

Abstract
Monotopic phosphoglycosyl transferases (monoPGTs) are an expansive superfamily of enzymes that catalyze the first membrane‐committed step in the biosynthesis of bacterial glycoconjugates. MonoPGTs show a strong preference for their cognate nucleotide diphospho‐sugar (NDP‐sugar) substrates. However, despite extensive characterization of the monoPGT superfamily through previous development of a sequence similarity network comprising >38,000 nonredundant sequences, the connection between monoPGT sequence and NDP‐sugar substrate specificity has remained elusive. In this work, we structurally characterize the C‐terminus of a prototypic monoPGT for the first time and show that 19 C‐terminal residues play a significant structural role in a subset of monoPGTs. This new structural information facilitated the identification of co‐conserved sequence “fingerprints” that predict NDP‐sugar substrate specificity for this subset of monoPGTs. A Hidden Markov model was generated that correctly assigned the substrate of previously unannotated monoPGTs. Together, these structural, sequence, and biochemical analyses have delivered new insight into the determinants guiding substrate specificity of monoPGTs and have provided a strategy for assigning the NDP‐sugar substrate of a subset of enzymes in the superfamily that use UDP‐di‐N‐acetyl bacillosamine. Moving forward, this approach may be applied to identify additional sequence motifs that serve as fingerprints for monoPGTs of differing UDP‐sugar substrate specificity.
Keywords: aromatic box, coevolution, di‐N‐acetylbacillosamine, glycoconjugate biosynthesis, membrane protein, multiple sequence alignment, undecaprenol phosphate
1. INTRODUCTION
Glycans and glycoconjugates are essential to living systems, yet the molecular structures of these complex biopolymers are not readily predicted from the sequences of the biosynthetic enzymes responsible for their biogenesis. A major pathway to glycoconjugate biosynthesis in bacteria involves the stepwise assembly of glycan onto a polyprenol phosphate (PrenP), commonly undecaprenol phosphate (UndP), which is initiated on the cytoplasmic face of cell membranes. The steps involve the action of an initiating phosphoglycosyl transferase (PGT) enzyme, followed by several glycosyl transferases (GTs). For pathways such as lipopolysaccharide and exopolysaccharide biosynthesis these translocated glycoconjugates are polymerized and finally transferred to lipid A or phospholipid carriers (Whitfield, Wear, & Sande, 2020; Whitfield, Williams, & Kelly, 2020). For pathways such as the protein glycosylation (Pgl) pathway of the Campylobacter genus, the ultimate glycoconjugates result from en bloc transfer of glycan to asparagine residues in a protein acceptor (Linton et al., 2005).
In many bacteria, the PGTs and GTs, in addition to modifying nucleoside diphospho‐sugar (NDP‐sugar) biosynthesis enzymes, flippase, and transferase enzymes are often colocalized in operons (Tytgat & Lebeer, 2014). The overall challenge is to “translate” the genomic data found in the operon into functional proteomic insight. Herein we demonstrate the identification, analysis, and application of co‐conserved sequence motifs for the prediction of substrate specificity for the catalytic domain of a prototypic monotopic PGT from the enzyme superfamily.
There are two superfamilies of PGTs, monotopic (monoPGT) and polytopic (polyPGT), which are structurally and mechanistically distinct but catalyze the same overall transformation (Al‐Dabbagh et al., 2008; Das et al., 2017; O'Toole, Bernstein et al., 2021). The monoPGT superfamily is further divided into small, large, or bifunctional classes based on their domain architecture: small monoPGTs (smPGTs) represent the smallest functional catalytic core, large monoPGTs (lgPGTs) have four putative transmembrane helices and a large loop region at their N‐terminus, and bifunctional PGTs have additional catalytic domains fused to the N‐ or C‐termini. Recently, a sequence similarity network (SSN) comprising ~38,000 nonredundant PGTs was constructed, yielding critical insights into domain expansion around the smPGT catalytic core (O'Toole, Imperiali et al., 2021). Experimental characterization of PGTs has revealed a high degree of specificity for the NDP‐sugar donors that correspond to the sugar found at the reducing end of the final, mature glycoconjugate produced by the biosynthetic pathway (Cartee et al., 2005; Glover et al., 2006; Merino et al., 2011; Patel et al., 2010; Patel et al., 2012). However, from sequence alone, the molecular determinants of this specificity remain unclear.
The PGT in the N‐linked protein glycosylation pathway (Pgl) of Campylobacter concisus, PglC, represents a prototypical smPGT. PglC transfers a phospho‐sugar from the UDP‐di‐N‐acetyl bacillosamine (UDP‐diNAcBac) donor onto the UndP acceptor (Morrison & Imperiali, 2014). The structure of this enzyme, reported in 2018, is the first and only high‐resolution monoPGT structure experimentally determined and reveals a unique membrane topology in which a re‐entrant membrane helix penetrates a single leaflet of the bilayer (Ray et al., 2018). However, in this structure a region of the C‐terminus representing ~8% of the sequence of the protein has unmodeled electron density in both polypeptide chains in the asymmetric unit. The location of this disordered C‐terminus is proximal to both the putative UndP and NDP‐sugar binding sites, thus limiting a full understanding of protein‐ligand interactions. Efforts to identify alternative crystal forms, as well as crystallization of PglC orthologs to address questions left by the crystal structure, have not yet resulted in new information. Thus, it remains a challenge to rationalize the specificity for UDP‐diNAcBac with the available data.
