

Abstract
Background and Aims
DNA mismatch repair deficiency (MMRD) drives microsatellite instability (MSI). Cells with MSI accumulate numerous frameshift mutations. Frameshift mutations affecting cancer-related genes may promote tumorigenesis and, therefore, are shared among independently arising MSI tumors. Consequently, such recurrent frameshift mutations can give rise to shared immunogenic frameshift peptides (FSPs) that represent ideal candidates for a vaccine against MSI cancer. Pathogenic germline variants of mismatch repair genes cause Lynch syndrome (LS), a hereditary cancer syndrome affecting approximately 20–25 million individuals worldwide. LS individuals are at high risk of developing MSI cancer. Previously, we demonstrated safety and immunogenicity of an FSP-based vaccine in a Phase I/IIa clinical trial in patients with a history of MSI colorectal cancer. However, the cancer-preventive effect of FSP vaccination in the scenario of LS has not been demonstrated so far.
Methods
A genome-wide database of 488,235 mouse coding mononucleotide repeats was established, from which a set of candidates was selected based on repeat length, gene expression and mutation frequency. In silico prediction, in vivo immunogenicity testing and epitope mapping was used to identify candidates for FSP vaccination.
Results
We identified four shared FSP neoantigens [Nacad(FSP-1), Maz(FSP-1), Senp6(FSP-1), Xirp1(FSP-1)] that induced CD4/CD8 T cell responses in naïve C57BL/6 mice. Using VCMsh2 mice, which have a conditional knockout of Msh2 in the intestinal tract and develop intestinal cancer, we showed vaccination with a combination of only four FSPs significantly increased FSP-specific adaptive immunity, reduced intestinal tumor burden and prolonged overall survival. Combination of FSP vaccination with daily naproxen treatment potentiated immune response, delayed tumor growth and prolonged survival even more effectively than FSP vaccination alone.
Conclusion
Our pre-clinical findings support a clinical strategy of recurrent FSP neoantigen vaccination for LS cancer immunoprevention.
Keywords: colorectal cancer, frameshift neoantigens, Lynch syndrome, mouse model, preventive cancer vaccine
Graphical Abstract
INTRODUCTION
Mismatch repair deficiency (MMRD) is an important mechanism driving mutagenesis and genomic instability in human cancers. MMR-deficient cells accumulate numerous somatic mutations including insertion/deletion (indel) mutations, predominantly altering repetitive microsatellite (MS) sequences1, 2. Indels in coding MS promote translational frameshifts, which also generate truncated frameshift peptide (FSP)-encoding neoproteins 3. Several studies have identified a large spectrum of genes affected by such frameshift mutations, thus demonstrating that indel mutations affecting key tumor suppressors such as the TGFBR2 are enriched in MMRD cancers 4–6.
MMRD cancers can develop sporadically or from hereditary predisposition as part of Lynch syndrome (LS). LS causes up to 2–5% of all colorectal cancers (CRCs)7 as well as endometrial, ovarian, uroepithelial and other cancers8. The estimated population incidence of LS is 1:225 – 1:360 9, 10. Affected individuals have an estimated 20–70% lifetime risk of developing cancer 11, 12. LS is caused mainly by heterozygous germline mutations in one of the MMR genes MSH2, MLH1, MSH6, or PMS2 13, 14. Inactivation of both alleles of an individual MMR gene is required to cause the MMRD, typically by somatic “second hit” inactivation of the functional MMR allele 15, 16.
MMRD CRCs display distinct clinico-histopathological features that are directly related to the high FSP load. Most importantly, these include increased infiltration with lymphocytes (TIL), memory T cells and improved survival compared to patients with low TIL CRC 17–19. Accordingly, MMRD tumors are among the most responsive to immune checkpoint inhibitors that enhance endogenous anti-tumor adaptive immunity, which is driven predominantly by FSPs20.
The existence of shared FSP neoantigens in MMRD cancers creates a mechanism-based framework for novel tumor immunopreventive approaches 3. Immunological studies performed by our group and others have identified several immunogenic MSI-associated FSPs in MMRD CRCs that are recognized by, and promote proliferation of, cytotoxic T cells 21, 22. Importantly, endogenous adaptive immunity against FSPs is detected in MMRD cancer patients, and also in tumor-free LS “previvors”, suggesting a role for immune surveillance in LS mutation carriers 23.
Vaccination with recurrent FSPs that are shared by multiple MMRD tumors of different patients is a promising approach to boost immune surveillance of MMRD pre-cancerous cell clones, and potentially immune-interception of sub-clinical MMRD tumors for effective immunoprevention 3. Recently, we performed a therapeutic phase IIa clinical trial demonstrating the safety and immunological efficacy of a tri-valent recurrent FSP-based vaccine in patients with a history of MMRD MSI CRC 24. However, whether recurrent FSP vaccination can reduce LS/sporadic MMRD tumor burden and prolong patient survival, in addition to boosting anti-tumor immunity, is unknown.
Non-steroidal anti-inflammatory drugs (NSAIDs), particularly aspirin (acetylsalicylic acid or ASA), have been intensively studied for GI cancer prevention. NSAIDs reduce Cyclooxygenase 1 and 2 production of prostaglandin E2 (PGE2), which binds to EP1–4 receptors 25. PGE2 drives intestinal tumorigenesis by both promoting pro-tumorigenic EP2/4 driven intestinal epithelial and stem cell proliferation and inhibiting immune surveillance and immune-interception of tumor neoantigens 26, 27. ASA reduces LS colorectal cancer incidence and is widely used for LS GI cancer prevention25, 28. However, a recent large-scale (approximately 20,000 participants) randomized clinical trial in community-dwelling older (>65 yo) men and women has raised questions whether ASA may actually increase overall pan-cancer rates and mortality, at least in elderly29. Recently, Naproxen (NAP), a propionic acid NSAID derivative, has shown pronounced cancer-preventive activity in LS mouse models, and increased immune surveillance in LS patients 30, 31.
LS mouse models have provided many important mechanistic and translational insights. Intestinal epithelial-specific MMRD mouse models such as VCMsh232 closely resemble the clinical phenotype seen in LS CRC patients because they specifically develop intestinal tumors, whereas constitutional MMRD mice (e.g. Msh2null) most frequently develop T cell lymphomas, which confounds survival analysis33. Previously we described a set of candidate coding mononucleotide repeat (cMNR) frameshift mutations in a small number of MSI mouse tumors and detected human/mouse orthologous conserved cMNR repeats34. Here, we used VCMsh2 mice to test the hypothesis that recurrent FSP vaccination alone or in combination with NSAID treatment can promote anti-FSP adaptive immunity sufficiently to reduce intestinal tumor burden and prolong overall survival.
MATERIALS & METHODS
Selection of frameshift peptides for vaccination
Computational Analysis of Mouse Genome to Identify Coding Microsatellites
Search tools and algorithms previously developed/applied for the human cMNR database (www.seltarbase.de)34–36 were adapted accordingly to detect all cMNRs in the mouse genome. Perl scripts were developed to use the ensembl API (www.ensembl.org/info/docs/api/) and a rigorous redundancy check at the 98% level was applied. All annotation-based cMNRs with a minimal repeat length of four mononucleotides were retrieved. Using several filters, repeat tracts within pseudogenes, vector sequences as well as homopolymeric nucleotide stretches at the most 5′ or 3′ ends of sequences were excluded. Candidate sequences were stored in a relational database for further analysis (http://www.bork.embl.de/Docu/yuan/rpt/). Processing through this analysis pipeline was based on gene sequence data of the Mus musculus Ensembl release version 77_38.
