

Abstract

Drug resistance mutations emerging during the treatment of non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) inhibitors represent a major challenge in personalized cancer treatment and require constant development of new inhibitors. For the covalent irreversible EGFR inhibitor osimertinib, the predominant resistance mechanism is the acquired C797S mutation, which abolishes the covalent anchor point and thus results in a dramatic loss in potency. In this study, we present next-generation reversible EGFR inhibitors with the potential to overcome this EGFR-C797S resistance mutation. For this, we combined the reversible methylindole-aminopyrimidine scaffold known from osimertinib with the affinity driving isopropyl ester of mobocertinib. By occupying the hydrophobic back pocket, we were able to generate reversible inhibitors with subnanomolar activity against EGFR-L858R/C797S and EGFR-L858R/T790M/C797S with cellular activity on EGFR-L858R/C797S dependent Ba/F3 cells. Additionally, we were able to resolve cocrystal structures of these reversible aminopyrimidines, which will guide further inhibitor design toward C797S-mutated EGFR.
Keywords: Cancer, Non-small cell lung cancer, Kinase inhibitor, Structure-based drug design, Epidermal growth factor receptor
The treatment of mutant EGFR with selective tyrosine kinase inhibitors (TKIs) in non-small cell lung cancer (NSCLC) has become well-established clinical practice because it provides improved survival rates for selected patient populations and is a paradigm for modern precision medicine in oncology. The emergence of resistance mutations upon treatment with TKIs, however, represents a major drawback and necessitates the constant development of next-generation TKIs. The covalent-irreversible inhibitor osimertinib (1) was developed as a third-generation TKI for the treatment of cancer patients that developed the EGFR-T790M resistance mutation.1−3 However, because of its safety and favorable pharmacokinetic profile, osimertinib has become the standard of care not only after the resistance mutation has developed but also for first-line treatment in patients with EGFR-mutated advanced NSCLC.4 Unfortunately, resistance mutations are observed in osimertinib treated patients, one of the most prevalent being the EGFR-C797S mutation, which abolishes the covalent anchor-point of osimertinib and results in a dramatic loss of efficacy in patients.5
Since the discovery of the C797S resistance mutation in osimertinib second-line treated patients, many approaches have been followed to develop inhibitors for the EGFR-L858R/T790M/C797S mutation.6,7 So far, no monotherapeutic agent has been approved by the FDA, making inhibitors for the multimutated version of EGFR an unmet medical need. Since the application of osimertinib as a first-line treatment against EGFR-activating mutations,4,8 the complete multitude of acquired resistances is not yet fully characterized. However, the EGFR-C797S mutation has been observed in several preclinical models as well as in patients9−11 and thus substantiates the demand for multiresistance EGFR inhibitors that are capable of overcoming both the T790M and C797S resistance mutations. From the different approaches targeting the C797S mutation,6,7 now the first reversibly binding inhibitors like BLU-525, BLU-701, BLU-945, BDTX-1535, and BAY2927088 are being investigated in a clinical setting.12−15
In this publication, we present reversible EGFR inhibitors that are based on the 2-aminopyrimidine hinge-binding motif known from osimertinib and additionally exploit the hydrophobic back pocket of the ATP-binding site with different substituents in the 5-position. The introduction of ester functions and growing the substituent into the back pocket showed significant impact on reversible binding affinity and cellular efficacy on osimertinib-resistant EGFR-L858R/C797S Ba/F3 cells.
In a screening campaign of known EGFR inhibitors against EGFR-L858R/C797S, we observed that the EGFR Exon20ins mutation inhibitor mobocertinib16,17 (2, IC50-LR/CS 25 nM) shows a more than 10-fold higher biochemical activity than osimertinib (IC50-LR/CS 354 nM). The only structural difference is the presence of an isopropyl ester in the 5-position of the 2-aminopyrimidine. Structural analysis of osimertinib bound to wild-type EGFR (PDB 4ZAU) showed an unoccupied hydrophobic back pocket in a position suitable for binding of the isopropyl ester of mobocertinib and thus potentially explaining the increased affinity. We previously published the cocrystal structure of mobocertinib in EGFR-T790M/V948R (PDB 7A6K) exposing additional interactions with the mutated kinase compared to osimertinib (PDB 6JX4).18 In addition, we were able to solve the cocrystal structure of the aminopyrimidine brigatinib (14), a repurposed ALK inhibitor,19 which exhibits good inhibition of T790M/C797S-mutated EGFR (PDB 7ZYM, Figure 1B, Supporting Information (SI), Figure S1) and has been reported to be beneficial in C797S-mutated NSCLC treatment in combination with the EGFR antibody cetuximab.20,21 Additionally, a therapeutic window for C797S-mutated EGFR, in terms of sparing wild-type EGFR, is unlikely to be achieved by an inhibitor bearing a covalent warhead addressing Cys797 present in wild-type EGFR. For these reasons and keeping in mind the unfavorable reorientation of the warhead in the C797S mutant (PDB 7ZYN, Figure 1C, SI, Figure S1), the elimination of the acrylamide moiety was the first step in our inhibitor design. The structural analysis of reversibly bound aminopyrimidines in C797S-mutated EGFR inspired us to create reversible 2-aminopyrimidine inhibitors that additionally exploit the hydrophobic back pocket. The elimination of the acrylamide from osimertinib and implementing different esters was the first step to investigate the effect of addressing the hydrophobic back pocket. Additionally, we chose WZ4002s (15) front pocket methylpiperazine substituent to exploit the observed ionic interaction with Asp800 (Figure 1C; for synthetic schemes and procedures, see SI).
