

Abstract
Short major histocompatibility complex (MHC) class I (MHC-I)-restricted peptides contain the minimal biochemical information to induce antigen (Ag)-specific CD8+ cytotoxic T cell responses, but are generally ineffective in doing so. To address this, we developed a cobalt-porphyrin (CoPoP) liposome vaccine adjuvant that induces rapid particleization of conventional, short synthetic MHC-I epitopes, leading to strong cellular immune responses at nanogram dosing. Along with CoPoP (to induce particle formation of peptides), synthetic monophosphoryl lipid A (PHAD) and QS-21 immunostimulatory molecules were included in the liposome bilayer to generate the “CPQ” adjuvant system. In mice, immunization with a short MHC-I-restricted peptide, derived from glycoprotein 70 (gp70), admixed with CPQ safely generated functional, Ag-specific CD8+ T cells, resulting in the rejection of multiple tumor cell lines, with durable immunity. When cobalt was omitted, the otherwise identical peptide and adjuvant components did not result in peptide binding and were incapable of inducing immune responses, demonstrating the importance of stable particle formation. Immunization with the liposomal vaccine was well-tolerated and could control local and metastatic disease in a therapeutic setting. Mechanistic studies showed that particle-based peptides were better taken up by antigen-presenting cells, where they were putatively released within endosomes and phagosomes for display on MHC-I surfaces. Based on the potency of the approach, the platform was demonstrated as a tool for in vivo epitope screening of peptide micro-libraries comprising a hundred peptides.
Keywords: peptide, CD8+ T cell, liposome, vaccine, cancer, immunotherapy
Graphical Abstract
Introduction
To kill cancer cells, the CD8+ T cell receptor (TCR) must recognize short tumor-derived peptides of 8–10 amino acids in association with major histocompatibility complex (MHC) class I (MHC-I) molecules. These short peptide epitopes are appealing for cancer vaccine development as they are simple to produce and provide, in theory, a direct method to induce CD8+ T cells against MHC-I antigen (Ag)-bearing target cells. Unfortunately, peptide-based cancer vaccine clinical trials have not produced compelling clinical responses in contrast to more recent immunotherapies, such as immune checkpoint blockade.1 While a number of reasons may account for this, one challenge for peptide-based cancer vaccines is the inability to potently generate Ag-specific CD8+ T cells with sufficient quantity and quality. Considerable efforts have been devoted to the development of improved peptide-based cancer vaccine systems. A variety of promising nanoscale materials have been utilized for cancer immunization including but not limited to nanodiscs,2 plant viruses,3–5 cancer cell-derived particles,6–8 self-assembled nanoparticles9 and liposomes.10, 11 Liposomes have been used frequently, as they are biocompatible and have a track record of use in pharmaceutical products.12–14 Peptide loading approaches with liposome include encapsulation,15, 16 covalent binding17–19 and non-covalent binding.20–23
Liposomes containing cobalt porphyrin–phospholipid (CoPoP) can bind peptides bearing an abbreviated polyhistidine-tag (his-tag) by simple admixing.24 The binding occurs spontaneously and is based on unusual intrabilayer coordination between the his-tag and the cobalt in the porphyrin bilayer, resulting in the generation of serum-stable Ag nanoparticles. Atomistic molecular dynamics simulations show that within bilayers, the metal center of CoPoP has 20-fold lesser exposure to water molecules compared to the metal center of headgroup-functionalized metal-chelating lipids, which accounts for the drastically improved stability of CoPoP-Ag spontaneous assemblies.25 We previously have shown that immunization with nanogram amounts of Ag, CoPoP liposomes can induce functional antibody (Ab) responses when admixed with recombinant proteins derived from various pathogens including malaria parasites,26–28 Lyme disease bacteria29 and SARS-CoV-2.30 In the current study, we developed the CoPoP system for use with short MHC-I-restricted peptides and show that strong, functional CD8+ T cell responses can be induced with nanogram amounts of peptide, reflecting dosing orders of magnitude lower than what is usually used in preclinical studies. As such, the approach described herein represents, to the best of our knowledge, one of the most potent approaches for short peptide immunization.
A challenge for cancer vaccine development is to identify short peptide immunogens that induce Ag-specific CD8+ T cell responses capable of recognizing the endogenously expressed target epitope on cancer cells. Neoantigens are mutated cancer-specific epitopes that provide a rich source of potential cancer vaccine targets. Several CD4+ T cell neoepitopes have been discovered that are immunogenic and have anti-tumor efficacy.31, 32 Identifying immunogenic CD8+ T cell neoepitopes that give rise to functional immune responses is more challenging. The practical implication is that the selection and design of short functional peptides (which, by virtue of their length, are restricted to binding MHC-I) derived from neoepitopes is very much an emerging practice. The use of synthetic long peptides that incorporate both CD8+ and CD4+ T cell epitopes can improve immunogenicity of peptide-based vaccines.33
The A5 peptide is derived from the AH1 epitope in the gp70 envelope protein (amino acids 423–431) of the murine leukemia virus (MLV).34 MLV sequences are endogenous in the genome of most laboratory mouse strains.35 gp70 is expressed in several tumor cell lines,36 and in this study the AH1 epitope served as a model CD8+ T cell epitope. Several studies have used the A5 epitope, which induces superior responses compared to AH1 itself, including the A5 epitope expressed as a recombinant peptide-MHC-I complex, which inhibited tumor growth in prophylactic settings.8, 37 Synthetic short A5 peptides conjugated to lipids and proteins have displayed some, albeit limited efficacy.38, 39 There may be ways to augment these responses, such as including chemotherapy treatment40 and immune checkpoint blockade.41 Here, together with the model A5 antigenic determinant, we report that CoPoP liposomes induced strong CD8+ T cell responses, leading to the rejection of CT26, CMS4, and 4T07 tumors in a prophylactic vaccine setting and also eliminated CT26 tumor growth and lung metastasis in an early therapeutic vaccine setting. A putative vaccine mechanism is proposed to account for the unexpected potency. We further demonstrate that because short, conventional MHC-I-restricted synthetic peptides (without unusual amino acids or covalent conjugation) can potently induce functional CD8+ T cells using simple short peptides with nanogram dosing in mice, this also enables epitope discovery via peptide micro-library screening in vivo.
Results and Discussion
Short peptides form particles when admixed with CoPoP/PHAD/QS-21 (CPQ) liposomes
As shown in Figure 1A, liposomes were formed with CoPoP, along with the immunostimulatory adjuvants QS-21, a saponin, and PHAD, a synthetic monophosphoryl lipid A (MPLA). The chemical structure of porphyrin–phospholipid (PoP, or 2HPoP since two hydrogens are present instead of cobalt), CoPoP, PHAD and QS-21 are shown in Figure 1A and the synthesis scheme and chemical characterization of PoP and CoPoP are shown in Figure S1–S8 and Table S1–S4. With 3 active components in the bilayer, these are referred to as “CPQ”. The role of CoPoP is simply to induce particle formation of the MHC-I- restricted peptides. QS-21 and monophosphoryl lipid A (MPLA) are components of AS01, a liposomal adjuvant used in licensed vaccines for malaria and herpes zoster.42 MPLA is a Toll-like receptor (TLR)-4 agonist glycolipid that can incorporate into bilayers. QS-21 is an amphipathic triterpene glycoside that can bind strongly to cholesterol and has strong vaccine adjuvant effects.43, 44 Alone, it can bind to local cellular cholesterol and cause necrosis at injection sites.45 It can also bind to cholesterol in the lipid bilayer of erythrocytes and causes pores. To reduce hemolysis and toxicity of QS-21, it is formulated with liposomes that contain cholesterol.43 Throughout this study, control liposomes that are identical to CPQ, but where two hydrogen atoms replace the cobalt in the porphyrin macrocycle are used as non-particle forming control liposomes and are termed “2HPQ”. Thus, comparison of CPQ and 2HPQ adjuvants enables examination of the impact of particle-based presentation of short peptides on immunogenicity.
Figure 1. CPQ liposomes rapidly and stably bind short MHC-I restricted peptides.
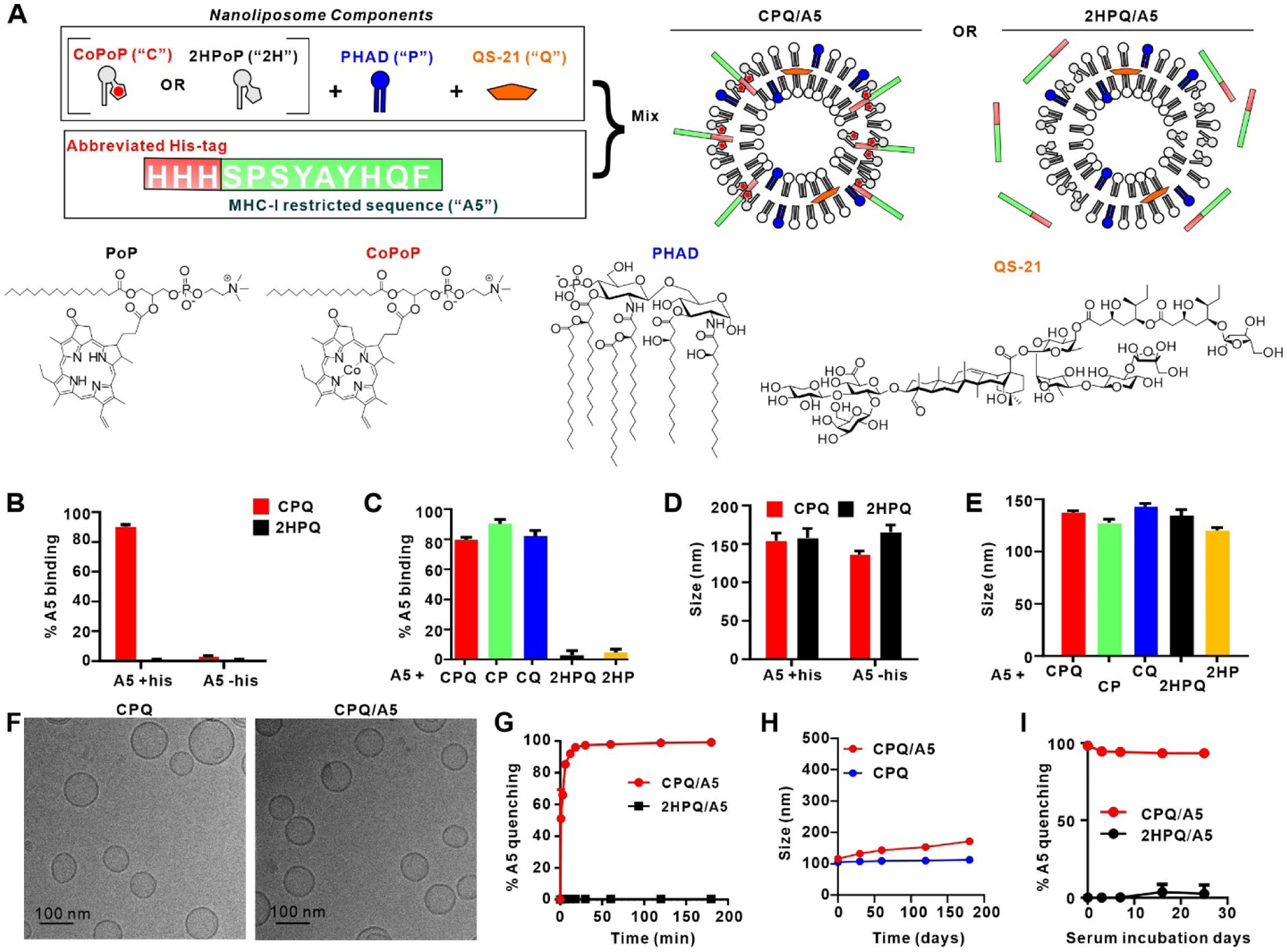
A) Components of the CPQ adjuvant system used in this study, with the model A5 peptide. B) Binding of the A5 peptide with or without his-tag to liposomes following 1 hr incubation. C) Binding of the A5 peptide (with his-tag) to indicated liposomes following 1 hr incubation. D) and (E) show hydrodynamic sizes of the liposomes in (B) and (C), respectively. F) Cryo-electron micrographs of CPQ liposomes with or without A5 peptide bound. G) Fluorescence quenching (indicative of peptide binding to liposomes) kinetics of HiLyte488-labelled A5 peptide to CPQ or 2HPQ liposomes. H) Refrigerated storage stability of CPQ and CPQ/A5 liposomes. I) Binding stability of HiLyte488-labelled A5 peptide with liposomes in the presence of 40 % human serum and incubation at 37 °C. Error bars show mean +/− std. dev. for triplicate experiments.
