


Abstract
Alterations of the DNA methylation pattern have been related to generalized chromosomal disruption and inactivation of multiple tumor suppressor genes in neoplasia. To screen for tumor-specific alterations and to make a global assessment of methylation status in cancer cells, we have modified the methylated CpG island amplification method to generate easily readable fingerprints representing the cell’s DNA methylation profile. The method is based on the differential cleavage of isoschizomers with distinct methylation sensitivity. Specific adaptors are ligated to the methylated ends of the digested genomic DNA. The ligated sequences are amplified by PCR using adaptor- specific primers extended at the 3′ end with two to four arbitrarily chosen nucleotidic residues to reduce the complexity of the product. Fingerprints consist of multiple anonymous bands, representing DNA sequences flanked by two methylated sites, which can be isolated and individually characterized. Hybridization of the whole product to metaphase chromosomes revealed that most bands originate from the isochore H3, which identifies the regions of the genome with the highest content of CpG islands and genes. Comparison of the fingerprints obtained from normal colon mucosa, colorectal carcinomas and cell lines revealed tumor-specific alterations that are putative recurrent markers of the disease and include tumor-specific hypo- and hypermethylations.
INTRODUCTION
Methylation of DNA is an epigenetic modification that plays an important role in the control of gene expression and chromosome structure in mammalian cells (reviewed in 1). Cytosine methylation of the CpG dinucleotide sequence is the most common form of post-replicative modification of genomic DNA in higher eukaryotes and is the driving force behind genomic imprinting (2) and X-chromosome inactivation (3). It also plays a crucial role in the earliest stages of embryogenesis (4). It has been proposed that the term methylome be used to refer to the complete set of DNA methylation modifications of a cell (5). Three to five percent of the cytosine residues in mammalian genomic DNA appear in the form of 5-methylcytosine (5mC) (4). Most of the 5mC (70–80%) is found in CpG-rich sequences (6), but neither 5mC distribution nor the spatial distribution of the CpG dinucleotide itself is random (7). In general, the CpG dinucleotide is greatly under-represented throughout the mammalian genome but it can be found at close to its expected frequency in small genomic regions (200 bp to a few kb), known as CpG islands (8). These areas are ‘protected’ from methylation [with the exception of CpG islands on the inactive X chromosome in females (9)] and are located in the proximal promoter regions of ∼40–50% of human genes (7).
Cancer cells are characterized by the accumulation of both genetic and epigenetic changes. Widespread genomic hypomethylation has been associated with genomic disruption and chromosomal instability (10). Alternatively, concomitant hypermethylation of specific DNA regions appears associated with the inactivation of common tumor suppressor genes (11,12). Therefore, the screening for differentially methylated sequences in tumors appears as a key tool to further understand the molecular mechanisms underlying malignant transformation of cells. Based on the methylated CpG island amplification (MCA) method (13), we have developed a modified approach to obtain DNA fingerprints representative of the methylome. This approach allows the adjustment of the fingerprint’s complexity and allows the identification of hypo- (loss of a band) and hypermethylations (appearance of a new band). This method, named amplification of inter-methylated sites (AIMS), has been evaluated for reproducibility and sensitivity and, in a preliminary setting, has been applied to the analysis of differential methylation in normal colon mucosa, paired colorectal carcinomas and colon cancer cell lines.
MATERIALS AND METHODS
Tissues and cell lines
Twenty colorectal carcinomas and their paired non-adjacent areas of normal colonic mucosa were collected simultaneously as fresh specimens and snap-frozen within 2 h of removal and then stored at –80°C. All samples were obtained from the Hospital de la Santa Creu i Sant Pau (Barcelona, Spain). Human colorectal carcinoma cell lines (HCT116, DLD-1, LoVo, SK-Co1, SW 480 and HT 29) were obtained from the American Type Culture Collection (ATCC; Manassas, VA).
