

Abstract
Pulsed electron−electron double resonance (PELDOR) spectroscopy, X-ray scattering interferometry (XSI), and single-molecule Förster resonance energy transfer (smFRET) are molecular rulers that provide inter- or intramolecular pair-wise distance distributions in the nanometer range, thus being ideally suitable for structural and dynamic studies of biomolecules including RNAs. The prerequisite for such applications requires site-specific labeling of biomolecules with spin labels, gold nanoparticles, and fluorescent tags, respectively. Recently, site-specific labeling of large RNAs has been achieved by a combination of transcription of an expanded genetic alphabet containing A-T/G-C base pairs and NaM-TPT3 unnatural base pair (UBP) with post-transcriptional modifications at UBP bases by click chemistry or amine−NHS ester reactions. However, due to the bulky sizes of functional groups or labeling probes used, such strategies might cause structural perturbation and decrease the accuracy of distance measurements. Here, we synthesize an α-thiophosphorylated variant of rTPT3TP (rTPT3αS), which allows for post-transcriptional site-specific labeling of large RNAs at the internal α-phosphate backbone via maleimide-modified probes. Subsequent PELDOR, XSI, and smFRET measurements result in narrower distance distributions than labeling at the TPT3 base. The presented strategy provides a new route to empower the molecular rulers for structural and dynamic studies of large RNA and its complex.
Graphical Abstract
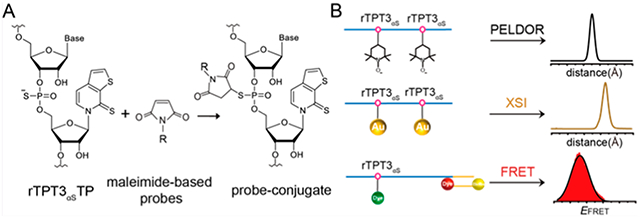
INTRODUCTION
RNAs are important biomolecules that play diverse roles in numerous cellular processes, including serving as templates for the synthesis of proteins, RNAs, or DNAs as catalytic centers for transcription, pre-mRNA splicing, translation, or as regulators of gene expression.1 The functional diversity of RNAs arises from their ability to form distinct three-dimensional (3D) structures and undergo conformational changes in response to a diverse array of cellular conditions.2 Knowledge about the RNA structure, interaction, and conformational dynamics is essential to understand their functions and to develop RNA-based or RNA-targeted therapeutics.3 Structural characterization of RNAs using techniques such as X-ray crystallography and cryoelectron microscopy is challenging due to RNAs’ inherent flexibility.4,5 Solution techniques, such as nuclear magnetic resonance (NMR) spectroscopy and small-angle X-ray and neutron scattering (SAXS/SANS), allow us to describe the flexibility and obtain unique structural and dynamics information of the RNA or RNA−ligand complex. However, NMR is limited to short RNA [on average 50−60 nucleotides (nts)] and SAXS/SANS has relatively low resolution.6,7 There is a growing trend in the field to characterize the structure, interaction, and conformational dynamics of RNAs using integrative methods, which combines experimental data of different resolutions and from multiple techniques with computational modeling. Among these techniques, pulsed electron−electron double resonance (PELDOR, also known as DEER) spectroscopy, X-ray scattering interferometry (XSI), and single-molecule Förster resonance energy transfer (smFRET) are molecular rulers that can measure intra- or intermolecular distance distributions in the nanometer range, provided that the biomolecules are site-specifically labeled with spin labels, gold nanoparticles (AuNPs), and fluorophores, respectively.8–11 Many studies have demonstrated that distance restraints, coupled with high-resolution structure information and computational modeling, are highly informative in investigating the global structure and conformational dynamics of RNA and RNA−protein complexes.12–14
In recent years, a plethora of strategies have been developed to achieve site-specific labeling of RNAs.15–18 RNAs can be prepared in sufficient amounts for structural studies by either solid-phase chemical synthesis or in vitro transcription (IVT) catalyzed by RNA polymerases. Different probes can be covalently attached at the nucleotide base, the sugar moiety, or the phosphate backbone of RNAs, either during RNA synthesis or postsynthetically. Chemical synthesis has been the most popular and versatile method for site-specific RNA labeling but is generally limited to RNAs smaller than 100 nts. The size limit might be mitigated by chemical or enzymatic ligation of the synthetic fragment with a polymerase-derived RNA, but these methods are generally laborious and restricted by ligation efficiency.19,20 Recently, site-specific labeling of very large RNAs (up to 700 nts) has been achieved by a combination of transcription of the unnatural base pair (UBP) systems and post-transcriptional modification through click chemistry or amine−NHS ester reactions.9–11,21,22 For example, by synthesis of alkyne- or amine-derivatized rTPT3TP and transcription of an expanded genetic alphabet containing NaM-TPT3 UBP system, spin labels, AuNPs, and fluorophores have been site-specifically attached to several large RNAs, thus facilitating pair-wise distance distribution measurements by PELDOR, XSI, and smFRET, respectively.9–11 These UBP-based methods overcome length limitations in conventional RNA labeling methods and can be efficiently performed under near-native conditions. Such functional modifications of UBPs are mainly introduced at the bases; however, due to the bulky sizes of functional groups or labeling probes used, such strategies might cause structural perturbations to the modified RNAs or decrease the accuracy of subsequent distance measurements.
An interesting functional modification of nucleic acids (DNA/RNA) is phosphorothioate (PS), which replaces one of the nonbridging oxygen atoms with sulfur in the phosphate backbone.23,24 PS-modified DNA and RNA have useful properties for various applications.25 For example, single-stranded PS-modified DNAs and RNAs have been effectively used to develop nuclease-resistant antisense drugs,24,26 mRNA with PS modification is shown to accelerate translation initiation.27 The PS modification on the backbone results in the formation of a chiral center; owing to its chiral nature, the PS substitution has been extensively used to probe metal binding and mechanisms of ribozymes.28 Furthermore, PS modification provides nucleophilic sites that are amenable to subsequent alkylation by labels containing iodoacetamide or maleimide.21,29 For example, multiple AuNPs and proteins can be precisely assembled on PS-modified DNA sequences by linking functional groups to the PS backbone.30,31 A number of potential advantages are available with backbone labeling in comparison to base-labeling procedures.21,29–31 First, compared to substitution at the nucleobase with functional groups such as alkyne, such single-atom substitution has fewer perturbations to the structure of nucleic acid. Second, as the phosphodiester bonds are generally solvent-exposed and not involved in base pairing, the presence of a label on the backbone may not drastically alter the stability and structure of duplex nucleic acids. Last but not the least, PS modifications are usually cost-effective. Nevertheless, PS-based site-specific labeling has been mainly performed for DNAs but less pursued for RNAs.
Here, we present the synthesis of an alpha-thiophosphorylated variant of rTPT3TP (rTPT3αSTP) and demonstrate its applications in post-transcriptional site-specific labeling of several large RNAs ranging from 97−719 nucleotides, including ribonuclease P (RNase P) from Bacillus stearothermophilus,10 3’stemloop (3′SL) element, and minigenomic (mini) RNA from dengue virus 2 (DENV2).9,32,33 RNase P is a ribozyme responsible for the 5′-end maturation of precursors of the transfer RNA (pre-tRNA). 3′SL and DENV-mini RNA represent the linear form and circular form of DENV2 genomic RNA, respectively. By combining transcription of the NaM-TPT3 UBP system and post-transcriptional thiol−maleimide reactions, we succeed in covalently attaching nitroxide spin label, AuNPs, and fluorophores into the PS backbones of such large RNAs in a site-specific manner. The labeled RNAs are further subject to PELDOR, XSI, and smFRET measurements, which result in narrower distant distributions than labeling at TPT3 bases. The presented strategy provides a new route for site-specific labeling of large RNAs and is expected to benefit the structural and dynamic studies of large RNAs by complementary biophysical techniques including PELDOR, XSI, smFRET, etc.
RESULTS
Chemical Synthesis of Alpha-Thiophosphorylated rTPT3TP.
The thiol−maleimide reactions have been widely used for bioconjugation and labeling of biomolecules such as cysteine-containing proteins.34 As natural RNAs do not contain thiols, to introduce a reactive thiol group at the phosphate backbone of RNA, we synthesized the alpha-thiophosphorylated ribonucleotide TPT3 triphosphate (rTPT3αSTP) by following the general procedures as described in the Supporting Information. The synthesized crude product of rTPT3αSTP can be readily separated into two diastereomeric (alpha-Sp) and (alpha-Rp) thiotriphosphates by reverse-phase HPLC (RP-HPLC) (Figure S1), which results from the replacement of nonbridging oxygen on the alpha-phosphorus atom of rTPT3TP by sulfur that makes the alpha-phosphorus atom stereogenic (Figure 1A). Because of the high efficiency of thiol−maleimide reactions under mild conditions, it is expected that RNAs containing one or multiple PS sites can be functionally derivatized with maleimide-modified labels in a single reaction (Figure 1B). The commercially available maleimide-based labels used in this study are shown in Figure 1C.
Figure 1.
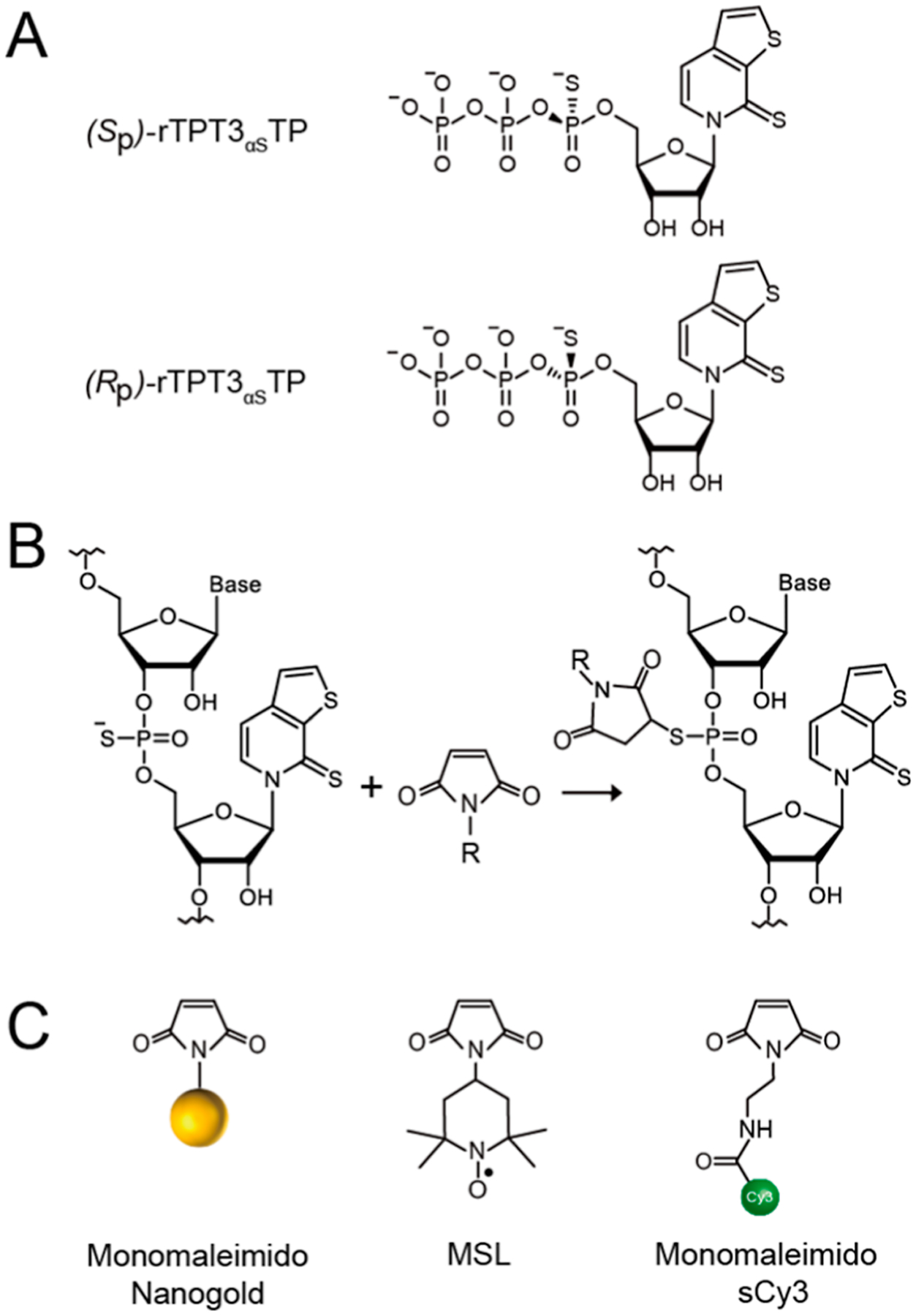
Compounds used in this study. (A) Chemical structures of the Sp and Rp diastereomers of rTPT3αSTP. (B) Schematic for covalent conjugation of rTPT3αS-modified RNAs with maleimide-modified probes via the thiol−maleimide reaction. (C) Chemical structures of the monomaleimido Nanogold, nitroxide spin label, and fluorescent dye (sCy3).