Mutation and expression analysis of cMNRs in VCMsh2 FFPE tumors
For MSI classification of identified tumors, four long non-coding microsatellite markers were used (mA24, mA27, mT27, mA33z34, 37). Marker MSI was defined by the occurrence of novel peaks in tumor compared to normal tissue. Tumor MSI was scored if at least 1/4 markers showed instability. For PCR-based indel mutation analysis, a candidate set of 56 cMNRs (Supplemental Figure 1) was selected. These cMNR candidates were chosen based on repeat length (≥ 8 nucleotides), representation of all 4 nucleotides, transcript isoform coverage, and robust cMNR amplification.
Predicted Computational NetMHC and SYFPETHI Immunogenicity of FSPs
To predict the potential immunogenicity of FSPs resulting from cMNR mutations, peptide sequences were submitted to online available epitope prediction tools SYFPEITHI (www.syfpeithi.de) and netMHC4.0 (http://www.cbs.dtu.dk/services/NetMHC/). Searches were performed for MHC class I antigens present in C57BL/6 mice (H2-Db and -Kb alleles). For SYFPEITHI, the calculated score was recorded, for netMHC4.0, the predicted percentage rank indicating MHC binding likelihood and the number of predicted strong binders and weak binders (corresponding to an affinity lower than 50 μM and 500 μM, respectively) were recorded for each peptide.
Evaluation of FSP immunogenicity in C57BL/6 mice
FSPs were synthesized by Genaxxon (Ulm, Germany). Details of peptide solution are described in Supplemental Material. For preparation of the vaccine formulations, 50 μg of 3 to 4 FSPs were mixed with 50 μg of OVA 257–264 and OVA 323–339 each and 20 μg CpG ODN 1826; the mixture was suspended in a final injection volume of 50 μl in PBS.
Control vaccine formulations without FSPs was prepared as described above, adding PBS in place of the FSPs. Vaccine mixes were administered four times in bi-weekly intervals to C57BL/6 mice. All vaccines were administered subcutaneously into the left or right flank of each mouse. One week after the last vaccination, cellular immune responses were measured using IFN-γ ELISpot assay from splenocytes. Humoral FSP-specific immune responses were measured using peptide ELISA. Reactions significantly above background after subtraction of no-peptide control spots and standard deviations of the peptide and the control spot counts were considered as positive. Assays for determining cellular and humoral immune responses were performed according to previously established protocols described in Supplemental Material. Detailed protocols for adjuvant optimization are provided in Supplemental Material.
Immunoprevention in VCMsh2 mice
Mouse Strains and Tissue Collection
Villin-Cre mice were bred to Msh2 LoxP/LoxP mice to generate VCMsh2 mice in the laboratory of Winfried Edelmann previously 32. Mice were genotyped to confirm the status of Cre transgene and floxed Msh2, respectively. Mice with the Villin promoter used to drive expression of Cre Recombinase and Msh2 exon 12 flanked by LoxP sites (VCMsh2) mice on the C57BL/6 genetic background32 were bred and housed in an SPF barrier facility at Weill Cornell Medical College according to Institutional Animal Care and Use Committee (IACUC) guidelines.
Cancer prevention and vaccination protocols
Control mice were given pelleted plain New Western Diet 1 (NWD1, Research Diets). ASA and NAP were purchased from Spectrum Chemicals Inc in powder form. Aspirin or Naproxen impregnated pelleted NWD1 (400 ppm ASA or 166 ppm NAP) at the age of weaning were given to mice assigned to those arms. For FSP vaccination, all peptides [Nacad(FSP-1), Xirp1(FSP-1), Maz(FSP-1) and Senp6(FSP-1)] were custom made by Thermo Fisher Scientific at >98% purity and the adjuvant CpG ODN1826 was purchased from InvivoGen. Vaccine components were prepared following manufacturer’s protocol in DMSO and combined fresh before each vaccination. The study consists of six arms: Controls, Vaccine only, NAP only, ASA only, Vaccine + NAP, and Vaccine + ASA. Starting at the age of 6–8 weeks, mice enrolled in FSP vaccine alone and FSP vaccine plus NSAID arms were vaccinated subcutaneously in either left or right hind 4 times bi-weekly followed by 4 times monthly. Per shot, each mouse received 50 μg of each FSP combined with 20 μg of the adjuvant.
Each treatment group was populated with 16–20 mice on a rolling basis for efficacy studies. Mice were monitored over their lifetime by Research Animal Resources and Compliance Veterinary Services and the investigator twice a week for the following signs to determine euthanasia; (1) weight loss (2) poor coat quality (3) hunched posture (4) pale limbs (anemia). Following the IACUC guidelines, mice presenting with the above signs were euthanized by CO2 inhalation.
Regulatory Compliance
All VCMsh2 mice were housed in isolation units approved by the Weill-Cornell Institutional Animal Care and Use Committee (Protocol number: AC-AAAN5700). The mice used in trials were allowed to run free in the cage, were fed the Western diet or NSAID-containing matched diet (Research Diets), and were provided water ad libitum. The Animal Care Facilities at DKFZ were utilized for housing C57BL/6 mice for frameshift peptide selection. The DKFZ facilities have been approved by FELASA and accredited. Adjuvant optimization studies were performed in the Frederic National lab according to IRB and ACUC regulations.
Immunohistochemistry (IHC)
Immunohistochemistry staining was performed on formalin-fixed, paraffin-embedded tissues from intestinal tumors taken from VCMsh2 mice treated with naproxen with or without the tetravalent FSPs vaccine according to standard protocols (see Supplementary Methods). Following primary antibodies were used: for CD3: clone SP7 abcam ab16669; for CD4: clone 4SM95 ThermoFisher Scientific #14–9766-82; for CD8: clone 4SM15 ThermoFisher Scientific #14–0808-82; for Foxp3: clone FJK-16s ThermoFisher Scientific #14–5773-82; for PD-1: clone EPR20665 abcam ab214421. Following biotinylated secondary antibodies were used: for CD3 and PD-1: Vector Laboratories BA-1100; for CD4, CD8 and Foxp3: Vector Laboratories BA-9401.
Immune cell quantification
The tissue slides were scanned with a NanoZoomer S210 slide scanner (Hamamatsu, Japan) with a scanning resolution of 0.23 μm/pixel in the 40x mode. The DAB chromogen positive stained cells were identified with QuPath (Version: 0.1.238) using the positive cell detection feature in at least one region of interest with an area of 0.25 mm2 within each tumor slide. The number of positive cells was counted and recorded. Tissue sections with insufficient quality or with no staining signal detectable in the entire section were excluded from the analysis.
Statistical evaluation
Mann-Whitney tests comparing the FSP vaccine group against the control group were performed using GraphPad Prism version 8.2.1 for Windows (GraphPad Software, San Diego, California USA, www.graphpad.com). GraphPad was also used to generate dot plots. Mann-Whitney comparisons of tumor burden (sum of all intestinal tumor weights per mouse) and Kaplan-Meier curve overall survival analyses were also performed using GraphPad Prism. Statistical analyses of adjuvant comparison results were conducted with GraphPad Prism 7 software using one-way ANOVA nonparametric analysis with the Kruskal-Wallis multiple comparisons test. P < 0.05 was considered significant.