Amplification of inter-methylated sites
A schematic diagram of the method is outlined in Figure 1. Procedures for DNA digestion and adaptor ligation were based on the MCA method (13). Briefly, 1 µg of genomic DNA was digested with 20 U of the methylation-sensitive restriction endonuclease SmaI (Amershan Pharmacia Biotech, Buckinghamshire, UK) for 16 h at 30°C, which cleaves leaving blunt ends (CCC/GGG). Subsequently, the DNA was digested with 4 U of PspAI (Stratagene, La Jolla, CA) for 6 h at 37°C, which leaves sticky ends (C/CCGGG). The adaptors were prepared by incubation of the oligonucleotides MCF (5′-CCGGTCAGAGCTTTGCGAAT-3′) and Blue (5′-ATTCGCAAAGCTCTGA-3′) at 65°C for 2 min, followed by cooling to room temperature for 30–60 min. One microgram of digested DNA was ligated to 2 nmol of adaptor using T4 DNA ligase (New England Biolabs, Boston, MA). The product of ligation was purified using the GFX Kit (Amershan Pharmacia Biotech) and eluted into 250 µl of water. In a second step, PCR amplification of sequences flanked by adaptors was performed using different primers or sets of primers corresponding to the Blue primer extended with the CCGGG (overhanging end) plus 1–4 arbitrarily chosen nucleotides.
Figure 1.

Schematic diagram of the AIMS technique. Genomic DNA is represented by a solid line, with seven CCCGGG recognition sites. Non-methylated (white boxes) and methylated sites (black boxes) are depicted. The non-methylated sites are cut in a first digestion using the methylation-sensitive SmaI restriction endonuclease, which leaves blunt ends. A second digestion is performed using the isoschizomer PspAI which leaves a CCGG overhang. Adaptors are ligated to the sticky ends. DNA fragments flanked by two ligated adaptors are amplified by PCR using specific primers that hybridize to the adaptor sequence plus the restriction site and 1 or more additional nucleotides that are arbitrarily chosen. This allows the amplification of a limited number of sequences that typically range from ∼200 to ∼2000 bp. Illustrative examples of fingerprints generated using primers extended with 1–3 nt are shown.
Primers extended with 2–4 arbitrary nucleotides produced AIMS fingerprints of moderate complexity and were selected for the analysis of samples. Readable band-rich fingerprints were also obtained by combining two different primers in each PCR. The following primer sets were used (3′ arbitrarily chosen nucleotides are shown underlined): Blue-CCGGG-CTA + Blue-CCGGG-TGG (set A), Blue-CCGGG-CTG + Blue-CCGGG-TGG (set B), Blue-CCGGG-CGCG + Blue-CCGGG-CAAC (set C), Blue-CCGGG-TG (set D).
PCR was performed using 1.5 µl of each ligated DNA (∼6 ng) as template in a 25 µl vol containing 1.1 µM of each primer, 2 U Taq polymerase (Boehringer Mannheim, Mannheim, Germany), 125 µM of each dNTP, 1 µCi [α-33P]dATP (Amersham) and PCR buffer (10 mM Tris–HCl pH 8.0, 50 mM potassium chloride, 1.5 mM magnesium chloride, 1.5 mM MgCl2). PCRs with primer sets A and B consisted of 30 two-step cycles (15 s at 94°C, 1 min 15 s at 74°C). PCR with primer set C consisted of 30 three-step cycles (15 s at 94°C, 45 s at 68°C and 1 min at 72°C). PCR cycles were preceded by denaturation for 1 min at 95°C and ended with an extension step of 5 min at 72°C. Reactions were performed in a Programmable Thermal Controller PTC100 (MJ Research Inc., Watertown, MA). The PCR products were diluted 1/4 in formamide dye buffer, denatured for 3 min at 95°C and 3 µl was run on a 6% polyacrylamide 8 M urea sequencing gel at 55 W for 5 h. The gels were dried under vacuum at 85°C and exposed to an X-ray film at room temperature without an intensifier screen for 3–6 days.
Differences in the intensity of bands between the tumor and its paired normal tissue were ascertained by direct eye inspection of the film. Densitometric analysis was considered unnecessary in normal–tumor comparisons since only reproducible and clear changes were taken into account. Bands appearing in the tumor but not in the normal tissue were interpreted as hypermethylations. Conversely, relative decreases in the intensity of a band in the tumor with respect to its paired normal tissue fingerprint were considered a symptom of hypomethylation. Homozygous loss and gene amplification may also produce dramatic changes in the display of a band. Therefore, this possibility, although uncommon, should also be considered in the interpretation of the differential representation. Assays for reproducibility were performed. The sensitivity and accuracy of the technique were determined by analysis of genomic DNA from a tumor tissue diluted in genomic DNA of normal tissue from the same patient. Variations in band intensity in the different DNA dilutions were assessed by densitometry using Phoretix 1D v.3.0 software (Nonlinear Dynamics, Newcastle upon Tyne, UK).
