

Abstract
Neocarzinostatin (NCS) is the most studied member of a family of chromoproteins secreted by a range of actinomycetes species. It has been proposed that in addition to their antitumoral activity related to the bound chromophores, this group of related proteins could be a secreted proteases superfamily. With the aim of dissecting the molecular basis of the proteolytic activity of NCS, an expression system allowing efficient expression of apo-NCS in Escherichia coli was constructed. The recombinant protein was properly folded and functional. Its histone-specific proteolytic activity was similar to the activity described for the natural protein. Further analyses unambiguously demonstrated that the proteolytic activity could be physically separated from NCS. This activity is therefore due not to NCS itself but to minor contaminating proteases, the nature of which differed in the recombinant and natural NCS preparations. The histone degradation test commonly used to monitor proteolytic activity is extremely sensitive and may easily generate false-positive results. These results strongly suggest that the possible proteolytic activity of the proteins of this family should be critically reconsidered.
Neocarzinostatin (NCS), isolated from Streptomyces carzinostaticus, is a complex consisting of a low-molecular-weight enediyne chromophore, tightly bound (Kd = 0.1 nM [8]) to a 113-amino-acid single chain protein (2, 13, 19). NCS belongs to a family of macromolecular chromoprotein antibiotics that also have antitumoral activity. The known members of this family are NCS, macromomycin (secreted by Streptomyces macromomyceticus), C-1027 and actinoxantin (secreted by Streptomyces globisporus), maduropeptin (secreted by Actinomadura madurae), and kedarcidin (secreted by an unidentified species of actinomycetes). With the exception of maduropeptin, the amino acid sequence of which has not yet been determined and the molecular weight of which is significantly higher than those of the other proteins, all proteins of this family have clearly related amino acid sequences. The antibiotic and antitumor activity of these compounds is due to the low-molecular-weight enediyne moiety. These enediyne-containing molecules form highly reactive biradical intermediates that cleave DNA at sequence-specific sites (3, 9, 10, 20). It has been suggested that the function of the apoprotein is essentially to stabilize and regulate the availability of the otherwise labile chromophore (11). However, studies with some proteins have detected proteolytic activity associated to the polypeptide moiety of the complex. Each chromoprotein seems to have a characteristic proteolytic specificity (22). Whereas C-1027 (18) and macromomycin (21) seem to have aminopeptidase activity, endopeptidase activity has been shown for kedarcidin (18), maduropeptin, and NCS (23, 24).
These activities have been detected with either histones (18, 21–24) or synthetic peptides (24). Histones were chosen as substrates because it was thought that these highly basic proteins would probably interact with the acid-rich apoproteins. It was indeed shown that histone H1, the richest in lysine, was the preferred substrate of NCS (23). Histones combine with DNA to form nucleosomes which are connected by histone H1. Based on the proteolytic capacity of the apoprotein, it has been suggested that the apoprotein is directly involved in the activity of the chromoprotein complex, providing active chromophores with easier access to their DNA target (23). These results have suggested that the protein component of these chromoproteins may provide a targeted delivery of the chromophores to the chromatine.
The association of the two unrelated properties, chromophore binding and enzymatic activity, in the same single domain protein rises important evolutionary questions. This might result from an evolutionary process that conferred a new proteolytic activity on a preexisting binding protein. Alternatively, a preexisting protease may have subsequently acquired strong and highly specific binding properties. As the various proteins belong to the same superfamily with a high level of identity between their amino acid sequences, and as all bind enediyne-containing chromophores, a divergent evolutionary process for chromophore binding appears to be the most likely origin. Furthermore, the different proteolytic activities observed in this chromoprotein family suggest that these activities may result from different adaptations of a preexisting binding protein. None of these proteins have sequence or structural similarity to any known protease family. This interesting situation, and the possible intriguing underlying evolutionary mechanism, led us to analyze the mechanistic basis for the activities of this potentially new family of proteases, starting with neocarzinostatin, the most studied family member.
A protein engineering strategy could be a powerful way to analyze the molecular basis of this catalytic activity. However, no expression system for producing recombinant apo-NCS had been described. We therefore constructed a synthetic gene and an expression vector for producing apo-NCS in Escherichia coli. We then compared the structural and functional properties of recombinant apo-NCS produced in E. coli with the same protein naturally produced by S. carzinostaticus. Full characterization of these proteins gave direct evidence that the proteolytic activity observed with both the recombinant and natural proteins was due not to the apoprotein but to minor contaminating proteases.
