

Abstract

Thiamine is metabolized into the coenzyme thiamine diphosphate (ThDP). Interrupting thiamine utilization leads to disease states. Oxythiamine, a thiamine analogue, is metabolized into oxythiamine diphosphate (OxThDP), which inhibits ThDP-dependent enzymes. Oxythiamine has been used to validate thiamine utilization as an anti-malarial drug target. However, high oxythiamine doses are needed in vivo because of its rapid clearance, and its potency decreases dramatically with thiamine levels. We report herein cell-permeable thiamine analogues possessing a triazole ring and a hydroxamate tail replacing the thiazolium ring and diphosphate groups of ThDP. We characterize their broad-spectrum competitive inhibition of ThDP-dependent enzymes and of Plasmodium falciparum proliferation. We demonstrate how the cellular thiamine-utilization pathway can be probed by using our compounds and oxythiamine in parallel.
Keywords: thiamine diphosphate, enzyme inhibition, metal-binding group, malaria
Thiamine 1 (vitamin B1) is essential for energy metabolism, and its deficiency leads to neurological disorders.1−3 Thiamine, being positively charged, requires transport into the cytoplasm, where it is converted into coenzyme thiamine diphosphate (ThDP) 2a by thiamine pyrophosphokinase (TPK) (Figure 1A).1−5 ThDP-dependent enzymes include pyruvate dehydrogenase complex E1-subunit (PDHc E1), pyruvate decarboxylase (PDC), oxoglutarate dehydrogenase complex E1-subunit (OGDHc E1), pyruvate oxidase (PO), and transketolase (TK). Individual enzymes differ in substrate preferences and reactions catalyzed, but they all share similar ThDP binding sites. In mammalian tissues, ThDP is the major form of thiamine, but lesser amounts of free thiamine 1, thiamine monophosphate, thiamine triphosphate 3 (ThTP), and adenosine thiamine triphosphate 4 (AThTP) are also found.1−3 These derivatives seem to have non-coenzyme roles:1−3 thiamine 1 is co-released with acetylcholine in acetylcholinergic neurons and seems to facilitate neurotransmission; ThTP 3 functions as a phosphate donor in a protein phosphorylation reaction; and AThTP 4 can inhibit poly-ADP-ribose polymerase 1, which is involved in DNA repair.6 Even ThDP may have non-coenzyme roles: elevated levels inhibit pyridoxal kinase and inhibit the binding of p53 to DNA. In bacteria, fungi, and plants ThDP also down-regulates its own biosynthesis by binding to riboswitches in the mRNA coding for biosynthetic enzymes.1−3
Figure 1.

(A) Thiamine-utilization pathways. (B) Binding mode of 2b and 5 in the ThDP pocket. (C) Comparison of the activities of thiamine/ThDP analogues. (D) Ligand design strategy.
A common method to study the coenzyme role of thiamine is to use thiamine/ThDP analogues (Figure 1B), both synthetic3,7−19 and naturally occurring,20,21 as ThDP-competitive inhibitors of ThDP-dependent enzymes. Figure 1C summarizes the three main types of thiamine/ThDP analogues that have been employed. The first type,7,8 represented by triazole-ThDP 5,8 features a neutral central ring in place of the ThDP’s positive thiazolium ring 2a, which abolishes the catalytic activity. These ThDP analogues are potent ThDP-dependent enzyme inhibitors as the neutral central ring captures the strong stabilizing interactions between the enzyme’s hydrophobic region and the catalytically active high-energy ThDP-ylide 2b (Figure 1B). They inhibit ThDP-dependent enzymes non-selectively (binding in place of the cofactor) but are not cell-permeable due to the polyanionic diphosphate.20 The second type,9−13 represented by furan 6,13 also features a neutral central ring but bears a neutral tail replacing the diphosphate. This neutral tail compensates for the loss of the ionic interaction between the diphosphate and the Mg2+ ion by forming interactions with surrounding residues in the diphosphate pocket of PDHc E1. Since these interactions are specific to PDHc E1, 6 is a potent and PDHc-selective inhibitor. It is membrane-permeable and can be used to study the cellular role of PDHc. The third type,14−21 represented by oxythiamine 7a, is a thiamine analogue featuring a modified pyrimidine ring. 7a is a prodrug; it enters cells, probably via thiamine transporters, and is activated by TPK to OxThDP 7b, which inhibits ThDP-dependent enzymes.1−3 Triazole-ThDP 5 and OxThDP 7b are known ligands for ThDP riboswitches which recognize both the aminopyrimidine and diphosphate moieties.22
Interruption of the thiamine-utilization pathway can result in diseases such as diabetes and neurodegeneration and is also found in many cancers.1−4 To understand these diseases, compounds causing thiamine deficiency within the cells can be used. Oxythiamine 7a is the most widely applied tool for inducing thiamine deficiency in cells and in vivo.1−3,14−18 We reported previously that oxythiamine inhibits the in vitro proliferation of Plasmodium falciparum.15 However, oxythiamine 7a has weaknesses as a probe: 1) its positively charged thiazolium ring can result in poor pharmacokinetic properties and degradation by thiaminases, so high oral doses are needed in vivo;15−17 2) the diphosphate moiety of 7b can be hydrolyzed by ThDP-phosphatases;23 3) its similarity to ThDP could allow it to participate in non-coenzyme roles of ThDP 2 or (after appropriate modification of the diphosphate) of 3 and 4 (Figure 1A);1,2 and 4) the required intracellular activation of 7a to 7b makes its action highly sensitive to the levels of thiamine. As an example of this last weakness, oxythiamine inhibits in vitro proliferation of the 3D7 strain of Plasmodium falciparum with an IC50 value of 11 μM in the absence of extracellular thiamine, but the IC50 value increased 470-fold (to 5.2 mM) with added thiamine (2.97 μM).15 We attribute the significant reduction of the anti-plasmodial effect with increasing thiamine levels to the competition with thiamine/ThDP at all stages of thiamine utilization, cell entry, pyrophosphorylation, and enzyme binding.
