

Abstract
Melanoma is the most aggressive of the skin cancers, with historically high rates of morbidity and mortality due to its resistance to traditional cytotoxic therapies. Recently, however, breakthroughs in new therapies have dramatically changed clinical outcomes of this disease. These advances emerged from an improved understanding of tumor oncogenesis and the interacting tumor microenvironment. Small molecules that target the oncogenic mitogen-activated protein kinase (MAPK) pathway, specifically the tyrosine kinase BRAF and its downstream signaling partner MEK, have demonstrated an improved overall survival and progression-free survival for BRAF-mutant melanoma. Additionally, manipulation of tumor immune surveillance by inhibitors of the immune suppressive programmed cell death 1 receptor (PD-1) and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) pathways have recently demonstrated durable responses in various cancers by promoting an anti-tumor immune response. Application of these targeted and immune-modulatory therapies has shown promising outcomes in melanoma. Combinations of these therapies may hold promise to enhance responses further. In this review, we will discuss the current targeted therapies and immunotherapies, and review the results of combination studies and speculate on future treatment paradigms.
1. Introduction
Melanoma is a common and aggressive skin cancer with historically limited treatment options for metastatic disease. While multi-faceted approaches to risk modification, screening, and chemotherapies have improved outcomes in other common cancers, such as breast, prostate, and lung, the incidence and mortality rates of melanoma have steadily increased in the USA [1]. The hallmark of melanoma is its propensity for early invasion with spread to organ sites such as, but not limited to, the brain, lung, bone, and liver [2]. This insidious quality, coupled with an innocuous appearance in early stages, results in some patients presenting with advanced disease on initial presentation or recurrence following surgery. Unfortunately, conventional therapies have limited success at late stages, with 5-year historical survival for stage III at 50% and stage IV at 5–10% [3, 4]. This urgent need has led to a new generation of therapies for melanoma.
2. Targeted Therapies
The aggressive nature of melanoma in part derives from mutations and other genomic alterations in key intrinsic cellular signaling pathways. Characterization of signaling molecules and proteins in these pathways has enabled the development of a new generation of highly selective therapeutic agents. The mitogen-activated protein kinase (MAPK) pathway was the first major proto-oncogenic pathway to be characterized in melanoma with the discovery of the mutant BRAF kinase in nearly 50% of tumor samples [5]. In addition, NRAS and NF1 mutations are present in a non-overlapping 15–20 and 10–15% of melanomas, respectively. Normally tightly regulated, the MAPK pathway plays a key role in propagating and modulating external growth signals to promote proliferation, growth, and division. The canonical MAPK pathway exerts its effects through the phosphorylation and activation of sequential proteins in its kinase cascade. The first component of the MAPK pathway is the Ras family of small GTPases, which act as ‘on’ or ‘off’ switches when stimulated by upstream signals [6]. Activation of transmembrane receptor proteins by external growth factors causes Ras to bind guanosine triphosphate (GTP), adopting its active conformation, allowing it to phosphorylate and activate the next kinase in the signaling cascade, Raf (a serine/threonine-specific protein kinase with three tissuespecific isoforms: ARAF, BRAF, and CRAF) [7]. This motif is repeated in successive steps of phosphorylation—kinase activation which amplifies the proliferative signal, leading from Ras → RAF → MEK and culminating in the activation of the effector kinase ERK. ERK ultimately acts on numerous cytoplasmic and nuclear targets to promote growth and cell division [6].
Given the importance of the MAPK pathway in cell growth and division, activating mutations in the various components of this pathway, including RAS, RAF, and MEK, have been found in a variety of cancers [8]. For example, RAS is found mutated in nearly a third of human cancers [9], and is especially prominent in pancreatic, colorectal, and lung cancers. NRAS, a Ras isoform, harbors mutations in 15–20% of melanomas.
BRAF, one of three RAF kinases, is mutated in ~8% of all cancers. A constitutively active variant, BRAF V600E (encoded by the BRAFT1799A missense mutation) is found in 40–50% of melanomas. To date, there are nearly 300 characterized mutations in BRAF [10], with those affecting BRAF V600 being the most common (primarily V600E, but also including V600K, V600R, in about 5% of melanomas) [11]. The non-V600E BRAF mutations, although clinically relevant and perhaps amenable to MEK inhibition, will not be addressed in this review. Nearly all oncogenic BRAF mutations, including the V600E mutation, result in a constitutively active kinase which activates MEK independently of an upstream signal or regulation.
The discovery of the mutant BRAF V600E kinase as an oncogenic driver in 2002 [12] unlocked the potential of employing small molecule inhibitors for targeting melanoma. Shortly after, the first BRAF inhibitor, sorafenib (BAY 43–9006), discovered in a screen against the similar protein CRAF as the target, was investigated for its activity against BRAF V600E melanoma cells. While sorafenib was more active against purified CRAF [half maximal inhibitory concentration (IC50) of 6 nM] than mutant BRAF (38 nM) [13], it nonetheless showed promise in mouse models of melanoma. In human trials, however, it failed to show efficacy as a single agent. Additionally, while sorafenib in combination with traditional chemotherapy was found to be superior to chemotherapy alone in several phase II randomized clinical trials, it did not ultimately improve overall survival (OS), and was not correlated with BRAF mutational status, suggesting the effects observed are independent of BRAF inhibition [14]. Sorafenib was shown later to have activity against other kinases, including vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF), arguing that any modest activity likely reflects its anti-angiogenic effects [15, 16]. Indeed, while ineffective in melanoma, sorafenib has since been approved for use in renal and hepatocellular carcinomas, two highly vascular malignancies [17, 18].
Despite the initial discovery of the BRAF V600E mutation in 2002, the development of a specific BRAF inhibitor was hampered by the difficulty of crystallizing the protein for rational drug design. After BRAF V600E was finally crystallized in 2004, the small molecule PLX4032 (Plexxikon), later known as vemurafenib, was formulated in 2005, with highly specific activity against mutant BRAF [19]. In fact, it was 12 times more specific for the BRAF V600E kinase than the wild-type BRAF kinase (IC50 of 13 and 160 nM, respectively) with relatively modest effects on non-V600E BRAF mutations [20].
This discovery was followed by the identification of GSK2118436 (GlaxoSmithKline), or dabrafenib, in 2009. Dabrafenib is a highly potent and mutant-specific BRAF inhibitor with 20 times more inhibitory activity against BRAF V600E than wild-type BRAF (IC50 of 0.6 vs 12 nM) [21, 22]. Additionally, it also demonstrated excellent activity against other V600 BRAF mutations (V600K/R, etc.) [21]. Lastly, LGX818 (Novartis, later Array), or encorafenib, began clinical evaluation in 2013. Preliminary in vitro data have demonstrated a high potency and promising clinical activity, and that it acts to induce autophagy leading to senescence in BRAF V600E melanoma cells [23].
3. Mutant BRAF Inhibitor Monotherapy Trials
Since the discovery of mutant BRAF inhibitors, there was a flurry of research to bring these targeted therapies into clinical use. Despite the initial preclinical data, sorafenib failed as a monotherapy in metastatic melanoma, likely due to suboptimal activity against BRAF oncoproteins. Since then, more specific BRAF inhibitors have been developed. The efficacy and safety of two mutant BRAF inhibitors, vemurafenib and dabrafenib, as monotherapies for BRAF-mutant metastatic melanoma, will be discussed in the following section (schematic in Fig. 1). Given its only recent entrance into the targeted therapy market, encorafenib has no published data as monotherapy. Instead, it is currently being evaluated in a phase III randomized clinical trial in combination with the MEK1/2 inhibitor binimetinib (COLUMBUS trial NCT01909453), with an estimated completion date in 2018.
Fig. 1.
