

Abstract
H2 relaxin is a peptide hormone that exerts its biological actions through the G protein-coupled receptor, RXFP1. The numerous important biological functions of H2 relaxin, including potent renal, vasodilatory, cardioprotective, and anti-fibrotic actions, have resulted in considerable interest in its use as a therapeutic for various cardiovascular diseases and other fibrotic indications. Interestingly though, H2 relaxin and RXFP1 have been shown to be overexpressed in prostate cancer, allowing for the downregulation or blocking of relaxin/RXFP1 to decrease prostate tumor growth. These findings suggest the application of an RXFP1 antagonist for the treatment of prostate cancer. However, these therapeutically relevant actions are still poorly understood and have been hindered by the lack of a high-affinity antagonist. In this study, we chemically synthesized three novel H2 relaxin analogues that have complex insulin-like structures with two chains (A and B) and three disulfide bridges. We report here the structure–activity relationship studies on H2 relaxin that resulted in the development of a novel high-affinity RXFP1 antagonist, H2 B-R13HR (∼40 nM), that has only one extra methylene group in the side chain of arginine 13 in the B-chain (ArgB13) of H2 relaxin. Most notably, the synthetic peptide was shown to be active in a mouse model of prostate tumor growth in vivo where it inhibited relaxin-mediated tumor growth. Our compound H2 B-R13HR will be an important research tool to understand relaxin actions through RXFP1 and may be a potential lead compound for the treatment of prostate cancer.
Keywords: relaxin, H2 relaxin, RXFP1 antagonist, GPCR, peptide
The peptide hormone relaxin is an important pregnancy hormone in most mammalian species (reviewed in refs (1, 2)). In humans, it is the RLN2 gene, encoding the peptide H2 relaxin, that is responsible for these pregnancy-related actions. The receptor for H2 relaxin is the G protein-coupled receptor, relaxin family peptide receptor 1 (RXFP1),3 which was originally called LGR7.4 H2 relaxin has important cardiovascular functions during pregnancy, including potent renal and vasodilatory actions.5,6 These actions together with the cardioprotective7 and anti-fibrotic actions8,9 of relaxin led to the use of H2 relaxin in clinical trials for the treatment of heart failure.10,11 There is still considerable interest in agonist drugs targeting RXFP1, especially chronic treatments for heart failure.12−14 Additionally, agonists targeting RXFP1 show promise as an anti-fibrotic therapy, based on relaxin’s combined ability to inhibit the impact of cell apoptosis, inflammation, hypertrophy, and oxidative stress to newly synthesized and secreted extracellular matrix (ECM) deposition while being able to stimulate the matrix metalloproteinase (MMP)-induced resolution of established ECM build-up.7−9
There is also a potential therapeutic utility for RXFP1 antagonists. Relaxin and/or RXFP1 expression has been demonstrated to be upregulated in numerous types of cancer.15−18 In prostate cancer, relaxin has been shown to increase tumor growth and invasiveness by virtue of its angiogenic (involving its MMP-promoting), vasodilatory, and ECM remodeling properties, which are critical for tumor viability.16−20 H2 relaxin signaling in prostate cancer may also drive androgen receptor signaling pathway activation via phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) and Wnt pathways through the stabilization of cytoplasmic β-catenin.21 Therefore, targeting the relaxin signaling by an RXFP1 antagonist could deliver a “dual hit” to prostate cancer cells by blocking the tumor growth-promoting activity of H2 relaxin as well as blocking the crosstalk between androgen receptor and relaxin-mediated androgen receptor signaling. An RXFP1 antagonist thus holds particular promise in developing treatments for late-stage, androgen-insensitive prostate cancer.
Despite the considerable interest in targeting RXFP1 for treating disease conditions, studies on the mechanism of action of H2 relaxin via RXFP1 have been hindered by the lack of a high-affinity antagonist. There is currently no structure of H2 relaxin bound to RXFP1 to guide antagonist development. However, various studies have highlighted that the mechanism by which relaxin binds to and activates RXFP1 is complex. RXFP1 contains a large extracellular domain (ECD) with a leucine-rich repeat (LRR) domain and linker region connected to a low-density lipoprotein Class A (LDLa) module.4 The H2 relaxin peptide is composed of two chains, an A- and a B-chain, linked by disulfide bonds (Figure 1). The primary site for H2 relaxin binding involves the B-chain residues ArgB13, ArgB17, and IleB20 interacting with the RXFP1 LRRs.22 Mutagenesis and modeling experiments suggest that ArgB13 and ArgB17 interact with acidic residues (Asp231, Glu233, Glu277, and Asp279) within LRR6 and LRR8 of RXFP1, whereas Ile20 interacts with hydrophobic residues (Trp180, Ile182, and Leu204) of LRR4-5.22 There is an additional binding site for H2 relaxin in the linker region that involves distinct residues in the linker, although the H2 relaxin residues involved in this interaction are unknown.23 Activation of RXFP1 also requires the N-terminal LDLa module,24 and the LDLa in combination with H2 relaxin bound to the ectodomain stabilizes and extends a helical region within the linker, enabling the motif to form a conformation that leads to receptor activation.25
Figure 1.
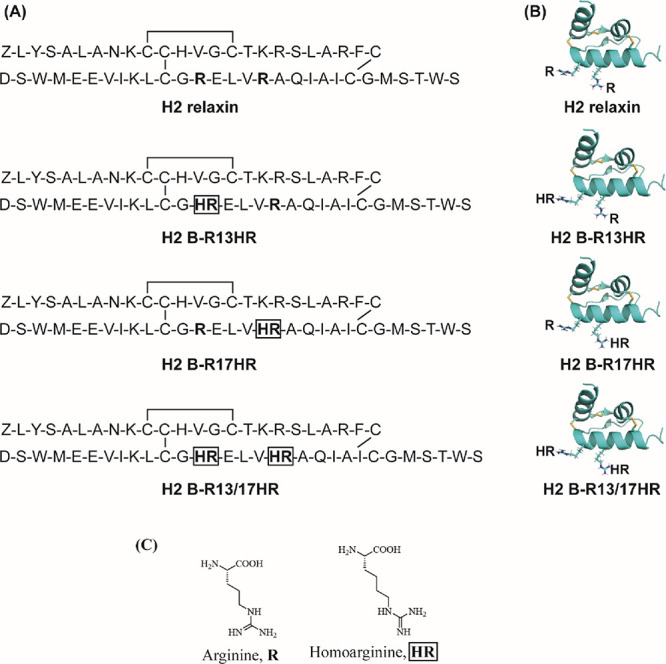
Amino acid sequences, structure of H2 relaxin, and predicted structures of H2 variants. (A) Primary sequence of human relaxin 2 (H2 relaxin) and its variants, H2 B-R13HR, H2 B-R17HR, and H2 B-R13/17HR. (B) Solution NMR structure of H2 relaxin (Protein Data Bank (PDB): 2MV1) and simulated structures of HR-incorporated H2 relaxin analogues. Structural estimation of HR substitution into H2 relaxin was achieved with PyMOL v 2.0 using the SwissSideChain plugin, and arginine residues were replaced with HR before the side chains were manually aligned to the original arginine side chain. (C) Structures of arginine (R) and homoarginine (HR).
This complex binding mode of H2 relaxin has hindered both agonist and antagonist development. However, a previous study suggested that the substitution of ArgB13 and ArgB17 with lysine led to the production of an RXFP1 antagonist.20 The authors of that study demonstrated that lentiviral production of an H2 relaxin prohormone with lysine substitutions at B13 and B17 successfully reduced H2 relaxin-mediated prostate tumor growth in PC3 luciferase cell line (PC3-Luc)-induced xenograft tumors.20 Chemical synthesis of the lysine-substituted H2 relaxin peptide (H2 B-R13/17K) demonstrated that this peptide was able to bind to RXFP1 and antagonize relaxin actions in vitro and in vivo.(26) However, as the affinity of H2 B-R13/17K for RXFP1 was very poor (0.4 μΜ), in this study, we investigated whether alternate amino acid substitutions at positions ArgB13 and ArgB17 could result in a high-affinity antagonist. Further, we investigated individual substitutions at ArgB13 and ArgB17 to determine which residue is responsible for antagonist activity. We report here the development of a high-affinity RXFP1 antagonist, which will not only be an important tool for understanding the mechanism of relaxin action but could also be useful as a potential lead for the treatment of prostate cancer.
Results
Design and Synthesis
To understand the roles of two key arginine residues, ArgB13 and ArgB17 of H2 relaxin, and develop an RXFP1 antagonist, we substituted the arginine residues with homoarginine (HR), an arginine isostere (Figure 1). This arginine structural mimic (Figure 1C) preserves the guanidinium group of arginine but has a side chain lengthened by one carbon. Three HR-incorporated H2 relaxin analogues were designed and then synthesized (Figure 1) by the total chemical synthesis method,27 which is solid phase peptide synthesis followed by regioselective disulfide bond formation strategies28 with very good purity (>95%, calculated from the HPLC trace, area under the curve (AUC) integration) (Supporting Information).