Selection of Model RNAs and Labeling Sites.
Previously, amine- and alkyne-functionalized modifications of rTPT3TP at the base (rTPT3ATP and rTPT3COTP) have been utilized for a site-specific spin and Nanogold labeling of several large RNAs including the 419-nt RNase P from B.stearothermophilus10 and the 97-nt 3′SL and 719-nt DENV-mini RNAs from DENV2.9 To demonstrate the applicability and advantages of PS-based RNA backbone labeling, the same labeling sites of the same RNase P (U67/U86) and 3′SL (A67/U97) RNAs were chosen to incorporate rTPT3αS for a subsequent site-specific spin and Nanogold labeling, respectively (Figure S2A,B). Based on the secondary and tertiary structures of these model RNAs, the labeling sites we selected are all located in the internal loops so that the introduction of rTPT3αS can avoid the potential interruptions of normal base pairing and will not cause drastic perturbation of the global architecture of the RNAs. Furthermore, the 3′SL and DENV-mini RNAs were chosen for site-specific fluorescent labeling and smFRET measurements based on total internal reflection fluorescence (TIRF) microscopy. The 3′-ends of 3′SL and DENV-mini, which represent the linear and circular conformations of flavivirus genomes, were cotranscribed with 30-nucleotide single-stranded RNA sequences (referred to 3′SL30 and DENV-mini30) (Figure S2C,D), which were annealed to a complementary DNA oligomer containing a 5′-end biotin and a 3′-end Cy5 for surface immobilization and fluorescent labeling. For orthogonal fluorophore labeling, suitable locations in 3′SL30 (U86) and DENV-mini30 (U708) were carefully chosen to incorporate rTPT3αS for subsequent Sulfo-cyanine3 (sCy3) labeling based on their secondary and tertiary structures. It is expected that the conformational changes of DENV2 genomic RNA due to linear-to-circular transition would result in evident FRET efficiency changes.
Preparation of PS-Containing Large RNAs.
For the transcription of singly (at the site of A67 or U97) or doubly (at sites of both A67 and U97) PS-modified 3′SL, or singly PS-modified 3′SL30 (at the site of U86) and DENV-mini30 (at the site of U708), as the labeling sites are close to the 3′ends, a single reverse primer containing two or one dNaMs was used to amplify the dsDNA templates by PCR and only one-step PCR was required. For a transcription of singly (at the site of U67 or U86) or doubly (at sites of both U67 and U86) PS-modified RNase P, a two-step overlap extension PCR was employed to generate the full-length UBP-modified dsDNA template (Figure 2A).
Figure 2.
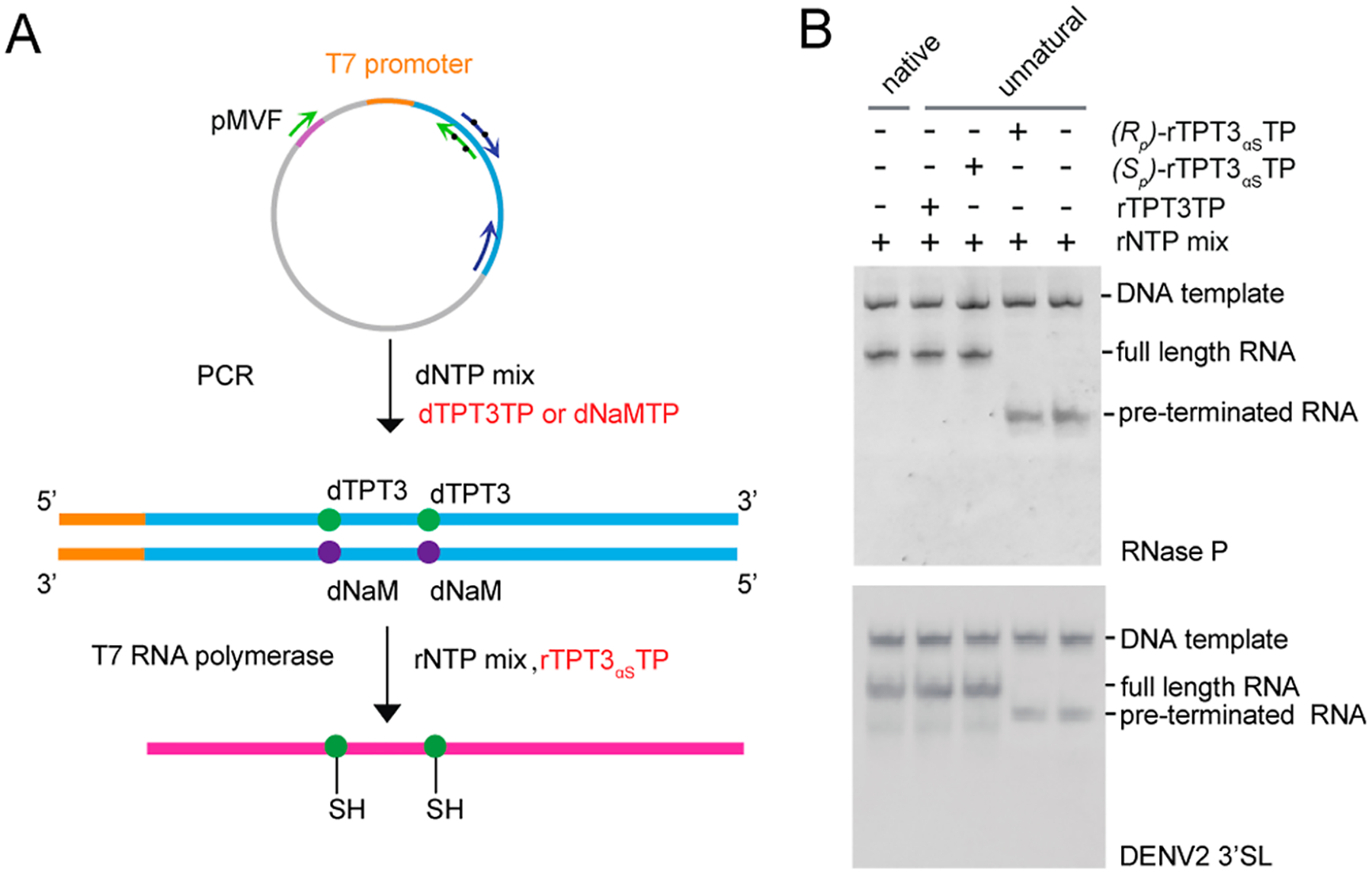
Site-specific PS modification of large RNAs by transcription of an expanded genetic alphabet containing TPT3-NaM UBP. (A) General procedure for site-specific PS modification of RNA. The double-stranded DNA template containing one or two dNaM modifications at the template strand can be prepared by PCR using a plasmid DNA template and single-stranded UBP-modified DNA primers. PSs can be site-specifically introduced into RNAs by IVT using an rNTP mix supplemented with (Sp)-rTPT3αSTP. (B) Native PAGE analysis of IVT of native or UBP-modified DNA templates encoding (upper) RNase P and (lower) DENV2 3′SL using Sp and Rp diastereomers of rTPT3αSTP. T7 RNAP only accepts (Sp)-rTPT3αSTP as a substrate for IVT. Transcription of native DNA template using rNTP mix results in one single band. rTPT3 or (Sp)-rTPT3αS can be efficiently incorporated into RNase P RNA and 3′SL by an IVT reaction catalyzed by T7 RNAP. If no rTPT3TP or only (Rp)-rTPT3αSTP is added to the IVT reaction, no full-length RNAs but only short abortive transcripts are observed.
T7 RNA polymerase (RNAP) was used to incorporate rTPT3αS into RNase P, 3′SL, 3′SL30, and DENV-mini30 RNAs. Studies have shown that T7 RNAP only accepts the Sp diastereomer as the substrate.35 To determine which diastereomer fraction is the substrate of T7 RNAP, we performed small-scale IVT for both RNase P and 3′SL RNAs using the unnatural dsDNA templates. As shown in Figure 2B, only one diastereomer can be efficiently utilized by T7 RNAP, which is then assigned as the Sp form. Transcriptions with the Sp form of rTPT3αSTP show major single bands for both RNase P and 3′SL (Figure 2B) RNAs; by contrast, only abortive RNAs were generated without Sp-rTPT3αSTP in the rNTP mix. These results indicate the successful incorporation of the Sp form of rTPT3αSTP into the specific sites of both RNase P and 3′SL RNAs, which were further purified by size-exclusion chromatography (SEC) under native conditions (Figure S3A,B). As only a small number of samples are required for smFRET measurements, both rTPT3αS-modified 3′SL30 and DENV-mini30 were purified by the native gel electrophoresis method (Figure S3C).
Reactivity and Stability of rTPT3αS-Containing RNAs.
There are three sulfur atoms in each rTPT3αS including one endocyclic and one exocyclic in the base and one in the phosphate backbone in rTPT3αS-modified RNA. To test their differences in reactivities to maleimide, singly rTPT3αS- or rTPT3-modified 3′SLs at the site of A67 were reacted with monomaleimido sCy3, but only rTPT3αS-modified RNA could be incorporated with the fluorophore, indicating that the PS is more nucleophile than the exocyclic and endocyclic sulfurs on the base (Figure S4).