Isolation and sequencing of DNA fragments
Bands appearing differentially methylated were excised from dried polyacrylamide gels, and DNA was reeluted in 50 µl of sterile H2O (80°C, 10 min) (14). PCR with the same primers and conditions used in the AIMS experiment was performed to amplify the fragment. PCR products were cloned into plasmid vectors using the TA cloning kit (Invitrogen, Groningen, The Netherlands). Automatic sequencing of multiple colonies was performed to ascertain the unique identity of the isolated band. Sequence homologies were searched using the BLAST engine available at www.ncbi.nlm.nih.gov.
Competitive hybridization of AIMS products to metaphase chromosomes
To determine the origin and chromosomal distribution of sequences generated by this approach, normal tissue DNA was used for an unrestrained AIMS performed with the Blue primer for amplification. AIMS product was labeled with SpectrumRed and whole genomic DNA was labeled with SpectrumGreen. The mixture was hybridized to metaphase chromosomes analogously to the comparative genomic hybridization method (15). Procedures were performed as described earlier (16). Genomic DNA was also digested with PspAI alone (without previous digestion with SmaI), ligated and ampified as in AIMS to determine the specificity of the AIMS technique for the amplification of inter-methylated sequences. The product was amplified with the Blue primer, labeled and hybridized to metaphase chromosomes (against whole genomic DNA) as described above.
Bisulfite genomic sequencing
Confirmation of differential methylation was performed by sequencing of PCR-amplified genomic DNA after bisulfite treatment as previously described (17). Templates for sequencing were generated by specific PCRs that included the AIMS bands and flanking regions. Primer sequences and assay conditions are available from the authors.
Calculation of the relative index of methylation
To determine the relative hypo- and hypermethylation of genomic DNA in tumor tissue, a ratio considering the number of bands with decreases and increases of intensity in the tumor with regard to the paired normal tissue was calculated and related to the total number of comparable bands. In cell lines the index of methylation was referred to the fingerprints obtained from six normal mucosas randomly selected. In this case, the analysis was limited to bands showing no variation among the normal tissue of different individuals.
RESULTS
Setting up and technical evaluation of AIMS
The usefulness of AIMS lies in two properties of the method: the generation of a high number of sequences representing two close methylated CpGs and the feasibility of reducing the complexity of the product to obtain readable fingerprints. Rich-band patterns resolved on sequencing gels are easily analyzed and interpreted, making suitable the study of a high number of arbitrarily chosen tags in large series of samples. The number of nucleotides added to the 3′ end of the primer hybridizing to the adaptor greatly conditions the complexity of the product as shown in Figure 1. Under the assay conditions described here, amplified bands ranged in size from ∼200 to ∼2000 bp, although the type of electrophoresis gels used in our setting precluded the resolution of fragments above ∼1200 bp. Primers offering the most readable fingerprints (that is more than 50 clearly identifiable bands) in conventional sequencing gels were those containing 3 or 4 additional nucleotides and when used in pairs. Assuming perfect matching, each additional nucleotide implies a reduction of complexity (1/5 for GC and 3/10 for AT according to frequency of the nucleotide in human genome), but to some point, even with the very stringent conditions used in our setting, AIMS is likely to behave as the arbitrarily primed PCR (18), and therefore some degree of flexibility in the initial cycles may allow the amplification of a higher number of sequences than expected.
The reproducibility of AIMS was examined by duplicate analysis of 19 normal–tumor DNA pairs obtained from colorectal cancer patients. An illustrative example is shown in Figure 2. The pattern was highly reproducible and most normal–tumor differences could be detected in both replicates. Some bands exhibited an individual-specific display (appeared in both normal and tumor tissue of some individuals but not in other) suggesting its polymorphic nature (data not shown). Fingerprints obtained with different amounts of DNA template (2–20 ng) were also comparable (data not shown), indicating that the PCR amplification normalizes the amplification ratio of bands within a wide range of initial template concentrations, probably due to the competitive nature of the AIMS.
Figure 2.

AIMS of normal–tumor (N and T, respectively) DNA pairs. Analysis was performed in duplicate. Two fingerprints generated with different sets of primers are shown. Band profiles were highly reproducible. Illustrative losses of band intensity (indicative of hypomethylation) and gains (indicative of hypermethylation) are marked with arrowheads pointing down and up, respectively.