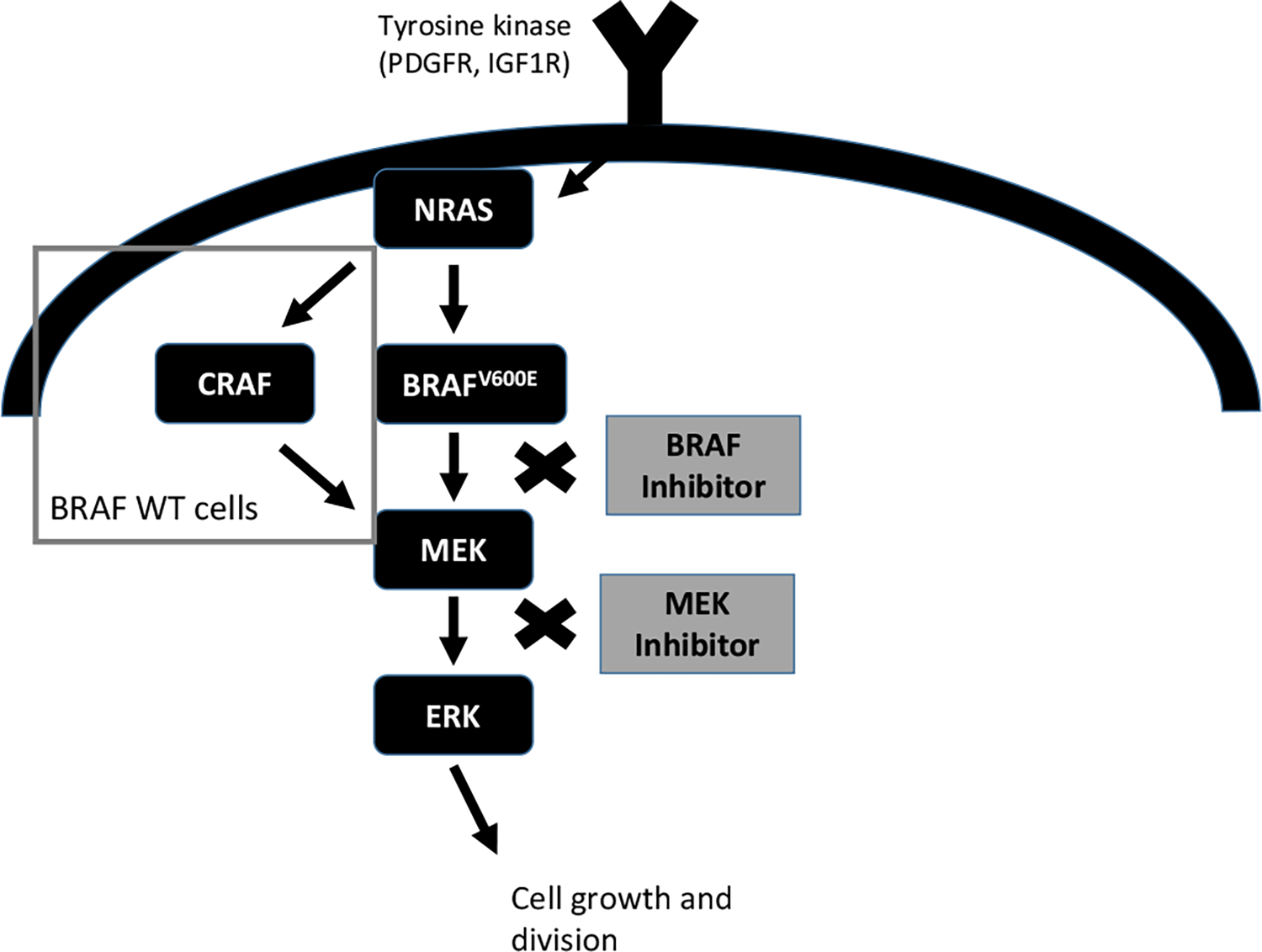
Mechanism of action of BRAF/MEK inhibitors. PDGFR platelet derived growth factor, IGF1R insulin growth factor 1 receptor, WT wild type
3.1. Vemurafenib: BRIM Trial
The initial phase I/II studies of vemurafenib demonstrated impressive clinical activity in patients with metastatic BRAF V600E-mutated melanoma. The phase I study extension evaluated 32 patients at the recommended phase II dosage of 960 mg twice daily, and reported an unconfirmed response rate of >80% [24]. The subsequent phase II study revealed a slightly more modest confirmed response rate of 53%, with a median progression-free survival (PFS) and OS of 6.8 and 15.9 months, respectively. This clinical activity was dramatically superior to that observed with cytotoxic chemotherapy, leading to a phase III study [25].
The phase III randomized clinical trial (BRIM-3) compared the efficacy and safety of vemurafenib monotherapy against dacarbazine monotherapy in treatment-naïve metastatic melanoma patients carrying BRAF V600 mutations. Vemurafenib monotherapy increased the median PSF (6.9 months vs 1.6 months) as well as OS (6 months OS 84 vs 64%). When these patients were subdivided based on their mutational status, both BRAF V600E (91% of the patients) and BRAF V600K (9% of the patients) benefitted similarly from vemurafenib therapy. While vemurafenib was well tolerated, with the majority of adverse events ruled as grade 1–2, all of the patients experienced at least one adverse event and 8% experienced grade 4 or worse adverse events. The majority of adverse events were low grade, including arthralgia, rash, fatigue, photosensitivity, and keratoacanthomas. Seven percent of patients in the vemurafenib arm discontinued therapy due to intolerance. The most common grade 3 or 4 adverse events were cutaneous squamous cell carcinomas, elevated liver function tests, keratoacanthomas, rash, and arthralgia. Cutaneous squamous cell carcinomas occurred in 19% of patients, usually in the first 2–3 months of therapy [26].
3.2. Dabrafenib: BREAK Trial
Following initial phase I and II data that supported the activity of dabrafenib [27, 28], this phase III clinical trial randomized treatment-naïve metastatic BRAF V600E melanoma patients to either dabrafenib monotherapy or dacarbazine chemotherapy (BREAK-3). PFS was significantly higher for the dabrafenib arm as compared to the dacarbazine arm (5.1 vs 2.7 months). Subsequently, patients who progressed on dacarbazine were crossed over to the dabrafenib arm. At the conclusion of the study at 2 years, 45% of those on the dabrafenib arm remained alive compared to 32% on the dacarbazine arm. Given the crossover and the study being powered for PFS, a difference in OS was not statistically detectable between the two groups. Grade 2 or higher adverse events were greater in the dabrafenib arm (53%:skin toxicity, fever, fatigue, arthralgia) when compared to the dacarbazine arm (44%: nausea, vomiting, neutropenia, fatigue). As compared with vemurafenib, fevers occurred more commonly, but phototoxicity occurred less often [29].
Patients with brain metastasis were specifically evaluated in the phase II BREAK-MB trial. This study demonstrated that 39.2% of newly diagnosed patients with BRAF V600E-mutated brain metastases achieved an overall intracranial response, as did 30.9% of patients with progressive brain metastases after local treatment failure (surgery or radiation). This study thus established the activity of BRAF inhibition in patients with brain involvement [30]. As with prior studies with BRAF inhibitors, the major severe adverse events were pyrexia and cutaneous squamous cell carcinomas; intracranial hemorrhage also occurred in 6% of patients [30].