Recent advances in machine learning have revolutionized our ability to computationally predict protein structures (Baek et al., 2021; Jumper et al., 2021; Mirdita et al., 2022). However, the utility of these tools is not limited to predicting protein structure (Akdel et al., 2022). Accurate models of protein targets can greatly assist crystallographic data processing, where the quality of the electron‐density map is intimately tied to the accuracy of the model. Herein, the machine learning‐based program AlphaFold was utilized to model full‐length PglC orthologs from several prokaryotes (Jumper et al., 2021). AlphaFold models of Campylobacter PGTs predicted the placement of the very C‐terminus with unexpectedly high confidence. Upon revisiting experimental maps with this model, we could confidently place residues 194–201 of the C‐terminus of C. concisus PglC in electron density previously assigned to the headgroup of a phosphatidylethanolamine phospholipid. The placement of these C‐terminal residues allowed the identification of an “aromatic box” motif comprising the side chains of five aromatic amino acids in known UDP‐diNAcBac‐specific PGTs (Burley & Petsko, 1985; Holliday et al., 2009; Lanzarotti et al., 2011; Makwana & Mahalakshmi, 2015). Mutagenesis of residues within this motif established its importance to catalysis. Analysis of a multiple sequence alignment (MSA) of >4000 smPGTs revealed clear conservation of this motif within diNAcBac‐specific PGTs. Examination of a similarity‐based alignment and predicted PGT models from the diNAcBac cluster and extant clusters demonstrate the existence of several sub‐types of PGTs, which are easily distinguished based on the predicted architecture around the highly conserved signature motifs. Together, these results demonstrate the application of AlphaFold to examine disordered regions of experimentally determined protein structures thereby allowing formulation of experimentally‐tractable hypotheses. The new structural analysis, together with sequence and biochemical analyses and knowledge of the biosynthetic pathways, have provided novel insight into the structure/function relationships determining substrate specificity in the mono PGTs.
2. RESULTS
2.1. Modeling smPGTs using AlphaFold
The structure of full‐length PglC from Campylobacter concisus was predicted using LocalColabFold (Figure 1a) (Mirdita et al., 2022). Three‐dimensional superposition revealed close agreement between predictions and experimentally‐determined structures, with <1.51 Å RMSD for models superposed to chain A, and <1.67 Å RMSD for models superposed to chain B (Table S1). Examination of the precision local‐distance difference test (pLDDT) score, which represents model confidence, for the predicted models revealed a region with unexpectedly high confidence at the C‐terminus (residues 194‐201) of all the predicted models (Figure S1). In the experimental structure, this region had been omitted as disordered. Close examination of the superpositions of the predicted and experimental models showed an overlap between the absolute C‐terminus of the AlphaFold models and the placement of the phosphatidyl ethanolamine (PE) headgroup in the experimental structure (Figure 1a,b). Re‐refining the crystallographic data, including the C‐terminal residues from the top AlphaFold prediction, revealed an excellent fit between the predicted model and experimental density previously occupied by the PE moiety (Figures 1b and S2, Table S2). This new insight into the placement of the C‐terminus of smPGTs provides critical information that may impact the understanding of substrate binding and specificity. The C‐terminal residues that can be defined in the new model comprise 4% of the smPGT sequence and represent an important structural feature of the enzyme that may contribute to the structure of the putative substrate binding pocket.
FIGURE 1.
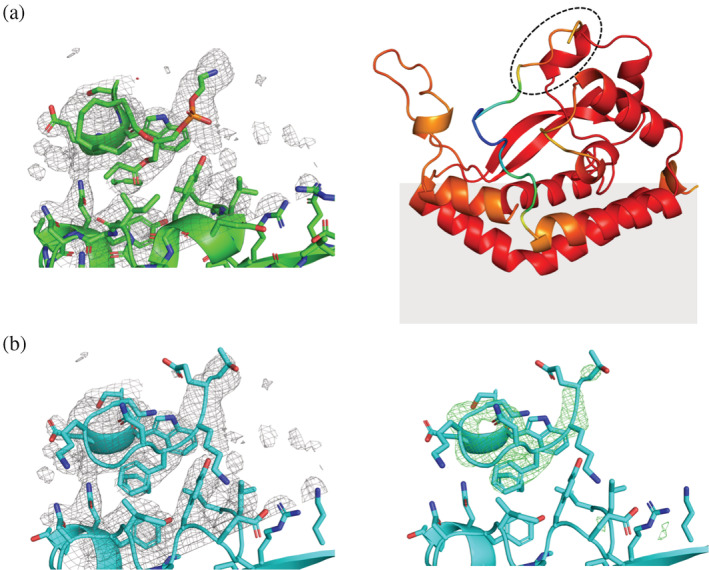
(a) Left: PDB 5W7L model (green) and 2mFO‐dFC map (gray mesh) contoured to 1.0 σ. Right: Alphafold model of C. concisus PglC, colored by pLDDT score. Red indicates areas of high confidence, while blue indicates areas of low confidence. The region occupied by the PE headgroup in the experimental structure is circled with dashes. The gray shading indicates the predicted position of the membrane. (b) Left: Re‐built model of C. concisus PglC (cyan). 2mFO‐dFC map contoured to 1.0 σ (gray mesh). Right: Simulated annealing with the residues 194–201 omitted displays a strong positive difference density (mFO‐dFC map contoured to 3.0 σ [green mesh]).
Complementary evolutionary covariance analysis of the smPGTs was also performed to obtain residue–residue contact predictions between the C‐terminus and the core soluble domain of PglC. Approximately 4000 smPGT sequences were extracted from the monoPGT SSN for coevolution analysis in GREMLIN (Kamisetty et al., 2013; O'Toole, Bernstein et al., 2021; Ovchinnikov et al., 2014). Several PglC residues were found to covary with C‐terminal residues (Figure 2a). The coevolution contact predictions coincided well with the AlphaFold model; residues that coevolve with the C‐terminus are all located within 12 Å of the C‐terminal backbone in the model (Figure 2b). Notably, the residue pair with the highest probability of covariance, E117 and K196, are in a position to form a salt bridge that may contribute to positioning the C‐terminus (Figure 2c).
FIGURE 2.
