

Abstract

Nuclear receptor related 1 (Nurr1) is a neuroprotective transcription factor and an emerging target in neurodegenerative diseases. Despite strong evidence for a role in Parkinson’s and Alzheimer’s disease, pharmacological control and validation of Nurr1 are hindered by a lack of suitable ligands. We have discovered considerable Nurr1 activation by the clinically studied dihydroorotate dehydrogenase (DHODH) inhibitor vidofludimus calcium and systematically optimized this scaffold to a Nurr1 agonist with nanomolar potency, strong activation efficacy, and pronounced preference over the highly related receptors Nur77 and NOR1. The optimized compound induced Nurr1-regulated gene expression in astrocytes and exhibited favorable pharmacokinetics in rats, thus emerging as a superior chemical tool to study Nurr1 activation in vitro and in vivo.
Introduction
Nuclear receptor related 1 (Nurr1, NR4A2) is a constitutively active ligand-activated transcription factor predominantly expressed in neurons.1−3 It has neuroprotective and antineuroinflammatory activity and emerges as an attractive target to treat neurodegenerative pathologies including Alzheimer’s disease (AD), Parkinson’s disease (PD), and multiple sclerosis (MS).2,3 Therapeutic potential of Nurr1 activation is supported by observations from several rodent models, demonstrating that Nurr1 knockout produces a PD-like phenotype and that diminished Nurr1 activity aggravates disease models of AD, PD, and MS.4−7 Moreover, human patients of these pathologies were found to exhibit diminished Nurr1 expression,6−9 and the neuroprotective effects of statin drugs could in part be referred to Nurr1 activation,10 further highlighting the receptor’s promise. However, validation of pharmacological Nurr1 activation as an approach to treat neurodegenerative diseases is pending, since selective chemical tools with high Nurr1 agonist potency are lacking.3,11 Although the anticipated therapeutic potential of Nurr1 as a neuroprotector has fueled efforts to develop Nurr1 agonists4,12−16 and inverse agonists,17,18 the micromolar potency of available Nurr1 modulators is insufficient.19 Here, we report the first Nurr1 agonist scaffold achieving nanomolar potency. We detected remarkable submicromolar Nurr1 agonism of the dihydroorotate dehydrogenase (DHODH) inhibitor vidofludimus (1)20,21 and systematically elucidated the scaffold’s structure–activity relationship to obtain the Nurr1 agonist 29, which exhibits nanomolar potency and selectivity over DHODH as well as the Nurr1 related receptors Nur77 and NOR1.
Results and Discussion
We have recently evaluated the DHODH inhibitor 1 (Figure 1a) as a weak partial farnesoid X receptor (FXR) agonist tool with unique modulatory characteristics.22 In follow-up experiments to reveal activity on related receptors (Figure 1b–d), 1 induced Nurr1 activation, suggesting potential as lead for Nurr1 agonist development. Full dose–response characterization of 1 in Gal4 hybrid reporter gene assays on Nurr1 (EC50 = 0.4 ± 0.2 μM, Figure 1b) and the related receptors Nur77 (NR4A1, EC50 = 3.1 ± 0.7 μM) and NOR1 (NR4A3, EC50 = 2.9 ± 0.9 μM) revealed considerable Nurr1 agonist potency and preference, which markedly exceeded available Nurr1 activators.31 did not affect the activity of the ligand-independent transcriptional inducer Gal4-VP1623,24 and was selective over constitutively active (Figure 1c) and lipid-binding nuclear receptors (Figure 1d) outside the NR4A family apart from weak peroxisome proliferator-activated receptor (PPAR) γ agonism (EC50 > 10 μM, Figure S1). 1 activated human Nurr1 also in its native full-length form as a monomer (NBRE, EC50 = 0.3 ± 0.1 μM Figure 1e) and RXR/Nurr1 heterodimer (DR5, EC50 = 0.4 ± 0.2 μM, Figure 1e) and bound to the recombinant Nurr1 LBD with submicromolar affinity (Kd = 0.7 μM) in isothermal titration calorimetry (ITC, Figure 1f). To further validate the effects of 1 as directly Nurr1-mediated and exclude artifacts of DHODH inhibition, we studied the effects of the structurally related DHODH inhibitors252, farudodstat,26 MEDS433,27 RP7214,28 and AG-63629 (Figures 1g and S2). 1 and its deuterated analogue 2 activated Nurr1 in the Gal4 hybrid assay and on the native NBRE. No other tested DHODH inhibitor caused activation of Gal4-Nurr1 or flNurr1, thus supporting the effects of 1 and 2 as directly mediated by interaction with Nurr1. Submicromolar Nurr1 agonism and binding affinity thus rendered 1 as a superior lead for Nurr1 agonist development, prompting us to elucidate its structure–activity relationship with the objective to enhance Nurr1 agonism and decrease DHODH inhibitory potency.
Figure 1.