3.3. Adverse Effects with Unilateral BRAF Inhibition
While the survival benefits of these new mutant BRAF inhibitors for patients with BRAF V600 melanoma compared to the traditional chemotherapies are undeniable, they also have a distinct toxicity profile. One significant adverse event common to both vemurafenib and dabrafenib is skin toxicity, particularly the risk of skin malignancies or low-grade non-malignant proliferative lesions. These BRAF inhibitor-associated cutaneous lesions are more prevalent, with earlier onset with vemurafenib (4–31%, median time 8 weeks) as compared to dabrafenib (6–11%, median time 16 weeks) across complied studies [31]. While vemurafenib and dabrafenib inhibit mutant BRAF signaling in metastatic melanomas, they promote oncogenic signaling in wild-type BRAF skin cells in a phenomenon termed paradoxical RAF signaling [32], exemplifying the complexity of the MAPK pathway. While the exact mechanism is not entirely clear; mutant BRAF inhibitors potentiate MAPK signaling in vitro, possibly through CRAF dimerization or through alterations in scaffolding protein conformation, leading to the activation of downstream ERK signaling [33]. Other mechanisms of this phenomenon, including RAF inhibitor-mediated reduction of BRAF auto-phosphorylation, have also been proposed [34]. Analysis of the cutaneous squamous cell carcinomas often demonstrated HRAS mutations, suggesting that BRAF inhibition in keratinocytes leads to paradoxical MAPK signaling in premalignant cells harboring a latent HRAS mutation [11]. Thus, BRAF inhibitors appear to ‘unmask’ these subsequent malignancies by promoting MAPK signaling in RAS mutant cells rather than truly inducing new malignancies. Further, non-cutaneous RAS-mutant malignancies have been rare [11].
While BRAF inhibitor monotherapy produced dramatic responses with tumor regression and improved survival, almost all patients eventually relapse with disease resistance to BRAF inhibition. This occurs through both MAPK-dependent and MAPK-independent mechanisms, but interestingly, does not include secondary BRAF mutations [11]. Instead, numerous genomic alterations elsewhere in the MAPK pathway have been found to confer resistance to mutant BRAF inhibitors, including NRAS mutations, BRAF amplifications, BRAF alternate splicing, increased CRAF levels, and MEK1/2 mutations [35, 36], representing the heterogeneity of melanoma genetics. Oncogenic BRAF inhibition has been documented in multiple studies to activate ERK, exemplifying the complexity of MAPK signaling. BRAF V600E inhibition has been shown to cooperate with oncogenic RAS to drive melanoma formation in mice by activating the RAF isoform CRAF [37]. Interestingly, recent evidence has shown that RAF dimers participate in an inhibitory auto-phosphyloration regulatory mechanism [38]. This auto-inhibitory process is disrupted when BRAF is mutated or if it is coupled to a BRAF inhibitor, leading to increased ERK activity [33, 39]. Furthermore, MAPK-independent mechanisms of resistance have been found in about 30% of relapsed melanoma [40]. Concurrent loss of the PTEN pathway renders melanomas resistant to oncogenic BRAF inhibition-induced cell death [41]. Up-regulation of survival proteins has also been implicated in BRAF inhibitor resistance [42].
4. MEK1/2 Inhibitor Monotherapy Trials
As mentioned, mutations in MAPK encoding genes occur in most melanomas, including BRAF, NRAS, NF1, and others. While the BRAF V600E mutation accounts for the majority of BRAF mutations, several other clinically relevant BRAF mutations also exist. Conceptually, inhibiting the MAPK pathway at a point downstream of BRAF could be an effective strategy in a broader spectrum of melanomas. The following section discusses MEK inhibitors as a monotherapy. Its activity in combination therapy will be discussed later in Sects. 5 and 8.
4.1. Trametinib
GSK1120212 (GlaxoSmithKline), or trametinib, is a highly specific inhibitor of MEK1/2. This agent was initially evaluated in a phase I study involving 81 patients with melanoma. Patients with BRAF V600 mutations who had not received prior mutant BRAF inhibitors had a 33% response rate. Four of 39 patients (10%) with wild-type BRAF melanoma also responded. None of the patients with an NRAS mutations responded to MEK1/2 inhibition. Interestingly, one of the responders was later found to have a BRAF L597 mutation, suggesting that trametinib may play a role in these non-V600 BRAF mutations. These results were limited to cutaneous or suspected cutaneous melanomas; none of the 16 patients with uveal melanomas responded [43].
4.2. Trametinib: METRIC Trial
The safety and efficacy of trametinib monotherapy was then compared to traditional chemotherapy in patients with BRAF V600 metastatic melanoma in the phase III METRIC-3 clinical trial. PFS was higher with trametinib than with chemotherapy (4.8 vs 1.5 months). OS at 6 months was also higher in the trametinib arm than the chemotherapy arm (81 vs 67%) despite allowing crossover from the chemotherapy arm to trametinib after progression. Adverse events were more prevalent in the trametinib group; the majority of which were rash, diarrhea, and peripheral edema. A decrease in ejection fraction was seen in 7% of the patients and was mostly asymptomatic and reversible, although 1% had serious cardiac events that led to drug discontinuation. Conspicuously, treatment-related skin malignancies were not observed [44].
4.3. Selumetinib and Binimetinib
Several other MEK inhibitors have been developed. Selumetinib (AZD6244) is a recently developed MEK inhibitor that, while not being superior to temozolomide in BRAF-mutant cutaneous melanoma [45], did show a modest superiority to chemotherapy in uveal melanoma [46]. However, selumetinib in combination with dacarbazine did not improve survival compared with dacarbazine alone in patients with BRAF-mutated melanoma [47]. Binimetinib (MEK162) has also been evaluated in NRAS-mutant melanoma, and demonstrated an improved PFS over dacarbazine (2.8 vs 1.5 months) [48].
5. Targeted Therapies: Combination Trials
For an aggressive cancer with high mutational load such as melanoma, the benefits of targeted monotherapies are muted by near inevitable disease progression and therapy resistance. Concurrently targeting multiple points in an oncogenic pathway could delay the development of resistance, improve response durability, and decrease adverse events. In melanoma, resistance often involves reactivation of MAPK signaling, suggesting that more complete pathway blockade could have clinical utility. As such, multiple clinical trials have evaluated the safety and efficacy of combining mutant BRAF inhibitors with MEK inhibitors in patients with mutant BRAF metastatic melanoma. The next section will review and discuss these trials.
5.1. Dabrafenib + Trametinib Versus Dabrafenib: COMBI-d Trial
Based on this rationale, dabrafenib and trametinib were combined to assess their efficacy and safety compared to dabrafenib alone in patients with BRAF-mutant melanoma. Following activity and safety demonstrations in initial phase I/II trials, two phase III randomized controlled trials were performed. In the COMBI-d trial, dabrafenib and trametinib were compared with dabrafenib alone, and demonstrated superior median PFS (11 vs 8.8 months) and response rate (69 vs 53%) (see Table 1). Median OS was similarly improved at (25.1 vs 18.7 months) [49]. The rate of serious adverse events was similar between the two groups, although combination therapy increased the likelihood of pyrexia. Interestingly, however, the addition of MEK inhibition significantly decreased the rates of cutaneous squamous cell carcinomas, likely by blunting paradoxical MAPK activation [50]. Notably, the 3-year follow-up from this study reported that outcomes remained improved for the combination, in terms of 3-year PFS (22 vs 12%) and OS (44 vs 32%). This study challenges the notion that acquired resistance is inevitable, and suggests that a subset of responding patients may have long-term benefit from combined MAPK blockade [51].
Table 1.
