

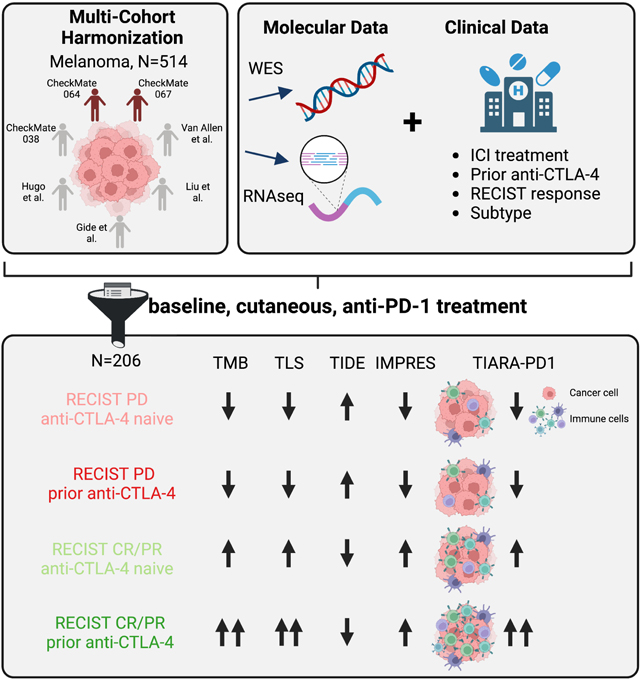
Summary
Immune checkpoint inhibitors (ICIs), including CTLA-4 and PD-1 blocking antibodies, can have profound effects on tumor immune cell infiltration that have not been consistent in biopsy series reported to date. Here, we analyze seven molecular datasets of samples from patients with advanced melanoma (N=514) treated with ICI agents to investigate clinical, genomic, and transcriptomic features of anti-PD-1 response in cutaneous melanoma. We find that prior anti-CTLA-4 therapy is associated with differences in genomic, individual gene, and gene signatures in anti-PD-1 responders. Anti-CTLA-4-experienced melanoma tumors that respond to PD-1 blockade exhibit increased tumor mutational burden, inflammatory signatures and altered cell cycle processes, compared to anti-CTLA-4-naive tumors or anti-CTLA-4-experienced, anti-PD-1-nonresponsive melanoma tumors. We report a harmonized, aggregate resource, and suggest that prior CTLA-4 blockade therapy is associated with marked differences in the tumor microenvironment that impact the predictive features of PD-1 blockade therapy response.
Keywords: Melanoma, immune checkpoint blockade, immunotherapy, meta-analysis
eTOC: Context and significance
Campbell et al. aggregate genomics and transcriptomics data across melanoma datasets, harmonizing molecular and clinical annotation across samples. Immune cell gene expression patterns and tumor mutational burden, as predictors of anti-PD-1 response, are modified by whether the patient previously received anti-CTLA-4 therapy.
Graphical Abstract
Introduction
Immune checkpoint inhibitors (ICIs) have revolutionized melanoma treatment. The anti-CTLA-4 ipilimumab was approved in 2011, followed by the approval of anti-PD-1 agents pembrolizumab and nivolumab in 2014.1,2 With five-year overall survival rates up to 52% in melanoma with combinatorial ICI approaches,3 there is substantial effort to identify predictive biomarkers for both response and resistance. Correlative studies have demonstrated patient stratification due to genomic and transcriptomic features; however, few discoveries, despite validation within and across cohorts, have been prospectively applied to clinical trials successfully.
Anti-PD-1 therapies have been approved for patients with tumors with mismatch repair deficiencies, leading to increased tumor mutational burden (TMB),4 or for solid tumors with high TMB (over 10 Mut/Mb), following progression on prior treatments and without additional treatment options.5 However, TMB and its inferred neoantigen burden have shown inconsistent concordance with response across and within tumor types and treatment settings.6–8 Bulk gene expression or spatial profiling for tumor immune infiltration or exclusion (e.g. TIDE, IMPRES), interferon gamma (IFNG) pathway activation (e.g. GEP), antigen presentation machinery, and antitumor immune cell populations have also been used to positively identify patients who will benefit from PD-1 blockade,9–15 and the combination of these patterns with high TMB have shown to be even more effective at predicting anti-PD-1 response.7,12
As more correlative datasets are generated, there have been challenges in both discovering and validating ICI biomarkers. Recent studies have attempted to overcome limitations in statistical power by aggregating molecular datasets.7,12,16,17 However, in doing so, it is important to note the level of heterogeneity in the dataset, and whether the annotation of technical, biological, and clinical variables is sufficient to delineate appropriate biomarkers and clinical contexts for application. Technical batch effects may include sample collection and preservation, nucleic acid extraction, sequencing approach, and methods for analysis. Additional biological challenges in these studies include the fundamental differences in the mechanism of each ICI, intrapatient or disease heterogeneity in genetic drivers, and immunogenicity within and across cancer types. Within melanoma, clinical trials are often inclusive of multiple melanoma subtypes, which have known differences in genomic profiles and observed differences in response to ICIs.8,18 Acral and mucosal melanomas have demonstrated reduced response and decreased survival, compared to cutaneous and occult (unknown) melanomas, and have reduced TMB, reduced UV-induced mutagenesis, fewer BRAF and NRAS hotspot mutations, and more frequent KIT mutations.8,18,19
Here, we present a harmonized dataset of whole exome sequencing (WES) and RNA sequencing (RNAseq) samples derived from melanoma tumor biopsies collected from 514 subjects treated with ICIs enrolled across seven clinical cohorts, two of which have not been previously reported that are from the Checkmate 064 and 067 clinical trials.20,21 We demonstrate that efforts in aggregating and harmonizing both molecular and clinical annotation of datasets facilitates statistical detection of genomic and expression-based correlates of response. Furthermore, exploration of clinical factors, such as melanoma subtype and prior immunotherapy treatment, reveal that previously reported predictors of anti-PD-1 response (e.g. TMB, types of immune infiltration) vary based on prior anti-CTLA-4 exposure. We summarize the collective immune infiltrate of biopsies using an aggregate metric, Tumor Immunogenicity Associated with Response to Anti-PD-1 (TIARA-PD-1), to further define tumor-intrinsic and microenvironment expression patterns related to the overall immune status of each biopsy. Our work provides a single, cohesive genomic reference dataset for uncovering molecular characteristics associated with ICI response and resistance in melanoma, and it may provide future utility for exploring molecular and clinical stratification of this disease.