Next we sought the sensitivity and accuracy of AIMS for detection of methylated sequences. Genomic DNA from a tumor tissue exhibiting a novel hypermethylated sequence was diluted in genomic DNA of normal tissue from the same patient. The AIMS assay clearly revealed the presence of the tumor-specific band at dilutions of up to 128-fold (Fig. 3). This implies that the technique is able to detect hypermethylated sequences even if they are present in <1% of the cells. The response was proportional to the dilution factor, demonstrating the quantitative nature of the method. Hypomethylations rarely appear as the complete absence of a band due to the almost unavoidable presence of contaminating normal cells in tumor specimens and band signal saturation. Besides this limitation, decreases of intensity attributable to hypomethylation were detectable in tumor DNA diluted 16-fold in normal tissue DNA (Fig. 3).
Figure 3.

To determine the sensitivity of the method, the DNA of a tumor was diluted in its paired normal DNA (dilution factor of tumor DNA is shown on top of each lane) and submitted to AIMS (left). Densitometric analysis of selected bands (right) demonstrated that amplification of the sequences with differential representation was proportional to the dilution factor. Band intensity is shown in arbitrary units (y-axis). According to this experiment, the sensitivity to detect hypermethylations was estimated in the range of 1/128th (up arrowhead). Hypomethylations (loss of band) also showed a quantitative response, but under the conditions described here, the sensitivity was limited to 16-fold (down arrowhead).
Chromosomal origin of AIMS products
Competitive hybridization to metaphase chromosomes of an AIMS product and whole genomic DNA yielded a characteristic pattern, indicative of the uneven distribution of amplified sequences (Fig. 4). Telomeric regions in most chromosomes and interstitial bands at 3p, 6p, 7q, 11 and 12 showed strong AIMS signal. It is noteworthy that chromosomes 17 and 19 exhibited the highest density of AIMS hybridization, in contrast with chromosome 18 that was mainly painted by genomic DNA. The hybridization of the amplified product obtained from a genomic DNA digested with PspAI alone (sequences flanked by CCCGGG sites irrespective of their methylation status) resulted in the homogeneous staining of all chromosomes (data not shown). These results indicate that the striking chromosome localization of AIMS products is determined by the distribution of methylated CCCGGG sites.
Figure 4.

Chromosomal procedence of AIMS products obtained from normal tissue DNA was ascertained by competitive hybridization to metaphase chromosomes. AIMS products (shown in red) displayed higher densities at most telomeric regions and some interstitial bands with chromosomes 17 and 19 showing very high signal. Competitive genomic DNA is shown in green.
Analysis of colorectal tumors and paired normal mucosa and cell lines
Nineteen of the twenty normal–tumor pairs produced readable fingerprints with all primers. The reiterative failure of a sample was attributed to poor DNA quality. Only AIMS performed with primers extended with 3–4 arbitrary nucleotides were considered for overall methylation analysis due to the ease band matching of fingerprints. Three different AIMS experiments yielded a total of 168 anonymous tags (set A, 64; set B, 52; set C, 52) that were evaluated for normal–tumor differential representation. Due to polymorphic display or variable resolution power of some gel electrophoreses, the total number of informative bands per case ranged from 157 to 167 (mean 164 ± 5). In this study we have considered only changes showing dramatic variations in band intensity, because these are more likely to reflect tumor-wide alterations. Twenty-nine bands displayed losses, sixteen gains, and six both types of changes in one or more tumors when compared with the paired normal mucosa. When data were analyzed by case, both losses and gains were observed in most tumors (Table 1) and appeared to be independently distributed (P = 0.682). On average, 1.35 ± 1.33 (range 0–3.73%) of the informative bands were lost in the tumor in contrast with 1.03 ± 0.95% (range 0–3.01%) that were gained.