Activity of Modified H2 Relaxin Peptides in HEK-RXFP1 Cells
The synthetic peptides were tested for binding and activity in human embryonic kidney (HEK)-293T cells stably over-expressing the RXFP1 receptor, in comparison to H2 relaxin and H2 B-R13/17K (Figure 2). Competition binding assays using europium-labeled H2 relaxin (Eu-H2 relaxin) demonstrated that H2 B-R13/17HR had higher affinity (∼65 nM) for RXFP1 than H2 B-R13/17K (∼0.4 μM) (H2 B-R13/17HR pKi = 7.20 ± 0.08, H2 B-R13/17K pKi = 6.44 ± 0.06; p < 0.001) but still much lower affinity than H2 relaxin (∼0.64 nM) (H2 relaxin pKi = 9.19 ± 0.13; p < 0.001: Figure 2A, Table 1). Interestingly, the single HR substituted peptides showed markedly different affinities with H2 B-R17HR having a similar affinity (∼1.3 nM) to H2 relaxin (∼0.64 nM) (H2 B-R17HR pKi = 8.90 ± 0.08, H2 relaxin pKi = 9.19 ± 0.13; ns) and H2 B-R13HR having an affinity (∼40 nM) slightly higher than H2 B-R13/17HR (∼65 nM) (pKi = 7.37 ± 0.17; Figure 2A, Table 1). The peptides were then tested for their ability to activate RXFP1 using a well-characterized cell line stably expressing RXFP1 and a pCRE reporter gene. As reported previously,26 H2 B-R13/17K was a weak partial agonist of RXFP1 (H2 B-R13/17K pEC50 = 6.82 ± 0.23, Emax = 69.6 ± 2.1, H2 relaxin: pEC50 = 10.60 ± 0.03, both p < 0.001; Figure 2B, Table 1). H2 B-R13/17HR and H2 B-R13HR were also partial agonists with higher activity and efficacy than H2 B-R13/17K (H2 B-R13/17HR pEC50 = 7.88 ± 0.05, Emax = 82.2 ± 5.1, H2 B-R13HR pEC50 = 8.33 ± 0.19, Emax = 82.7 ± 0.7; pEC50p < 0.001, Emax p < 0.05). Conversely, H2 B-R17HR was a full agonist with similar activity and efficacy to H2 relaxin (H2 B-R17HR pEC50 = 10.66 ± 0.24, Emax = 89.5 ± 2.9, ns).
Figure 2.

Activity of B-R13/17 modified peptides in comparison to H2 relaxin in HEK-RXFP1 cells. (A) Whole cell Eu-H2 relaxin competition binding assays. (B) cAMP activity expressed as percent maximum H2 relaxin response from CRE reporter gene assays. Data are expressed as the mean ± SEM of at least three experiments performed in triplicate.
Table 1. Pooled Binding Affinity (pKi) and cAMP Activity (Peptide Potency: pEC50 and Efficacy: Emax) Dataa.
| cAMP
activity |
Eu-H2 relaxin binding | ||
|---|---|---|---|
| ligand | pEC50 | Emax | affinity pKi |
| H2 relaxin | 10.60 ± 0.03 (7)# | 100 (7)# | 9.19 ± 0.13 (5)# |
| H2 B-R13/17K | 6.82 ± 0.23 (4)** | 69.6 ± 2.1 (4)** | 6.44 ± 0.06 (3)** |
| H2 B-R13/17HR | 7.88 ± 0.05 (7)**,# | 82.2 ± 5.1 (7)*,^ | 7.20 ± 0.08 (4)**,# |
| H2 B-R17HR | 10.66 ± 0.24 (6)# | 89.5 ± 2.9 (6)θ | 8.90 ± 0.08 (4)# |
| H2 B-R13HR | 8.33 ± 0.19 (5)**,# | 82.7 ± 0.7 (5)*,^ | 7.37 ± 0.17 (4)**,# |
*p < 0.05; **p < 0.001 vs H2 relaxin; ^p < 0.05, θp > 0.01, #p < 0.001 vs H2 B-R13/17K.
Activity of Modified H2 Relaxin Peptides in THP1 Cells
The HR-modified peptides were then tested for their ability to activate cyclic adenosine monophosphate (cAMP) accumulation in THP1 cell line that natively expresses low levels of RXFP1. (Figure 3, Table 2). Interestingly, all of the peptides demonstrated full agonist activity, although the potency of H2 B-R13/17HR (pEC50 = 7.47 ± 0.15, p < 0.001) and H2 B-R13HR (pEC50 = 7.67 ± 0.16, p < 0.001) was lower than H2 relaxin (pEC50 = 9.29 ± 0.11). H2 B-R17HR was a full agonist for cAMP accumulation in THP1 cells with a similar potency to H2 relaxin (pEC50 = 9.21 ± 0.13).
Figure 3.

cAMP accumulation activity of B-R13/17HR modified peptides in comparison to H2 relaxin in THP1 cells. Data are expressed as the percent maximum H2 relaxin response and represent the mean ± SEM of at least three experiments performed in triplicate.
Table 2. Pooled cAMP Activity (Peptide Potency: pEC50 and Efficacy: Emax) Data for THP1 Cell Activation.
| THP1
cAMP activity |
||
|---|---|---|
| ligand | pEC50 | Emax |
| H2 relaxin | 9.29 ± 0.11 (7) | 100 (7) |
| H2 B-R17HR | 9.21 ± 0.13 (3) | 101.5 ± 0.6 (3) |
| H2 B-R13HR | 7.67 ± 0.16 (4)* | 100.2 ± 3.9 (4) |
| H2 B-R13/17HR | 7.47 ± 0.15 (3)* | 85.6 ± 10.6 (3) |
Activity of Modified H2 Relaxin Peptides in Human Cardiac Fibroblasts
The effects of H2 B-R13/17K, H2 B-R13/17HR, and H2 B-R13HR were then evaluated in transforming growth factor beta 1 (TGF-β1) (2 ng/mL)-stimulated human cardiac myofibroblasts (HCMFs), which were previously shown to express RXFP1 and respond to relaxin.29 Consistent with its MMP-promoting effects in other human myofibroblast culture models,30−33 H2 relaxin (100 ng/mL/16.8 nM) consistently promoted MMP-2 levels by ∼65–70% above corresponding levels measured in TGF-β1 alone-stimulated HCMFs, after 72 h in culture (p < 0.01 vs TGF-β1 alone; Figure 4A). When co-administered with the same dose of H2 relaxin over the same culture period, 1 μM H2 B-R13/17K, H2 B-R13/17HR, or H2 B-R13HR significantly and equivalently prevented the MMP-2-promoting effects of H2 relaxin such that MMP-2 levels in these co-treated groups were similar to those measured from TGF-β1 alone-stimulated cells (all p < 0.05 vs H2 relaxin alone; all no different to TGF-β1 alone; Figure 4A). To determine if any of these modified H2 relaxin peptides had improved antagonist efficacy over the others at a 10-fold lower dose, they were also each co-administered with H2 relaxin at 0.1 μM and equivalently induced a trend toward inhibiting the MMP-promoting actions of H2 relaxin (all by ∼50–55%), although not to a statistically significant extent (Figure 4A). Importantly, none of the modified peptides evaluated (at 0.1 μM) affected MMP-2 expression in the absence of H2 relaxin. These findings suggested that modifications to the H2 relaxin B-chain involving HR substitutions were able to effectively antagonize the MMP-promoting effects of H2 relaxin to the same extent as when a lysine (K) substitution was made.
Figure 4.

Evaluation of the effects of H2 B-R13/17K, H2 B-R13/17HR, and H2 B-R13HR on the MMP-promoting effects of H2 relaxin and effects of H2 B-R13/17HR and H2 B-R13HR on the collagen-inhibitory effects of H2 relaxin in human cardiac myofibroblasts (HCMFs). (A) Representative gelatin zymograph of MMP-2 levels (gelatinase A; 72 kDa) that were secreted into the cell media from TGF-β1 (2 ng/mL)-stimulated HCMFs alone (lane 1) and TGF-β1-stimulated HCMFs that were co-treated with H2 relaxin (16.8 nM) alone (lane 2) or H2 relaxin and two different concentrations (1 or 0.1 μM) of H2 B-R13/17K (lanes 3 and 4, respectively), H2 B-R13/17HR (lanes 5 and 6, respectively) or H2 B-R13HR (lanes 7 and 8, respectively) or 0.1 μM H2 B-R13/17K (lanes 9), H2 B-R13/17HR (lanes 10) or H2 B-R13HR (lane 3) alone for 72 h in culture. Also shown is the relative mean ± SEM OD of MMP-2 from each of the groups evaluated, from n = 3 separate experiments conducted in duplicate. (B) Mean ± SEM collagen content from 500,000 TGF-β1-stimulated HCMFs as well as TGF-β1-stimulated HCMFs that were co-treated with H2 relaxin (16.8 nM) alone, H2 B-R13/17HR (1 μM) or H2 B-R13HR (1 μM) alone, or H2 relaxin+H2 B-R13/17HR or H2 relaxin+H2 B-R13HR for 72 h in culture, from n = 4 separate experiments conducted in duplicate. *p < 0.05, **p < 0.01 vs TGF-β1 alone; #p < 0.05, ##p < 0.01 vs (TGF-β1+)H2 relaxin-treated group.