Results
Cohort summary
We aggregated seven clinical cohorts of patients with melanoma treated with ICI regimens, comprising 514 patients with either whole exome sequencing (WES, N=339), RNA sequencing (RNAseq, N=403), or matched WES and RNAseq (N=258, Figure 1A) derived from melanoma tumor biopsies.6,8,20–24 While some of these datasets were previously published, 22% of the patients enrolled in these trials (N=115 patients)20,21 with correlative WES (N=184 tumors) or RNAseq (N=95; N=58 paired) have never been interrogated, providing an additional rich source of data. The following demographics were assembled for all patients and corresponding tumor specimens: RECIST 1.1 response to therapy, melanoma subtype, sex, age, treatment regimen, prior CTLA-4 blockade therapy status, originating study cohort, and time point with respect to therapy (Figure 1B, Table 1). Treatment regimens included anti-PD-1 monotherapy, anti-CTLA-4 monotherapy, the concurrent combination of these two therapies, or the sequential treatment with these two therapies (i.e. anti-PD-1-to-anti-CTLA4 or anti-CTLA4-to-anti-PD-1).
Figure 1. Project Overview.
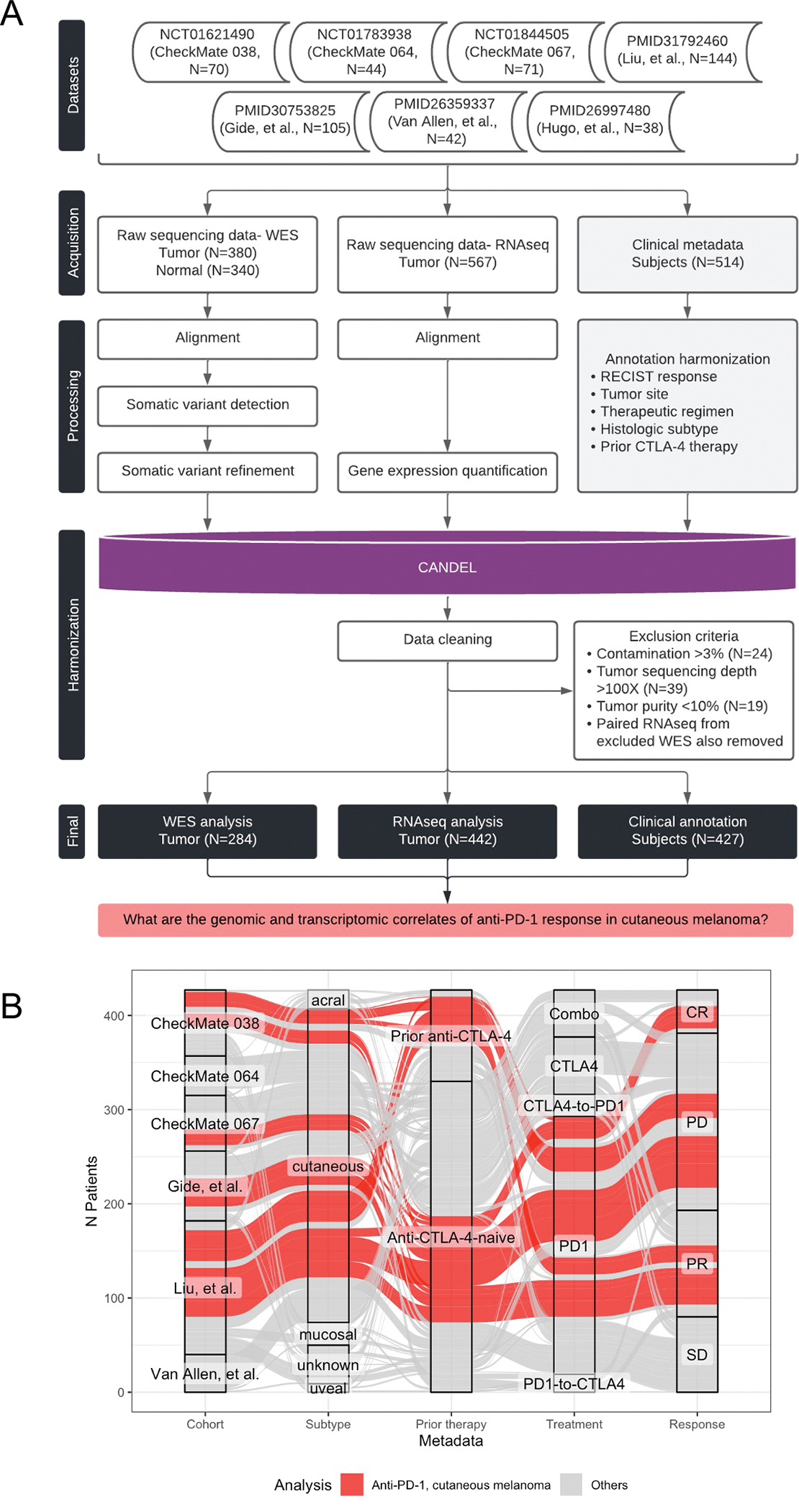
(A) Overview of the datasets and pipeline for harmonized data processing and analysis. (B) Alluvial plot depicts the demographics of patients with melanoma tumor samples in the final dataset (x-axis; cohort, subtype, prior ICI therapy, treatment regimen, and RECIST response). Each individual (alluvium) is colored by whether the corresponding sample was included in analysis of cutaneous melanoma tumors treated with anti-PD-1. See Table 1 and Figure S1.
Table 1.