MATERIALS AND METHODS
NCS apoprotein produced from S. carzinostaticus.
A 200-ml culture of S. carzinostaticus was grown for 48 h as described by Kikushi et al. (12). NCS apoprotein was purified as described by Favaudon (6). In this protocol, the naturally produced apoprotein is separated from the holoprotein by ion-exchange chromatography on carboxymethyl cellulose.
Expression system.
A synthetic gene coding for NCS was synthesized by assembling eight overlapping oligonucleotides by PCR. The nucleotide sequence was designed to incorporate several unique restriction sites, and E. coli codon usage was taken into account. This gene was inserted into the expression vector pET12a (NOVAGEN), to give the expression plasmid pNCS.sec. In this construct, the coding sequence is fused to the ompT signal sequence, to direct secretion of the target protein into the periplasm. The E. coli strain used for expression was BLR (DE3)pLysS.
Purification of the neocarzinostatin apoprotein secreted by E. coli.
Cells freshly transformed with the expression vector were grown on 2YT medium containing ampicillin, tetracycline, and chloramphenicol at 30°C. The culture medium was separated from the bacteria, and soluble proteins directly secreted into the culture medium were precipitated with 650 g of ammonium sulfate per liter. The proteins were collected by centrifugation for 20 min at 17,000 × g. The precipitate was dialyzed first against double-distilled water and then against 20 mM ammonium acetate buffer (pH 5). The protein solution was applied to a Q Sepharose Fast Flow column equilibrated with 20 mM ammonium acetate buffer (pH 5). Proteins were eluted with 500 mM ammonium acetate (pH 5) at a flow rate of 4 ml/min. The eluate was then applied to a Sephadex G-50 column equilibrated with 50 mM phosphate buffer (pH 7) and eluted at a flow rate of 0.5 ml/min.
Purification of protein substrates.
Histone H1 was purified from calf thymus as described by de Nooij et al. (5), and apocytochrome c was purified by the silver sulfate method of Paul, as modified by Fisher et al. (7).
Physicochemical properties of the recombinant apo-NCS.
The amino acid sequence of the recombinant was analyzed on an Applied Biosystems model 473A microsequencer, and the molecular weight of the recombinant protein was determined by electrospray and matrix-assisted laser desorption ionization–time-of-flight mass spectrometry using standard methods.
Circular dichroism (CD) spectra were recorded from 180 to 260 nm on a Mark V dichrograph (Jobin-Yvon) equipped with a thermostatically controlled cell holder and connected to a computer for data acquisition. Data were acquired from 15 μM sample solutions in phosphate buffer using quartz cells with a 0.1-mm path length. One and two-dimensional nuclear magnetic resonance (NMR) spectra were recorded on a 500-MHz Varian spectrometer, using the conditions described elsewhere (1, 2).
Ethidium bromide (EtBr) binding to apo-NCS (15) was studied by fluorimetry with an Aminco SLM 8000 fluorimeter, by monitoring the intrinsic fluorescence of a 1.75 μM EtBr solution (λexc = 479 nm, λem = 620 nm) as a function of apo-NCS concentration. Saturation curve data was analyzed by using the following equation:
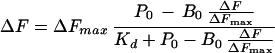 |
1 |
where ΔFmax equals Fmax − F0, ΔF equals F − F0, and F and F0 are the fluorescence intensities measured in the presence and absence of the protein, respectively, P0 is the total protein concentration, B0 is the total EtBr concentration, and Kd is the dissociation constant.
Proteolytic activity measurements.
Apo-NCS (0.1 mg/ml) was incubated with protein substrate (1 mg/ml) in 50 mM Tris-HCl buffer (pH 7.5) at 37°C in a total volume of 100 μl. The mixture was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) in a 12% gel, and the protein bands were stained with Coomassie blue. For the synthetic peptide, (WAGKTPVKKASGPW; supplied by NEOSYSTEM), the mixture was analyzed by high-pressure liquid chromatography on a Vydac C18 column equilibrated in 0.1% trifluoroacetic acid in water with elution by a 0 to 80% acetonitryl–0.1% trifluoroacetic acid gradient.
Purification of apo-NCS antibody.