Figure 1.

Cocrystal structures of EGFR with different inhibitors were used for a structure-guided design approach of reversible aminopyrimidine-based inhibitors. (A) Comparison of osimertinib (gray, PDB 6JX4) and mobocertinib (white, PDB 7A6K) bound to EGFR-T790M. Hydrogen bond network of mobocertinib is in red, and the reorientation of Met790 (green surface) by the isopropyl ester is highlighted (red circle). (B) The ALK inhibitor brigatinib bound to EGFR-T790M/C797S (PDB 7ZYM). Ionic interactions of the front pocket substituent with Asp800 highlighted in blue. (C) WZ4002 (white) bound to EGFR-T790M/C797S (PDB 7ZYN) in comparison to WZ4002 (gray) bound to EGFR-T790M (PDB 3IKA) showing an additional ionic interaction with Asp800. The unfavorable reorientation of the acrylamide is highlighted (red arrow). All |2Fo–Fc| maps are contoured at a rmsd of 1. Simulated annealing |FoFc| omit electron density maps are shown in SI, Figure S1.
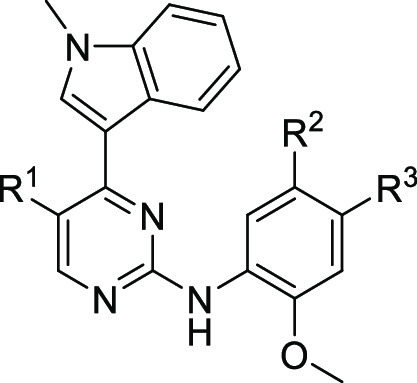
Compared to osimertinib, a small improvement toward the L858R/C797S mutant could be observed for the unsubstituted derivative 3 (Table 1). However, implementation of the ester functionality and growing its size from a methyl to the isopropyl ester was necessary to increase the biochemical activity to a low nanomolar level (6, IC50 = 1.6 nM). To our delight, the success in biochemical optimization of inhibitor 6 could also be transferred to a cellular environment. The proliferative inhibition of EGFR-L858R/C797S dependent Ba/F3 cells could be improved to a submicromolar level with 0.6 μM for the isopropyl ester 6. To further investigate the impact of addressing the hydrophobic back pocket on the biochemical and cellular activity and the compatibility toward T790M-mutated EGFR, a comprehensive structure-activity investigation including different esters, amides, inverse amides, and heterocyclic bioisosters was performed, alongside with derivatizations of the front pocket substituent and the indole. With the cocrystal structure of covalently bound mobocertinib with EGFR-T790M in hand, we designed and synthesized a library including diverse substituents for the hydrophobic back pocket to obtain further insights into the nature of this pocket. Within the library consisting of esters, amides, and heterocycles (R1, for complete SAR see SI, Table S2), the larger aromatic esters 7 and 8 proved to be the most suitable for the inhibition of C797S-mutated EGFR and showed a 10-fold increase in potency (Table 2).
Table 1. Biochemical and Cellular Evaluation of First Subset of 2-Aminopyrimidines Based EGFR Inhibitorsa.
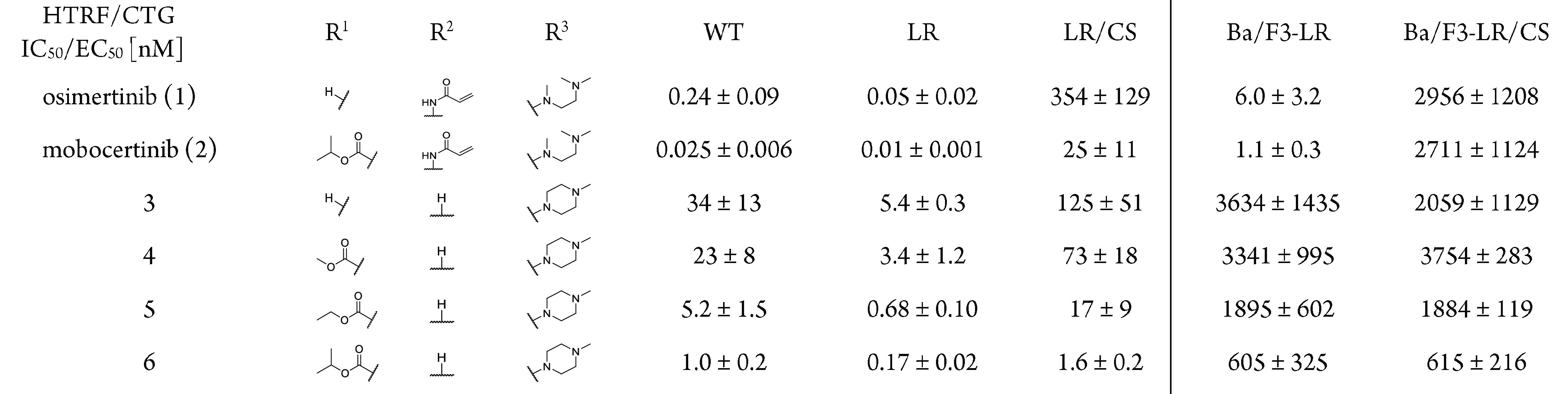
EGFR wild-type (WT), L858R (LR), and L858R/C797S (LR/CS) were measured by HTRF-Assay. EGFR-L858R and EGFR-L858R/C797S dependent Ba/F3 cells were measured by CTG assay.
Table 2. Biochemical and Cellular SAR of 2-Aminopyrimidine Based EGFR Inhibitorsa.