Vidofludimus (1) is a potent Nurr1 agonist. (a) Chemical structure of 1. (b) 1 activated Gal4-Nurr1 with an EC50 value of 0.4 ± 0.2 μM. Data are the mean ± S.E.M., n ≥ 3. (c) Apart from NR4A receptors, 1 (10 μM) had no effect on the activity of constitutively active nuclear receptors. Data are the mean ± S.E.M., n = 3. (d) 1 (10 μM) showed no activity on lipid-binding nuclear receptors apart from weak PPARγ agonism (EC50 > 10 μM). Heatmap shows mean relative activation vs reference agonists; n = 3. (e) 1 activated full-length human Nurr1 on the response elements for the Nurr1 monomer (NBRE, EC50 = 0.3 ± 0.1 μM) and the RXR/Nurr1 heterodimer (DR5, EC50 = 0.4 ± 0.2 μM). Data are the mean ± S.E.M., n ≥ 3. (f) Binding of 1 to Nurr1 LBD was confirmed by isothermal titration calorimetry (ITC) with a Kd value of 0.7 μM. The upper panel shows the isotherm of the 1-protein titration; the lower panel shows the fitting of the heat of binding. (g) Activity of DHODH inhibitors related to 1 on Nurr1 in the hybrid Gal4-Nurr1 and a full-length Nurr1 (NBRE) reporter gene assay. All compounds were tested at 10 μM; data are the mean ± S.E.M., n = 3. Chemical structures are given in Figure S2.
The next-generation DHODH inhibitor analogue 2 of 1 comprising a fully deuterated methoxy substituent interestingly exhibited five-fold higher potency as Nurr1 agonist than 1. The exchange of hydrogen by deuterium is considered a conservative (bio-)isosteric replacement and may offer an opportunity to address pharmacokinetic issues; but also an increased on-target potency of deuterated analogues is not unprecedented.30 Based on the favorably increased Nurr1 agonism of the deuterated analogue 2, we retained this modification for further SAR evaluation.
To identify regions offering potential for optimization toward potent Nurr1 agonists with selectivity over DHODH, we broadly probed the SAR with structural variations on all parts of the scaffold commencing with the central fluorobenzene motif (Table 1). Removal of the fluorine atom (3) or introduction of a second fluorine substituent on this central aromatic ring (4) was tolerated but incorporation of a nitrogen atom leading to pyridine derivative 5 markedly diminished Nurr1 agonism. Extension of the central benzene to naphthalene (6) or indane (7) or its replacement by a bicyclo[2.2.2]octane (8) completely abolished activity on Nurr1. The central fluorinated benzene moiety of the original scaffold thus appeared favored for Nurr1 agonist development. Moreover, lack of Nurr1 agonism of 9 comprising an N-methyl amide indicated importance of the secondary amide linkage.
Table 1. Variation of the Central Amidobenzene Motif.
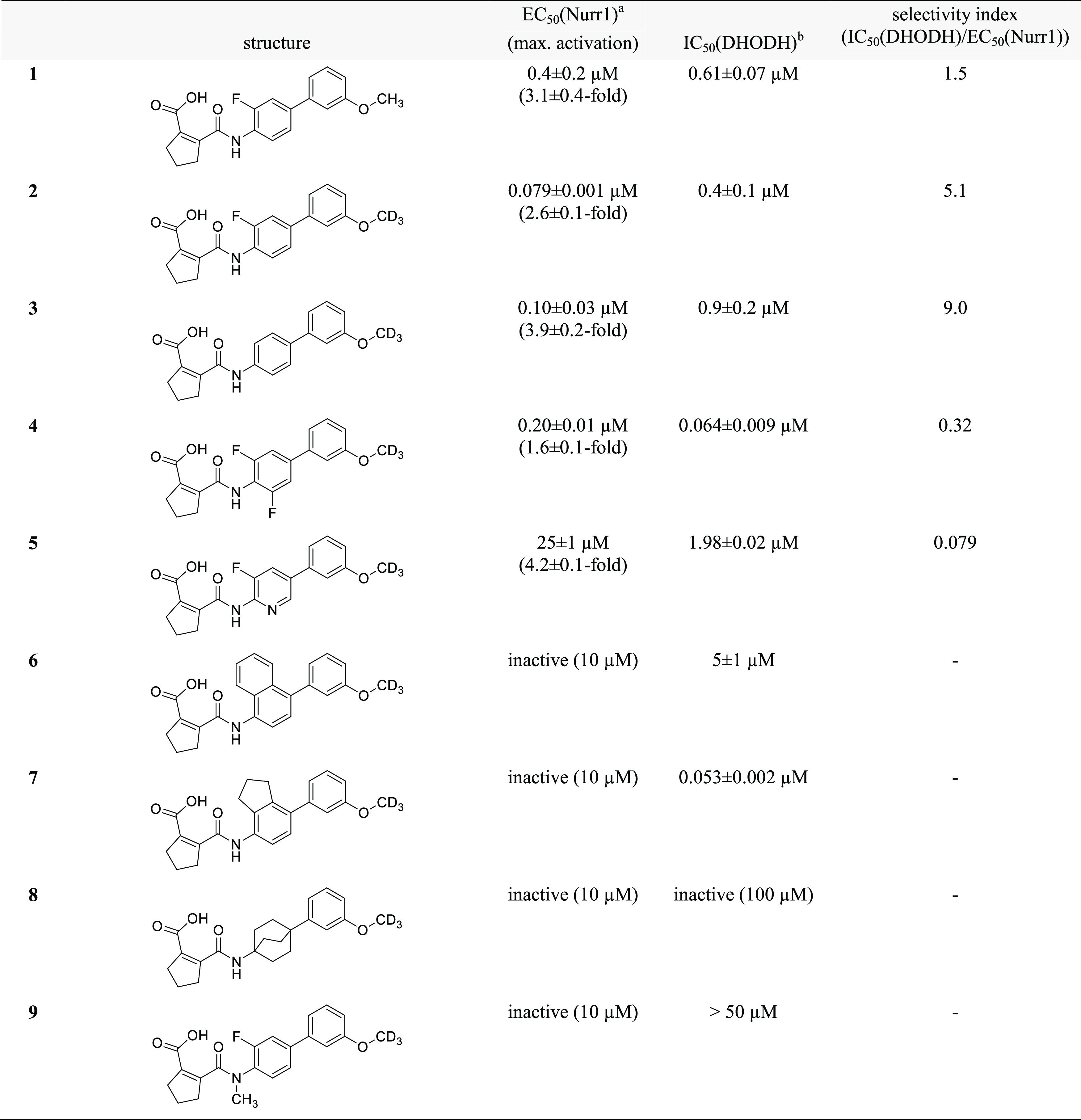
As the N-phenyl amide region appeared to offer no major potential to optimize Nurr1 agonist potency and selectivity, we focused our attention on variations in the cyclopentene carboxylic acid part of the scaffold (Table 2). Incorporation of oxygen (10) and sulfur (11) in the cyclopentene motif caused up to a five-fold loss in potency, indicating that the original cyclopentene skeleton was favored. The 4,4-difluorocyclopentene analogue 12 was slightly less potent as a Nurr1 agonist than 4 while gaining potency on DHODH and was hence not favored in terms of selectivity for Nurr1. For the carboxylic acid motif, steric rather than acidic features appeared to be critical, since the corresponding primary amide 13 and the small hydroxylamide 14 retained Nurr1 agonism with slightly reduced potency. Albeit this loss in Nurr1 agonist potency, 13 and 14 contributed important SAR insights, as their DHODH inhibitory potency was strongly reduced, opening a potential avenue to selective Nurr1 agonists. N-Methylhydroxylamide 15 despite following the trend to lower DHODH inhibition was also substantially less active on Nurr1 than 14, indicating potential involvement of an H-bond donor interaction in Nurr1 activation. Amide-derived carboxylic acid bioisosteres with bulkier substituents like N-methoxy amide 16, cyanamide 17, and methyl sulfonamide 18 were inactive on Nurr1.
Table 2. Variation of the Cyclopentene Carboxylic Acid Motif.
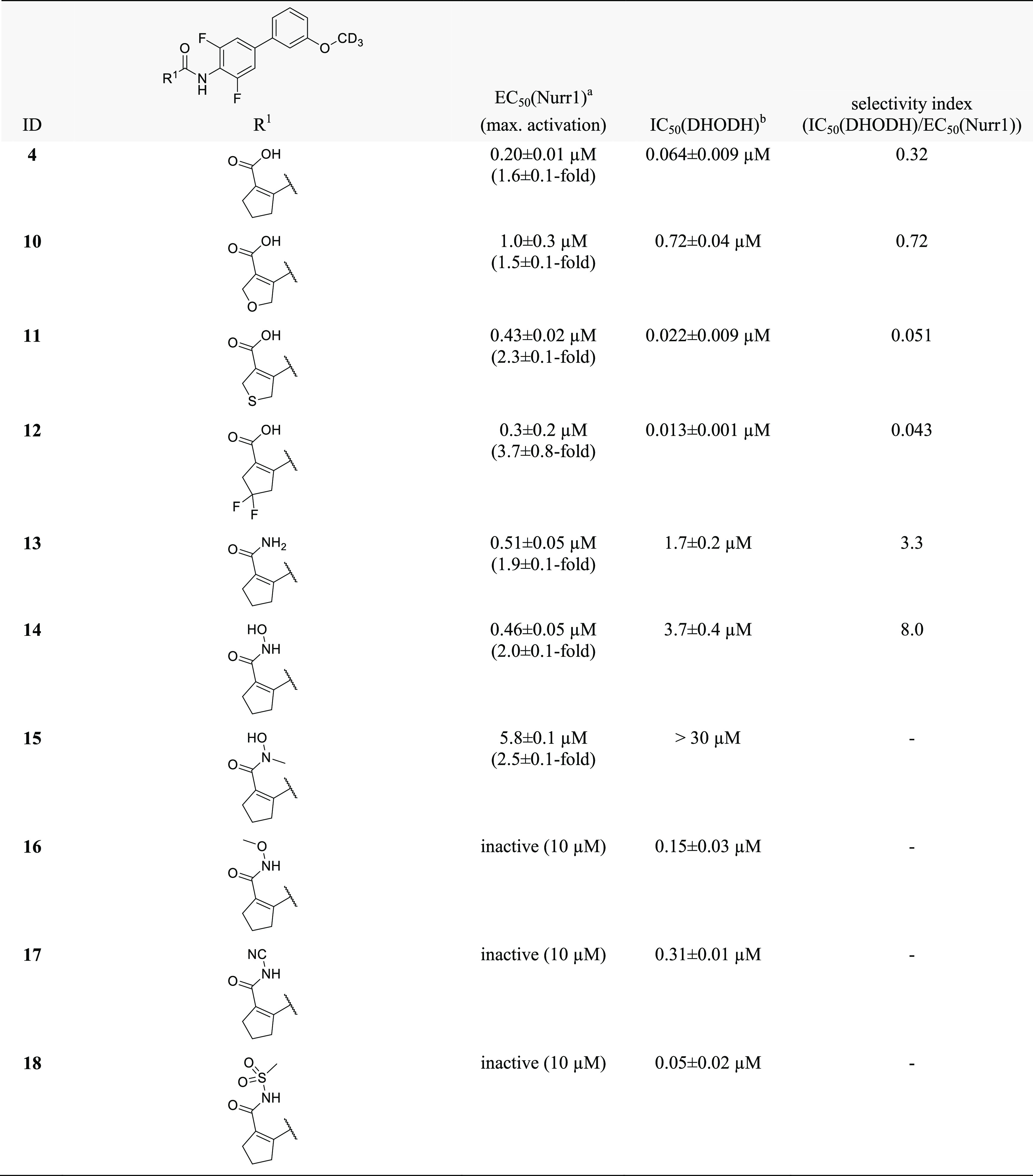
As remaining substructure of the scaffold to be evaluated, we moved our attention to variations of the terminal alkoxyphenyl motif (Table 3). Changing the regiochemistry of this phenyl residue from meta (4) to ortho (19) substitution was tolerated with a slight loss in potency. Apart from this, the SAR of the alkoxybenzene was steep and allowed no improvement of Nurr1 agonism. Replacement of the benzene ring (4) by a thiazole (20) markedly diminished Nurr1 agonism despite similar geometry, and introduction of nitrogen atoms in the benzene ring next to the alkoxy substituent (21, 22) was also not tolerated. Incorporation of the alkoxy substituent in a two-ring system like benzodioxol (23) and benzoxazole (24) disrupted Nurr1 agonism, too.