Apo-NCS serum was obtained by hyperimmunization of a rabbit by intradermal injections of apo-NCS emulsified within complete Freund's adjuvant. Subsequent booster injections were administrated at intervals of 3 weeks under the same conditions. The rabbit was bled 1 week after each booster injection. Apo-NCS antibodies were purified from the serum by affinity chromatography on an immobilized apo-NCS column. The column used was a Hitrap N-hydroxysuccinimide-activated column grafted with pure recombinant apo-NCS as recommended by the supplier. The rabbit serum was applied to this column, and elution was performed with 50 mM glycine–100 mM NaCl (pH 2.5). Fractions were neutralized immediately after elution by adding one-fifth the volume of 1 M Tris-HCl buffer at pH 9.
Thermal stability of apo-NCS.
The thermal stability and reversibility of heat denaturation were studied by differential scanning calorimetry (DSC) on a Microcal model MC2. DSC measurements were made with a 3-mg/ml apo-NCS solution dialyzed overnight against phosphate buffer (pH 7.5). Buffer solution from the dialysis bath was used as a reference. All solutions were degassed just before loading into the calorimeter. Scanning was performed at 1 K/min, and the reversibility of the thermal transition was checked by rescanning the sample.
RESULTS
Growing conditions.
The initial aim of the experiments reported was to produce recombinant apo-NCS to enable us to define clearly the role of the apoprotein in proteolytic activity and to identify, by mutagenesis, the residues involved in this activity. The first step was to obtain a large amount of recombinant functional apoprotein. Growth conditions and purification methods were optimized so as to overproduce soluble protein. Our first attempts showed that growing the cells at 37°C led mostly to the production of apo-NCS in the form of inclusion bodies. When the temperature was decreased to 30°C, the recombinant protein was mostly secreted into the culture medium (Fig. 1) in a soluble, folded form.
FIG. 1.

SDS-PAGE of cells crude lysate of a culture incubated at 37°C (lane 1), culture supernatant after growth at 37°C (lane 2), cells crude lysate of a culture incubated at 30°C (lane 3), and culture supernatant after growth at 30°C (lane 4). The arrows show the band corresponding to apo-NCS.
Characterization of the recombinant protein.
The recombinant protein was fully characterized to check that it is strictly equivalent to the natural apo-NCS. We first checked that the mature product is completely devoid of the signal peptide encoded by the synthetic gene. The N-terminal sequence was AAPTATVP, identical to that of the natural protein, indicating that the peptide signal was correctly processed. Mass spectrometry indicated the presence of a single species with a molecular mass of 11,100 Da. Thus, the recombinant protein was homogeneous, of the expected size, and without any proteolysis on the N- or C-terminal end, excepted for cleavage of the synthetic signal peptide. SDS-PAGE showed one well-resolved band with no visible contaminants on an overloaded gel.
CD spectra for natural apo-NCS and the recombinant protein were recorded in identical conditions. The two spectra are fully superimposable (Fig. 2). NCS is an all-β protein, but the general shape of the CD spectra is somewhat atypical, with a positive maximum at 225 nm. This CD signal may result from constraints in the polypeptide backbone. The presence of disulfide bridges connecting cysteine residues very close together at positions 37 to 47 and 88 to 93 may account for such signal reported as “not typical” secondary structures (16).
FIG. 2.

CD spectra of recombinant (solid line) and natural (dotted line) apo-NCS.
One-dimensional 1H NMR spectra of natural and recombinant apo-NCS are very similar, the peaks having similar chemical shifts and relative intensities (Fig. 3), illustrating the perfect identity of natural and recombinant apo-NCS. It has previously been shown that EtBr binds to apo-NCS at the natural chromophore site (15). Therefore, EtBr binding provides a convenient way to monitor the functional integrity of the binding site of the recombinant apoprotein. A Kd of 2 μM was obtained for recombinant apo-NCS, similar to the value previously reported (15) for the natural apoprotein (1 μM), indicating that the recombinant protein is fully functional.
FIG. 3.

Comparison of the 1H NMR spectra acquired at 500 MHz, in H2O at pH 5.5 and 35°C, of natural (a) and recombinant (b) proteins.
Histone H1 cleavage by NCS.
Incubation of calf thymus histone H1 with natural or recombinant apo-NCS leads to the formation of low-molecular-weight species (Fig. 4) within 2 h of incubation. This result is similar to those previously reported (23, 24). However, a noticeable difference is observed in the proteolytic activity of the recombinant and natural proteins, because although the concentration of the recombinant protein was higher, the time required to obtain full proteolysis of the sample was 10 times longer than for natural NCS. Thus, the proteolytic activity of apo-NCS purified from E. coli is lower than the proteolytic activity of the apoprotein purified from S. neocarzinostaticus.