Based on these findings, the antagonistic effects of H2 B-R13/17HR or H2 B-R13HR were further evaluated on the collagen-modulating effects of H2 relaxin. Consistent with the well-documented collagen-inhibitory effects of H2 relaxin when applied to TGF-β1-stimulated human myofibroblasts,30,31 H2 relaxin (100 ng/mL) significantly prevented the TGF-β1-stimulated collagen deposition in HCMFs over 72 h by ∼30% (p < 0.01 vs TGF-β1 alone; Figure 4B). The co-administration of H2 B-R13/17HR (1 μM) or H2 B-R13HR (1 μM) with the same dose of H2 relaxin completely prevented these collagen-inhibitory effects of H2 relaxin over 72 h to levels that were similar to that measured from TGF-β1 alone stimulated cells (both p < 0.01 vs H2 relaxin alone; Figure 4B). However, neither of these modified peptides (at 1 μM) affected the TGF-β1-stimulated collagen deposition on their own (Figure 4B).
Activity of Modified H2 Relaxin Peptides in a Mouse Model of Prostate Cancer
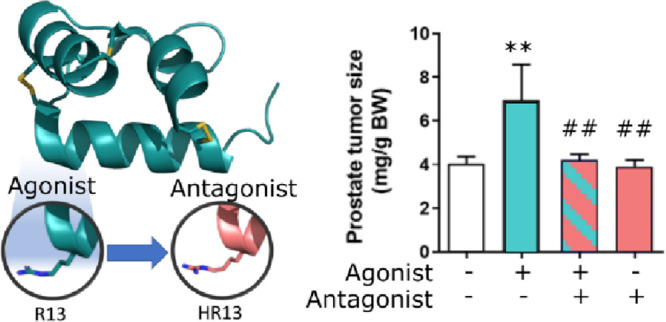
As the proof-of-principle studies conducted in HCMFs (Figure 4) demonstrated that the two HR-substituted H2 relaxin peptides could effectively block the RXFP1-mediated effects of H2 relaxin, the antagonistic potential of H2 B-R13/17HR and H2 B-R13HR was further evaluated in a murine model of prostate cancer in vivo.(34) Consistent with the angiogenic and prostate tumor growth-promoting effects of H2 relaxin,20,35 mice that received H2 relaxin (0.15 mg/kg/day; from days 2–10 post-RM1 cell administration) alone had significantly increased prostate tumor weights (by ∼73%; p < 0.01 vs untreated group) after 8 days of treatment, compared to their untreated counterparts (Figure 5). However, mice co-treated with H2 relaxin and a 10-fold higher dose of either H2 B-R13/17HR (1.5 mg/kg/day) or H2 B-R13HR (1.5 mg/kg/day) had equivalent prostate tumor weights to their untreated counterparts (both p < 0.01 vs H2 relaxin treatment alone), indicating that these modified H2 relaxin peptides had inhibited the prostate tumor growth-promoting effects of H2 relaxin (Figure 5). On the other hand, no differences in prostate tumor growth were measured from mice that were treated with the modified H2 relaxin peptides alone compared to untreated mice (Figure 5), suggesting that these modified peptides did not have any growth-promoting effects on their own.
Figure 5.

Evaluation of the effects of H2 B-R13/17HR and H2 B-R13/HR on the prostate tumor growth-promoting effects of H2 relaxin. Shown is the prostate tumor size (mg) of RM1 cell-injected mice that were left untreated for 10 days versus RM1 cell-injected mice that were treated with H2 relaxin (0.15 mg/kg/day) alone, H2 B-R13/17HR (1.5 mg/kg/day) or H2 B-R13HR (1.5 mg/kg/day) alone, or H2 relaxin+H2 B-R13/17HR or H2 relaxin+H2 B-R13HR from days 2–10 post-RM1 cell administration; from n = 4–6 mice/group. **p < 0.01 vs untreated group; ##p < 0.01 vs H2 relaxin alone-treated group.
To further assess the antagonistic effects of H2 B-R13/17HR or H2 B-R13HR, the MMP-promoting effects of H2 relaxin were assessed from protein extracts isolated from the six groups evaluated (Figure 6). Consistent with the prostate tumor-promoting effects of H2 relaxin (Figure 5), mice treated with H2 relaxin alone had significantly increased levels of prostate MMP-9 (by ∼1.3-fold) and MMP-2 (by ∼0.55-fold) after 8 days of treatment (both p < 0.01 vs corresponding levels from untreated mice), compared to respective measurements from untreated mice (Figure 6). The co-administration of a 10-fold higher either H2 B-R13/17HR (1.5 mg/kg/day) or H2 B-R13HR (1.5 mg/kg/day) with H2 relaxin was found to completely suppress the MMP-promoting effects of H2 relaxin within the prostate of mice (both p < 0.05 vs H2 relaxin treatment alone), whereas neither modified peptide alone affected gelatinase levels within the mouse prostate compared to respective levels measured from untreated mice (Figure 6).
Figure 6.

Evaluation of the effects of H2 B-R13/17HR and H2 B-R13HR on the MMP-promoting effects of H2 relaxin in mouse prostate tumors. (A) Representative zymograph of MMP-9 (gelatinase B; 92 kDa) and MMP-2 (gelatinase A; 72 kDa) levels from mouse prostate tumor-derived protein extracts from untreated mice (lanes 1 and 2) and mice treated with 0.15 mg/kg/day H2 relaxin alone (lanes 3 and 4); 0.15 mg/kg/day H2 relaxin+1.5 mg/kg/day H2 B-R13/17HR (lanes 5 and 6); 1.5 mg/kg/day H2 B-R13/17HR alone (lanes 7 and 8); 0.15 mg/kg/day H2 relaxin+1.5 mg/kg/day H2 B-R13HR (lanes 9 and 10); or 1.5 mg/kg/day H2 B-R13HR alone (lanes 11 and 12). Three separate zymographs analyzing 4–6 samples per group produced similar results. Relative mean ± SEM OD of (B) MMP-9 and (C) MMP-2 from each of the six groups analyzed, from n = 4–6 mice/group. **p < 0.01 vs untreated group; #p < 0.05, ##p < 0.01 vs H2 relaxin alone-treated group.
Circular Dichroism Study
We studied the secondary structure of HR-variants using circular dichroism (CD) spectroscopy and compared it with native H2 relaxin. The CD spectra (Figure 7) demonstrated that all peptides possessed a typical α-helical pattern, where the double minima were observed at 208 and 222 nm. The α-helix content for H2 relaxin and its variants, calculated using the mean residual ellipticity at 222 nm ([θ]222),36 was 42% for H2 relaxin, 33% for H2 B-R17HR, 28% for H2 B-R13HR, and 22% for H2 B-R13/17HR.
Figure 7.

Circular dichroism spectra of H2 relaxin and HR-incorporated variants in phosphate buffer (pH 7.5) at 25 °C.
Discussion and Conclusions
Over the past years, there has been great interest in the peptide hormone H2 relaxin and its cognate receptor, RXFP1, due to its assessment in clinical trials for the treatment of scleroderma, orthodontic tooth movement, and acute heart failure. However, studies on the biology of RXFP1 have been hindered by the lack of a high-affinity antagonist.
We have previously synthesized an H2 relaxin analogue, H2 BR13/17K, that demonstrated antagonist activity in some cells and partial agonist activity in HEK cells overexpressing RXFP1.26 In this study, we investigated alternative modifications of the key ArgB13 and ArgB17 residues and also produced single arginine-modified versions to determine which of the residues is critical for antagonist activity.
Bullesbach and colleagues37 first reported the importance of the arginine residues at positions 13 and 17 of the H2 relaxin B-chain in relaxin receptor binding. Since Bullesbach et al. demonstrated that the analogue H2 B-R13/17K had abolished bioactivity in the pubic symphysis bioassay with partial binding affinity for RXFP1, Silvertown et al.20 was the first to hypothesize that the H2 B-R13/17K analogue could act as an antagonist and developed a prorelaxin lysine substituted mutant (H2 B-R13/17K) for lentiviral-driven prohormone expression. Extending this effort, we chemically synthesized the mature lysine-substituted H2 relaxin peptide (H2 B-R13/17K)26 and demonstrated that it was able to bind to RXFP1 and antagonize relaxin actions in vitro and in vivo. However, as the affinity of H2 B-R13/17K for RXFP1 was very poor (∼0.4 μM), in this study, we investigated whether H2 relaxin analogues with HR-substitutions at positions 13 and 17 would improve the binding affinity while retaining antagonist activity. It was hypothesized that retaining the guanidinium group in the side chain of HR variants would improve the affinity for RXFP1 as it is predicted that these side chains are involved in hydrogen bonding interactions with glutamic acid and aspartic acid residues in the LRR region of the RXFP1 ectodomain.37 Further, it was predicted that the addition of an extra methylene group (CH2) in the HR structure would mimic the lysine effect, which resulted in RXFP1 antagonism. Therefore, we produced H2 B-R13/17HR as well as the individual mutants H2 B-R13HR and H2 B-R17HR and tested their activity on RXFP1 binding, cAMP signaling in RXFP1 expressing cells and human cardiac fibroblasts (HCFs) in vitro, and in an experimental model of prostate cancer in vivo.