In this paper we prepare a series of neutral triazole-based thiamine analogues (Figure 1D), resulting in the discovery of thiamine analogues with inhibitory activity against multiple ThDP-dependent enzymes. The anti-plasmodial activity of the analogues are assessed and compared to that of oxythiamine. Lacking the diphosphate, they are unlikely to be recognized by other ThDP-binding proteins or riboswitches. Also, they are not dependent on thiamine transporters or pyrophosphorylation by TPK, which reduces their sensitivity to the extracellular level of thiamine. We envision that these compounds will be useful tools to study the coenzyme roles of ThDP, complementing oxythiamine in inducing the effects of thiamine deficiency.
Triazole-ThDP 5 is one of the most potent inhibitors of ThDP-dependent enzymes, with KI for PDC from Zymomonas mobilis of 30 pM.8,24 Based on this, our pharmacophore model of thiamine/ThDP analogues (Figure 1D) consists of the aminopyrimidine-CH2–triazole moiety joined to a neutral metal-binding group (MBG) by a linker of variable length for optimal positioning of the MBG. The use of a neutral MBG in place of the polyanionic diphosphate was for membrane-permeability.25 Seven different MBGs were tested in this study, adapted from other reported metalloenzyme inhibitors (Scheme 1).26 Most MBGs were attached by linkers of three different lengths. All ligands were efficiently synthesized in 2–5 steps from a common precursor azide 8, in turn obtained in a single-step from inexpensive thiamine.8 OxThDP was synthesized as reported.18
Scheme 1. Synthesis of Triazole-Based Thiamine Analogues.

Reagents and conditions: (i) CuSO4·5H2O, sodium ascorbate, t-BuOH, H2O, RT; (ii) TsCl, pyridine, RT; (iii) NaN3, DMF, RT; (iv) H2(g), 10% Pd/C, MeOH, RT; (v) carboxylic acid, DCC, DMAP, DMF, RT; (vi) Tf2O, pyridine, 35 °C; (vii) NH2OBn.HCl, CDI, DMF, THF, RT; (viii) BCl3, DCM, RT; or H2(g), 10% Pd/C, MeOH, RT; (ix) N-Boc-1,2-phenylenediamine, DCC, DMAP, DMF, 40 °C; (x) TFA, DCM, RT. For all compounds: a, n = 1; b, n = 2; c, n = 3. Potential metal-binding atoms of each metal-binding group (MBG) are highlighted.
To demonstrate widespread inhibition of ThDP-dependent enzymes, we chose four enzymes across three kingdoms—porcine PDHc E1, Saccharomyces cerevisiae PDC, E. coli OGDHc E1, and Aerococcus viridans PO. The different reactions catalyzed, preferred substrates (Table 1), and primary sequences reflect the structurally and functionally diverse members of the family.
Table 1. Comparison of Assay Enzymes and Preliminary Screening Data.


Data are the means of measurements in three technical replicates.
[Compound] = 250 μM, [ThDP] = 50 μM.
[Compound] = 1.5 mM, [ThDP] = 0.3 mM; compounds insufficiently soluble were excluded. (The KM of yeast PDC for ThDP was found to be 11–23 μM, so a high [ThDP] was needed to fully saturate the enzyme.)
The 32 compounds shown in Table 1 were subjected to preliminary screening for inhibition of the four enzymes. Free alcohols 9a–c, which lack the MBG but retain the aminopyridine-CH2–triazole moiety, were included as reference compounds. OxThDP 7b was used as a positive control. The compounds showed a wide range of percentage inhibition on PDHc E1, PDC, and OGDHc E1; they were mostly more potent than the alcohols 9a–c, indicating that the modified tail can contribute to binding. Surprisingly, most ligands were inactive on PO, with only 7b, 15a, 18c, 24b, and 24c inhibiting weakly. Five inhibitors particularly attracted our attention: bis-triazoles 15–17c and hydroxamates 24b,c. The two hydroxamates showed inhibitory activities comparable to that of OxThDP 7b and markedly greater than those of 9a–c for all four enzymes; thus they are truly multi-targeting ligands, warranting further characterization. On the other hand, 15–17c were very selective, potent inhibitors of PDHc E1.
