranslational modification of certain cysteines on these proteins has been shown to play an important role in several hormone signal transduction pathways, including the βAR system. For example, labile thioesterification via palmitoylation of a specific cysteine in the β2ARs (and many other G protein–coupled receptors), as well as Gαs, and the agonist-promoted dynamic turnover of the binding of this moiety, has been shown to influence the ability of these effectors to regulate the activation of AC.53,54 Thus, competition for these thiol sites by NO could exert significant regulatory effects on AC activation independently of, or in cooperation with, the usual agonist induction mechanisms.
Pinsky et al11 showed that endogenous cardiac NO levels achieve dynamic, beat-to-beat oscillations on the order of micromolar magnitude, and that this phenomenon is amplified by ventricular loading, which could potentially serve an autoregulatory function. Indeed, it has recently been shown that intracardiac production and release of NO significantly augments the Frank-Starling response.26 These physiological NO levels in heart are thus comparable to that achieved in the present experiments using 0.1 to 1 μmol/L SNAP or 1 μmol/L NOC5 (≈0.1 to 0.5 μmol/L NO) and associated with a positive inotropic effect. If the rate of denitrosylation of R-S-NO is sufficiently slow (ie, on the time scale of the cardiac cycle), then it is possible that these physiological NO oscillations achieve a functional integration or steady-state bound level, which in turn, could proportionately regulate the function of the effector protein. Such proportionate control of protein function via the degree of protein S-nitrosylation has been demonstrated for the cardiac ryanodine receptor, for example.29 It is thus possible that the activation of AC demonstrated in these experiments at levels of NO compatible with that achieved by the beating heart could indeed serve a physiological autoregulatory role. Notably, it has been shown that both myocardial cAMP55-58 and PKA55 activities oscillate during the cardiac cycle (their peaks coinciding with mechanical systole), and this phenomenon could thus be due, in part, to the NO-AC activation mechanism discussed here. Still, a potential caveat should be raised insofar as the current experiments were conducted at 25°C (and the contraction experiments at 0.5-Hz electrical stimulation rate) and thus may not be directly relevant to normal heart.
The exact nature for the biphasic activation pattern of AC by SNAP cannot be elucidated from our study. However, it seems reasonable to speculate that in isolated membranes, in which the NO buffering capacity of myoglobin59,60 is absent, NO released by high doses of SNAP would achieve substantially higher levels in the local environment of the membrane-bound AC than in intact cells (yielding an apparent leftward shift in the dose-response curve in isolated membranes compared with in vivo) and thus have nonspecific toxic (or inactivating) rather than activating effects on AC activity. Lander et al,50 when measuring GTPase activity induced by NO in isolated human peripheral blood mononuclear cell membranes, observed a similar biphasic response of enzyme activity (ie, activation with low concentrations of NO and inhibition with high concentrations of NO), attributing the inhibition that occurred at high doses to alteration of target proteins by nonspecific oxidation of thiols and tyrosine residues leading to enzyme inactivation.
In summary, the present results demonstrate that exogenously applied NO generated spontaneously by the donor SNAP, when given at high concentrations, can produce a negative inotropic effect that is mediated by a cGMP-dependent PKG activation resulting in the reduction in myofilament responsiveness to Ca2+ but not by decreased Ca2+ availability. Low concentrations of NO, on the contrary, evoke a positive inotropic effect determined primarily by a cAMP-dependent increase in the Cai by a novel mechanism whereby NO increases cAMP levels in intact rat ventricular myocytes by a cGMP-independent activation of AC.
Table 1.
Effect of SNAP on Contraction and Ca2+ Transient Parameters of Single Rat Myocytes
| TA (% of Resting Cell Length) |
t1/2 Contraction, ms |
IFT Amplitude (410/490 nm) |
t1/2 IFT, ms |
|
|---|---|---|---|---|
| n | 5 | 5 | 5 | 5 |
| Control | 5.7±0.3 | 354±14 | 0.168 ±0.01 | 260±15 |
| SNAP 1 μmol/L | 7.9±0.61 | 350 ±14 | 0.211±0.021 | 266±11 |
| --------------------------------------------------------------------------- | ||||
| Control | 6.1 ±0.4 | 340±10 | 0.155±0.03 | 264±18 |
| SNAP 100 μmol/L | 4.3±0.31 | 311±121 | 0.153±0.02 | 256±15 |
TA indicates twitch amplitude; t1/2 contraction, half-relaxation time of contraction; IFT amplitude, indo 1 fluorescent transient, an index of the Ca2+ transient amplitude; and t1/2 IFT, 50% relaxation of indo 1 fluorescent transient.