As anticipated, H2 B-R13/17HR demonstrated improved binding affinity at RXFP1 compared to H2 B-R13/17K. Interestingly, the single HR mutants displayed markedly different affinities with H2 B-R17HR showing only a slightly lower affinity to H2 relaxin and H2 B-R13HR having a similar affinity to H2 B-R13/17HR. These higher binding affinities were associated with more potent actions on cAMP activation in HEK-RXFP1 cells with H2 B-R17HR showing similar full agonist potency to H2 relaxin. In contrast, H2 B-R13HR demonstrated partial agonist activity, although it was more potent and efficacious than H2 B-R17K. The peptides were also tested for their ability to activate cAMP in THP1 cells that natively express RXFP1 at levels ∼250-fold lower than HEK-RXFP1 cells. All of the HR-substituted peptides acted as full agonists of cAMP accumulation in THP1 cells with H2 B-R13/17HR and H2 B-R13HR demonstrating potencies 500-fold lower than H2 relaxin and H2 B-R17HR consistent with their lower binding affinities for RXFP1. Importantly, we have demonstrated that substitution at ArgB17 had no effect on H2 relaxin binding; hence, further testing was only performed on H2 B-R13/17HR and H2 B-R13HR.
The anti-fibrotic actions of relaxin are one of its major organ-protective properties, which involve its ability to regulate MMPs, as part of its ability to provoke collagen remodeling [reviewed in refs (7, 9)]. Importantly, we have demonstrated that these effects do not involve cAMP activation.33 In proof-of-principle studies, H2 B-R13/17HR and H2 B-R13HR were first tested in HCMFs-expressing RXFP1 in comparison to the lysine-substituted H2 relaxin analogue (H2 B-R13/17K) to determine their antagonistic efficacy in blocking the anti-fibrotic effect of H2 relaxin that were mediated via RXFP1. The HR- and K-substituted peptides showed similar antagonistic efficacy in blocking the MMP-2-promoting effects of H2 relaxin (at 1 μM) when co-administered with H2 relaxin (Figure 4A). Additionally, H2 B-R13/17HR and H2 B-R13HR were able to equivalently antagonize the collagen-inhibitory effects of H2 relaxin when co-administered at 1 μM (Figure 4B), whereas these modified peptides did not induce any MMP-promoting or collagen-inhibitory effects on their own.
Previous studies had shown using RXFP1-expressing myofibroblast culture models that H2 relaxin directly signaled through cyclic monophosphate guanosine (cGMP)38 or an extracellular signal-regulated kinase (ERK)1/2-neuronal oxide (NO) synthase (nNOS)-NO-cGMP-dependent pathway33 to inhibit TGF-β1 signal transduction at the level of intracellular Smad2 and Smad3 phosphorylation. This in turn affected the ability of TGF-β1 to promote fibroblast differentiation into myofibroblasts and myofibroblast-mediated collagen synthesis. Additionally, by suppressing the TGF-β1/pSmad2/pSmad3 axis, H2 relaxin appeared to release iNOS activity (which is inhibited by TGF-β1) to additionally contribute to the MMP-promoting actions of the hormone.33 The results of this study are consistent with our previous study where H2 B-R13/17K was able to antagonize the actions of H2 relaxin on renal myofibroblast differentiation while having no effect in the absence of H2 relaxin.26 Hence, it is likely that H2 B-R13/17HR and H2 B-R13HR act in a similar manner to block H2 relaxin-induced ERK1/2 or cGMP-mediated effects on myofibroblasts.
Relaxin peptide17 or its mRNA16 expression have been shown to be significantly higher in recurrent prostate cancer samples. Stimulation with H2 relaxin increased cell proliferation, invasiveness, and adhesion of LNCaP and PC3 cells.16,17 The suppression of relaxin/RXFP1 via short interfering RNAs decreases cell invasiveness by 90 to 95%.16 In two other studies, downregulation of RXFP1 expression using small interfering RNA (siRNA) was shown to reduce tumor growth and metastasis in a xenograft model of prostate cancer,17,18 suggesting the potential therapeutic benefits of an RXFP1 antagonist in prostate cancer. Therefore, we have tested both antagonist candidates, H2 B-R13/17HR and H2 B-R13HR, in a mouse model of prostate tumor growth in vivo. Consistent with previous findings in mice,20 the administration of H2 relaxin to mice at a dose of 0.15 mg/kg/day (which was expected to produce circulating relaxin levels of 5–10 ng/mL, when administered via osmotic mini-pumps), significantly promoted prostate tumor growth 10 days after mice were injected with RM1 cells and when corrected for the body weight of mice (Figure 5). Additionally, H2 relaxin significantly promoted MMP-2 and MMP-9 levels in prostate tumors (Figure 6) within this time frame. For the first time, this study demonstrated that co-administering a 10-fold higher dose of H2 B-R13/17HR or H2 B-R13HR (1.5 mg/kg/day) again completely suppressed the tumor-promoting (Figure 5) and MMP-promoting (Figure 6) effects of H2 relaxin, without having any effects on these measures in the absence of H2 relaxin. These findings are somewhat consistent with a previous study conducted by Silvertown et al.20 using the prohormone K-substituted mutant, which also successfully reduced H2 relaxin-mediated prostate tumor growth in PC3-Luc-induced xenograft tumors. These findings further validated that the MMP-promoting effects of H2 relaxin have the potential to worsen prostate cancer progression39 as MMPs have been shown to contribute to tumor neovascularization and subsequent metasis.40 They also suggest that the mechanism of H2 relaxin-mediated MMP promotion and tumor growth did not involve cAMP activation and that both H2 B-R13/17HR or H2 B-R13HR could potentially block the tumor growth-related angiogenic effects of relaxin.
Previous studies have demonstrated that H2 relaxin-mediated prostate tumor growth involves G protein βΥ subunit-mediated activation of Akt via PI3K activation21,41 and also by cAMP-mediated activation of protein kinase A (PKA).41 Hence, it is possible that the modified H2 relaxin peptides are blocking the βΥ subunit-mediated pathways in our studies. Notably, H2 relaxin has been demonstrated to activate PI3K by a Gi-βΥ-mediated mechanism in various cell types42,43 and PI3K and Gi/o inhibition in human cardiac fibroblasts markedly reduced cGMP activation.29 Hence, it is possible that the modified H2 relaxin peptides are mediating their effects in myofibroblasts and prostate cells independent of cAMP by blocking H2 relaxin stimulated Gi-βΥ-mediated mechanisms. Further studies are required to fully elucidate the mechanisms of H2 relaxin action in myofibroblasts and prostate cells for which the antagonists developed in this study will be useful tools.
The secondary structural data, measured by CD spectroscopy, is consistent with the binding and activation data in that the most helical peptides, H2 relaxin, and H2 B-R17HR, have the highest affinity and potency for RXFP1. Notably, the extra methylene group in the side chain of ArgB17 has limited impact on either binding or activation, highlighting that both the length of the side chain and the slightly reduced helicity do not impact on the interaction and activation of RXFP1. In contrast, the reduced affinity of H2 B-R13HR and H2 B-R13/17HR compared with H2 relaxin is likely due to slight disruptions in the core α-helical structure of the peptides due to the extra methylene group at the side chain of ArgB13. Most importantly, the antagonist activity of H2 B-R13HR highlights that the interaction of ArgB13 with Asp231 and Glu233 is involved with key conformational changes associated with receptor activation. Further structural studies are necessary to fully appreciate the mechanism of H2 B-R13HR binding and antagonism at RXFP1.
In conclusion, in this study, we have developed a novel RXFP1 antagonist, H2 B-R13HR, with very high affinity (Ki ∼40 nM) by the addition of only one extra methylene group in the side chain of ArgB13. Our compound will be an important research tool to understand the complex mode of H2-relaxin/RXFP1 interaction as well as to dissect their physiological and pathological roles. A large proportion of prostate cancer patients develop androgen-independent prostate cancer with an average survival of just two years. Therefore, there is a significant unmet need for new, non-androgen, anti-hormonal strategies for the management of prostate cancer. Our peptide H2 B-R13HR was shown to be active in a mouse model of prostate tumor growth in vivo where it inhibited H2 relaxin-mediated tumor growth. Therefore, H2 B-R13HR might be a potential lead to developing therapeutics for the treatment of prostate cancer.
Experimental Section
Materials
Rink amide resin (0.36 mmol/g), Nα-Fmoc-protected amino acids, and O-(1H-6-chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HCTU) were purchased from GL Biochem (China). 2,2′-Dipyridyldisulfide (DPDS), anisole, 3,6-dioxa-1,8-octanedithiol (DODt), triisopropylsilane (TIPS), guanidine hydrochloride, and trifluoromethanesulfonic acid (TFMSA) were purchased from Sigma-Aldrich (Australia). Trifluoroacetic acid (TFA), acetic acid, piperidine, N,N-diisopropylamine (DIEA), and all solvents were purchased from Merck Millipore (Australia). All solvents were of analytical grade. Recombinant H2 relaxin (H2 relaxin) was kindly provided by Corthera Inc. (San Carlos, CA, USA; a subsidiary of Novartis AG, Basel, Switzerland).
Peptide Synthesis
The three relaxin variants (Figure 1) were chemically produced, as described previously.26 In brief, the linear A- and B-chain of each analogue was synthesized using an optimized Fmoc-based solid phase peptide synthesis (SPPS) protocol.44 The Fmoc deprotection of the peptides was carried out using 20% piperidine in DMF. The peptides were then cleaved from the resin by trifluoroacetic acid (TFA).