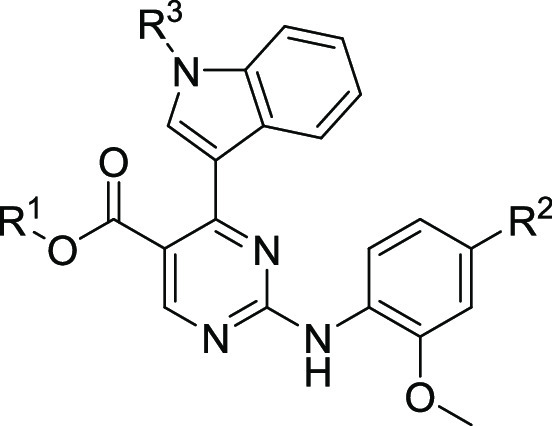
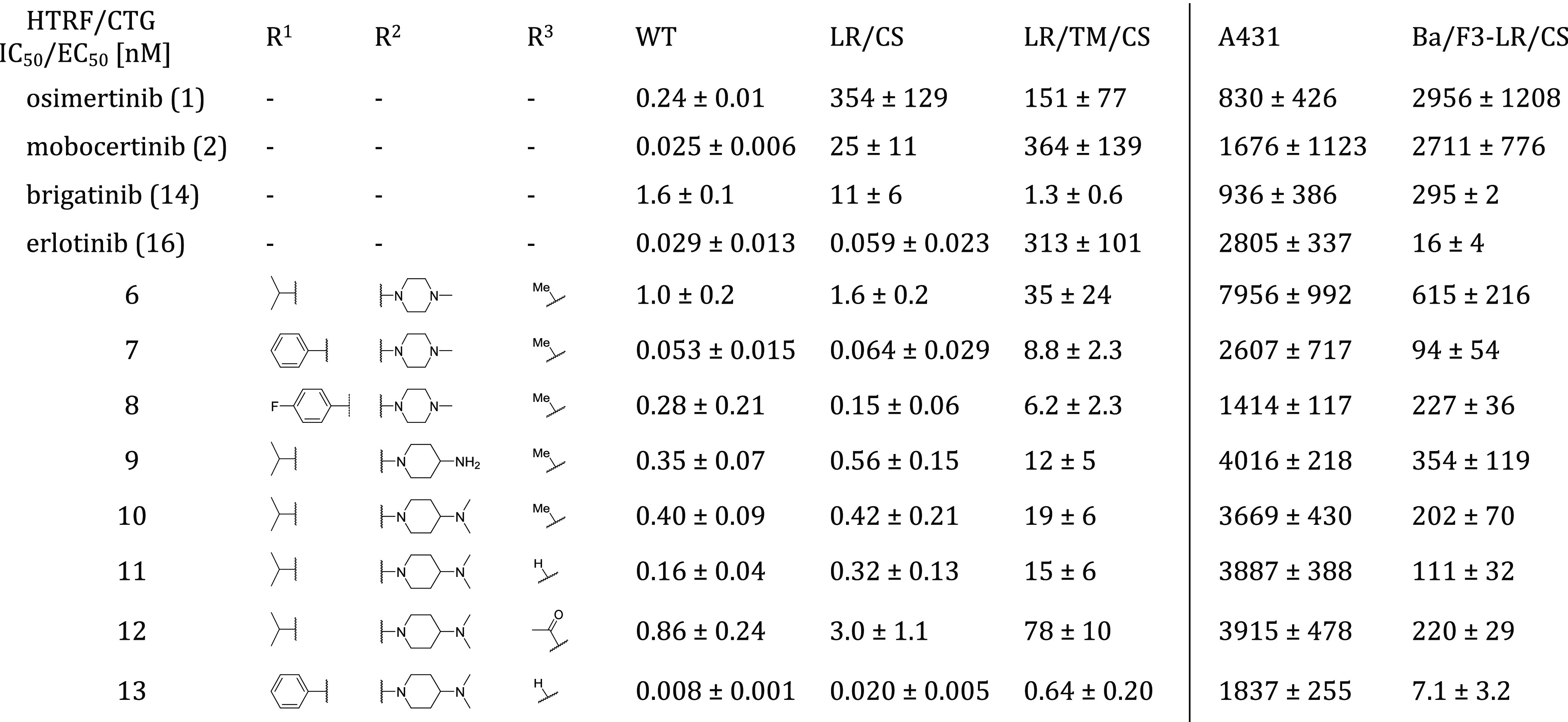
EGFR wild-type (WT), L858R (LR), L858R/C797S (LR/CS), and L858R/T790M/C797S (LR/TM/CS) were measured by HTRF-Assay. EGFR wild-type dependent A431 cells and EGFR-L858R/C797S dependent Ba/F3 cells were measured by CTG assay.
To improve the inhibitors interaction with the acidic side chain of Asp800, we investigated several front pocket substituents (R2). We gained inhibitor 9, which we were able to cocrystallize with EGFR-T790M/C797S, confirming our proposed mode of binding of the reversible bound aminopyrimidines (PDB 7ZYP, Figure 2A, SI, Figure S1). The 4-aminopiperidine substituted inhibitor 9 and its dimethylated variant 10 showed improved biochemical and cellular activity toward C797S-mutated EGFR compared to the methylpiperazine 6 (Table 2). However, further structural analysis of the occupied back pocket with the Met790 gatekeeper showed a suboptimal interaction profile of the isopropyl ester. Comparison with osimertinib bound to EGFR-T790M revealed a necessary rearrangement of the methionine side chain, which is also observable in the cocrystal structure of mobocertinib in EGFR (Figure 1A) and explains an unfavorable influence of the isopropyl esters toward T790M-mutated EGFR with respect to wild-type selectivity.