FIG. 4.

SDS-PAGE analysis of the reaction of apo-NCS with calf thymus histones. Reaction conditions: 50 mM Tris-HCl buffer (pH 7.5), histones (1 mg/ml), and recombinant (500 μg/ml) (A) or natural (100 μg/ml) apo-NCS (B) in a total volume of 100 μl. Numbers are incubation times (hours) for each sample.
Specificity of the proteolytic activity.
Previous studies concerning the proteolytic activity of apo-NCS have shown higher levels of proteolytic activity for apo-NCS with histone H1 than with other proteins. We used various proteins as substrates and determined the specificity of recombinant and natural apo-NCS. Of the proteins other than histone H1 tested (RNase, β-lactalbumin, albumin, carbonic anhydrase, cytochrome c, and apo-cytochrome c), only apo-cytochrome c was cleaved by both types of NCS. Cytochrome c and apo-cytochrome c are highly basic proteins. Apo-cytochrome c is known to be a very flexible and loosely folded protein (17). Histone H1 is also a highly flexible protein (4). Thus, in addition to its basic character, the main reason for the apparent specificity of apo-NCS for histone H1 seems to be the high degree of flexibility of histone H1 rather than its specific tertiary structure.
Proteolytic activity was also tested against a synthetic peptide, the sequence of which (WAGKTPVKKASGPW) was derived from the peptides used in previous studies (23). We monitored hydrolysis products by adding two tryptophans, one at each extremity of the peptide. According to Zein et al. (23), there are two cleavage sites, between Lys-Lys and Lys-Ala. As expected, the apo-NCS from Streptomyces cleaves this peptide (Fig. 5). Although proteolysis was not complete, the number of peptides detected at 220 and 280 nm clearly indicates that the peptide was cleaved at at least two positions. However, in the same experimental conditions, the apo-NCS from E. coli has no proteolytic activity against this peptide (data not shown).
FIG. 5.

Chromatography elution of proteolysate of the synthetic peptide treated with apo-NCS from Streptomyces, monitored at 215 (A) and 280 (B) nm.
Inhibition of proteolytic activity.
We investigated the possible involvement of the chromophore binding site residues in catalytic activity by studying apo-NCS proteolytic activity in the presence of 50 μM EtBr. No significant effect was observed for either of the two proteins, suggesting that the amino acids involved in proteolytic activity are located outside the binding site of the chromophore. Similarly, serine protease inhibitors such as phenylmethylsulfonyl fluoride and 4-2-aminoethyl-benzosulfonyl-fluorohydrochloride (EABSF) did not affect proteolytic activity. EDTA, however, strongly inhibited the proteolytic activity associated with apo-NCS from S. carzinostaticus but did not inhibit the proteolytic activity associated with the recombinant form of apo-NCS (Fig. 6). These results suggest that the proteolytic activities detected in the two preparations are due to different compounds.
FIG. 6.
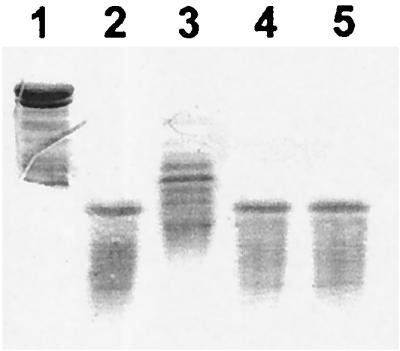
SDS-PAGE analysis of the inhibition of proteolytic activity by EDTA. Lane 1, histone; lane 2, histone plus apo-NCS from Streptomyces; lane 3, histone + apo-NCS from Streptomyces plus EDTA; lane 4, histone plus apo-NCS from E. coli; lane 5, histone + apo-NCS from E. coli plus EDTA.
Thermostability of apo-NCS and proteolytic activity.
NCS is a thermostable protein with a melting transition at 68°C. Figure 7 shows the buffer-corrected partial molar excess heat capacity data for a 3-mg/ml recombinant apo-NCS solution in phosphate buffer (pH 7.5) at a scan rate of 1 K/min. Under these conditions, the transition occurs with a ΔH of 100 kcal/mol and a melting temperature of 68°C, as for natural apoprotein. The reversibility of the thermal transition was checked by rescanning the sample. Three successive scans were performed, showing a high degree of reversibility, with 90% recovery of the denaturation enthalpy. It should be emphasized that the DSC profile is a highly sensitive indicator of the folded structure and that the observed reversibility unambiguously demonstrates that NCS thermal denaturation is a highly reversible process. In contrast, a single heating at 80°C, in the conditions used for DSC measurements, of both types of apo-NCS sample (from S. neocarzinostaticus and from E. coli) led to full inactivation of the associated proteolytic activity.