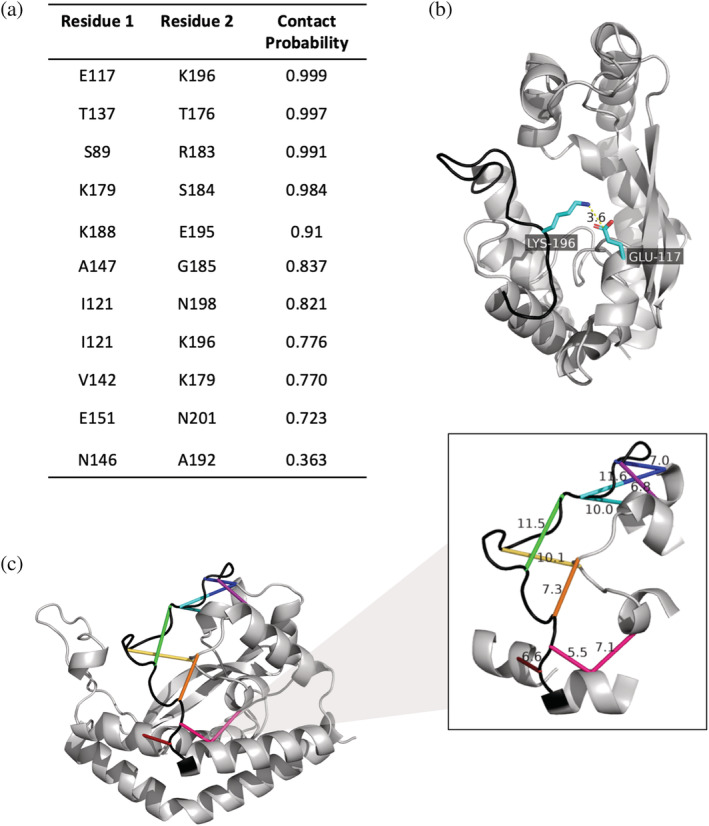
(a) Contact probabilities of residues in the C‐terminal loop determined through coevolution analysis in GREMLIN. Contact probability is defined as the probability of the residue pair being in contact, given the scaled score and the number of sequences per length. (b) The residue pair with the highest contact probability is placed onto the AlphaFold Cc PglC model. Lys196 and Glu 117 likely form a salt bridge to position the C‐terminal loop. (c) Residues that covary with C‐terminal residues are connected in the same color on the Cc PglC AlphaFold model. Distances (in Å) between the alpha carbons of the coevolving residues are shown.
2.2. Identification and characterization of a conserved aromatic box motif in PglC
Upon placement of the C‐terminus, it was apparent that a previously unidentified aromatic box motif comprising Tyr118, Tyr122, Trp150, Phe154, and Phe197 was present proximal to the putative NDP‐sugar binding site (Figure 3a,b). The GREMLIN coevolution analysis of monoPGTs described above supports the presence of the aromatic box motif, as contacts were predicted between residues in the aromatic box (Figure 3c).
FIGURE 3.
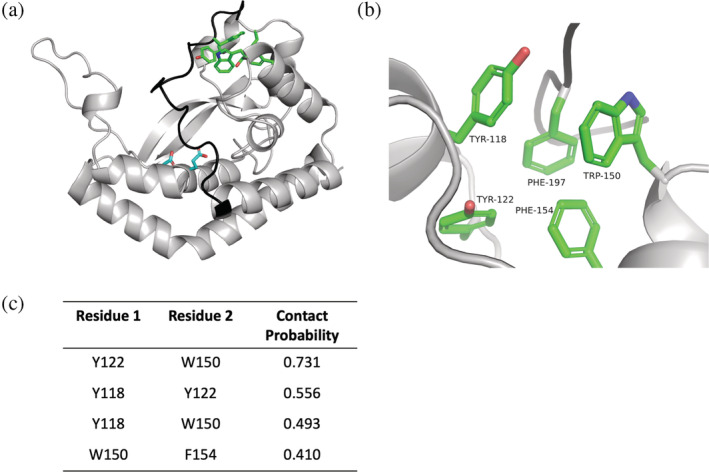
Identification of an aromatic box motif in C. concisus PglC. (a). AlphaFold model of Cc PglC with aromatic box residues highlighted in green, the Asp‐Glu catalytic dyad in cyan, and the C‐terminal loop in black. (b). Close‐up view of the aromatic box motif. (c). GREMLIN coevolution contact probabilities between residues in the aromatic box. Contact probability is defined as the probability of the residue pair being in contact, given the scaled score and the number of sequences per length.
Mutagenesis of the C. concisus PglC Trp150 and Phe197 was performed to test the functional importance of the aromatic box in PglC. Variants were expressed and purified, followed by subsequent evaluation of activity using the UMP‐Glo® assay that monitors UMP‐release (Table 1, Figures S3 and S4) (Das et al., 2016). Disruption of the core of the aromatic box through the introduction of a Trp150Leu mutation reduced protein stability and resulted in a complete loss of activity. Similarly, the introduction of a Phe197Ala mutation was not well tolerated and caused a 100‐fold loss in activity compared to wild‐type PglC. Clearly, aromatic box residues are vital for PglC structure and function. The conservative mutation of Trp150 and Phe197 to other aromatic residues, however, was well tolerated and variants were stable under room temperature assay conditions (Figure S5). Trp150 and Phe197 aromatic variants showed a 10‐fold drop in k cat but no significant change in the UDP‐diNAcBac KM (Table 1). The 10‐fold loss of activity observed in the aromatic variants highlights the preference for certain aromatic residues over others at each site within the aromatic box. Prior analysis of published structures with aromatic boxes identified preferred distances and dihedral angles for the aromatic interactions that make up aromatic box motifs (Burley & Petsko, 1985; Lanzarotti et al., 2011; Makwana & Mahalakshmi, 2015). Our results are consistent with the concept that aromatic residues within the box are not interchangeable but rather have evolved to interplay with one another and surrounding residues. The aromatic box is positioned at a distance from the biochemically identified active site of PglC, which is centered at the Asp93‐Glu94 dyad and is therefore not likely to be directly involved in catalysis. Rather, the reduction in turnover observed is most likely due to the aromatic box playing a structural role in the catalytically competent conformation of PglC.