The A5 peptide was found to only bind with CPQ liposome when it contained a his-tag (Figure 1B). Shortly after mixing A5 with liposomes, ~80 % was converted into particle form, as assessed by a microcentrifugal filtration assay. QS-21 and PHAD did not impact the binding between the liposome and peptide (Figure 1C). However, the corresponding liposomes that contained the porphyrin-lipid but lacked cobalt displayed minimal binding to A5. Following particle formation with the peptide, the size of all the liposomes (CPQ, CoPoP/PHAD (“CP”), CoPoP/QS-21 (“CQ”), 2HPQ and 2HPoP/PHAD (“2HP”)) remained ~100–150 nm based on dynamic light scattering (Figure 1D and Figure 1E). Cryo-electron microscopy revealed that both CPQ liposomes with or without peptide bound were spherical and unilamellar, with sizes close to 100 nm (Figure 1F and Figure S9, which depicts a larger field of view). Peptide binding to CPQ liposomes did not impact their polydispersity or surface charge, and 2HPQ liposomes were also unaffected by incubation with the peptides (Table S5). To test peptide binding kinetics and serum stability of the resulting vaccine, A5 was fluorescently labeled. Upon liposome binding, fluorescence energy transfer from the fluorophore to CPQ or 2HPQ liposomes results in a decrease in fluorescence emission, which can be measured. Within 20 min of incubation, the fluorescence of the labeled A5 peptide was fully quenched, reflecting rapid binding to CPQ liposomes (Figure 1G). 2HPQ did not quench any fluorescence of A5, reflecting a lack of peptide binding. The maximum loading efficacy of the peptide was observed when approximately a 4-fold mass ratio of CoPoP was used (Figure S10). These results are consistent with the microcentrifugal filtration assay using the unlabeled peptide. In refrigerated storage, CPQ with or without the A5 peptide bound was stable for at least three months, with sizes from 100–150 nm (Figure 1H). In 40 % human serum, the binding of A5 and CPQ liposomes remained mostly intact for weeks, reflecting peptide-particle formation is highly stable in biological fluids (Figure 1I).
Immunization with A5/CPQ induces robust Ag-specific CD8+ T cell responses
BALB/c mice were immunized on days 0 and 7 with 500 ng A5 admixed with CPQ, and peripheral blood was collected on day 13. Based on tetramer (tet) staining (Figure S11), mice immunized with CPQ admixed with A5 (CPQ/A5) induced ~20 % Ag-specific T cells within the CD8+ T cell population (Figure 2A and Figure 2B). Vaccination using CoPoP liposomes without QS-21 (CP/A5) did not produce detectable Ag-specific CD8+ T cells in the blood or spleen (Figure S12). Without PHAD in the liposome, CQ/A5 produced less Ag-specific CD8+ T cells compared to CPQ/A5. The vast majority of these AH1+ CD8+ T cells (89 %) were of the effector-memory T cell (TEM) phenotype based on differential expression of CD44 and CD62L (Figure 2C). The overall percentage of TEM cells in the CD8+ T cell population increased significantly after CPQ/A5 vaccination (Figure 2D).
Figure 2. A5 peptide admixed with CPQ liposome induces robust Ag-specific CD8+ T cell responses.
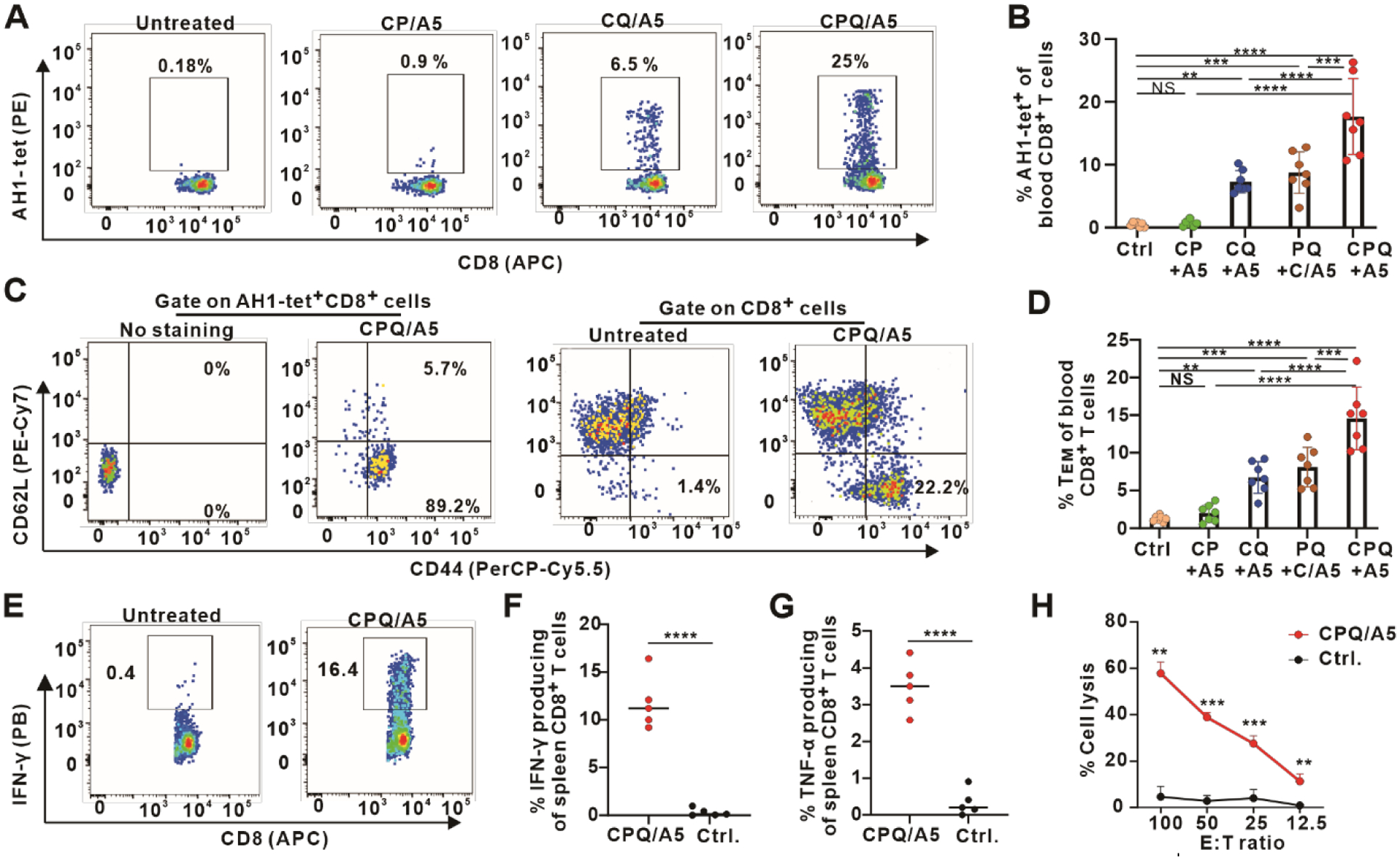
BALB/c mice were immunized intramuscularly on days 0 and 7 with 500 ng A5 admixed with the indicated adjuvants. Blood and spleen were collected on day 14 for antibody staining. Flow cytometry gating (A) and percentage (B) of AH1-tet+ cells in the CD8+ T cell population. T cell phenotype gating (C) and percentage (D) of TEM in the CD8+ T cell population. Flow cytometry gating (E) and percentage (F) of IFN-γ producing cells in the CD8+ T cell population in splenocytes from immunized or control mice after peptide restimulation in vitro. G) Percentage of TNF-α producing cells in the CD8+ T cell population in splenocytes after antigen restimulation in vitro. H) In vitro lysis of CT26 target cells (T) by effector cells (E) from splenocytes of CPQ/A5-vaccinated mice or untreated mice at various E:T ratios. Error bars show mean +/− std. dev. for n=5–7 mice per group or n=3 independent experiments (H). Significance shown with ** p < 0.01, *** p < 0.001, and **** p < 0.0001, as analyzed by one-way ANOVA followed by Bonferroni’s post hoc analysis (B, D) or comparing different groups with two-tailed unpaired Student’s t-test (F, G, H).
We also tested whether presentation of the A5 peptides on the same particle as QS-21 and PHAD was important. To do so, mice were immunized with CPQ/A5, or alternatively with CoPoP liposomes with A5 (C/A5) admixed with 2HPQ liposomes, with equivalent doses of peptide and adjuvant. While particle presentation of A5 with QS-21 and PHAD in separate liposomes could still induce Ag-specific CD8+ T cells, presentation of all components on the same particle was significantly more effective. This improvement was maintained on days 21 and 27 (Figure S13). This implies co-delivery of the peptide and adjuvant to the same cells in the draining lymph node benefits the subsequent immune response.
Splenocytes were collected for intracellular staining of interferon-gamma (IFN-γ) and tumor-necrosis factor alpha (TNF-α) 7 days after the boost immunization. Splenocytes from untreated mice or mice vaccinated with CPQ/A5 were prepared for in vitro restimulation with the A5 peptide. Based on the flow cytometry gating (Figure S14), ~12 % of the CD8+ T cells produced IFN-γ (Figure 2E and Figure 2F) and ~4 % of the CD8+ T cells produced TNF-α (Figure 2G). This indicates that CPQ/A5 induced a strong CD8+ T cell response, as measured by cytokine production. Splenocytes were stimulated by the A5 peptide and cultured together with Interleukin-2 (IL-2) for 5 days and then used as effector cells (E) to be combined with CT26 cells that were pulsed with the A5 peptide as target cells (T). These effector T cells lysed ~60 % of the tumor cells in 4 hr at an effector-to-target (E:T) cell number ratio of 100 (Figure 2H). Initial studies with the A5 peptide included a zwitterionic charged C-terminus tripeptide comprising the charged amino acids “ERR” with the aim to improve solubility, since MHC-I binding epitopes tend to be hydrophobic. However, this was later found to be non-essential. Other peptides might stand to benefit from solubilization approaches, which facilitate controlled peptide dissolution and dosing. The peptide sequences used for all experiments in this study are listed in Table S6 and S7. Other short, charged peptide solubilization motifs (EEE and RRR) were also assessed with A5. Overall, all the peptides, which also included a his-tag, formed particles with CPQ with simple mixing (Figure S15A). The RRR-terminated peptide bound non-specifically to 2HPQ liposomes, likely due to ionic interaction with the phospholipid headgroups. The sizes of liposomes after peptide binding remained ~100–200 nm (Figure S15B). The polydispersity (PDI) was ~0.1–0.2 (Figure S15C). No significant impact was observed on the induction of Ag-specific CD8+ T cell frequency using A5 peptides with or without any of the three solubilization motifs (Figure S15D).
CPQ/A5 as a prophylactic cancer vaccine
To test the function of the vaccine-induced, Ag-specific CD8+ T cells, BALB/c mice immunized with varying nanogram doses of CPQ/A5 were challenged with CT26 cells, which express gp70. Seven days after the first vaccination, there was an increase in the frequency of Ag-specific CD8+ T cells. After the booster vaccination, even as little as 20 ng of A5 (along with just 80 ng CoPoP, 80 ng PHAD and 80 ng QS-21) induced 5 % of peripheral CD8+ T cells to be Ag-specific (Figure 3A). A general dose response was followed, with increasing CPQ/A5 doses inducing a stronger CD8+ T cell response, up to a plateau of 500 ng A5. Higher doses did not increase the percentage of Ag-specific cells in CD8+ T cells, and actually resulted in a lower frequency. Since the peptide to adjuvant ratio was fixed in all cases, at higher peptide doses, more QS-21 was also administrated, which may have induced toxicity in the immune cells in the draining lymph nodes. Indeed, when immunized with 1000 ng A5 peptide with liposomes formulated to provide 1000 ng QS-21 instead of 4000 ng (which was used with the fixed Ag to adjuvant ratio), Ag-specific CD8+ T cells increased significantly (Figure S16).
Figure 3. Durable and robust protection from diverse tumor model challenges with CPQ/A5 immunization.