RESULTS
Computational Identification of Mouse Genome Coding Microsatellites
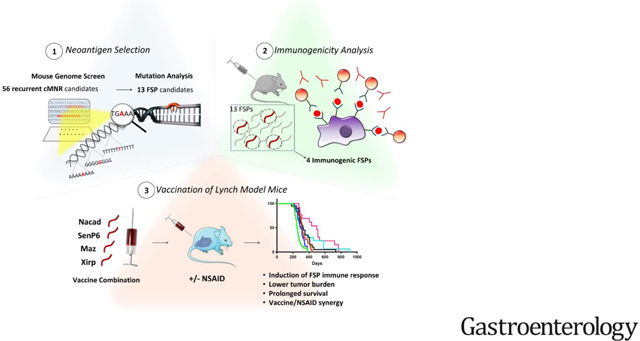
A multistep strategy was employed to develop a mouse vaccine comprised of the most immunogenic FSP candidates and to test their efficacy in immunocompetent VCMsh2 mice (Figure 1).
Figure 1. Experimental Strategy for Developing a Murine FSP Vaccine.

Using a computational approach, we established a genome-wide database of coding microsatellites in the mouse genome. This database comprises 488,235 cMNRs consisting of ≥ 4 nucleotides in length (Figure 2). Since increased repeat length correlates with increased cMNR mutation probability5, 6, we focused our subsequent analyses on cMNRs with a repeat length of ≥ 8 nucleotides.
Figure 2. Distribution of coding mononucleotide repeats (cMNRs) in the mouse genome.

The occurrence of cMNRs is shown according to repeat type and length(≥ 8 nucleotides).
cMNR Frameshift Mutation Patterns in Intestinal Tumors of VCMsh2 mice
For mutational analyses of candidate cMNRs, intestinal adenomas and carcinomas as well as normal mucosae from the same animals were collected from VCMsh2 mice (n=25; age between 7 and 15 months, one tumor per animal). Tumors were confirmed as MSI/MMRD when analyzed by a panel of mononucleotide repeats (mA24, mA27, mT27, mA33) previously established as sensitive and specific for MSI detection in mouse tumors 5. Frameshift mutation analyses were performed on a selected subset of 56 cMNR candidates that showed increased repeat length (≥ 8 nucleotides), represented all four nucleobases, covered different transcript isoforms and allowed robust amplification from FFPE tissue specimens (Supplemental Figure 1). The majority of these cMNR candidates (36/56, 64.3%) carried frameshift mutations in one or more tumors, with mutation frequencies ranging from 6.5%−75% (Supplemental Figure 1). After eliminating candidates polymorphic in normal tissue and/or showing a mutation frequency of <15% in tumors, we further evaluated 13 remaining cMNR candidate genes and confirmed their expression in normal and tumor tissues by RT-PCR.
In silico prediction of Frameshift Peptide Immunogenicity
We next searched for potential immunogenic MHC binding motifs in 26 FSP neopeptides derived from (−1) and (−2) indel mutations affecting these 13 cMNRs (Table 1). Using epitope prediction tools netMHC4.0 and SYFPEITHI 1.0, a subset of 10 FSPs turned up as potentially immunogenic candidates and were selected for subsequent immunological analyses. Based on our mutation data, these 10 FSPs covered most of the analyzed tumors (15/16; 93%)
Table 1.
Mutation frequency of cMNR and FSP sequences
| Gene | cMNR | Intestinal Expression | Mutation Frequency | WT FSP Sequence | SYFPEITHI Score | netMHC | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Type | Monomorphic | Normal | Tumor | Total | FS-Specific | Db | Kb | % rank | ||||
|
| ||||||||||||
| Nacad | C14 | yes | yes | yes | 75% | FS(−1) | 56.25% | VIYAPPPPAEGRWPCWLLRAH* | 15 | s0 | s0 | 3 |
| w0 | w0 | |||||||||||
| FS(−2) | 37.5% | DVIYAPPPQQRGGGRAGYSERIDGQRDRETGVSAGTRPGHARGGCGR* | 14 | s0 | s0 | 6 | ||||||
| w0 | w0 | |||||||||||
|
| ||||||||||||
| Xirp1 | C9 | yes | yes | yes | 37.5% | FS(−1) | 31.25% | GKGPGGPPLSSPKRVMYRLSVGCLRPTL* | 19 | s0 | s1 | 0.4 |
| w2 | w4 | |||||||||||
| FS(−2) | 6.25% | GKGPGGPP* | - | - | - | - | ||||||
|
| ||||||||||||
| 5730596B20Rik | C12 | yes | yes | yes | 37.5% | FS(−1) | 37.5% | GTLPPPPPTQH* | 6 | s0 | s0 | 39 |
| w0 | w0 | |||||||||||
| FS(−2) | 0.00% | LGTLPPPPQPSTEQSGWKHHQ* | 7 | s0 | s0 | 55 | ||||||
| w0 | w0 | |||||||||||
|
| ||||||||||||
| Rif1 | A12 | yes | yes | yes | 33.3% | FS(−1) | 20.00% | AHTDKKKK* | - | - | - | - |
| FS(−2) | 13.33% | AHTKDKKKSETVGQTETRIFISKNKEW* | 15 | s0 | s0 | 4.5 | ||||||
| w0 | w0 | |||||||||||
|
| ||||||||||||
| Maz | C8 | yes | yes | yes | 33.3% | FS(−1) | 20.00% | PCTLLAPPSPCWAWTPGGWAAS* | 7 | s0 | s0 | 9.5 |
| w0 | w0 | |||||||||||
| FS(−2) | 13.33% | FPCTLLAPLPRAGPGLPGGGRPHELLPATSGSRPEPPAGRG* | 17 | s0 | s0 | 3.5 | ||||||
| w0 | w0 | |||||||||||
|
| ||||||||||||
| Hic1 | C10 | yes | yes | yes | 31.25% | FS(−1) | 18.75% | DRTFPSPPRIGAI* | 14 | s0 | s0 | 5 |
| w0 | w0 | |||||||||||
| FS(−2) | 12.5% | DRTFPSPPELARYNI* | 18 | s0 | s0 | 4.5 | ||||||
| w0 | w0 | |||||||||||
|
| ||||||||||||
| Sdccag1 | A11 | yes | yes | yes | 25% | FS(−1) | 25.00% | EAPKGKKKSKRTSSCRSRRRTSRCL* | 12 | s0 | s0 | 8 |
| w0 | w0 | |||||||||||
| FS(−2) | 6.25% | EAPKGKKKAKEQAAAEAAEEQAAACRCGSQPVSLCQCQKIL* | 16 | s0 | s0 | 2.5 | ||||||
| w0 | w0 | |||||||||||
|
| ||||||||||||
| Tmem107 | G9 | yes | yes | yes | 25% | FS(−1) | 25.00% | TQYFGMGGVVENRSQI* | 24 | s2 | s0 | 0.5 |
| w1 | w0 | |||||||||||
| FS(−2) | 6.25% | TQYFGMGGWWKIDPKSEGFPHLDLSCTCEIGRVLKSHTHPNPPAYKVWRFKCLGWKGL* | 21 | s0 | s1 | 0.25 | ||||||
| w0 | w2 | |||||||||||
|
| ||||||||||||
| Srcin1 | C9 | yes | yes | yes | 25% | FS(−1) | 25.00% | DEGMWPPPTTS* | 6 | s0 | s0 | 31 |
| w0 | w0 | |||||||||||
| FS(−2) | 0.00% | VDEGMWPPQQPPEPVPQEGGS* | 12 | s0 | s0 | 22 | ||||||
| w0 | w0 | |||||||||||
|
| ||||||||||||
| Marcks | A11 | yes | yes | yes | 20% | FS(−1) | 20.00% | SSETPKKKRSAFPSRSPSS* | 12 | s0 | s0 | 5.5 |
| w0 | w0 | |||||||||||
| FS(−2) | 0.00% | SSETPKKKEALFLQEVLQAERLLLQEEQEGVGRGR* | 16 | s0 | s0 | 0.8 | ||||||
| w3 | w0 | |||||||||||
|
| ||||||||||||
| Senp6 | A11 | yes | yes | yes | 18.75% | FS(−1) | 18.75% | VKCSMKKKIMLSMKMKNQVTENLRARTFVIEPKVRMASGMNASVLYIIQMP* | 27 | s10 | s0 | 0.03 |
| w12 | w2 | |||||||||||
| FS(−2) | 0.00% | VKCSMKKKSCYQ* | 1 | s0 | s0 | 20 | ||||||
| w0 | w0 | |||||||||||
|
| ||||||||||||
| Phactr4 | A10 | yes | yes | yes | 18.75% | FS(−1) | 18.75% | PWKWRKKKAVISSKRHQKF* | 12 | s0 | s0 | 3.5 |
| w0 | w0 | |||||||||||
| FS(−2) | 0.00% | PWKWRKKKQ* | 2 | s0 | s0 | 75 | ||||||