Published clinical trial data for first-line regimens for advanced melanoma
| Agent | Target | Response rate (%) | PFS | OS | Grade 3–4 toxicities (%) | References |
|---|---|---|---|---|---|---|
|
| ||||||
| Vemurafenib + cobimetinib | BRAF + MEK | 68 | Median 12.3 months | Median 22.3 months | 60 | PMID: 27480103 |
| Dabrafenib + trametinib | BRAF + MEK | 64–69 | Median 11.0–11.4 months | Median 25.1 months, 72% 12-month OS | 32–48 | PMID: 26037941, 25399551 |
| Pembrolizumab | PD-1 | 33–45a | 47% 6-month PFS | 74% 12-month OS | 10–14 | PMID: 27092830, 25891173 |
| Nivolumab | PD-1 | 40–44% | Median 5.1–6.9 months | 73% 12-month OS | 12–16 | PMID: 25399552, 26027431 |
| Ipilimumab + nivolumab | PD-1 + CTLA-4 | 58–61 | Median 11.5 months | 64% 24-month OS | 54–55 | PMID: 27622997, 26027431 |
CTLA-4 cytotoxic T-lymphocyte-associated protein 4, OS overall survival, PD-1 programmed cell death protein 1, PFS progression-free survival
Higher response rate in 123 patients with no prior treatments in a large phase I trial
5.2. Dabrafenib + Trametinib Versus Vemurafenib: COMBI-v Trial
The efficacy and safety of the dabrafenib and trametinib combination was also compared against vemurafenib monotherapy in the phase III COMBI-v clinical trial. The initial report showed the median PFS to be superior in the combination group as compared to vemurafenib (11.4 vs 7.3 months), and OS was also improved (12-month OS 72 vs 65%) [52]. As with prior studies, adverse events were comparable between the two study groups, although pyrexia was higher with combination therapy (53 vs 21%). Cutaneous malignancies were vastly decreased with combination therapy compared to vemurafenib monotherapy (1 vs 18%) [52].
5.3. Vemurafenib + Cobimetinib Versus Vemurafenib: CoBRIM Trial
In a phase III randomized clinical trial, cobimetinib, a highly specific MEK1/2 inhibitor, was combined with vemurafenib and compared with vemurafenib monotherapy. The results are comparable to those observed with dabrafenib and trametinib, as vemurafenib and cobimetinib increased PFS compared to vemurafenib alone (12.3 vs 7.2 months). Median OS was also equally encouraging for the combination therapy vs. vemurafenib monotherapy (22.3 vs 17.4 months). The risk of disease progression or death was significantly reduced by 42% by the combination therapy at the final analysis. Serious adverse events were more prevalent in the combination group (37 vs 28%). Specifically, combination therapy increased the risk of cardiovascular adverse events, serous retinopathy, elevated liver enzymes, and elevated creatine phosphokinase. By contrast, combination therapy decreased the incidence of skin malignancies compared to mutant BRAF inhibitor alone [53].
5.4. Encorafenib + Binimetinib Versus Vemurafenib or Encorafenib: COLUMBUS Trial
Encorafenib is a recently developed BRAF inhibitor, and its use in melanoma is currently being evaluated in combination with the MEK1/2 inhibitor binimetinib. In a two-part, phase III randomized clinical trial, investigators sought to compare the efficacy of encorafenib and binimetinib against either vemurafenib or encorafenib alone, as well as to tease out the contribution of binimetinib by decreasing the dose of encorafenib. Results from part I of the trial revealed improved PFS with combination encorafenib and binimetinib when compared to either vemurafenib or encorafenib alone (14.9 vs. 7.3 or 9.6 months). Adverse events were comparable between the combination and monotherapies, although patients on the combination trial are more likely to have elevated creatinine phosphokinase and retinal pigment epithelial detachment [54]. Part II of the COLUMBUS trial is currently underway, and results are expected to be released in the near future.
6. Conclusions of Targeted Therapy Trials
The development of MAPK inhibitors is a success story in translational medicine and rational drug design, and heralded a paradigm shift in the treatment of metastatic melanoma. These early studies clearly demonstrated the efficacy of MAPK inhibitors compared to traditional chemotherapy. The following section will discuss the lessons learned from these studies.
6.1. Quality of Life
The primary endpoints of these studies focused on PFS or OS. While mortality is arguably the most objective goal of any treatment regimen, morbidity is much harder to quantify, but remains critically important. A patient’s perception of their disease is a complex blend of mortality measures, toxicity, pain, functionality, and symptoms, and is influenced by emotional, personal, and spiritual factors. In the absence of a cure and in the prolongation of a disease state, the quality of life permitted by a treatment becomes an important consideration for therapy selection. Quality-of-life scores capture a multi-dimensional appreciation that objectively characterizes the treatment response from a patient’s perspective.
In the BREAK-3 trial, patients on dabrafenib experienced stabilization or only minor deterioration of functionality and symptoms over time, whereas patients on dacarbazine deteriorated steadily. This divergence was further demonstrated with patients who crossed over from the dacarbazine arm to the dabrafenib arm. While the BREAK-3 trial was not powered to detect a pre-specified difference as defined by the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire (EORTC QLQ-C30) survey, the mean differences show a favorable quality-of-life profile for the BRAF inhibitor. Further, patients who crossed over from the chemotherapy to targeted-therapy arm experienced a meaningful improvement in functionality [55]. These effects were similarly observed for the MEK1/2 inhibitor trametinib in the METRIC trial [56].
As discussed previously, MAPK inhibitor combination regimens are consistently superior to BRAF inhibitor monotherapy in prolonging survival. With multi-agent therapy, toxicity and tolerability become particularly important. This is especially true in oncology pharmacology, where the balance between tolerability and benefit is a fine line. This was first examined in the COMBI-d trial where dabrafenib plus trametinib was compared against dabrafenib alone. Patients on the combination regimen experienced improved global health scores, functionality, pain, and symptoms compared to monotherapy. One exception was gastrointestinal symptoms, which worsened with combination therapy. Although the trial was designed with survival as an endpoint, it demonstrated clinically and statistically significant improvements in many quality-of-life metrics [57].
6.2. Effect of Combination Therapy on Non-melanoma Skin Cancers
A noteworthy collection of adverse effects that plague BRAF inhibitors is the risk of developing keratoacanthomas. This side effect is reflective of the underlying molecular mechanism of action, leading to paradoxical BRAF activation. Mutant BRAF inhibitors have been shown to lead to auto- or hetero-dimerization of RAF molecules in vitro, which can activate downstream signaling either through autophosphorylation or by preferential activation of ERK [33] (while bypassing MEK). This paradoxical RAF activation appears to be tissue specific and more robust in keratinocytes [58], as these skin lesions are primarily cutaneous squamous cell carcinomas or keratoacanthomas. Paradoxical RAF activation, however, is not sufficient for oncogenesis. Analysis of keratoacanthomas in patients treated with mutant BRAF inhibitor revealed HRAS mutations, suggesting that mutant BRAF inhibitors activate a pro-proliferative cascade in keratinocytes with latent RAS mutations [59]. One intriguing, non-cutaneous malignancy has been reported with vemurafenib therapy. A patient on vemurafenib developed clinically apparent chronic myelomonocytic leukemia (CMML) with an NRAS mutation. Interestingly, the CMML responded to vemurafenib withdrawal and later the addition of a MEK inhibitor [60, 61]. Pre-clinical and early clinical studies are evaluating the next-generation BRAF inhibitors PLX7904 and PLX8394, which do not activate RAS [62].
MEK1/2 inhibitors as single agents do not increase the rate of subsequent skin malignancies. Importantly, concomitant BRAF and MEK inhibition causes fewer cutaneous malignancies than BRAF inhibitor monotherapy, suggesting that the oncogenic signal promoted by mutant BRAF inhibition is countered by MEK1/2 inhibition. This finding further supports the use of combination therapy over monotherapy.