Peptide Characterization
After the purification and characterization of both chains by reversed-phase high-performance liquid chromatography (RP-HPLC) and matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry (MS) (MALDI TOF MS), two interchain disulfide bonds were formed in a stepwise manner.27,45−47 The direct detect assay-free cards and the direct detect spectrometer were used to measure the actual peptide contents. The analysis of the purified final products was carried out on a Gemini 5 μm C18 LC column (250 × 4.6 mm, pore size 110 Å, particle size 5 μm) using a Shimadzu (Japan) Nexera analytical RP-HPLC that incorporates an SPD-M40 UV detector, at a constant flow rate of 1.5 mL/min, in the best appropriate gradient mode with buffer A: 0.1% aq. TFA and buffer B: 0.1% TFA in acetonitrile. The detection wavelength was set at 214 nm (characteristic of the amide bond). The molecular weight of the final peptides was confirmed by a Shimadzu MALDI-8020 MALDI-TOF mass spectrometer using sinapinic acid as the matrix. The purity of the H2 relaxin analogues was over 95% calculated from the HPLC AUC integration (Supporting Information).
HEK-RXFP1 Binding Assays
HEK-293T cells stably transfected with RXFP1 (HEK-RXFP1) were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal calf serum, 100 μg/mL penicillin, 100 μg/mL streptomycin, and 2 mM l-glutamine and plated into 96-well plates pre-coated with poly-l-lysine for whole cell binding assays. Competition binding experiments utilizing europium-labeled H2 relaxin were performed as described48 in the absence or presence of increasing concentrations of peptide analogues. Specific binding was determined by the addition of 1 μM H2 relaxin. Data are presented as the mean ± standard error of the mean(S.E.M.) of the % specific binding of triplicate wells, repeated in at least three separate experiments. Data are fitted with one-site binding curves and pKi values determined using GraphPad Prism 9 (GraphPad Inc., San Diego, CA). Statistical differences in pKi values were analyzed using a one-way analysis of variance with Uncorrected Fisher’s least significant difference (LSD) post-hoc analysis using GraphPad Prism 9.
HEK-RXFP1 cAMP Reporter Gene Assays
The evaluation of the ability of native H2 relaxin and HR relaxin analogues to stimulate a cAMP response in HEK-RXFP1 cells was conducted using a cAMP reporter gene assay as described previously.24 HEK-RXFP1 cells stably expressing a pCRE β-galactosidase reporter plasmid were plated in 96-well plates. Cells were incubated with increasing concentrations of H2 relaxin or relaxin analogues. The amount of cAMP-driven β-galactosidase expression in each well was assessed with a colorimetric assay measuring absorbance at 570 nm on a Benchmark Plus microplate spectrophotometer (BioRad). Ligand-induced cAMP stimulation was expressed as a percentage of maximal response of relaxin. Each data point was measured in triplicate and each experiment conducted independently at least three separate times. Statistical analysis was performed using a one-way analysis of variance with Uncorrected Fisher’s LSD post-hoc analysis using GraphPad Prism 9.
THP1 Cell Direct cAMP Assays
The ability of the peptide analogs to stimulate cAMP accumulation in RXFP1-expressing THP1 cells49 was assessed as described previously50 using direct cAMP measurement (cAMP dynamic 2 HTRF Kit, Cisbio). Cells were stimulated with peptides for 30 min in the presence of 50 μM of the phosphodiesterase inhibitor IBMX. Ligand-induced cAMP stimulation was expressed as a % of maximal response to H2 relaxin. Each data point was measured in triplicate, and each experiment was conducted independently at least three times. Statistical analysis was performed using a one-way analysis of variance with Uncorrected Fisher’s LSD post-hoc analysis using GraphPad Prism 9.
Circular Dichroism (CD) Spectroscopy
The CD spectra of H2 relaxin and variants were obtained at 25 °C with a 1 mm path-length cell. The peptides were dissolved in phosphate-buffered saline (PBS) solution (pH 7.4). The wavelength of scanning was 190–280 nm, and the data pitch was 0.1 nm. Five accumulations were taken for each peptide during continuous scanning at a speed of 0.5 times per point. The concentration of all four peptides was 0.2 μg/μL, which was converted to molar concentration for calculating the mean residue ellipticity. The signal was recorded as millidegree and normalized to give units of mean-residue ellipticity (MRE or q) or [θ] according to the following equation:
MRE = mdeg/(c × l × N), where mdeg is the CD output in millidegrees, c is the molar peptide concentration, l is the light path length in mm, and N is the number of amino acid residues.
Evaluation of Modified H2 Relaxin Peptides in Human Cardiac Fibroblasts
To investigate the effects of H2 B-R13/17K, H2 B-R13/17HR, and H2 B-R13HR on the MMP-promoting actions of H2 relaxin in vitro, primary human cardiac fibroblasts (HCFs; which contained a mixture of atrial and ventricular fibroblasts; ScienCell Research Laboratories, San Diego, CA, USA) that expressed RXFP129 were used. These cells were maintained as described previously,29 passaged when they reached 70–80% confluence, and were used between passages 3 and 5 for all studies. Cells were plated in 12-well plates at an equal density of 100,000 cells/cm2 (per well). All wells were then immediately stimulated with transforming growth factor-beta1 (TGF-β1; 2 ng/mL; to promote myofibroblast (HCMF) differentiation), given that H2 relaxin mediates its anti-fibrotic actions in response to TGF-β1 stimulation.33,51,52 Sub-groups of duplicate wells treated with TGF-β1 were additionally treated with H2 relaxin alone (100 ng/mL; equivalent to 16.8 nM). In turn, sub-groups of TGF-β1+H2 relaxin-treated duplicate wells were co-administered with either H2 B-R13/17K, H2 B-R13/17HR, and H2 B-R13HR (at either 1 μM or 0.1 μM). Additional duplicate wells stimulated with TGF-β1 alone were also treated with either H2 B-R13/17K, H2 B-R13/17HR, and H2 B-R13HR (at 0.1 μM) in the absence of H2 relaxin. All treatment groups were maintained in growth media containing 10% fetal bovine serum (FBS) for 48 h and then in media containing 0.5% FBS for a further 24 h as FBS can interfere with the zymographic analysis of MMP expression/activity. At the completion of the 72 h experiments, the conditioned media from each well was collected and stored in −20 °C (for analysis of MMPs by gelatin zymography). These studies were completed n = 3 separate times.
To investigate the effects of H2 B-R13/17HR and H2 B-R13HR on the collagen-inhibitory effects of H2 relaxin, HCFs were plated in 12-well plates at an equal density of 500,000 cells/cm2 (per well). Duplicate wells were again stimulated with TGF-β1 (2 ng/mL; to generate HCMFs) in the absence or presence of H2 relaxin (100 ng/mL) and the absence or presence of H2 B-R13/17HR (1 μM) or H2 B-R13HR (1 μM). As additional controls, TGF-β1-stimulated cells were separately co-administered with H2 B-R13/17HR (1 μM) or H2 B-R13HR (1 μM) in the absence of H2 relaxin. All treatment groups were maintained in growth media containing 10% for FBS 72 h. The media from each well was then removed, and 0.5 mL of 6 M hydrochloric acid (HCl) was added to each cell layer before the cell layers were scraped off each well and placed in 9 mL Kimax screw-capped glass tubes (Kimble/Kontes Laboratories, Pasadena, TX, USA). Samples were hydrolyzed in 6 M HCl for 18–20 h at 110 °C (in a hot plate) before being cooled and lyophilized in a freeze-dryer to dry weight (to remove HCl). Each sample was then re-suspended in 25 μL of 0.1 M HCl so that duplicate 10 μL aliquots could be assayed for hydroxyproline content.53 The studies detailed above were completed n = 4 separate times.
Mouse Prostate Tumor Model
RM1 cells, a murine prostate carcinoma androgen-insensitive cell line, were derived by transformation from the genital ridge of C57BL6/J mice. Cells were grown in 75 cm2 flasks in Dulbecco’s Modified Eagle Medium (DMEM; Invitrogen, New York, NY, USA) with glucose (1000 mg/L) and 10% fetal bovine serum (FBS; Sigma, Australia) and incubated with 5% CO2 at 37 ° C. Five-week-old male C57BL/6 mice were obtained from Monash Animal Services, housed and maintained under standard conditions. Mice were anesthetized via inhalation of a 5% isoflurane/95% oxygen air mixture. An incision was made through the skin and muscle of the abdomen to expose the bladder and the prostate. A cell suspension (10 μL) containing 5 × 103 RM1 cells in DMEM media plus 10% FBS was injected orthotopically into the prostate to induce prostate tumor growth,34 and the wound was then closed. The sub-group of mice (n = 6) was left untreated until day 10 post-RM1 cell administration (a time-point at which these mice developed small but significantly increased prostate tumors34). Additional sub-groups of mice (n = 5–6/group) were subcutaneously implanted with osmotic mini-pumps (model 1007D, which have flow rates of 0.5 μL/h; Durect Corp., Cupertino, CA, USA) containing H2 relaxin alone (0.15 mg/kg/day), H2 B-R13/17HR alone (1.5 mg/kg/day), H2 B-R13HR alone (1.5 mg/kg/day), or a combination of H2 relaxin+H2 B-R13/17HR or H2 relaxin+H2 B-R13HR, from 2 days post-RM1 cell administration until day 10 post-cell administration. Each pump had a reservoir that allowed it to continuously infuse the peptides administered to mice for 8 days. These experiments were approved by Monash University Animal Ethics Committees, which adheres to the Australian Guidelines for the Care and Use of Laboratory Animal for Scientific Purposes.