Table 1. Rates of differential methylation in colorectal carcinomas.
| Case ID | Age | Sex | Dukes’ stage | Informative bands | Gainsa | Lossesb | No. of changes |
|---|---|---|---|---|---|---|---|
| 53 | 70 | F | B | 167 | 0 | 0 | 0 |
| 65 | 72 | M | B | 167 | 0 | 0 | 0 |
| 100 | 66 | F | C | 167 | 1 | 6 | 7 |
| 101 | 81 | M | B | 161 | 4 | 6 | 10 |
| 110 | 68 | F | C | 166 | 5 | 0 | 5 |
| 122 | 52 | M | A | 168 | 4 | 3 | 7 |
| 134 | 74 | F | C | 168 | 3 | 1 | 4 |
| 163 | 85 | F | B | 161 | 3 | 1 | 4 |
| 164 | 83 | M | D | 168 | 2 | 1 | 3 |
| 190 | 35 | F | C | 168 | 1 | 1 | 2 |
| 194 | 81 | M | B | 168 | 0 | 3 | 3 |
| 195 | 81 | M | B | 168 | 0 | 3 | 3 |
| 253 | 53 | F | A | 165 | 1 | 1 | 2 |
| 259 | 75 | M | B | 167 | 3 | 6 | 9 |
| 326 | 96 | F | B | 157 | 1 | 0 | 1 |
| 332 | 72 | F | A | 157 | 1 | 0 | 1 |
| 333 | 58 | M | B | 157 | 2 | 2 | 4 |
| 336 | 65 | F | A | 157 | 0 | 5 | 5 |
| 340 | 70 | M | C | 157 | 1 | 3 | 4 |
aNo. of bands appearing in the tumor but not in the paired normal tissue.
bNo. of bands with diminished intensity in the tumor as compared with the paired normal tissue.
AIMS profiles of cell lines were compared against consensus fingerprints obtained from five normal colon mucosa tissues (Fig. 5). Comparisons are not as accurate as in primary tumors because inter-individual polymorphisms may be confounded with tumor-specific changes. Therefore, only bands appearing in all normal tissues were considered for analysis (n = 136). All cell lines showed losses (hypomethylations) that affected up to one-third (LoVo, 32%; HT29, 21%; HCT116, 16%; DLD-1, 25%; SW480, 21%) of the bands present in normal tissues. Novel bands also appeared in all cell lines and some of them were coincidental with that showing gains in primary tumors, but due to the lack of matching normal tissue potential hypermethylations have not been considered.
Figure 5.
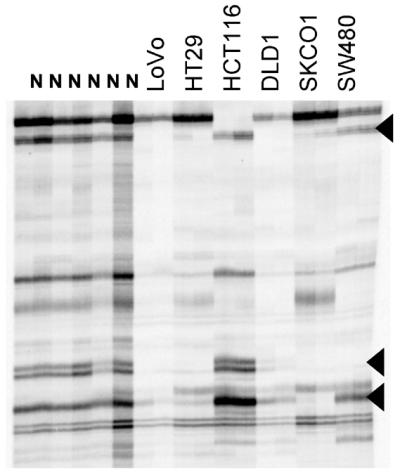
AIMS profiles of six normal tissue and six human colon cancer cell lines (duplicate analysis). Arrowheads indicate some of the bands present in all normal tissues and lost in one or more cell lines.
Identification of DNA fragments showing differential methylation in colorectal carcinomas
Eleven bands showing the different status of methylation between normal and tumor pairs were cloned and sequenced. A homology search revealed the putative identity of the sequence of DNA containing the inter-methylated CpG sites. The restriction site CCCGGG and the arbitrary nucleotides added to the 3′ end of the amplification primer were identified in all sequences and the size of the predicted DNA fragment was coincidental with that obtained in AIMS. Many, but not all, cloned bands displayed a high CpG content and a predicted CpG island lay within or very near the amplified sequence (Table 2). Five sequences contained an Alu or part of an Alu repeat, but in all cases non-repetitive sequences were also present. Therefore, it was always possible the assignment of a unique origin in the human genome map. Bisulfite sequencing of DNA fragments containing some of the bands detected by AIMS confirmed the identity of the band display and methylation of the SmaI site (Fig. 6).