TABLE 1.
Kinetic parameters of C. concisus PglC aromatic box variants. Steady‐state kinetic measurements were performed as described in the experimental section and the data were fit using the Michaelis–Menten equation.
| Variant | Activity a | Km (μM) | Km 95% CI (μM) | kcat (s−1) | kcat 95% CI (s−1) | kcat/Km (μM−1 s−1) |
|---|---|---|---|---|---|---|
| WT | +++ | 26.5 | (17.6, 40.3) | 13.8 | (11.8, 16.5) | 0.52 |
| W150F | ++ | 24.8 | (18.0, 34.3) | 1.48 | (1.35, 1.63) | 0.060 |
| W150Y | ++ | 24.7 | (19.8, 30.8) | 1.94 | (1.81, 2.08) | 0.078 |
| W150L | − | n.d. | n.d. | n.d. | n.d. | n.d. |
| F197W | ++ | 25.7 | (17.4, 37.8) | 1.32 | (1.17, 1.50) | 0.052 |
| F197Y | ++ | 20.2 | (16.8, 24.3) | 1.25 | (1.18, 1.33) | 0.062 |
| F197A | + | n.d. | n.d. | n.d. | n.d. | n.d. |
Note: +++ Activity at 0.3 nM; ++ Activity at 3 nM; + Activity at 30 nM; − No activity at 30 nM.
Abbreviations: n.d., not determined; Cl., confidence limits.
The wild‐type level of activity in the presence of 0.3 nM enzyme, to which all variant enzymes are compared, is defined as (+++). A designation of (++) represents the activity of a variant enzyme that attains ~85% of wild‐type activity with 3 nM enzyme. A designation of (+) represents the activity of a variant enzyme that attains ~85% of wild‐type activity with 30 nM enzyme. A designation of (−) is used to describe the activity of a variant enzyme that attains <20% of wild‐type activity with 30 nM enzyme.
2.3. Analysis of smPGTs sub‐family
To better understand the context of the aromatic box motif within the entire superfamily of monoPGTs, an alignment and cladogram were constructed (Figure 4). The 4684 nonredundant smPGT sequences from the co‐evolution analysis were seeded with nine well‐characterized smPGTs of known substrate specificity as well as catalytic domains of Bi/Lg‐PGTs from Campylobacter sp, Salmonella enterica, Escherichia coli, and Staphylococcus aureus, among others (Figure 4a). This augmented dataset was then used to generate a multiple sequence alignment (MSA) and corresponding cladogram revealing clear clustering based on UDP‐sugar specificity (Figure 4b,c). All of the known UDP‐diNAcBac‐utilizing PGTs cluster together, and the aromatic box motif is highly conserved within this branch. This conservation is even more striking when compared to the same positions in the PGTs outside of the diNAcBac cluster (Table S3). Additionally, a co‐conserved motif from Gly71 to Pro75 (GLLLP) was identified on a loop distal to the aromatic box (Figure 4c). In the C. concisus PglC, this mobile loop region has been hypothesized to close upon substrate binding based on covariance analysis (Lukose et al., 2015; Ray et al., 2018).
FIGURE 4.
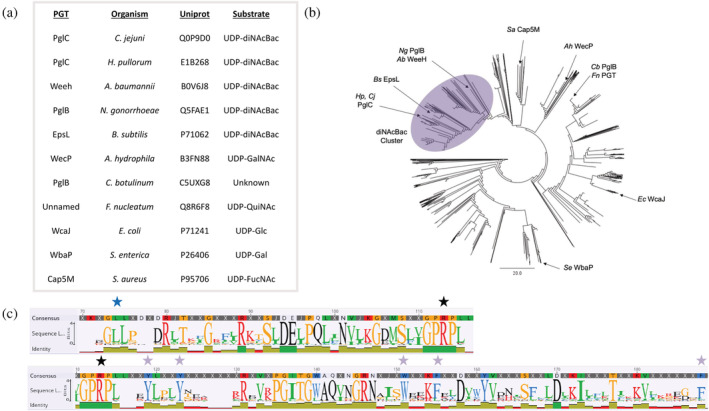
Sequence analysis and clustering of smPGTs. (a). Table of characterized PGTs and their annotated preferred substrate assigned based on mature glycoconjugate composition. (b). Cladogram created from a multiple sequence alignment of a nonredundant set of 4684 smPGT sequences seeded with characterized PGT sequences. The cladogram reveals clear clustering based on UDP‐sugar specificity. The UDP‐diNAcBac cluster is highlighted in purple. (c). Sequence logo created from an MSA of the UDP‐diNacBac PGT cluster. Aromatic box residues are starred in purple, the “GLLLP” motif is starred in blue, and the conserved “PRP” nucleotide‐binding motif is starred in black.
We reasoned that the apparent substrate‐based clustering may facilitate computational prediction of other uncharacterized UDP‐diNAcBac‐utilizing PGTs. The UDP‐diNAcBac cluster of sm‐PGTs was extracted from the phylogenetic tree and used to build a profile hidden Markov model (HMM) using HMMer. Using characterized PGTs as markers, a score cutoff of 200 was identified as sufficient to separate UDP‐diNAcBac‐utilizing PGTs from other exemplar core sequences from the superfamily (Table 2). To assess the predictive nature of this HMM, we analyzed EpsL from the exopolysaccharide (EPS) biosynthetic pathway of Bacillus subtilis (Arnaouteli et al., 2021). This PGT was assigned a score of 242.5 by the profile HMM, indicating its native substrate is UDP‐diNAcBac, and implying that the sugar at the reducing end of the B. subtilis EPS is diNAcBac. Experimental validation of this result will be reported separately.
TABLE 2.