Although it has been reported that incorporation of the thiol group in the internal phosphate backbone of RNA could result in a hydrolysis reaction with the previous 2′ hydroxy group,36 the rTPT3αS-modified RNAs used in this study are stable in near natural pH conditions (pH = 7). As shown in Figure S5, even after storage at 4 °C for 48 h, still no hydrolysis or degradation is observed for rTPT3αS-modified RNase P, 3′SL, and 3′SL30 RNAs. Besides, IVT is performed under alkalescent conditions, and the rTPT3αS-modified full-length RNA still could be synthesized.
We further characterize the hydrolytic stability of the above sCy3-labeled 3′SL under different buffer conditions. The rTPT3-modified 3′SL is used as control. rTPT3-modified and sCy3-labeled 3′SLs in various buffers with pHs ranging from 4−10 were heated at 65 °C for 10 min before being assayed by denaturing PAGE gels (Figure S6A–B). When the buffers are Mg2+-free, no significant hydrolysis can be observed for both RNAs at all pHs; by contrast, when the buffers are supplemented with 5 mM Mg2+, both rTPT3-modified and sCy3-labeled 3′SLs are still stable below pH 10 but are completely hydrolyzed at pH 10 (Figure S6B). These results are consistent with the phenomenon of nonenzymatic RNA hydrolysis promoted by buffer and Mg2+ at high temperatures.37 The time dependence of Mg2+-promoted RNA hydrolysis was also assayed. As shown in Figure S6C, rTPT3-modified 3′SL was partially hydrolyzed after being heated at 65 °C for 15 min in buffer containing 5 mM Mg2+ and at pH 9, but sCy3-labeled 3′SL was completely hydrolyzed under the same condition, indicating that rTPT3αS-modified RNA (sCy3-labeled 3′SL) is more hydrolytically sensitive than rTPT3-modified RNA under alkaline conditions. Taken together, the rTPT3αS-containing RNA can be kept stable under certain conditions.
Effects of PS Modification on the RNA Structure and Function.
To investigate the effects of PS modification on the structure and function of RNAs, we first characterized the rTPT3αS-modified RNase P and 3′SL RNAs using SAXS. The scattering profiles of singly or doubly rTPT3αS-modified RNase P RNAs and 3′SL RNAs are superimposable to the scattering profiles of the corresponding wild-type RNAs. The Guinier regions of all the scattering profiles are linear, indicating that all RNA samples are monodispersed and homogeneous in solution. The paired distance distribution functions of the singly or doubly rTPT3αS-modified RNase P RNAs and 3′SL RNAs are similar to those of the corresponding wild-type RNAs (Figure 3A–C). The radius of gyration (Rg), maximum diameter (Dmax), and molecular weights are summarized in Table S1. The similar Rg and Dmax of the rTPT3αS-modified and the corresponding wild-type RNAs indicated that PS modification did not cause significant structure perturbations (Table S1).
Figure 3.
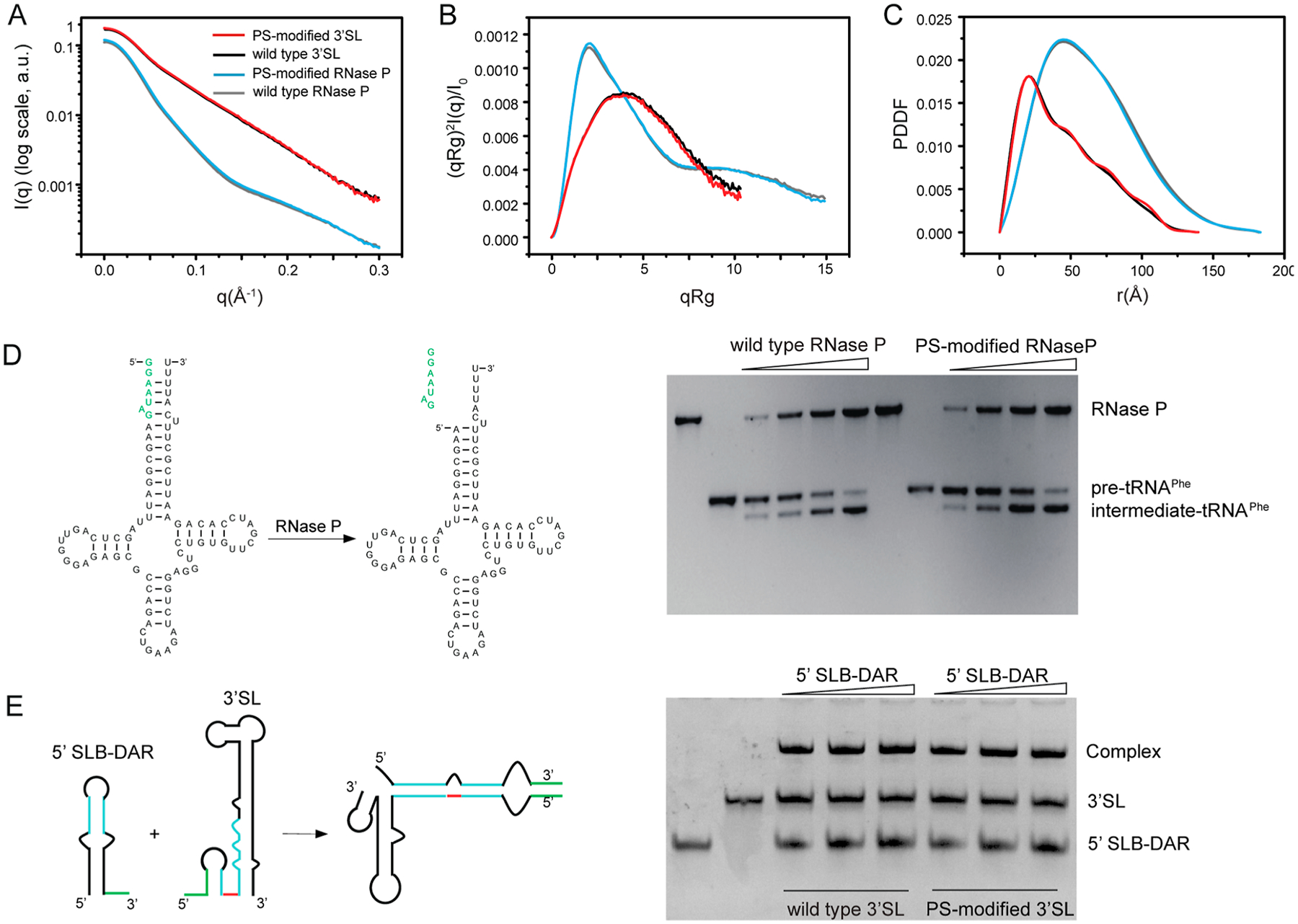
Phosphorythionate modifications have minimal effects on the global structure and function of RNAs. (A−C) The experimental scattering profiles (A), dimensionless Kratky plots (B), and PDDFs (C) of PS-modified RNase P (blue) and 3′SL (red) RNAs are superimposable to those of the corresponding wild-type RNase P (gray) and 3′SL (black) RNAs. (D) Pre-tRNA processing assay. Left: secondary structure of the yeast pre-tRNAPhe and the intermediate tRNAPhe product. Right: PS-modified and wild-type RNase P show a similar processing activity. The concentration of both wild-type and PS-modified RNase P RNAs increase from 10, 25, 50, to 100 nM, while the concentration of pre-tRNAPhe is constant. No obvious differences in concentration dependence of enzymatic activities can be observed between wild-type and PS-modified RNase P RNAs. (E) EMSA of 5′SLB-DAR in the absence or presence of wild-type or PS-modified 3′SL RNAs. The concentrations of 3′SLs are constant. The 5′SLB-DAR:3′SL molar ratio is 0.5:1, 1:1, and 1.5:1, respectively.
We next investigated whether rTPT3αS-modification affects the RNase P catalytic activity. RNase P is a ribozyme that catalyzes the hydrolysis of a phosphodiester bond in precursors of transfer RNA (pre-tRNA), resulting in the formation of the 5′-phosphorylated intermediate tRNA and the release of a 5′-precursor fragment. As shown in Figure 3D, using a yeast pre-tRNAPhe as a substrate, we observed the formations of intermediate tRNAPhe products in the presence of both wild-type and doubly rTPT3αS-modified RNase P RNA, suggesting that PS modification does not affect the catalytic activity of RNase P RNA. We further assess whether PS modification disturbs 3′SL function. We applied an in vitro electrophoretic mobility shift assay (EMSA) between doubly rTPT3αS-modified 3′SL and DENV 5′SLB (stem-loop B)-DAR (downstream of the AUG region), which contains two complementary sequences, 5′UAR (upstream of AUG region) and DAR to the 3′SL.9 As shown in Figure 3E, both the wild-type and doubly rTPT3αS-modified 3′SL interact with its complementary sequence in 5′SLB-DAR to form hybrid RNAs, indicating that PS modification does not impair the folding and binding activity of 3′SL RNA. Taken together, PS modification minimally perturbs the structure and function of RNase P and 3′SL RNAs.
Distance Measurement of RNase P by PELDOR Spectroscopy.
The purified singly or doubly rTPT3αS-modified RNase P RNAs were subjected to spin labeling via the thiol−maleimide reaction (Figure 4A). The maleimide spin-labeled RNase P RNAs were purified with HiTrap Q anion-exchange chromatography column followed by SEC (Figure S7). The kinetics of the pre-tRNA processing assay indicated that spin labeling did not impair the catalytic activity by the RNase P (Figure S8).
Figure 4.