Values are mean±SEM.
Significant change vs control value (P<0.05).
We acknowledge the expert technical assistance of Dr Xiaolin Wang, who performed the cAMP and cGMP assays.
Acknowledgment
We acknowledge the expert technical assistance of Dr Xiaolin Wang, who performed the cAMP and cGMP assays.
Contributor Information
Martin G. Vila-Petroff, Laboratory of Cardiovascular Science, Intramural Research Program, National Institute on Aging, Gerontology Research Center, Baltimore, Md.
Antoine Younes, Laboratory of Cardiovascular Science, Intramural Research Program, National Institute on Aging, Gerontology Research Center, Baltimore, Md..
Josephine Egan, Laboratory of Clinical Investigation, Intramural Research Program, National Institute on Aging, Gerontology Research Center, Baltimore, Md..
Edward G. Lakatta, Laboratory of Cardiovascular Science, Intramural Research Program, National Institute on Aging, Gerontology Research Center, Baltimore, Md.
Steven J. Sollott, Laboratory of Cardiovascular Science, Intramural Research Program, National Institute on Aging, Gerontology Research Center, Baltimore, Md.
References
- 1.Moncada S, Palmer RMJ, Higgs EA. Nitric oxide: physiology, pathology, and pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 2.Ignarro LJ, Kadoitz PJ. The pharmacological role of cyclic GMP in vascular smooth muscle relaxation. Annu Rev Pharmacol Toxicol. 1985;25:171–191. [DOI] [PubMed] [Google Scholar]
- 3.Brady AJB, Warren JB, Poole-Wilson PA, Williams TJ, Harding SE. Nitric oxide attenuates cardiac myocyte contraction. Am J Physiol. 1993;265:H176–H182. [DOI] [PubMed] [Google Scholar]
- 4.Meulemans AL, Sipido KR, Sys SU, Brutsaert DL. Atriopeptin III induces early relaxation of ventricular cardiac muscle. Circ Res. 1988;62:1171–1174. [DOI] [PubMed] [Google Scholar]
- 5.Smith JA, Shah AM, Lewis MJ. Factors released from endocardium of the ferret and pig modulate myocardial contraction. J Physiol (Lond). 1991;439:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weyrich AS, Ma XL, Buerke M, Murohara T, Armstead VE, Lefer AM, Nicolas JM, Thomas AP, Lefer DJ, Vinten-Johansen J. Physiological concentrations of nitric oxide do not elicit an acute negative inotropic effect in unstimulated cardiac muscle. Circ Res. 1994;75:692–700. [DOI] [PubMed] [Google Scholar]
- 7.Crystal GJ, Gurevicius J. Nitric oxide does not modulate myocardial contractility acutely in situ canine hearts. Am J Physiol. 1996;270:H156–H157. [DOI] [PubMed] [Google Scholar]
- 8.Mohan P, Brutsaert DL, Paulus WJ, Stanislas U. Myocardial contractile response to nitric oxide and cGMP. Circulation. 1996;93:1223–1229. [DOI] [PubMed] [Google Scholar]
- 9.Kodja G, Kottenberg K, Nix P, Schluter KD, Piper HM, Noack E. Low increases in cGMP induced by organic nitrates and nitrovasodilators improves contractile response of rat ventricular myocytes. Circ Res. 1996;78:91–101. [DOI] [PubMed] [Google Scholar]
- 10.Shah AM. Paracrine modulation of heart cell function by endothelial cells. Cardiovasc Res. 1996;31:847–867. [PubMed] [Google Scholar]
- 11.Pinsky DJ, Patton S, Mesaros S, Brovkovych V, Kubaszewski E, Grunfeld S, Malinski T. Mechanical transduction of nitric oxide synthesis in the beating heart. Circ Res. 1997;81:372–379. [DOI] [PubMed] [Google Scholar]