Figure 2.

(A) Cocrystal structure of reversible aminopyrimidine 9 in EGFR-T790M/C797S (PDB 7ZYP). Hinge binding ishighlighted in red. The |2Fo–Fc| map is contoured at a rmsd of 1. The simulated annealing |FoFc| omit electron density map is shown in SI, Figure S1. (B) Modeling of the indole using Ligandscout (MMFF94 energy minimization). Hydrogen bond of the free indole with the side chain of Asp855 and hinge binding is highlighted in red. Ionic interaction of aminopiperidine with the side chain of Asp800 is highlighted in blue. (C) Modeling of the acetyl indole using Ligandscout (MMFF94 energy minimization). Hydrogen bond of the carbonyl oxygen with the side chain of Lys745 and hinge binding are highlighted in red.
Finally, with the obtained cocrystal structure of 9 bound to EGFR-T790M/C797S, we also wanted to investigate the effects of derivatization of the indole moiety (R3) and potential modifications were initially investigated using LigandScout(22) (Figure 2), indicating that the acetylated and the free indole might form additional interactions. Whereas modification of the methylindole toward the free indole 11 showed a potential interaction with the side chain of Asp855 of the DFG motif (Figure 2B), the acetylated indole 12 revealed a potential interaction of the carbonyl-oxygen with the catalytic Lys745 similarly to previously described EGFR inhibitors (Figure 2C).6,12,23−26
Comparison of the three synthesized derivatives finally showed that the free indole 11 had superior activity toward EGFR-L858R/C797S in biochemical and cellular settings (Table 2) compared to the methylated and the acetylated inhibitors. As described in the discovery of osimertinib,1 the methylindole is driving wild-type selectivity toward T790M-mutated EGFR while lowering overall activity against wild-type and mutated EGFR in comparison with the free indole. Our inhibitor series also shows the methylindole to be slightly more selective against C797S- and T790M-mutated EGFR in a biochemical setting. However, the three indole derivatives show equipotency toward the wild-type dependent A431 cell line, while the free indole shows increased activity against targeted Ba/F3 EGFR-L858R/C797S cells. With these findings in hand, we combined our best features within the inhibitor 13: As anticipated, it showed highly improved biochemical and cellular efficacy toward L858R/C797S-mutated EGFR. With picomolar affinities against the EGFR-L8585R activating and EGFR-L858R/C797S resistance mutation, inhibitor 13 showed a higher activity than the investigated reference compounds brigatinib (14) and erlotinib (16). Inhibitor 13 displayed a favorable cellular activity profile with low nanomolar activities against EGFR-L858R and EGFR-L858R/C797S dependent Ba/F3 cell lines, while simultaneously having low activity toward the EGFR wild-type dependent A431 cell line (EC50, 1.8 μM) and the parental Ba/F3 cell line (EC50 – 1.2 μM; for complete biochemical and cellular data, see SI, Table S6). Additionally, looking at the T790M resistance mutation, we were able to generate an inhibitor which has an advantage over first-generation EGFR inhibitors regarding T790M tolerability. Inhibitor 13 exhibits low picomolar inhibition against the EGFR-L858R/T790M resistance mutation and subnanomolar activity against the EGFR-L858R/T790M/C797S triple mutant, surpassing both erlotinib and the repurposed ALK inhibitor brigatinib. In preclinical mutagenesis experiments, erlotinib is shown to bring up the T790M resistance mutation when treating Ba/F3 EGFR-L858R/C797S osimertinib-resistant cells.9 With our improved activity toward the T790M-mutated EGFR variants, our inhibitor might avoid the uprising of T790M-mutated cells in comparison to erlotinib. Although the potency of inhibitor 13 toward T790M-mutated EGFR variants could be increased significantly, the accompanying increase in EGFR wild-type inhibition might hinder the application toward the first-generation EGFR inhibitor T790M resistance. Our optimized inhibitors show improved, but only moderate inhibition against the corresponding Ba/F3-L858R/T790M/C797S cell line with an EC50 of 0.47 μM for phenol ester 13 and an EC50 of 0.32 μM for its p-fluorophenol analogue (61, for complete SAR, see SI, Table S2). For a more successful treatment of T790M/C797S-mutated EGFR, we propose reversible aminopyrimidines bearing no substituent or a halogen in 5-position. Especially aminopyrimidines substituted at the 5-position with a halogen allow an additional halogen bond with the Met790 side chain, which we were able to observe in our crystal structure of brigatinib in EGFR-T790M/C797S (Figure 1B), which shifts the activity in favor of T790M-mutated EGFR (Table 2). Brigatinib-inspired 5-chloro- and 5-bromo-aminopyrimidines exploiting this halogen bond have recently been described. However, these compounds show only slight improvement in their activity toward triple-mutant Ba/F3 cells in comparison to brigatinib.27
To our satisfaction, we were able to solve a cocrystal structure with inhibitor 13 bound to EGFR-T790M, which allowed us to visualize and verify the inhibitor design and the anticipated optimization steps (PDB 7ZYQ, Figure 3, SI, Figure S1). As expected, inhibitor 13 displayed the ionic interaction of the front pocket substituent with the acidic side chain of Asp800. Additionally, the hydrogen bond between the DFG motifs Asp855 side chain and the free indole could be confirmed as predicted by LigandScout.22 Moreover, we identified the typically hydrophobic interactions with the hydrophobic clamp (Val726, Leu844, and Leu718) and the side chains of Ala743 and Leu792 and stapled interaction of the phenol ester with Lys745 and Met790. A similar stapling of an aromatic substituent between the catalytic lysine and the methionine gatekeeper is present in several T790M inhibitors,28,29 which seems a more favorable orientation than the aforementioned repulsion of the Met790 gatekeeper, present in our crystal structures of the isopropyl esters mobocertinib (Figure 1A) and 9 (Figure 2A). Although this stapled interaction seems to be driving the potency of our optimized inhibitor against T790M mutants, unfortunately, no improvement in wild-type selectivity can be observed.