6.3. MEK Inhibition Toxicities
MEK1/2 inhibitors have characteristic adverse effects as single agents and with combination therapy. They can cause cardiotoxicity leading to a decrease in ejection fraction in patients on MEK1/2 monotherapy [44] and combination regimens, an effect not seen with mutant BRAF inhibitors alone [63]. These episodes of cardiac dysfunction are mostly asymptomatic and reversible with dose reduction, although in the METRIC trial, two patients had grade 3 serious cardiac events [44]. The underlying mechanism of cardiotoxicity is not well understood. In light of this, patients with pre-existing decreased cardiac ejection fraction may not tolerate regimens that include MEK1/2 inhibitors and are likely more appropriate for BRAF inhibitor monotherapy. Some patients with mild cardiac dysfunction may tolerate combination therapy, although extremely close monitoring should be implemented.
6.4. Ongoing Questions
With these recent trials, there is ample evidence for using MAPK inhibitors, especially in combination, in metastatic melanoma. However, much remains unanswered. The three existing combination MAPK kinase trials clearly demonstrate that combination therapies offer many benefits over monotherapy alone. It remains unclear what advantages, if any, various combinations have over one another, as no head-to-head trials have been performed. In the absence of those studies, selection is based on provider discretion and is often based on experience, toxicities, and dosing schedule.
Other questions also remain. First, a small subset of patients clearly demonstrates durable benefits. Pre-emptively identifying these patients with molecular or clinical biomarkers has been a major challenge. Recent studies have identified that patients with fewer than three disease sites and normal LDH have a higher likelihood of durable benefit, but this is not universal. Circulating tumor DNA has been shown to prognosticate between BRAF responders as well as predicate disease progression, but the technology is not fully developed and clinical usage is yet to be implemented [64, 65]. Second, overcoming resistance has been a vexing problem. Heterogeneity at resistance has impeded development of active subsequent therapies. Studies that have attempted crossover from single-agent therapy to combination MAPK blockade have been unimpressive [66]. Animal studies have suggested that the development of drug resistance occurs through oncogene addiction whereby drug withdrawal in an intermittent dosing schedule improves the duration of response [67]. This has been supported by various clinical case reports [60] and is currently being investigated in several clinical trials (NCT01894672, NCT02263898). Finally, much work remains to extend the benefits of MAPK blockade to non-BRAF-mutant melanomas. Several studies are ongoing with MEK inhibitor monotherapy (for NRAS and atypical BRAF mutations) and with other targeted therapy agents (e.g., CDK4/6 inhibitors) [68].
7. Immunotherapy
Immune checkpoint inhibitors have dramatically shifted the landscape of many cancers, including melanoma. The notion of host anti-tumor immune defense is not new, but development of therapies to exploit this concept have not reached fruition until recently.
Malignant cells, despite harboring many genetic aberrations associated with neoplastic transformation, often mask their immunologic presence by down-regulating tumor immunosurveillance and expressing or exploiting so-called immune checkpoints. These immune checkpoints include the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death 1 receptor (PD-1) and its ligands (PD-L1, PD-L2), targets of these newer therapies [69]. The immune checkpoints function normally to maintain self-tolerance to protect normal tissue when the immune system is responding to an infection.
7.1. Immune Checkpoints: Physiology and Pathology
CTLA-4 is a transmembrane receptor found normally on T cells and is important in dampening T-cell activation and response. It achieves this by sequestering the activating CD80 and CD86 ligands away from the costimulatory CD28 receptor (which normally engages with the T-cell receptor (TCR) to induce an activating response) (Fig. 2). Additionally, CTLA-4 also sets off an intracellular inhibitory signaling cascade in T cells to counteract any activated TCR signaling. While CTLA-4 is expressed on cytotoxic CD8 effector cells, its immunodulatory effect is accomplished mostly by down-regulating activating CD4 T helper 1 cells and promoting the suppressive effects of T-regulatory cells [69, 70].
Fig. 2.

Mechanism of checkpoint inhibition. CTLA-4 cytotoxic T-lymphocyte-associated protein 4, MHC major histocompatibility complex, PD-1 programmed cell death protein 1, PD-L1 programmed death-ligand 1, TCR T cell receptor
Similarly, PD-1, in concert with PD-L1 and PD-L2, suppresses T-cell activation, acting mostly at the interface between normal tissue and inflammation to prevent autoimmunity and to promote peripheral tolerance. PD-L1 is a cell-bound receptor broadly expressed on T cells, B cells, macrophages, dendritic cells, mast cells, as well as vascular endothelial cells. Binding of PD-L1 to its cognate receptor PD-1 inhibits kinases involved in the T-cell cascade [69].
Despite harboring many genetic aberrations, which would identify them as foreign, cancer cells have hijacked these immune checkpoint processes to evade immune destruction. Furthermore, the tumor microenvironment is a complex structure with multiple cell types and paracrine interactions. Intermingling with tumor cells are tumor infiltrating lymphocytes (TIL) that have complex and diverse implications in the neoplastic process, but generally promote a suppressive immune state, leading to T-cell anergy [70, 71] (Fig. 2).
7.2. Immune Checkpoint Inhibitors in Melanoma
Blocking these checkpoint inhibitors with monoclonal antibodies re-educates the native immune system to recognize previously concealed targets. The CTLA-4 blocking antibody ipilimumab was approved for use in melanoma in 2011 after a three-arm, randomized controlled trial showed an improved survival benefit of 3.5 months with ipilimumab over the gp100 vaccine [72]. Importantly, this was the first therapy shown to increase survival in metastatic melanoma. In addition to an overall favorable response, a subset of patients (19–25%) had remarkable survival beyond 2 years [73], exceeding the historical rate of 5–10% [3]. Currently, ipilimumab is primarily used as salvage therapy for patients who fail anti-PD-1, or in combination with nivolumab.
Anti-PD-1 agents have provided even more impressive clinical efficacy and have changed the outlook for patients with melanoma as well as other cancers. Early results suggest these agents produce durable responses in at least one third of patients, and produce tolerable toxicity profiles. The efficacy of the PD1 blocking antibody nivolumab in metastatic melanoma was superior to chemotherapy, with a greater overall response rate (40 vs 14%), median PFS (5.1 vs 2.2 months) and OS at 1 year (72.9 vs 42.1%) in patients with untreated BRAF wild-type melanoma [74]. It was additionally effective in patients who were previously treated with ipilimumab in another randomized controlled trial and with greater OS at 6 months (30 vs 13.3%) as well as less severe adverse events [75]. Nivolumab was approved as a single agent in 2015 for metastatic melanoma.
Pembrolizumab is another PD-1 antibody that has been studied in patients with metastatic melanoma. The KEYNOTE-006 trial demonstrated the utility of pembrolizumab in treatment-naïve metastatic melanoma [76], and resulted in more patients who were progression free at 2 years when compared to ipilimumab (at two different doses of pembrolizumab; 31 and 28 vs 14%). At the 2-year update, pembrolizumab was superior to ipilimumab with regard to OS (55.1 and 55.3 vs 43%) and PFS (31.2 and 27.8 vs 13.5%); indeed, the median OS was not reached for patients on any dosage of pembrolizumab compared to 16 months for patients on ipilimumab [77]. In untreated patients, the objective response rate was >40% in a large phase I study [78]. Pembrolizumab was FDA approved in 2014 for metastatic melanoma and is now widely used.
The activity of these agents led to interest in combination immunotherapy. When combined together, nivolumab plus ipilimumab produces superior activity compared to either agent as a monotherapy [79], as shown by the CHECKMATE-067 study. Combined checkpoint inhibitors resulted in a superior PFS of 11.5 months compared to nivolumab alone (6.9 months) or ipilimumab alone (2.9 months), as well as superior response rates (59 vs 44 vs 19%) [79]. Long-term comparisons of OS, however, are not yet published. Combination therapy results in a significantly higher toxicity rate as well. The practice patterns of using single-agent anti-PD-1 vs. combination ipilimumab/nivolumab are quite varied. Many experts favor combination therapy for patients with poor prognostic features, including brain or liver metastases, elevated LDH, symptomatic/bulky disease, or non-cutaneous melanomas.