Table 3. Variation of the Alkoxyphenyl Motif.
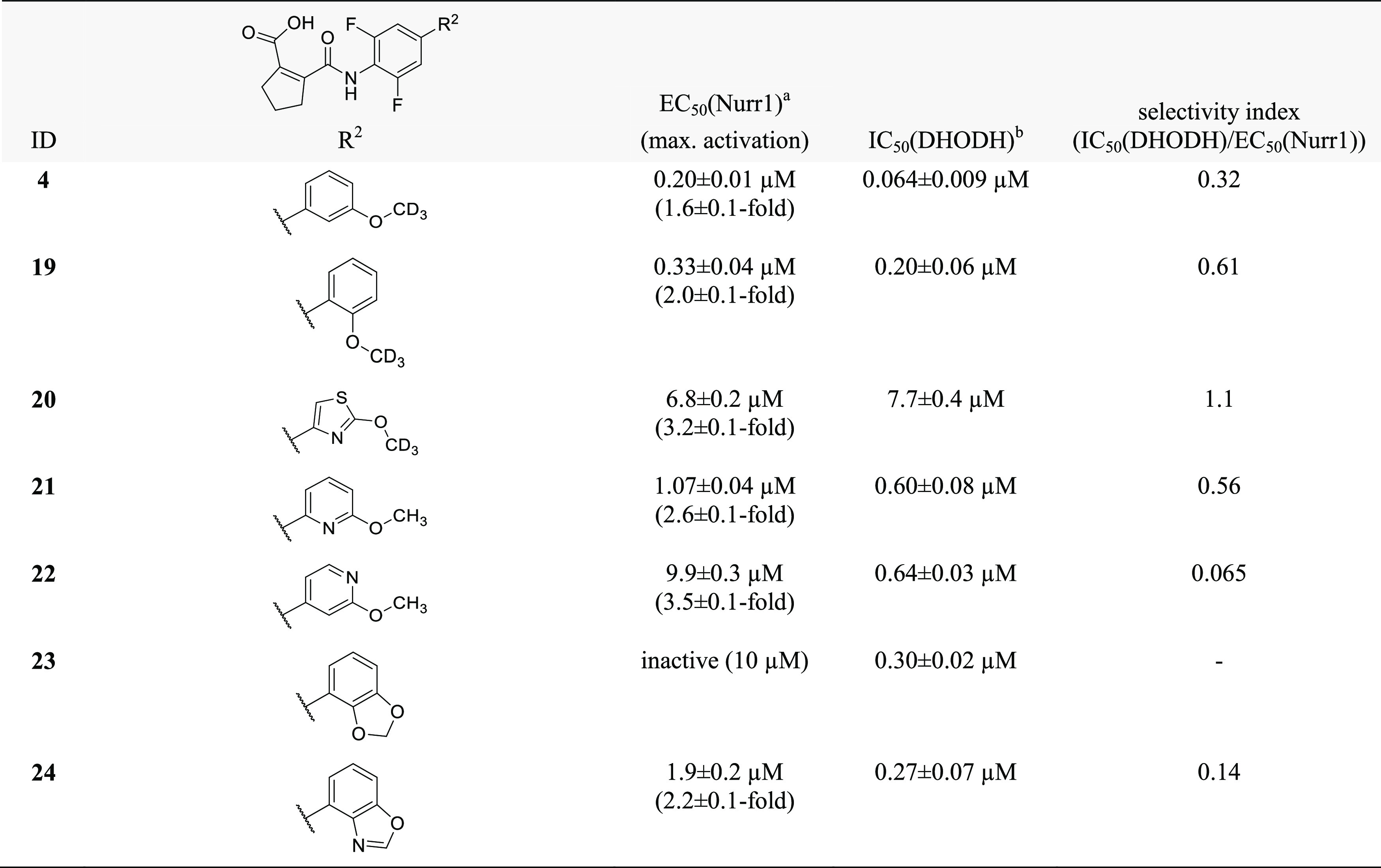
Albeit revealing a few structural modifications that enhanced Nurr1 agonism, the SAR of the central benzene ring (Table 1), the cyclopentene carboxylic acid motif (Table 2), and the terminal alkoxybenzene (Table 3) indicated that the original scaffold was overall favored. To gain potency on Nurr1 and selectivity over DHODH, the alkoxy substituent appeared most promising as already the minor change from methoxy (1) to the deuterated analogue 2 considerably promoted Nurr1 agonism. Thus, we set out to elucidate the SAR of the alkoxy substituent further (Table 4).
Table 4. Variation of the Alkylether Motif.
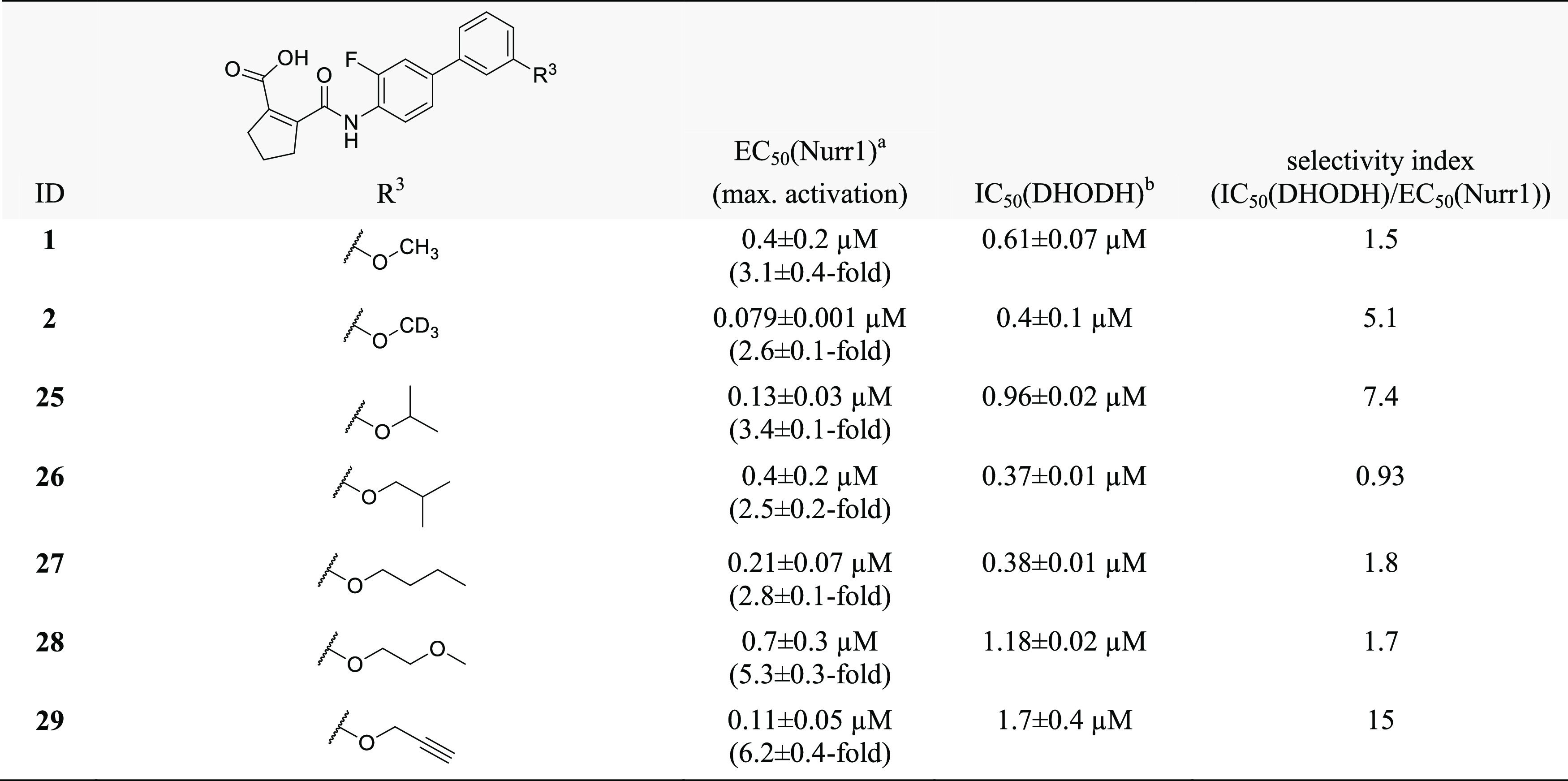
The isopropylether 25 was slightly less active on Nurr1 than the deuterated methoxy analogue 2 but markedly improved selectivity over DHODH. iso-Butyl derivative 26 revealed further reduced potency on Nurr1 and less selectivity, indicating a preference for smaller alkyl groups to obtain potent and selective Nurr1 agonists. Moreover, the n-butylether 27 outmatched the branched analogue 26 in terms of Nurr1 agonist potency, indicating that linear groups were favored. Introduction of an additional heteroatom in methoxyethyl derivative 28 resulted in strong Nurr1 agonist efficacy but a loss in potency compared to alkyl ethers 25–27. A propynyl ether (29) emerged as a highly favored motif, boosting potency (EC50 = 0.11 ± 0.05 μM) and efficacy (6.2-fold activation) on Nurr1 while reducing DHODH inhibitory potency (IC50 = 1.7 ± 0.4 μM). The marked drop in DHODH inhibition observed for 29 may be due to an unfavorable entropic contribution of the hydrophobic propynyl substituent, as the alkoxy group is solvent exposed in the binding mode of this scaffold to DHODH31 (Figure S3).
The optimized Nurr1 agonist 29 (Figure 2a) exhibited high-affinity binding to the Nurr1 LBD in ITC (Kd = 0.3 μM, Figure 2b) and activated full-length human Nurr1 on the monomer (NBRE, EC50 = 0.22 ± 0.08 μM) and the RXR-heterodimer (DR5, EC50 = 0.36 ± 0.08 μM) response elements. Like the lead compound 1, 29 had a favorable preference for Nurr1 (EC50 = 0.11 ± 0.05 μM, Figure 2c) over the related Nur77 (EC50 = 1.4 ± 0.6 μM) and NOR1 (EC50 = 1.3 ± 0.3 μM) and was selective over other constitutively active (Figure 2d) and lipid-activated nuclear receptors (Figure 2e). In Nurr1-expressing human astrocytes (T98G),1029 induced mRNA expression of the Nurr1-regulated genes tyrosine hydroxylase (TH) and vesicular amino acid transporter 2 (VMAT2) (Figure 2f), demonstrating cellular target engagement. In addition to this favorable cellular activity profile, 29 showed no cytotoxic effects in a multiplex toxicity assay in COS-7 cells (Figure S4). 29 hence emerges as a superior Nurr1 agonist and as a next-generation chemical tool for this receptor.
Figure 2.
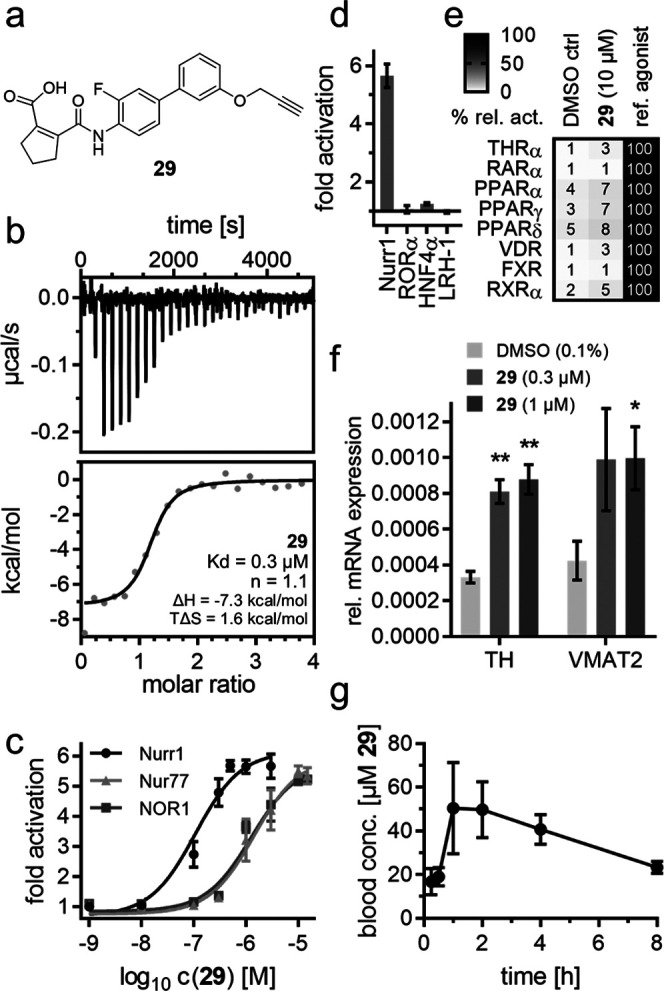
In vitro profiling of 29 as a Nurr1 agonist tool. (a) Chemical structure of 29. (b) Binding of 29 to the Nurr1 LBD was confirmed by ITC with a Kd value of 0.3 μM. The upper panel shows the isotherm of the 29-protein titration; the lower panel shows the fitting of the heat of binding. (c) 29 exhibited >10-fold selectivity for Nurr1 (EC50 = 0.11 ± 0.05 μM) over the related receptors Nur77 (EC50 = 1.4 ± 0.6 μM) and NOR1 (EC50 = 1.3 ± 0.3 μM). Data are the mean ± S.E.M. fold activation from Gal4 hybrid reporter gene assays; n ≥ 3. (d, e) 29 revealed no relevant activity on related constitutively active (d) and lipid-binding (e) nuclear receptors at 10 μM. Data are the mean ± S.E.M. fold activation (d) or mean relative activation (e); n ≥ 2. (f) 29 induced mRNA expression of the Nurr1-regulated genes tyrosine hydroxylase (TH) and vesicular amino acid transporter 2 (VMAT2). Data are the mean ± S.E.M., n = 4. *p < 0.05, **p < 0.01 (two-sided t-test vs DMSO-treated cells). (g) 29 revealed a favorable PK profile in rats after a single 5 mg/kg p.o. dose. Data are the mean ± S.E.M.; n = 3.
In addition, 29 holds potential to enable in vivo studies on Nurr1 activation. As there is no species difference for the Nurr1 LBD between humans and rats, while the murine receptor differs,32 we profiled 29 for potential in vivo application in rats. Compared to hDHODH (IC50 = 1.7 ± 0.4 μM), 29 exhibited slightly lower DHODH inhibitory potency on the rat enzyme (IC50 = 6 ± 1 μM), providing even improved selectivity for Nurr1 in this species. A single oral dose of 29 (5 mg/kg) produced a very favorable pharmacokinetic profile in rats with a 4.4 h half-life, 56 μM peak plasma concentration, and 89% bioavailability (Figure 2g and Table 5), thus rendering Nurr1 agonist 29 suitable for in vivo experiments.
Table 5. Pharmacological Properties of Nurr1 Agonist 29.