Table 2. Characterized AIMS bands.
| Band ID | AIMS primersa | Band size (bp) | %GC | CpG/GpC ratio | Chromosome map | Blast homology (region)b | Gene | Alu repeat | Methylation status in tumorsc |
|---|---|---|---|---|---|---|---|---|---|
| 33.1 | TG | 332 | 59 | 0.17 | 5q35 | NT 023244 (10736–11067) | LOC133632 | No | Hypermethylated |
| 33.4 | TG | 994 | 49 | 0.19 | 18q22 | NT 025028 (665126–666119) | LOC125710 | No | Hypermethylated |
| 33.7 | TG | 984 | 56 | 0.22d | 19q13 | NT 011128 (188906–189889) | GRIK5 | No | Hypermethylated |
| Z | CTG-TGG | 197 | 47 | 0.64d | 2q14 | NT 005112 (1496215–1496411) | None | No | Hypermethylated |
| S7 | CTG-TGG | 260 | 56 | 0.35 | 13q12 | NT 009888 (1372721–1372980) | LOC121757 | Yes | Hypermethylated |
| T8 | CTG-TGG | 232 | 63 | 0.31 | 17q21 | NT 010748 (707670–707901) | LOC124789 | No | Variable |
| O | CTG-TGG | 351 | 57 | 0.43d | 19p13 | NT 030863 (53094–53444) | None | Yes | Variable |
| LL1 | CTA-TGG | 355 | 54 | 0.74d | 9q33 | NT 017568 (4599694–4600050) | LOC138090 | Yes | Variable |
| R | CTA-TGG | 287 | 55 | 0.37 | 11q13 | NT 008992 (1630079–1630365) | None | No | Variable |
| D2 | CGCG-CAAC | 296 | 45 | 0.40d | 18q11 | NT 010966 (1001383–1001678) | FLJ21610 | Yes | Hypomethylated |
| N3 | CGCG-CAAC | 349 | 43 | 0.38d | Xq13 | NT 030884 (340496–340844) | LOC139929 | Yes | Hypomethylated |
aPrimers used for restricted amplification consisted of the Blue sequence extended at the 3′ end with CCGGG and 2–4 nucleotides to restrain product complexity. A single primer (with 2 extended nucleotides) or combinations of two primers (with 3 or 4 additional nucleotides) were used.
bBuild 27 of human genome. Nucleotide position within the contig (strand +) is shown in parentheses.
cAs compared with the paired normal tissue.
dThe whole sequence or a fragment of the sequence lay within a predicted CpG island.
Figure 6.

(Top) The differential display of a band generated by AIMS in two cell lines (analyzed in duplicate). (Bottom) The bisulfite sequencing of a DNA fragment containing this sequence and demonstrating the methylation status of the CpG dinucleotide at the SmaI site in HCT116 cells as opposed to SW480 cells.
DISCUSSION
AIMS appears as a sensitive approach to screen for the differential methylation of DNA in cancer cells. Moreover, its application to a large series of samples appears feasible because of its technical simplicity and low requirement for starting material. The method is specially powerful in the detection of hypermethylated sequences, which are often associated with the inactivation of tumor suppressor genes (12,19). Hypomethylations are also detectable although contaminating cells maintaining the methylation status may limit the sensitivity to ∼10%. To be amplified, a sequence must fulfill two requirements: (i) to contain two closely spaced methylated SmaI sites and (ii) to show homology to the nucleotides extended at the 3′ end of the primer. The restricted amplification nature of AIMS renders fingerprints of adjustable complexity that are readily interpretable. Complementary representations may be obtained in experiments using different primers or combinations of primers.
The distribution of AIMS bands along chromosomes clearly shows that most of them originate from a subset of R bands, including T bands, that correspond to the CpG island and gene-rich regions (20), which are also identified as the H3 isochore (21). This is reinforced by the striking coincidence of chromosome hybridization patterns of AIMS products with that reported for H3 isochore (22). While this concordance does not indicate the nature of the amplified sequences, it can be concluded that the AIMS screening for methylated regions is specially intensive in the chromosomal areas with a higher functional interest. This bias in genome screening jointly with the high sensitivity of AIMS to detect hypermethylations provide the most important advantages of this technique.