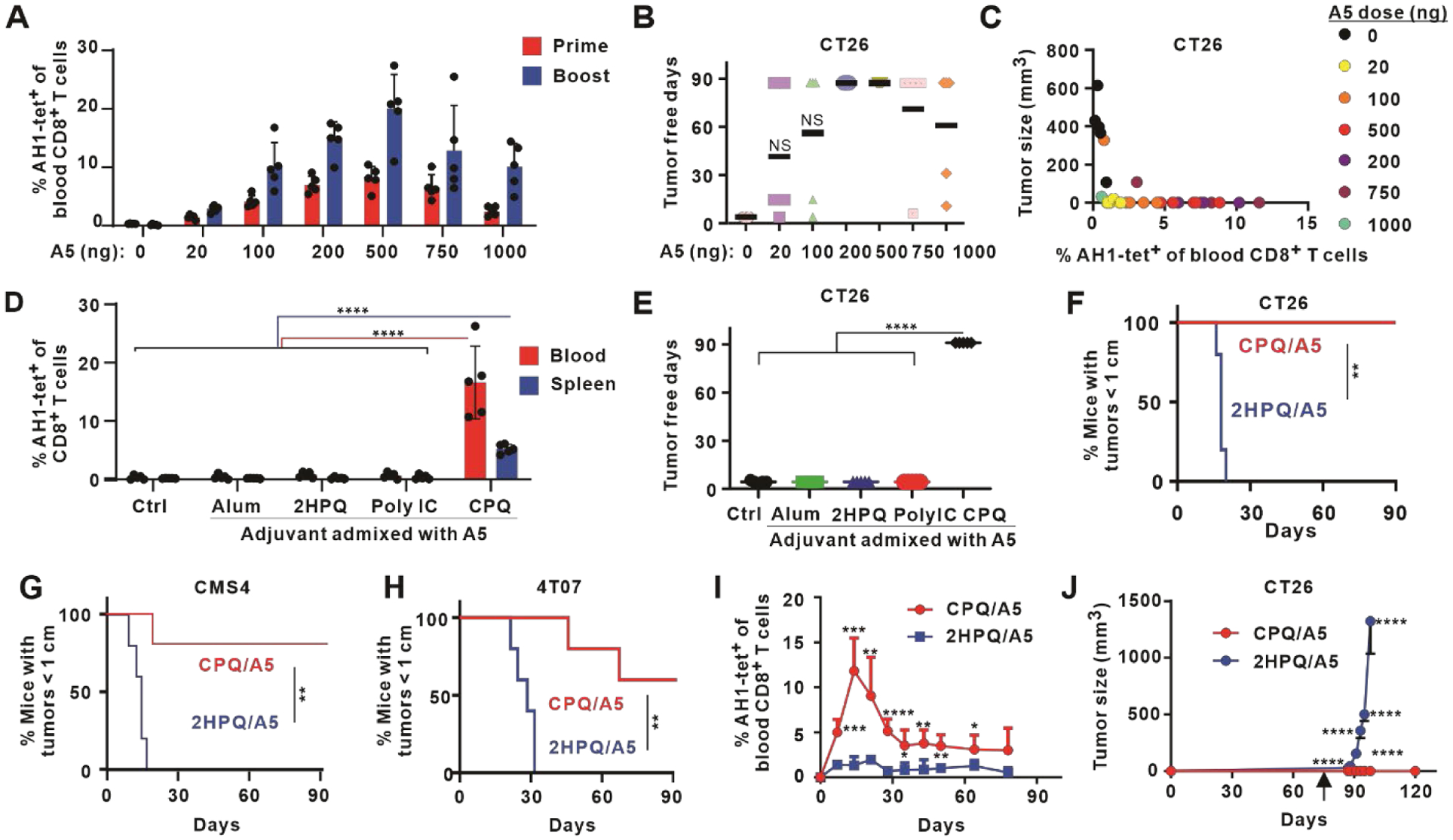
Mice were immunized on days 0 and 7 with the indicated vaccine dose, blood were collected on days 7 and 13, and tumor cells were inoculated on day14. A) Percentage of AH1-tet+ cells in the CD8+ T cell population in blood. B) Tumor-free days following CT26 challenge. The study duration was 90 days. C) Correlation of Ag-specific CD8+ T cells and tumor sizes 2 weeks after tumor inoculation. D) Percentage of AH1-tet+ cells in the CD8+ T cell population in the blood and spleen on day 13, and E) tumor-free days after challenge with CT26 cells, for mice immunized with 500 ng A5 admixed with the indicated adjuvant. F) Percentage of mice with tumor sizes smaller than 1 cm after immunization with 500 ng A5 peptide admixed with CPQ or 2HPQ liposomes and challenged subcutaneously with CT26 (F) or CMS4 (G) cells, or orthotopically with 4T07 (H) cells. I) Kinetics of AH1-tet+ CD8+ T cells in the the blood of mice vaccinated with 500 ng A5 peptide admixed with CPQ or 2HPQ on day 0 and 7 J) Tumor growth of mice challenged with CT26 cells subcutaneously on day 80 (indicated by arrow). Bars show mean for n=5 mice per group, and std dev. where indicated. * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001, analyzed by one-way ANOVA with Bonferroni’s post hoc test (D, E), log rank test (F, G, H), or two-tailed unpaired Student’s t-test (I, J). Asterisks in I and J indicate statistically significant differences between CPQ/A5 and 2HPQ/A5 at the same time point.
Within four days of tumor challenge, all control mice developed palpable tumors. However, following vaccination with just 20 ng of A5 peptide, 40 % of the immunized mice remained tumor-free for 90 days post-tumor challenge (Figure 3B). Immunization with 200 or 500 ng of A5 led to complete tumor rejection in all mice. Mice immunized with 750 or 1000 ng actually had lower tumor rejection rates, although the average tumor-free period was still 70 and 60 days after tumor challenge, respectively. The lower protection rate is in accordance to the Ag-specific CD8+ T cell frequency data. Among the mice that developed tumors, higher induction of Ag-specific CD8+ T cells was associated with smaller tumor sizes (Figure 3C).
Next, we compared the immunogenicity of 500 ng A5 peptide admixed with CPQ or other vaccine adjuvants including 2HPQ (lacking cobalt), Alhydrogel (Alum), or polyinosinic polycytidylic acid (poly(I:C)). Only mice immunized with CPQ admixed with 500 ng A5 produced AH1-specific CD8+ T cells in both the blood and spleen (Figure 3D), which was accompanied by complete rejection of a CT26 tumor challenge (Figure 3E). In contrast, mice immunized with 500 ng A5 admixed with other adjuvants did not produce detectable AH1-specific CD8+ T cells and developed tumors within four days of challenge. Thus, CPQ was required for effective immunization using the short A5 peptide.
Although gp70 is a shared biomarker expressed in several murine cancer cell lines,36 its use in cancer vaccines has generally been focused on CT26 tumors. To assess whether the CPQ/A5 could offer protection in various tumor models, mice immunized with CPQ/A5 and non-particle forming 2HPQ/A5 were challenged with CT26 cells (Figure 3F), as well as other tumor models that express gp70; the CMS4 murine sarcoma (Figure 3G) and the orthotopic 4T07 breast cancer (Figure 3H) model.46 Immunization with CPQ/A5 significantly prevented tumor growth, resulting in much lower percentages of mice with tumor sizes reaching 1 cm following tumor challenge in all three cancer models, with 60–100 % of mice showing complete tumor rejection.
The durability of CPQ/A5 immunization was next assessed. Mice were immunized with 500 ng A5 peptide admixed with CPQ or 2HPQ on days 0 and 7, and the Ag-specific CD8+ T cell response was assessed in the peripheral blood. The frequency of AH1-tet+ cells in the CD8+ T cell population increased following the initial immunization and boosting, with a maximum frequency observed on day 14 (Figure 3I). Mice immunized with 2HPQ/A5 had minimal percentage of AH1-tet+ cells in the CD8+ T cell population at all time points tested. In the CPQ/A5 group, after day 14, the frequency of Ag-specific T cells in the CD8+ T cell population gradually subsided but was sustained around 5 % of all CD8+ T cells by day 80. On day 80, over two months after the final boosting with 500 ng A5 peptide, mice were challenged with CT26 cancer cells. As shown in Figure 3J, even at this time point, all CPQ/A5 immunized mice fully rejected the tumor challenge without any sign of growth for at least 40 days. All mice vaccinated with A5 peptide admixed with cobalt-free liposomes developed rapidly growing tumors.
Safety studies were carried out in CD-1 mice, which are an outbred strain that may capture a broader range of potential toxicity responses. Mice were vaccinated on days 0 and 7 with CPQ/A5 at the functional dose of 500 ng of peptide. Mice exhibited normal weight gain (Figure 4A). No obvious differences in the heart, liver, spleen, lung or kidney were observed with histology (Figure 4B). A complete blood cell count (Figure 4C) and serum chemistry panel (Figure 4D) revealed that all parameters of vaccinated mice were in the normal range of healthy mice.
Figure 4. Safety of CPQ/A5.
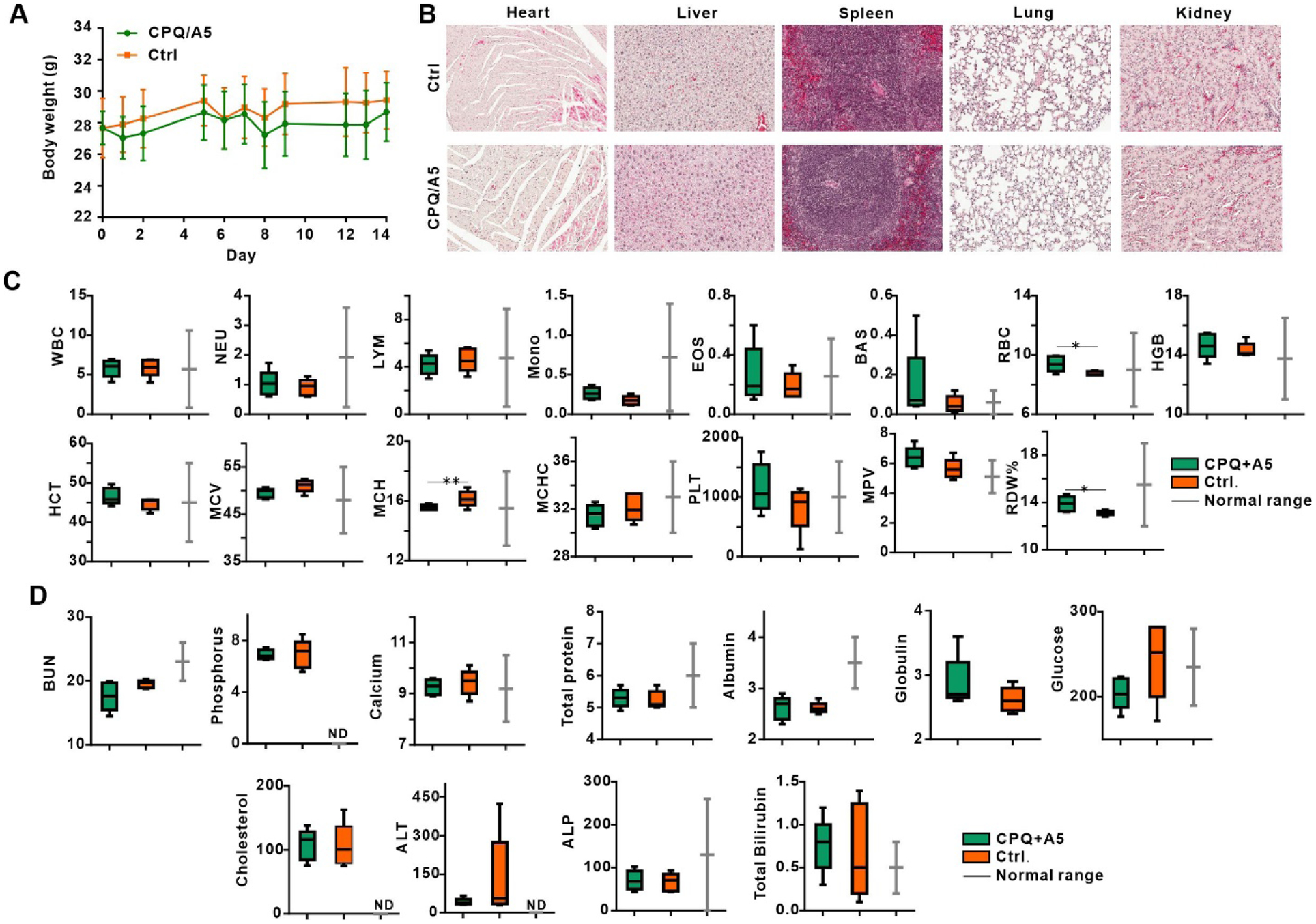
Female CD-1 mice were vaccinated intramuscularly with CPQ/A5 on days 0 and 7 with 500 ng A5 peptide admixed with CPQ liposomes (also containing 2 μg each of CoPoP, QS-21 and PHAD) and compared to untreated control mice. Blood and organs were collected on day 14. A) Body weight of CD-1 mice. B) Embedded hematoxylin and eosin stained slices of indicated organs. C) Complete blood count parameters as follows: WBC (white blood cells), NEU (neutrophils), LYM (lymphocytes), MONO (monocytes), EOS (eosinophils), BAS (basophils), RBC (red blood cell count), HGB (hemoglobin), HCT (hematocrit), MCV (mean cell volume), MCH (mean cell hemoglobin), MCHC (mean cell hemoglobin concentration), PLT (platelet), MPV (mean platelet volume), RDW (red cell distribution width). D) Serum markers with their general description as follows: BUN (blood urea nitrogen), phosphorus, calcium, total protein, albumin, globulin, glucose, cholesterol, ALT (alanine aminotransferase), ALP (alkaline phosphatase) and total bilirubin. Values show mean +/− std. dev for n=5 mice per group. “ND”; no data provided for normal range. Data show box-and-whiskers plots, the line represents the median, the whiskers show the data range and the box shows the interquartile range. * p < 0.05, ** p < 0.01, analyzed by two-tailed unpaired Student’s t-test.
CPQ/A5 as a therapeutic cancer vaccine
While CPQ/A5 was shown to be potent in a prophylactic setting, most cancer vaccines would be initially tested in patients with advanced or metastatic disease. To address this, CPQ/A5 was assessed in mice after tumor implantation or in settings of experimental lung metastasis. In the former setting, mice were inoculated with CT26 tumors on day 0 then immunized on day 5 with 500 ng A5 peptide, a time point at which tumors first became measurable and started rapidly growing (Figure 5A). Mice were boosted with 500 ng A5 peptide a week later, and in the intervening period, tumors grew to ~3 mm by day 12. However, within days following the second immunization with CPQ/A5, all tumors shrank and disappeared without evidence of regrowth for 90 days (Figure 5B). Control mice or mice that were immunized with non-particle forming 2HPQ/A5 had continued rapid tumor growth and no mice had tumors less than 1 cm by day 25 (Figure 5C, Figure 5D and Figure 5E).
Figure 5. Therapeutic efficacy of CPQ/A5 vaccination in early-stage CT26 cancers.