EC50 values from Gal4 hybrid reporter gene assays.
PK data from female rats after a single 5 mg/kg p.o. dose.
Conclusions
The neuroprotective and antineuroinflammatory characteristics of Nurr1 suggest strong therapeutic potential for pharmacological activation of this transcription factor in neurodegenerative pathologies but potent and selective Nurr1 agonists were lacking as chemical tools for target validation.3 We have discovered considerable Nurr1 agonism of the DHODH inhibitor 1, which is clinically studied as a new therapeutic option in MS treatment.21 While further studies will have to evaluate whether Nurr1-mediated effects contribute to the therapeutic benefit from 1 in MS,10,211 emerged as a highly attractive lead to develop Nurr1 agonists with improved potency and selectivity. Our systematic structural variation revealed a rather steep SAR for the scaffold as Nurr1 agonist with only few regions tolerating modifications. While the overall skeleton evolved as favored for Nurr1 agonism, variation of the alkoxy substituent from a methoxy (1) to a propynyloxy group (29) enabled an improvement in Nurr1 agonist potency, especially concerning activation efficacy, and considerably enhanced selectivity for Nurr1 over the original target DHODH. Compared to available Nurr1 agonists, 29 provides superior chemical tool characteristics and is suitable for in vivo studies on Nurr1 activation in health and disease.
Chemistry
Compounds 2–9 and 19–24 were prepared by reacting the corresponding amines 2a–9a and 19a–24a with 5,6-dihydro-1H-cyclopenta[c]furan-1,3(4H)-dione (30a), respectively, according to Scheme 1.
Scheme 1. Synthesis of 2–9 and 19–24.