The remaining compounds were not potent enough to warrant further biological testing. However, some docking studies were performed to explain the observed inhibitory effects (Figures S1–3). The ThDP-competitive relationship of the inhibitors was validated by repeating the assays at increased [ThDP]:[compound] ratio (Table 2). As expected, the percentage inhibition by 7b, 15–17, and 24b and c all decreased, consistent with competitive inhibition. These assays showed that 17c is the strongest binder among the bis-triazole series 15–17, selective to PDHc.
Table 2. Inhibitory Data of Bis-triazoles 15–17 and Hydroxamates 24b,c under Increased ThDP Levels.
|
Inhibition (%)a |
||||
|---|---|---|---|---|
| Compound | PDHc E1b | Yeast PDCc | OGDHc E1b | POb |
| OxThDP 7b | 75 ± 3 | 63 ± 3 | 41 ± 4 | 32 ± 5 |
| 15a | <20 | Insoluble | <20 | <20 |
| 15b | 40 ± 4 | Insoluble | <20 | <20 |
| 15c | 75 ± 4 | Insoluble | <20 | <20 |
| 16a | <20 | Insoluble | <20 | <20 |
| 16b | 43 ± 2 | Insoluble | <20 | <20 |
| 16c | 79 ± 2 | Insoluble | <20 | <20 |
| 17a | <20 | Insoluble | <20 | <20 |
| 17b | 45 ± 3 | Insoluble | <20 | <20 |
| 17c | 82 ± 4 | Insoluble | <20 | <20 |
| 24b | 70 ± 3 | 42 ± 4 | 78 ± 3 | 27 ± 4 |
| 24c | 73 ± 4 | 37 ± 4 | 82 ± 2 | 24 ± 4 |
Data are the means of measurements in three technical replicates. Compared to Table 1, the assays here were conducted with an increase in [ThDP]:[inhibitor] from 1:5 to 1:2.
[Compound] = 250 μM, [ThDP] = 125 μM.
[Compound] = 1500 μM, [ThDP] = 750 μM; compounds insufficiently soluble were excluded.
In silico docking studies into the ThDP-binding pocket of human PDHc E1 (Figures 2 and S4–S7) suggested 7b and 24b and c overlay well with ThDP, with the same V-shaped conformation of the aminopyridine-CH2–thiazole moiety. Hydroxamates 24b and c showed a non-ionic, bidentate metal-binding pose (Figure 2C). For bis-triazole 17c, docking studies suggested that two of the triazole nitrogen atoms interact with the Mg2+ (Figure 2D). 17c, with the p-OMe group, seemed to be a better inhibitor than 15c or 16c because the O of the OMe group hydrogen bonds to the side chain of a glutamine residue. The in silico studies also suggested that the selectivity of 15–17 for PDHc E1 is because the longer side-chains are too bulky to be accommodated in the smaller diphosphate pockets in OGDHc E1, PDC, and PO (Figure S7).
Figure 2.
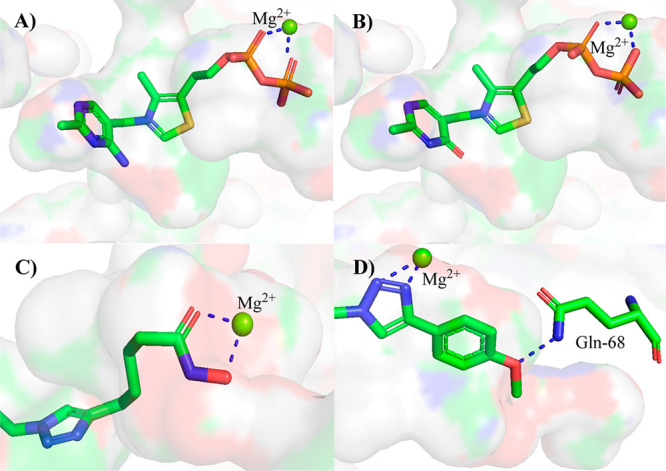
In silicoprediction of interactions with human PDHc E1. (A) ThDP 2a. (B) OxThDP 7b. (C) Hydroxamate 24c. (D) Bis-triazole 17c.