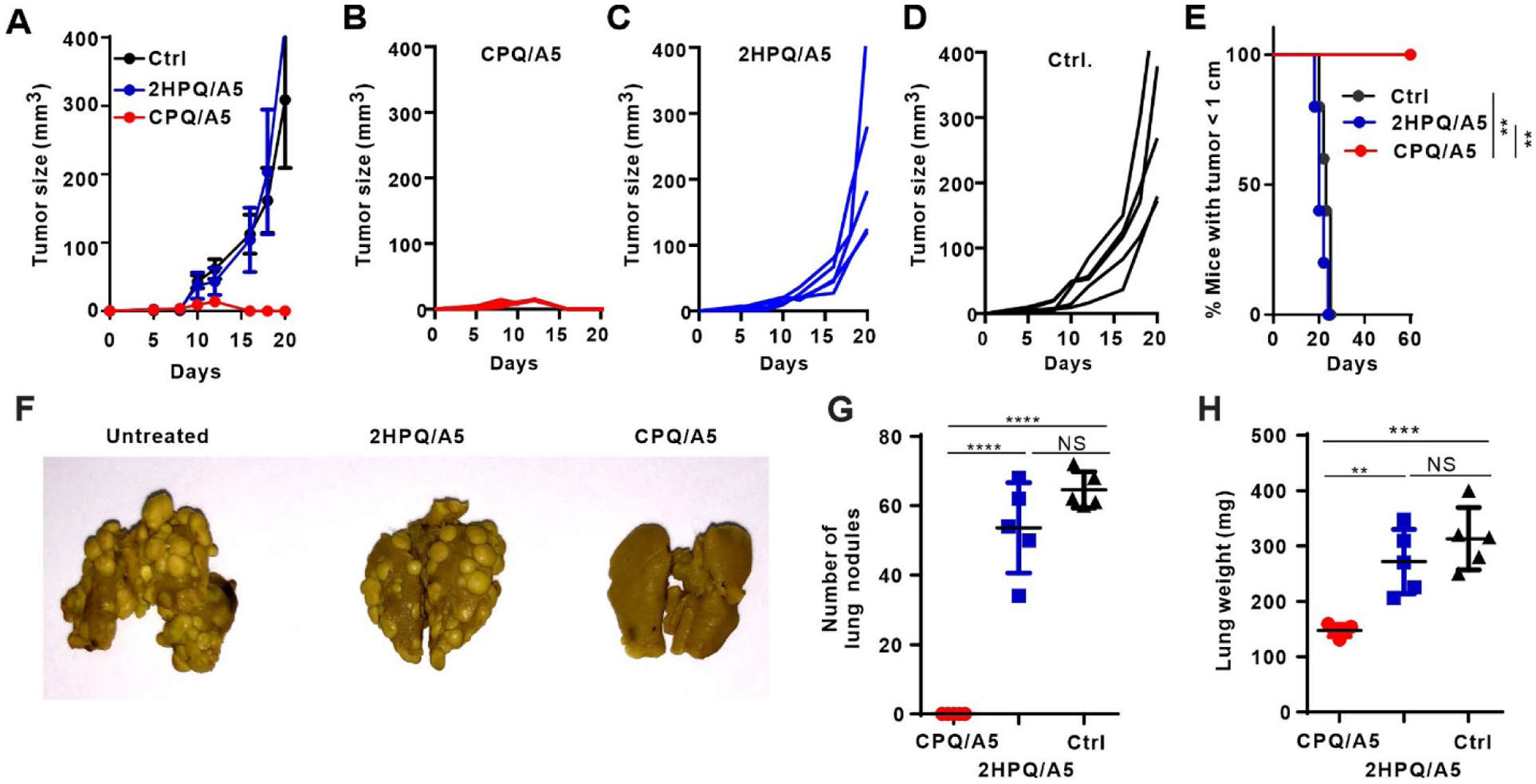
BALB/c mice were inoculated with CT26 cells subcutaneously on day 0, then immunized with CPQ/A5 or 2HPQ/A5 (500 ng peptide) on days 5 and 12. Average tumor growth of mice in each group (A). Tumor growth of individual mice vaccinated with CPQ/A5 (B), 2HPQ/A5 (C) or untreated (D) and percentage of mice with tumor sizes smaller than 1 cm (E), for n=5 mice per group. For the metastasis model, mice were injected intravenously with CT26 tumor cells, and then immunized 2 and 9 days later. Lungs were assessed for metastases on day 18. Metastases were present in the untreated and 2HPQ/A5 groups, but not in the CPQ/A5 group, as shown by a representative photograph (F), the count of lung metastasis nodules (G) and the lung weights (H). Error bars show mean +/−std. dev. for n=5 mice per group. ** p < 0.01, *** p < 0.001, and **** p < 0.0001, analyzed by log-rank test (E) or one-way ANOVA with Bonferroni’s post hoc test (G, H).
To test CPQ immunization in a metastatic setting, an experimental CT26 lung metastasis model was established. Mice were injected intravenously with CT26 tumor cells on day 0 and vaccinated on day 2 and 9 with 500 ng A5 peptide. Lung nodules were recorded on day 18. In untreated mice or in mice receiving the non-particleizing 2HPQ/A5 vaccine, dozens of nodules were observed (Figure 5F). In stark contrast, nodules were not detectable in mice that were immunized with CPQ/A5. Control mice or mice injected with 2HPQ/A5 had on average more than 50 lung nodules per mouse (Figure 5G). Lung metastasis was confirmed by increased lung weights; mice without any treatment or injected with 2HPQ/A5 had nearly double the lung weight compared to mice immunized with CPQ/A5 (Figure 5H).
CPQ mechanistic features
Next, we sought to determine the immunologic basis for vaccine potency of CPQ/A5. After admixing with CPQ, short peptides form particles that are stable in serum (Figure 1), which can be transported to the draining lymph nodes. A schematic of the overall putative mechanism is shown in Figure 6A. Immune cells are recruited to the lymph nodes, where antigen-presenting cells (APCs) take up the liposomes into phagosomes and endosomes. There, peptides are putatively released from the CPQ liposome where they are presented on MHC-I molecules expressed prior to externalization and activation of T cells. Evidence in support of such a model of immunization is presented here.
Figure 6. Putative mechanism of CPQ/A5 immunization.
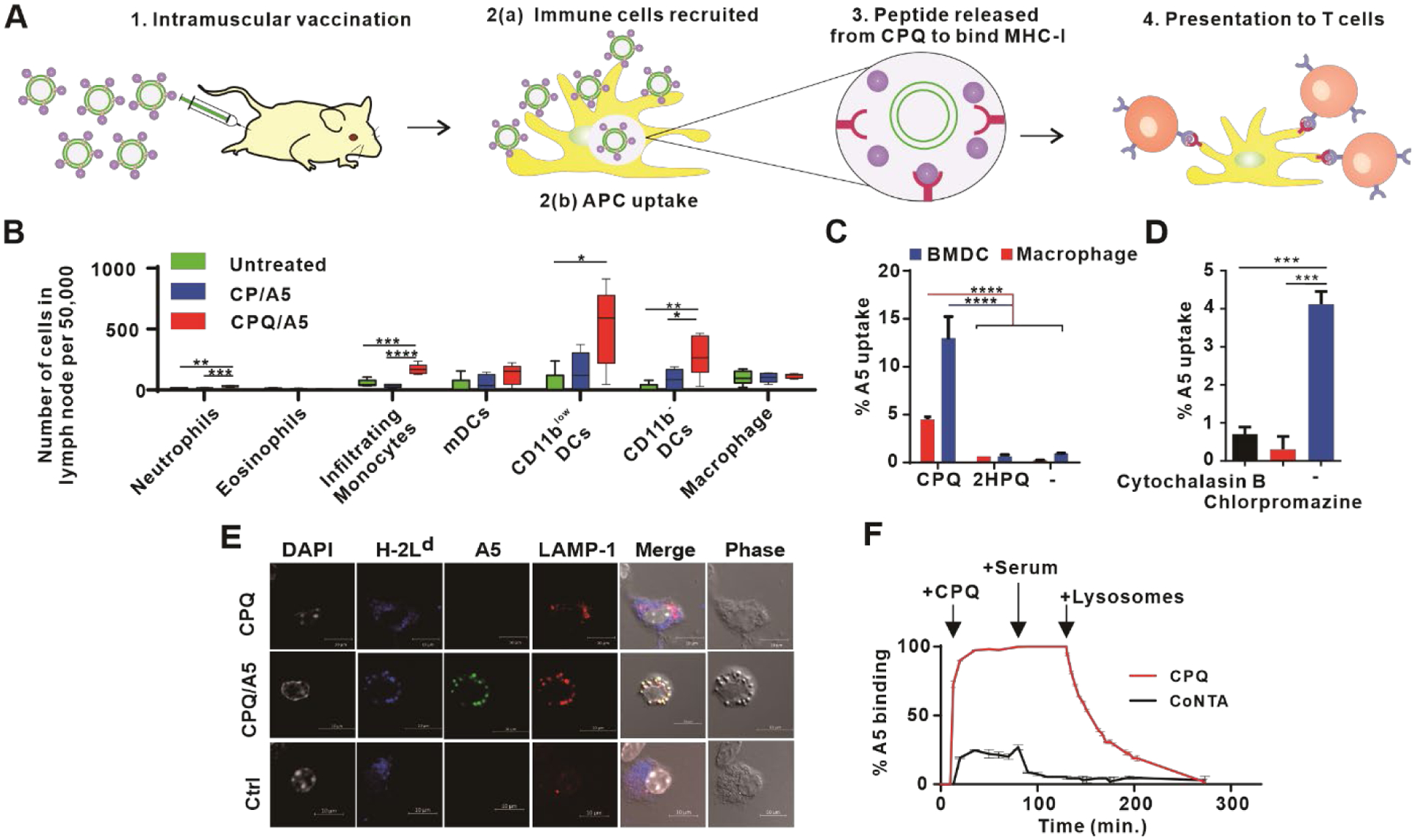
A) Schematic illustration of T cell activation following immunization with CPQ/A5. B) Immune cell populations in draining lymph nodes harvested two days after immunization with the indicated vaccination. Data show box-and-whiskers plots, the line represents the median, the whiskers show the data range and the box shows the interquartile range. C) Uptake of A5 in murine macrophages or BMDCs following 1 hr incubation. D) Uptake of A5 peptide in macrophages in the presence of the phagocytosis and endocytosis inhibitors. E) Confocal micrographs of murine BMDCs incubated with beads coated with CPQ or CPQ/A5-HiLyte488 showing colocalization with H-2Ld (MHC-I) and LAMP-1. Scale bar, 10 μm. F) Binding of A5 to liposomes containing CoPoP or CoNTA, followed by the addition of serum and lysosome extract as indicated. Error bars show mean +/−std. dev. for n=5 mice for lymph node studies and n=3 replicate experiments for other studies. * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001, analyzed by one-way ANOVA with Bonferroni’s post hoc test (B-D).
Following intramuscular administration, liposomes migrated to 1–3 lymph nodes close to the injection site in 1 hr and 3–4 lymph nodes in 4 hr (Figure S17). Twenty-four hr after a single intramuscular immunization, liposomes could be detected in at least 3 separate lymph nodes of the upper and lower limbs (Figure S18A, B). Immunization at 2 or 3 separate injection sites led to detectable liposome distribution in up to 9 lymph nodes; however, there was no increase Ag-specific CD8+ T cell induction when the same dose was provided with multiple injection sites (Figure S18C). To investigate immune cell recruitment, the draining inguinal lymph nodes were collected, and the cells were analyzed by flow cytometry 48 hr after intramuscular immunization with CPQ/A5 or CP/A5 (the difference between these being the inclusion of QS-21 in the CPQ liposomes). Compared to CP/A5-vaccinated mice and untreated mice, higher numbers of Neutrophils, infiltrating monocytes and CD11b− DCs were recruited in the lymph nodes of mice vaccinated with CPQ/A5 (Figure 6B). MPLA and QS-21 has been shown to facilitate immune cell recruitment to the injection site.47 Other immune cell types (e.g., eosinophils, myeloid DCs (mDCs), CD11blow DCs and macrophages) were not observed to significantly increase in draining lymph nodes.
The modest increase of certain immune cells in the lymph nodes following immunization does not likely fully account for the robust enhancement in the Ag-specific CD8+ T cell response by CPQ. The uptake of the peptide Ag was examined in vitro using fluorescence microscopy with a labeled A5 peptide. Macrophages and bone marrow derived dendritic cells (BMDCs) were incubated with CPQ/A5 or 2HPQ/A5 and uptake was assessed. When admixed with CPQ, 5 % of the total A5 peptide in the incubation mixture was taken up by macrophages, and 13 % were taken up by BMDCs (Figure 6C). However, there was no Ag uptake when A5 alone or 2HPQ/A5 were incubated with the cells. Kinetics revealed that in macrophages, ~3 % of the CPQ/A5 peptide is taken up within 20 min, increasing to 4–5 % in 1 hr (Figure S19A). Fluorescence microscopy confirmed these results; the A5 peptide was only taken up in macrophages after admixing with CPQ liposomes (Figure S19B). Interestingly, as CPQ quenches the fluorescence of the labeled A5 peptide upon binding, the strong fluorescence of labeled A5 peptide in the micrographs suggest that the peptide becomes released from the liposomes after cellular uptake. Release of the peptide was detected in cells, based on a fluorometric quenching assay, that suggested there was greater intracellular peptide release with a shorter his-tag length (Figure S19C). In the presence of cytochalasin B (a phagocytosis inhibitor) and chlorpromazine (an inhibitor of clathrin-mediated endocytosis), significantly less A5 peptide was taken up by macrophages (Figure 6D). Taken together, these data showed that the CPQ/A5 complex is taken up by immune cells via phagocytic and endocytosis.