Reagents and conditions: (a) CH3CN (rt/40 °C for 2, 4, 6, 19–24) or CH2Cl2 (rt/30 °C for 3, 5, 7, 9) or THF (60 °C for 8), 2 h to 5 days, 14–82%.
Compounds 10–12 with variations in the five-membered ring were similarly obtained by using alternative dicarboxylic acid anhydrides (30b–d) and amine 4a (Scheme 2).
Scheme 2. Synthesis of 10–12.

Reagents and conditions: (a) CH3CN, rt, 6 h, 25–78%.
Amines 3a and 5a–7a were prepared by Suzuki reaction of the corresponding aryl bromides 3b and 5b–7b with 3-(2H3)methoxybenzene boronic acid (31a) according to Scheme 3.
Scheme 3. Synthesis of Amine Building Blocks 3a and 5a–7a.

Reagents and conditions: (a) Pd(PPh3)4 (3a) or Pd(dppf)Cl2 (5a–7a), Cs2CO3 (3a, 5a) or Na2CO3 (6a, 7a), 1,4-dioxane/H2O, 90 °C, 2–3 h, 57–91%.
Similarly, amines 2a, 4a, and 19a–24a were prepared by Suzuki reaction of the corresponding aryl bromides 2b, 4b, and 19b–24b with the single or double fluorinated aminobenzene boronic acid pinacol esters 31b or 31c, respectively (Scheme 4).
Scheme 4. Synthesis of Amine Building Blocks 2a, 4a, and 19a–24a.

Reagents and conditions: (a) Pd(dppf)Cl2 (2a, 4a), Pd(PPh3)4 (19a–22a) or Pd(PPh3)2Cl2 (23a, 24a), Cs2CO3 (19a–22a) or Na2CO3 (2a, 4a, 23a, 24a), 1,4-dioxane/H2O, 90 °C, 2–3 h, 60–96%.
Aryl bromides 20b–22b were obtained from aryldihalogenides 20c–22c by nucleophilic aromatic substitution with deuterated methanol according to Scheme 5.
Scheme 5. Synthesis of Aryl Bromides 20b–22b.

Reagents and conditions: (a) NaOH (20b, 21b) or NaH (22b), CD3OD, THF, 0 °C (22b) or rt (20b, 21b), 2 h, 69–91%.
The benzene bioisosteric phenylbicyclo[2.2.2]octaneamine 8a was prepared from 4-(methoxycarbonyl)bicyclo[2.2.2]octane-1-carboxylic acid (8b) over seven steps according to Scheme 6. 8b was esterified with phthaloxime (8c) and treated with freshly prepared bis(3-methoxyphenyl)zinc to give methoxyphenylbicyclo[2.2.2]octane 8d. Demethylation with boron tribromide to 8e and subsequent Willamson ether synthesis with iodo(2H3)methane afforded the deuterated analogue 8f. Ester hydrolysis (8g) and Curtius rearrangement provided carbamate 8h, and removal of the N-Boc protecting group yielded the amine building block 8a.
Scheme 6. Synthesis of Phenylbicyclo[2.2.2]octaneamine Building Block 8a.

Reagents and conditions: (a) DIC, 2-hydroxyisoindoline-1,3-dione, 4-DMAP, CH2Cl2, rt, overnight, 55%; (b) (3-methoxyphenyl)magnesium bromide, LiCl, ZnCl2, THF, rt, 1 h; then 6,6′-dimethyl-2,2′-bipyridine, Ni(acac)2, CH3CN, 80 °C, overnight, 83%; (c) BBr3, CH2Cl2, rt, 4 h, 89%; (d) CD3I, K2CO3, CH3CN, 60 °C, 12 h, 94%; (e) LiOH, H2O, MeOH, rt, 12 h, 99%; (f) (Boc)2O, diphenylphosphoryl azide, NEt3, toluene, 80 °C, 12 h, 27%; (g) HCl, MeOH, rt, 3 h, 100%.
N-Methylbiarylamine building block 9a was obtained from 2a according to Scheme 7. Boc-protection of 2a yielded 9b, which was deprotonated with sodium hydride and subsequently treated with methyl iodide to afford 9c before Boc cleavage provided 9a.
Scheme 7. Synthesis of N-Methylamine Building Block 9a.

Reagents and conditions: (a) (Boc)2O, N,N-diisopropylethylamine, dimethylformamide (DMF), rt, 2 h, 77%; (b) NaH, THF, 0 °C, 1 h; then CH3I, rt, 4 h, 72%; (c) HCl, 1,4-dioxane, rt, 1 h.
Dicarboxylic acid anhydrides 30a and 30d were commercially available, 30c was prepared as described previously,20 and 30b was obtained from 32a according to Scheme 8. For this, 32a was reacted with carbon monoxide in the presence of 1,1′-bis(diphenylphosphino)ferrocene, Pd2(dba)3, and methanol to obtain dimethyl dicarboxylate 32b. Acidic ester hydrolysis to 32c and subsequent treatment with acetyl chloride afforded anhydride 30b.
Scheme 8. Synthesis of Dicarboxylic Acid Anhydride 30b.

Reagents and conditions: (a) CO, Pd2(dba)3, dppf, MeOH, DMF, 50 °C, overnight, 82%; (b) HCl, HOAc, 100 °C, 2 h, 80%; (c) acetyl chloride, PhCH3, 110 °C, 4 h, 84%.
The carboxylic acid bioisosteres 13–18 were prepared from 4 according to Scheme 9 by amide coupling with various amine derivatives and appropriate coupling reagents. Treatment of 4 with ammonium chloride in the presence of EDC and 4-DMAP produced the primary amide 13. N-Hydroxylamide 14 and N-methyl-N-hydroxylamide 15 were obtained from 4with hydroxylamine hydrochloride or N-methylhydroxylamine hydrochloride in the presence of CDI. The reaction of 4 with O-methylhydroxylamine hydrochloride in the presence of DCC and 4-DMAP yielded 16, treatment of 4 with cyanamide in the presence of HATU and base produced 17, and 18 was synthesized from 4 with methanesulfonamide, DCC, and 4-DMAP.
Scheme 9. Synthesis of Carboxylic Acid Bioisosteres 13–18.

Reagents and conditions: (a) NH4Cl, EDC, 4-DMAP, DMF, 70 °C, 8 h, 35%; (b) NH2OH·HCl, CDI, THF, rt, 8 h, 74%; (c) NHMeOH·HCl, CDI, THF, rt, 1 h, 8%; (d) NH2OMe, DCC, DMAP, NEt3, CH2Cl2, 60 °C, 8 h, 14%; (e) NH2CN, HATU, NiPr2Et, DMF, rt, 8 h, 22%; (f) NH2SO2Me, DCC, DMAP, NEt3, CH2Cl2, 60 °C, 8 h, 12%.
Compounds 25–28 comprising variations of the alkoxy group were prepared from methyl 2-oxocyclopentane-1-carboxylate (33a) according to Scheme 10. 33a was transformed to triflate 33b by treatment with sodium hydride and triflic anhydride. The reaction of 33b with sodium formate in the presence of LiCl, acetic anhydride, N,N-diisopropylamine, and XPhos-Pd-G233 afforded cyclopentenedicarboxylic acid monoester 33c. Amide coupling with 4-bromo-2-fluoroaniline (33d) to 33e and subsequent ester hydrolysis provided 33f as a substrate for Suzuki reactions with boronic acids 25a–28a to the final compounds 25–28.
Scheme 10. Synthesis of 25–28.