The isolation and characterization of bands showing differential display between two samples is readily achievable. In a preliminary setting, we have identified 11 bands showing a differential methylation status between normal and tumor tissue. Novel bands appearing in the tumor tissue are indicative of sequence hypermethylation. Bisulfite genomic sequencing of some of these sequences confirmed this postulate. Recurrent hypermethylation of specific sequences might be considered an indicator of the putative inactivation of tumor suppressor genes (11,12). Nevertheless, AIMS raw data should be interpreted cautiously. Age-related methylation (23) is also likely to be observed and, due to the high sensitivity of this approach, hypermethylations affecting a small subset of cells may also arise. Additionally, some apparent normal–tumor changes may reflect clonal expansions of heterogeneous methylation profiles in normal colonic mucosa (24). Spurious amplification of bands due to incomplete digestion with SmaI cannot be totally excluded, but the robustness of the competitive amplification and the characteristic patterns of the artifacts (smears for poor digestions and bands appearing sporadically; data not shown) can be easily distinguished from recurrent changes. Nevertheless, as for most screening approaches, functional interpretation of the methylation changes revealed by AIMS requires confirmatory and specific investigations. While analysis of a larger series of samples is needed before attempting a thorough study of each one of the loci showing differential methylation, the preliminary results unveil the power of AIMS to track novel targets of abnormal methylation in cancer.
The fingerprints analyzed show clear normal–tumor changes and also little intensity variations between paired samples. In this setting, we have taken into consideration only the definite variations and, therefore, we are probably underestimating the extent of aberrant methylation in cancer cells. According to our data, hypomethylations are slightly more frequent than hypermethylations in most primary tumors. Hypomethylation is considered as a general trend in most colorectal cancers (25,26), which is accompanied by specific hypermethylation of a limited number of loci (reviewed in 11). Nevertheless, the skewness of our approach toward complex sequences may skip a high proportion of hypomethylations occurring at repetitive elements, which are the 5mC-richest regions in mammalian genome (27).
In addition to the discovery of novel targets, due to its arbitrary nature AIMS allows the assessment of global methylation profiles. The small number of cases analyzed precludes further interpretations, but the arbitrary nature of the approach offering a genome-wide representation of the methylome may be essential to investigate the nature and extent of the postulated CpG island methylator phenotype (28).
In conclusion, we have developed and evaluated a novel method to screen for differential DNA methylation. The approach is feasible for the analysis of large series of samples. Moreover, a high number of sequence tags may be generated in a few experiments. Represented tags arise from the chromosomal regions that are richest in CpG islands and genes. AIMS appears to be a powerful tool for identifying new genes critical to carcinogenesis.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Gemma Aiza and Silvia Beà for technical support. This work was supported by grants from the Ministerio de Ciencia y Tecnologia (SAF 00/81). J.F. is a fellow of the FPU program from the Spanish Ministry of Education. R.-A.R. was a fellow of Comissió Interdepartamental de Recerca i Innovació Tecnològica (CIRIT).
REFERENCES
- 1.Jones P.A. and Takai,D. (2001) The role of DNA methylation in mammalian epigenetics. Science, 293, 1068–1070. [DOI] [PubMed] [Google Scholar]
- 2.Falls J.G., Pulford,D.J., Wylie,A.A. and Jirtle,R.L. (1999) Genomic imprinting: implications for human disease. Am. J. Pathol., 154, 635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riggs A.D. and Pfeifer,G.P. (1992) X-chromosome inactivation and cell memory. Trends Genet., 8, 169–174. [DOI] [PubMed] [Google Scholar]
- 4.Kafri T., Ariel,M., Brandeis,M., Shemer,R., Urven,L., McCarrey,J., Cedar,H. and Razin,A. (1992) Developmental pattern of gene-specific DNA methylation in the mouse embryo and germ line. Genes Dev., 6, 705–714. [DOI] [PubMed] [Google Scholar]
- 5.Feinberg A.P. (2001) Methylation meets genomics. Nature Genet., 27, 9–10. [DOI] [PubMed] [Google Scholar]
- 6.Ehrlich M., Gama-Sosa,M.A., Huang,L.H., Midgett,R.M., Kuo,K.C., McCune,R.A. and Gehrke,C. (1982) Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res., 10, 2709–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bird A.P. (1986) CpG-rich islands and the function of DNA methylation. Nature, 321, 209–213. [DOI] [PubMed] [Google Scholar]
- 8.Cooper D.N. and Krawczak,M. (1989) Cytosine methylation and the fate of CpG dinucleotides in vertebrate genomes. Hum. Genet., 83, 181–188. [DOI] [PubMed] [Google Scholar]
- 9.Antequera F. and Bird,A. (1993) Number of CpG islands and genes in human and mouse. Proc. Natl Acad. Sci. USA, 90, 11995–11999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lengauer C., Kinzler,K.W. and Vogelstein,B. (1997) DNA methylation and genetic instability in colorectal cancer cells. Proc. Natl Acad. Sci. USA, 94, 2545–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baylin S.B. and Herman,J.G. (2000) DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet., 16, 168–174. [DOI] [PubMed] [Google Scholar]
- 12.Jones P.A. and Laird,P.W. (1999) Cancer epigenetics comes of age. Nature Genet., 21, 163–167. [DOI] [PubMed] [Google Scholar]
- 13.Toyota M., Ho,C., Ahuja,N., Jair,K.W., Li,Q., Ohe-Toyota,M., Baylin,S.B. and Issa,J.P. (1999) Identification of differentially methylated sequences in colorectal cancer by methylated CpG island amplification. Cancer Res., 59, 2307–2312. [PubMed] [Google Scholar]
- 14.Arribas R., Tortola,S., Welsh,J., McClelland,M. and Peinado,M.A. (1997) Arbitrarily primed PCR and RAPDs. In Micheli,M.R. and Bova,R. (eds), Fingerprinting Methods Based on Arbitrarily Primed PCR. Springer, Heidelberg, pp. 47–53.