Tissue Collection
At 10 days post-RM1 cell administration, all untreated and treated mice were weighed before being killed, and their prostate tumors and attached seminal vesicles were dissected out, photographed, and weighed. The prostate tumors were then separated from the seminal vesicle and weighed again. Prostate tumors were then frozen at −80 °C for protein isolation.
Protein Extraction and Quantification
The total protein content from prostate tumors was isolated by homogenizing tissues in 0.25% (w/v) Triton-X100 containing 10 mM calcium chloride (CaCl2) (at a ratio of 20 mL per gram of tissue wet weight). The homogenates were then centrifuged at 4000 rpm for 20 min before the supernatants were discarded and the pellet (containing most of the protein) resuspended in an equal volume of 0.1 M CaCl2. Samples were then heated at 60 °C in a shaking water bath for 4 min before being cooled and transferred into Millipore concentrators (Merck Millipore, Darmstadt, Germany) with a 10 kDa cutoff. Samples were concentrated by centrifugation, washed with 1 mL of Tris–HCl, pH 6.8, and further concentrated until they were reduced to 100–150 μL in volume, and quantified for protein content using the Bradford protein assay.54
Gelatin Zymography
To determine the effects of H2 relaxin in the absence or presence of H2 B-R13/17K, H2 B-R13/17HR, or H2 B-R13HR on MMP expression and activity, gelatin zymography of conditioned media or concentrated prostate tumor homogenates was performed. Fixed volumes of media (2.5-3 μL of 1:10 diluted samples for the detection of gelatinase A/MMP-2; 6 μL for the detection of gelatinase B/MMP-9) or 1.5 μg of total protein from prostate tumors was loaded onto 7.5% of acrylamide gels containing 1 mg/mL porcine gelatin, and 3.5% acrylamide stacking gels, and assessed by gelatin zymography as described previously.33,55 Zymographs were stained with 1% Coomassie Blue containing 40% isopropanol for 1 h before being destained with 7% acetic acid for approximately 45 min. The clear bands on each gel indicated sites at which MMP-2 (∼72 kDa) and MMP-9 (∼92 kDa) cleaved the gelatin that was in each separating gel. The optical density (OD) of MMP-2 and MMP-9 expression was then determined with the Bio-Rad ChemicDoc XRS (Bio-Rad, Hercules, CA, USA) and Quantity-One software.
Statistical Analysis
Unless otherwise stated, all data are presented as the mean ± standard error of the mean (SEM) and were statistically analyzed with GraphPad Prism 9.0.1 (GraphPad Software Inc.; San Diego, CA, USA). In most cases, the data was analyzed using a one-way ANOVA. Post hoc tests were only conducted if the overall ANOVA p value achieved statistical significance (i.e., p < 0.05), and there was no significant variance in homogeneity. A Tukey’s post-hoc test was used where multiple groups were compared to each other. A non-parametric (Kruskal–Wallis) test was conducted for data that were expressed relative to the control group, which was expressed as 1 in each case (relative MMP data). All data were included unless any values were >2 standard deviations (SDs) from the mean. No approaches were used to reduce unwanted sources of variation by data normalization or to generate normal data. Differences were considered statistically significant at p < 0.05.
Acknowledgments
This work was supported by the National Health & Medical Research Council (NHMRC) of Australia Grants to M.A.H. and C.S.S. (GNT2001178) and R.A.D.B. (GNT2001027); an NHMRC Senior Research Fellowship to R.A.D.B. (GNT1135837); and a Monash Biomedicine Discovery Institute Senior Research Fellowship to C.S.S. Studies at the Florey Institute were supported by the Victorian Government’s Operational Infrastructure Support Program. We thank Sharon Layfield and Tania Ferraro for their technical assistance.
Glossary
ABBREVIATIONS
- RXFP1
relaxin family peptide receptor 1
- ECM
extracellular matrix
- MMP
matrix metalloproteinase
- ECD
extracellular domain
- LRR
leucine-rich repeat
- LDLa
low-density lipoprotein class A
- Arg/R
arginine
- Ile
isoleucine
- Asp
aspartic acid
- Glu
glutamic acid
- Trp
tryptophan
- Leu
leucine
- PC3-Luc
PC3 luciferase cell line
- HR
homoarginine
- Eu-H2 relaxin
europium-labeled H2 relaxin
- TGF-β1
transforming growth factor beta 1
- HCMF
human cardiac myofibroblasts
- K
lysine
- CD
circular dichroism
- CH2
methylene group
- HCFs
human cardiac fibroblasts
- cGMP
cyclic monophosphate guanosine
- cAMP
cyclic adenosine monophosphate
- ERK
extracellular signal-regulated kinase
- NO
neuronal oxide
- nNOS
neuronal oxide synthase
- Akt
protein kinase B
- PI3K
phosphatidylinositol 3-kinase
- PKA
protein kinase A
- HCTU
O-(1H-6-chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- DPDS
2,2′-dipyridyldisulfide
- DODt
3,6-dioxa-1,8-octanedithiol
- TIPS
triisopropylsilane
- TFMSA
trifluoromethanesulfonic acid
- TFA
trifluoroacetic acid
- DIEA
N,N-diisopropylamine
- SPPS
solid-phase peptide synthesis
- RP-HPLC
reversed-phase high-performance liquid chromatography
- MALDI TOF MS
matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry (MS)
- HEK-293T
human embryonic kidney
- RPMI
Roswell Park Memorial Institute
- S.E.M.
standard error of the mean
- LSD
least significant difference
- PBS
phosphate-buffered saline
- MRE
mean-residue ellipticity
- FBS
fetal bovine serum
- HCl
hydrochloric acid
- CaCl2
calcium chloride
- OD
optical density
- SDs
standard deviations
- AUC
area under the curve
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.3c00053.
Characterization of peptides by MALDI-TOF MS and analytical HPLC (PDF)
Author Contributions
All authors have given approval for the final version of the manuscript. Conceived and designed the experiments: M.A.H., C.S.S., S.S., and R.A.D.B. Performed the experiments and analyzed the data: P.P., N.A.N., I.P.H., H.W., and T.H. Contributed reagents/materials: M.A.H., C.S.S., S.S., and R.A.D.B. Wrote the paper: R.A.D.B., C.S.S., and M.A.H.
The authors declare no competing financial interest.
Supplementary Material
References
- Bathgate R. A.; Hsueh A. J.; Sherwood O. D.. Physiology and Molecular Biology of the Relaxin Peptide Family. In Physiology of Reproduction, 3rd ed.; Neill J. D., Ed. Elsevier: San Diego, 2006; pp. 679–770, 10.1016/B978-012515400-0/50021-X [DOI] [Google Scholar]
- Patil N. A.; Rosengren K. J.; Separovic F.; Wade J. D.; Bathgate R. A. D.; Hossain M. A. Relaxin family peptides: structure-activity relationship studies. Br. J. Pharmacol. 2017, 174, 950–961. 10.1111/bph.13684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bathgate R. A.; Ivell R.; Sanborn B. M.; Sherwood O. D.; Summers R. J. International Union of Pharmacology: Recommendations for the nomenclature of receptors for relaxin family peptides. Pharmacol. Rev. 2006, 58, 7–31. 10.1124/pr.58.1.9. [DOI] [PubMed] [Google Scholar]
- Hsu S. Y.; Nakabayashi K.; Nishi S.; Kumagai J.; Kudo M.; Sherwood O. D.; Hsueh A. J. Activation of orphan receptors by the hormone relaxin. Science 2002, 295, 671–674. 10.1126/science.1065654. [DOI] [PubMed] [Google Scholar]
- Conrad K. P.; Novak J. Emerging role of relaxin in renal and cardiovascular function. Am. J. Physiol. Regul Integr. Comp. Physiol. 2004, 287, R250–R261. 10.1152/ajpregu.00672.2003. [DOI] [PubMed] [Google Scholar]
- Samuel C. S.; Du X. J.; Bathgate R. A.; Summers R. J. ’Relaxin’ the stiffened heart and arteries: the therapeutic potential for relaxin in the treatment of cardiovascular disease. Pharmacol. Ther. 2006, 112, 529–552. 10.1016/j.pharmthera.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Martin B.; Romero G.; Salama G. Cardioprotective actions of relaxin. Mol. Cell. Endocrinol. 2019, 487, 45–53. 10.1016/j.mce.2018.12.016. [DOI] [PubMed] [Google Scholar]