Profile HMM trained on diNAcBac cluster can distinguish UDP‐diNAcBac specific PGTs.
| HMM scoring | |||||
|---|---|---|---|---|---|
| E‐value | Score | PGT | Organism | Uniprot ID | Substrate |
| 7.20E‐75 | 246.2 | PglC | C. jejuni | Q0P9D0 | UDP‐diNAcBac |
| 2.00E‐75 | 248.2 | PglC | H. pullorum | E1B268 | UDP‐diNAcBac |
| 8.70E‐74 | 242.7 | WeeH | A. baumannii | B0V6J8 | UDP‐diNAcBac |
| 8.50E‐73 | 239.5 | PglB | N. gonorrhoeae | Q5FAE1 | UDP‐diNAcBac |
| 1.00E‐73 | 242.5 | EpsL | B. subtilis | P71062 | UDP‐diNAcBac |
| 8.00E‐51 | 169.6 | WecP | A. hydrophila | B3FN88 | UDP‐GalNAc |
| 2.60E‐48 | 161.4 | PglB | C. botulinum | C5UXG8 | Unknown |
| 2.70E‐41 | 138.6 | Unnamed | F. nucleatum | Q8R6F8 | UDP‐QuiNAc |
| 1.00E‐51 | 172.5 | WcaJ | E. coli | P71241 | UDP‐Glc |
| 1.60E‐54 | 181.7 | WbaP | S. enterica | P26406 | UDP‐Gal |
| 3.90E‐56 | 186.8 | Cap5M | S. aureus | P95706 | UDP‐FucNAc |
We also examined smPGT tree branches outside of the diNAcBac cluster. Analysis of the amino‐acid sequences demonstrated that, in general, monoPGTs that do not use UDP‐diNAcBac are truncated at the C‐terminus by ~14 residues relative to Campylobacter PglC. In addition, both the GLLLP motif and the aromatic box motif are degenerate in non‐diNAcBac PGTs (Table S3). We chose a representative set of non‐diNAcBac PGT sequences and predicted their structure in an analogous manner to the Campylobacter PGTs. Although the signature re‐entrant membrane helix motif, membrane‐associated helices, and position of putative catalytic residues are highly similar to those in PglC in these predictions, a surprising degree of structural variability was observed in regions in the vicinity of the substrate binding pocket (Figure 5).
FIGURE 5.
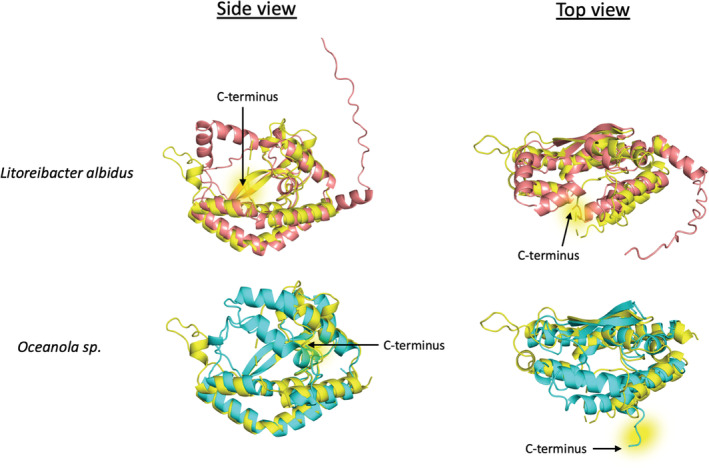
AlphaFold predicted structures of monoPGTs from Litoreibacter albidus (top, salmon) and Oceanicola sp. (bottom, cyan) aligned with the rebuilt Campylobacter concisus PglC structure (yellow). C‐terminal residues of the structures are highlighted in yellow. RMSD C. concisus PglC to Litoreibacter albidus PGT 1.839 Å. RMSD C. concisus PglC to Oceanicola sp. PGT 1.827 Å.
3. DISCUSSION
3.1. Using Alphafold to re‐interpret experimental data
In many experimental 2mFo‐dFc‐weighted electron density maps, density comprises discontinuous regions of order and disorder. In such cases, maintaining the register of the polypeptide chain during model building is challenging, especially for moderate‐resolution maps or maps with long disordered regions (Headd et al., 2012; Karmali et al., 2009). Improvement of structural models at moderate resolution is critically important in establishing structure–function relationships and remains a significant hurdle for the structural biology community. The advent of machine learning‐based methods for protein structure prediction provides a powerful approach for addressing this limiting issue (Akdel et al., 2022). Indeed, crystallographic software packages such as PHENIX have begun directly incorporating application programming interfaces (APIs) into prediction software such as ROSETTA and AlphaFold (Baek et al., 2021; Jumper et al., 2021; Liebschner et al., 2019).
3.2. Updated model provides new insight into monoPGTs
In the original X‐ray crystal structure analysis of PglC from C. concisus, discontinuous electron density prevented the placement of 19 C‐terminal residues from the 201‐residue protein. Here, we show that augmenting a high‐quality dataset with a prediction from AlphaFold can significantly change the interpretation of the experimental map. The placement of C‐terminal residues in PglC has allowed a new understanding of monotopic PGT structure and function. The C‐terminal residues of PglC are stabilized by a salt bridge between E117 and K196 and by interactions between F197 and the other aromatic residues that make up an aromatic box motif. Evolutionary covariance analysis has established the importance and conservation of these interactions in the UDP‐diNAcBac‐utilizing members of the superfamily. Without computational prediction, the presence of this conserved aromatic box motif comprising residues distant in linear sequence would not have been possible.