Overview of patient and sample demographics in final dataset
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| CheckMate 038 | CheckMate 064 | CheckMate 067 | Gide, et al. | Liu, et al. | Van Allen, et al. | Aggregate | ||
| Patients with melanoma | ||||||||
| Sex | N | |||||||
| female | 30 | 9 | 18 | 26 | 59 | 13 | 155 | |
| male | 35 | 33 | 41 | 48 | 83 | 27 | 267 | |
| Age | median (min-max) | 56 (22–89) | 62.5 (30–84) | 58 (34–85) | 61.5 (24–90) | NA | 61 (22–83) | 59 (22–90) |
| Melanoma subtype | N | |||||||
| cutaneous | 44 | 34 | 55 | 60 | 104 | 36 | 333 | |
| unknown | 12 | 0 | 2 | 7 | 18 | 2 | 41 | |
| mucosal | 7 | 3 | 0 | 2 | 10 | 2 | 24 | |
| acral | 2 | 1 | 2 | 5 | 10 | 0 | 20 | |
| uveal | 5 | 4 | 0 | 0 | 0 | 0 | 9 | |
| Treatment | N | |||||||
| anti-PD-1 | 70 | 0 | 21 | 41 | 142 | 0 | 274 | |
| anti-CTLA-4 | 0 | 0 | 21 | 0 | 0 | 40 | 61 | |
| Combo (anti-PD-1 and anti-CTLA-4) | 0 | 0 | 17 | 33 | 0 | 0 | 50 | |
| anti-CTLA-4 to anti-PD-1 | 0 | 23 | 0 | 0 | 0 | 0 | 23 | |
| anti-PD-1 to anti-CTLA-4 | 0 | 19 | 0 | 0 | 0 | 0 | 19 | |
| Patients with tumors treated with anti-PD-1 therapy | ||||||||
| Prior ICI | N | |||||||
| anti-CTLA-4 experience | 39 | 0 | 0 | 0 | 58 | 0 | 97 | |
| anti-CTLA-4 naive | 31 | 0 | 21 | 41 | 84 | 0 | 177 | |
| Response | N | |||||||
| CR | 3 | 0 | 5 | 4 | 17 | 0 | 29 | |
| PR | 12 | 0 | 5 | 15 | 37 | 0 | 69 | |
| SD | 24 | 0 | 4 | 6 | 23 | 0 | 57 | |
| PD | 31 | 0 | 7 | 16 | 65 | 0 | 119 | |
| Timepoint | N | |||||||
| Baseline | 68 | 0 | 21 | 41 | 142 | 0 | 272 | |
| On-therapy | 61 | 0 | 0 | 9 | 0 | 0 | 70 | |
Somatic variant calling and gene expression quantification were harmonized across all cohorts (Figure S1), and tumor-normal WES pairs were evaluated for a range of quality control metrics (Figure S1–S2, Methods). Once samples were excluded due to low sequencing coverage, low estimated tumor purity, or the presence of unmatched sample contamination, the final cohort presented in the following analyses included 427 patients, 284 tumor biopsies with WES, and 442 biopsies with RNAseq (188 biopsies had matched WES and RNAseq data). With these efforts, we aimed to establish a publicly available resource summarizing these data, but to utilize the scale of this dataset to specifically evaluate genomic and transcriptomic correlates of anti-PD-1 response in cutaneous melanoma.
Tumor mutational burden is associated with anti-PD-1 benefit in anti-CTLA-4-experienced patients
Studies have described the positive association between nonsilent TMB and clinical benefit with ICIs, with discrepant results in the statistical significance within and across tumor types.17,25,26 We evaluated baseline tumor WES samples (N=246), comparing TMB across clinical demographics. There was no difference in baseline TMB across treatment regimens (Figure S2). Concordant with previous reports, TMB was significantly higher in melanoma biopsies of cutaneous origin (median 7.55, 0.046–276 mut/Mb), compared to either mucosal (median 1.83, 0.96–15.8), acral (median 1.43, 0.047–56.4), or uveal (median 0.41, 0.097–0.95) origin (Wilcoxon test, p<0.01, FDR<0.01) (Figure 2A). Tumors with unknown site of origin had ranges of TMB (median 8.11, 0.16–51.9; Wilcoxon test p=0.74, FDR 0.74) that were similar to cutaneous melanoma. There were no significant differences in baseline TMB across individual cohorts or by treatment regimen (Figure S2).
Figure 2. Genomic correlates of anti-PD-1 response.
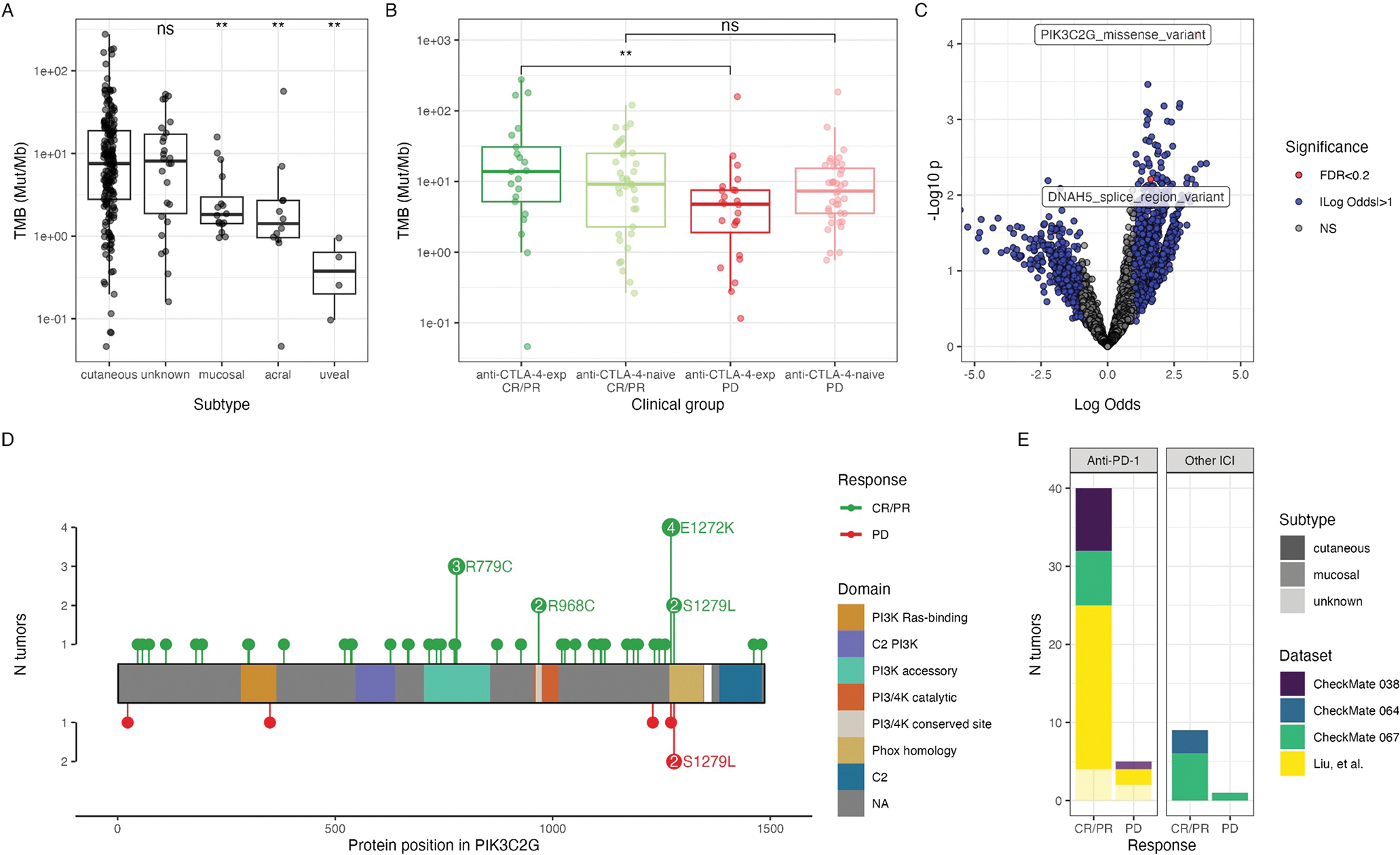
Comparison of TMB (Mut/Mb), quantified by the number of nonsilent mutations and normalized by tumor purity and sequencing coverage, across (A) subtypes or (B) clinical groups, defined by anti-PD-1 response and prior anti-CTLA-4 treatment. Groups are compared using a Wilcoxon test (**, p<0.01; ns, p>0.05). In (A), each subtype is compared to the cutaneous group. (C) Logistic regression comparing genes and mutation types between anti-PD-1 responsive (CR/PR) and nonresponsive (PD) tumors. Significance and log-odds ratio (x-axis) are indicated. (D) Lolliplot of PIK3C2G mutations in all baseline tumors, stratified by CR/PR (top) and PD (bottom). (E) Corresponding cohorts of PIK3C2G-mutant tumors. See Figure S2.