8. Combining Immunotherapy with MAPK Inhibition in Melanoma
Both classes of agents, developed in parallel, have demonstrated impressive survival and quality-of-life benefits in patients with melanoma. MAPK inhibitors produce significant clinical responses for patients with metastatic melanomas, but are usually associated with resistance and disease progression. Immune checkpoint inhibitors do not produce universal responses, but in subsets of patients durable clinical benefit occurs, likely due to ensuring tumor immunosurveillance.
One could postulate that combining these agents could produce synergistic efficacy. The value of pairing a tumoricidal drug (MAPK inhibitors) with checkpoint inhibitors could produce cooperative effects by releasing tumor antigens to prime immune cells against the now unmasked tumors. A number of pre-clinical studies have demonstrated enhanced immune infiltration with BRAF and/or MEK inhibition [80–82]. Clinical analysis of melanomas in patients treated with BRAF inhibitors showed greater lymphocyte infiltration in tumor samples and was associated with decreased tumor burden and increased tumor necrosis [83]. These studies provide further evidence for combination therapy. Clinically, the goal of combining MAPK inhibitors with immune checkpoint inhibitors is to promote a consistent and long-lasting clinical response.
Although in early phases, initial investigations have demonstrated cautiously optimistic results in combining both MAPK inhibition and checkpoint inhibitors. The following section will discuss and highlight the results of some of these studies.
The first reported phase I study of vemurafenib and ipilimumab reported excessive hepatotoxicity [84]. Although asymptomatic and reversible with drug discontinuation, those results cautioned the toxicity profile of combination therapy. A subsequent study released in 2014 examined the use of ipilimumab + dabrafenib with or without trametinib in metastatic melanoma. Unfortunately, the triple combination arm produced a number of grade 3 and 4 toxicities, including colitis complicated by intestinal perforation [85]. The accrual for the triple combination arm was consequently halted, although the dabrafenib and ipilimumab combination arm appears more tolerable.
Given these toxicities, successive combination therapy trials have carefully evaluated dosages and monitored for adverse events. Also of note, there are numerous and potentially promising combination immunotherapy trials currently underway. For the sake of brevity, however, we have chosen to focus on those involving combinations of targeted therapy (e.g., kinase inhibitors) and immune checkpoint inhibitors.
8.1. Durvalumab + Dabrafenib: NCT02027961
An ongoing trial is investigating the PD-L1 antibody MEDI4736 (durvalumab) with dabrafenib and trametinib. Adverse effects have correlated with increased durvalumab dosage, but not with combination therapy, and were mostly tolerable and acceptable [86]. Biopsies collected from these patients showed evidence of increased immune infiltration (CD8 + effector T cells and interferon-γ) post treatment. Clinical responses are ongoing, and the primary data collection closed April 2017. Results are expected to be released soon.
8.2. Atezolizumab + Vemurafenib + Cobimetinib: NCT01656642 and NCT02908672
The PD-L1 antibody atezolizumab is currently being evaluated in combination with vemurafenib or vemurafenib plus cobimetinib in a phase Ib clinical trial. Grade 3 or 4 adverse events were seen at about a similar rate between the triple combination therapy and MAPK inhibitors (5/14 vs 6/14) and were manageable and reversible [86]. At the time of the last study update, clinical responses are unconfirmed given limited follow-up. Study results are expected to be released in the near future with T-cell activation markers and molecular studies. The phase III TRIOLOGY trial compares the triple combination atezolizumab with vemurafenib plus cobinmetinib against vemurafenib and cobinmetinib and will be pivotal in determining the role of combining immunotherapy with MAPK inhibitors. The study was launched in June 2017, and preliminary results with PFS as the primary endpoint are expected to be released in Nov 2019.
8.3. Pembrolizumab + Dabrafenib + Trametinib: NCT02625337 and NCT02130466
Pembrolizumab is approved for metastatic melanoma, with an acceptable safety profile and response rate. It is currently being investigated in combination with MAPK inhibitors in two separate clinical trials. The phase II IMPemBra trial compares pembrolizumab, dabrafenib, and trametinib in three different doses with pembrolizumab monotherapy. This trial started in January 2016, with an expected primary endpoint in December 2017. More notable is the KEYNOTE-022 trial, a five-part series designed to evaluate pembrolizumab with dabrafenib and trametinib combination. In this study, patients with metastatic melanoma are randomized to either pembrolizumab with dabrafenib and trametinib or dabrafenib and trametinib in a randomized phase II trial [87, 88]. This study is currently accruing and study completion is estimated to be 2021.
8.4. PDR001 + Dabrafenib + Trametinib: NCT02967692
The anti-PD1 IgG4 antibody PDR001, developed by Novartis, is currently being investigated in combination with dabrafenib and trametinib in a randomized phase III trial. While preclinical data on PDR001 is scarce, its efficacy is under evaluation for a variety of solid tumors, including non-small cell lung cancer, hepatocellular carcinoma, neuro-endocrine tumors, and advanced melanomas. The NCT02967692 trial is the first study assessing PDR001 exclusively in melanomas. The study began in Feb 2017 and results are expected to be released in late 2019.
8.5. Atezolizumab + Cobimetinib in Colon Cancer: NCT01988896
While not a melanoma study, extremely provocative results were identified from this early study of atezolizumab and cobimetinib in colon cancer. While either agent has minimal efficacy in mismatch repair proficient colon cancer as monotherapy, the combination was associated with a response rate of 17% [89]. This study demonstrates proof of principle that combination MAPK blockade and immune checkpoint inhibition can produce synergistic benefits.
9. Conclusions and Future Perspectives
Both targeted and immune therapies have transformed melanoma from a malignancy with a dismal prognosis and limited treatment options to a cancer with multiple active therapies. Moving forward, one could project that immune therapy will likely move into the first-line option for most patients given the durability of responses, with BRAF/MEK inhibition as salvage therapy. Combining immune therapy with BRAF and/or MEK inhibitors may enhance these responses, and provide a preferred option moving forward, although the data are too limited at this time to make confident projections. A biomarker-driven approach may also alter treatment paradigms. One could envision biomarkers of response to both targeted and immune therapies (or combined therapy) which could drive treatment selection. Ultimately, understanding the most optimal treatment sequences and combinations will maximize the utility of currently available therapies. Combining MAPK inhibition and immune checkpoint inhibitors is one such approach, and remains unproven but promising.
Key Points.
Molecularly targeted therapies blocking BRAF and MEK have transformed the management of BRAF-mutated melanoma as monotherapies and, more importantly, as combination therapy.
These agents are encumbered, however, by high rates of resistance.
Developing further combinations with immunotherapy strategies may provide additional therapeutic benefits.
Funding
Dr. Johnson receives funding from the National Institutes of Health/National Cancer Institute (NIH/NCI) (K23 CA204726).
Footnotes
Conflict of interest Dr. Johnson is on advisory boards for BMS, Genoptix, and Merck, and receives research funding from Incyte. Alice Yao Zhou declares no conflict of interest.