MPLA, which mimics components of the bacterial membrane, has been associated with MHC-I expression within phagosomes.48 The expression of the H-2Ld MHC-I haplotype (which is the restriction element for the A5 peptide) was assessed following incubation with CPQ/A5 in BMDCs. To assist phagosome visualization, a protocol was developed to coat silica microbeads with CPQ or CPQ/A5 (Figure S20). Immunofluorescence microscopy was carried out using antibodies (Abs) against the phagosome marker lysosomal-associated membrane protein 1 (LAMP-1), and H-2Ld. BMDCs incubated with CPQ beads showed co-localized fluorescence of both H-2Ld and LAMP-1, as expected. BMDCs that were incubated with CPQ/A5 showed co-localized fluorescence of all the components; A5 peptide, H-2Ld and LAMP-1 (Figure 6E). H-2Ld expression was also observed in cells incubated with CPQ, CP, CQ and CoPoP liposome (Figure S21).
The detectable fluorescence signal of the A5 peptide within microphages and BMDCs implies it was released from the liposomes and became unquenched following cellular uptake. Indeed, when incubated with commercial lysosome extract in vitro, A5 peptide release from CoPoP liposomes was detected (Figure 6F). Almost all the peptide was released from CPQ within 2 hr. Liposomes that contained cobalt-nitrilotriacetic acid (NTA) on the headgroup of lipids could not stably bind the A5 peptide, likely due to the much greater water exposure of the metal center. Shorter his-tag sequences (i.e., 2 histidine residues instead of 6) led to faster release from CPQ liposomes in vitro upon exposure to lysosome extracts (Figure S22). The released peptides from macrophage cell extracts could be detected intact by high performance liquid chromatography (HPLC). As shown in Figure S23, the elution time of A5 peptide in the cell lysate was the same as the pure A5 peptide. Taken together, these data suggest that peptides bound to CPQ liposomes are preferentially taken up by APCs, and possibly are released intact to MHC-I directly into cell phagosomes and lysosomes for presentation on their surface. This mechanism would be compatible with the vacuolar pathway for MHC-I peptide presentation, which avoids the necessity for the peptide to be transported first through the cytosol.49 We note that some studies within this work used the ERR tripeptide on the C terminus to address potential solubility issues, although we later found that this was generally not required. The presence of this charged tag inhibited peptide binding to recombinant H-2Ld in vitro using an enzyme-linked immunosorbent assay (ELISA) assay (Figure S24). However, the presence or absence of the charged tripeptide did not impact the induction of Ag-specific CD8+ T cells. Thus, immunization did not appear overly sensitive to the flanking residues of the MHC-I epitope. Further proteolytic processing of the peptide to remove the small number of adjacent residues to the MHC-I epitope is possible and should be further investigated. It should be noted that shorter abbreviated his-tags on the N terminus resulted in a greater induction of Ag-specific CD8+ T cells (Figure S25). The reason may be related to the enhanced release of the peptide following intracellular uptake as described above.
Antigen screening using CPQ
Modern genomic, proteomic, and bioinformatic approaches can rapidly identify extensive lists of coding mutations (i.e. neoantigens) in cancer cells, which have been reported for the murine CT26 and 4T1 cell lines.50–52 However, reliably determining which of these are immunogenic and can produce functional responses is not yet realized. Given the potency of the CPQ system, we assessed low-cost peptide micro-libraries to screen candidate peptides. Based on published work,52 we selected 100 predicted neoantigens on the basis of the strongest MHC-I affinity, as well as several neoantigens that were shared between both CT26 and 4T1 cell lines (Table S8).
Mice were immunized with 5 library peptides at a time, along with the A5 peptide, with all peptides combined and admixed with CPQ. After two intramuscular injections, splenocytes were collected, and then restimulated with each of the synthetic micro-library peptides, individually. The overview of the screening process is shown in Figure 7A. Production of IFN-γ was measured and peptide immunogenicity was determined relative to the A5 peptide, which served as in internal control that could induce strong Ag-specific CD8+ T cell responses (Figure S26A) and high levels of IFN-γ production (Figure S26B) throughout a range of peptide doses and Ag densities on the liposome. Multiplexing and immunizing with 5 peptides simultaneously was found to be effective for inducing Ag-specific CD8+ T cells (Figure S27A) with readily detectable levels of IFN-γ (Figure S27B), whereas multiplexing with more peptides did not provide consistent IFN-γ production in all splenocyte preparations tested.
Figure 7. Short peptide micro-library screening with CPQ reveals the RragcL385P 9mer peptide as a functional vaccine epitope to inhibit CT26 and 4T1 lung metastasis.
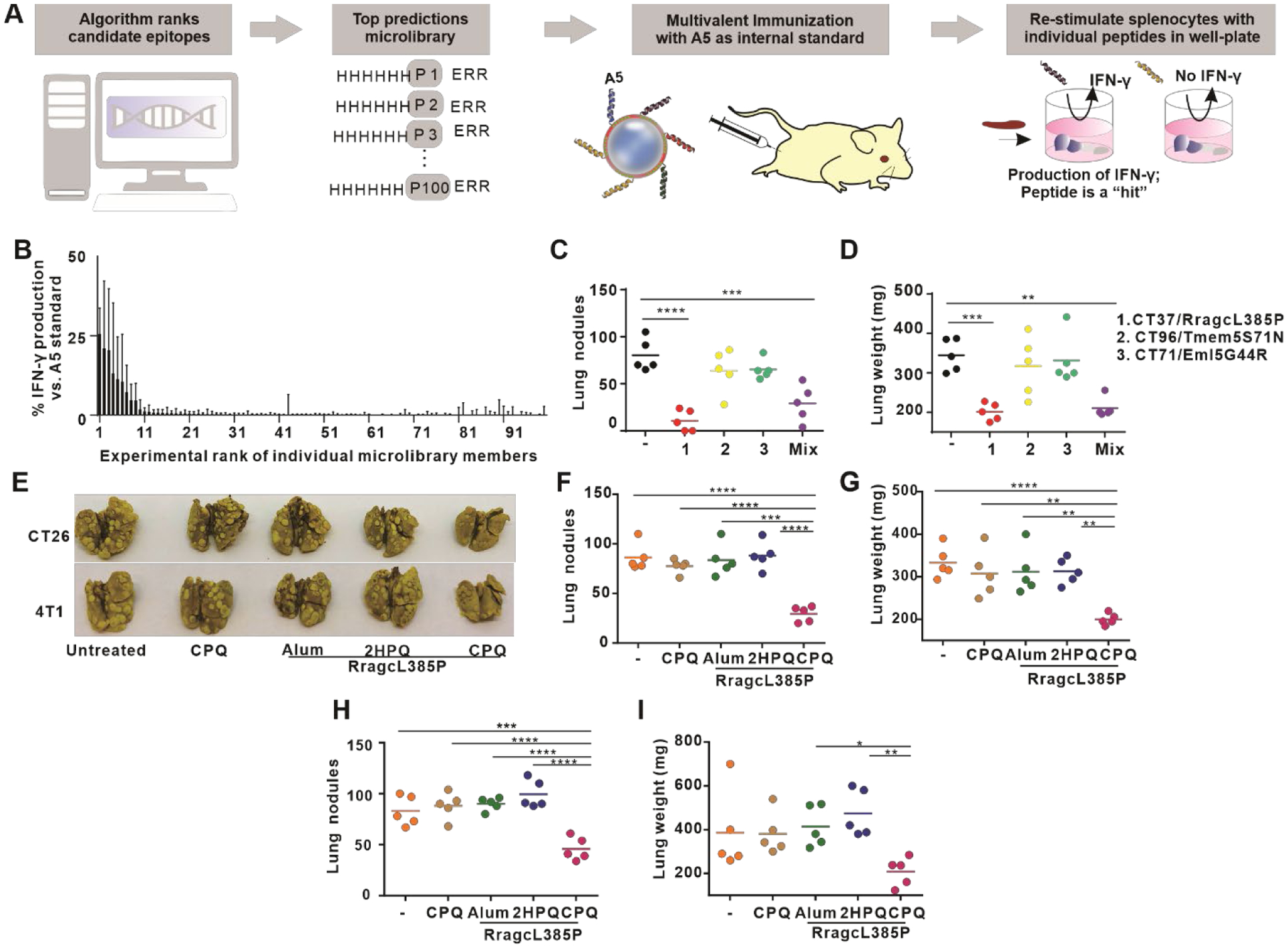
A) Approach used for in vivo screening of a 100 peptide micro-library. Mice were immunized with pooled micro-library peptides (5 peptides at a time, along with A5 serving as an internal control). Collected splenocytes were then restimulated with individual short peptides and IFN-γ was measured relative to A5 restimulation to indicate Ag-specific T cell presence. B) Identification of immunogenic peptides. Error bars show data range of triplicate wells from n=2 mice per group, expressed relative to the IFN-γ produced by A5 in the same immunization group. C) Mice were intravenously challenged with CT26 cells on day 0, and then immunized with CPQ with RragcL385P, Eml5G44R, Tmem5S71N or the combination 1 and 8 days later (1000 ng total peptide). Lung nodules (C) and weight (D) were assessed on day 18. The challenge was repeated but immunization was with 500 ng RragL385P with the indicated adjuvants, and lung metastases were assessed following challenge with CT26 (F) or 4T1 (H) cells. Images of lungs from different groups were taken (E). Lung weights also were assessed for CT26 (G) and 4T1 (I) tumor-bearing mice. Lines show mean for n=5 mice per group. * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001, analyzed by one-way ANOVA with Bonferroni’s post hoc test.
Peptides formed particles with CoPoP liposomes with sizes smaller than 150 nm and low polydispersity (Figure S28). For Ag screening, multivalent vaccines composed of peptides with the same MHC-I haplotypes (H-2Ld, H-2Dd, or H-2Kd) and the internal A5 peptide were admixed with CPQ, Peptide cocktails bound with CPQ liposome and formed nanoparticles with negative zeta-potentials (Figure S29). After immunization, only ~10 % of MHC-I binding peptides induced T cells in splenocytes that produced IFN-γ with peptide restimulation, and none were nearly as effective as the A5 peptide (Figure 7B). The most highly immunogenic peptides only produced about a quarter of the level of IFN-γ relative to the A5 peptide. This result underscores the challenges in identifying immunogenic CD8+ T cell epitopes from predicted MHC-I binding peptides, even using a strong adjuvant system. One caveat of these results is that we did not determine whether the immunodominance of the A5 peptide had a deleterious impact on the immunogenicity of the other peptides within the cocktail. It is also feasible that immunization with higher antigen doses of single individual peptides could have induced stronger immunogenic responses, although this would decrease the in vivo peptide micro-library screening throughput.
The three most immunogenic 9-mer peptides (RragcL385P, Tmem5S71N and Eml5G44R) were then assessed in the CT26 lung metastasis model. Vaccines were prepared by admixing peptides with CPQ liposomes (Figure S30), followed by two immunizations of 1000 ng (total) of peptides with CPQ, 1 and 8 days after intravenous administration of the tumor cells. Eighteen days following challenge, untreated mice had ~75 lung nodules (Figure 7C). However, mice vaccinated with CPQ/RragcL385P had just 15 lung nodules. Mice vaccinated with the other two neoantigens showed no significant difference in lung metastasis compared to the untreated mice. Mice vaccinated with a combination of all three of the neoantigens had an average of 25 lung nodules, which was likely attributed to the presence of the RragcL385P Ag. The lung weight confirmed the efficiency of immunization with the RragcL385P vaccine in reducing lung metastases (Figure 7D).
Anti-tumor efficacy of the short RragcL385P peptide (SPKALAHNG) admixed with CPQ was assessed in a therapeutic lung metastasis model. Mice were inoculated with tumor cells on day 0 and vaccinated on day 1 and 8. Lungs from mice inoculated with 4T1 tumor cells were collected on day 16 and lungs from mice inoculated with CT26 were collected on day 18. With a 500 ng peptide immunization dose, CPQ/RragcL385P vaccine significantly inhibited lung tumor growth in BALB/c mice (Figure 7E). For mice inoculated with CT26 cells, lungs from control groups (CPQ alone, Alum/RragcL385P, 2HPQ/RragcL385P) had an average of 90 lung nodules and lungs from CPQ/RragcL385P had an average of 30 lung nodules (Figure 7F). The lung weights reflected the result of the nodule counts (Figure 7G). For mice inoculated with 4T1 cells, lungs from control groups (CPQ alone, and the peptide mixed with Alum or 2HPQ) had an average of 80 nodules, while lungs from CPQ/RragcL385P had an average of 40 (Figure 7H); again, the lung weights reflected the results of the nodule counts (Figure 7I). However, in vitro cell cytotoxicity experiments showed that splenocytes from CPQ/RragcL385P immunized mice were not cytotoxic against CT26 cells, compared to splenocytes from CPQ/A5 immunized mice (Figure S31). Surprisingly, based on genomic DNA sequencing, the RragcL385P mutation present in both CT26 and 4T1 cell lines was also found in the genomic DNA of the BALB/cAnNCrl mouse strain from Charles River that was used in the immunization studies (Figure S32). On the other hand, BALB/cJ mice from Jackson Laboratories, the parent mouse strain that was used as a reference to identify the mutation in the murine cell lines,52 did not harbor the mutation. Thus, even though the effect of inhibition of lung metastasis after vaccination with CPQ/RragcL385P was confirmed and reproducible (Figure S33) in BALB/cAnNCrl mice challenged with 4T1 and CT26 tumor cells, the outcome appeared to be due to an off-target effect that requires further investigation. Thus, although the screening of MHC-I peptides successfully identified a functional epitope, the mechanism of action could not be confirmed to be from tumor cell lysis, so further investigation is warranted to determine the role of immunization with the RragcL385P epitope. In future work, we still anticipate this screening approach will be able to identify peptide epitopes that induce CD8+ cytotoxic T lymphocytes (CTLs) capable of epitope-specific tumor cell lysis.