- Samuel C. S.; Royce S. G.; Hewitson T. D.; Denton K. M.; Cooney T. E.; Bennett R. G. Anti-fibrotic actions of relaxin. Br. J. Pharmacol. 2017, 174, 962–976. 10.1111/bph.13529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel C. S.; Bennett R. G. Relaxin as an anti-fibrotic treatment: Perspectives, challenges and future directions. Biochem. Pharmacol. 2022, 197, 114884 10.1016/j.bcp.2021.114884. [DOI] [PubMed] [Google Scholar]
- Teerlink J. R.; Cotter G.; Davison B. A.; Felker G. M.; Filippatos G.; Greenberg B. H.; Ponikowski P.; Unemori E.; Voors A. A.; Adams K. F. Jr.; Dorobantu M. I.; Grinfeld L. R.; Jondeau G.; Marmor A.; Masip J.; Pang P. S.; Werdan K.; Teichman S. L.; Trapani A.; Bush C. A.; Saini R.; Schumacher C.; Severin T. M.; Metra M. Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): a randomised, placebo-controlled trial. Lancet 2013, 381, 29–39. 10.1016/S0140-6736(12)61855-8. [DOI] [PubMed] [Google Scholar]
- Metra M.; Teerlink J. R.; Cotter G.; Davison B. A.; Felker G. M.; Filippatos G.; Greenberg B. H.; Pang P. S.; Ponikowski P.; Voors A. A.; Adams K. F.; Anker S. D.; Arias-Mendoza A.; Avendano P.; Bacal F.; Bohm M.; Bortman G.; Cleland J. G. F.; Cohen-Solal A.; Crespo-Leiro M. G.; Dorobantu M.; Echeverria L. E.; Ferrari R.; Goland S.; Goncalvesova E.; Goudev A.; Kober L.; Lema-Osores J.; Levy P. D.; McDonald K.; Manga P.; Merkely B.; Mueller C.; Pieske B.; Silva-Cardoso J.; Spinar J.; Squire I.; Stepinska J.; Van Mieghem W.; von Lewinski D.; Wikstrom G.; Yilmaz M. B.; Hagner N.; Holbro T.; Hua T. A.; Sabarwal S. V.; Severin T.; Szecsody P.; Gimpelewicz C.; Investigators R.-A.-C. Effects of Serelaxin in Patients with Acute Heart Failure. N. Engl. J. Med. 2019, 381, 716–726. 10.1056/NEJMoa1801291. [DOI] [PubMed] [Google Scholar]
- Illiano S.; Poirier B.; Minoletti C.; Pasquier O.; Riva L.; Chenede X.; Menguy I.; Guillotel M.; Prigent P.; Le Claire S.; Gillot F.; Thill G.; Lo Presti F.; Corbier A.; Le Bail J. C.; Grailhe P.; Monteagudo E.; Ingenito R.; Bianchi E.; Philippo C.; Duclos O.; Mallart S.; Bathgate R.; Janiak P. Characterization of a new potent and long-lasting single chain peptide agonist of RXFP1 in cells and in vivo translational models. Sci. Rep. 2022, 12, 20435. 10.1038/s41598-022-24716-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dschietzig T. B. Relaxin-2 for heart failure with preserved ejection fraction (HFpEF): Rationale for future clinical trials. Mol. Cell. Endocrinol. 2019, 487, 54–58. 10.1016/j.mce.2019.01.013. [DOI] [PubMed] [Google Scholar]
- Sun J.; Hao W.; Fillmore N.; Ma H.; Springer D.; Yu Z. X.; Sadowska A.; Garcia A.; Chen R.; Muniz-Medina V.; Rosenthal K.; Lin J.; Kuruvilla D.; Osbourn J.; Karathanasis S. K.; Walker J.; Murphy E. Human Relaxin-2 Fusion Protein Treatment Prevents and Reverses Isoproterenol-Induced Hypertrophy and Fibrosis in Mouse Heart. J. Am. Heart Assoc. 2019, 8, e013465 10.1161/JAHA.119.013465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burston H. E.; Kent O. A.; Communal L.; Udaskin M. L.; Sun R. X.; Brown K. R.; Jung E.; Francis K. E.; La Rose J.; Lowitz J.; Drapkin R.; Mes-Masson A. M.; Rottapel R.. Inhibition of relaxin autocrine signaling confers therapeutic vulnerability in ovarian cancer. J. Clin. Invest., 2021, 131, (7), , 10.1172/JCI142677. [DOI] [PMC free article] [PubMed]
- Feng S.; Agoulnik I. U.; Bogatcheva N. V.; Kamat A. A.; Kwabi-Addo B.; Li R.; Ayala G.; Ittmann M. M.; Agoulnik A. I. Relaxin promotes prostate cancer progression. Clin. Cancer Res. 2007, 13, 1695–1702. 10.1158/1078-0432.ccr-06-2492. [DOI] [PubMed] [Google Scholar]
- Feng S.; Agoulnik I. U.; Li Z.; Han H. D.; Lopez-Berestein G.; Sood A.; Ittmann M. M.; Agoulnik A. I. Relaxin/RXFP1 signaling in prostate cancer progression. Ann. N. Y. Acad. Sci. 2009, 1160, 379–380. 10.1111/j.1749-6632.2008.03793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S.; Agoulnik I. U.; Truong A.; Li Z.; Creighton C. J.; Kaftanovskaya E. M.; Pereira R.; Han H. D.; Lopez-Berestein G.; Klonisch T.; Ittmann M. M.; Sood A. K.; Agoulnik A. I. Suppression of relaxin receptor RXFP1 decreases prostate cancer growth and metastasis. Endocr.-Relat. Cancer 2010, 17, 1021–1033. 10.1677/erc-10-0073. [DOI] [PubMed] [Google Scholar]
- Nair V. B.; Samuel C. S.; Separovic F.; Hossain M. A.; Wade J. D. Human relaxin-2: historical perspectives and role in cancer biology. Amino Acids 2012, 43, 1131–1140. 10.1007/s00726-012-1375-y. [DOI] [PubMed] [Google Scholar]
- Silvertown J. D.; Symes J. C.; Neschadim A.; Nonaka T.; Kao J. C.; Summerlee A. J.; Medin J. A. Analog of H2 relaxin exhibits antagonistic properties and impairs prostate tumor growth. FASEB J 2007, 21, 754–765. 10.1096/fj.06-6847com. [DOI] [PubMed] [Google Scholar]
- Liu S.; Vinall R. L.; Tepper C.; Shi X. B.; Xue L. R.; Ma A. H.; Wang L. Y.; Fitzgerald L. D.; Wu Z.; Gandour-Edwards R.; deVere White R. W.; Kung H. J. Inappropriate activation of androgen receptor by relaxin via beta-catenin pathway. Oncogene 2008, 27, 499–505. 10.1038/sj.onc.1210671. [DOI] [PubMed] [Google Scholar]
- Bullesbach E. E.; Schwabe C. The relaxin receptor-binding site geometry suggests a novel gripping mode of interaction. J. Biol. Chem. 2000, 275, 35276–35280. 10.1074/jbc.m005728200. [DOI] [PubMed] [Google Scholar]
- Sethi A.; Bruell S.; Patil N.; Hossain M. A.; Scott D. J.; Petrie E. J.; Bathgate R. A. D.; Gooley P. R. The complex binding mode of the peptide hormone H2 relaxin to its receptor RXFP1. Nat. Commun. 2016, 7, 11344–11344. 10.1038/ncomms11344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott D. J.; Layfield S.; Yan Y.; Sudo S.; Hsueh A. J.; Tregear G. W.; Bathgate R. A. Characterization of novel splice variants of LGR7 and LGR8 reveals that receptor signaling is mediated by their unique LDLa modules. J. Biol. Chem. 2006, 281, 34942–34954. 10.1074/jbc.M602728200. [DOI] [PubMed] [Google Scholar]
- Sethi A.; Bruell S.; Ryan T.; Yan F.; Tanipour M. H.; Mok Y. F.; Draper-Joyce C.; Khandokar Y.; Metcalfe R. D.; Griffin M. D. W.; Scott D. J.; Hossain M. A.; Petrie E. J.; Bathgate R. A. D.; Gooley P. R. Structural Insights into the Unique Modes of Relaxin-Binding and Tethered-Agonist Mediated Activation of RXFP1 and RXFP2. J. Mol. Biol. 2021, 433, 167217 10.1016/j.jmb.2021.167217. [DOI] [PubMed] [Google Scholar]
- Hossain M. A.; Samuel C. S.; Binder C.; Hewitson T. D.; Tregear G. W.; Wade J. D.; Bathgate R. A. The chemically synthesized human relaxin-2 analog, B-R13/17K H2, is an RXFP1 antagonist. Amino Acids 2010, 39, 409–416. 10.1007/s00726-009-0454-1. [DOI] [PubMed] [Google Scholar]
- Hossain M. A.; Rosengren K. J.; Haugaard-Jonsson L. M.; Zhang S.; Layfield S.; Ferraro T.; Daly N. L.; Tregear G. W.; Wade J. D.; Bathgate R. A. The A-chain of human relaxin family peptides has distinct roles in the binding and activation of the different relaxin family peptide receptors. J. Biol. Chem. 2008, 283, 17287–17297. 10.1074/jbc.M801911200. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Lin F.; Hossain M. A.; Shabanpoor F.; Tregear G. W.; Wade J. D. Simultaneous post-cysteine(S-Acm) group removal quenching of iodine and isolation of peptide by one step ether precipitation. Int. J. Pept. Res. Ther. 2008, 14, 301–305. 10.1007/s10989-008-9148-x. [DOI] [Google Scholar]