- 12.Balligand JL, Kelly RA, Marsden PA, Smith TW, Michel T. Control of cardiac muscle cell function by an endogenous nitric oxide signaling system. Proc Natl Acad Sci U S A. 1993;90:347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balligand JL, Kobzic L, Han X, Kaye DM, Belhassen L, O’Hara DS, Kelly RA, Smith TW, Michel T. Nitric oxide-dependent parasympathetic signaling is due to activation of constitutive endothelial (type III) nitric oxide synthase in cardiac myocytes. J Biol Chem. 1995;270:14582–14586. [DOI] [PubMed] [Google Scholar]
- 14.Fischmeister R, Shier A. Interactive effects of isoprenaline, forskolin, and acetylcholine on Ca2+ current in frog ventricular myocytes. J Physiol (Lond). 1989;417:213–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Méry PF, Pavoine C, Belhassen L, Pecker F, Fischmeister R. Nitric oxide regulates cardiac Ca2+ current: involvement of cGMP-inhibited and cGMP-stimulated phosphodiesterases through guanylyl cyclase activation. J Biol Chem. 1993;268:26286–26295. [PubMed] [Google Scholar]
- 16.Han X, Shimoni Y, Giles WR. A cellular mechanism for nitric oxide-mediated cholinergic control of mammalian heart rate. J Gen Physiol. 1995;106:45–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levi RC, Alloatti G, Fischmeister R. Cyclic GMP regulates the Ca-channel current in guinea pig ventricular myocytes. Pflugers Arch. 1989;413:685–687. [DOI] [PubMed] [Google Scholar]
- 18.Méry PF, Lohman SM, Walter U, Fischmeister R. Ca2+ current is regulated by cyclic GMP-dependent protein kinase in mammalian cardiac myocytes. Proc Natl Acad Sci U S A. 1991;88:1197–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herzig S, Patil P, Neumann J, Staschen CM, Yue DT. Mechanisms of β-adrenergic stimulation of cardiac Ca2+ channels revealed by discrete-time Markov analysis of slow gating. Biophys J. 1993;5:1599–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wahler GM, Dollinger SJ. Nitric oxide donor SIN-1 inhibits mammalian cardiac calcium current through cGMP-dependent protein kinase. Am J Physiol 1995;268:C45–C54. [DOI] [PubMed] [Google Scholar]
- 21.Shah AM, Spurgeon HA, Sollott SJ, Talo A, Lakatta EG. 8-Bromo-cGMP reduces the myofilament response to Ca2+ in intact cardiac myocytes. Circ Res. 1994;74:970–978. [DOI] [PubMed] [Google Scholar]
- 22.Watanabe AM, Besch HR Jr. Interaction between cyclic adenosine monophosphate and cyclic guanosine monophosphate in guinea pig ventricular myocardium. Circ Res. 1975;37:309–317. [DOI] [PubMed] [Google Scholar]
- 23.Linden J, Brooker G. The questionable role of cyclic guanosine 3′:5′-monophosphate in heart. Biochem Pharmacol. 1979;28:3351–3360. [DOI] [PubMed] [Google Scholar]
- 24.Brooker G. Dissociation of cyclic GMP from the negative inotropic action of carbachol in guinea pig atria. J Cyclic Nucleotide Res. 1977;3:407–413. [PubMed] [Google Scholar]
- 25.Diamond J, Chu EB. A novel cyclic GMP-lowering agent, LY83583, blocks carbachol-induced cyclic GMP elevation in rabbit atrial strips without blocking the negative inotropic effects of carbachol. Can J Physiol Pharmacol. 1985;63:908–911. [DOI] [PubMed] [Google Scholar]
- 26.Prendergast BD, Sagach VF, Shah AM. Basal release of nitric oxide augments the Frank-Starling response in the isolated heart. Circulation. 1997;96:1320–1329. [DOI] [PubMed] [Google Scholar]
- 27.Stamler JS. Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell. 1994;78:931–936. [DOI] [PubMed] [Google Scholar]
- 28.Stoyanovsky D, Murphy T, Anno PR, Kim YM, Salama G. Nitric oxide activates skeletal and cardiac ryanodine receptors. Cell Calcium. 1997;21:19–29. [DOI] [PubMed] [Google Scholar]