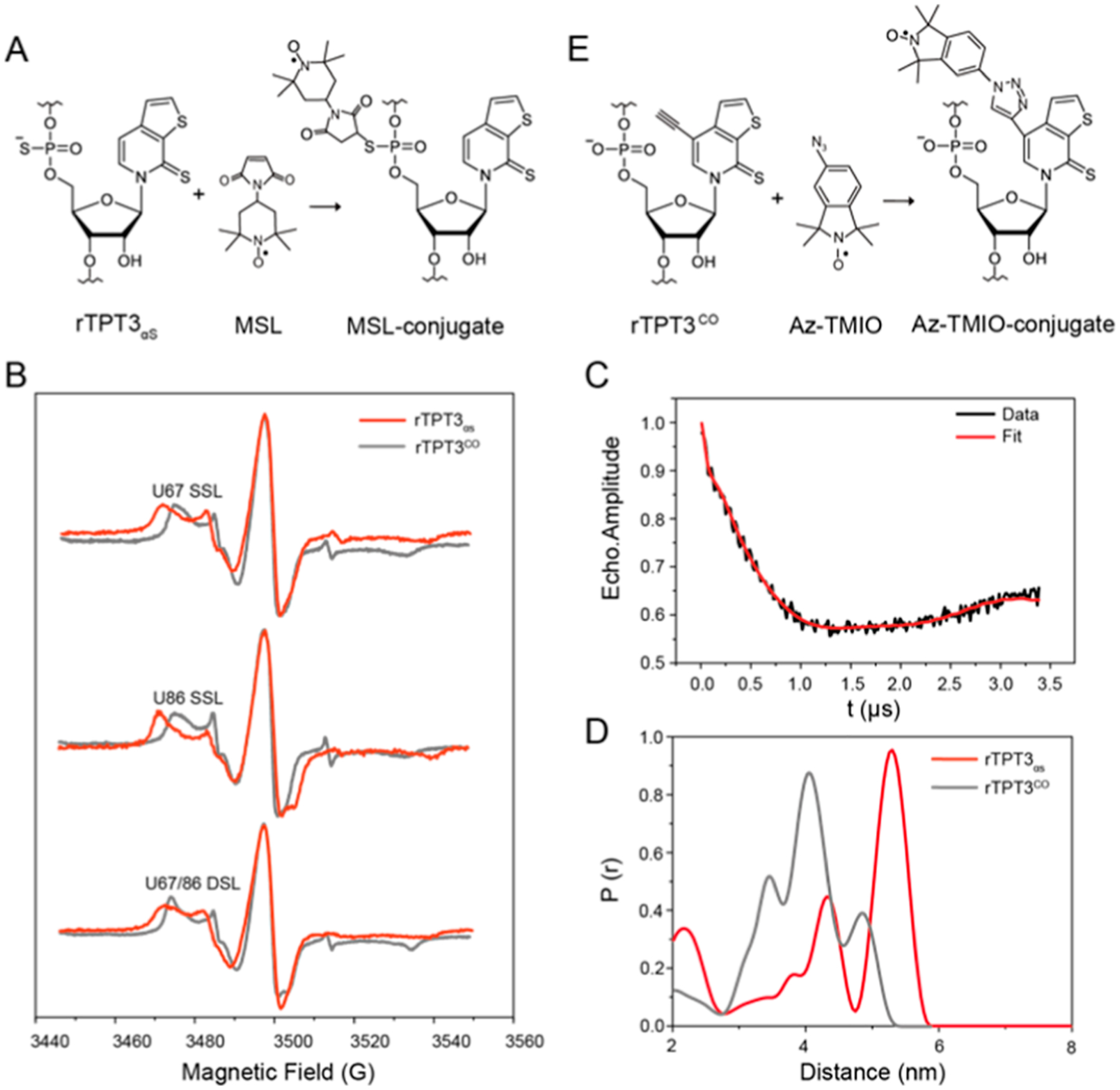
PS-based site-specific spin labeling of RNase P RNA and distance distribution measurement by PELDOR. See also Figure S7 for additional PELDOR data and analysis. (A) Schematic for covalent conjugation of MSL to rTPT3αS-modified RNA via the thiol−maleimide reaction. (B) CW-EPR spectra of the rTPT3αS-based (red) U67 (top) or U86 (middle) SSL and U67/U86 (bottom) DSL RNase P RNAs. CW-EPR spectra of rTPT3CO-based (gray) singly and DSL RNase P RNAs at the same sites were overlaid for comparison. (C) Background-corrected time trace of DSL RNase P. (D) Interspin distance distributions of the rTPT3αS-based (red) and rTPT3CO-based (gray) U67/U86 DSL RNase P RNAs obtained using the Tikhonov regularization approach. (E) Schematic for the click chemistry reaction between RNA containing alkyne-functionalized rTPT3 and azide-modified nitroxide.
To confirm the successful coupling of nitroxide−maleimide to PS-modified RNase P RNAs, we measured the X-band continuous-wave (CW) electron paramagnetic resonance (EPR) spectra of the spin-labeled RNAs at room temperature and compared them with that of the free nitroxide standard. The line shape of a CW-EPR spectrum is dictated by the reorientation dynamics (rotational motions) of the nitroxide. As nitroxide motions reduce, averaging of its g- and hyperfine tensors becomes incomplete, resulting in line broadening and the appearance of extra features at the low-field and high-field regions.38 As shown in Figure 4B, all MSL-labeled RNA spectra show broad lines, indicating a large reduction of the nitroxide motility that is consistent with what is expected from the successful covalent attachment of nitroxide to the high molecular-weight RNase P RNAs. Spin-counting was carried out for the measured CW-EPR spectra, and the labeling efficiencies for singly spin-labeled (SSL: at sites of U67 or U86) and doubly spin-labeled (DSL: at sites of both U67 and U86) RNase P RNAs are 47, 48, and 50%, respectively (Table S2).
The DSL RNase P sample was used for distance measurements by PELDOR. Figure 4C shows the background-corrected X-band PELDOR trace, which clearly demonstrates the occurrence of dipolar oscillation. The absolute modulation depth is ~35%, consistent with the measured high efficiency of labeling. The distance distributions obtained using the Tikhonov regularization approach39 are broad (Figure 4D), which gives a most populated and sharp peak at 5.3 nm, and two minor peaks at 4.6 and 2.3 nm, respectively. As a control, no dipolar decay and oscillation was observed from the SSL RNase P RNA samples, indicating that the signal of DSL RNase P RNA is intramolecular, rather than intermolecular, arising from the aggregation of the labeled RNA at high concentrations (Figure S9). According to the crystal structure of RNase P (PDB ID: 2a64), the distance between the respective N1 atoms of U67 and U86 (labeling sites) is 3.32 nm. To account for the measured distance, both U67 and U86 in the crystal structure are replaced with rTPT3, and the attached MSL was modeled using the ALLNOX program.40 The program generates rotamers of the MSL label by stepwise variations of the rotatable torsional angles in the linker, identifies the rotamers without steric collision within the macromolecule, and then outputs the interlabel distances. It appears that some of the rotamers fit within the loops of the RNA, which gives rise to a configuration with a longer distance of 4.9 nm (Figure S10). Considering the inherent flexibility of the large RNase P RNA, the local dynamics of the PS triester backbone, and the size of MSL nitroxide including the linker, the measured distance distribution is reasonable.
The present EPR data were further compared with the previous EPR data of RNase P RNA, to which the nitroxide was covalently attached at the base of rTPT3CO (Figure 4E). As shown in Figure 4B, a broader △Hpp and 2Aeff are observed in the CW-EPR spectra of the present RNase P RNAs, indicating that at these labeling sites, the nitroxide spin label attached to the RNA backbone is more rigid than that attached to the base of rTPT3. These results are consistent with the sharp distance distributions for the present DSL RNase P RNA (Figure 4D).41 Nevertheless, both rTPT3CO-based and rTPT3αS-based DSL RNase P show three peaks in the distance distributions, which could result from the local dynamics of the substituted residues (Figure S10).
Distance Measurement of 3′SL by XSI.
Purified singly and doubly rTPT3αS-modified 3′SL were subjected to Nanogold labeling in the presence of two- or fivefold excess of 1.4 nm monomaleimido Nanogold (Figure 5A). The excess free Nanogold and Nanogold-labeled RNAs were separated by using a HiTrap Q anion-exchange chromatography column (Figure S11). The successful conjugation of monomaleimido Nanogold to PS-modified 3′SL RNAs can be verified by the absorbance at 420 nm of the Nanogold. The Nanogold labeling efficiency can be determined by measuring the UV−vis absorbance of the Nanogold-labeled RNAs at 260 and 420 nm, respectively, as previously reported.9 The efficiencies of the singly Nanogold-labeled (at sites of A67 or U97) and the doubly Nanogold-labeled (at sites of both A67 and U97) 3′SL RNAs are 54, 60, and 61%, respectively (Table S2).
Figure 5.
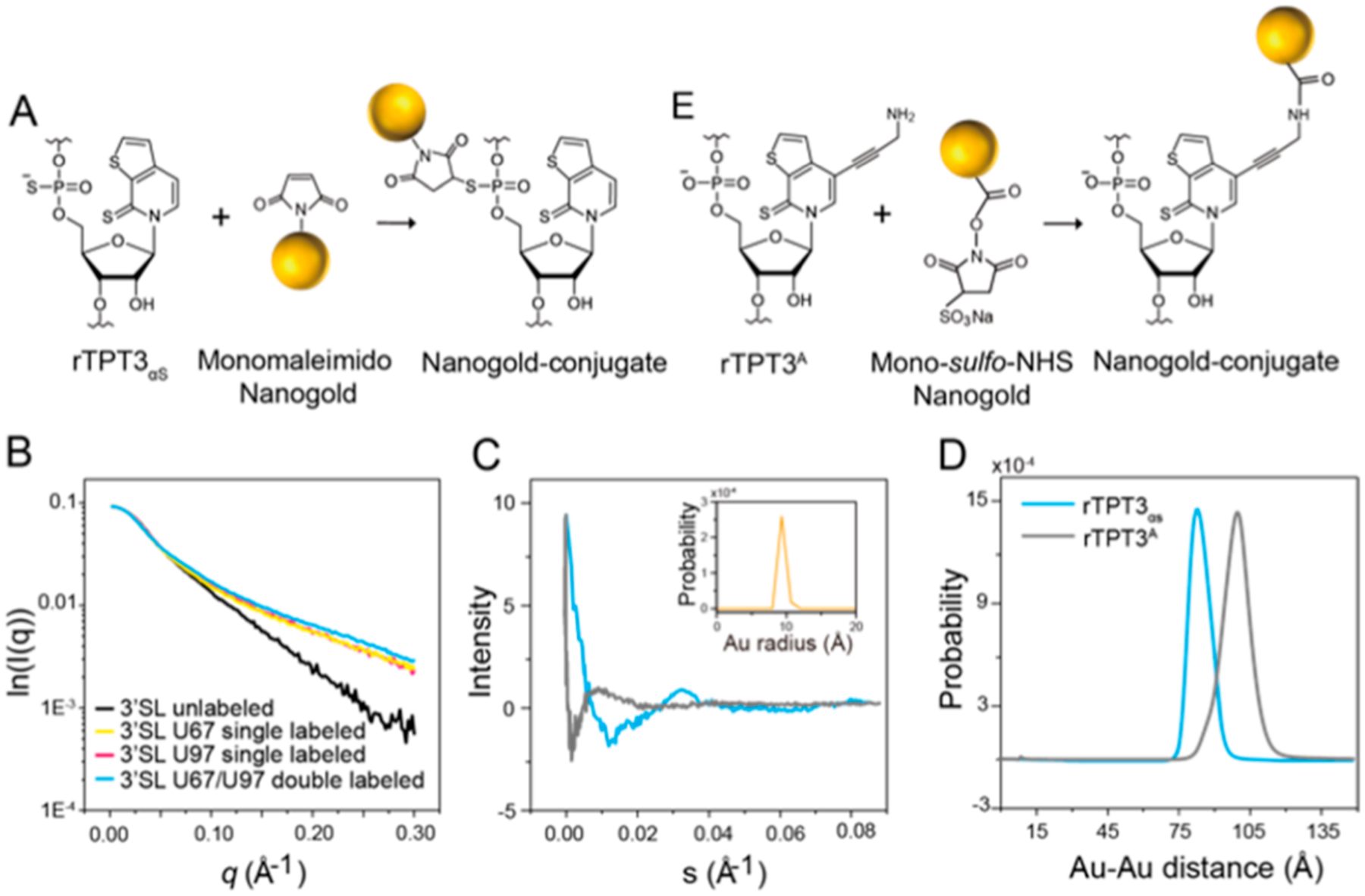
PS-based site-specific Nanogold labeling of 3′SL RNA and distance distribution measurement by XSI. (A) Schematic for covalent conjugation of monomaleimido Nanogold to rTPT3αS-modified RNA via the thiol−maleimide reaction. (B) I0-normalized scattering profiles of U67 singly Nanogold-labeled (yellow), U97 singly Nanogold-labeled (red), U67/U97 doubly Nanogold-labeled (blue), and unlabeled 3′SLs (black). (C) Scattering interference patterns of the two rTPT3αS-conjugated (blue) or rTPT3A-conjugated (gray) Nanogold labels in 3′SL RNAs. (D) Probability distribution of the center-to-center distance distributions between the two rTPT3αS-conjugated (blue) or rTPT3A-conjugated (gray) Nanogold labels in 3′SL RNAs. (E) Schematic for covalent conjugation of mono-sulfo-NHS-Nanogold to rTPT3A-modified RNA via the amine-N HS ester reaction.