| w0 | w0 | |||||||||||
|
| ||||||||||||
| Chrnb2 | C10 | yes | yes | yes | 18.75% | FS(−1) | 12.5% | VRTRPSPPHLSPASWVLKPFAINAKGIFLILCGNWQQGCLCHLGMHLRHRQVGL* | 21 | s1 | s0 | 0.5 |
| w3 | w1 | |||||||||||
| FS(−2) | 6.25% | VRTRPSPPISLQPHGS* | 16 | s0 | s0 | 11 | ||||||
| w0 | w0 | |||||||||||
Genes affected by coding mononucleotide (cMNR) frameshift (FS) mutations and derived frameshift peptides (FSPs). Predicted immunoscores for FSPs (bold letters) including an 8 amino acid wild type (WT) sequence are indicated for C57BL/6 alleles H2-Db and H2-Kb (SYFPEITHI 1.0 and netMHC4.0). The numbers of strong (s) and weak (w) binders are shown. Low numbers of percent rank (% rank) as well as SYFPEITHI scores >12 predict strong MHC binding. FSPs used for further analysis are indicated by grey boxes.
Induction of FSP-specific cellular and humoral immune responses in C57BL/6 mice
The immunogenicity of 10 FSPs with highest in silico prediction scores was experimentally evaluated in C57BL/6 mice (Figure 3A). To determine induction of antigen-specific T cell responses, ex vivo IFN-γ ELISpot assays were performed. Four out of 10 FSP neoantigens, i.e. Maz(FSP-1), Nacad(FSP-1), Xirp1(FSP-1) and Senp6(FSP-1) triggered the induction of T cell responses significantly above background in vaccinated C57BL/6 mice (Figure 3B). Vaccination was repeated using a mixture of the four positive candidates, and ELISpot was repeated for each peptide separately to validate the immunogenicity of FSP neoantigens when administered mixed together. Separate analysis of CD4 and CD8 T cell responses demonstrated that Maz(FSP-1) and Senp6(FSP-1) elicited CD4 T cell responses, whereas Xirp1(FSP-1) predominantly induced a CD8 T cell response. Nacad(FSP-1) induced both, CD4 and CD8 T cell responses (Figure 3C). Characterization of the immunogenic regions of the FSPs by utilizing shorter peptide fragments from the N- and C-terminal part of the FSPs demonstrated that the immunogenic parts of Xirp1(FSP-1) and Senp6(FSP-1) are located at the C-terminus of their FSPs in contrast to Nacad(FSP-1) whose immunogenic region resides within the N-terminus of this FSP. A mixed pattern was observed for Maz(FSP-1) which is immunogenic as a whole in both parts of the FSP. Epitope location was correlated with in silico predictions (Figure 3D). Potential immunogenicity of wildtype peptide stretches at the N-terminus was excluded, because cross-reactivity with the respective wildtype protein sequences was observed (Supplemental Figure 2). Three FSPs Nacad(FSP-1), Senp6(FSP-1) and Maz(FSP-1) also elicited a humoral immune responses detectable by peptide ELISA (Supplemental Figure 3).
Figure 3. FSP Immunogenicity.

(A) FSP vaccination scheme of C57BL/6 mice. FSP mixes or OVA mix (50μg each) were injected subcutaneously biweekly for four times using CpG ODN 1826 (20μg) as an adjuvant. The vaccine schedule was chosen to ensure life-long robust FSP-specific immune responses, including a starting point in early adulthood and booster vaccinations after the initial priming phase. (B) IFN-γ ELISpot analysis. The average number of spot forming units (SFUs) for each mouse is shown for each peptide. OVA mix corresponds to the mixture of the OVA peptide CD4 and CD8 epitope, which was used as a control in this experimental setup. (C) CD4 and CD8 T cell responses against four FSPs. Average SFU numbers are presented for each peptide. (D) Epitope mapping of immunogenic peptides. Wildtype peptides (grey) as well as FSPs with overlapping N/C-terminal sequences (blue boxes, red amino acids) were synthesized and immunogenicity for the regions where epitopes might be located was determined by IFN-γ ELISpot. The average number of SFUs per mouse are shown in the graph. (E) Endogenous FSP reactivity in Lynch Mice. IFN-γ ELISpot analysis. Each dot represents the number of spot forming units (SFUs) of splenocytes per mouse. VCMsh2 mice were pulsed with a mixture of 4 peptides (FSPs). C57BL/6 (B6) mice pulsed with either FSPs or OVA peptide served as control. The median, interquartile range/standard deviation and significance level is indicated.
Early occurrence of recurrent FSP mutations in VCMsh2 intestinal tumors
We evaluated a separate non-overlapping cohort of 7 month old VCMsh2 mice (n=16 mice, 25 tumors) fed on regular chow diet to test if recurrent Maz, Nacad, Senp6 or Xirp1 FSP are early mutations that could serve as candidate vaccine neoantigens. VCMsh2 mice develop intestinal tumors starting at age 6–9 months 32. When MSI tumors (n=16) were tested for FS mutations, moderate to high frequencies were observed for Nacad(−1) (50%), Senp6(−1) (25%) and Maz(−1) (6%). Thus, even tumors occuring in VCMsh2 mice at young ages have already acquired FSP mutations. These FSP mutations most likely represent early mutation events. FS mutations in Xirp1 were not detected in MSI tumors of mice at the age of 7 months, but at a high frequency in tumors from older mice (Table 1).
VCMsh2 endogenous adaptive immunity against recurrent FSP mutations
To test if VCMsh2 mice have endogenous adaptive immunity directed against recurrent FSP neoantigens that could be boosted by vaccination, we performed splenocyte IFN-γ ELISpot for Maz(FSP-1), Nacad(FSP-1), Senp6(FSP-1) and Xirp1(FSP-1) in 8–15 month old C57BL/6 and VCMsh2 mice. This showed that compared to C57BL/6 control or OVA peptide-immunized mice (N=5 and 6 respectively), VCMsh2 splenocytes pulsed with Maz(FSP-1), Nacad(FSP-1), Senp6(FSP-1) and Xirp1(FSP-1) peptides in vitro had significantly higher numbers of activated IFN-γ+ T cells (Figure 3E). Thus, similar to our previous findings that LS mutation carriers have endogenous adaptive immunity against recurrent FSP neoantigens 23, VCMsh2 mice have endogenous adaptive immunity against recurrent tumor FSP neoantigens.