Full IC50 determinations for bis-triazole 17c and hydroxamates 24b,c showed that they inhibited ThDP-dependent enzymes in a dose-dependent manner (Figure 3A–D). As ThDP-competitive ligands, their KI value, affinity relative to ThDP, and ligand efficiency (L.E.) are shown in Figure 3E. L.E., measuring the binding energy of a ligand to its target (in kcal mol–1 derived from the KI value) per heavy atom of the ligand, is a widely applied metric in medicinal chemistry;27 drug discovery efforts often aim to develop clinical candidates with L.E. > 0.3.27,28 Bis-triazole 17c showed the best inhibition of PDHc E1, with a KI of 30 nM; as it lacks activity on the other three enzymes, 17c is an efficient, selective inhibitor of PDHc. Both hydroxamates, 24b (L.E. = 0.50) and 24c (L.E. = 0.48), are extremely efficient inhibitors of PDHc and very good inhibitors of the other three enzymes, too. The affinities of hydroxamates 24b and c and OxThDP 7b for PDHc E1 are all comparable to that of ThDP (Table 2 and Figure 3), which is in line with the findings of others on OxThDP.2,3 The molecular properties of hydroxamates 24b and c (Table S1) are predicted to be in the range preferred for drugs,27,28 so they should be active on intact live cells.
Figure 3.

Inhibition of ThDP-dependent enzymes by 17c and 24b,c. IC50 values determined at [ThDP] (A, B) 10 μM, (C) 30 μM, and (D) 50 μM. Data are the means of measurements in three technical replicates. Where error bars are not visible they are smaller than the symbol used. The PDC used here was bacterial (from Zymomonas mobilis) due to the better assay conditions that avoid solubility issues. (E) KI values and ligand efficiencies (L.E.). KI values are determined according to the KM for ThDP of the respective enzymes. *Affinity of the inhibitor versus that of ThDP (i.e. [ThDP]/IC50 or KM(ThDP)/KI).
Next the effects of the thiamine/ThDP analogues on in vitro proliferation of the 3D7 strain of P. falciparum were determined. Infected erythrocytes were treated with 16a–c, 17a–c, and 24b,c. Parasite proliferation was measured by SYBR-Safe assay of parasite DNA.11,15 The IC50 (concentration at which the compound suppresses parasite proliferation by 50%) was determined at various levels of extracellular thiamine (Table 3) and is compared to our previous data on oxythiamine 7a.15
Table 3. IC50 Values (μM) for the Anti-plasmodial Activity, and Cytotoxicity of Selected Compounds.
|
IC50of Suppression ofP. falciparumproliferation
(μM) |
|||||
|---|---|---|---|---|---|
| Compound | Thiamine-free | [Thiamine] = 2.97 μM | [Thiamine] = 297 μM | HFF Cytotoxicity (μM) | Selectivity Indexa |
| 7ab | 11 ± 4 | 5200 ± 300 | 5500 ± 500 | ND | ND |
| 16a | >200 | >200 | >200 | ND | ND |
| 16b | >200 | >200 | >200 | >200 | ND |
| 16c | 67 ± 5 | 70 ± 8 | 89 ± 19 | 48 ± 5 | 0.7 |
| 17a | 129 ± 9 | 156 ± 13 | 169 ± 11 | >200 | ND |
| 17b | 51 ± 3 | 78 ± 8 | 82 ± 11 | 90 ± 5 | 1.8 |
| 17c | 44 ± 2 | 62 ± 7 | 69 ± 9 | 46 ± 7 | 1.1 |
| 24b | 0.9 ± 0.1 | 1.3 ± 0.2 | 1.4 ± 0.4 | 15 ± 3 | 16.7 |
| 24c | 3.0 ± 1.2 | 3.8 ± 2 | 4.0 ± 1.8 | 44 ± 4 | 14.7 |
Selectivity index is IC50 (thiamine-free) vs HFF cytotoxicity; ND, not determined. Refer to Figures S8 and S9 for details.
Ref (17).
Most of the thiamine analogues tested inhibited proliferation of the parasite (Table 3) in a dose-dependent manner (Figures 4A and S8). In thiamine-free culture medium, the PDHc-selective 16 and 17 (IC50 ≥ 44 μM) were considerably weaker than the multi-targeting hydroxamates 24b,c (IC50 = 0.9–3 μM) and oxythiamine 7a (IC50 = 11 μM). The comparable enzyme inhibition by 24b,c and 7b (Table 2) translated into similar low-micromolar anti-plasmodial activities (Table 3), though 24b is 3-fold more active than 24c and 11-fold more active than 7a. This suggests that the passive diffusion of 24b,c results in a greater concentration in the parasites than the presumed active transport and pyrophosphorylation of 7a.
Figure 4.

Anti-plasmodial activities of 24b and 17c. (A) Activities against P. falciparum 3D7 in medium containing thiamine at 0 μM (○), 2.97 (●), and 297 μM (△). (B) Cytotoxicities on HFF cells. (C) Activities against P. falciparum 3D7 transfected with an empty plasmid (●), and expressing PfTPK-GFP (○) in thiamine-free medium. Data are the means of measurements from three independent experiments, each performed in three technical replicates.