FIG. 7.

Corrected partial molar excess heat capacity for a 3-mg/ml apo-NCS solution in phosphate buffer (pH 7.5) at a scan rate of 1 K/min. The profiles obtained for two consecutive scans are shown.
Separation of apoprotein and proteolytic activity.
We attempted to separate proteolytic activity from apo-NCS in two ways. First, ion-exchange chromatography on a Mono Q FPLC column equilibrated with 50 mM amonium acetate buffer (pH 7.5) was performed on a stock solution of NCS displaying proteolytic activity. Elution with a gradient of NaCl gave a single peak corresponding to apo-NCS (Fig. 8). Although no proteolytic activity was found elsewhere in the gradient, nor eluted by a step at 1 M NaCl, the protein recovered was found to have a relative activity corresponding to less than 5% of that of the same sample before chromatography. This suggests that activity could be minimized by repeated purification.
FIG. 8.

Elution profile of an ion-exchange chromatography on a Mono Q FPLC column (0.5 by 5 cm) of a stock solution of NCS with a gradient of 0 to 0.3 M NaCl (flow rate, 1 ml/min). Proteolytic activity was observed only in the fraction containing protein (+). The apparent specific activity of this fraction was 5% of the apparent specific activity before chromatography, as estimated from the time required to proteolyze 50% of histone H1.
In the second method, we used polyclonal antibodies to separate proteolytic activity from apo-NCS. In a first step, stock solutions of apo-NCS from S. neocarzinostaticus or E. coli were incubated with affinity-purified anti-NCS rabbit polyclonal antibodies. The antibody-NCS complex was then trapped by adding agarose beads cross-linked with protein A which were removed by centrifugation. As expected, NCS was no longer detectable in the supernatant by SDS-PAGE, indicating that all apo-NCS molecules were trapped by antibodies. However, the supernatant, which did not contain apo-NCS, had the same level of proteolytic activity as the original preparations, indicating that proteolytic activity can be physically separated from apo-NCS molecules (Fig. 9). Control experiments conducted with the same reagents but with no added apo-NCS showed that the sole source of proteolytic activity in these experiments was the apo-NCS preparations. The clear conclusion of this experiment is that the observed proteolytic activity of recombinant and natural apo-NCS, similar in all respects to previously published observations, can be physically separated from apoprotein and is due to a molecular species other than apo-NCS.
FIG. 9.
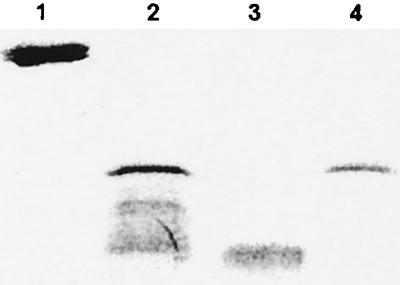
SDS-PAGE analysis of the inhibition of proteolytic activity by specific anti-NCS antibodies. Lane 1, histone; lane 2, histone plus apo-NCS; lane 3, histone plus supernatant from immunoprecipitation of apo-NCS (see text); lane 4, purified apo-NCS.
DISCUSSION
Since the discovery of NCS and the DNA cleavage activity associated with the chromophore, many studies have been performed to elucidate and optimize antitumoral activity. In the last decade, related proteins have been purified; in the course of these studies, it has been suggested that a proteolytic activity is associated with the apoproteins, with proteolytic specificity apparently unique to each chromoprotein (23). The proteolytic activity appeared to be selective for histones, particularly histone H1. It was therefore suggested that these highly acidic apoproteins protected their chromophores from deactivation and delivered them to the spacer DNA after cleaving histone H1 (22). We have reinvestigated this proteolytic activity by using a recombinant apo-NCS produced by expression of a synthetic gene in E. coli. The recombinant apo-NCS is strictly devoid of the chromophore, and this method makes it possible to construct any mutant required to characterize the amino acids directly involved in the proteolytic process.