Reagents and conditions: (a) NaH, Tf2O, 0 °C to rt, 1 h, 70%; (b) HCOONa, DMF, LiCl, Ac2O, DIPA, XPhos-Pd-G2, 0 °C then 50 °C, 18 h, 57%; (c) EDC·HCl, CHCl3, rt, 2 h, then 33d, NEt3, reflux, 18 h, 13%; (d) LiOH, THF/H2O, rt, 20 min, 95%; (e) XPhos-Pd-G2, Cs2CO3, PhCH3/EtOH/DMF/H2O, 100 °C, 18 h, 59–99%.
Propynyl ether derivative 29 was obtained from phenol analogue 29a(34) by treatment with 3-bromoprop-1-yne to obtain the double substituted 29b from which ester hydrolysis afforded 29 (Scheme 11).
Scheme 11. Synthesis of 29.

Reagents and conditions: (a) 3-bromoprop-1-yne, K2CO3, DMF, 60 °C, 16 h, 74%; (b) NaOH, MeOH/THF/H2O, 0 °C, 3 h, 83%.
Experimental Section
Chemistry
General
All chemicals were of reagent grade, purchased from commercial sources (e.g., Sigma-Aldrich, abcr, Enamine, and BLDpharm), and used without further purification unless otherwise specified. All reactions were conducted in oven-dried Schlenk glassware under an Ar atmosphere and in absolute solvents. Other solvents, especially for work-up procedures, were of reagent grade or purified by distillation (i-hexane, EtOAc, EtOH). Reactions were monitored by thin-layer chromatography (TLC) on TLC Silica gel 60 F254 aluminum sheets by Merck and visualized under ultraviolet light (254 nm) or by in-process liquid chromatography–mass spectrometry (LC/MS). Purification of compounds 25–32 by column chromatography (CC) was performed on a puriFlash XS520Plus system (Advion, Ithaca, NY) using high-performance spherical silica columns (SIHP, 50 μM) by Interchim and a gradient of i-hexane to EtOAc; reversed-phase CC was performed on a puriFlash 5.250 system (Advion) using C18HP columns (SIHP, 15 μM) by Interchim and a gradient of 0.1% FA in H2O, 10–100% acetonitrile (high-performance liquid chromatography (HPLC) gradient grade). All other compounds were purified with a Biotage Isolera One combiflash chromatography system (SEPAFLASH, 40–63 Å) with the solvent mixtures specified in the corresponding experiment. Preparative HPLC was performed using a combiflash reversed-phase chromatography (C18) Boston ODS 40 g Flash 35–50 mL/min at 200 psi with gradient A: 0.1% NH4HCO3 in water, 10–100% ACN, or with gradient B: 0.1% TFA in water, 10–100% ACN. Melting points (uncorrected) were determined using glass capillaries on an M3000 melting point meter (Krüss). Mass spectra were obtained on a puriFlash-CMS system (Advion) using atmospheric pressure chemical ionization (APCI) or on an Ion Trap Esquire 3000 + instrument (Bruker Corporation, Billerica, MA) using electrospray ionization (ESI-LCMS). High-resolution mass spectrometry (HRMS) was obtained with a Thermo Finnigan LTQ FT instrument for electrospray ionization (ESI). NMR spectra were recorded on Bruker Avance III HD 400 or 500 MHz spectrometers equipped with a CryoProbe Prodigy broadband probe (Bruker). Chemical shifts are reported in δ values (ppm), and coupling constants (J) in hertz (Hz). Signals are described as br for broad. The purity of 25–28 was analyzed on a system consisting of a Sciex API 3200 QTrap triple quadrupole mass spectrometer, a quarternary Agilent 1100 pump (G1311A) with a degasser (G1322A), an Agilent 1100 oven (G1316A), an Agilent 1100 DAD (G1315B), and a Shimadzu SIL 20A HT autosampler under the control of Analyst 1.6 (Sciex). As a stationary phase, a Zorbax SBAq (3.5 μm, 100 mm × 3 mm, Agilent, protected with a 0.5 μm and a 0.2 μm frit) was used in combination with 5 mM ammonium formate (A) and acetonitrile (B) as a mobile phase at a flow rate of 500 μL/min. Compounds were investigated by 10 μL injections of 1 and 10 μM sample solutions (100 mM stock solutions in DMSO diluted in the mobile phase) under isocratic conditions (A/B 50:50 or 60:40, v/v). MS detection was done under negative ESI conditions, recording corresponding [(M – H)]− ions in the SIM mode and mass transitions based on the loss of 44 Da (CO2) in the multiple reaction monitoring (MRM) modes. Ultraviolet (UV) detection was done by recording spectra in a range from 210 to 360 nm. The purity of all other compounds was analyzed on an Agilent Technologies 1200 Series machine under the following conditions: LC-Mass Method 1: column: Sunfire C18, 4.6 mm × 50 mm, 3.5 μm; mobile phase: A: water (0.01% TFA), B: ACN (0.01% TFA); gradient: 5–95% B in 1.5 min; flow rate: 2.0 mL/min; oven temperature: 50 °C; mass range: 110–1000; detection: UV (214, 254 nm) or LC-Mass Method 2: column: Xbridge C18(2) (4.6 mm × 50 mm, 3.5 μm); mobile phase: A: H2O (10 mmol NH4HCO3), B: ACN; elution program: gradient from 10 to 95% of B in 1.5 min at 1.8 mL/min; temperature: 50 °C; detection: UV (214, 254 nm) and MS (ESI, Pos mode, 103 to 800 amu). CD3I used for deuteration had ≥99% isotopic purity, and the deuterated products had ≥98% isotopic purity according to NMR. All compounds for biological testing had a purity of >95% based on the 254 nm UV trace. NMR and LCMS/UV data are provided in the Supporting Information. General procedures as well as synthesis and analytical characterization of 2–28 and precursors are described in the Supporting Information.
2-[(3-Fluoro-3′-prop-2-yn-1-yloxy[1,1′-biphenyl]-4-yl)carbamoyl]cyclopent-1-ene-1-carboxylic acid (29)
2 N NaOH (3 mL) was added to a solution of compound 29b (0.45 g, 1.1 mmol) in MeOH (1 mL) and THF (3 mL) at 0 °C. After stirring for 3 h at rt, the pH was adjusted to 5–6. This mixture was concentrated, and the resulting residue was purified by reversed-phase column chromatography (0.1% TFA in H2O, 10–100% MeCN as mobile phase) to give compound 29 (0.34 g, yield: 83%) as a yellow solid. Mp 190 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.51–13.55 (br s, 1H), 10.69 (br s, 1H), 8.09 (t, J = 8.4 Hz, 1H), 7.63 (dd, J = 12.4, 1.6 Hz, 1H), 7.53 (dd, J = 8.4, 1.3 Hz, 1H), 7.40 (t, J = 7.9 Hz, 1H), 7.35–7.27 (m, 2H), 7.00 (dd, J = 8.1, 1.9 Hz, 1H), 4.90 (d, J = 2.4 Hz, 2H), 3.61–3.56 (t, J = 2.1 Hz, 1H), 2.81 (t, J = 7.0 Hz, 2H), 2.70 (t, J = 7.2 Hz, 2H), 1.90 (p, J = 7.7 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 166.7, 165.2, 158.2, 154.2 (d, 1JCF = 245.6 Hz), 147.3, 140.3, 137.4 (d, 3JCF = 7.0 Hz), 135.6, 130.5, 125.9 (d, 2JCF = 11.9 Hz), 124.4, 123.0 (d, 4JCF = 2.8 Hz), 119.9, 114.8, 114.1 (d, 2JCF = 20.5 Hz), 113.3, 79.7, 78.8, 55.9, 36.9, 34.9, 21.6. LCMS (ESI): m/z 380.2 ([M + H]+). HRMS (ESI): m/z calculated 380.12926 for C22H18FNO4 + H+, found 380.12894 ([M + H]+).
Prop-2-yn-1-yl 2-[(3-fluoro-3′-prop-2-yn-1-yloxy[1,1′-biphenyl]-4-yl)carbamoyl]cyclopent-1-ene-1-carboxylate (29b)
3-Bromoprop-1-yne (0.35 g, 2.9 mmol) and K2CO3 (0.61 g, 4.4 mmol) were added to a solution of 2-[(3-fluoro-3′-hydroxy-[1,1′-biphenyl]-4-yl)carbamoyl]cyclopent-1-ene-1-carboxylic acid (29a, 0.50 g, 1.5 mmol; prepared according to ref (34)) in DMF (10 mL). The mixture was heated to 60 °C for 16 h, then cooled to rt, diluted with H2O, and extracted with EtOAc (3×). The combined organic layer was washed with brine, dried with Na2SO4, and concentrated. The resulting residue was purified by column chromatography (PE/EtOAc = 2:1) to afford compound 29b (0.45 g, yield: 74%) as a yellow solid. LCMS (ESI): m/z 418.2 ([M + H]+).
Biological Characterization
Hybrid Reporter Gene Assays
Nurr1 modulation was determined in a Gal4 hybrid reporter gene assays in HEK293T cells (German Collection of Microorganisms and Cell Culture GmbH, DSMZ) using pFR-Luc (Stratagene, La Jolla, CA; reporter), pRL-SV40 (Promega, Madison, WI; internal control), and pFA-CMV-hNurr1-LBD, coding for the hinge region and ligand binding domain of the canonical isoform of human Nurr1. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM), high glucose supplemented with 10% fetal calf serum (FCS), sodium pyruvate (1 mM), penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37 °C and 5% CO2 and seeded in 96-well plates (3 × 104 cells/well). After 24 h, the medium was changed to Opti-MEM without supplements, and the cells were transiently transfected using the Lipofectamine LTX reagent (Invitrogen) according to the manufacturer’s protocol. Five hours after transfection, the cells were incubated with the test compounds in Opti-MEM supplemented with penicillin (100 U/mL), streptomycin (100 μg/mL), and 0.1% DMSO for 16 h before luciferase activity was measured using the Dual-Glo Luciferase Assay System (Promega) according to the manufacturer’s protocol on a Tecan Spark luminometer (Tecan Deutschland GmbH, Germany). Firefly luminescence was divided by Renilla luminescence and multiplied by 1000, resulting in relative light units (RLUs) to normalize for transfection efficiency and cell growth. Fold activation was obtained by dividing the mean RLU of the test compound by the mean RLU of the untreated control. All samples were tested in at least three biologically independent experiments in duplicates. For dose–response curve fitting and calculation of EC50 values, the equation “[Agonist] vs response (three parameters)” was used in GraphPad Prism (version 7.00, GraphPad Software, La Jolla, CA). Nuclear receptor selectivity profiling was performed with corresponding pFA-CMV-hNR-LBD clones and suitable reference compounds on THRα (pFA-CMV-hTHRα-LBD,35 1 μM T3), RARα (pFA-CMV-hRARα-LBD,36 1 μM tretinoin), PPARα (pFA-CMV-hPPARα-LBD,37 1 μM GW7647), PPARγ (pFA-CMV-hPPARγ-LBD,37 1 μM rosiglitazone), PPARδ (pFA-CMV-hPPARδ-LBD,37 1 μM L165,041), RORα (pFA-CMV-RORα-LBD,38 1 μM SR1001), VDR (pFA-CMV-hVDR-LBD,39 1 μM calcitriol), FXR (pFA-CMV-hFXR-LBD,40 1 μM GW4064), RXRα (pFA-CMV-hRXRα-LBD,41 1 μM bexarotene), HNF4α (pFA-CMV-hHNF4α-LBD42), and LRH-1 (pFA-CMV-hLRH-1-LBD).
Reporter Gene Assays for Full-Length Human Nurr1
Activation of full-length human Nurr1 was studied in transiently transfected HEK293T cells using the reporter plasmids pFR-Luc-NBRE17 or pFR-Luc-DR517 each containing one copy of the respective human Nurr1 response element NBRE Nl3 (TGA TAT CGA AAA CAA AAG GTC A) or DR5 (TGA TAG GTT CAC CGA AAG GTC A). The full-length human nuclear receptor Nurr1 (pcDNA3.1-hNurr1-NE; Addgene plasmid #102363) and, for DR5, RXRα (pSG5-hRXR) were overexpressed. pRL-SV40 (Promega) was used for normalization of transfection efficacy and to observe test compound toxicity. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM), high glucose supplemented with 10% fetal calf serum (FCS), sodium pyruvate (1 mM), penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37 °C and 5% CO2 and seeded in 96-well plates (3 × 104 cells/well). After 24 h, the medium was changed to Opti-MEM without supplements, and the cells were transiently transfected using the Lipofectamine LTX reagent (Invitrogen) according to the manufacturer’s protocol. Five hours after transfection, the cells were incubated with the test compounds in Opti-MEM supplemented with penicillin (100 U/mL), streptomycin (100 μg/mL), and 0.1% DMSO for 16 h before luciferase activity was measured using the Dual-Glo Luciferase Assay System (Promega) according to the manufacturer’s protocol on a Tecan Spark luminometer (Tecan Deutschland GmbH, Germany). Firefly luminescence was divided by Renilla luminescence and multiplied by 1000 resulting in relative light units (RLU) to normalize for transfection efficiency and cell growth. Fold activation was obtained by dividing the mean RLU of the test compound by the mean RLU of the untreated control. All samples were tested in at least three biologically independent experiments in duplicates. For dose–response curve fitting and calculation of EC50 values, the equation “[Agonist] vs response (three parameters)” was used in GraphPad Prism (version 7.00, GraphPad Software, La Jolla, CA).