With 2.97 μM thiamine in the culture medium (presumably leading to higher levels of ThDP within the parasites), the IC50 of the better performing hydroxamate 24b increased from 0.9 ± 0.1 μM to 1.3 ± 0.2 μM (n = 3; P = 0.0188; T-test), and the IC50 of the best-performing bis-triazole 17c increased from 44 ± 2 μM to 62 ± 7 μM (n = 3; P = 0.0136; T-test) (Figure 4A). This is consistent with their competitive relationship with thiamine/ThDP in cell-based assays. Interestingly, the drop in the inhibitory effect of 17c, and 24b was much less than that of oxythiamine 7a.15 This suggests that the lowered inhibitory effect of oxythiamine is more due to competition with thiamine for transport into the cell and/or pyrophosphorylation by TPK (resulting in much less OxThDP being formed) than due to competition of OxThDP with ThDP for binding to the enzymes. Increasing the thiamine concentration in the culture medium 100-fold to 297 μM resulted in a statistically insignificant further change in the anti-plasmodial potency of all compounds (including 7a) (Table 3). Probably one or more of the parasite’s thiamine-utilization steps become saturated even at 2.97 μM thiamine, so adding more extracellular thiamine no longer significantly increases the intracellular thiamine/ThDP levels.17
Some compounds were also tested on human foreskin fibroblast (HFF) cells. Most exhibited dose-dependent cytotoxicity (Table 3, and Figures 4B and S9), as expected (because thiamine is essential in all organisms). In the bis-triazole series, the longer derivatives (16c and 17c) showed higher potencies than their shorter counterparts on both P. falciparum and human cells. Hydroxamate 24b was the strongest inhibitor of proliferation of HFF cells, consistent with its potent inhibition of multiple ThDP-dependent enzymes. Although 7a was not tested on HFF cells in this study, numerous in vitro and in vivo studies have confirmed that 7a is toxic.14−17 Most of the compounds were non-selective, but 24b and 24c were 16.7 and 14.7 times more selective against the parasite than the HFF cells (Table 3).
Although 17c, 24b and 24c are very similar in their inhibition of PDHc E1 (Figure 3A), 24b is almost 50-fold more potent than 17c at inhibiting proliferation of the parasites (Figure 4A). This suggests that some other ThDP-dependent enzyme(s) may be more critical than PDHc for proliferation. Possibilities include transketolase, which is required for synthesis of ribose 5-phosphate by the non-oxidative pentose phosphate pathway, and 1-deoxyxylulose 5-phosphate synthase, the first and rate-determining enzyme of terpene biosynthesis via the non-mevalonate pathway. Previous studies have shown that inhibition of the non-mevalonate pathway in P. falciparum does have an anti-proliferative effect.29 The greater activity on the parasites than on the HFF cells (Table 3) would be consistent with this, as human cells do not use the non-mevalonate pathway. The low anti-proliferative effect of 17c might also be because PDHc and OGDHc enzymes can, to some extent, accept each other’s substrates, and so OGDHc could compensate for the lack of activity of the inhibited PDHc.15
We also tested 17c and 24b on a parasite line expressing extra copies of TPK (with a GFP-tag).11 Our previous studies showed that in the thiamine-free culture medium, the parasite over-expressing TPK was 70-fold more sensitive to oxythiamine 7a than a parasite bearing an empty plasmid (the negative control).15 This is because the extra copies of TPK significantly improve the conversion of extracellular 7a to intracellular 7b.11,15 By contrast, in thiamine-free culture medium, the sensitivities of the two parasite lines to 17c and to 24b were almost identical (Figures 4C and S10, the small differences were statistically insignificant). This insensitivity toward TPK over-expression not only confirms that the analogues do not require the action of TPK for enzyme binding but also indicates that TPK is not their target.
These studies collectively suggest that hydroxamate 24b is as competent as oxythiamine to cause thiamine deficiency in cells. However, with the triazole scaffold replacing the thiazolium ring, 24b is uncharged under physiological conditions, so it is more drug-like19,27,28 and unlikely to be degraded by thiaminases.16 Moreover, without the diphosphate moiety, 24b would likely retain activity on oxythiamine-resistant organisms that over-express the SLC19A1 transporter14 (which removes OxThDP from the cell) or ThDP hydrolases23 (which hydrolyze OxThDP to OxThMP). Also, the neutral triazole ring of 24b is expected to improve the selectivity (relative to OxThDP) to the coenzyme role of ThDP over non-coenzyme roles. ThDP riboswitches are known to prefer a charged central ring,22 and the other (unknown) non-coenzyme targets probably do as well, as they are unlikely to be optimized for binding the neutral ThDP ylide 2b, as the enzymes are. Furthermore, it most unlikely that 24b, lacking the diphosphate, would be converted into derivatives equivalent to ThTP 3 or AThTP 4, whereas OxThDP may well be.