Full characterization of the recombinant apoprotein showed that it is strictly identical to the natural apo-NCS. Chemical characterization by microsequencing and mass spectrometry showed that it is uniformly processed. Structural properties, as monitored by far-UV CD and NMR, indicate that the recombinant protein is correctly folded. EtBr bound efficiently to recombinant apo-NCS. These data clearly indicate that the recombinant apo-NCS is correctly folded.
The recombinant and natural proteins were indistinguishable with respect to physicochemical properties but differed in proteolytic activities. As previously shown (23), histone H1 is cleaved by both natural and recombinant apo-NCS, but this activity of the recombinant apoprotein is weaker. However, the first data to indicate a clear difference in the proteolytic activities of the two forms of neocarzinostatin came from the hydrolysis of the synthetic peptide. The peptide is fully cleaved by the apo-NCS prepared from S. carzinostaticus, but the apo-NCS extracted from E. coli does not cleave it at all. This suggests that the amino acid sequences recognized are different in the two cases. Moreover, the effect of EDTA on proteolytic activity indicates that the activity associated with apo-NCS from S. carzinostaticus was dependent on divalent cations. In contrast, EDTA does not affect the proteolytic activity associated with apo-NCS from E. coli. These results provide strong evidence for there being two different origins of proteolytic activities according to the source of apo-NCS. Taking into account the high level of identity of the natural and recombinant apoproteins, this suggests that the proteolytic activity is not directly related to apoprotein structure but may result from minor protease contaminants different in nature for the two sources. This possibility was raised in previous studies, but the authors proposed that their data suggested, within reasonable limits, that the proteolytic activity observed was not due to minor protease activity (22).
It should be noted that according to standard purity criteria, the apo-NCS preparations used here and in previous studies were classed as pure proteins. Staining with Coomassie blue detected a single band on the polyacrylamide gel, even if the gel was overloaded. Similarly, only one species was detected by mass spectrometry.
Other observations suggested that the proteolytic activity is actually due to a protein other than the apo-NCS. Ion-exchange chromatography reduced significantly the specific proteolytic activity of apo-NCS indicating that functional apo-NCS could be separated from the proteolytically active component by repeated purification. The addition of polyclonal antibodies to the reaction mixture did not affect the proteolytic activity (data not shown).
Although consistent, these data are not fully conclusive: although the specific proteolytic activity is strongly reduced by ion-exchange chromatography, it is not completely abolished, and it is therefore possible that various isoforms of the protein are present or that there is an indispensable cofactor required for proteolytic activity. Similarly, the lack of an effect of polyclonal antibodies on proteolytic activity may result from the absence of an inhibitory antibody, although such hypothesis seems unlikely with polyclonal antibodies.
The definitive data come from thermal denaturation experiments and trapping of the apo-NCS–antibody complex. Thermal denaturation studies showed that unfolding is reversible for apo-NCS but causes irreversible inactivation of proteolytic activity for both apo-NCS (recombinant and natural). Finally, the use of specific anti-NCS antibodies made it possible to physically separate apo-NCS from the proteolytic activity present in the preparations. Apo-NCS was very specifically removed from the incubation mixture by trapping the apo-NCS–anti-NCS antibody complex on protein A-agarose beads, and the levels of proteolytic activity were similar in the NCS preparation and in the supernatant devoid of apo-NCS. Clearly, if proteolysis was due to a weakly active apo-NCS or to a small proportion of apo-NCS molecules with proteolytic activity, a decrease in activity proportional to the quantity of NCS in the medium would have been observed.
The simplest interpretation of these results is that a minor contaminant with proteolytic activity is present. This would also explain why proteins with similar structures and sequences are associated with various proteolytic activities, from exopeptidase to endopeptidases (18, 21, 22, 24). The activity being due to a contaminant would also account for the very low specific activity: the experimental conditions used (apoprotein/substrate ratio, incubation time) indicate a specific activity at least 3 orders of magnitude lower than that of typical proteases like trypsin. The observed specificity for histone H1, which was thought to be a biologically relevant property, can be due not only to the amino acid sequence of the substrate but also to its flexibility. NCS proteolytic activity was measured for various proteins possessing at least one cleavage site, according to Zein et al. (23). These experiments suggested that the specificity could be related to the structure of histone H1. This protein has a structured domain and a flexible domain; the flexible part of the protein could be responsible for the high susceptibility to proteolysis of histone H1. This hypothesis is supported by the difference in the susceptibility to proteolysis of cytochrome c and apo-cytochrome c. Histone H1, due to its high flexibility, is a highly sensitive proteolytic substrate, as is apo-cytochrome c. It may be possible to detect minor proteolytically active contaminant with histone H1 that would probably not be detected with other substrates.