- 29.Xu L, Eu JP, Meissner G, Stamler JS. Activation of cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. [DOI] [PubMed] [Google Scholar]
- 30.Sollott SJ, Ziman B, Lakatta EG. Nitric oxide (NO) modulation of cardiac myocyte signaling and contraction. Circulation. 1995;92(suppl S):2412. Abstract. [Google Scholar]
- 31.Sollott SJ, Ziman B, Lakatta EG. Activation of distinct cAMP- and cGMP-dependent pathways by nitric oxide (NO) in cardiac myocytes. J Mol Cell Cardiol. 1996;28:A126. Abstract. [Google Scholar]
- 32.Capogrossi MC, Kort AA, Spurgeon HA, Lakatta EG. Single adult rabbit and rat cardiac myocytes retain the Ca2+- and species-dependent systolic and diastolic contractile properties of intact muscle. J Gen Physiol. 1986;88:589–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spurgeon HA, Stern MD, Baartz G, Raffaeli S, Hansford RG, Talo A, Lakatta EG, Capogrossi MC. Simultaneous measurement of Ca2+, contraction, and potential in cardiac myocytes. Am J Physiol. 1990;258:574–586. [DOI] [PubMed] [Google Scholar]
- 34.Sollott SJ, Lakatta EG. Secondary [Ca2+]i-dependent modulation of contractility in single cardiac myocytes. Circulation. 1992;86:1–749. Abstract.1617762 [Google Scholar]
- 35.Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc Natl Acad Sci U S A. 1990;87:2466–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garthwaite J, Southam E, Boulton CL, Nielsen EB, Schmidt K, Mayer B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol Pharmacol. 1995;48:184–188. [PubMed] [Google Scholar]
- 37.Zhou YY, Cheng H, Bogdanov KY, Hohl C, Altschuld R, Lakatta EG, Xiao RP. Localized cAMP-dependent signaling mediates β2-adrenergic modulation of cardiac excitation-contraction coupling. Am J Physiol. 1997;273:H1611–H16118. [DOI] [PubMed] [Google Scholar]
- 38.Lohmann SM, Fischmeister R, Walter U. Signal transduction by cGMP in heart. Basic Res Cardiol. 1991;86:503–514. [DOI] [PubMed] [Google Scholar]
- 39.Kirstein M, Rivet-Bastide M, Hatem S, Bernardeau A, Mercadier JJ, Fischmeister R. Nitric oxide regulates the calcium current in isolated human atrial myocytes. J Clin Invest. 1995;95:794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walter U. Physiological role of cGMP- and cGMP-dependent protein kinase in the cardiovascular system. Rev Physiol Biochem Pharmacol. 1989;113:41–88. [DOI] [PubMed] [Google Scholar]
- 41.Wahler GM, Dollinger SJ. Nitric oxide donor SIN-1 inhibits mammalian cardiac calcium current through cGMP-dependent protein kinase. Am J Physiol. 1995;268:C45–C54. [DOI] [PubMed] [Google Scholar]
- 42.Pfitzer G, Rüegg JC, Flockerzi V, Hofmann F. cGMP-dependent protein kinase decreases calcium sensitivity of skinned cardiac fibers. FEBS Lett. 1982;149:171–175. [DOI] [PubMed] [Google Scholar]
- 43.Clement O, Puceat M, Vassort G. Protein kinases modulate Ca sensitivity of cardiac myofilaments in rat skinned cells. J Physiol (Lond). 1991;438:96P. Abstract. [Google Scholar]
- 44.Blumenthal DK, Stull JT, Gill GN. Phosphorylation of cardiac troponin by guanosine 3′:5′-monophosphate-dependent protein kinase. J Biol Chem. 1978;253:334–336. [PubMed] [Google Scholar]
- 45.Lincoln TM, Corbin JD. Purified cyclic GMP-dependent protein kinase catalyzes the phosphorylation of cardiac troponin inhibitory subunit (TN-I). J Biol Chem. 1978;253:337–339. [PubMed] [Google Scholar]