To demonstrate the applicability of PS-based Nanogold labeling of large RNA, we measured the distance distributions between sites of A67 and U97 in 3′SL by XSI. The scattering profiles of the quartets of 3′SL and the free AuNP were normalized against I0 and are shown in Figure 5B. When compared with the native 3′SL sample, the two singly labeled and doubly labeled 3′SL showed a prominent increase in scattering intensity at an elevated q range from 0.05 to 0.30 Å, which is consistent with stable conjugation of the RNAs with one or two Nanogolds that scatter X-ray strongly. The overall structural parameters derived from scattering profiles are shown in Table S1. The full set of scattering profiles of the RNA quartets and the free Nanogold were combined to derive the Nanogold−Nanogold scattering interference profile and the center-to-center distance distribution between the pairs of Nanogold for the 3′SL by following the previously described protocols.42,43 The Nanogold−Nanogold scattering interference profile shows obvious oscillation, suggesting a rigid distance distribution (Figure 5C). Transformation of the scattering interference profiles results in distance distribution with mean values of 75.8 Å and a variance of 48.5 Å2 (Figure 5D). The distance between P1 atoms of A67 and U97 is 84.0 Å in the atomic model of 3′SL.44 Given the radius of Nanogold and the length of the linkers, the mean AuNP−AuNP distance of 75.8 Å in 3′SL RNA is reasonably consistent with the atomic model. The Nanogold−Nanogold scattering interference profiles and distance distributions of the 3′SL RNAs prepared with the rTPT3αS-based and rTPT3A-based labeling schemes were further compared (Figure 5E). Clearly, the PS-based backbone Nanogold labeling scheme results in a larger first oscillation dip in the interference profile and shorter but sharp (covariance: 48.5 vs 52.7 Å2) distance distribution than the previous base-based Nanogold labeling scheme (Figure 5B,D).
Conformational Dynamics of the 3′SL RNA Characterized by smFRET.
The single-stranded positive-sense RNA genome of flavivirus undergoes conformational transitions between the linear and circular forms to fulfill its diverse roles in translation, replication, and packaging.32,33 The circular form of genomic RNA is mediated by long-range RNA−RNA interaction, and according to the current model, 3′SL is supposed to undergo significant conformational change during genome cyclization (Figure S2C–D). To demonstrate the transition between the two conformations of the flavivirus genome, the 97-nt 3′SL and the 719-nt DENV-mini RNAs from dengue virus 2, which represent the linear and circular conformations of the flavivirus genomic RNA, have been site-specifically Nanogold-labeled and characterized by XSI.9 To demonstrate the applicability of PS-based fluorophore labeling in the characterization of conformational dynamics of large RNAs by smFRET, purified rTPT3αS-modified 3′SL30 at the site of U86 (3′SL30-αS) and DENV-mini30 at the site of U708 (DENV-mini30-αS) (Figure S12) were subjected to site-specific labeling with sCy3 maleimide (Figure 6A). The labeling efficiencies were estimated to be 34 and 40%, respectively, by quantifying the UV−visible absorption at 260 and 549 nm of sCy3-labeled RNAs (Table S2). The EMSA assay indicated that both wild-type and sCy3-labeled 3′SL30 integrated with 5′SLB-DAR to form the complex, showing that sCy3 labeling had minimal perturbation on the function of 3′SL30 (Figure S13).
Figure 6.
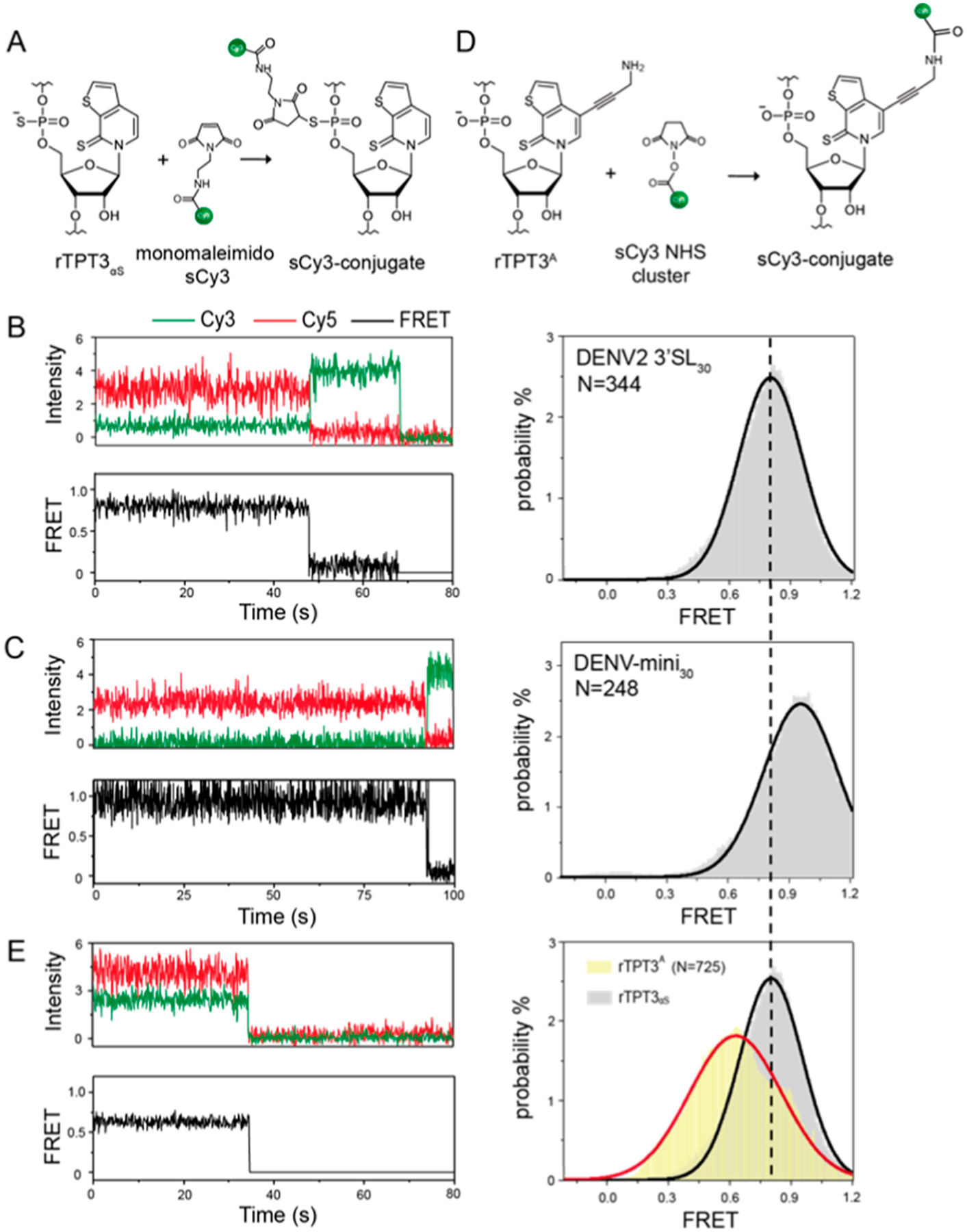
PS-based site-specific sCy3 fluorophore labeling of 3′SL30 and DENV-mini30 RNAs and conformational dynamics measurement by smFRET. (A) Schematic for covalent conjugation of maleimide-modified sCy3 to rTPT3αS-modified RNAs via the thiol−maleimide reaction. (B,C) Representative smFRET traces (left) and FRET histogram (right) of (B) DENV2 3′SL30 and (C) DENV-mini30 RNAs with rTPT3αS-conjugated sCy3. The corresponding FRET intensity trajectories are shown by the black trace. N denotes the total number of traces used to generate the histograms. (D) Schematic for covalent conjugation of rTPT3A-modified RNA with the sCy3 NHS ester via the amine−NHS ester reaction (right). (E) Representative smFRET trace (left) and FRET histogram (right) of DENV2 3′SL30-base with rTPT3A-conjugated sCy3. The FRET distribution of 3′SL30 with rTPT3αS-conjugated sCy3 is displayed for comparison.
The sCy3-labeled RNAs were further annealed with 5′-biotin- and 3′-Cy5-labeled DNA oligonucleotides to generate orthogonally labeled RNAs and achieve surface immobilization. The time-dependent sCy3 and Cy5 fluorescence signals emitted from the respective dye-labeled RNAs were collected and are shown in Figure 6B,C. The FRET efficiencies (EFRET) were calculated from the sCy3 and Cy5 intensities of hundreds of individual molecules, which reflects the intramolecular distance between sCy3 and Cy5 labeling sites.45 The FRET efficiency histograms, in which the number of FRET events was plotted against FRET values, display a major peak for both 3′SL30-αS and DENV-mini30-αS, whose peak centers and populations were further extracted via Gaussian fitting. The peak EFRET values for 3′SL30-αS and DENV-mini30-αS are 0.80 ± 0.01 and 0.99 ± 0.01, respectively. In addition, the 90% traces of the EFRET value of 0.99 have Cy5 bleached first accompanied by sCy3 increase, followed by sCy3 bleaching later, which means that there may be little or no dye-dye quenching between Cy5 and sCy3. Therefore, the change in FRET efficiency indicated that 3′SL undergoes a significant conformational change and the distance between the labeling sites in 3′SL becomes shorter upon genome cyclization. These results are consistent with the directly measured distances between the P1 atoms of residues in the atomic models of 3′SL (U86 and U97: 2.89 nm) and DENV-mini (U708 and U719: 1.85 nm), respectively, since U97 and U719 are expected to be close to the Cy5 labeling sites.
Previously, an amine-derivatized rTPT3TP (rTPT3ATP) has been utilized to achieve site-specific Nanogold labeling of large RNAs at the rTPT3 base through the UBP system.9 To compare the effects of different RNA fluorescent labeling schemes on the FRET efficiency histograms, a new 3′SL30 RNA with sCy3 labeled at the rTPT3 base (3′SL30-base) was prepared using rTPT3ATP and the UBP system by following similar procedures (Figure 6D). The 3′SL30-base is further annealed with 5′-biotin- and 3′-Cy5-labeled DNA oligonucleotides to generate orthogonally labeled RNAs and achieve surface immobilization. As shown in Figure 6E, the FRET efficiency histogram of 3′SL30-base RNA under the same condition shows a broader distribution with peak EFRET values of 0.63 ± 0.01. Given the bulky size of rTPT3A and its longer linker, it is reasonable that the distance between sCy3 and Cy5 labeling sites of 3′SL30-base becomes longer. Furthermore, the FRET efficiency distribution for 3′SL30-base RNA is broader than that for 3′SL30-αS RNA. These results are consistent with the above PELDOR and XSI observations that functional probes labeled at the internal phosphate backbone results in narrower distance distributions than that at the base.