These results exclude previous hypotheses concerning the possible physiological role of this activity. As apo-NCS has no intrinsic proteolytic activity, it does not necessarily have to reach DNA to cause its cleavage; thus, the primary function of the apoprotein is probably to stabilize and regulate the availability of the labile chromophore. It is not clear whether the holo-NCS enters the cell and releases the chromophore at its site of action or whether it releases the chromophore at the cell surface to be carried inside. It has been shown that holo-NCS can be taken up by the cells, but Lazarus et al. (14), by using holo-NCS covalently bond to agarose, have shown that the chromophore released at the cell surface can be taken up by the cell. If holoprotein uptake is not required for chromophore activity, these experiments do not determine whether holo-NCS is ordinarily taken up intact if cells are treated with the drug. Our results are consistent with holo-NCS-induced chromatin cleavage experiments showing that histone H1 is not specifically cleaved by this treatment (3). We believe that the protocol used by us and others to detect apo-NCS proteolytic activity is extremely sensitive and consequently very likely to generate false-positive artifacts.
The results presented herein clearly raise the possibility that low levels of various proteolytic activities described for other chromoproteins may also be due to the proteolytic activities of minor contaminants. They strongly suggest that more definitive evidence, for example, the abolition of activity by directed mutations, is required for such activities to be considered reliable and biologically relevant.
ACKNOWLEDGMENTS
This work was supported by the Centre National de la Recherche Scientifique, by the Ministère de l'Enseignement Supérieur et de la Recherche, and by a contract with the Institut Curie.
Bernadette Heyd and Guilhem Lerat contributed equally to this work and thus should be considered co-first authors.
REFERENCES
- 1.Adjadj E, Mispelter J, Quiniou E, Dimicoli J-L, Favaudon V, Lhoste J-M. Proton NMR studies of apo-neocarzinostatin from Streptomyces carzinostaticus. Sequence-specific assignment and secondary structure. Eur J Biochem. 1990;190:263–271. doi: 10.1111/j.1432-1033.1990.tb15571.x. [DOI] [PubMed] [Google Scholar]
- 2.Adjadj E, Quiniou E, Mispelter J, Favaudon V, Lhoste J M. Three-dimensional solution structure of apo-neocarzinostatin from Streptomyces carzinostaticus determined by NMR spectroscopy. Eur J Biochem. 1991;203:505–511. doi: 10.1111/j.1432-1033.1992.tb16576.x. [DOI] [PubMed] [Google Scholar]
- 3.Beerman T A, Mueller G, Grimmond H. Effects of neocarzinostatin on chromatin in HeLa S3 nuclei. Mol Pharmacol. 1982;23:493–499. [PubMed] [Google Scholar]
- 4.Bradbury E M. Conformations and flexibilities of histones and high mobility group (HMG) proteins in chromatin structure and function. Ciba Found Symp. 1983;93:246–270. doi: 10.1002/9780470720752.ch14. [DOI] [PubMed] [Google Scholar]
- 5.de Nooij E H, Westenbrink H G K. Isolation of a homogenous Lys-rich histone from calf thymus. Biochim Biophys Acta. 1962;62:608–609. doi: 10.1016/0006-3002(62)90254-8. [DOI] [PubMed] [Google Scholar]
- 6.Favaudon V. Gamma-radiolysis study of the reductive activation of neocarzinostatin by the carboxyl radical. Biochimie. 1983;65:593–607. doi: 10.1016/s0300-9084(84)80023-1. [DOI] [PubMed] [Google Scholar]
- 7.Fisher W R, Taniuchi H, Anfinsen C B. On the role of heme in the formation of the structure of cytochrome c. J Biol Chem. 1973;248:3188–3193. [PubMed] [Google Scholar]