We were specifically interested in the association between TMB and response to anti-PD-1 therapy in patients with cutaneous melanoma, leveraging the statistical power of our cohort. When TMB was compared across biopsies of cutaneous melanoma origin treated with anti-PD-1 that responded (RECIST CR/PR) or progressed (RECIST PD), TMB was significantly greater in biopsies of patients with anti-PD-1 clinical response compared to no response (Wilcoxon test, p=0.024). This association only remained significant, however, in tumors from patients that were previously treated with anti-CTLA-4 therapy (Wilcoxon test, p=0.0062, FDR=0.037) and not in anti-CTLA-4-naive tumors (Wilcoxon test, p=0.515, FDR=0.51) (Figure 2B), suggesting an interaction effect between these two variables. This was consistent with what was previously reported,8 while other subsets were not of sufficient size to identify these differences (Figure S2).
Single nucleotide variants in PIK3C2G are positively associated with clinical benefit to checkpoint inhibitors
While studies have attempted to discover associations between response to immune checkpoint blockade and single gene mutational statuses, success has been partially limited by the larger patient numbers required for statistical power following multiple hypothesis correction.27 Tumor sampling and purity, prior treatment, and melanoma subtype, which are also associated with prognosis following ICI and the genomic landscape,8 add complexity to this problem. We explored whether our aggregation and harmonization may overcome these challenges by applying logistic regression to genes and mutation types in cutaneous melanoma tumors treated with anti-PD-1, comparing results from biopsies of patients with response (CR/PR) to no response (PD). Gene-mutation combinations were filtered to those which occurred at least 10 times (out of 151 [6.6%] tumors evaluated), resulting in 7,579 unique gene-mutation type pairs evaluated. There were two nonsilent alterations that met the statistical thresholds, following multiple testing correction (p<0.05, FDR<0.2), enrichment of PIK3C2G missense mutations (p=7.5e-5, FDR=0.15) and DNAH5 splice region variants (p=6.2e-3, FDR=0.18) in cutaneous tumors from patients with response to anti-PD-1 (Figure 2C). When we repeated this analysis across all melanoma subtypes and ICI regimens, PIK3C2G missense mutations remained associated with anti-PD-1 response across all melanoma subtypes (p=1.3e-5, FDR=0.033) as well as response to any ICI regimen across all subtypes (p=3.2e-6, FDR=0.011).
The most common alterations in PIK3C2G included recurrent missense mutations R779C and E1272K (Figure 2D). Of the 49 tumors with PIK3C2G mutations, 44 had matched RNAseq data; however, when mutations were queried in RNAseq, both the gene and the mutation were infrequently expressed. The expression of PIK3C2G was low across all available melanoma tumors with RNAseq data (0–14.68 FPKM; median 0). Notably, the link between PIK3C2G mutations and anti-PD-1 response was previously demonstrated but was not specifically reported since it was not significant following multiple hypothesis correction.8 The inclusion of additional harmonized cohorts (CheckMate 038, 064, and 067) (Figure 2E) provided additional key support of this finding.
Prior CTLA-4 blockade differentially stratifies the expression landscape of melanoma tumors with and without response to anti-PD-1
Previous studies have described remodeling of the tumor microenvironment by anti-CTLA-4 blockade.28,29 Due to the differences observed in TMB with respect to prior anti-CTLA-4 therapy and anti-PD-1 response, we performed supervised differential expression analysis between baseline cutaneous melanoma biopsies from anti-CTLA-4-experienced patients (N=39; N=17 CR/PR, N=22 PD) and anti-CTLA-4-naive patients (N=92; N=44 CR/PR, N=48 PD) treated with anti-PD-1 monotherapy (Figure 3A). When patients were stratified by their response to anti-PD-1, we found more significant differences across biopsies of patients with response to anti-PD-1 (CR/PR; N=1,025 significant genes), than across biopsies of patients without response to anti-PD-1 (PD; N=113 significant genes), when comparing anti-CTLA-4 experience (Figure 3B–C).
Figure 3. Prior CTLA-4 blockade differentially stratifies the expression landscape of melanoma tumors responding or not to anti-PD-1.