Optimization of FSP-specific T cell activity with CpG adjuvant
Several adjuvants recognized for their induction of CD8 T cell responses in mice were tested for compatibility with the FSP vaccine and for capacity to induce broad T cell responses across the panel of FSPs, including TLR9 agonists, STING agonists and Montanide. Classes of novel adjuvant compounds have demonstrated the ability to activate type I interferon from dendritic cells, promote cross-presentation of soluble antigen to the MHC class I pathway, and have induced the development of tumor antigen-specific cytolytic T cells in murine models 39–41. In addition, Montanide is a water-in-oil emulsion formulation that has a long track record of being paired with peptide vaccines in preclinical cancer vaccine investigations as well as clinical trials 42, 43. To assess the FSP-specific T cell induction capability of these adjuvants, the 4-peptide FSP pool was combined with either the CpG-C ODN 2395, the CpG-B ODN 1826, the STING agonist 2’3’-cGAMP, or formulated with Montanide as an emulsion. The oil-in-water emulsion AddaVax was combined with the CpG ODNs and 2’3’-cGAMP to enhance adjuvant efficacy 44. We observed robust T cell responses specific to the panel of FSPs as measured by ELISpot detection of IFN-γ spot-forming units (SFUs) to Nacad(FSP-1), Xirp1(FSP-1), Maz(FSP-1), and Senp6(FSP-1) peptides with CpG/AddaVax adjuvant formulations (Supplemental Figure 4). However, 2’3’-cGAMP/AddaVax adjuvant yielded substantially lower T cell responses, while the Montanide formulation resulted in negligible SFUs. Further investigation of the FSP vaccine in mice was conducted with CpG-B ODN 1826.
Reduced tumor burden and Increased Survival of FSP Vaccinated VCMsh2 LS Mice
Next we examined whether these four immunogenic FSPs might affect the survival of immuno-competent VCMsh2 mice that develop small intestinal and colon tumors at the age of 6 months and have a mean life expectancy of about 14 months 32. 6–8 weeks old VCMsh2 mice were either vaccinated with 4 FSPs or ovalbumin control and CpG adjuvant and boosted 3X (Figure 4, Table 2). Tumor burden and survival analysis of vaccinated (n=39) and control mice (n=36) revealed that recurrent FSP neoantigen vaccination reduced tumor burden (mean control 71.84 mg vs vaccinated 47.26 mg, P=0.0024, Mann-Whitney) and significantly increased survival (control median age 256 vs vaccinated 327 days, control mean 263 vs vaccinated 351 days, p<0.0001, Log-rank test).
Figure 4. Cancer-Immunoprevention in Lynch Mice.

Reduced tumor burden from combination NSAID and recurrent FSP neoantigen vaccination. (A) Recurrent FSP neoantigen vaccination in combination with NSAID prolongs overall survival of VCMsh2 mice. Kaplan-Meier survival curves of VCMsh2 mice treated with control (untreated), ASA, NAP, FSP vaccine, FSP vaccine with ASA or FSP vaccine with NAP as described in the methods.
(B) FSP vaccination and combination with NSAID treatment decreases tumor burden in VCMsh2 mice. Scatter dot plot representing tumor burden (sum of all intestinal tumor weights per mouse, [mg]) per mouse in cohorts of control (untreated), treated with ASA, NAP, FSP vaccine, FSP vaccine plus ASA or FSP vaccine plus NAP.Both panels present data updated from that originally presented in Figure S3 of PMID: 32641470, here including a larger cohort of control and naproxen treated VCMsh2 mice.
Table 2.
Tumor burden and survival in different treatment groups
| Tumor Burden (mg) | Control | Aspirin (400 mg/kg) | Naproxen (166 mg/kg) | FSP Vaccine alone | FSP Vaccine + Aspirin | FSP vaccine + Naproxen |
|---|---|---|---|---|---|---|
| Mean | 71.84 | 57.63 | 37.32 | 47.26 | 33.40 | 20.18 |
| Sample Size (n) | 11 | 12 | 14 | 19 | 12 | 14 |
| Survival (days) | Control | Aspirin (400 mg/kg) | Naproxen (166 mg/kg) | FSP Vaccine | FSP vaccine + Aspirin | FSP vaccine + Naproxen |
| Mean | 263.04 | 297.68 | 322.13 | 350.95 | 385.94 | 468 |
| Median | 256 | 297 | 309 | 327 | 311 | 464.5 |
| Sample Size (n) | 25 | 25 | 23 | 20 | 17 | 14 |
Further reduced tumor burden and increased survival of naproxen treated and FSP vaccinated VCMsh2 LS mice
FSP vaccination was also tested in combination with ASA and NAP. VCMsh2 mice were fed on a diet including NAP (166 ppm) or ASA (400 ppm) as previously performed31. As expected, levels of PGE2 and other inflammatory prostaglandins PGD2 and PGF2a were significantly reduced in intestinal mucosa from these mice (Supplemental Figure 5). Similar to the previous findings 31, both NAP and ASA exposure affected intestinal tumor development: NAP significantly reduced tumor burden and prolonged survival (P=0.0099, Mann-Whitney and P=0.0005, log-rank test), while ASA trended in this direction (P=0.23 and P=0.081 respectively, Figure 4). FSP vaccination more effectively prolonged survival compared with ASA (P=0.011). The combination of FSP vaccination and NAP significantly prolonged survival compared to either intervention alone (P=0.0016 and P=0.0005, log-rank test, respectively) or ASA alone (P=0.0001).
Quantification of immune cells in manifest tumors
In total, 22 tumor tissue specimens could be analyzed for immune cell infiltration (12 from the FSP vaccine group and 10 from the control group). Highest numbers of positively stained cells were observed for CD4-positive cells. Significantly elevated CD4-positive cell counts were recorded in tumors from the FSP vaccine group compared to tumors from the control group (P=0.048). Similarly, CD8 T cell counts were significantly higher in the FSP vaccine vs. control group (P=0.031). No significant differences were observed for infiltrating Foxp3-positive or PD-1-positive cells (Figure 5).
Figure 5. Quantitative evaluation of immune cell subtypes.

(A) Representative IHC stainings. (B) Quantitative evaluation. Immune cell densities are shown for the antibodies detecting CD4, CD8, Foxp3, PD-1. Black lines indicate median values. P values are provided for all comparisons.
NAP or ASA exposure did not have any significant effects on immune cell densities. Among tumors from vaccinated mice, CD8 and CD4 TIL counts tended to be lower in animals exposed to NAP or ASA, although sample sizes were small.