Conclusions
CPQ liposomes induced stable particle formation of short peptides and were highly effective for inducing Ag-specific CD8+ T cells that inhibited tumor growth in multiple mouse tumor models in both local and metastatic settings using the A5 model epitope. Immunization was well-tolerated in mice. The putative mechanism of potency is related to encouraging infiltration of APCs into draining lymph nodes, enhanced delivery of the short peptide to APCs, followed by the release of the peptide for binding to MHC-I expressed within endosomes and phagosomes. Based on this potency, micro-libraries were screened to identify a shared epitope, RragcL385P, that reproducibly reduced metastatic disease when vaccinated together with CPQ in both CT26 and 4T1 cell lines, although this epitope appeared to operate from an off-target effect, which requires further study to understand the basis for this observation. In future work, we anticipate using the CPQ system with other MHC-I epitopes, as well as for additional studies dedicated to functional epitope screening and discovery.
Experimental
Materials
Co(II)PoP was synthesized and characterized as described in the Supporting Information. The following other lipids were used: Dioleoylphosphatidylcholine (DOPC; Corden; catalog number: LP-R4–070), 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid)succinyl (Co-NTA-lipid; Avanti; catalog number: 791113), cholesterol (PhytoChol; Wilshire Technologies), synthetic PHAD (Avanti; catalog number: 699800P), and QS-21 (Desert King; catalog number: NC0949192). The following adjuvants were obtained: Alhydrogel 2 % aluminium gel (Accurate Chemical and Scientific Corporation; catalog number: A1090BS). Poly (I:C) (Sigma; catalog number: P1530). Granulocyte–macrophage colony-stimulating factor (GM-CSF) was obtained from Shenandoah Biotechnology (catalog number: 200–15-AF). Chlorpromazine hydrochloride was obtained from VWR (catalog number: TCC2481). Cytochalasin B was obtained from Acros (catalog number: 228090010). Lysosomes was obtained from Xeno tech (catalog number: H0610.L). 10x catabolic buffer was obtained from Xeno tech (catalog number: K5200). The following antibodies were obtained from BioLegend, APC-CD8a antibody (catalog number:100712), FITC-I-A/I-E antibody (catalog number:107605), FITC-B220 (catalog number:103206), FITC-CD4 antibody (catalog number:100405), PerCP/Cyanine5.5-CD44 (catalog number:103031), PE/Cy7-CD62L antibody (catalog number:104417), pacific blue IFN-γ (catalog number: 505818), PE-TNF-α (catalog number: 506305), Alexa Fluor 488-Ly6C (catalog number: 128021), PE/Cy7-CD11b (catalog number: 101215), PE-Ly6G (catalog number: 127607), APC/Cy7-CD11c (catalog number: 117323), PerCP/Cyanine5.5-CD3 (catalog number: 100217), Alex Fluor 700-I-A/I-E (catalog number: 107621). Brefeldin A (BD, catalog number: 555029), live/dead fixable dye (Invitrogen; catalog number: L34965), fixation/permeabilization kit (BD; catalog number: 554714). Cell lysis buffer was obtained from BioVision (catalog number: 5830). A5-HiLyte488 was synthesized by Anaspec. Other peptides were selected and synthesized by Genscript and characterized as described in the Supporting Information.
Vaccine preparation and characterization
Liposomes were prepared by ethanol injection and lipid extrusion as reported previously.26 The prepared liposomes were dialyzed in phosphate buffered saline (PBS) at 4 °C to remove ethanol and passed through a 0.2 μm sterile filter. For liposomes containing QS-21, QS-21 (1 mg/mL) was added to liposomes overnight at 4 °C with the [DOPC: Chol: CoPoP/PoP: PHAD: QS-21] mass ratio of [20:5:1:1:1]. The final liposome concentration was adjusted to 320 μg/mL CoPoP; we did not actually measure individual lipid concentrations, but operated on the assumption that the input concentration was maintained.
To prepare CPQ, CP, CQ, 2HP and 2HPQ vaccine, liposome and peptides were incubated at mass ratio of 4:1 for 1 hr at room temperature. To prepare PQ + C/A5 vaccine, A5 peptide was incubated with CoPoP liposomes (lacking PHAD or QS-21) for 1 hr, then 2HPQ liposome was added to the sample immediately before injection. For desired Ag dosing, liposomes were incubated with Ag, as described above, then diluted in PBS. To prepare Alhydrogel (Alum) vaccines, A5 was mixed with 2 % Alum for 1 hr and diluted with 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer before injection. Each vaccine contained 500 ng of peptide and 75 μg Alum. To prepare the poly(I:C) vaccine, the peptide was mixed with poly(I:C) for 1 hr and then further diluted in PBS for a dose of 500 ng peptide and 50 μg poly(I:C).
To characterize binding of liposomes and peptides, peptides were incubated with liposomes or PBS for 1 hr at room temperature and subjected to micro-centrifugal filtration tube with a 100 kDa cutoff (PALL; catalog number: 29300) to separate free peptide from liposomes. Micro bicinchoninic acid (BCA) (Thermo; catalog number: 23235) assay was used to determine the amount of free peptide in the filtrate. Dynamic light scattering with a NanoBrook 90 plus PALS instrument was used to measure sizes and polydispersity index of 500-fold diluted samples in PBS.
To measure fluorescent peptide binding, the fluorescence of the A5-HiLyte488 peptide was monitored with 491 nm excitation and 527 nm emission in a microplate reader (TECAN Safire). Upon binding to the liposomes, the fluorescence becomes quenched due to energy transfer from HiLyte488 to porphyrin of CPQ or 2HPQ. The percentage of fluorescence quenching of peptide was assessed by the fluorescence intensity in the test conditions compared to the free peptide. Binding kinetics were ascertained by monitoring the changes in fluorescence after incubating the same volume of peptide (80 μg/mL) with liposomes in PBS (320 μg/mL) or PBS. Samples were diluted 50 fold prior to measurement in the plate reader. To characterize the serum stability of vaccine, pre-prepared A5-HiLyte488 and liposome particles were incubated in 40 % human serum in PBS at 37 °C (0.8 μg/mL peptide concentration) and sample fluorescence was measured directly at different time points. For the in vitro release of peptide in lysosome study, lysosome solutions were prepared as described in the manufacture’s instruction. Briefly, in a 96 wells plate, 10 μL 10 × catabolic buffer was mixed with 50ul 1 × lysosome and 40 μL water. Prepared CPQ/A5-HiLyte488, CoNTA/A5-HiLyte488 or PBS/A5-HiLyte488 were added to lysosome solution and incubated at 37 °C at a final concentration of 0.8 μg/mL peptide. The fluorescence of the mixture was measured at indicated time points.
Cryo-electron microscopy
To analyze the morphology of CPQ liposomes before and after binding of A5 peptide, approximately 3.6 μL of each sample was applied to the holey carbon grids and manually blotted using the Vitrobot blotting paper (Standard Vitrobot Filter Paper, Ø55/20mm, Grade 595). Right after blotting, a new drop of the sample was applied to the EM grid and blotted again using the standard routine with the two blotting pads in the Vitrobot Mark IV (Thermo Fisher Scientific) for 3 sec and a blot force +1. The grid was then immediately plunged into liquid ethane. The Vitrobot was set at 25 °C and 100 % relative humidity. For all samples, we used c-flat grids (C-Flat 2/2–3Cu-T), which were washed with chloroform for 2 hr negative glow discharge in air at 5mA for 15 seconds right before the sample was applied for vitrification. Samples were imaged in a Tecnai F20 electron microscope operated at 200 kV using a side-entry Gatan 626 single tilt cryo-holder. Images were collected in a TVIPS XF416 CMOS camera at a magnification of 50,000x, which produced images with a calibrated pixel size of 2.145Å. Images were collected with a total dose of ~ 10 e−/Å2 using a defocus ranging from −1.75 to − 2.50 μm.
Cell studies
RAW264.7 murine macrophage cells were obtained from the American Type Culture Collection (ATCC) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10 % fetal bovine serum (FBS) and 1 % penicillin/streptomycin (pen/strep). CT26 colon cells were obtained from ATCC and cultured in RPMI 1640 with 10 % FBS and 1 % pen/strep. The 4T07 cell line was kindly provided by Dr. Josh Gamble (Karmanos Cancer Institute, Detroit, MI) and cultured in DMEM containing 10 % FBS and 1× Glutamine and 1 % pen/strep. CMS4-met cells were kindly provided by Dr. Abrams (Roswell Park, Buffalo, NY) and cultured in Roswell Park Memorial Institute (RPMI) 1640 media containing 10 % FBS and 1 % pen/strep. 4T1 cells were kindly provided by Dr. Yun Wu (University at Buffalo, Buffalo, NY) and cultured in RPMI 1640 with 10 % FBS and 1 % pen/strep. BMDCs were derived from bone marrow from the femurs and tibia of BALB/c mice. 107 cells/mL were cultured in 10 mL RPMI 1640 culture medium with 10 % FBS, 1 % pen/strep, and 20 ng/mL of recombinant murine GM-CSF. On day 3, an additional 10 mL media containing GM-CSF was added, so the final volume was 20 mL. On day 6, non-adherent cells were collected and cultured in a 24-well plate at 5 × 105 cell/mL in RPMI 1640 culture medium containing 10 % FBS and 1 % pen/strep. For the splenocyte studies, freshly isolated spleens were dissociated and filtered through a 70 μm cell strainer. The plunger from a sterile 3 mL syringe was used to dissociate tissue through the strainer, 5 mL of cold PBS was used to wash cells into a 50 mL tube. Cells were centrifuged at 500 × g for 5 min, the supernatants were discarded. Red blood cells were lysed with a 5 mL red blood cell lysis buffer for 5 min, then 35 mL PBS was added to the tube. Cells were centrifuged again and the cell pellets were collected for further use. Splenocytes were cultured in RPMI 1640 supplemented with 10 % FBS, 1 % pen/strep, 2 mM glutamine, 1 mM sodium pyruvate, 1× non-essential amino acids solution and 50 μM β-mercapethanol. Cells were cultured in 5 % CO2 / 95 % air at 37 °C in a humidified chamber.
For in vitro cell uptake studies, RAW264.7 cells (2.5 × 105 per well) and BMDCs (2.5 × 105 per well) were cultured in a 24-well plates overnight, then treated with CPQ/A5-HiLyte488, 2HPQ/A5-HiLyte488 and PBS/A5-HiLyte488 (peptide concentration of 1 μg/mL) for 10 min, 30 min or 1 hr. For phagocytosis and endocytosis inhibitor study, cells were first pre-incubated with cytochalasin B (10 μg/mL) or chlorpromazine (10 μg/mL) for 1 hr before the cell uptake study. Cells were washed and lysed with 0.1 % Triton X-100 and 10 mM dithiothreitol (DTT). The fluorescence signals were measured before and after adding DTT. Cellular A5-HiLyte488 uptakes were calculated by preparing an A5-HiLyte488 standard curve.
For HPLC of cell lysate, 1 × 106 RAW 264.7 murine macrophages were seeded in a T25 cell culture flask until confluent. CPQ/A5-HiLyte488 (peptide concentration of 2 μg/mL) or PBS was added to cell culture medium for the indicated hr. Cells were washed, lysed and centrifuged. Supernatant was collected and injected to a reversed phase HPLC column Agilent poroshell 120 EC-C18 (2.7 μm packing, 4.6×50 mm length). The mobile phase consisted of acetonitrile and 0.1 % Trifluoroacetic acid (TFA) in water and the method was 5 % to 60 % acetonitrile for 10 min at 1mL/min. The HPLC system consist of Agilent Technologies 1260 Infinity and a Diode-array detector (G1315C DAD VL+) set at 475 nm.
Murine studies
In vivo immunization: Murine studies were performed according to protocols approved by the University at Buffalo IACUC. 5–6 week-old female BALB/c mice (Charles River Laboratories, strain BALB/cAnNCrl) were immunized intramuscularly on the right hind leg. BALB/cJ mice (Jackson Laboratories) were used in this study only for DNA sequencing where indicated.
Tumor challenge: For the prophylactic vaccine tumor model, mice were vaccinated on day 0 and 7, and challenged on day 14. For testing the long-term protection of vaccine, mice were challenged on day 80. For the therapeutic vaccine tumor model, mice were inoculated with tumor cells subcutaneously on day 0, and then vaccinated with indicated vaccine on days 5 and 12. Tumor growth was monitored three times a week and tumor sizes were calculated by equation: Tumor volume = length × width2/2. Animals were euthanized when the tumor sizes reached 1 cm in diameter or when animals developed an ulceration. For the experimental lung metastasis tumor model, animals were injected intravenously via tail vein with tumor cells on day 0, then were left untreated or treated with intramuscular injection with the indicated vaccines on day 2 and 9 for the A5 vaccine studies or day 1 and 8 for the RragcL385P, Tmem5S71N or EML5G44R peptide screening studies. Lungs were excised and stained with Bouin’s solution (Sigma Catalog: HT10132) on day 18 for mice injected with CT26 cells and on day 16 for mice injected with 4T1 cells. Tumor nodules were counted manually and lung weights were measured.