Although the structural and mechanistic differences may make hydroxamate 24b superior to oxythiamine 7a in some ways, its utility in understanding the roles of the thiamine-utilization pathway in disease states is maximized when applied alongside 7a, as in this study. We previously attributed the anti-plasmodial activity of oxythiamine to inhibition of multiple ThDP-dependent enzymes in the parasite,15 but it is possible that action on some non-coenzyme role of thiamine may have also contributed. In this study, however, 24b, almost certainly lacking any activity on non-coenzyme roles, exhibited comparable activity to oxythiamine 7a (in thiamine-free culture medium), which suggests that inhibition of ThDP-dependent enzymes is indeed the main target of oxythiamine. Thus, using 7a and 24b together helps assign the observed effects to the coenzyme or non-coenzyme role more confidently.
Bis-triazoles 17a–c are potent and selective inhibitors of PDHc E1, which plays a vital role in bioenergetics, linking glycolysis with the Krebs cycle.4 The relative activities on cells (17c > b > a, Table 3) is likely to be a function of the PDHc E1 inhibition (17c > b > a, Table 2). It is, however, possible that the higher hydrophobicity of the longest derivative 17c led to better cellular uptake. Further cell-based characterization of 17a–c was not pursued due to their narrow-spectrum inhibition. However, recent studies have shown that selective PDHc E1 inhibitors have potential against certain cancers,30,31 so 17c may be of interest in this field. The ligand efficiency of 17c is good (0.34), so it could be a good starting point for the development of anti-cancer drugs.
In summary, this study has demonstrated hydroxamate 24b is an antagonist of thiamine through inhibiting the coenzyme role of ThDP. Its utility is broad: active on both P. falciparum and human cells, and capable of inhibiting different ThDP-dependent enzymes from various species. We have discussed its use alongside oxythiamine 7a in inducing thiamine deficiency in cell-based systems; this can help avoid misinterpretation of the role of these multi-targeting coenzyme antagonists and indicate whether coenzyme or non-coenzyme roles of thiamine are the main target. Based on its affinity and ligand efficiency on target enzymes as well as its potent and selective anti-plasmodial action, hydroxamate 24b is a potential anti-malarial agent.
Acknowledgments
A.H.Y.C. and T.C.S.H. were supported by K.M. Medhealth. I.F. was supported by a Research Training Program scholarship from the Australian government. We are grateful to the Canberra Branch of the Australian Red Cross Lifeblood for red blood cells and members of the van Dorren Lab (ANU) for HFF cells.
Glossary
Abbreviations
- AThTP
adenosine thiamine triphosphate
- HFF
human foreskin fibroblast
- L.E.
ligand efficiency
- MBG
metal-binding group
- OGDHc E1
oxoglutarate dehydrogenase complex E1 subunit
- OxThDP
oxythiamine diphosphate
- PDC
pyruvate decarboxylase
- PDHc E1
pyruvate dehydrogenase complex E1 subunit
- PO
pyruvate oxidase
- ThDP
thiamine diphosphate
- ThTP
thiamine triphosphate
- TK
transketolase
- TPK
thiamine pyrophosphokinase
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.3c00047.
Supporting figures and tables, protocols for biological assays, computational studies, compound synthesis and characterization, and NMR spectra (PDF)
Author Contributions
A.H.Y.C. conceived the project, synthesized most compounds. and performed some enzyme assays. T.C.S.H. performed most enzyme assays and the computational dockings. R.P. synthesized some compounds. F.J.L. supervised this work. I.F. performed cell assays under the supervision of K.J.S.
The authors declare no competing financial interest.
Supplementary Material
References
- Bunik V. I.; Tylicki A.; Lukashev N. V. Thiamin Diphosphate-Dependent Enzymes: From Enzymology to Metabolic Regulation, Drug Design and Disease Models. FEBS J. 2013, 280 (24), 6412–6442. 10.1111/febs.12512. [DOI] [PubMed] [Google Scholar]