References
- 1.SEER Cancer Statistics Review, 1975–2014 [Internet]. National Cancer Institute. Bethesda, MD. 2017. https://seer.cancer.gov/csr/1975_2014/. [Google Scholar]
- 2.Shain AH, Bastian BC. From melanocytes to melanomas. Nat Rev Cancer. 2016;16(6):345–58. [DOI] [PubMed] [Google Scholar]
- 3.Bhatia S, Tykodi SS, Thompson JA. Treatment of metastatic melanoma: an overview. Oncology. 2009;23(6):488–96. [PMC free article] [PubMed] [Google Scholar]
- 4.Dickson PV, Gershenwald JE. Staging and prognosis of cutaneous melanoma. Surg Onclogy. 2011;20(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson DB, Sosman JA. Therapeutic advances and treatment options in metastatic melanoma. JAMA Oncol. 2015;1(3):380–6. [DOI] [PubMed] [Google Scholar]
- 6.Gaestel M MAPKAP kinases—MKs—two’s company, three’s a crowd. Nat Rev Mol Cell Biol. 2006;7(February):120–30. [DOI] [PubMed] [Google Scholar]
- 7.Kwong LN, Chin L. The brothers RAF. Cell. 2010;140(2):180–2. [DOI] [PubMed] [Google Scholar]
- 8.Burotto M, Chiou VL, Lee J-M, Kohn EC. The MAPK pathway across different malignancies: a new perspective. Cancer. 2015;120(22):3446–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 2008;27(2):253–61. [DOI] [PubMed] [Google Scholar]
- 10.Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, et al. COSMIC: mining complete cancer genomes in the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2011;39(SUPPL. 1):945–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer. 2014;14(7):455–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–54. [DOI] [PubMed] [Google Scholar]
- 13.Adnane L, Trail PA, Taylor I, Wilhelm SM. Sorafenib (BAY-43–9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 2005;407(5):597–612. [DOI] [PubMed] [Google Scholar]
- 14.Halilovic E, Solit DB. Therapeutic strategies for inhibiting oncogenic BRAF signaling. Curr Opin Pharmacol. 2008;8(4):419–26. [DOI] [PubMed] [Google Scholar]
- 15.Mangana J, Levesque MP, Karpova MB, Dummer R. Sorafenib in melanoma. Expert Opin Investig Drugs. 2012;21(4):557–68. [DOI] [PubMed] [Google Scholar]
- 16.Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5(10):835–44. [DOI] [PubMed] [Google Scholar]
- 17.Fishman MN, Tomshine J, Fulp WJ, Foreman PK. A systematic review of the efficacy and safety experience reported for sorafenib in advanced renal cell carcinoma (RCC) in the post-approval setting. PLoS One. 2015;10(4):1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stotz M, Gerger A, Haybaeck J, Kiesslich T, Bullock MD, Pichler M. Molecular targeted therapies in hepatocellular carcinoma: past, present and future. Anticancer Res. 2015;35(11):5737–44. [PubMed] [Google Scholar]
- 19.Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K, et al.Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat Rev Drug Discov. 2012;11(11):873–86. [DOI] [PubMed] [Google Scholar]
- 20.Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci. 2008;105(8):3041–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kainthla R, Kim KB, Falchook GS. Dabrafenib for treatment of BRAF-mutant melanoma. Pharmgenom Pers Med. 2013;7(1):21–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Menzies AM, Long GV, Murali R. Dabrafenib and its potential for the treatment of metastatic melanoma. Drug Des Dev Ther. 2012;6:391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Z, Jiang K, Zhu X, Lin G, Song F, Zhao Y, et al. Encorafenib(LGX818), a potent BRAF inhibitor, induces senescence accompanied by autophagy in BRAFV600E melanoma cells. Cancer Lett. 2016;370(2):332–44. [DOI] [PubMed] [Google Scholar]
- 24.Sullivan RJ, Flaherty K. MAP kinase signaling and inhibition inmelanoma. Oncogene. 2012;32(19):2373–9. [DOI] [PubMed] [Google Scholar]
- 25.Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600–mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366(8):707–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McArthur GA, Chapman PB, Robert C, Larkin J, Haanen JB, Dummer R, et al. Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15(3):323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ascierto PA, Minor D, Ribas A, Lebbe C, O’Hagan A, Arya N, et al. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J Clin Oncol. 2013;31(26):3205–11. [DOI] [PubMed] [Google Scholar]
- 28.Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012;379(9829):1893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–65. [DOI] [PubMed] [Google Scholar]
- 30.Long GV, Trefzer U, Davies MA, Kefford RF, Ascierto PA, Chapman PB, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): A multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(11):1087–95. [DOI] [PubMed] [Google Scholar]
- 31.Anforth R, Fernandez-Peñas P, Long GV. Cutaneous toxicities of RAF inhibitors. Lancet Oncol. 2013;14(1):e11–8. [DOI] [PubMed] [Google Scholar]
- 32.Samatar AA, Poulikakos PI. Targeting RAS–ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014;13(12):928–42. [DOI] [PubMed] [Google Scholar]
- 33.Holderfield M, Nagel TE, Stuart DD. Mechanism and consequences of RAF kinase activation by small-molecule inhibitors. Br J Cancer. 2014;111(4):640–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tse A, Verkhivker GM. Exploring molecular mechanisms of paradoxical activation in the BRAF kinase dimers: atomistic simulations of conformational dynamics and modeling of allosteric communication networks and signaling pathways. PLoS One. 2016;11(11):1–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manzano JL, Layos L, Bugés C, de los Llanos Gi M, Vila L, Martínez-Balibrea E, et al. Resistant mechanisms to BRAF inhibitors in melanoma. Ann Transl Med. 2016;4(12):237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Montagut C, Sharma SV, Shioda T, McDermott U, Ulman M, Ulkus LE, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68(12):4853–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-DuvasI, Dhomen N, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140(2):209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu J, Stites EC, Yu H, Germino EA, Meharena HS, Stork PJS, et al. Allosteric activation of functionally asymmetric RAF kinase dimers. Cell. 2013;154(5):1036–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holderfield M,Merritt H,Chan J, WallrothM, Tandeske L, ZhaiH, et al. RAF inhibitors activate the MAPK pathway by relieving inhibitory autophosphorylation. Cancer Cell. 2013;23(5):594–602. [DOI] [PubMed] [Google Scholar]
- 40.Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, et al.Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4(1):80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paraiso KHT, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71(7):2750–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wellbrock C MAPK pathway inhibition in melanoma: resistance three ways. Biochem Soc Trans. 2014;42(4):727–32. [DOI] [PubMed] [Google Scholar]
- 43.Falchook GS, Lewis KD, Infante JR, Gordon MS, Vogelzang NJ, DeMarini DJ, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(8):782–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M,et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–14. [DOI] [PubMed] [Google Scholar]
- 45.Kirkwood JM, Bastholt L, Robert C, Sosman J, Larkin J, HerseyP, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res. 2012;18(2):555–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carvajal RD, Sosman JA, Quevedo JF, Milhem MM, Joshua AM, Kudchadkar RR, et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma. JAMA. 2014;311(23):2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robert C, Dummer R, Gutzmer R, Lorigan P, Kim KB, Nyakas M, et al. Selumetinib plus dacarbazine versus placebo plus dacarbazine as first-line treatment for BRAF-mutant metastatic melanoma: a phase 2 double-blind randomised study. Lancet Oncol. 2013;14(8):733–40. [DOI] [PubMed] [Google Scholar]
- 48.Dummer R, Schadendorf D, Ascierto PA, Arance A, Dutriaux C, Di Giacomo AM, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017;18(4):435–45. [DOI] [PubMed] [Google Scholar]
- 49.Long GV, Stroyakovskiy D, Gogas H, Levchenko E, De Braud F, Larkin J, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet. 2015;386(9992):444–51. [DOI] [PubMed] [Google Scholar]