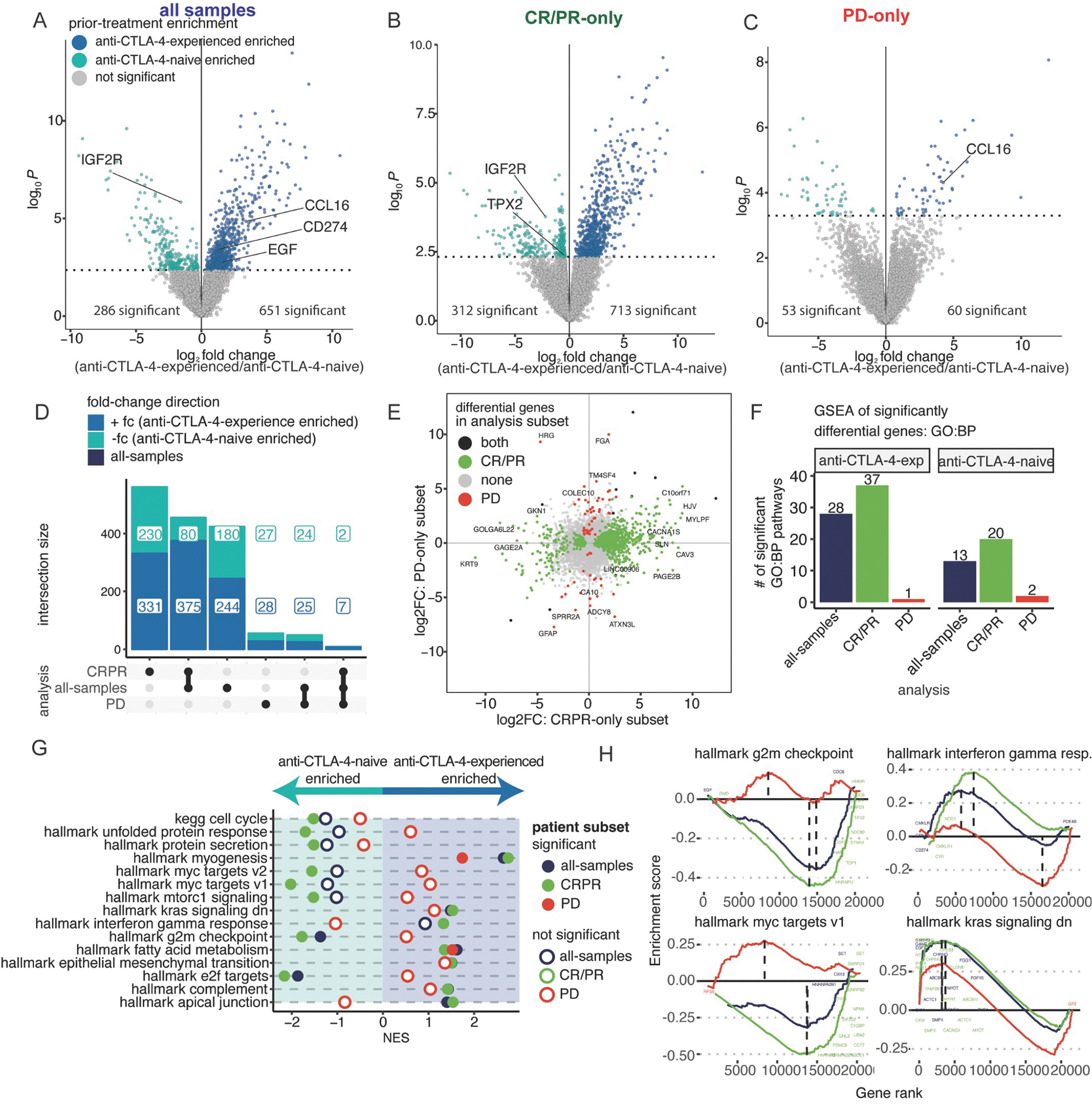
(A-C) Differential expression analysis comparing baseline anti-CTLA-4-experienced to anti-CTLA-4-naive, cutaneous melanoma tumors (N=131): (A) all-samples, (anti-CTLA-4-naive=92, anti-CTLA-4-experienced=39), (B) CR/PR-only (anti-CTLA-4-naive=48, anti-CTLA-4experienced=17), and (C) PD-only (anti-CTLA-4-naive=44, anti-CTLA-4-experienced=22). (D) UpSet plot of differential genes comparing anti-CTLA-4-experienced to anti-CTLA-4-naive patients. (E) Scatter plot of each gene comparing the log fold-change difference between anti-CTLA-4-experienced and anti-CTLA-4-naive patients. Color indicates significance of a gene in both, neither, CR/PR-only, or PD-only subsets. (F) Barplots showing the number of statistically significant pathways using differential genes. (G) Normalized Enrichment Score (NES) results from ranked-based GSEA of anti-CTLA-4-experienced versus anti-CTLA-4-naive tumors in each patient subset. Filled circles indicate statistical significance, hollow circles indicate no statistical significance. (H) GSEA rank plots showing differential or shared enrichment directions between anti-CTLA-4-experienced vs anti-CTLA-4-naive patients, colored as in (G). See Figure S3 and Table S1–2.
Genes that were differentially expressed between anti-CTLA-4-experienced and anti-CTLA-4-naive tumors were mostly unique to either response or no response to anti-PD-1 therapy, with 63.5% and 48.7% of genes specific to either subset, respectively (Figure 3D). The lack of overlap in these results suggested that there were biological differences between melanomas responsive and nonresponsive to PD-1 blockade, based upon their prior treatment with anti-CTLA-4 (Figure 3E). Only 11 genes were shared by both analyses; nine out of 11 genes had shared direction of effect.