Aromatic box motifs are common structural elements. These networks of overlapping π‐systems can range from simple stacking interactions to complex networks involving five or more aromatic amino acids (Burley & Petsko, 1985; Holliday et al., 2009; Lanzarotti et al., 2011; Makwana & Mahalakshmi, 2015). Aromatic box motifs increase protein stability, and disruption of any one residue in an aromatic cluster may significantly destabilize a protein (Frank et al., 2002; Kannan & Vishveshwara, 2000). Further, these motifs are involved in ligand binding in a number of protein families—notable examples include Cereblon binding of thalidomide (Hartmann et al., 2014), as well as UDP‐sugar utilizing glycosyl transferases (Park et al., 2018). In our study, analysis of PglC aromatic box variants shows that the aromatic box motif is indeed critical for PglC stability and activity. The replacement of one of the five aromatic residues with an aliphatic residue results in a significant loss of protein stability and activity, and the replacement of one residue with a different aromatic residue results in a substantial reduction in catalytic turnover. To explain this loss in activity, we hypothesize that the C‐terminal residues are important for establishing the active site of PglC; mutation of the aromatic box may alter the interaction of the C‐terminus with the remainder of the structure, thereby distorting the active site and impacting catalysis. Further experimentation is needed to test this model.
3.3. Application of substrate specificity prediction to the broader PGT superfamily
Selectivity for a particular NDP‐sugar substrate is a defining characteristic of the monotopic PGT superfamily. Despite extensive biochemical characterization and structural information, the link between specificity and sequence has remained ambiguous (Lukose et al., 2015; O'Toole, Imperiali et al., 2021; Ray et al., 2018). By leveraging a diverse set of several thousand PGT sequences and our improved structural model, we have identified both sequence and structure fingerprints of UDP‐diNAcBac‐utilizing PGTs. Although previous attempts to connect sequence and NDP‐glycan selectivity in the Acinetobacter baumannii identified single amino acids associated with smPGT substrate specificity, these results were difficult to reconcile with the subsequent PglC structure (Harding et al., 2018). The co‐conservation of the aromatic box with the GLLLP motif of the mobile loop portion of PglC paints a clearer picture of regions of the protein which may affect substrate binding and positioning during the course of catalysis. Differential conservation of these regions in PGTs known to utilize substrates other than UDP‐diNAcBac leads to the intriguing hypothesis that subtle structural differences in several regions of PGTs distal to the catalytic site may work in concert to drive substrate selectivity. As more structural and biochemical characterization of monoPGT enzymes becomes available, we envision similar substrate specificity predictions may be created to describe monoPGTs of differing substrate selectivity.
3.4. Predicted structures of non‐diNAcBac PGTs
Owing to the ability of AlphaFold to accurately predict the structure of Campylobacter PglC, we leveraged this tool to generate structures of PGTs outside of the diNAcBac cluster. AlphaFold can also be applied to model the structures of non‐diNAcBac PGTs with high confidence and predicts intriguing structural variability across PGT clades (Figure S6). One example of a structurally divergent PGT is the smPGT from Litoreibacter albidus (Uniprot: J0URD9), which lacks the aromatic box motif, has a truncated C‐terminus and is predicted to encode a helix‐turn‐helix motif in place of the loop containing GLLLP motif in PglC (Figure 5). Another example is the smPGT from Oceanicola sp. (Uniprot: A0A254R773). This PGT is predicted to encode a 10 amino‐acid extension to helix 2, forming a structured lid over the active site which occupies the same region as the absolute C‐terminus of PglC. This PGT also lacks the GLLLP motif of the diNAcBac PGTs (Figure 5). While neither of these PGTs has been expressed or characterized, it is tempting to hypothesize a general theme of structural variation above the active site dictating substrate specificity, or interactions with additional proteins in the biosynthetic pathway.
4. MATERIALS AND METHODS
4.1. Protein expression and purification
Expression and purification of both the wild type and variants of PglC from C. concisus were carried out using previously published protocols (Ray et al., 2018). Detailed experimental procedures are provided in the supplement.
Activity assays. UMP/CMP‐Glo (Promega) was used for measuring PglC activity by monitoring UMP byproduct release, as described previously (Mirdita et al., 2022). All assays were performed at room temperature in assay buffer containing 50 mM HEPES at pH 7.5, 150 mM NaCl, 5 mM MgCl2, 0.1% Triton X‐100, and 10% DMSO. To measure the relative activity of each variant, PglC at concentrations ranging from 0.3 to 30 nM was reacted with 20 μM UDP‐diNAcBac and 20 μM UndP and quenched at 5 and 10 min. The reaction was predetermined to be linear over 10 min at the given concentrations. Control experiments were performed in the absence of PglC and UDP‐diNAcBac. At the end of the reaction, a 20 μL aliquot was quenched with an equal volume of the UMP‐Glo reagent, mixed gently, and transferred to a 96‐well plate (Corning; white, flat bottom, nonbinding surface, half area). A SynergyH1 multimode plate reader (Biotek) was used to measure luminescence. The 96‐well plate was shaken inside the plate reader chamber at 237 cpm at 25°C in the double orbital mode for 16 min, followed by 44 min incubation at the same temperature, after which time the luminescence was recorded (gain: 200, integration time: 0.5 s). Conversion of luminescence to UMP concentration was carried out using a standard curve.
For Michaelis–Menten kinetic analysis, similar assays were carried out using various concentrations of UDP‐diNAcBac (2.5–200 μM) with 0.3 nM PglC for WT and 3 nM for PglC variants. Reactions were quenched at several time points (3, 6, 9, 12, and 15 min), and reaction rates were calculated in Excel. Rates in the linear range with less than 10% substrate turnover were used for steady‐state kinetic analysis. Kinetic parameters were calculated using nonlinear regression in GraphPad Prism.
4.2. Structure modeling
Alphafold models for PGTs were built using LocalColabFold v1.3.0 (Mirdita et al., 2022) with flags ‐‐amber, ‐‐templates, ‐‐num‐recycle 3, ‐‐use‐gpu‐relax activated.
4.3. X‐ray crystallographic data processing
Refinement of C. concisus PglC with the additional C‐terminal residues was performed using PHENIX (Liebschner et al., 2019) and model building with Coot (Emsley et al., 2010) using structure factors from PDBid 5W7L. The resulting model was validated using MolProbity (Chen et al., 2010). All structure figures were generated using PyMOL (Schrodinger, LLC, 2015). X‐ray crystallographic structure factors and coordinates have been deposited in the Protein Data Bank under accession code PDBid 8G1N.