Figure 3.

(A) Cocrystal structure of 13 bound to EGFR-T790M/V948R (PDB 7ZYQ). Hinge binding and hydrogen bond with Asp855 side chain are highlighted in red, and ionic interaction with Asp800 side chain is highlighted in blue. The |2Fo–Fc| map is contoured at a rmsd of 1. The simulated annealing |FoFc| omit electron density map is shown in SI, Figure S1. (B) View of stapled interaction of the phenol ester with Lys745 and Met790. (C) Schematic hydrophobic interaction profile of 13 within EGFR-T790M/V948R.
To gain further insight into the compound optimization effect beyond the biochemical and cellular level, we performed initial investigations regarding in vitro metabolic stability and kinome selectivity of the isopropyl ester 11 and the phenol ester 13. To our satisfaction, no significant decrease in microsomal stability (mouse, phase I; SI, Table S6) could be observed for the phenol ester 13 (CLint: 55 μL/min/mg) compared to the isopropyl ester 11 (CLint: 49 μL/min/mg). Additionally, both compounds showed higher microsomal stability than osimertinib (CLint: 81 μL/min/mg) and mobocertinib (CLint: 149 μL/min/mg). To elucidate the kinome selectivity of our compound class, we tested the compounds against a subset of 100 kinases spread across the human kinome at a concentration of 200 nM (SI, Figure S2 and Table S4). The isopropyl ester 11 showed a good kinome selectivity with seven non-ErbB off-targets (inhibition >40%), including ABL1, ALK, BLK, HCK, LCK, LYN, and PEAK1. The phenol ester 13 showed increased inhibition against several kinases with 16 non-ErbB off-targets. The activity toward the well-known EGFR inhibitor off-target BTK as well as other members of the tyrosine kinase subfamily as FES, FGFR2, FYN, PDGFRα, PDGFRβ, TXK, VEGFR2, and VEGFR3 increased above the threshold of 40% inhibition. Interestingly, typical off-targets for covalent EGFR inhibitors bearing a Cys797 analogue like BLK (Cys319), BTK (Cys481), and TXK (Cys350) are inhibited despite the lacking covalent mode-of-action.30 In contrast, other Cys797 analogue kinase off-targets like BMX (Cys496), ITK (Cys442), and JAK3 (Cys909) benefit from our reversible binding mode. Keeping in mind the over 10-fold increase in biochemical activity and a 15-fold increase in cellular activity against the targeted EGFR-L858R/C797S mutant, the concomitant increase toward tyrosine kinases seems tolerable when changing from the isopropyl ester to the phenol ester.
In this work, we demonstrated the improvement of 2-aminoyprimidine inhibitors against EGFR-L858R/C797S osimertinib first-line and EGFR-L858R/T790M/C797S osimertinib second-line resistance mutations based on the structural features of the Exon20ins mutation inhibitor mobocertinib. We were able to improve the biochemical activity from osimertinib to a highly potent reversible inhibitor 13. Additionally, we were able to restore cellular activity against EGFR-L858R/C797S dependent Ba/F3 cells to a low nanomolar level (13, 7 nM) with high selectivity over EGFR wild-type dependent A431 cells (13, 1.8 μM). To exploit the hydrophobic back pocket even more effectively, we were able to resolve cocrystal structures of the isopropyl ester 9 and phenol ester 13 in C797S- and T790M-mutated EGFR. The distinct sandwich motif of the phenol being locked between Met790 and Lys745 gives insights, how the initially unsatisfactory inhibitory activity against T790M-mutated EGFR can be improved. With this, we generated an inhibitor, which shows high inhibition against several NSCLC driving EGFR activating and resistance mutations. We are confident that this work will guide and stimulate further development of reversible inhibitors targeting C797S resistance mutations, which inevitable will be a frequently observed resistance mechanism in Cys797 reliant NSCLC treatment.
Acknowledgments
This work was co-funded by the Mercator Research Center Ruhr (MERCUR), the German Federal Ministry for Education and Research (NGFNPlus, InCa and e:Med) (grant no. BMBF 01GS08104, 01ZX2201B, 01ZX1303C), the Deutsche Forschungsgemeinschaft (DFG) (435700499), the German federal state North Rhine-Westphalia (NRW), the European Union (European Regional Development Fund: Investing In Your Future) (EFRE-800400), NEGECA (PerMed NRW), EMODI, IGNITE (Ex-2021-0033), Drug Discovery Hub Dortmund (DDHD), and has received funding from the program"Netzwerke 2021", an initiative of the Ministry of Culture and Science of the State of North Rhine Westphalia (CANcer TARgeting, NW21-062C). We thank the staff at the PXII X10SA beamline for the possibility of collecting diffraction data. Thanks also go to the LDC, namely Anke Unger and Mia Zischinsky, for providing and measuring the CLint-data.