RNAseq analysis reveals increased immune response in the VCMsh2 intestinal tumor microenvironment
Next, to explore the molecular mechanisms of recurrent neoantigen vaccination and NSAID, we performed RNAseq using FFPE blocks from tumor and normal intestinal tissue from VCMsh2 mice. As expected, this revealed substantial numbers of differentially regulated genes from FSP vaccination, NSAID cancer prevention or their combination, in both tumors and normal intestinal tissue (Supplemental Figures 6 and 7, GSEA accession GSE175744). Only one differentially expressed gene, Ptmap1/Ptma-ps1, was shared among tumors from LS mice that were FSP vaccinated, treated with NSAIDs, or both (Supplemental Figure 6 B). Ptmap1 is a poorly characterized gene that is ubiquitously expressed, encodes an open-reading frame with 76% amino acid identify to Ptma and may be an antagonist of Ptma. Re-analysis of previously published RNAseq data 45, 46 showed that PTMA is upregulated in LS patient colorectal cancers and premalignant lesions compared to normal mucosa (P=0.00015, Wilcoxon two-tailed) (Supplemental Figure 6 C), suggesting the PTMA/PTMAP1 axis as a potential novel candidate mechanism in LS CRC tumorigenesis. There were no significant changes in histologically normal intestinal mucosa for immune checkpoint-encoding genes PD1/PDL1, CTLA4 or LAG3 upon FSP vaccination or NSAID treatment (Supplemental Table 2). In terms of pathway analysis, as expected, VCMsh2 intestinal tumors compared to adjacent normal tissue were characterized by upregulation of WNT, NOTCH and MYC signaling, epithelial to mesenchymal transition (EMT), angiogenesis, hypoxia, stem cell and proliferation gene pathways, among others (Supplemental Figure 7). Additionally, these tumors had downregulation of Th1 Interferon-gamma (IFN-γ) signaling and Interferon-alpha (IFN-α) signaling, and upregulation of Th2 CD4+ T cell mediated humoral B cell immunity. By comparison, tumors in FSP-vaccinated mice had notably increased IFN-γ and p53 signaling, apoptosis and reversal of IFN-α downregulation. Thus, overall in the tumor microenvironment from FSP vaccinated mice, evidence for a relative upregulation of Th1 compared to Th2 signaling pathways was detected (Supplemental Table 2 and Supplemental Figure 7). Interestingly, NSAIDs significantly downregulated Th2 humoral immune response pathways, including B cell proliferation, B cell activation and IgA production, thereby increasing the relative Th1:Th2 levels in the tumor microenvironment. In summary, both FSP vaccination and NSAIDs increased the relative Th1 vs Th2 immune response in the VCMsh2 intestinal tumor microenvironment, although presumably via different mechanisms, while not significantly impacting immune checkpoint gene expression levels.
DISCUSSION
LS is an important model to study immunopreventive cancer vaccination, because tumors accumulate a predictable set of recurrent immunogenic neoantigens. This study provides the first robust evidence of tumor-preventive potential for a FSP neoantigen-based vaccine in a LS mouse model. Our experimental approach combined a series of methodical steps of in silico, in vitro and in vivo analyses consisting of (i) computational identification of murine cMNR candidates, (ii) molecular identification of shared tumor FS mutation targets, (iii) in silico prediction of FSP MHC binding motifs, (iv) FSP immunogenicity testing and (v) integrated survival, tumor burden and adaptive immune analyses in FSP-vaccinated LS mice.
As we have shown in a previous clinical phase I/II trial, FSP neoantigen vaccination is safe, non-toxic and can elicit pronounced cellular and humoral immune response in advanced-stage MSI colorectal cancer patients 24. However, data supporting the potential tumor-preventive effect of a FSP-based vaccine has been lacking. To evaluate FSP-based vaccination in the preventive setting, we here used the VCMsh2 LS mouse model 32 and compared survival of vaccinated mice with controls. We were able to demonstrate enhanced anti-FSP immunity, reduced tumor burden and significantly prolonged survival of VCMsh2 mice receiving the FSP vaccine compared to unvaccinated mice.
The VCMsh2 model recapitulates human LS as these mice develop MMRD intestinal tumors. These mice have an intestinal-specific exon 12 deletion of the Msh2 gene by Villin-Cre and inactivate MMR functions similar to MMRD found in LS patients. As an additional advantage, and in contrast to constitutive MMR-knockout models, the conditional ablation of Msh2 in VCMsh2 mice promotes tumor development specifically in the intestinal epithelium. This model also provides benefits in terms of NSAID cancer prevention studies because dietary ASA can suppress tumorigenesis in these mice similar to the tumor-protective effect of long-term ASA observed in humans (CAPP2 trial of LS patients28). Despite these similarities, tumor localization appears to be different because VCMsh2 mice usually develop tumors in the small intestine, whereas colonic tumors predominate in LS patients. Also, a direct transfer of human FSPs to the murine model is not feasible due to (1) genomic differences, in terms of the location of cMNRs in gene coding regions and the nucleotide sequence of the respective genes, and (2) differences in the MHC molecules responsible for antigen presentation. Therefore, we established a comprehensive database of murine cMNRs, providing a unique source of murine candidate genes and derived FSPs.
Using this genome-wide computational approach, we were able to select four candidate FSPs, which are shared by different murine MSI intestinal tumors, and validated their immunogenicity in vivo. The four FSPs Nacad(FSP-1), Maz(FSP-1), Senp6(FSP-1), and Xirp1(FSP-1) generated cellular and humoral immune responses in naïve mice. ELISpot analyses showed CD8 T cell-specific response for Xirp1(FSP-1), a mixed CD4/CD8 T cell response for Nacad(FSP-1), and a CD4 T cell-specific response for Maz1(FSP-1) and Senp6(FSP-1), partially overlapping with in silico prediction of MHC binding motifs. Three out of these four FSPs also elicited humoral immune responses, which were only absent for Xirp1(FSP-1), consistent with the observed restriction of cellular immune responses to CD8, but not CD4 T cells, which play a major role in mediation of humoral immune responses. When analyzing immune cell infiltration of tumors in VCMsh2 mice receiving the FSP vaccine, we observed a significantly elevated density of CD4-positive and CD8-positive T cells in tumors compared to non-vaccinated control tumors; densities for CD4-positive T cells were generally higher than the density of CD8-positive T cells. These findings are consistent with a predominance of CD4 responses triggered by FSP vaccination as observed in human patients with MSI CRC who were vaccinated with a trivalent FSP vaccine 24. Previously, CD4-positive T cell responses were shown to be predominant and associated with clinical response in “personalized” melanoma and glioblastoma patient neoantigen and tumor associated antigen vaccine trials 47–49. Thus, our findings support important roles for CD4-positive (in addition to CD8-positive) T cells in durable clinical responses to tumor vaccines.
The fact that cMNR mutations affecting the genes Nacad, Maz, Senp6, and Xirp1 were found to be recurrent across tumors may suggest a functional role of these mutations as drivers of tumor development. Although some evidence of involvement in tumor development exists for the genes Maz and Senp6 50–53, very limited functional data is available for Nacad and Xirp1. Thus, further studies are required to elucidate a potential functional contribution of these mutations to MSI tumor progression.
Although, as outlined above, the precise amino acid sequence differs between murine and human FSP neoantigens, the observed immunopreventive effects support the hypothesis that vaccination with shared FSP neoantigens may be a powerful approach for cancer prevention in LS. In particular, it is notable that vaccination with only a limited number of FSPs combined with relatively low dose of naproxen significantly delayed tumor formation and prolonged overall survival in LS mice providing a rational approach for combination cancer- and Immuno-prevention.
Our study for the first time addresses the important question as to whether FSP vaccination and NSAID are synergistic or antagonistic in LS cancer prevention. The increased survival and reduced tumor burden of mice receiving both FSP vaccination and NSAID treatment is consistent with a role for combined chemo- and immunoprevention to reduce tumor burden in a practical manner. Our observations suggest that the combination of FSP vaccination and NSAID treatment is beneficial, possibly by partially complementary mechanisms, which need to be elucidated in future studies.