- 46.Endoh M, Shimizu T. Failure of dibutyryl-cAMP and 8-bromo-cGMP to mimic the antagonistic actions of carbachol on the positive inotropic effects of sympathomimetic amines in the canine isolated ventricular myocardium. Jpn J Pharmacol. 1979;29:423–433. [DOI] [PubMed] [Google Scholar]
- 47.Kodja G, Kottenberg K, Stasch JP. Positive inotropic effect of exogenous and endogenous NO in hypertrophic rat hearts. Br J Pharmacol. 1997;122:813–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ono K, Trautwein W. Potentiation by cyclic GMP of β-adrenergic effect on Ca2+ current in guinea-pig ventricular cells. J Physiol (Lond). 1991;443:387–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heuze-Joubert I, Mennecier P, Simonet S, Laubie M, Verbeuren TJ. Effect of vasodilators, including nitric oxide, on the release of cGMP and cAMP in the isolated perfused rat kidney. Eur J Pharmacol. 1992;220:161–171. [DOI] [PubMed] [Google Scholar]
- 50.Lander HM, Sehajpal PK, Novogrodsky A. Nitric oxide signaling: a possible role for G proteins. J Immunol. 1993;151:7182–7187. [PubMed] [Google Scholar]
- 51.Miyamoto A, Laufs U, Pardo C, Liao JK. Modulation of bradykinin receptor ligand binding affinity and its coupled G-proteins by nitric oxide. J Biol Chem. 1997;272:19601–19608. [DOI] [PubMed] [Google Scholar]
- 52.Xiao R-P, Lakatta EG. β1-Adrenoceptor stimulation and β2-adrenoceptor stimulation differ in their effect on contraction, cytosolic Ca2+, and Ca2+ current in single rat ventricular cells. Circ Res. 1993;73:286–300. [DOI] [PubMed] [Google Scholar]
- 53.Moffett S, Adam L, Bonin H, Loisel TP, Bouvier M, Mouillac B. Palmitoylated cysteine 341 modulates phosphorylation of the β2-adrenergic receptor by the cAMP-dependent protein kinase. J Biol Chem. 1996;271:21490–21497. [DOI] [PubMed] [Google Scholar]
- 54.Ammer H, Schulz R. Enhanced stimulatory adenylyl cyclase signaling during opioid dependence is associated with a reduction in palmitoylated Gsα. Mol Pharmacol. 1997;52:993–999. [DOI] [PubMed] [Google Scholar]
- 55.Krause EG, Bartel S, Beyerdorfer I, Freier W, Gerber K, Obst D. Transient changes in cyclic AMP and in the enzymic activity of protein kinase and phosphorylase during the cardiac cycle in the canine myocardium and the effect of propranolol. Mol Cell Biochem. 1989;89:181–186. [DOI] [PubMed] [Google Scholar]
- 56.Wollenberger A, Babskii EB, Krause EG, Genz S, Blohm D, Bogdanova EV. Cyclic changes in levels of cyclic AMP and cyclic GMP in frog myocardium during the cardiac cycle. Biochem Biophys Res Commun. 1973;55:446–452. [DOI] [PubMed] [Google Scholar]
- 57.Brooker G. Change in myocardial adenosine 3′,5′ cyclic monophosphate during the contraction cycle. Recent Adv Stud Cardiac Struct Metab. 1973;3:207–212. [PubMed] [Google Scholar]
- 58.Brooker G. Oscillation of cyclic adenosine monophosphate concentration during the myocardial contraction cycle. Science. 1973;182:933–934. [DOI] [PubMed] [Google Scholar]
- 59.Ignarro LJ. Heme-dependent activation of guanylate cyclase by nitric oxide: a novel signal transduction mechanism. Blood Vessels. 1991;28:67–73. [DOI] [PubMed] [Google Scholar]
- 60.Ishibashi T, Hamaguchi M, Kato K, Kawada T, Ohta H, Sasage H, Imai S. Relationship between myoglobin contents and increases in cyclic GMP produced by glyceryl trinitrate and nitric oxide in rabbit aorta, right atrium and papillary muscle. Naunyn Schmiedebergs Arch Pharmacol. 1993;347:553–561. [DOI] [PubMed] [Google Scholar]