Glossary
Abbreviations
- ALK
anaplastic lymphoma kinase
- CTG
CellTiter-Glo
- EGFR
epidermal growth factor receptor
- HTRF
homogeneous time-resolved fluorescence
- NSCLC
nonsmall cell lung cancer
- SAR
structure–activity relationship
- TKI
tyrosine kinase inhibitor
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00514.
Experimental procedures, synthetic schemes, compound analysis, complete SAR-table, kinome-selectivity screen (PDF)
Author Present Address
† Michael Beck Department Computational Life Science, Bayer AG, Division Cropscience, 40789 Monheim am Rhein, (Germany)
Author Contributions
# K.J. and J.N. contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): D.R. is founder and consultant of PearlRiver Bio GmbH and shareholder of Centessa Pharmaceuticals plc. PearlRiver Bio GmbH is part of Centessa Pharmaceuticals plc.
Supplementary Material
References
- Finlay M. R. V.; Anderton M.; Ashton S.; Ballard P.; Bethel P. A.; Box M. R.; Bradbury R. H.; Brown S. J.; Butterworth S.; Campbell A.; Chorley C.; Colclough N.; Cross D. A. E.; Currie G. S.; Grist M.; Hassall L.; Hill G. B.; James D.; James M.; Kemmitt P.; Klinowska T.; Lamont G.; Lamont S. G.; Martin N.; McFarland H. L.; Mellor M. J.; Orme J. P.; Perkins D.; Perkins P.; Richmond G.; Smith P.; Ward R. A.; Waring M. J.; Whittaker D.; Wells S.; Wrigley G. L. Discovery of a Potent and Selective EGFR Inhibitor (AZD9291) of Both Sensitizing and T790M Resistance Mutations That Spares the Wild Type Form of the Receptor. J. Med. Chem. 2014, 57 (20), 8249–8267. 10.1021/jm500973a. [DOI] [PubMed] [Google Scholar]
- Cross D. A. E.; Ashton S. E.; Ghiorghiu S.; Eberlein C.; Nebhan C. A.; Spitzler P. J.; Orme J. P.; Finlay M. R. V.; Ward R. A.; Mellor M. J.; Hughes G.; Rahi A.; Jacobs V. N.; Brewer M. R.; Ichihara E.; Sun J.; Jin H.; Ballard P.; Al-Kadhimi K.; Rowlinson R.; Klinowska T.; Richmond G. H. P.; Cantarini M.; Kim D. W.; Ranson M. R.; Pao W. AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer Discov. 2014, 4 (9), 1046–1061. 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok T. S.; Wu Y.-L.; Ahn M.-J.; Garassino M. C.; Kim H. R.; Ramalingam S. S.; Shepherd F. A.; He Y.; Akamatsu H.; Theelen W. S. M. E.; Lee C. K.; Sebastian M.; Templeton A.; Mann H.; Marotti M.; Ghiorghiu S.; Papadimitrakopoulou V. A. Osimertinib or Platinum–Pemetrexed in EGFR T790M–Positive Lung Cancer. NEJM 2017, 376 (7), 629–640. 10.1056/NEJMoa1612674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria J.-C.; Ohe Y.; Vansteenkiste J.; Reungwetwattana T.; Chewaskulyong B.; Lee K. H.; Dechaphunkul A.; Imamura F.; Nogami N.; Kurata T.; Okamoto I.; Zhou C.; Cho B. C.; Cheng Y.; Cho E. K.; Voon P. J.; Planchard D.; Su W.-C.; Gray J. E.; Lee S.-M.; Hodge R.; Marotti M.; Rukazenkov Y.; Ramalingam S. S. Osimertinib in Untreated EGFR -Mutated Advanced Non–Small-Cell Lung Cancer. NEJM 2018, 378 (2), 113–125. 10.1056/NEJMoa1713137. [DOI] [PubMed] [Google Scholar]
- Thress K. S.; Paweletz C. P.; Felip E.; Cho B. C.; Stetson D.; Dougherty B.; Lai Z.; Markovets A.; Vivancos A.; Kuang Y.; Ercan D.; Matthews S. E.; Cantarini M.; Barrett J. C.; Jänne P. A.; Oxnard G. R. Acquired EGFR C797S Mutation Mediates Resistance to AZD9291 in Non-Small Cell Lung Cancer Harboring EGFR T790M. Nat. Med. 2015, 21 (6), 560–562. 10.1038/nm.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabe T.; Lategahn J.; Rauh D. C797S Resistance: The Undruggable EGFR Mutation in Non-Small Cell Lung Cancer?. ACS Med. Chem. Lett. 2018, 9 (8), 779–782. 10.1021/acsmedchemlett.8b00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J.; Zhou Z.; Sun X.; Yang Z.; Zheng P.; Xu S.; Zhu W. The New Opportunities in Medicinal Chemistry of Fourth-Generation EGFR Inhibitors to Overcome C797S Mutation. Eur. J. Med. Chem. 2021, 210, 112995. 10.1016/j.ejmech.2020.112995. [DOI] [PubMed] [Google Scholar]
- Ramalingam S. S.; Yang J. C.-H.; Lee C. K.; Kurata T.; Kim D.-W.; John T.; Nogami N.; Ohe Y.; Mann H.; Rukazenkov Y.; et al. Osimertinib As First-Line Treatment of EGFR Mutation-Positive Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 841–849. 10.1200/JCO.2017.74.7576. [DOI] [PubMed] [Google Scholar]