Given the divergence in gene expression patterns between anti-CTLA-4-experienced and anti-CTLA-4-naive tumors within each anti-PD-1 response group, we utilized two methods for gene set enrichment analysis (GSEA) to more broadly characterize molecular processes across these comparisons. First, GSEA was performed within each response group using only significantly different genes (FDR<0.10). Consistent with our results at the gene level, there were stronger differences at the gene set level when comparing anti-CTLA-4 experience in anti-PD-1-responsive tumors using GO:BP gene sets (Figure 3F). In anti-PD-1-responsive tumors, those with anti-CTLA-4 experience were significantly enriched for processes related to MAPK signaling, hypoxia response, and inflammatory signaling pathways (e.g. Ig production, interferon signaling, adaptive immune response), while anti-CTLA-4-naive tumors were enriched for cell-cycle pathways. This same analysis revealed very few significantly enriched pathways in biopsies of patients with no response to anti-PD-1, with two exceptions: the adaptive immune response gene set was enriched in anti-CTLA-4-experienced tumors, while the innate immune system gene set was enriched in anti-CTLA-4-naive tumors.
In our second GSEA method, we performed a rank-based GSEA in Hallmark gene pathways30 across all genes from the prior differential gene expression analysis. Within the anti-PD-1-responsive subset, anti-CTLA-4-experienced tumors were enriched for gene sets associated with interferon gamma response and genes downregulated by KRAS activation, while anti-CTLA-4-naive tumors were enriched for cell cycle, E2F, G2M checkpoint, and mTORC1 signaling pathways (Figure 3G–H). Nearly all of the significant gene sets in anti-PD-1-responsive tumors showed opposing, but not significant, trends in anti-PD-1-nonresponsive tumors (e.g. gene sets enriched in anti-CTLA-4-naive tumors in the anti-PD-1-responsive subset were enriched in anti-CTLA-4-experienced tumors in the anti-PD-1-nonresponsive subset) (Figure 3G–H). The only two shared significant processes across both anti-PD-1 responding and nonresponding tumors were the enrichment of fatty acid metabolism and myogenesis in anti-CTLA-4-experienced tumors.
Collectively, these two approaches revealed interactions between anti-CTLA-4 experience and anti-PD-1 response, particularly in responsive tumors. We highlight that the majority of pathways from GSEA would not have achieved statistical significance without the stratification of anti-PD-1 baseline tumors by clinical response, supporting the continued stratification of patients by both anti-PD-1 response and prior anti-CTLA-4 therapy for the subsequent analysis.
Strength of inflammatory gene expression patterns in anti-PD-1 responding biopsies is associated with prior anti-CTLA-4 experience
To more specifically explore gene expression patterns associated with anti-PD-1 response, we stratified cutaneous melanoma tumors with RNAseq data (N=131) into four subsets, based upon anti-PD-1 response (CR/PR, PD) and prior anti-CTLA-4 experience (experienced, naive). Unsupervised principal component analysis of the top 15% most variable genes did not reveal separation across clinical demographics (Figure 4A, Figure S3A–B). We utilized four models for supervised differential expression analysis to compare biopsies of anti-PD-1 response with no response: comparison of all samples, comparison of all samples controlling for anti-CTLA-4 experience, comparison of response within anti-CTLA-4-experienced tumors, and comparison of response within anti-CTLA-4-naive tumors (Figure 4B–D). By comparing the results of these approaches, we corroborated our previous results indicating interactions between anti-CTLA-4 experience and anti-PD-1 response (Figure 4E). This was supported by analysis in each patient subset, where there were no significantly different genes comparing anti-PD-1 response in anti-CTLA-4-experienced tumors.
Figure 4. Strength of inflammatory gene expression patterns in anti-PD-1 responding biopsies is associated with anti-CTLA-4 experience.
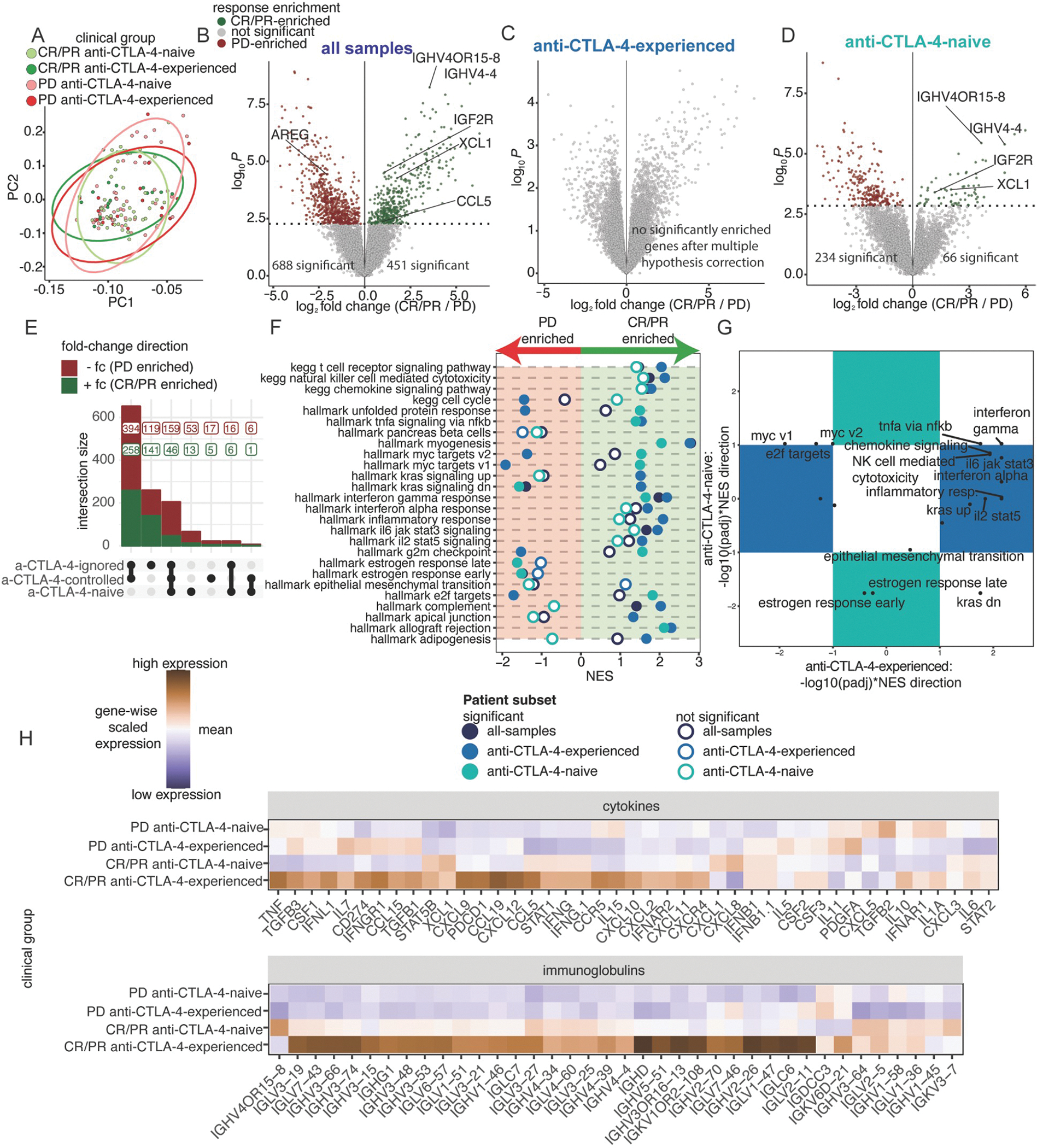