DISCUSSION
In this work, we synthesize an α-thiophosphorylated triphosphate of ribonucleotide TPT3 (rTPT3αSTP) and have demonstrated its applicability to empower the molecular rulers of PELDOR, XSI, and smFRET for accurate distance distribution measurements, which are fundamental to understanding the structure, conformational dynamics, and function of large RNAs. Our results demonstrate that by site-specific incorporation of one and/or two PS triesters into the backbone of large RNAs, commercially available maleimide-based spin labels, gold nanoparticles, and fluorescent dyes can be covalently attached to large RNAs via the thiol−maleimide reactions in an efficient manner. Compared with the nucleotide base-based covalent labeling schemes, in which the functional groups are usually large, RNA labeling schemes through the PS triester backbone modifications cause minimal perturbations to the structure and function of RNAs and result in narrower distance distributions and thus can increase the accuracy of the distance distribution measurements. PELDOR, XSI, and smFRET are complementary techniques. For example, both PELDOR and XSI generally provide ensemble-averaged distance distributions, but FRET can be performed at both the ensemble and single-molecule levels. While PELDOR and smFRET can measure a distance in the typical range of 20−80 Å, XSI can provide information about distance distribution for a considerable range of distances ranging from 50 up to 400 Å. Thus, starting from the same DNA templates, different types of labeling can be easily achieved and allow for comprehensive structural and dynamic characterization for the same RNA using complementary molecular rulers.
Our work also demonstrates the potential for future applications of PS-modified RNA through covalent modifications. Currently, there are a few examples of applications of PS-modified DNAs. For example, proteins, gold nanoparticles, photosensitive molecules, and functional tags can be precisely assembled to the PS sites of DNAs for various applications.25 By contrast, applications of PS-RNAs are even less pursued. One of the reasons could be that it is challenging to site-specifically incorporate PS triesters into large RNAs. Facilitated by transcription of the expanded genetic alphabet containing NaM-TPT3 UBP system, our RNA polymerase-catalyzed strategy overcomes the size limits in conventional chemical synthesis methods (in this work: 97−719 nts), and it is expected that the demonstrated strategy can have wide applications in RNA nanotechnology or surface sciences; for example, functional groups can be covalently attached to the PS sites in RNA origami or nanostructures of several hundred to thousands of nucleotides. Since T7 RNAP only accepts Sp diastereomer as a substrate, chiral pure PS-RNAs can be efficiently prepared. This differs from the solid-phase chemical synthesis approach, which usually requires postsynthesis processing to obtain chiral pure PS-RNA.46 This will not only benefit biophysical studies presented in this work but also could enable additional applications, for example, investigating the role of metal coordination in RNA folding and catalysis. Nevertheless, the coupling efficiencies via thiol−maleimide reactions reported in this work are somewhat lower than the previous “click chemistry” method. This could be explained by the relatively lower reaction activity between the thiol group and the maleimide group when compared with the “click chemistry” reaction.47,48
MATERIALS AND METHODS
Materials.
The phosphoramidites of dNaM and dTPT3, triphosphorylated nucleotides of dNaM, dTPT3, rTPT3, and amine-derivatized rTPT3 (rTPT3A) were custom-synthesized by WuXi AppTec, China, as previously reported.9 The maleimide spin label (MSL) N-(1-Oxyl-2,2,6,6-tetramethyl-4-piperidinyl) maleimide was purchased from Toronto Research Chemicals Inc. The 1.4 nm monomaleimido Nanogold was purchased from Nanoprobes, Inc. The sCy3 maleimide and sCy3 NHS ester were purchased from Lumiprobe Cooperation. 2× Golden mix of Taq DNA polymerase (supplied with buffer and natural dNTPs mix) was purchased from TSINGKE Biological Technology Co., Ltd. (Beijing, China). The T7 RNAP was homemade as previously described.9 Plasmids encoding the RNase P RNA from B. stearothermophilus, the pre-tRNAPhe from yeast, the 3′SL, and the DENV-mini RNAs of Dengue Virus 2 with an upstream T7 promoter were the same as previously reported.9,10 All DNA oligonucleotide primers (containing natural and/or unnatural nucleotides) were synthesized by regular solid-phase chemical synthesis and purified by OPC purification. The 5′-biotinylated and 3′-Cy5-modified DNA oligonucleotide (5′ -biotin-CCTGGTCCGGTGGTCCGCCTGCTGGTC-Cy5-3′) was synthesized by Sangon Biotech (Shanghai, China).
Preparation of Native or UBP-Modified DNAs.
The procedures to generate double-stranded DNA (dsDNA) templates for IVT are similar to those previously reported.9–11 In brief, to generate dsDNA templates for IVT of native RNAs (RNase P, tRNAPhe, 3′SL, and 3′SL30), PCRs were performed using a forward primer (pMVF) targeting a common sequence 624 bp upstream of the T7 promoter and a reverse primer specific to the respective cDNAs in the plasmids. To prepare unnatural dsDNA templates containing one or two NaMs at the template strands for IVT of singly or doubly UBP-modified RNAs, two-step overlap extension PCRs were performed. In step I, reverse primers containing dNaM nucleotides were used to introduce the dNaM and dTPT3 into the intermediate DNA fragments by PCR. The intermediate DNA fragments were recovered and mixed to assemble the full-length DNA templates by PCR in step II, which can be further PCR-amplified to produce the respective dsDNA templates for subsequent large-scale IVTs.
Preparation of Native or UBP-Modified RNAs.
The IVT systems for native or UBP-modified RNAs are similar as previously reported.9–11 Prior to large-scale RNA preparations, the optimal MgCl2 concentrations for the respective RNAs were screened in a 20 μL IVT reaction system. As T7 RNAP only accepts Sp diastereomer of rNTP as a substrate (see Results), the final concentrations of Sp-rTPT3αsTP and rTPT3ATP in the reactions are 1 mM. The native dsDNA templates were used for the preparation of the respective wild-type RNAs (RNase P RNA, pre-tRNAPhe, 3′SL, and 3′SL30). The unnatural dsDNA templates containing one or two NaMs at the template strands were used for the preparation of site-specifically TPT3αs- or TPT3A-modified RNAs.
The purification protocols for the respective RNAs are similar to those previously reported.9,10 For native or rTPT3αs-modified RNase P and 3′SL RNAs which were subject to subsequent spin or Nanogold labeling, since the transcription products migrate as a single major band on native PAGE, the transcription supernatants were directly applied to Superdex 200 and Superdex 75 Increase 10/300 GL columns, respectively, and purified by SEC. The SEC buffer contains 100 mM KH2PO4/K2HPO4, 1 mM EDTA, pH 7.0. For rTPT3αs- or rTPT3A-modified 3′SL30 and DENV-mini30 RNAs which were subject to subsequent fluorophores labeling, as only little RNAs were required, the RNAs were purified by native polyacrylamide gel electrophoresis using the “crush and soak” method and recovered with FRET buffer containing 20 mM Na2HPO4/NaH2PO4, pH 6.7, 100 mM KCl, 1 mM EDTA, and 1 mM TCEP (Tris (2-carboxyethyl) phosphine hydrochloride). The purified RNAs were collected and concentrated with the Amicon Ultra centrifugal filter devices (Sigma-Aldrich) and stored at −80 °C for further use.
Hydrolytic Stability of PS-Containing RNA.
rTPT3-modified, rTPT3αS-modified, or rTPT3αS-labeled RNAs (5 pmol) were incubated at 4 or 65 °C at different buffer conditions and then assayed on 12% denaturing PAGE gel.
Small-Angle X-ray Scattering.
All the natural and unnatural RNA samples were exchanged into the final SAXS buffer containing 20 mM HEPES, 150 mM NaCl, 1 mM MgCl2, 5 mM DTT, and 5% (v/v) glycerol, pH 7.0, using the Amicon Ultra centrifugal filter devices (Millipore). All natural and unnatural RNA samples without Nanogold labeling were diluted to final concentration series of 0.75−3 mg/mL. The data collection and processing procedures are similar to those described before.44 In brief, SAXS data were collected at room temperature at the Beamline 12-ID-B of the Advanced Photon Source, Argonne National Laboratory. The setup was adjusted to achieve scattering q values of 0.005 < q < 0.089 Å−1, where q = (4π/λ) sin θ and 2θ is the scattering angle. 30 2D images for each buffer or sample were recorded using a flow cell with an exposure time of 1 s for natural or unnatural RNAs so as to minimize radiation damage and obtain a good signal-to-noise ratio. The 2D images were reduced to one-dimensional scattering profiles by MATLAB script onsite. The scattering profile, the forward scattering intensity (I0), the radius of gyration (Rg), the pair distance distribution function (PDDF) p(r), and the maximum dimension, Dmax, of the RNA samples were calculated using the same procedures as described before.44 The volume of correlation (Vc) was calculated by using the program Scatter, and the molecular weights of natural RNAs were calculated on a relative scale using the Rg/Vc power law.49
Pre-tRNA Processing Assay.
The pre-tRNA processing assay was performed as previously reported.10,50 In brief, the enzymatic activity of the native and PS-modified RNase P RNAs was tested by performing the pre-tRNA processing assay, in which the yeast pre-tRNAPhe substrate (40 nM) was mixed with native or PS-modified RNase P RNAs (10, 25, 50, and 100 nM, respectively). The reactions were carried out in the SEC buffer with the addition of 15 mM MgCl2 and 400 mM KCl. The mixtures were incubated at 50 °C for 30 min, and subsequently, the reactions were quenched by adding 8 M urea and 5 mM EDTA. All reaction mixtures were denatured at 95 °C for 10 min and loaded directly on a 15% denaturing polyacrylamide gel. The polyacrylamide gel was stained with Gelsafe (YPR-Bio) and imaged with a Tanon 2500 Gel Imaging Systems. The kinetics of pre-tRNA processing by RNase P was performed by incubating 20 nM native or spin-labeled RNase P RNAs with a 40 nM pre-tRNAPhe substrate under the same condition as just mentioned; sampling was performed at 5, 10, 20, and 30 min for analysis.
Electrophoretic Mobility Shift Assay.
The EMSA was performed by following the previously published protocol.9 In brief, 2 pmol of wild-type, rTPT3αS-modified, or sCy3-labeled 3′SL RNAs was mixed with 1/2/3 pmol of 5′ SLB-DAR RNA, and the mixtures were incubated at 37 °C for 30 min. The reaction mixtures were then analyzed using 10% nondenaturing PAGE. After electrophoresis, the gels were stained with 1:1000 diluted Gelsafe (YPR-Bio) for 7 min and imaged with a Tanon 2500 Gel Imaging Systems. The grayscale was analyzed with ImageJ.
Site-Specific Incorporation of Nitroxide Spin Labels into RNase P RNA.
The MSL nitroxide spin label was freshly dissolved with DMSO to a final concentration of 200 mM. Prior to nitroxide labeling, the singly (at the site of U67 or U86) or doubly (at the sites of both U67 and U86) rTPT3αS-modified RNase P was supplemented with freshly prepared 1 mM TCEP and incubated at room temperature for 10 min. Then, the RNAs were precipitated with ethanol and redissolved with buffer containing 100 mM KH2PO4/K2HPO4, pH 7.0, 50% (v/v) DMSO. The singly or doubly rTPT3αS-modified RNase P RNAs were mixed with MSL with a molar ratio of 1:200 and 1:500, respectively; then, the reactions were carried out at room temperature overnight.51 The mixtures were further purified with HiTrap Q column pre-equilibrated with buffer A (20 mM HEPES pH 7.0, 20 mM KCl, 20% (v/v) acetonitrile), and then eluted with a salt gradient to 100% buffer B (20 mM HEPES pH 7.0, 1 M KCl, 20% (v/v) acetonitrile). The fractions of MSL-labeled RNAs were combined and concentrated.