- Rangachari D.; To C.; Shpilsky J. E.; VanderLaan P. A.; Kobayashi S. S.; Mushajiang M.; Lau C. J.; Paweletz C. P.; Oxnard G. R.; Jänne P. A.; Costa D. B. EGFR-Mutated Lung Cancers Resistant to Osimertinib through EGFR C797S Respond to First-Generation Reversible EGFR Inhibitors but Eventually Acquire EGFR T790M/C797S in Preclinical Models and Clinical Samples. J. Thorac. Oncol. 2019, 14 (11), 1995–2002. 10.1016/j.jtho.2019.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchibori K.; Inase N.; Nishio M.; Fujita N.; Katayama R. Identification of Mutation Accumulation as Resistance Mechanism Emerging in First-Line Osimertinib Treatment. J. Thorac. Oncol. 2018, 13 (7), 915–925. 10.1016/j.jtho.2018.04.005. [DOI] [PubMed] [Google Scholar]
- Wang C.; Zhao K.; Hu S.; Li M.; Song Y. Patterns and Treatment Strategies of Osimertinib Resistance in T790M-Positive Non-Small Cell Lung Cancer: A Pooled Analysis. Front. Oncol. 2021, 11, 600844. 10.3389/fonc.2021.600844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eno M. S.; Brubaker J. D.; Campbell J. E.; De Savi C.; Guzi T. J.; Williams B. D.; Wilson D.; Wilson K.; Brooijmans N.; Kim J.; Özen A.; Perola E.; Hsieh J.; Brown V.; Fetalvero K.; Garner A.; Zhang Z.; Stevison F.; Woessner R.; Singh J.; Timsit Y.; Kinkema C.; Medendorp C.; Lee C.; Albayya F.; Zalutskaya A.; Schalm S.; Dineen T. A. Discovery of BLU-945, a Reversible, Potent, and Wild-Type-Sparing Next-Generation EGFR Mutant Inhibitor for Treatment-Resistant Non-Small-Cell Lung Cancer. J. Med. Chem. 2022, 65 (14), 9662–9677. 10.1021/acs.jmedchem.2c00704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel F.; Siegel S.; Graham K.; Karsli-Uzunbas G.; Korr D.; Schroeder J.; Boemer U.; Hillig R. C.; Mortier J.; Niehues M.; Golfier S.; Schulze V.; Menz S.; Kamburov A.; Hermsen M.; Cherniak A.; Eis K.; Eheim A.; Meyerson M.; Greulich H. BAY 2927088: The First Non-Covalent, Potent, and Selective Tyrosine Kinase Inhibitor Targeting EGFR Exon 20 Insertions and C797S Resistance Mutations in NSCLC. Eur. J. Cancer 2022, 174, S9–S10. 10.1016/S0959-8049(22)00827-9. [DOI] [Google Scholar]
- Lucas M.; Merchant M.; O’Connor M.; Smith S.; Trombino A.; Waters N.; Eathiraj S.; Buck E. BDTX-1535, a Fourth Generation EGFR Inhibitor, Targeting Intrinsic and Acquired Resistance Mutations in NSCLC. Eur. J. Cancer 2022, 174, S22. 10.1016/S0959-8049(22)00862-0. [DOI] [Google Scholar]
- Tavera L.; Campbell J.; Dhande A.; Dineen T.; Iliou M.; Rouskin-Faust T.; Rozsahegyi E.; Conti C. BLU-945 or BLU-701 as Single Agents versus Their Combination with Osimertinib in EGFR L858R Driven Tumor Models. Eur. J. Cancer 2022, 174, S63. 10.1016/S0959-8049(22)00971-6. [DOI] [Google Scholar]
- Gonzalvez F.; Vincent S.; Baker T. E.; Gould A. E.; Li S.; Wardwell S. D.; Nadworny S.; Ning Y.; Zhang S.; Huang W.-S.; Hu Y.; Li F.; Greenfield M. T.; Zech S. G.; Das B.; Narasimhan N. I.; Clackson T.; Dalgarno D.; Shakespeare W. C.; Fitzgerald M.; Chouitar J.; Griffin R. J.; Liu S.; Wong K.; Zhu X.; Rivera V. M. Mobocertinib (TAK-788): A Targeted Inhibitor of EGFR Exon 20 Insertion Mutants in Non–Small Cell Lung Cancer. Cancer Discov. 2021, 11 (7), 1672–1687. 10.1158/2159-8290.CD-20-1683. [DOI] [PubMed] [Google Scholar]
- Riely G. J.; Neal J. W.; Camidge D. R.; Spira A. I.; Piotrowska Z.; Costa D. B.; Tsao A. S.; Patel J. D.; Gadgeel S. M.; Bazhenova L.; Zhu V. W.; West H. L.; Mekhail T.; Gentzler R. D.; Nguyen D.; Vincent S.; Zhang S.; Lin J.; Bunn V.; Jin S.; Li S.; Jänne P. A. Activity and Safety of Mobocertinib (TAK-788) in Previously Treated Non–Small Cell Lung Cancer with EGFR Exon 20 Insertion Mutations from a Phase I/II Trial. Cancer Discov. 2021, 11 (7), 1688–1699. 10.1158/2159-8290.CD-20-1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lategahn J.; Tumbrink H. L.; Schultz-Fademrecht C.; Heimsoeth A.; Werr L.; Niggenaber J.; Keul M.; Parmaksiz F.; Baumann M.; Menninger S.; Zent E.; Landel I.; Weisner J.; Jeyakumar K.; Heyden L.; Russ N.; Müller F.; Lorenz C.; Brägelmann J.; Spille I.; Grabe T.; Müller M. P.; Heuckmann J. M.; Klebl B. M.; Nussbaumer P.; Sos M. L.; Rauh D. Insight into Targeting Exon20 Insertion Mutations of the Epidermal Growth Factor Receptor with Wild Type-Sparing Inhibitors. J. Med. Chem. 2022, 65 (9), 6643–6655. 10.1021/acs.jmedchem.1c02080. [DOI] [PubMed] [Google Scholar]