- 15.Kallioniemi A., Kallioniemi,O.P., Sudar,D., Rutovitz,D., Gray,J.W., Waldman,F. and Pinkel,D. (1992) Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science, 258, 818–821. [DOI] [PubMed] [Google Scholar]
- 16.Masramon L., Ribas,M., Cifuentes,P., Arribas,R., Garcia,F., Egozcue,J., Peinado,M.A. and Miro,R. (2000) Cytogenetic characterization of two colon cell lines by using conventional G-banding, comparative genomic hybridization and whole chromosome painting. Cancer Genet. Cytogenet., 121, 17–21. [DOI] [PubMed] [Google Scholar]
- 17.Rein T., Zorbas,H. and DePamphilis,M.L. (1997) Active mammalian replication origins are associated with a high-density cluster of mCpG dinucleotides. Mol. Cell. Biol., 17, 416–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Welsh J. and McClelland,M. (1990) Fingerprinting genomes using PCR with arbitrary primers. Nucleic Acids Res., 18, 7213–7218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baylin S.B., Herman,J.G., Graff,J.R., Vertino,P.M. and Issa,J.P. (1998) Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv. Cancer Res., 72, 141–196. [PubMed] [Google Scholar]
- 20.Craig J.M. and Bickmore,W.A. (1994) The distribution of CpG islands in mammalian chromosomes. Nature Genet., 7, 376–382. [DOI] [PubMed] [Google Scholar]
- 21.Saccone S., De Sario,A., Della Valle,G. and Bernardi,G. (1992) The highest gene concentrations in the human genome are in telomeric bands of metaphase chromosomes. Proc. Natl Acad. Sci. USA, 89, 4913–4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saccone S., Caccio,S., Kusuda,J., Andreozzi,L. and Bernardi,G. (1996) Identification of the gene-richest bands in human chromosomes. Gene, 174, 85–94. [DOI] [PubMed] [Google Scholar]
- 23.Ahuja N., Li,Q., Mohan,A.L., Baylin,S.B. and Issa,J.P. (1998) Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res., 58, 5489–5494. [PubMed] [Google Scholar]
- 24.Yatabe Y., Tavare,S. and Shibata,D. (2001) Investigating stem cells in human colon by using methylation patterns. Proc. Natl Acad. Sci. USA, 98, 10839–10844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goelz S.E., Vogelstein,B., Hamilton,S.R. and Feinberg,A.P. (1985) Hypomethylation of DNA from benign and malignant human colon neoplasms. Science, 228, 187–190. [DOI] [PubMed] [Google Scholar]
- 26.Feinberg A.P. and Vogelstein,B. (1987) Alterations in DNA methylation in human colon neoplasia. Semin. Surg. Oncol., 3, 149–151. [DOI] [PubMed] [Google Scholar]
- 27.Yoder J.A., Walsh,C.P. and Bestor,T.H. (1997) Cytosine methylation and the ecology of intragenomic parasites. Trends Genet., 13, 335–340. [DOI] [PubMed] [Google Scholar]
- 28.Toyota M., Ahuja,N., Ohe-Toyota,M., Herman,J.G., Baylin,S.B. and Issa,J.P. (1999) CpG island methylator phenotype in colorectal cancer. Proc. Natl Acad. Sci. USA, 96, 8681–8686. [DOI] [PMC free article] [PubMed] [Google Scholar]