By analyzing transcriptomic pathways altered by FSP vaccination and NSAIDs, we identified a differentially expressed gene, Ptmap1, that was differentially regulated in tumors from LS mice that were FSP vaccinated, treated with NSAIDs, and then further upregulated by combined treatment. PTMA is a nuclear oncoprotein-transcription factor that is upregulated in solid tumors including CRC, is functionally associated with CRC poor prognosis 54. PTMAP1 overall remains poorly characterized. Because PTMAP1 is highly homologous to PTMA at the protein level, it may antagonize PTMA oncogenic transcriptional program functions. However, thse findings are exploratory and further studies are needed to assess the mechanistic role of the PTMA/PTMAP1 axis in Lynch syndrome CRC tumorigenesis.
In terms of transcriptomic pathways, both FSP vaccination and NSAIDs increased the relative Th1 vs Th2 T cell immune response in the VCMsh2 intestinal tumor microenvironment, while not significantly impacting critical immune checkpoint gene expression levels, which for PD-1 was confirmed by quantitative immunohistochemistry (Figure 5). Thus, elevated Th1 T cell activation in the tumor microenvironment is the most likely immune surveillance mechanism to account for the observed reduced tumor burden in FSP vaccinated, NSAID treated, and combination treated mice.
The study has several limitations. Our study was conducted in mice that have defined MHC molecules, whereas LS patients have much broader HLA (class I and class II MHC) diversity. It remains to be demonstrated, whether a broader spectrum of shared FSPs in combination with other cancer prevention approaches might represent an even more effective strategy to prevent LS tumors in patients with greater HLA diversity. Second, although CpG1826 showed best results among the 4 adjuvants screened in LS mice, it is unclear whether alternative adjuvants might be more effective for FSP vaccination in human LS patients. In the same context, the most effective boosting regimen is still unknown. Additional pre-clinical studies addressing these questions will be required before large scale LS immunoprevention trials are carried out.
In summary, our results strongly support the concept of FSP neoantigen vaccination as a promising strategy for immunoprevention of intestinal MMR-deficient tumors, particularly in the setting of LS. We provide first evidence that vaccination with a small number of shared FSP neoantigens alone effectively delays formation of naturally occurring tumors and prolonged survival in a LS mouse model. Furthermore, as NSAID use is the current choice of care for LS patients, we show that this benefit can significantly be enhanced in a cooperative manner by combining FSP vaccination with NSAID treatment. Clinical translation of this concept of neoantigen-based tumor prevention is encouraging, and our studies support further preclinical and clinical studies to translate this benefit for LS immunoprevention.
Supplementary Material
Short Summary
Vaccination of Lynch syndrome mice with shared immunogenic frameshift peptide neoantigens derived from recurrent frameshift mutations reduced intestinal tumor formation and prolonged survival, even more efficiently upon parallel NSAID treatment.
„What you need to know“ Box
Background and context
Lynch syndrome tumors are characterized by DNA mismatch repair deficiency and microsatellite instability, which gives rise to tumor-specific, mutation-induced frameshift peptide neoantigens.
New findings
We for the first time demonstrate in a hereditary cancer model that tumor prevention by vaccination with mutation-induced neoantigens is feasible and effective.
Limitations
The results are restricted to a mouse model, and clinical effectiveness of a tumor-preventive neoantigen-based vaccine needs to be demonstrated in a clinical trial.
Impact
Vaccination against microsatellite instability-induced frameshift peptide neoantigens prevents tumors in a mouse model. The vaccine strategy therefore holds great promise for individuals with Lynch syndrome.
Grant support:
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (GE 592-9/1) and a contract from the PREVENT Preclinical Development Program, Division Cancer Prevention, National Cancer Institute (HHSN2612015000391), U01CA233056 and U24CA232979. This work was supported by grant R01 CA219463, a gift from the Feinberg Family Foundation, the MD Anderson Colorectal Cancer Moonshot to E.V; and P30 CA016672 (US National Institutes of Health/National Cancer Institute) to the University of Texas MD Anderson Cancer Center Core Support Grant.
Disclosures:
Dr. Vilar has a consulting or advisory role with Janssen Research and Development and Recursion Pharma. He has received research support from Janssen Research and Development.
Abbreviations:
- ASA
aspirin
- cMNR
coding mononucleotide repeat
- CRC
colorectal cancer
- IACUC
Institutional Animal Care and Use Committee
- IFN-α
Interferon-alpha
- IFN-γ
Interferon-gamma
- FFPE
formalin-fixed paraffin embedded
- FSP
frameshift peptide
- LS
Lynch syndrome
- MMRD
mismatch repair deficiency
- MS
microsatellite
- MSI
microsatellite instability
- NAP
naproxen
- NSAID
non-steroidal anti-inflammatory drug
- PGE2
prostaglandin E2
- SFU
spot-forming unit
- TIL
tumor infiltrating lymphocytes
Footnotes
Transcript profiling:
GEO accession GSE175744
Data transparency statement: All relevant data are contained in the manuscript or the Supplemental Material. Transcript profiling data are available under the following link: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE175744.
The rest of the authors do not report a conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ionov Y, Peinado MA, Malkhosyan S, et al. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993;363:558–61. [DOI] [PubMed] [Google Scholar]
- 2.Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science 1993;260:816–9. [DOI] [PubMed] [Google Scholar]
- 3.Kloor M, von Knebel Doeberitz, M. The immune biology of microsatellite-unstable cancer. Trends in Cancer 2016;2:121–131. [DOI] [PubMed] [Google Scholar]
- 4.Markowitz S, Wang J, Myeroff L, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 1995;268:1336–8. [DOI] [PubMed] [Google Scholar]
- 5.Woerner SM, Yuan YP, Benner A, et al. SelTarbase, a database of human mononucleotide- microsatellite mutations and their potential impact to tumorigenesis and immunology. Nucleic Acids Res 2010;38:D682–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jonchere V, Marisa L, Greene M, et al. Identification of Positively and Negatively Selected Driver Gene Mutations Associated With Colorectal Cancer With Microsatellite Instability. Cell Mol Gastroenterol Hepatol 2018;6:277–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Colas C, Coulet F, Svrcek M, et al. Lynch or not Lynch? Is that always a question? Adv Cancer Res 2012;113:121–66. [DOI] [PubMed] [Google Scholar]
- 8.Latham A, Srinivasan P, Kemel Y, et al. Microsatellite Instability Is Associated With the Presence of Lynch Syndrome Pan-Cancer. J Clin Oncol 2019;37:286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Win AK, Jenkins MA, Dowty JG, et al. Prevalence and Penetrance of Major Genes and Polygenes for Colorectal Cancer. Cancer Epidemiol Biomarkers Prev 2017;26:404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haraldsdottir S, Rafnar T, Frankel WL, et al. Comprehensive population-wide analysis of Lynch syndrome in Iceland reveals founder mutations in MSH6 and PMS2. Nat Commun 2017;8:14755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moller P, Seppala TT, Bernstein I, et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: a report from the Prospective Lynch Syndrome Database. Gut 2018;67:1306–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seppala T, Pylvanainen K, Evans DG, et al. Colorectal cancer incidence in path_MLH1 carriers subjected to different follow-up protocols: a Prospective Lynch Syndrome Database report. Hered Cancer Clin Pract 2017;15:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fishel R, Lescoe MK, Rao MR, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993;75:1027–38. [DOI] [PubMed] [Google Scholar]