Acute toxicity studies: 8-week-old female CD-1 mice (Envigo) were either untreated or injected with CPQ/A5 on days 0 and 7 intramuscularly, with doses of 0.5 μg A5 peptide, 2 μg CoPoP, 2 μg PHAD and 2 μg QS-21 per mouse. On day 14, anticoagulated blood and serum were collected for standard complete blood cell count and serum panel, 15 μL of blood was assessed by Heska Element HT5 Hematology Analyzer for complete blood cell count within 4 hr of blood collection. Serum was assessed by the Heska Element DC Chemistry Analyzer. Organs (heart, liver, spleen, lung, kidney) were fixed in formalin, stored in 70 % ethanol and subject to hematoxylin and eosin (H&E) staining and imaging as previously reported.26
IFN-γ ELISA: 2.5 × 105 splenocytes were seeded in a 96 wells plate and stimulated with 10 μg/mL antigens for 72 hr. 50 μL of supernatant was collected from each well and subjected to IFN-γ ELISA (Thermofisher; catalog: BMS606TEN) according to manufacturer’s protocol.
Antibody staining
For tetramer staining, H-2Ld-restricted AH1 (SPSYVYHQF) peptide was complexed with MHC-I (H-2Ld) and conjugated with PE by the NIH Tetramer core facility. For tetramer staining of PBMC, 60 μL of blood incubated with the AH1 tetramer for 1 hr at 4 °C (100 × dilution), then incubated with CD8a, MHC-II (I-A/I-E), B220, CD4, CD44 and CD62L antibodies for 30 min at 4 °C (1000 × dilution). Red blood cells were lysed by cell lysis buffer for 5 min then cells were centrifuged at 500 × g for another 5 min. The cell pellets were washed twice for flow cytometry analysis. For tetramer staining of splenocytes, 1 × 106 cells were incubated with tetramer and antibodies in the same condition as blood, and then washed twice for flow cytometry analysis. Flow cytometry studies were carried out using a BD LSRFortessa™ X-20 cytometer. Flowjo (version 10) software was used for data analysis.
For intracellular staining, 100 μL of 1 × 106 splenocytes were seeded in a flat bottom 96 wells plate and stimulated with 10 μg/mL antigen for 15–18 hr in the cell culture incubator. Then Brefeldin A was added to the plates with a dilution of 1000 × for another 5 hr. Cells were transferred to a round bottom 96 wells plate and centrifuged at 1350 rpm for 3 min, the cell pellets were washed twice and stained with 500 × live/dead fixable dye, 200 × diluted CD8a, 200 × CD4, 200 × diluted CD62L and 200 × diluted CD44 for 25 min at 4 °C with shaking. Cells were washed twice and fixed and permeabilized by fixation/permeabilization buffer for 20 min at 4 °C. Cells were washed twice with perm wash buffer and stained with 200 × diluted IFN-γ and 200 × diluted TNF-α for 30 min on ice. Cells were washed twice by perm/wash buffer for flow cytometry.
For cell recruitment studies, CD-1 mice were either untreated or injected intramuscularly with CPQ/A5 or CP/A5. 48 hr later, mice were euthanized and lymph nodes were collected for cell extraction. Cells were fixed with 4 % paraformaldehyde and washed, then stained with combination antibodies against Ly6C, CD11b, Ly6G, CD11c, CD3, I-A/I-E and F4/80 for 1 hr on ice. Cells were identified as reported previously.26
For immunofluorescence microscopy, silica beads were coated by shaking liposomes (containing 320 μg/mL PoP or CoPoP in addition to other components including fluorescent A5) with 25 mg/mL beads (Spherotech Silica Particles, 1.5–1.9 μm; catalog: SIP-15–10) for 10 min at 2000 rpm, followed shaking at 1200 rpm for 45 min. Free liposomes (in the supernatant) were removed by centrifugation at 1200 rcf for 2 min, and beads were washed twice with PBS in this manner. Glass coverslips were treated with 1 % Alcian blue for 10 min at 37 °C in the incubator, followed by 3 washes with PBS. 5 × 105 BMDCs were seeded on Alcian blue-treated glass coverslips for 30 min, then incubated with liposome-coated silica beads or uncoated silica beads for 3 hr. Cells were washed with PBS 3 times, then fixed with 4% paraformaldehyde (VWR; catalog: 30525-89-4) for 20 min at 4 °C. Slides were washed with PBS 3 times, followed by incubation with 5 % bovine serum albumin (BSA) in PBS with 0.1 % Triton X-100 (PBST) for 30 min at room temperature. Cells were incubated with 500 × diluted anti-mouse H-2Ld (Invitrogen; catalog number: PIMA170109) at 4 °C overnight, and washed with 5 % BSA in PBST 3 times, followed by incubating with 1000 × diluted Alexa Flour 555 anti-mouse secondary (Invitrogen; catalog number: A21137) for 30 min at room temperature. The slides were wash with PBST for 3 times, and stained with 500 × diluted anti-mouse LAMP-1 (Invitrogen; catalog number: 50-128-11) for 30 min at room temperature. After incubation, the slides were washed with PBST 3 times, then stained with 1000 × diluted Alexa Flour 647 chicken anti-rat IgG (Invitrogen catalog number: A21472) for 30 min at room temperature, then washed with PBST 3 times. Slides were stained with 4′,6-diamidino-2-phenylindole (DAPI) in anti-fade mounting medium (Vectashield; catalog number: H-1200). Images were acquired on a Zeiss LSM 710 Confocal Microscope.
For cytotoxic T lymphocyte (CTL) cytotoxicity assays, isolated splenocytes were cultured in cell culture medium and stimulated with murine IL-2 (Pepro Tech; catalog number: 212–12; 10 IU/mL) and antigens (10 μg/mL) for 5 days to use them as effector cells. 5000 CT26 cells were seeded in a 96 well plate and pulsed with 10 μg/mL antigens for 1 hr, then splenocytes were added to the plate at different E:T ratios for 5 hr. The cytotoxicity of splenocytes on CT26 cells was assessed by lactate dehydrogenase (LDH) release using a Non-Radioactive Cytotoxicity Assay Kit (Promega; catalog number: G1780) according to manufacturer instructions.
DNA sequencing of RragcL385P in CT26 and 4T1 cells
DNA was extracted using DNeasy Blood & Tissue kit (QIAGEN; catalog number: 69504) and PCR-amplified using forward primer TCACTGTTCACGTCTGTCCT and reverse primer ACTGAGTTCTGAGGTCTCT. 1.5 % agarose gels were used to purify DNA, and the DNA bands were cut and extracted using QIAquick Gel Extraction Kit (QIAGEN; catalog number: 28706). The quality and concentration of the isolated PCR products were measured using NanoDrop One (ThermoFisher). The purified DNA was sequenced by the Sanger sequencing method at the DNA Sequencing Core, Baylor College of Medicine, Houston, TX. Data was analyzed with Snapgene software.
Statistical analysis
Data were analyzed with Prism 8 (GraphPad Software) using the tests described in the figure captions. P values less than 0.05 were considered statistically significant. Values are generally reported as mean ± S.D. with the indicated sample size unless otherwise indicated.
Supplementary Material
Acknowledgements
The authors acknowledge assistance from Hsiao-Ju Lee with DNA sequencing. This study was supported by the National Institutes of Health grant # R01 CA247771 (to both JFL and SIA). Confocal microscopy and biological imaging core facility at the University at Buffalo supported by National Science Foundation major research instrumentation Grant # DBI 0923133. Cryo-EM images were collected at the Facility for Electron Microscopy Research (FEMR) at McGill University. FEMR is supported by the Canadian Foundation for Innovation, the Quebec government and McGill University. The NIH Tetramer Core Facility provided MHC-I tetramers.
Footnotes
Supporting Information Available:
Additional experimental data are in the supporting information.
Supporting Methods; Figure S1: Scheme for PoP synthesis. Figure S2: Scheme for CoPoP synthesis. Figure S3: 1H NMR of PoP. Table S1: 1H shifts and assignments for 1H NMR of PoP. Figure S4: 13C NMR of PoP. Figure S5: 1H NMR spectra of CoPoP. Table S2: 1H shifts and assignments for 1H NMR of CoPoP. Figure S6: 13C NMR spectra of CoPoP. Figure S7: Mass spectra of CoPoP. Figure S8: HPLC traces of PPa, PoP and CoPoP. Table S3: ICP analysis of CoPoP powder. Table S4: Representative UV-Vis absorption of PPa, PoP, and CoPoP. Table S5: Polydispersity and zeta potential of liposomes. Figure S9: Cryo-electron micrographs of CPQ liposomes with or without A5 peptide bound. Figure S10: Peptide binding to liposomes with different mass ratios. Figure S11: Gating of AH1-tet+ T cells in blood, TEM cells in AH1-tet+ cells and TEM cells in CD8+ T cells. Figure S12: CPQ/A5 vaccination elicits robust AH1 specific CD8+ T cell production in the spleen. Figure S13: The impact of PHAD, QS-21 and antigens in the same liposome. Figure S14: Gating of IFN-γ and TNF-α producing cells in the CD8+ T cell population in splenocytes. Figure S15: The effect of optional solubility tag EEE, RRR and ERR on 6his-A5 peptide. Figure S16: The effect of QS21 and antigen dosage to T cell production. Figure S17: Liposomes drain to lymph nodes rapidly. Figure S18: Vaccine injection at multiple sites increases number of lymph nodes targeted but does not increase T cell production. Figure S19: Macrophage uptake of CPQ/A5-HiLyte488 liposome. Figure S20: PoP lipids coated on silica beads. Figure S21: Liposome/peptide complexes were uptaken in BMDC and colocalize with LAMP-1 and H-2Ld. Figure S22: A5 with 2 his-tag released in lysosomes in vitro. Figure S23: Integrity of fluorescent A5 peptide uptaken by macrophages. Figure S24: Impact of EEE, RRR, ERR- tag and his-tag length on A5 on H-2Ld binding and inducing CD8+ T cells. Figure S25: Impact of his-tag length of A5 peptide on binding to liposome and immunogenicity. Figure S26: Effect of QS-21 and peptide dose on AH1-tet+ CD8+ T cells and IFN-γ production with CPQ/A5 immunization. Figure S27: CPQ as a platform to multiplex antigens. Figure S28: Library peptides bind to CPQ and 2HPQ. Figure S29: 5 library peptides and A5 peptide bind to CPQ and 2HPQ as multivalent vaccines. Figure 30: RragcL385P, Teme5S71N and Eml5G44R peptides bind to CPQ and 2HPQ liposomes. Figure S31: Cytotoxicity of splenocytes from CPQ/A5 and CPQ/RragcL385P vaccinated mice. Figure S32: DNA sequencing of Rragc385. Figure S33: The anti-tumor efficacy of CPQ/RragcL385P was confirmed again in BALB/c mice from Charles River Laboratory. Table S6: A5 peptides used in each figure. Table S7: Properties of A5 related peptides. Table S8: Properties of synthetic peptides for CT26 epitope screening.