4.4. Sequence alignments and phylogeny
Sequences encoding smPGTs were extracted from a previously described SSN (O'Toole, Bernstein et al., 2021). This set of PGTs was then seeded with characterized smPGTs as well as the catalytic domains of characterized bi/lgPGTs. An MSA and corresponding phylogenetic tree were generated using Geneious Prime (Version February 2, 2022).
Coevolution analysis. Coevolution analysis was performed in Gremlin (https://gremlin.bakerlab.org) (Kamisetty et al., 2013; Ovchinnikov et al., 2014). The sequence alignment of smPGTs was used as the input. Iterations were set to zero to use the given alignment without MSA enrichment. All other parameters were set to default.
4.5. Profile hidden Markov model generation
The cluster of UDP‐diNAcBac utilizing PGTs was extracted from the overall alignment and randomly split 1:1 into a training set and a test set. The profile HMM was trained on the training set of diNAcBac PGTs using HMMer (Version 3.2.2) with default options (Orwick‐Rydmark et al., 2016). The resulting model was applied to both the test set of diNAcBac PGTs and the non‐diNAcBac sequences from the parent alignment. A cutoff score of 200 (E value ≤ 1E‐72) was sufficient to select for characterized diNAcBac‐utilizing PGTs over characterized non‐diNAcBac‐utilizing PGTs.
AUTHOR CONTRBUTIONS
Alyssa J. Anderson: Conceptualization, methodology, validation, formal analysis, investigation, writing—original draft, writing—review & editing, visualization. Greg J. Dodge: Conceptualization, methodology, validation, formal analysis, investigation, writing—original draft, writing—review & editing, visualization. Karen N. Allen: Conceptualization, writing—review & editing, supervision, funding acquisition. Barbara Imperiali: Conceptualization, writing—review & editing, supervision, funding acquisition.
FUNDING INFORMATION
This work was funded by National Institutes of Health grants R01 GM131627 and GM039334 (to B.I. and K.N.A.) and F32 GM134576 (G.J.D.).
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supporting information
Data S1. Supplementary materials: AROBOX‐SI‐4‐14‐23.
Detailed experimental procedures for protein purification and protein stability analysis. Additional tables and figures to support conclusions in the main text of the article.
ACKNOWLEDGMENTS
The authors thank Dr. Leah Seebald for her assistance with preliminary PglC purifications and activity assays.
Anderson AJ, Dodge GJ, Allen KN, Imperiali B. Co‐conserved sequence motifs are predictive of substrate specificity in a family of monotopic phosphoglycosyl transferases. Protein Science. 2023;32(6):e4646. 10.1002/pro.4646
Review Editor: Nir Ben‐Tal
DATA AVAILABILITY STATEMENT
Sequence alignment, sequence tree, and hidden Markov model are available for download from Mendeley Data. DOI: 10.17632/b57wtpx78y.1. Updated C. concisus PglC will supersede the original model on the PDB using the accession code 5W7L. X‐ray crystallographic structure factors and coordinates have been deposited in the PDB under accession code PDB 8G1N.
REFERENCES
- Akdel M, Pires DE, Pardo EP, Jänes J, Zalevsky AO, Mészáros B, et al. A structural biology community assessment of AlphaFold2 applications. Nat Struct Mol Biol. 2022;29(11):1056–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Dabbagh B, Mengin‐Lecreulx D, Bouhss A. Purification and characterization of the bacterial UDP‐glcnac: Undecaprenyl‐phosphate GlcNAc‐1‐phosphate transferase WecA. J Bacteriol. 2008;190(21):7141–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaouteli S, Bamford NC, Stanley‐Wall NR, Kovács ÁT. Bacillus subtilis biofilm formation and social interactions. Nat Rev Microbiol. 2021;19(9):600–14. [DOI] [PubMed] [Google Scholar]
- Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, et al. Accurate prediction of protein structures and interactions using a three‐track neural network. Science. 2021;373(6557):871–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burley S, Petsko GA. Aromatic‐aromatic interaction: a mechanism of protein structure stabilization. Science. 1985;229(4708):23–8. [DOI] [PubMed] [Google Scholar]
- Cartee RT, Forsee WT, Bender MH, Ambrose KD, Yother J. CpsE from type 2 Streptococcus pneumoniae catalyzes the reversible addition of glucose‐1‐phosphate to a polyprenyl phosphate acceptor, initiating type 2 capsule repeat unit formation. J Bacteriol. 2005;187(21):7425–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, et al. Molprobity: all‐atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(1):12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das D, Kuzmic P, Imperiali B. Analysis of a dual domain phosphoglycosyl transferase reveals a ping‐pong mechanism with a covalent enzyme intermediate. Proc Natl Acad Sci. 2017;114(27):7019–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das D, Walvoort M, Lukose V, Imperiali B. A rapid and efficient luminescence‐based method for assaying phosphoglycosyltransferase enzymes. Sci Rep. 2016;6(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(4):486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank BS, Vardar D, Buckley DA, McKnight CJ. The role of aromatic residues in the hydrophobic core of the villin headpiece subdomain. Protein Sci. 2002;11(3):680–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover KJ, Weerapana E, Chen MM, Imperiali B. Direct biochemical evidence for the utilization of UDP‐bacillosamine by PglC, an essential glycosyl‐1‐phosphate transferase in the Campylobacter jejuni N‐linked glycosylation pathway. Biochemistry. 2006;45(16):5343–50. [DOI] [PubMed] [Google Scholar]
- Harding CM, Haurat MF, Vinogradov E, Feldman MF. Distinct amino acid residues confer one of three UDP‐sugar substrate specificities in Acinetobacter baumannii PglC phosphoglycosyltransferases. Glycobiology. 2018;28(7):522–33. [DOI] [PubMed] [Google Scholar]
- Hartmann MD, Boichenko I, Coles M, Zanini F, Lupas AN, Alvarez BH. Thalidomide mimics uridine binding to an aromatic cage in cereblon. J Struct Biol. 2014;188(3):225–32. [DOI] [PubMed] [Google Scholar]