- Sarwar M.; Samuel C. S.; Bathgate R. A.; Stewart D. R.; Summers R. J. Serelaxin-mediated signal transduction in human vascular cells: bell-shaped concentration-response curves reflect differential coupling to G proteins. Br. J. Pharmacol. 2015, 172, 1005–1019. 10.1111/bph.12964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unemori E. N.; Pickford L. B.; Salles A. L.; Piercy C. E.; Grove B. H.; Erikson M. E.; Amento E. P. Relaxin induces an extracellular matrix-degrading phenotype in human lung fibroblasts in vitro and inhibits lung fibrosis in a murine model in vivo. J. Clin. Invest. 1996, 98, 2739–2745. 10.1172/JCI119099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain M. A.; Rosengren K. J.; Samuel C. S.; Shabanpoor F.; Chan L. J.; Bathgate R. A.; Wade J. D. The minimal active structure of human relaxin-2. J. Biol. Chem. 2011, 286, 37555–37565. 10.1074/jbc.M111.282194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W. J.; Choi I. K.; Lee J. H.; Lee J. S.; Kim Y. O.; Rah D. K.; Yun C. O. Relaxin-expressing adenovirus decreases collagen synthesis and up-regulates matrix metalloproteinase expression in keloid fibroblasts: in vitro experiments. Plast. Reconstr. Surg. 2012, 130, 407e–417e. 10.1097/PRS.0b013e31825dbf56. [DOI] [PubMed] [Google Scholar]
- Chow B. S.; Chew E. G.; Zhao C.; Bathgate R. A.; Hewitson T. D.; Samuel C. S. Relaxin signals through a RXFP1-pERK-nNOS-NO-cGMP-dependent pathway to up-regulate matrix metalloproteinases: the additional involvement of iNOS. PLoS One 2012, 7, e42714 10.1371/journal.pone.0042714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison I. P.; Vinh A.; Johnson I. R. D.; Luong R.; Drummond G. R.; Sobey C. G.; Tiganis T.; Williams E. D.; JJ O. L.; Brooks D. A.; Selemidis S. NOX2 oxidase expressed in endosomes promotes cell proliferation and prostate tumour development. Oncotarget 2018, 9, 35378–35393. 10.18632/oncotarget.26237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain M. A.; Kocan M.; Yao S. T.; Royce S. G.; Nair V. B.; Siwek C.; Patil N. A.; Harrison I. P.; Rosengren K. J.; Selemidis S.; Summers R. J.; Wade J. D.; Bathgate R. A. D.; Samuel C. S. A single-chain derivative of the relaxin hormone is a functionally selective agonist of the G protein-coupled receptor, RXFP1. Chem. Sci. 2016, 7, 3805–3819. 10.1039/C5SC04754D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholtz J. M.; Qian H.; York E. J.; Stewart J. M.; Baldwin R. L. Parameters of helix-coil transition theory for alanine-based peptides of varying chain lengths in water. Biopolymers 1991, 31, 1463–1470. 10.1002/bip.360311304. [DOI] [PubMed] [Google Scholar]
- Bullesbach E. E.; Yang S.; Schwabe C. The receptor-binding site of human relaxin II. A dual prong-binding mechanism. J. Biol. Chem. 1992, 267, 22957–22960. 10.1016/S0021-9258(18)50040-5. [DOI] [PubMed] [Google Scholar]
- Kocan M.; Sarwar M.; Ang S. Y.; Xiao J.; Marugan J. J.; Hossain M. A.; Wang C.; Hutchinson D. S.; Samuel C. S.; Agoulnik A. I.; Bathgate R. A. D.; Summers R. J. ML290 is a biased allosteric agonist at the relaxin receptor RXFP1. Sci. Rep. 2017, 7, 2968. 10.1038/s41598-017-02916-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominska K.; Ochedalski T.; Kowalska K.; Matysiak-Burzynska Z. E.; Pluciennik E.; Piastowska-Ciesielska A. W. Interaction between angiotensin II and relaxin 2 in the progress of growth and spread of prostate cancer cells. Int. J. Oncol. 2016, 48, 2619–2628. 10.3892/ijo.2016.3458. [DOI] [PubMed] [Google Scholar]
- Quintero-Fabian S.; Arreola R.; Becerril-Villanueva E.; Torres-Romero J. C.; Arana-Argaez V.; Lara-Riegos J.; Ramirez-Camacho M. A.; Alvarez-Sanchez M. E. Role of matrix metalloproteinases in angiogenesis and cancer. Front. Oncol. 2019, 9, 1370. 10.3389/fonc.2019.01370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinall R. L.; Mahaffey C. M.; Davis R. R.; Luo Z.; Gandour-Edwards R.; Ghosh P. M.; Tepper C. G.; de Vere White R. W. Dual blockade of PKA and NF-kappaB inhibits H2 relaxin-mediated castrate-resistant growth of prostate cancer sublines and induces apoptosis. Horm. Cancer 2011, 2, 224–238. 10.1007/s12672-011-0076-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halls M. L.; Bathgate R. A.; Summers R. J. Relaxin family peptide receptors, RXFP1 and RXFP2, modulate cAMP signalling by distinct mechanisms. Mol. Pharmacol. 2006, 70, 214–226. 10.1124/mol.105.021691. [DOI] [PubMed] [Google Scholar]
- Nguyen B. T.; Yang L.; Sanborn B. M.; Dessauer C. W. Phosphoinositide 3-kinase activity is required for biphasic stimulation of cyclic adenosine 3′,5′-monophosphate by relaxin. Mol. Endocrinol. 2003, 17, 1075–1084. 10.1210/me.2002-0284. [DOI] [PubMed] [Google Scholar]
- Li W.; O’Brien-Simpson N. M.; Hossain M. A.; Wade J. D. The 9-Fluorenylmethoxycarbonyl (Fmoc) Group in Chemical Peptide Synthesis – Its Past, Present, and Future. Aust. J. Chem. 2020, 73, 271–276. 10.1071/CH19427. [DOI] [Google Scholar]
- Bathgate R. A. D.; Lin F.; Hanson N. F.; Otvos L. Jr.; Guidolin A.; Giannakis C.; Bastiras S.; Layfield S. L.; Ferraro T.; Ma S.; Zhao C.; Gundlach A. L.; Samuel C. S.; Tregear G. W.; Wade J. D. Relaxin-3: Improved synthesis strategy and demonstration of its high affinity interaction with the relaxin receptor LGR7 both in vitro and in vivo. Biochemistry 2006, 45, 1043–1053. 10.1021/bi052233e. [DOI] [PubMed] [Google Scholar]
- Hossain M. A.; Bathgate R. A.; Kong C. K.; Shabanpoor F.; Zhang S.; Haugaard-Jonsson L. M.; Rosengren K. J.; Tregear G. W.; Wade J. D. Synthesis, conformation, and activity of human insulin-like peptide 5 (INSL5). ChemBioChem 2008, 9, 1816–1822. 10.1002/cbic.200800113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain M. A.; Zhang S.; Lin F.; Ferraro T.; Bathgate R. A. D.; Tregear G. W.; Wade J.D. Regioselective disulfide solid phase synthesis, chemical characterization and in vitro receptor binding activity of equine relaxin. Int. J. Pept. Res. Ther. 2006, 12, 211–215. 10.1007/s10989-006-9020-9. [DOI] [Google Scholar]
- Shabanpoor F.; Akhter Hossain M.; Ryan P. J.; Belgi A.; Layfield S.; Kocan M.; Zhang S.; Samuel C. S.; Gundlach A. L.; Bathgate R. A.; Separovic F.; Wade J. D. Minimization of Human Relaxin-3 Leading to High-Affinity Analogues with Increased Selectivity for Relaxin-Family Peptide 3 Receptor (RXFP3) over RXFP1. J. Med. Chem. 2012, 55, 1671–1681. 10.1021/jm201505p. [DOI] [PubMed] [Google Scholar]
- Mookerjee I.; Hewitson T. D.; Halls M. L.; Summers R. J.; Mathai M. L.; Bathgate R. A.; Tregear G. W.; Samuel C. S. Relaxin inhibits renal myofibroblast differentiation via RXFP1, the nitric oxide pathway, and Smad2. FASEB J 2009, 23, 1219–1229. 10.1096/fj.08-120857. [DOI] [PubMed] [Google Scholar]
- Bathgate R. A.; Samuel C. S.; Burazin T. C.; Layfield S.; Claasz A. A.; Reytomas I. G.; Dawson N. F.; Zhao C.; Bond C.; Summers R. J.; Parry L. J.; Wade J. D.; Tregear G. W. Human relaxin gene 3 (H3) and the equivalent mouse relaxin (M3) gene. Novel members of the relaxin peptide family. J. Biol. Chem. 2002, 277, 1148–1157. 10.1074/jbc.M107882200. [DOI] [PubMed] [Google Scholar]
- Samuel C. S.; Unemori E. N.; Mookerjee I.; Bathgate R. A.; Layfield S. L.; Mak J.; Tregear G. W.; Du X. J. Relaxin modulates cardiac fibroblast proliferation, differentiation and collagen production and reverses cardiac fibrosis in vivo. Endocrinology 2004, 145, 4125–4133. 10.1210/en.2004-0209. [DOI] [PubMed] [Google Scholar]
- Hossain M. A.; Man B. C.; Zhao C.; Xu Q.; Du X. J.; Wade J. D.; Samuel C. S. H3 relaxin demonstrates antifibrotic properties via the RXFP1 receptor. Biochemistry 2011, 50, 1368–1375. 10.1021/bi1013968. [DOI] [PubMed] [Google Scholar]
- Samuel C. S. Determination of collagen content, concentration, and sub-types in kidney tissue. Methods Mol. Biol. 2009, 466, 223–235. 10.1007/978-1-59745-352-3_16. [DOI] [PubMed] [Google Scholar]
- Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Woessner J. F. Jr. Quantification of matrix metalloproteinases in tissue samples. Methods Enzymol. 1995, 248, 510–528. 10.1016/0076-6879(95)48033-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.