Isothermal Titration Calorimetry (ITC)
ITC experiments were conducted on an Affinity ITC instrument (TA Instruments, New Castle, DE) at 25 °C with a stirring rate of 75 rpm. Nurr1 LBD protein (10 or 30 μM) in buffer (20 mM Tris pH 7.5, 100 mM NaCl, 5% glycerol) containing 3 or 5% DMSO was titrated with test compounds 1 or 29 (100 μM in the same buffer containing 3 or 5% DMSO) in 25 injections (1 × 1 μL and 24 × 3 μL or 1 × 1 μL and 24 × 4 μL) with an injection interval of 180 or 300 s. As control experiments, the test compounds were titrated to the buffer, and the buffer was titrated to the Nurr1 LBD protein under otherwise identical conditions. The heat rates of the compound–Nurr1 LBD titrations were analyzed using NanoAnalyze software (TA Instruments, New Castle, DE) with an independent binding model.
Human and Rat DHODH Inhibition Assay
Inhibition of human and rat DHODH was measured in vitro using an N-terminally truncated recombinant human or rat DHODH enzyme, respectively, as described in ref (31). The final assay mixture contained 60 μM 2,6-dichloroindophenol, 50 μM decylubiquinone, 100 μM dihydroorotate, and the DHODH protein whose concentration was adjusted in a way that an average slope of approx. 0.2 AU/min served as the positive control (no inhibitor). Measurements were performed in 50 mM TrisHCl, 150 mM KCl, and 0.1% Triton X-100 at pH 8.0 and at 30 °C with at least six different concentrations of a test compound. The reaction was started by adding dihydroorotate and measuring the absorption at 600 nm for 2 min. Each test compound concentration used for IC50 calculation was tested in at least three independent experiments.
Evaluation of Nurr1-Regulated Gene Expression
T98G (ATCC CRL-1690) was grown in DMEM, high glucose supplemented with 10% FCS, sodium pyruvate (1 mM), penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37 °C and 5% CO2 and seeded at a density of 250,000 cells per well in a 12-well plate. After 24 h, the medium was changed to DMEM, high glucose supplemented with 0.2% fetal calf serum (FCS), penicillin (100 U/mL), and streptomycin (100 μg/mL), and the cells were incubated for another 24 h before stimulation with 29 (0.3 and 1 μM). After 16 h of incubation, the medium was removed, cells were washed with phosphate-buffered saline (PBS), and after full aspiration of residual liquids immediately frozen at −80 °C until further procession. Total RNA was isolated using E.Z.N.A. Total RNA Kit I (Omega Bio-tek, Norcross) following the manufacturer’s instructions. RNA concentration and purity were assessed using a NanoDrop One UV–vis spectrophotometer (Thermo Fisher Scientific, Waltham) at 260/280 nm. Right before reverse transcription (RT), RNA was linearized at a concentration of 133 ng/μL at 65 °C for 10 min and then immediately incubated on ice for at least 1 min. Reverse transcription was performed using 2 μg of total RNA, 20 U Recombinant RNasin Ribonuclease Inhibitor (Promega, Mannheim, Germany), 100 U SuperScript IV Reverse Transcriptase including 5× First Strand Buffer and 0.1 M dithiothreitol (Thermo Fisher Scientific, Waltham), 3.75 ng of linear acrylamide, 625 ng of random hexamer primers (#11277081001, Merck, Darmstadt, Germany), and 11.25 nmol of deoxynucleoside triphosphate mix (2.8 nmol each ATP, TTP, CTP, GTP; #R0186, Thermo Fisher Scientific, Waltham) at a volume of 22.45 μL at 50 °C for 10 min and 80 °C for 10 min using a Thermal cycler XT96 (VWR International, Darmstadt, Germany). A quantitative polymerase chain reaction (qPCR) was conducted using an Applied Biosystems QuantStudio 1 (Waltham) and an SYBR green-based detection method. 0.2 μL of prepared cDNA was added to 6 pmol of forward and reverse primers, separately, 0.8 U Taq DNA Polymerase (#M0267, New England Biolabs, Ipswich), 40 ppm SYBR Green I (#S9430, Sigma-Aldrich, St. Louis), 15 nmol of deoxynucleoside triphosphate mix (as indicated above), 60 nmol of MgCl2, 4 μg of bovine serum albumin (#B14, Thermo Fisher Scientific, Waltham), 20% BioStab PCR Optimizer II (#53833, Merck, Darmstadt, Germany), and 10% Taq buffer without detergents (#B55, Thermo Fisher Scientific, Waltham) topped up at a final volume of 20 μL with ddH2O. Samples underwent 40 cycles of 15 s denaturation at 95 °C, 15 s of primer annealing at 62.4 °C, and 20 s of elongation at 68 °C. PCR product specificity was evaluated using a melting curve analysis ranging from 65 to 95 °C. TH and VMAT2 mRNA expression was normalized to GAPDH mRNA expression per sample using the ΔCt-method. The following primers were used: VMAT2 (SLC18A2): 5′-GCT ATG CCT TCC TGC TGA TTG C-3′ (fw) and 5′-CCA AGG CGA TTC CCA TGA CGT T-3′ (rev); TH: 5′-GCT GGA CAA GTG TCA TCA CCT G-3′ (fw) and 5′-CCT GTA CTG GAA GGC GAT CTC A-3′ (rev); GAPDH: 5′-AGG TCG GAG TCA ACG GAT TT-3′ (fw) and 5′-TTC CCG TTC TCA GCC TTG AC-3′ (rev).
Multiplex Toxicity Assay
COS-7 cells (DSMZ #ACC 60) were grown in DMEM high glucose, supplemented with 10% FCS, sodium pyruvate (1 × 10–3 M), penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37 °C and 5% CO2. The day before the experiment, cells were seeded in 96-well plates (5 × 104 cells per well) in a culture medium with reduced serum content (0.2%). The next day, the medium was changed, maintaining the low serum content and additionally containing 0.1% DMSO with 29 (0.1, 0.3, 1, 3 or 10 μM), 0.1% DMSO with bexarotene (100 μM) or flavopiridol (100 μM) as positive controls, or 0.1% DMSO alone as untreated control. After incubation for 24 h, the medium was aspirated, and the cells were washed once with 100 μL of PBS and incubated for 30 min with PBS containing either 1 μM NucView 405 fluorogenic caspase-3 substrate (#10405, Biotium, Fremont) or 0.05× Biotium Live-or-Dye NucFix Red Staining (#BOT-32010-T, Biozol, Eching, Germany) to detect apoptosis and nonregulated forms of cell death, respectively. After incubation, a total of six fluorescence images per well at 10× magnification were taken to detect NucView (Ex: 381–400 nm, Em: 414–450 nm) and NucFix Red (Ex: 543–566 nm, Em: 580–611 nm), using on a Tecan Spark Cyto (Tecan Group AG). Reference readings for background correction and detection of autofluorescence were taken at the given wavelength prior to staining. Thereafter, the medium was aspirated, and the cells were incubated for 3 h with 90 μL of culture medium (0.2% FCS) and 10 μL of Cell Counting Kit-8 solution (CCK-8, MedChem Express #HY-K0301), and absorbance was measured at 2 and 24 h of incubation at 450 nm on a Tecan Spark Cyto to determine the metabolic activity of the cells. Additionally, confluence was assessed using the Tecan Spark Cyto before drug administration, after the first medium exchange, 24 h after drug administration, and after fluorescence imaging.
Pharmacokinetics Analysis in Rats
The pharmacokinetic profile of 29 was evaluated in three female Sprague Dawley rats (8 week old; Janvier Labs, France). All experimental procedures were approved by and conducted in accordance with the regulations of the local Animal Welfare authorities (Landesamt für Gesundheit und Verbraucherschutz, Abteilung Lebensmittel- und Veterinärwesen, Saarbrücken). At six time points (0.25, 0.5, 1, 2, 4, and 8 h) after oral cassette dosing of 5 mg/kg (vehicle: 5% solutol/95% NaCl solution (at 0.9% saline concentration); application volume: 5 mL/kg), blood (20 μL) was collected from the tail vein into Li-heparin tubes, cooled on dry ice within 1–2 min of sampling, and stored at −20 °C until further processing. 29 was quantified by LCMS, and pharmacokinetic analysis was performed using Kinetica 5.0 software (Thermo Scientific, Waltham).
Acknowledgments
This research was co-funded by the European Union (ERC, NeuRoPROBE, 101040355). Views and opinions expressed are, however, those of the author(s) only and do not necessarily reflect those of the European Union or the European Research Council. Neither the European Union nor the granting authority can be held responsible for them. This project was financially supported by Immunic AG, Gräfelfing, Germany. Gal4-VP16 was a gift from Lea Sistonen (Addgene plasmid #71728).
Glossary
Abbreviations Used
- AD
Alzheimer’s disease
- DHODH
dihydroorotate dehydrogenase
- FXR
farnesoid X receptor
- ITC
isothermal titration calorimetry
- LBD
ligand binding domain
- MS
multiple sclerosis
- Nurr1
nuclear receptor related 1
- PD
Parkinson’s disease
- PPAR
peroxisome proliferator-activated receptor
- RXR
retinoid X receptor
- TH
tyrosine hydroxylase
- VMAT2
vesicular amino acid transporter 2
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c00415.
Chemical structures of DHODH inhibitors related to 1; co-crystal structure of hDHODH in complex with A; synthetic procedures and analytical data of 2–28 and their precursors; NMR spectroscopic data and purity analysis of 2–29 (PDF)
Molecular formula strings contain molecular structures of 1–29 and associated activity data (CSV)
The authors declare the following competing financial interest(s): C.G. and H.K. are employees of Immunic AG and own shares and/or stock-options of the parent company of Immunic AG, Immunic Inc. The Immunic AG employees also hold patent applications for some compounds described in this manuscript (WO2022/214691 and PCT/EP2022/087752). There are no further competing interests to declare.
Supplementary Material
References
- Wang Z.; Benoit G.; Liu J.; Prasad S.; Aarnisalo P.; Liu X.; Xu H.; Walker N. P. C.; Perlmann T. Structure and Function of Nurr1 Identifies a Class of Ligand- Independent Nuclear Receptors. Nature 2003, 423, 555–560. 10.1038/nature01645. [DOI] [PubMed] [Google Scholar]
- Decressac M.; Volakakis N.; Björklund A.; Perlmann T. NURR1 in Parkinson Disease - From Pathogenesis to Therapeutic Potential. Nat. Rev. Neurol. 2013, 9, 629–636. 10.1038/nrneurol.2013.209. [DOI] [PubMed] [Google Scholar]
- Willems S.; Merk D. Medicinal Chemistry and Chemical Biology of Nurr1 Modulators: An Emerging Strategy in Neurodegeneration. J. Med. Chem. 2022, 65, 9548–9563. 10.1021/acs.jmedchem.2c00585. [DOI] [PubMed] [Google Scholar]
- Kim C.-H.; Han B.-S.; Moon J.; Kim D.-J.; Shin J.; Rajan S.; Nguyen Q. T.; Sohn M.; Kim W.-G.; Han M.; Jeong I.; Kim K.-S.; Lee E.-H.; Tu Y.; Naffin-Olivos J. L.; Park C.-H.; Ringe D.; Yoon H. S.; Petsko G. A.; et al. Nuclear Receptor Nurr1 Agonists Enhance Its Dual Functions and Improve Behavioral Deficits in an Animal Model of Parkinson’s Disease. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 8756–8761. 10.1073/pnas.1509742112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montarolo F.; Perga S.; Martire S.; Bertolotto A. Nurr1 Reduction Influences the Onset of Chronic EAE in Mice. Inflammation Res. 2015, 64, 841–844. 10.1007/s00011-015-0871-4. [DOI] [PubMed] [Google Scholar]