(A) PC1 and PC2 from PCA of the top 15% most variable genes across 131 baseline, cutaneous melanoma samples, following batch effect correction by cohort. Colors indicate four clinical groups stratified across response and prior treatment. Ellipses indicate the distribution of clinical groups. (B-D) Differential expression analysis comparing tumors between CR/PR and PD patients across patient subsets: (B) all-samples (CR/PR=65, PD=66), (C) anti-CTLA-4-experienced-only (CR/PR=17, PD=22), and (D) anti-CTLA-4-naive-only (CR/PR=48, PD=44). (E) UpSet plot of differential genes comparing CR/PR to PD patients across each patient subset. (F) NES results from ranked-based GSEA of CR/PR versus PD patients in each patient subset. Hollow circles indicate no statistical significance. (G) Scatterplot of adjusted p-values with NES direction for GSEA results comparing CR/PR versus PD patients in the anti-CTLA-4-experienced and anti-CTLA-4-naive subsets. Pathways in the colored boxes indicate statistical significance in only the anti-CTLA-4-experienced or anti-CTLA-4-naive subset. (H) Heatmaps showing scaled mean expression of cytokine (top) and immunoglobulin-related genes (bottom). See Figure S3 and Tables S3–4.
We explored whether differences were driven by one or a subset of clinical cohorts, and we found that the differential expression analysis within subgroups was poorly powered (Figure S3C). When the ranks of significant genes were compared between the analysis of the aggregate dataset with each individual cohort, positive correlations were significant but weak (Pearson correlations: Checkmate 038, 0.32; Checkmate 064–067, 0.085; Gide, 0.49; Liu, 0.33) for all cohorts (Figure S3D). Notably, the Liu cohort did not have the strongest rank-based correlation despite having the largest sample contribution. This sensitivity analysis underscores the importance of statistical power provided by our data harmonization and aggregation efforts to understand associations between molecular and clinical features.
We further explored these gene patterns by performing GSEA across all genes of curated Hallmark and KEGG pathways, revealing striking differences between anti-CTLA-4-experienced and anti-CTLA-4-naive tumors when comparing biopsies of patients with and without response to anti-PD-1 (Figure 4F–G). Within anti-CTLA-4-experienced tumors, anti-PD-1 responding tumors were enriched for genes regulated by KRAS activation, while anti-PD-1 non-responding tumors were enriched for cell cycle and MYC target genes. However, these gene sets displayed the reverse patterns in anti-CTLA-4-naive tumors (Figure 4G). Gene sets related to inflammation, cytotoxicity, and immune cell activation were positively enriched in anti-PD-1 responding tumors, but enrichment was significant only in anti-CTLA-4-experienced patients. The expression of cytokines and immunoglobulin regions was also variable, with the highest expression in antiCTLA-4-experienced tumors of patients who responded to anti-PD-1, followed by anti-CTLA-4-naive without a response, with the lowest expression in biopsies of patients with no response to anti-PD-1 (Figure 4H). Consistent with prior studies, these results suggest that biopsies of patients with response to anti-PD-1 have increased immune infiltration and activity compared to non-responding biopsies, but these features are more strongly enriched in biopsies of patients who are anti-CTLA-4 experienced and responded to anti-PD-1.
Anti-CTLA-4 experience is associated with globally increased adaptive immune infiltration signatures
While these analyses revealed differences in expression or enrichment for inflammatory processes associated with anti-CTLA-4 experience and anti-PD-1 response, we were interested in which immune cell-types may be contributing to these signals. Previous studies have described the association between tertiary lymphoid structures (TLS) and the infiltration of antitumor CD8 T-cells and anti-PD-1 response,9 as well as the mechanism by which anti-CTLA-4 mediates T-cell trafficking from the lymph node to the tumor site.28,29 To interrogate immune cell-types in the tumor microenvironment, we computed a single-sample immune signature score using gene signatures from previously published and curated gene sets to estimate immune cell-type enrichment and activity from bulk tumor RNAseq data (see Methods). Concordant with the gene expression analysis, anti-CTLA-4-experienced, anti-PD-1-responsive tumors had the greatest average enrichment across all immune cell signatures, with the strongest enrichment for lymphocyte, cDC1, and TLS gene signatures (Figure 5A). Cell signature estimation using EPIC demonstrated similar trends with anti-CTLA-4 experience. The EPIC Endothelial signature also suggests increased blood vessels in the CR/PR anti-CTLA-4 experienced group compared to PD groups (Figure S4A–B).
Figure 5. Anti-CTLA-4 experience is associated with globally increased adaptive immune infiltration signatures.