- 50.Long GV, Grob J-J, Nathan P, Ribas A, Robert C, Schadendorf D, et al. Factors predictive of response, disease progression, and overall survival after dabrafenib and trametinib combination treatment: a pooled analysis of individual patient data from randomised trials. Lancet Oncol. 2016;17(12):1743–54. [DOI] [PubMed] [Google Scholar]
- 51.Long GV, Flaherty KT, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K–mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grob JJ, Amonkar MM, Karaszewska B, Schachter J, Dummer R, Mackiewicz A, et al. Comparison of dabrafenib and trametinib combination therapy with vemurafenib monotherapy on health-related quality of life in patients with unresectable or metastatic cutaneous BRAF Val600-mutation-positive melanoma (COMBI-v): results of a phase 3, open-label, randomised trial. Lancet Oncol. 2015;16(13):1389–98. [DOI] [PubMed] [Google Scholar]
- 53.Larkin J, Ascierto PA, Dréno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867–76. [DOI] [PubMed] [Google Scholar]
- 54.Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, et al. Results of COLUMBUS part 1: a phase 3 trial of encorafenib (ENCO) plus binimetinib (BINI) versus vemurafenib (VEM) or ENCO in BRAF-mutant melanoma. In: Society for Melanoma Research. 2016. [Google Scholar]
- 55.Grob JJ, Amonkar MM, Martin-Algarra S, Demidov LV, Goodman V, Grotzinger K, et al. Patient perception of the benefit of a BRAF inhibitor in metastatic melanoma: quality-of-life analyses of the BREAK-3 study comparing dabrafenib with dacarbazine. Ann Oncol. 2014;25(7):1428–36. [DOI] [PubMed] [Google Scholar]
- 56.Schadendorf D, Amonkar MM, Milhem M, Grotzinger K, Demidov LV, Rutkowski P, et al. Functional and symptom impact of trametinib versus chemotherapy in BRAF V600E advanced or metastatic melanoma: quality-of-life analyses of the METRIC study. Ann Oncol. 2014;25(3):700–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schadendorf D, Amonkar MM, Stroyakovskiy D, Levchenko E, Gogas H, De Braud F, et al. Health-related quality of life impact in a randomised phase III study of the combination of dabrafenib and trametinib versus dabrafenib monotherapy in patients with BRAF V600 metastatic melanoma. Eur J Cancer. 2015;51(7):833–40. [DOI] [PubMed] [Google Scholar]
- 58.Doma E, Rupp C, Varga A, Kern F, Riegler B, Baccarini M. Skin tumorigenesis stimulated by Raf inhibitors relies upon Raf functions that are dependent and independent of ERK. Cancer Res. 2013;73(23):6926–37. [DOI] [PubMed] [Google Scholar]
- 59.Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366(3):207–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Callahan MK, Rampal R, Harding JJ, Klimek VM, Chung YR, Merghoub T, et al. Progression of RAS-mutant leukemia during RAF inhibitor treatment. N Engl J Med. 2012;367(24):2316–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abdel-Wahab O, Klimek VM, Gaskell AA, Viale A, Cheng D, Kim E, et al. Efficacy of intermittent combined RAF and MEK inhibition in a patient with concurrent BRAF- and NRAS-mutant malignancies. Cancer Discov. 2014;4(5):538–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang C, Spevak W, Zhang Y, Burton EA, Ma Y, Habets G, et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature. 2015;526(7574):583–6. [DOI] [PubMed] [Google Scholar]
- 63.Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2014;372(1):141116004513004. [DOI] [PubMed] [Google Scholar]
- 64.Schreuer M, Meersseman G, Van Den Herrewegen S, Jansen Y, Chevolet I, Bott A, et al. Quantitative assessment of BRAF V600 mutant circulating cell-free tumor DNA as a tool for therapeutic monitoring in metastatic melanoma patients treated with BRAF/MEK inhibitors. J Transl Med. 2016;14(1):95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Busser B, Lupo J, Sancey L, Mouret S, Faure P, Plumas J, et al. Plasma circulating tumor DNA levels for the monitoring of melanoma patients: landscape of available technologies and clinical applications. Biomed Res Int. 2017;2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Johnson DB, Flaherty KT, Weber JS, Infante JR, Kim KB, Kefford RF, et al. Combined BRAF (dabrafenib) and MEK inhibition (trametinib) in patients with BRAFV600-mutant melanoma experiencing progression with single-agent BRAF inhibitor. J Clin Oncol. 2014;32(33):3697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494(7436):251–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Johnson DB, Puzanov I. Treatment of NRAS-mutant melanoma. Curr Treat Options Oncol. 2015;16(4):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mougiakakos D, Choudhury A, Lladser A, Kiessling R, Johansson CC. Regulatory T cells in cancer. Adv Cancer Res. 2010;107(10):57–117. [DOI] [PubMed] [Google Scholar]
- 71.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565–70. [DOI] [PubMed] [Google Scholar]
- 72.Hodi FS, Day SJO, Mcdermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McDermott D, Haanen J, Chen TT, Lorigan P, O’Day S. Efficacy and safety of ipilimumab in metastatic melanoma patients surviving more than 2 years following treatment in a phase III trial (MDX010–20). Ann Oncol. 2013;24(10):2694–8. [DOI] [PubMed] [Google Scholar]
- 74.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–30. [DOI] [PubMed] [Google Scholar]
- 75.Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16(4):375–84. [DOI] [PubMed] [Google Scholar]
- 76.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372(26):2521–32. [DOI] [PubMed] [Google Scholar]
- 77.Schachter J, Ribas A, Long G, Arance A, Grob J, Mortier L, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival analysis of KEYNOTE- 006. J Clin Oncol. 2016;34(15_suppl):9504. [Google Scholar]
- 78.Ribas A, Hamid O, Daud A, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA. 2016;315(15):1600–9. [DOI] [PubMed] [Google Scholar]
- 79.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, et al. MAP kinase inhibition promotes T cell and antitumor activity in combination with PD-L1 checkpoint blockade. Immunity. 2016;44(3):609–21. [DOI] [PubMed] [Google Scholar]
- 81.Hu-Lieskovan S, Mok S, Homet Moreno B, Tsoi J, Robert L, Goedert L, et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAFV600E melanoma. Sci Transl Med. 2015;7(279):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Loi S, Dushyanthen S, Beavis PA, Salgado R, Denkert C, Savas P, et al. RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin Cancer Res. 2016;22(6):1499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res. 2012;18(5):1386–94. [DOI] [PubMed] [Google Scholar]
- 84.Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368(14):1365–6. [DOI] [PubMed] [Google Scholar]
- 85.Puzanov I, Callahan MK, Linette GP, Patel SP, Luke JJ, Sosman JA, et al. Phase 1 study of the BRAF inhibitor dabrafenib (D) with or without the MEK inhibitor trametinib (T) in combination with ipilimumab (Ipi) for V600E/K mutation–positive unresectable or metastatic melanoma (MM). J Clin Oncol. 2014;32(15_suppl):2511. [Google Scholar]
- 86.Ribas A, Butler M, Lutzky J, Lawrence DP, Robert C, Miller W, et al. Phase I study combining anti-PD-L1 (MEDI4736) with BRAF (dabrafenib) and/or MEK (trametinib) inhibitors in advanced melanoma. J Clin Oncol. 2015;33(15_suppl):3003. [Google Scholar]
- 87.Long GV, Hamid O, Hodi FS, Lawrence DP, Atkinson V, Starodub A, et al. Phase 2 study of the safety and efficacy of pembrolizumab (pembro) in combination with dabrafenib (D) and trametinib (T) for advanced melanoma (KEYNOTE-022). J Clin Oncol. 2016;34(15_suppl):9596. [Google Scholar]
- 88.Ribas A, Hodi FS, Lawrence DP, Atkinson V, Starodub A, Carlino MS, et al. Pembrolizumab (pembro) in combination with dabrafenib (D) and trametinib (T) for BRAF-mutant advanced melanoma: phase 1 KEYNOTE-022 study. J Clin Oncol. 2016;34(15_suppl):3014.27325863 [Google Scholar]
- 89.Bendell JC, Kim TW, Goh BC, Wallin J, Oh D-Y, Han S-W, et al. Clinical activity and safety of cobimetinib (cobi) and atezolizumab in colorectal cancer (CRC). J Clin Oncol. 2016;34(15_suppl):3502.27458302 [Google Scholar]