- 8.Goldberg I H, Hatayama T, Kappen L S, Napier M A, Povirk L F. Molecular actions and targets for cancer chemiotherapeutic agents. Vol. 1. New York, N.Y: Academic Press; 1981. pp. 163–191. [Google Scholar]
- 9.Hatayama T, Yukioka M. Action of neocarzinostatin on cell nuclei: release of specific chromatin. Biochem Biophy Res Commun. 1982;104:889–896. doi: 10.1016/0006-291x(82)91332-8. [DOI] [PubMed] [Google Scholar]
- 10.Kappen L S, Ellenberger T E, Goldberg I H. Mechanism and base specificity of DNA breakage in intact cells by neocarzinostatin. Biochemistry. 1987;26:384–390. doi: 10.1021/bi00376a008. [DOI] [PubMed] [Google Scholar]
- 11.Kappen L S, Napier M A, Goldberg I H. Roles of chromophore and apoprotein in neocarzinostatin action. Proc Natl Acad Sci USA. 1980;7:1970–1974. doi: 10.1073/pnas.77.4.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kikushi M, Shoji M, Ishida N. Pre-neocarzinostatin, a specific antagonist of neocarzinostatin. J Antibiot. 1974;27:766–774. doi: 10.7164/antibiotics.27.766. [DOI] [PubMed] [Google Scholar]
- 13.Kim K-H, Kwon B-M, Myers A G, Rees D R. Crystal structure of neocarzinostatin, an antitumor protein-chromophore complex. Science. 1993;262:1042–1046. doi: 10.1126/science.8235619. [DOI] [PubMed] [Google Scholar]
- 14.Lazarus H, Raso V, Samy T S A. In vitro inhibition of human leukemic cells CCRF-CEM) by agarose-immobilized neocarzinostatin. Cancer Res. 1977;37:3731–3138. [PubMed] [Google Scholar]
- 15.Mohanty S, Sieker L C, Drobny G P. Sequential 1H NMR assignment of the complex of aponeocarzinostatin with ethidium bromide and investigation of protein-drug interactions in the chromophore binding site. Biochemistry. 1994;33:10579–10590. doi: 10.1021/bi00201a003. [DOI] [PubMed] [Google Scholar]
- 16.Perczel A, Hollosi M. Turns. In: Fasman G, editor. Circular dichroism and the conformational analysis of biomolecules. New York, N.Y: Plenum Press; 1996. pp. 285–381. [Google Scholar]
- 17.Privalov P L, Tiktopulo E I, Venyaminov S, Griko Y V, Makhatadze G I, Khechinashvili N N. Heat capacity and conformation of proteins in the denatured state. J Mol Biol. 1989;205:737–750. doi: 10.1016/0022-2836(89)90318-5. [DOI] [PubMed] [Google Scholar]
- 18.Sakata N, Tsuchiya K S, Moriya Y, Hayashi H, Hori M, Otani T, Nagai M, Aoyagi T. Aminopeptidase activity of an antitumor antibiotic, C-1027. J Antibiot. 1991;45:113–117. doi: 10.7164/antibiotics.45.113. [DOI] [PubMed] [Google Scholar]
- 19.Teplyakov A, Obmolova G, Wilson K, Kuromizu K. Crystal structure of apo-neocarzinostatin at 0.15-nm resolution. Eur J Biochem. 1993;213:737–741. doi: 10.1111/j.1432-1033.1993.tb17814.x. [DOI] [PubMed] [Google Scholar]
- 20.Tien K M, Samy T S A. Effects of neocarzinostatin on mammalian nuclei: release of nucleosomes. Biochim Biophys Acta. 1978;518:186–190. doi: 10.1016/0005-2787(78)90129-6. [DOI] [PubMed] [Google Scholar]
- 21.Zaheer A, Zaheer S, Montgomery R. Peptidase activity of macromomycin apoprotein. J Biol Chem. 1985;260:11787–11792. [PubMed] [Google Scholar]
- 22.Zein N, Casazza A M, Doyle T W, Leet J E, Schroeder D R, Solomon W, Nadler S G. Selective proteolytic activity of the antitumor agent kedarcidin. Proc Natl Acad Sci USA. 1993;90:8009–8012. doi: 10.1073/pnas.90.17.8009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zein N, Solomon W, Colson K L, Schroeder D R. Maduropeptin: an antitumor chromoprotein with selective protease activity and DNA cleaving properties. Biochemistry. 1995;34:11591–11597. doi: 10.1021/bi00036a035. [DOI] [PubMed] [Google Scholar]
- 24.Zein N, Reiss P, Bernatowicz M, Bolgar M. The proteolytic specificity of the natural enediyne-containing chromoproteins is unique to each chromoprotein. Chem Biol. 1995;7:451–455. doi: 10.1016/1074-5521(95)90262-7. [DOI] [PubMed] [Google Scholar]