- 14.Jasperson KW, Tuohy TM, Neklason DW, et al. Hereditary and familial colon cancer. Gastroenterology 2010;138:2044–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hemminki A, Peltomaki P, Mecklin JP, et al. Loss of the wild type MLH1 gene is a feature of hereditary nonpolyposis colorectal cancer. Nat Genet 1994;8:405–10. [DOI] [PubMed] [Google Scholar]
- 16.Veigl ML, Kasturi L, Olechnowicz J, et al. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci U S A 1998;95:8698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dolcetti R, Viel A, Doglioni C, et al. High prevalence of activated intraepithelial cytotoxic T lymphocytes and increased neoplastic cell apoptosis in colorectal carcinomas with microsatellite instability. Am J Pathol 1999;154:1805–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buckowitz A, Knaebel HP, Benner A, et al. Microsatellite instability in colorectal cancer is associated with local lymphocyte infiltration and low frequency of distant metastases. Br J Cancer 2005;92:1746–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giannakis M, Mu XJ, Shukla SA, et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep 2016;17:1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mandal R, Samstein RM, Lee KW, et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 2019;364:485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Linnebacher M, Gebert J, Rudy W, et al. Frameshift peptide-derived T-cell epitopes: a source of novel tumor-specific antigens. Int J Cancer 2001;93:6–11. [DOI] [PubMed] [Google Scholar]
- 22.Saeterdal I, Bjorheim J, Lislerud K, et al. Frameshift-mutation-derived peptides as tumor- specific antigens in inherited and spontaneous colorectal cancer. Proc Natl Acad Sci U S A 2001;98:13255–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwitalle Y, Kloor M, Eiermann S, et al. Immune response against frameshift-induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology 2008;134:988–97. [DOI] [PubMed] [Google Scholar]
- 24.Kloor M, Reuschenbach M, Pauligk C, et al. A Frameshift Peptide Neoantigen-Based Vaccine for Mismatch Repair-Deficient Cancers: A Phase I/IIa Clinical Trial. Clin Cancer Res 2020. [DOI] [PubMed] [Google Scholar]
- 25.Burn J, Gerdes AM, Macrae F, et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet 2011;378:2081–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer 2010;10:181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roulis M, Kaklamanos A, Schernthanner M, et al. Paracrine orchestration of intestinal tumorigenesis by a mesenchymal niche. Nature 2020;580:524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burn J, Sheth H, Elliott F, et al. Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year follow-up and registry-based 20-year data in the CAPP2 study: a double-blind, randomised, placebo-controlled trial. Lancet 2020;395:1855–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McNeil JJ, Nelson MR, Woods RL, et al. Effect of Aspirin on All-Cause Mortality in the Healthy Elderly. N Engl J Med 2018;379:1519–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reyes-Uribe L, Wu W, Gelincik O, et al. Naproxen chemoprevention promotes immune activation in Lynch syndrome colorectal mucosa. Gut 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin-Lopez J, Gasparini P, Coombes K, et al. Mutation of TGFbeta-RII eliminates NSAID cancer chemoprevention. Oncotarget 2018;9:12554–12561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kucherlapati MH, Lee K, Nguyen AA, et al. An Msh2 conditional knockout mouse for studying intestinal cancer and testing anticancer agents. Gastroenterology 2010;138:993–1002.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heyer J, Yang K, Lipkin M, et al. Mouse models for colorectal cancer. Oncogene 1999;18:5325–33. [DOI] [PubMed] [Google Scholar]
- 34.Woerner SM, Tosti E, Yuan YP, et al. Detection of coding microsatellite frameshift mutations in DNA mismatch repair-deficient mouse intestinal tumors. Mol Carcinog 2015;54:1376–86. [DOI] [PubMed] [Google Scholar]
- 35.Woerner SM, Kloor M, von Knebel Doeberitz M, et al. Microsatellite instability in the development of DNA mismatch repair deficient tumors. Cancer Biomark 2006;2:69–86. [DOI] [PubMed] [Google Scholar]
- 36.Woerner SM, Kloor M, Mueller A, et al. Microsatellite instability of selective target genes in HNPCC-associated colon adenomas. Oncogene 2005;24:2525–35. [DOI] [PubMed] [Google Scholar]
- 37.Kabbarah O, Mallon MA, Pfeifer JD, et al. A panel of repeat markers for detection of microsatellite instability in murine tumors. Mol Carcinog 2003;38:155–9. [DOI] [PubMed] [Google Scholar]
- 38.Bankhead P, Loughrey MB, Fernandez JA, et al. QuPath: Open source software for digital pathology image analysis. Sci Rep 2017;7:16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Su T, Zhang Y, Valerie K, et al. STING activation in cancer immunotherapy. Theranostics 2019;9:7759–7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shirota H, Tross D, Klinman DM. CpG Oligonucleotides as Cancer Vaccine Adjuvants. Vaccines (Basel) 2015;3:390–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gogoi H, Mansouri S, Jin L. The Age of Cyclic Dinucleotide Vaccine Adjuvants. Vaccines (Basel) 2020;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hailemichael Y, Dai Z, Jaffarzad N, et al. Persistent antigen at vaccination sites induces tumor-specific CD8(+) T cell sequestration, dysfunction and deletion. Nat Med 2013;19:465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iwasa S, Yamada Y, Heike Y, et al. Phase I study of a new cancer vaccine of ten mixed peptides for advanced cancer patients. Cancer Sci 2016;107:590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maynard SK, Marshall JD, MacGill RS, et al. Vaccination with synthetic long peptide formulated with CpG in an oil-in-water emulsion induces robust E7-specific CD8 T cell responses and TC-1 tumor eradication. BMC Cancer 2019;19:540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bommi PV, Bowen CM, Reyes-Uribe L, et al. The Transcriptomic Landscape of Mismatch Repair-Deficient Intestinal Stem Cells. Cancer Res 2021;81:2760–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang K, Taggart MW, Reyes-Uribe L, et al. Immune Profiling of Premalignant Lesions in Patients With Lynch Syndrome. JAMA Oncol 2018;4:1085–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keskin DB, Anandappa AJ, Sun J, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019;565:234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sahin U, Oehm P, Derhovanessian E, et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020;585:107–112. [DOI] [PubMed] [Google Scholar]
- 49.Sahin U, Derhovanessian E, Miller M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017;547:222–226. [DOI] [PubMed] [Google Scholar]
- 50.Chen T, Xue H, Lin R, et al. MiR-34c and PlncRNA1 mediated the function of intestinal epithelial barrier by regulating tight junction proteins in inflammatory bowel disease. Biochem Biophys Res Commun 2017;486:6–13. [DOI] [PubMed] [Google Scholar]
- 51.Yu ZH, Lun SM, He R, et al. Dual function of MAZ mediated by FOXF2 in basal-like breast cancer: Promotion of proliferation and suppression of progression. Cancer Lett 2017;402:142–152. [DOI] [PubMed] [Google Scholar]
- 52.Bologna S, Altmannova V, Valtorta E, et al. Sumoylation regulates EXO1 stability and processing of DNA damage. Cell Cycle 2015;14:2439–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tosti E, Katakowski JA, Schaetzlein S, et al. Evolutionarily conserved genetic interactions with budding and fission yeast MutS identify orthologous relationships in mismatch repair-deficient cancer cells. Genome Med 2014;6:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang M, Cui F, Lu S, et al. Increased expression of prothymosin-alpha, independently or combined with TP53, correlates with poor prognosis in colorectal cancer. Int J Clin Exp Pathol 2014;7:4867–76. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.