- Tylicki A.; Łotowski Z.; Siemieniuk M.; Ratkiewicz A. Thiamine and Selected Thiamine Antivitamins - Biological Activity and Methods of Synthesis. Biosci. Rep. 2018, 38 (1), BSR20171148. 10.1042/BSR20171148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleshin V. A.; Mkrtchyan G. V.; Bunik V. I. Mechanisms of Non-Coenzyme Action of Thiamine: Protein Targets and Medical Significance. Biochemistry Moscow 2019, 84 (8), 829–850. 10.1134/S0006297919080017. [DOI] [PubMed] [Google Scholar]
- Frank R. A. W.; Leeper F. J.; Luisi B. F. Structure, Mechanism and Catalytic Duality of Thiamine-Dependent Enzymes. Cell. Mol. Life Sci. 2007, 64 (7–8), 892–905. 10.1007/s00018-007-6423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prajapati S.; Rabe von Pappenheim F.; Tittmann K. Frontiers in the Enzymology of Thiamin Diphosphate-Dependent Enzymes. Curr. Opin. Struct. Biol. 2022, 76, 102441. 10.1016/j.sbi.2022.102441. [DOI] [PubMed] [Google Scholar]
- Tanaka T.; Yamamoto D.; Sato T.; Tanaka S.; Usui K.; Manabe M.; Aoki Y.; Iwashima Y.; Saito Y.; Mino Y.; Deguchi H. Adenosine Thiamine Triphosphate (AThTP) Inhibits Poly(ADP-Ribose) Polymerase-1 (PARP-1) Activity. J. Nutr. Sci. Vitaminol. 2011, 57 (2), 192–196. 10.3177/jnsv.57.192. [DOI] [PubMed] [Google Scholar]
- Leeper F. J.; Hawksley D.; Mann S.; Perez Melero C.; Wood M. D. H. Studies on Thiamine Diphosphate-Dependent Enzymes. Biochem. Soc. Trans. 2005, 33 (4), 772–775. 10.1042/BST0330772. [DOI] [PubMed] [Google Scholar]
- Erixon K. M.; Dabalos C. L.; Leeper F. J. Synthesis and Biological Evaluation of Pyrophosphate Mimics of Thiamine Pyrophosphate Based on a Triazole Scaffold. Org. Biomol. Chem. 2008, 6 (19), 3561. 10.1039/b806580b. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Zhang S.; He H.; Jiang W.; Hou L.; Xie D.; Cai M.; Peng H.; Feng L. Design and Synthesis of Highly Selective Pyruvate Dehydrogenase Complex E1 Inhibitors as Bactericides. Bioorg. Med. Chem. 2018, 26 (1), 84–95. 10.1016/j.bmc.2017.11.021. [DOI] [PubMed] [Google Scholar]
- Feng J.; He H.; Zhou Y.; Cai M.; Peng H.; Liu H.; Liu L.; Feng L.; He H. Structure Optimization and Bioactivity Evaluation of ThDP Analogs Targeting Cyanobacterial Pyruvate Dehydrogenase E1. Bioorg. Med. Chem. 2019, 27 (24), 115159. 10.1016/j.bmc.2019.115159. [DOI] [PubMed] [Google Scholar]
- Chan A. H. Y.; Fathoni I.; Ho T. C. S.; Saliba K. J.; Leeper F. J. Thiamine Analogues as Inhibitors of Pyruvate Dehydrogenase and Discovery of a Thiamine Analogue with Non-Thiamine Related Antiplasmodial Activity. RSC Med. Chem. 2022, 13 (7), 817–821. 10.1039/D2MD00085G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan A. H. Y.; Ho T. C. S.; Agyei-Owusu K.; Leeper F. J. Synthesis of Pyrrothiamine, a Novel Thiamine Analogue, and Evaluation of Derivatives as Potent and Selective Inhibitors of Pyruvate Dehydrogenase. Org. Biomol. Chem. 2022, 20 (45), 8855–8858. 10.1039/D2OB01819E. [DOI] [PubMed] [Google Scholar]
- Chan A. H. Y.; Ho T. C. S.; Parle D.; Leeper F. J. Furan-based inhibitors of pyruvate dehydrogenase: SAR study, biochemical evaluation and computational analysis. Org. Biomol. Chem. 2023, 21 (8), 1755–1763. 10.1039/D2OB02272A. [DOI] [PubMed] [Google Scholar]
- Mendoza R.; Miller A. D.; Overbaugh J. Disruption of Thiamine Uptake and Growth of Cells by Feline Leukemia Virus Subgroup A. J. Virol. 2013, 87 (5), 2412–2419. 10.1128/JVI.03203-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan X. W. A.; Wrenger C.; Stahl K.; Bergmann B.; Winterberg M.; Müller I. B.; Saliba K. J. Chemical and Genetic Validation of Thiamine Utilization as an Antimalarial Drug Target. Nat. Commun. 2013, 4 (1), 2060. 10.1038/ncomms3060. [DOI] [PubMed] [Google Scholar]
- Liu S.; Miriyala S.; Keaton M. A.; Jordan C. T.; Wiedl C.; Clair D. K. S.; Moscow J. A. Metabolic Effects of Acute Thiamine Depletion Are Reversed by Rapamycin in Breast and Leukemia Cells. PLoS One 2014, 9 (1), e85702. 10.1371/journal.pone.0085702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M.; Smeets R.; Gosau M.; Vollkommer T.; Fuest S.; Stetzer E.; Kluwe L.; Coy J. F.; Burg S. Tolerance of Human Fibroblasts to Benfo-Oxythiamine in vitro. IJERPH 2022, 19 (7), 4112. 