References
- 1.Bezu L; Kepp O; Cerrato G; Pol J; Fucikova J; Spisek R; Zitvogel L; Kroemer G; Galluzzi L, Trial Watch: Peptide-Based Vaccines in Anticancer Therapy. Oncoimmunology 2018, 7, e1511506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuai R; Ochyl LJ; Bahjat KS; Schwendeman A; Moon JJ, Designer Vaccine Nanodiscs for Personalized Cancer Immunotherapy. Nat. Mater 2017, 16, 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murray AA; Wang C; Fiering S; Steinmetz NF, In Situ Vaccination with Cowpea vs Tobacco Mosaic Virus against Melanoma. Mol. Pharm 2018, 15, 3700–3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yildiz I; Shukla S; Steinmetz NF, Applications of Viral Nanoparticles in Medicine. Curr. Opin. Biotechnol 2011, 22, 901–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steinmetz NF, Viral Nanoparticles as Platforms for Next-Generation Therapeutics and Imaging Devices. Nanomedicine 2010, 6, 634–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fang RH; Hu C-MJ; Luk BT; Gao W; Copp JA; Tai Y; O’Connor DE; Zhang L, Cancer Cell Membrane-Coated Nanoparticles for Anticancer Vaccination and Drug Delivery. Nano Lett 2014, 14, 2181–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fang RH; Hu C-MJ; Zhang L, Nanoparticles Disguised as Red Blood Cells to Evade the Immune System. Expert Opin. Biol. Ther 2012, 12, 385–389. [DOI] [PubMed] [Google Scholar]
- 8.Jordan KR; McMahan RH; Kemmler CB; Kappler JW; Slansky JE, Peptide Vaccines Prevent Tumor Growth by Activating T Cells That Respond to Native Tumor Antigens. Proc. Natl. Acad. Sci. U.S.A 2010, 107, 4652–4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lynn GM; Sedlik C; Baharom F; Zhu Y; Ramirez-Valdez RA; Coble VL; Tobin K; Nichols SR; Itzkowitz Y; Zaidi N, Peptide–TLR-7/8a Conjugate Vaccines Chemically Programmed for Nanoparticle Self-Assembly Enhance CD8 T-Cell Immunity to Tumor Antigens. Nat. Biotechnol 2020, 38, 320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watson DS; Endsley AN; Huang L, Design Considerations for Liposomal Vaccines: Influence of Formulation Parameters on Antibody and Cell-Mediated Immune Responses to Liposome Associated Antigens. Vaccine 2012, 30, 2256–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cui Z; Han S-J; Vangasseri DP; Huang L, Immunostimulation Mechanism of LPD Nanoparticle as a Vaccine Carrier. Mol. Pharm 2005, 2, 22–28. [DOI] [PubMed] [Google Scholar]
- 12.Kraft JC; Freeling JP; Wang Z; Ho RJ, Emerging Research and Clinical Development Trends of Liposome and Lipid Nanoparticle Drug Delivery Systems. J. Pharm. Sci 2014, 103, 29–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwendener RA, Liposomes as Vaccine Delivery Systems: A Review of the Recent Advances. Ther. Adv. Vaccines 2014, 2, 159–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He X; Abrams SI; Lovell JF, Peptide Delivery Systems for Cancer Vaccines. Adv. Ther 2018, 1, 1800060. [Google Scholar]
- 15.Heuts J; Varypataki EM; van der Maaden K; Romeijn S; Drijfhout JW; van Scheltinga AT; Ossendorp F; Jiskoot W, Cationic Liposomes: A Flexible Vaccine Delivery System for Physicochemically Diverse Antigenic Peptides. Pharm. Res 2018, 35, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vasievich EA; Ramishetti S; Zhang Y; Huang L, Trp2 Peptide Vaccine Adjuvanted with (R)-DOTAP Inhibits Tumor Growth in an Advanced Melanoma Model. Mol. Pharm 2012, 9, 261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taneichi M; Ishida H; Kajino K; Ogasawara K; Tanaka Y; Kasai M; Mori M; Nishida M; Yamamura H; Mizuguchi J, Antigen Chemically Coupled to the Surface of Liposomes Are Cross-Presented to CD8+ T Cells and Induce Potent Antitumor Immunity. J. Immunol 2006, 177, 2324–2330. [DOI] [PubMed] [Google Scholar]
- 18.Farzad N; Barati N; Momtazi-Borojeni AA; Yazdani M; Arab A; Razazan A; Shariat S; Mansourian M; Abbasi A; Saberi Z, P435 HER2/neu-Derived Peptide Conjugated to Liposomes Containing DOPE as an Effective Prophylactic Vaccine Formulation for Breast Cancer. Artif. Cells Nanomed. Biotechnol 2019, 47, 664–672. [DOI] [PubMed] [Google Scholar]
- 19.Jacoberger-Foissac C; Saliba H; Seguin C; Brion A; Kakhi Z; Frisch B; Fournel S; Heurtault B, Optimization of Peptide-Based Cancer Vaccine Compositions, by Sequential Screening, Using Versatile Liposomal Platform. Int. J. Pharm 2019, 562, 342–350. [DOI] [PubMed] [Google Scholar]
- 20.Ghaffar KA; Marasini N; Giddam AK; Batzloff MR; Good MF; Skwarczynski M; Toth I, Liposome-Based Intranasal Delivery of Lipopeptide Vaccine Candidates Against Group A Streptococcus. Acta Biomater 2016, 41, 161–168. [DOI] [PubMed] [Google Scholar]
- 21.Khongkow M; Liu T-Y; Bartlett S; Hussein WM; Nevagi R; Jia Z; Monteiro MJ; Wells J; Ruktanonchai UR; Skwarczynski M, Liposomal Formulation of Polyacrylate-Peptide Conjugate as a New Vaccine Candidate against Cervical Cancer. Prec. Nanomed 2018, 1, 183–193. [Google Scholar]
- 22.Altin JG; van Broekhoven CL; Parish CR, Targeting Dendritic Cells with Antigen-Containing Liposomes: Antitumour Immunity. Expert Opin. Biol. Ther 2004, 4 (11), 1735–1747. [DOI] [PubMed] [Google Scholar]
- 23.Gargett T; Abbas MN; Rolan P; Price JD; Gosling KM; Ferrante A; Ruszkiewicz A; Atmosukarto II; Altin J; Parish CR, Phase I Trial of Lipovaxin-MM, a Novel Dendritic Cell-Targeted Liposomal Vaccine for Malignant Melanoma. Cancer Immunol. Immunother 2018, 67, 1461–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shao S; Huang W-C; Lin C; Hicar MD; LaBranche CC; Montefiori DC; Lovell JF, An Engineered Biomimetic MPER Peptide Vaccine Induces Weakly HIV Neutralizing Antibodies in Mice. Ann. Biomed. Eng 2019, 48, 1991–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Federizon J; Feugmo CGT; Huang W-C; He X; Miura K; Razi A; Ortega J; Karttunen M; Lovell JF, Experimental and Computational Observations of Immunogenic Cobalt Porphyrin Lipid Bilayers: Nanodomain-Enhanced Antigen Association. Pharmaceutics 2021, 13 (1), 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang W-C; Deng B; Lin C; Carter KA; Geng J; Razi A; He X; Chitgupi U; Federizon J; Sun B; Long CA; Ortega J; Dutta S; King CR; Miura K; Lee S-M; Lovell JF, A Malaria Vaccine Adjuvant Based on Recombinant Antigen Binding to Liposomes. Nat. Nanotechnol 2018, 13, 1174–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang W-C; Deng B; Seffouh A; Ortega J; Long CA; Suresh RV; He X; Miura K; Lee S-M; Wu Y, Antibody Response of a Particle-Inducing, Liposome Vaccine Adjuvant Admixed with a Pfs230 Fragment. NPJ vaccines 2020, 5, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang W-C; Deng B; Mabrouk MT; Seffouh A; Ortega J; Long C; Miura K; Wu Y; Lovell JF, Particle-Based, Pfs230 and Pfs25 Immunization Is Effective, but Not Improved by Duplexing at Fixed Total Antigen Dose. Malaria J 2020, 19 (1), 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Federizon J; Frye A; Huang W-C; Hart TM; He X; Beltran C; Marcinkiewicz AL; Mainprize IL; Wills MK; Lin Y-P, Immunogenicity of the Lyme Disease Antigen OspA, Particleized by Cobalt Porphyrin-Phospholipid Liposomes. Vaccine 2020, 38, 942–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang W-C; Zhou S; He X; Chiem K; Mabrouk MT; Nissly RH; Bird IM; Strauss M; Sambhara S; Ortega J; Wohlfert EA; Martinez-Sobrido L; Kuchipudi SV; Davidson BA; Lovell JF, SARS-CoV-2 RBD Neutralizing Antibody Induction Is Enhanced by Particulate Vaccination. Adv. Mater 2020, 32, 2005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tran E; Turcotte S; Gros A; Robbins PF; Lu Y-C; Dudley ME; Wunderlich JR; Somerville RP; Hogan K; Hinrichs CS, Cancer Immunotherapy Based on Mutation-Specific CD4+ T Cells in a Patient with Epithelial Cancer. Science 2014, 344, 641–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kreiter S; Vormehr M; van de Roemer N; Diken M; Löwer M; Diekmann J; Boegel S; Schrörs B; Vascotto F; Castle JC; Tadmor AD; Schoenberger SP; Huber C; Türeci Ö; Sahin U, Mutant MHC Class II Epitopes Drive Therapeutic Immune Responses to Cancer. Nature 2015, 520 (7549), 692–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Melief CJM; van der Burg SH, Immunotherapy of Established (Pre) Malignant Disease by Synthetic Long Peptide Vaccines. Nat. Rev. Cancer 2008, 8 (5), 351–360. [DOI] [PubMed] [Google Scholar]
- 34.Slansky JE; Rattis FM; Boyd LF; Fahmy T; Jaffee EM; Schneck JP; Margulies DH; Pardoll DM, Enhanced Antigen-Specific Antitumor Immunity with Altered Peptide Ligands That Stabilize the MHC-Peptide-TCR Complex. Immunity 2000, 13, 529–538. [DOI] [PubMed] [Google Scholar]
- 35.Jenkins NA; Copeland N; Taylor B; Lee B, Organization, Distribution, and Stability of Endogenous Ecotropic Murine Leukemia Virus DNA Sequences in Chromosomes of Mus Musculus. J. Virol 1982, 43, 26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scrimieri F; Askew D; Corn DJ; Eid S; Bobanga ID; Bjelac JA; Tsao ML; Allen F; Othman YS; Wang S-CG; Huang AY, Murine Leukemia Virus Envelope Gp70 Is a Shared Biomarker for the High-Sensitivity Quantification of Murine Tumor Burden. Oncoimmunology 2013, 2, e26889–e26889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jordan KR; McMahan RH; Oh JZ; Pipeling MR; Pardoll DM; Kedl RM; Kappler JW; Slansky JE, Baculovirus-Infected Insect Cells Expressing Peptide-MHC Complexes Elicit Protective Antitumor Immunity. J. Immunol 2008, 180 (1), 188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goodwin TJ; Huang L, Investigation of Phosphorylated Adjuvants Co-Encapsulated with a Model Cancer Peptide Antigen for the Treatment of Colorectal Cancer and Liver Metastasis. Vaccine 2017, 35, 2550–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang W; Xu L; Park H-B; Hwang J; Kwak M; Lee PCW; Liang G; Zhang X; Xu J; Jin J-O, Escherichia Coli Adhesion Portion Fimh Functions as an Adjuvant for Cancer Immunotherapy. Nat. Commun 2020, 11, 1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kang TH; Mao CP; Lee SY; Chen A; Lee JH; Kim TW; Alvarez RD; Roden RB; Pardoll D; Hung CF; Wu TC, Chemotherapy Acts as an Adjuvant to Convert the Tumor Microenvironment into a Highly Permissive State for Vaccination-Induced Antitumor Immunity. Cancer Res 2013, 73, 2493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moynihan KD; Opel CF; Szeto GL; Tzeng A; Zhu EF; Engreitz JM; Williams RT; Rakhra K; Zhang MH; Rothschilds AM; Kumari S; Kelly RL; Kwan BH; Abraham W; Hu K; Mehta NK; Kauke MJ; Suh H; Cochran JR; Lauffenburger DA et al. , Eradication of Large Established Tumors in Mice by Combination Immunotherapy That Engages Innate and Adaptive Immune Responses. Nat. Med 2016, 22, 1402–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Didierlaurent AM; Laupèze B; Di Pasquale A; Hergli N; Collignon C; Garçon N, Adjuvant System AS01: Helping to Overcome the Challenges of Modern Vaccines. Expert Rev. Vaccines 2017, 16, 55–63. [DOI] [PubMed] [Google Scholar]
- 43.Beck Z; Matyas GR; Alving CR, Detection of Liposomal Cholesterol and Monophosphoryl Lipid a by QS-21 Saponin and Limulus Polyphemus Amebocyte Lysate. Biochim. Biophys. Acta 2015, 1848, 775–780. [DOI] [PubMed] [Google Scholar]
- 44.Liu J; Harms M; Garamus VM; Müller-Goymann CC, Reentrant Structural Phase Transition in Amphiphilic Self-Assembly. Soft Matter 2013, 9, 6371–6375. [Google Scholar]
- 45.Garçon N; Chomez P; Van Mechelen M, Glaxosmithkline Adjuvant Systems in Vaccines: Concepts, Achievements and Perspectives. Expert Rev. Vaccines 2007, 6, 723–739. [DOI] [PubMed] [Google Scholar]
- 46.Ryan MH; Bristol JA; McDuffie E; Abrams SI, Regression of Extensive Pulmonary Metastases in Mice by Adoptive Transfer of Antigen-Specific CD8(+) CTL Reactive against Tumor Cells Expressing a Naturally Occurring Rejection Epitope. J. Immunol 2001, 167, 4286–92. [DOI] [PubMed] [Google Scholar]
- 47.Didierlaurent AM; Collignon C; Bourguignon P; Wouters S; Fierens K; Fochesato M; Dendouga N; Langlet C; Malissen B; Lambrecht BN, Enhancement of Adaptive Immunity by the Human Vaccine Adjuvant AS01 Depends on Activated Dendritic Cells. J. Immunol 2014, 193, 1920–1930. [DOI] [PubMed] [Google Scholar]
- 48.Nair-Gupta P; Baccarini A; Tung N; Seyffer F; Florey O; Huang Y; Banerjee M; Overholtzer M; Roche PA; Tampé R, TLR Signals Induce Phagosomal MHC-I Delivery from the Endosomal Recycling Compartment to Allow Cross-Presentation. Cell 2014, 158, 506–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mantegazza AR; Magalhaes JG; Amigorena S; Marks MSJT, Presentation of Phagocytosed Antigens by MHC Class I and II. Traffic 2013, 14, 135–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Castle JC; Loewer M; Boegel S; de Graaf J; Bender C; Tadmor AD; Boisguerin V; Bukur T; Sorn P; Paret C, Immunomic, Genomic and Transcriptomic Characterization of CT26 Colorectal Carcinoma. BMC genomics 2014, 15, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim K; Skora AD; Li Z; Liu Q; Tam AJ; Blosser RL; Diaz LA; Papadopoulos N; Kinzler KW; Vogelstein B, Eradication of Metastatic Mouse Cancers Resistant to Immune Checkpoint Blockade by Suppression of Myeloid-Derived Cells. Proc. Natl. Acad. Sci. U.S.A 2014, 111, 11774–11779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Castle JC; Loewer M; Boegel S; Tadmor AD; Boisguerin V; De Graaf J; Paret C; Diken M; Kreiter S; Türeci Ö, Mutated Tumor Alleles Are Expressed According to Their DNA Frequency. Sci. Rep 2014, 4, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.