EPR Spectroscopy.
The protocols for sample preparation, data collection, and processing of EPR spectroscopy were similar to those previously described.10 In brief, the MSL-modified RNase P RNAs were first lyophilized and then redissolved with 50% (v/v) glycerol/water prior to EPR data collection. For CW-EPR measurement, 10 μL samples of MSL-labeled RNA with concentrations of 100 μM were loaded into a borosilicate glass capillary sealed at one end. CW-EPR spectra were obtained on a Bruker EMX X-band spectrometer. The experimental parameters were the same as previously reported. The CW-EPR spectra were processed and counted as a previous procedure.10,41 For PELDOR EPR measurement, 30 μL of doubly MSL-labeled samples with a concentration around 200 μM were loaded into a round quartz capillary sealed at one end, and were flash-frozen in liquid nitrogen. The PELDOR EPR parameters were the same as previously described.10 The accumulation time was set to 20 h with 100 shots per point. Raw time domain PELDOR data was background-corrected using LongDistances1020 (developed by Christian Altenbach, available at www.biochemistry.ucla.edu/Faculty/Hubbell).52 The interspin distance distributions were computed from the resulting dipolar evolution data using the model-free regularization analysis in LongDistances.
Site-Specific Gold Nanoparticle Labeling of DENV2 3′SL.
The 1.4 nm monomaleimido Nanogold containing a single reactive maleimide was dissolved in 40% (v/v) DMSO/DEPC H2O solution following the product instruction manual (http://www.nanoprobes.com/). Prior to Nanogold labeling, singly or doubly rTPT3αS-modified 3′SL RNAs were treated with freshly prepared 1 mM TCEP at room temperature for 10 min. TCEP was removed by ethanol precipitation, and the RNAs were redissolved with 100 mM KH2PO4/K2HPO4, pH 7.0. By mixing 8 nmol of singly or doubly rTPT3αS-modified 3′SL RNAs with 16 or 40 nmol of monomaleimido Nanogold, respectively, the coupling reactions were carried out at room temperature overnight. The Nanogold-labeled 3′SL RNAs were further purified with HiTrap Q column (GE Healthcare) as previously reported.9
XSI Data Collection and Processing.
The protocols for XSI sample preparation, data collection, and processing are similar to those previously reported.9 In brief, all the Nanogold-labeled RNAs were conditioned into XSI buffer containing 20 mM HEPES, 100 mM KCl, 5 mM MgCl2, 0.5 mM TCEP, 5% (v/v) glycerol, pH 7.0, and diluted into final concentrations of 50 μM. The free Nanogold sample was concentrated to 100 μM. The exposure time for Nanogold-labeled RNA samples is 0.1 s for each image. The scattering profiles of the RNA quartets of 3′SL including the native 3′SL, two singly Nanogold-labeled and one doubly Nanogold-labeled 3′SL, and the free Nanogold was used to calculate the Nanogold−Nanogold scattering interference profiles (IAu−Au(s)), from which the Nanogold−Nanogold distance distributions were inferred with the MATLAB scripts in AuSAXSGUI (https://github.com/thomas836/AuSAXSGUI) shared by the Jan Lipfert group.42,43
Site-Specific Fluorescent Labeling of RNAs.
The purified rTPT3αS-modified (at the site of U86) 3′SL30 and DENV-mini30 (at the site of U708) products were precipitated with ethanol and then dissolved with FRET buffer to a final concentration of 150 μM. The sCy3-maleimide (dissolved in 100% DMSO to improve the solubility and maintain the properties) was added with a molar ratio of 1:30 for fluorescent labeling. The reacting mixtures were incubated at room temperature overnight. Then, the sCy3-labeled RNAs were precipitated by ethanol three times as described above and resuspended in FRET buffer. Similarly, the rTPT3A-modified (at the site of U86) 3′SL RNA was buffer-exchanged into 0.1 M NaHCO3 (pH 8.0) and subjected to fluorescent labeling. The sCy3 NHS ester (dissolved in 100% DMSO) was added with a molar ratio of 1:15 for rTPT3A-modified 3′SL RNA. The coupling reaction was incubated at room temperature overnight and then precipitated by ethanol three times and resuspended in FRET buffer.
smFRET Data Collection and Processing.
For single-molecule experiments, sCy3-labeled 3′SL30 and DENV-mini30 were annealed with 5′-biotin- and 3′-Cy5-modified DNA substrates in FRET buffer with a molar ratio of 1.3:1, and the mixture was then incubated at 95 °C for 2 min and cooled fast on ice. Samples were diluted 1000 times in the FRET buffer and immobilized on the glass slides by biotin−streptavidin interactions. The smFRET experiments were performed at 25 °C in FRET buffer with an oxygen-scavenging system containing 3 mg/mL glucose, 100 μg/mL glucose oxidase (Sigma-Aldrich), 40 μg/mL catalase (Roche), 1 mM cyclooctatetraene (COT, Sigma-Aldrich), 1 mM 4-nitrobenzylalcohol (NBA, Sigma-Aldrich), and 1.5 mM 6-hydroxy-2,5,7,8-tetramethyl-chromane-2-carboxylic acid (Tro-lox, Sigma-Aldrich).53 Single-molecule fluorescence and FRET measurements were carried out on a home-built objective-type TIRF microscope, based on a Nikon Eclipse Ti-E with an EMCCD camera (Andor iXon Ultra 897), and solid-state 532 and 640 nm excitation lasers (Coherent Inc. OBIS Smart Lasers). Fluorescence emission from the probes was collected by the microscope and spectrally separated by interference dichroic (T635lpxr, Chroma).
The smFRET movies were collected using Cell Vision software (Beijing Coolight Technology). Collected movies were analyzed by a custom-made software program developed as an ImageJ plugin (http://rsb.info.nih.gov/ij). Fluorescence spots were fitted by a 2-D Gaussian function, matching the donor and acceptor spots using a variant of the Hough transform.7 The background-subtracted total volume of the 2-D Gaussian peak was used as raw fluorescence intensity I. FRET traces displaying stable fluorescent signals were picked and further analyzed. The FRET efficiency was calculated via the equation
where ID and IA are the raw fluorescence intensity of the donor and acceptor, respectively, and χ is the cross-talk of the donor emission into the acceptor channel.
Supplementary Material
ACKNOWLEDGMENTS
We thank the staff at the beamline 12-ID-B, Advanced Photon Source, Argonne National Laboratory, for assistance during XSI data collection.
Funding
This work was supported by grants from the National Key Research and Development Project of China (2021YFA1301500 to X.F.), the National Natural Science Foundation of China (nos. U1832215 to X.F., 21877069, 21922704, and 22061160466 to C.C.), the US National Science Foundation (MCB-1818107, P.Z.Q.), the US National Institute of Health [S10 RR028992 (P.Z.Q.), T32-GM118289 (J.S.)], and the Anton B. Burg Foundation (P.Z.Q.).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.2c00199.
Synthetic procedures and characterizations of rTPT3αSTP, secondary structure of RNAs, RNA purification and labeling, additional EPR data, SAXS-related data, and coupling efficiency data (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acschembio.2c00199
The authors declare no competing financial interest.
Contributor Information
Yanping Hu, Beijing Advanced Innovation Center for Structural Biology, Beijing Frontier Research Center for Biological Structure, School of Life Sciences, Tsinghua University, Beijing 100084, China.
Yan Wang, Beijing Advanced Innovation Center for Structural Biology, Beijing Frontier Research Center for Biological Structure, School of Life Sciences, Tsinghua University, Beijing 100084, China;.
Jaideep Singh, Department of Chemistry, University of Southern California, Los Angeles, California 90089, United States;.
Ruirui Sun, Beijing Advanced Innovation Center for Structural Biology, Beijing Frontier Research Center for Biological Structure, School of Life Sciences, Tsinghua University, Beijing 100084, China.
Lilei Xu, Beijing Advanced Innovation Center for Structural Biology, Beijing Frontier Research Center for Biological Structure, School of Life Sciences, Tsinghua University, Beijing 100084, China.
Xiaolin Niu, Beijing Advanced Innovation Center for Structural Biology, Beijing Frontier Research Center for Biological Structure, School of Life Sciences, Tsinghua University, Beijing 100084, China.
Keyun Huang, Beijing Advanced Innovation Center for Structural Biology, Beijing Frontier Research Center for Biological Structure, School of Life Sciences, Tsinghua University, Beijing 100084, China.
Guangcan Bai, State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, Beijing 100191, China.
Guoquan Liu, State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, Beijing 100191, China;.
Xiaobing Zuo, X-ray Science Division, Argonne National Laboratory, Lemont, Illinois 60439, United States.
Chunlai Chen, Beijing Advanced Innovation Center for Structural Biology, Beijing Frontier Research Center for Biological Structure, School of Life Sciences, Tsinghua University, Beijing 100084, China;.
Peter Z. Qin, Department of Chemistry, University of Southern California, Los Angeles, California 90089, United States;
Xianyang Fang, Beijing Advanced Innovation Center for Structural Biology, Beijing Frontier Research Center for Biological Structure, School of Life Sciences, Tsinghua University, Beijing 100084, China;.