10.3390/ijerph19074112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowska E.; Czerniecka M.; Czyżewska U.; Zambrzycka A.; Łotowski Z.; Tylicki A. Differences in the Efficiency of 3-Deazathiamine and Oxythiamine Pyrophosphates as Inhibitors of Pyruvate Dehydrogenase Complex and Growth of HeLa Cells in vitro. J. Enz. Inhib. Med. Chem. 2021, 36 (1), 122–129. 10.1080/14756366.2020.1844681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A. A.; Le Huerou Y.; De Meese J.; Gunawardana I.; Kaplan T.; Romoff T. T.; Gonzales S. S.; Condroski K.; Boyd S. A.; Ballard J.; Bernat B.; DeWolf W.; Han M.; Lee P.; Lemieux C.; Pedersen R.; Pheneger J.; Poch G.; Smith D.; Sullivan F.; Weiler S.; Wright S. K.; Lin J.; Brandhuber B.; Vigers G. Synthesis, in Vitro and in Vivo Activity of Thiamine Antagonist Transketolase Inhibitors. Bioorg. Med. Chem. Lett. 2008, 18 (6), 2206–2210. 10.1016/j.bmcl.2007.11.101. [DOI] [PubMed] [Google Scholar]
- Reddick J. J.; Saha S.; Lee J. M.; Melnick J. S.; Perkins J.; Begley T. P. The mechanism of action of bacimethrin, a naturally occurring thiamin antimetabolite. Bioorg. Med. Chem. Lett. 2001, 11 (17), 2245–2248. 10.1016/S0960-894X(01)00373-0. [DOI] [PubMed] [Google Scholar]
- Rabe von Pappenheim F.; Aldeghi M.; Shome B.; Begley T.; de Groot B. L.; Tittmann K. (2020). Structural basis for antibiotic action of the B1 antivitamin 2′-methoxy-thiamine. Nat. Chem. Biol. 2020, 16 (11), 1237–1245. 10.1038/s41589-020-0628-4. [DOI] [PubMed] [Google Scholar]
- Chen L.; Cressina E.; Dixon N.; Erixon K.; Agyei-Owusu K.; Micklefield J.; Smith A. G.; Abell C.; Leeper F. J. Probing Riboswitch–Ligand Interactions Using Thiamine Pyrophosphate Analogues. Org. Biomol. Chem. 2012, 10 (30), 5924–5931. 10.1039/c2ob07116a. [DOI] [PubMed] [Google Scholar]
- Goyer A.; Hasnain G.; Frelin O.; Ralat M. A.; Gregory J. F.; Hanson A. D. A Cross-Kingdom Nudix Enzyme That Pre-Empts Damage in Thiamin Metabolism. Biochem. J. 2013, 454 (3), 533–542. 10.1042/BJ20130516. [DOI] [PubMed] [Google Scholar]
- Pei X.-Y.; Erixon K. M.; Luisi B. F.; Leeper F. J. Structural Insights into the Prereaction State of Pyruvate Decarboxylase from Zymomonas mobilis. Biochemistry 2010, 49 (8), 1727–1736. 10.1021/bi901864j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudge E. S.; Chan A. H. Y.; Leeper F. J. Prodrugs of Pyrophosphates and Bisphosphonates: Disguising Phosphorus Oxyanions. RSC Med. Chem. 2022, 13 (4), 375–391. 10.1039/D1MD00297J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho T. C. S.; Chan A. H. Y.; Ganesan A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63 (21), 12460–12484. 10.1021/acs.jmedchem.0c00830. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Keserü G. M.; Leeson P. D.; Rees D. C.; Reynolds C. H. The Role of Ligand Efficiency Metrics in Drug Discovery. Nat. Rev. Drug Discovery 2014, 13 (2), 105–121. 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- Meanwell N. A. Improving Drug Candidates by Design: a Focus on Physicochemical Properties as a Means of Improving Compound Disposition and Safety. Chem. Res. Toxicol. 2011, 24 (9), 1420–1456. 10.1021/tx200211v. [DOI] [PubMed] [Google Scholar]
- Jomaa H.; Wiesner J.; Sanderbrand S.; Altincicek B.; Weidemeyer C.; Hintz M.; Türbachova I.; Eberl M.; Zeidler J.; Lichtenthaler H. K.; Soldati D.; Beck E. Inhibitors of the Nonmevalonate Pathway of Isoprenoid Biosynthesis as Antimalarial Drugs. Science 1999, 285 (5433), 1573–1576. 10.1126/science.285.5433.1573. [DOI] [PubMed] [Google Scholar]
- Hensley C. T.; Faubert B.; Yuan Q.; Lev-Cohain N.; Jin E.; Kim J.; Jiang L.; Ko B.; Skelton R.; Loudat L.; Wodzak M.; Klimko C.; McMillan E.; Butt Y.; Ni M.; Oliver D.; Torrealba J.; Malloy C. R.; Kernstine K.; Lenkinski R. E.; DeBerardinis R. J. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164 (4), 681–694. 10.1016/j.cell.2015.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee T. S.; Anderson R. G.; Pladna K. M.; Isom S.; Ghiraldeli L. P.; Miller L. D.; Chou J. W.; Jin G.; Zhang W.; Ellis L. R.; Berenzon D.; Howard D. S.; Hurd D. D.; Manuel M.; Dralle S.; Lyerly S.; Powell B. L. A Phase I Study of CPI-613 in Combination with High-Dose Cytarabine and Mitoxantrone for Relapsed or Refractory Acute Myeloid Leukemia. Clin. Cancer Res. 2018, 24 (9), 2060–2073. 10.1158/1078-0432.CCR-17-2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.