- Uchibori K.; Inase N.; Araki M.; Kamada M.; Sato S.; Okuno Y.; Fujita N.; Katayama R. Brigatinib Combined with Anti-EGFR Antibody Overcomes Osimertinib Resistance in EGFR-Mutated Non-Small-Cell Lung Cancer. Nat. Commun. 2017, 8, 14768. 10.1038/ncomms14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Zhou L.; Yin J. C.; Wu X.; Shao Y. W.; Gao B. Lung Adenocarcinoma Harboring EGFR 19del/C797S/T790M Triple Mutations Responds to Brigatinib and Anti-EGFR Antibody Combination Therapy. J. Thorac. Oncol. 2019, 14 (5), e85–e88. 10.1016/j.jtho.2019.01.015. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Yang N.; Zhang Y.; Li L.; Han R.; Zhu M.; Feng M.; Chen H.; Lizaso A.; Qin T.; Liu X.; He Y. Effective Treatment of Lung Adenocarcinoma Harboring EGFR-Activating Mutation, T790M, and Cis-C797S Triple Mutations by Brigatinib and Cetuximab Combination Therapy. J. Thorac. Oncol. 2020, 15, 1369–1375. 10.1016/j.jtho.2020.04.014. [DOI] [PubMed] [Google Scholar]
- Wolber G.; Langer T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45 (1), 160–169. 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Wu J.; Wang A.; Qi Z.; Jiang T.; Chen C.; Zou F.; Hu C.; Wang W.; Wu H.; Hu Z.; Wang W.; Wang B.; Wang L.; Ren T.; Zhang S.; Liu Q.; Liu J. Discovery of N -(5-((5-Chloro-4-((2-(Isopropylsulfonyl)Phenyl)Amino)Pyrimidin-2-Yl)Amino)-4-Methoxy-2-(4-Methyl-1,4-Diazepan-1-Yl)Phenyl)Acrylamide (CHMFL-ALK/EGFR-050) as a Potent ALK/EGFR Dual Kinase Inhibitor Capable of Overcoming a Variety of ALK/EGFR Associated Drug Resistant Mutants in NSCLC. Eur. J. Med. Chem. 2017, 139, 674–697. 10.1016/j.ejmech.2017.08.035. [DOI] [PubMed] [Google Scholar]
- Romu A. A.; Lei Z.; Zhou B.; Chen Z.-S.; Korlipara V. Design, Synthesis and Biological Evaluation of WZ4002 Analogues as EGFR Inhibitors. Bioorg. Med. Chem. Lett. 2017, 27 (21), 4832–4837. 10.1016/j.bmcl.2017.09.048. [DOI] [PubMed] [Google Scholar]
- Heppner D. E.; Günther M.; Wittlinger F.; Laufer S. A.; Eck M. J. Structural Basis for EGFR Mutant Inhibition by Trisubstituted Imidazole Inhibitors. J. Med. Chem. 2020, 63 (8), 4293–4305. 10.1021/acs.jmedchem.0c00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima K.; Kawauchi H.; Tanimura H.; Tachibana Y.; Chiba T.; Torizawa T.; Sakamoto H. CH7233163 Overcomes Osimertinib-Resistant EGFR-Del19/T790M/C797S Mutation. Mol. Cancer Ther. 2020, 19 (11), 2288–2297. 10.1158/1535-7163.MCT-20-0229. [DOI] [PubMed] [Google Scholar]
- Li S.; Zhang T.; Zhu S.-J.; Lei C.; Lai M.; Peng L.; Tong L.; Pang Z.; Lu X.; Ding J.; Ren X.; Yun C.-H.; Xie H.; Ding K. Optimization of Brigatinib as New Wild-Type Sparing Inhibitors of EGFR T790M/C797S Mutants. ACS Med. Chem. Lett. 2022, 13 (2), 196–202. 10.1021/acsmedchemlett.1c00555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel J.; Smith S.; Lategahn J.; Tumbrink H. L.; Goebel L.; Becker C.; Hennes E.; Keul M.; Unger A.; Müller H.; Baumann M.; Schultz-Fademrecht C.; Günther G.; Hengstler J. G.; Rauh D. Structure-Guided Development of Covalent and Mutant-Selective Pyrazolopyrimidines to Target T790M Drug Resistance in Epidermal Growth Factor Receptor. J. Med. Chem. 2017, 60 (18), 7725–7744. 10.1021/acs.jmedchem.7b00515. [DOI] [PubMed] [Google Scholar]
- Ito K.; Nishio M.; Kato M.; Murakami H.; Aoyagi Y.; Ohe Y.; Okayama T.; Hashimoto A.; Ohsawa H.; Tanaka G.; Nonoshita K.; Ito S.; Matsuo K.; Miyadera K. TAS-121, A Selective Mutant EGFR Inhibitor, Shows Activity Against Tumors Expressing Various EGFR Mutations Including T790M and Uncommon Mutations G719X. Mol. Cancer Ther. 2019, 18 (5), 920–928. 10.1158/1535-7163.MCT-18-0645. [DOI] [PubMed] [Google Scholar]
- Liu R.; Zhan S.; Che Y.; Shen J. Reactivities of the Front Pocket N-Terminal Cap Cysteines in Human Kinases. J. Med. Chem. 2022, 65 (2), 1525–1535. 10.1021/acs.jmedchem.1c01186. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.