REFERENCES
- (1).Cech TR; Steitz JA The noncoding RNA revolution-trashing old rules to forge new ones. Cell 2014, 157, 77–94. [DOI] [PubMed] [Google Scholar]
- (2).Ganser LR; Kelly ML; Herschlag D; Al-Hashimi HM The roles of structural dynamics in the cellular functions of RNAs. Nat. Rev. Mol. Cell Biol 2019, 20, 474–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Falese JP; Donlic A; Hargrove AE Targeting RNA with small molecules: from fundamental principles towards the clinic. Chem. Soc. Rev 2021, 50, 2224–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Reyes FE; Garst AD; Batey RT Strategies in RNA crystallography. Methods Enzymol. 2009, 469, 119–139. [DOI] [PubMed] [Google Scholar]
- (5).Kappel K; Zhang K; Su Z; Watkins AM; Kladwang W; Li S; Pintilie G; Topkar VV; Rangan R; Zheludev IN; Yesselman JD; Chiu W; Das R Accelerated cryo-EM-guided determination of three-dimensional RNA-only structures. Nat. Methods 2020, 17, 699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Fang X; Stagno JR; Bhandari YR; Zuo X; Wang Y-X Small-angle X-ray scattering: a bridge between RNA secondary structures and three-dimensional topological structures. Curr. Opin. Struct. Biol 2015, 30, 147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Barnwal RP; Yang F; Varani G Applications of NMR to structure determination of RNAs large and small. Arch. Biochem. Biophys 2017, 628, 42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Dimura M; Peulen TO; Hanke CA; Prakash A; Gohlke H; Seidel CA Quantitative FRET studies and integrative modeling unravel the structure and dynamics of biomolecular systems. Curr. Opin. Struct. Biol 2016, 40, 163–185. [DOI] [PubMed] [Google Scholar]
- (9).Wang Y; Chen Y; Hu Y; Fang X Site-specific covalent labeling of large RNAs with nanoparticles empowered by expanded genetic alphabet transcription. Proc. Natl. Acad. Sci. U.S.A 2020, 117, 22823–22832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wang Y; Kathiresan V; Chen Y; Hu Y; Jiang W; Bai G; Liu G; Qin PZ; Fang X Posttranscriptional site-directed spin labeling of large RNAs with an unnatural base pair system under nondenaturing conditions. Chem. Sci 2020, 11, 9655–9664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Niu X; Sun R; Chen Z; Yao Y; Zuo X; Chen C; Fang X Pseudoknot length modulates the folding, conformational dynamics, and robustness of Xrn1 resistance of flaviviral xrRNAs. Nat. Commun 2021, 12, 6417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Duss O; Yulikov M; Jeschke G; Allain FH EPR-aided approach for solution structure determination of large RNAs or protein-RNA complexes. Nat. Commun 2014, 5, 3669. [DOI] [PubMed] [Google Scholar]
- (13).Shi X; Huang L; Lilley DM; Harbury PB; Herschlag D The solution structural ensembles of RNA kink-turn motifs and their protein complexes. Nat. Chem. Biol 2016, 12, 146–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zhang X; Tung CS; Sowa GZ; Hatmal MM; Haworth IS; Qin PZ Global structure of a three-way junction in a phi29 packaging RNA dimer determined using site-directed spin labeling. J. Am. Chem. Soc 2012, 134, 2644–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Paredes E; Evans M; Das SR RNA labeling, conjugation and ligation. Methods 2011, 54, 251–259. [DOI] [PubMed] [Google Scholar]
- (16).Depmeier H; Hoffmann E; Bornewasser L; Kath-Schorr S Strategies for covalent labeling of long RNAs. ChemBioChem 2021, 22, 2826–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Egloff D; Oleinich IA; Zhao M; König SL; Sigel RK; Freisinger E Sequence-specific post-synthetic oligonucleotide labeling for single-molecule fluorescence applications. ACS Chem. Biol 2016, 11, 2558–2567. [DOI] [PubMed] [Google Scholar]
- (18).Büttner L; Javadi-Zarnaghi F; Höbartner C Site-specific labeling of RNA at internal ribose hydroxyl groups: terbium-assisted deoxyribozymes at work. J. Am. Chem. Soc 2014, 136, 8131–8137. [DOI] [PubMed] [Google Scholar]
- (19).Hanspach G; Trucks S; Hengesbach M Strategic labelling approaches for RNA single-molecule spectroscopy. RNA Biol. 2019, 16, 1119–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lebars I; Vileno B; Bourbigot S; Turek P; Wolff P; Kieffer B A fully enzymatic method for site-directed spin labeling of long RNA. Nucleic Acids Res. 2014, 42, No. e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Seo YJ; Malyshev DA; Lavergne T; Ordoukhanian P; Romesberg FE Site-specific labeling of DNA and RNA using an efficiently replicated and transcribed class of unnatural base pairs. J. Am. Chem. Soc 2011, 133, 19878–19888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lavergne T; Lamichhane R; Malyshev DA; Li Z; Li L; Sperling E; Williamson JR; Millar DP; Romesberg FE FRET Characterization of Complex Conformational Changes in a Large 16S Ribosomal RNA Fragment Site-Specifically Labeled Using Unnatural Base Pairs. ACS Chem. Biol 2016, 11, 1347–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wang L; Jiang S; Deng Z; Dedon PC; Chen S DNA phosphorothioate modification-a new multi-functional epigenetic system in bacteria. FEMS Microbiol. Rev 2019, 43, 109–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zheng YY; Wu Y; Begley TJ; Sheng J Sulfur modification in natural RNA and therapeutic oligonucleotides. RSC Chem. Biol 2021, 2, 990–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Saran R; Huang Z; Liu J Phosphorothioate nucleic acids for probing metal binding, biosensing and nanotechnology. Coord. Chem. Rev 2021, 428, 213624. [Google Scholar]
- (26).Crooke ST; Baker BF; Crooke RM; Liang XH Antisense technology: an overview and prospectus. Nat. Rev. Drug Discovery 2021, 20, 427–453. [DOI] [PubMed] [Google Scholar]
- (27).Kawaguchi D; Kodama A; Abe N; Takebuchi K; Hashiya F; Tomoike F; Nakamoto K; Kimura Y; Shimizu Y; Abe H Phosphorothioate Modification of mRNA Accelerates the Rate of Translation Initiation to Provide More Efficient Protein Synthesis. Angew. Chem., Int. Ed. Engl 2020, 59, 17403–17407. [DOI] [PubMed] [Google Scholar]
- (28).Forconi M; Herschlag D Use of phosphorothioates to identify sites of metal-ion binding in RNA. Methods Enzymol. 2009, 468, 311–333. [DOI] [PubMed] [Google Scholar]
- (29).Popova AM; Qin PZ A nucleotide-independent nitroxide probe reports on site-specific stereomeric environment in DNA. Biophys. J 2010, 99, 2180–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Lee JH; Wernette DP; Yigit MV; Liu J; Wang Z; Lu Y Site-specific control of distances between gold nanoparticles using phosphorothioate anchors on DNA and a short bifunctional molecular fastener. Angew. Chem., Int. Ed. Engl 2007, 46, 9006–9010. [DOI] [PubMed] [Google Scholar]
- (31).Lee JH; Wong NY; Tan LH; Wang Z; Lu Y Controlled alignment of multiple proteins and nanoparticles with nanometer resolution via backbone-modified phosphorothioate DNA and bifunctional linkers. J. Am. Chem. Soc 2010, 132, 8906–8908. [DOI] [PubMed] [Google Scholar]
- (32).Iglesias NG; Gamarnik AV Dynamic RNA structures in the dengue virus genome. RNA Biol. 2011, 8, 249–257. [DOI] [PubMed] [Google Scholar]
- (33).Gebhard LG; Filomatori CV; Gamarnik AV Functional RNA elements in the dengue virus genome. Viruses 2011, 3, 1739–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Ravasco J; Faustino H; Trindade A; Gois PMP Bioconjugation with Maleimides: A Useful Tool for Chemical Biology. Chemistry 2019, 25, 43–59. [DOI] [PubMed] [Google Scholar]
- (35).Griffiths AD; Potter BV; Eperon IC Stereospecificity of nucleases towards phosphorothioate-substituted RNA: stereochemistry of transcription by T7 RNA polymerase. Nucleic Acids Res. 1987, 15, 4145–4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Gish G; Eckstein F DNA and RNA sequence determination based on phosphorothioate chemistry. Science 1988, 240, 1520–1522. [DOI] [PubMed] [Google Scholar]
- (37).AbouHaidar MG; Ivanov IG Non-enzymatic RNA hydrolysis promoted by the combined catalytic activity of buffers and magnesium ions. Z. Naturforsch., C: J. Biosci 1999, 54, 542–548. [DOI] [PubMed] [Google Scholar]
- (38).Zhang X; Cekan P; Sigurdsson ST; Qin PZ, Studying RNA Using Site-Directed Spin-Labeling and Continuous-Wave Electron Paramagnetic Resonance Spectroscopy. In Biophysical, Chemical, and Functional Probes of RNA Structure, Interactions and Folding: Part B, 2009; pp 303–328. DOI: 10.1016/s0076-6879(09)69015-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Dalmas O; Hyde HC; Hulse RE; Perozo E Symmetry-constrained analysis of pulsed double electron-electron resonance (DEER) spectroscopy reveals the dynamic nature of the KcsA activation gate. J. Am. Chem. Soc 2012, 134, 16360–16369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Beasley KN; Sutch BT; Hatmal MH; Langen R; Qin PZ; Haworth IS, Computer modeling of spin labels: NASNOX, PRONOX, and ALLNOX. In Methods Enzymol, Elsevier: 2015; Vol. 563, pp 569–593. DOI: 10.1016/bs.mie.2015.07.021 [DOI] [PubMed] [Google Scholar]
- (41).Zhang X; Cekan P; Sigurdsson ST; Qin PZ Studying RNA using site-directed spin-labeling and continuous-wave electron paramagnetic resonance spectroscopy. Methods Enzymol. 2009, 469, 303–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Shi X; Bonilla S; Herschlag D; Harbury P Quantifying Nucleic Acid Ensembles with X-ray Scattering Interferometry. Methods Enzymol. 2015, 558, 75–97. [DOI] [PubMed] [Google Scholar]
- (43).Zettl T; Das R; Harbury PAB; Herschlag D; Lipfert J; Mathew RS; Shi X Recording and Analyzing Nucleic Acid Distance Distributions with X-Ray Scattering Interferometry (XSI). Curr. Protoc. Nucleic Acid Chem 2018, 73, No. e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Zhang Y; Zhang Y; Liu ZY; Cheng ML; Ma J; Wang Y; Qin CF; Fang X Long non-coding subgenomic flavivirus RNAs have extended 3D structures and are flexible in solution. EMBO Rep. 2019, 20, No. e47016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Helm M; Kobitski AY; Nienhaus GU Single-molecule Forster resonance energy transfer studies of RNA structure, dynamics and function. Biophys. Rev 2009, 1, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Grant GPG; Popova A; Qin PZ Diastereomer characterizations of nitroxide-labeled nucleic acids. Biochem. Biophys. Res. Commun 2008, 371, 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Mattson G; Conklin E; Desai S; Nielander G; Savage MD; Morgensen S A practical approach to crosslinking. Mol. Biol. Rep 1993, 17, 167–183. [DOI] [PubMed] [Google Scholar]
- (48).Martínez-Jothar L; Doulkeridou S; Schiffelers RM; Sastre Torano J; Oliveira S; van Nostrum CF; Hennink WE Insights into maleimide-thiol conjugation chemistry: Conditions for efficient surface functionalization of nanoparticles for receptor targeting. J. Controlled Release 2018, 282, 101–109. [DOI] [PubMed] [Google Scholar]
- (49).Rambo RP; Tainer JA Accurate assessment of mass, models and resolution by small-angle scattering. Nature 2013, 496, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Lan P; Tan M; Zhang Y; Niu S; Chen J; Shi S; Qiu S; Wang X; Peng X; Cai G; Cheng H; Wu J; Li G; Lei M Structural insight into precursor tRNA processing by yeast ribonuclease P. Science 2018, 362, No. eaat6678. [DOI] [PubMed] [Google Scholar]
- (51).Sjodt M; Clubb RT Nitroxide Labeling of Proteins and the Determination of Paramagnetic Relaxation Derived Distance Restraints for NMR Studies. Bio-Protoc. 2017, 7, No. e2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Wingler LM; Elgeti M; Hilger D; Latorraca NR; Lerch MT; Staus DP; Dror RO; Kobilka BK; Hubbell WL; Lefkowitz RJ Angiotensin analogs with divergent bias stabilize distinct receptor conformations. Cell 2019, 176, 468–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Peng S; Sun R; Wang W; Chen C Single-Molecule Photoactivation FRET: A General and Easy-To-Implement Approach To Break the Concentration Barrier. Angew. Chem., Int. Ed 2017, 56, 6882–6885. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.