- Moon M.; Jung E. S.; Jeon S. G.; Cha M.-Y.; Jang Y.; Kim W.; Lopes C.; Mook-Jung I.; Kim K.-S. Nurr1 (NR4A2) Regulates Alzheimer’s Disease-Related Pathogenesis and Cognitive Function in the 5XFAD Mouse Model. Aging Cell 2019, 18, e12866 10.1111/acel.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon M.; Jeong I.; Kim C.-H. H.; Kim J.; Lee P. K. J. J.; Mook-Jung I.; Leblanc P.; Kim K.-S. S. Correlation between Orphan Nuclear Receptor Nurr1 Expression and Amyloid Deposition in 5XFAD Mice, an Animal Model of Alzheimer’s Disease. J. Neurochem. 2015, 132, 254–262. 10.1111/jnc.12935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Gao Y.; Chang N. Nurr1 Overexpression Exerts Neuroprotective and Anti-Inflammatory Roles via down-Regulating CCL2 Expression in Both in Vivo and in Vitro Parkinson’s Disease Models. Biochem. Biophys. Res. Commun. 2017, 482, 1312–1319. 10.1016/j.bbrc.2016.12.034. [DOI] [PubMed] [Google Scholar]
- Satoh J.-i.; Nakanishi M.; Koike F.; Miyake S.; Yamamoto T.; Kawai M.; Kikuchi S.; Nomura K.; Yokoyama K.; Ota K.; Kanda T.; Fukazawa T.; Yamamura T. Microarray Analysis Identifies an Aberrant Expression of Apoptosis and DNA Damage-Regulatory Genes in Multiple Sclerosis. Neurobiol. Dis. 2005, 18, 537–550. 10.1016/j.nbd.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Willems S.; Marschner J.; Kilu W.; Faudone G.; Busch R.; Duensing-Kropp S.; Heering J.; Merk D. Nurr1 Modulation Mediates Neuroprotective Effects of Statins. Adv. Sci. 2022, 9, 2104640 10.1002/advs.202104640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Tello P.; Lin H.; Khan P.; de Vera I. M. S.; Kamenecka T. M.; Kojetin D. J. Assessment of NR4A Ligands That Directly Bind and Modulate the Orphan Nuclear Receptor Nurr1. J. Med. Chem. 2020, 63, 15639–15654. 10.1021/acs.jmedchem.0c00894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruning J. M.; Wang Y.; Oltrabella F.; Tian B.; Kholodar S. A.; Liu H.; Bhattacharya P.; Guo S.; Holton J. M.; Fletterick R. J.; Jacobson M. P.; England P. M. Covalent Modification and Regulation of the Nuclear Receptor Nurr1 by a Dopamine Metabolite. Cell Chem. Biol. 2019, 26, 674–685. 10.1016/j.chembiol.2019.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kholodar S. A.; Lang G.; Cortopassi W. A.; Iizuka Y.; Brah H. S.; Jacobson M. P.; England P. M. Analogs of the Dopamine Metabolite 5,6-Dihydroxyindole Bind Directly to and Activate the Nuclear Receptor Nurr1. ACS Chem. Biol. 2021, 16, 1159–1163. 10.1021/acschembio.1c00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan S.; Jang Y.; Kim C. H.; Kim W.; Toh H. T.; Jeon J.; Song B.; Serra A.; Lescar J.; Yoo J. Y.; Beldar S.; Ye H.; Kang C.; Liu X. W.; Feitosa M.; Kim Y.; Hwang D.; Goh G.; Lim K. L.; et al. PGE1 and PGA1 Bind to Nurr1 and Activate Its Transcriptional Function. Nat. Chem. Biol. 2020, 16, 876–886. 10.1038/s41589-020-0553-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems S.; Ohrndorf J.; Kilu W.; Heering J.; Merk D. Fragment-like Chloroquinolineamines Activate the Orphan Nuclear Receptor Nurr1 and Elucidate Activation Mechanisms. J. Med. Chem. 2021, 64, 2659–2668. 10.1021/acs.jmedchem.0c01779. [DOI] [PubMed] [Google Scholar]
- Willems S.; Müller M.; Ohrndorf J.; Heering J.; Proschak E.; Merk D. Scaffold Hopping from Amodiaquine to Novel Nurr1 Agonist Chemotypes via Microscale Analogue Libraries. ChemMedChem 2022, 17, e2022000 10.1002/cmdc.202200026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems S.; Kilu W.; Ni X.; Chaikuad A.; Knapp S.; Heering J.; Merk D. The Orphan Nuclear Receptor Nurr1 Is Responsive to Non-Steroidal Anti-Inflammatory Drugs. Commun. Chem. 2020, 3, 85 10.1038/s42004-020-0331-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaienne D.; Willems S.; Schierle S.; Heering J.; Merk D. Development and Profiling of Inverse Agonist Tools for the Neuroprotective Transcription Factor Nurr1. J. Med. Chem. 2021, 64, 15126–15140. 10.1021/acs.jmedchem.1c01077. [DOI] [PubMed] [Google Scholar]
- Isigkeit L.; Merk D. Opportunities and Challenges in Targeting Orphan Nuclear Receptors. Chem. Commun. 2023, 59, 4551–4561. 10.1039/D3CC00954H. [DOI] [PubMed] [Google Scholar]
- Leban J.; Kralik M.; Mies J.; Gassen M.; Tentschert K.; Baumgartner R. SAR, Species Specificity, and Cellular Activity of Cyclopentene Dicarboxylic Acid Amides as DHODH Inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 4854–4857. 10.1016/j.bmcl.2005.07.053. [DOI] [PubMed] [Google Scholar]
- Muehler A.; Peelen E.; Kohlhof H.; Gröppel M.; Vitt D. Vidofludimus Calcium, a next Generation DHODH Inhibitor for the Treatment of Relapsing-Remitting Multiple Sclerosis. Mult. Scler. Relat. Disord. 2020, 43, 102129 10.1016/j.msard.2020.102129. [DOI] [PubMed] [Google Scholar]
- Heering J.; Jores N.; Kilu W.; Schallmayer E.; Peelen E.; Muehler A.; Kohlhof H.; Vitt D.; Linhard V.; Gande S. L.; Chaikuad A.; Sreeramulu S.; Schwalbe H.; Merk D. Mechanistic Impact of Different Ligand Scaffolds on FXR Modulation Suggests Avenues to Selective Modulators. ACS Chem. Biol. 2022, 17, 3159–3168. 10.1021/acschembio.2c00599. [DOI] [PubMed] [Google Scholar]
- Sadowski I.; Ma J.; Triezenberg S.; Ptashne M. GAL4-VP16 Is an Unusually Potent Transcriptional Activator. Nature 1988, 335, 563–564. 10.1038/335563a0. [DOI] [PubMed] [Google Scholar]
- Budzyński M. A.; Puustinen M. C.; Joutsen J.; Sistonen L. Uncoupling Stress-Inducible Phosphorylation of Heat Shock Factor 1 from Its Activation. Mol. Cell. Biol. 2015, 35, 2530–2540. 10.1128/MCB.00816-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Zhang J.; Wang J.; Ren C.; Tang P.; Ouyang L.; Wang Y. Recent Advances of Human Dihydroorotate Dehydrogenase Inhibitors for Cancer Therapy: Current Development and Future Perspectives. Eur. J. Med. Chem. 2022, 232, 114176 10.1016/j.ejmech.2022.114176. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Quah J. Y.; Ng Y.; Chooi J. Y.; Toh S. H. M.; Lin B.; Tan T. Z.; Hosoi H.; Osato M.; Seet Q.; Lisa Ooi A. G.; Lindmark B.; McHale M.; Chng W. J. ASLAN003, a Potent Dihydroorotate Dehydrogenase Inhibitor for Differentiation of Acute Myeloid Leukemia. Haematologica 2020, 105, 2286–2297. 10.3324/haematol.2019.230482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainas S.; Pippione A. C.; Lupino E.; Giorgis M.; Circosta P.; Gaidano V.; Goyal P.; Bonanni D.; Rolando B.; Cignetti A.; Ducime A.; Andersson M.; Järvå M.; Friemann R.; Piccinini M.; Ramondetti C.; Buccinnà B.; Al-Karadaghi S.; Boschi D.; et al. Targeting Myeloid Differentiation Using Potent 2-Hydroxypyrazolo[1,5-a]Pyridine Scaffold-Based Human Dihydroorotate Dehydrogenase Inhibitors. J. Med. Chem. 2018, 61, 6034–6055. 10.1021/acs.jmedchem.8b00373. [DOI] [PubMed] [Google Scholar]
- Nair A.; Barde P. J.; Routhu K. V.; Viswanadha S.; Veeraraghavan S.; Pak S.; Peterson J. A.; Vakkalanka S. A First in Man Study to Evaluate the Safety, Pharmacokinetics and Pharmacodynamics of RP7214, a Dihydroorotate Dehydrogenase Inhibitor in Healthy Subjects. Br. J. Clin. Pharmacol. 2022, 89, 1127–1138. 10.1111/bcp.15562. [DOI] [PubMed] [Google Scholar]
- McDonald G.; Chubukov V.; Coco J.; Truskowski K.; Narayanaswamy R.; Choe S.; Steadman M.; Artin E.; Padyana A. K.; Jin L.; Ronseaux S.; Locuson C.; Fan Z. P.; Erdmann T.; Mann A.; Hayes S.; Fletcher M.; Nellore K.; Rao S. S.; et al. Selective Vulnerability to Pyrimidine Starvation in Hematologic Malignancies Revealed by AG-636, a Novel Clinical-Stage Inhibitor of Dihydroorotate Dehydrogenase. Mol. Cancer Ther. 2020, 19, 2502–2515. 10.1158/1535-7163.MCT-20-0550. [DOI] [PubMed] [Google Scholar]
- Pirali T.; Serafini M.; Cargnin S.; Genazzani A. A. Applications of Deuterium in Medicinal Chemistry. J. Med. Chem. 2019, 62, 5276–5297. 10.1021/acs.jmedchem.8b01808. [DOI] [PubMed] [Google Scholar]
- Baumgartner R.; Walloschek M.; Kralik M.; Gotschlich A.; Tasler S.; Mies J.; Leban J. Dual Binding Mode of a Novel Series of DHODH Inhibitors. J. Med. Chem. 2006, 49, 1239–1247. 10.1021/jm0506975. [DOI] [PubMed] [Google Scholar]
- Bateman A.; Martin M. J.; Orchard S.; Magrane M.; Agivetova R.; Ahmad S.; Alpi E.; Bowler-Barnett E. H.; Britto R.; Bursteinas B.; Bye-A-Jee H.; Coetzee R.; Cukura A.; da Silva A.; Denny P.; Dogan T.; Ebenezer T. G.; Fan J.; Castro L. G.; et al. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. 10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacchi S.; Fabrizi G.; Goggiamani A. Palladium-Catalyzed Hydroxycarbonylation of Aryl and Vinyl Halides or Triflates by Acetic Anhydride and Formate Anions †. Org. Lett. 2003, 5, 4269–4272. 10.1021/ol0354371. [DOI] [PubMed] [Google Scholar]
- Leban J.; Saeb W.; Garcia G.; Baumgartner R.; Kramer B. Discovery of a Novel Series of DHODH Inhibitors by a Docking Procedure and QSAR Refinement. Bioorg. Med. Chem. Lett. 2004, 14, 55–58. 10.1016/j.bmcl.2003.10.021. [DOI] [PubMed] [Google Scholar]
- Gellrich L.; Heitel P.; Heering J.; Kilu W.; Pollinger J.; Goebel T.; Kahnt A.; Arifi S.; Pogoda W.; Paulke A.; Steinhilber D.; Proschak E.; Wurglics M.; Schubert-Zsilavecz M.; Chaikuad A.; Knapp S.; Bischoff I.; Fürst R.; Merk D. L-Thyroxin and the Nonclassical Thyroid Hormone TETRAC Are Potent Activators of PPARγ. J. Med. Chem. 2020, 63, 6727–6740. 10.1021/acs.jmedchem.9b02150. [DOI] [PubMed] [Google Scholar]
- Pollinger J.; Gellrich L.; Schierle S.; Kilu W.; Schmidt J.; Kalinowsky L.; Ohrndorf J.; Kaiser A.; Heering J.; Proschak E.; Merk D. Tuning Nuclear Receptor Selectivity of Wy14,643 towards Selective Retinoid X Receptor Modulation. J. Med. Chem. 2019, 62, 2112–2126. 10.1021/acs.jmedchem.8b01848. [DOI] [PubMed] [Google Scholar]
- Rau O.; Wurglics M.; Paulke A.; Zitzkowski J.; Meindl N.; Bock A.; Dingermann T.; Abdel-Tawab M.; Schubert-Zsilavecz M. Carnosic Acid and Carnosol, Phenolic Diterpene Compounds of the Labiate Herbs Rosemary and Sage, Are Activators of the Human Peroxisome Proliferator-Activated Receptor Gamma. Planta Med. 2006, 72, 881–887. 10.1055/s-2006-946680. [DOI] [PubMed] [Google Scholar]
- Moret M.; Helmstädter M.; Grisoni F.; Schneider G.; Merk D. Beam Search for Automated Design and Scoring of Novel ROR Ligands with Machine Intelligence**. Angew. Chem., Int. Ed. 2021, 60, 19477–19482. 10.1002/anie.202104405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flesch D.; Cheung S.-Y.; Schmidt J.; Gabler M.; Heitel P.; Kramer J. S.; Kaiser A.; Hartmann M.; Lindner M.; Lüddens-Dämgen K.; Heering J.; Lamers C.; Lüddens H.; Wurglics M.; Proschak E.; Schubert-Zsilavecz M.; Merk D. Nonacidic Farnesoid X Receptor Modulators. J. Med. Chem. 2017, 60, 7199–7205. 10.1021/acs.jmedchem.7b00903. [DOI] [PubMed] [Google Scholar]
- Schmidt J.; Klingler F.-M.; Proschak E.; Steinhilber D.; Schubert-Zsilavecz M.; Merk D. NSAIDs Ibuprofen, Indometacin, and Diclofenac Do Not Interact with Farnesoid X Receptor. Sci. Rep. 2015, 5, 14782 10.1038/srep14782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitel P.; Gellrich L.; Kalinowsky L.; Heering J.; Kaiser A.; Ohrndorf J.; Proschak E.; Merk D. Computer-Assisted Discovery and Structural Optimization of a Novel Retinoid X Receptor Agonist Chemotype. ACS Med. Chem. Lett. 2019, 10, 203–208. 10.1021/acsmedchemlett.8b00551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer I.; Willems S.; Ni X.; Heering J.; Chaikuad A.; Merk D. Chemical Starting Matter for HNF4α Ligand Discovery and Chemogenomics. Int. J. Mol. Sci. 2020, 21, 7895 10.3390/ijms21217895. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.