- Headd JJ, Echols N, Afonine PV, Grosse‐Kunstleve RW, Chen VB, Moriarty NW, et al. Use of knowledge‐based restraints in Phenix. Refine to improve macromolecular refinement at low resolution. Acta Crystallogr D Biol Crystallogr. 2012;68(4):381–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday GL, Mitchell JB, Thornton JM. Understanding the functional roles of amino acid residues in enzyme catalysis. J Mol Biol. 2009;390(3):560–77. [DOI] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamisetty H, Ovchinnikov S, Baker D. Assessing the utility of coevolution‐based residue–residue contact predictions in a sequence‐and structure‐rich era. Proc Natl Acad Sci. 2013;110(39):15674–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan N, Vishveshwara S. Aromatic clusters: a determinant of thermal stability of thermophilic proteins. Protein Eng. 2000;13(11):753–61. [DOI] [PubMed] [Google Scholar]
- Karmali AM, Blundell TL, Furnham N. Model‐building strategies for low‐resolution x‐ray crystallographic data. Acta Crystallogr D Biol Crystallogr. 2009;65(2):121–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzarotti E, Biekofsky RR, Estrin DA, Marti MA, Turjanski AG. Aromatic–aromatic interactions in proteins: beyond the dimer. J Chem Inf Model. 2011;51(7):1623–33. [DOI] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkóczi G, Chen VB, Croll TI, et al. Macromolecular structure determination using x‐rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol. 2019;75(10):861–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linton D, Dorrell N, Hitchen PG, Amber S, Karlyshev AV, Morris HR, et al. Functional analysis of the Campylobacter jejuni N‐linked protein glycosylation pathway. Mol Microbiol. 2005;55(6):1695–703. [DOI] [PubMed] [Google Scholar]
- Lukose V, Luo L, Kozakov D, Vajda S, Allen KN, Imperiali B. Conservation and covariance in small bacterial phosphoglycosyltransferases identify the functional catalytic core. Biochemistry. 2015;54(50):7326–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makwana KM, Mahalakshmi R. Implications of aromatic–aromatic interactions: from protein structures to peptide models. Protein Sci. 2015;24(12):1920–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino S, Jimenez N, Molero R, Bouamama L, Regué M, Tomás JM. A UDP‐HexNAc: Polyprenol‐P‐GalNAc‐1‐P‐transferase (WecP) representing a new subgroup of the enzyme family. J Bacteriol. 2011;193(8):1943–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M. ColabFold: making protein folding accessible to all. Nat Methods. 2022;19(6):679–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison MJ, Imperiali B. The renaissance of bacillosamine and its derivatives: pathway characterization and implications in pathogenicity. Biochemistry. 2014;53(4):624–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole KH, Bernstein HM, Allen KN, Imperiali B. The surprising structural and mechanistic dichotomy of membrane‐associated phosphoglycosyl transferases. Biochem Soc Trans. 2021;49(3):1189–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole KH, Imperiali B, Allen KN. Glycoconjugate pathway connections revealed by sequence similarity network analysis of the monotopic phosphoglycosyl transferases. Proc Natl Acad Sci. 2021;118(4):e2018289118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orwick‐Rydmark M, Arnold T, Linke D. The use of detergents to purify membrane proteins. Curr Protoc Protein Sci. 2016;84(1):4.8.1–4.8.35. [DOI] [PubMed] [Google Scholar]
- Ovchinnikov S, Kamisetty H, Baker D. Robust and accurate prediction of residue–residue interactions across protein interfaces using evolutionary information. eLife. 2014;3:e02030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JB, Kim YH, Yoo Y, Kim J, Jun S‐H, Cho JW, et al. Structural basis for arginine glycosylation of host substrates by bacterial effector proteins. Nat Commun. 2018;9(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel KB, Furlong SE, Valvano MA. Functional analysis of the c‐terminal domain of the WbaP protein that mediates initiation of O‐Antigen synthesis in Salmonella enterica . Glycobiology. 2010;20(11):1389–401. [DOI] [PubMed] [Google Scholar]
- Patel KB, Toh E, Fernandez XB, Hanuszkiewicz A, Hardy GG, Brun YV, et al. Functional characterization of UDP‐glucose: undecaprenyl‐phosphate glucose‐1‐phosphate transferases of Escherichia coli and Caulobacter crescentus . J Bacteriol. 2012;194(10):2646–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray LC, Das D, Entova S, Lukose V, Lynch AJ, Imperiali B, et al. Membrane association of monotopic phosphoglycosyl transferase underpins function. Nat Chem Biol. 2018;14(6):538–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodinger, LLC . The PyMol molecular graphics system, version 1.8. Forthcoming, November. 2015.
- Tytgat HL, Lebeer S. The sweet tooth of bacteria: common themes in bacterial glycoconjugates. Microbiol Mol Biol Rev. 2014;78(3):372–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield C, Wear SS, Sande C. Assembly of bacterial capsular polysaccharides and exopolysaccharides. Annu Rev Microbiol. 2020;74:521–43. [DOI] [PubMed] [Google Scholar]
- Whitfield C, Williams DM, Kelly SD. Lipopolysaccharide O‐antigens—bacterial glycans made to measure. J Biol Chem. 2020;295(31):10593–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplementary materials: AROBOX‐SI‐4‐14‐23.
Detailed experimental procedures for protein purification and protein stability analysis. Additional tables and figures to support conclusions in the main text of the article.
Data Availability Statement
Sequence alignment, sequence tree, and hidden Markov model are available for download from Mendeley Data. DOI: 10.17632/b57wtpx78y.1. Updated C. concisus PglC will supersede the original model on the PDB using the accession code 5W7L. X‐ray crystallographic structure factors and coordinates have been deposited in the PDB under accession code PDB 8G1N.