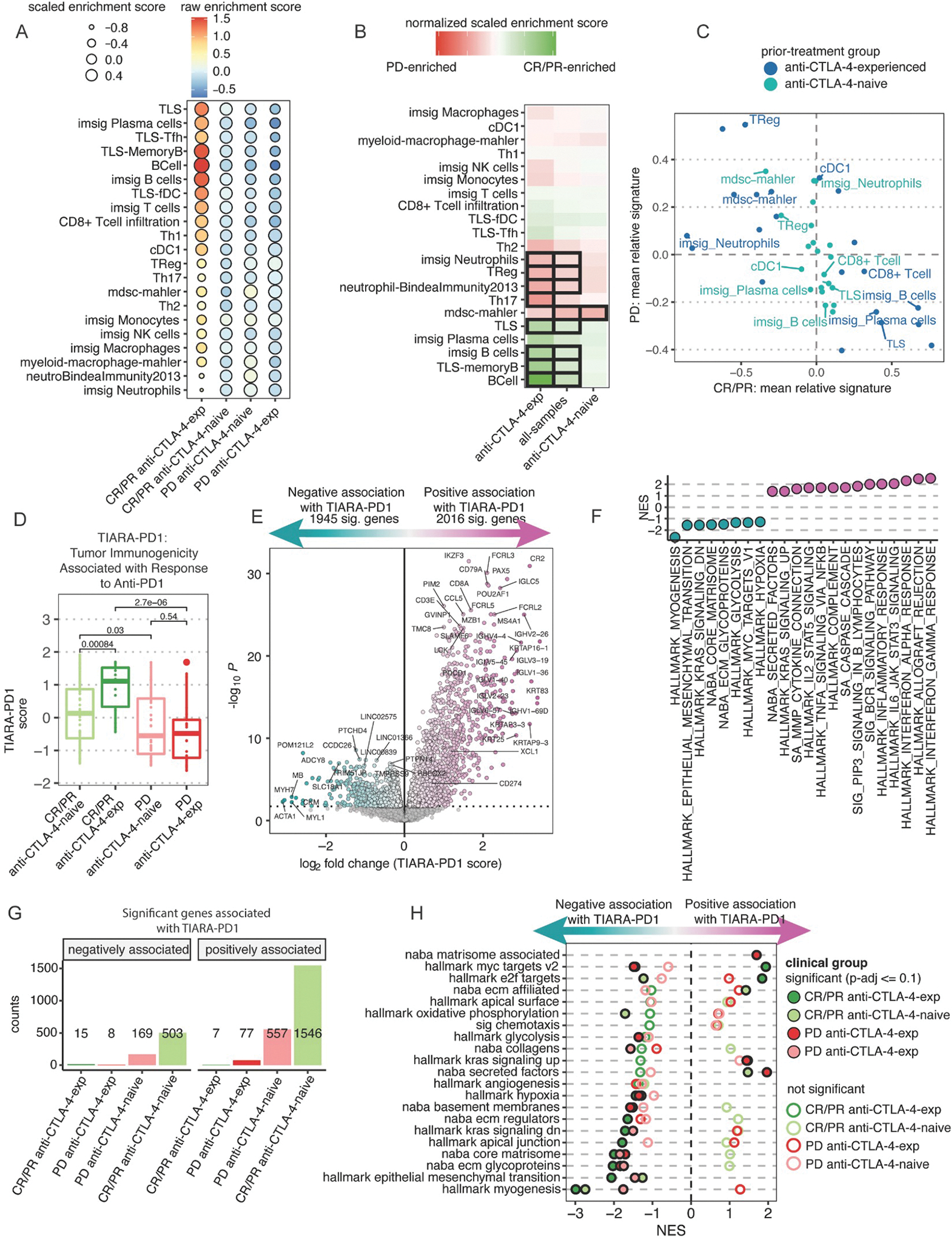
(A) Heatmap of immune cell-type scores computed using published gene signatures. Colors represent the averaged, raw score from patients in each clinical group. Size of each point represents the averaged, relative score within each clinical group (see STAR Methods). (B) Heatmap of the mean score difference of each cell-type between CR/PR and PD tumors from 3 analyses either observing all samples together or stratified by prior treatment. Black tile outlines indicate statistical significance by Wilcoxon signed-rank test with BH-adjusted p-values <=0.1. (C) Scatter plot comparing the averaged cell-type signature score of CR/PR and PD patients, colored by prior treatment. (D) Boxplots of TIARA-PD-1 score compared between response and prior treatment clinical groups (see STAR Methods). Statistical significance determined by Wilcoxon signed-rank test. (E) Differential expression analysis as a function of TIARA-PD-1. The x-axis is the expression difference associated with a unit change in TIARA-PD-1. (F) NES of ranked-based GSEA for statistically significant pathways curated from HALLMARK and NABA (Table S5). (G) Barplots of the number of genes significantly different as a function of TIARA-PD-1 within patient subsets. (H) Rank-based GSEA results for significant pathways curated from HALLMARK and NABA across each clinical group. Filled circles indicate statistical significance, hollow circles indicate no statistical significance. See Figure S4–6 and Tables S4–7.
To determine which immune cell-types may be cooperative within the tumor microenvironment, we normalized the raw cell-type signatures for each immune cell-type within each sample and summarized the average score for each cell-type within each clinically defined subset. This revealed that anti-CTLA-4-experienced, anti-PD-1 responding biopsies had the strongest enrichment of CD8 T cell, B cell, Th1, TLS, and cDC1 signatures, along with the poorest enrichment of neutrophils, Tregs, Th2, Th17, MDSCs, and NK cells (Figure 5A–B). Conversely, anti-PD-1 non-responding biopsies had greater enrichment of Treg, neutrophil, and MDSC signatures. While anti-CTLA-4-experienced tumors appear to have stronger overall enrichment of immune cell signatures, the anti-PD-1-non-responding biopsies also showed reduced lymphocyte infiltration and TLS signatures, along with increased NK cell signatures. When we compared the average enrichment score of each cell-type between anti-PD-1 responding and non-responding biopsies, there was an overall inverse association in cell-type signatures, across all cell-types (Figure 5C). Importantly, this difference across anti-PD-1 response groups was more substantial in anti-CTLA-4-experienced tumors.
Tumor infiltration is differentially associated with MYC and E2F targets in biopsies that responded to anti-PD-1 after anti-CTLA-4 treatment
Gene expression patterns associated with immune infiltration, inflammatory signaling, and antitumor immune cytotoxicity have been shown to positively predict response to anti-PD-1 from baseline tumor specimens. Several methods have been established to aggregate expression metrics across gene sets from bulk RNA data, leading to successful stratification of anti-PD-1 or ICI responders.10,13–15 However, we aimed to understand how tumor-intrinsic patterns may coordinate with immune cell-types responsible for anti-PD-1 response. To better stratify tumors based upon the immune infiltrate, we generated an interpretable, aggregate gene expression score, using immune cell-type signatures that were increased or decreased in biopsies of patients with response to anti-PD-1, to summarize the Tumor Infiltration Associated with Response to Anti-PD-1 in melanoma (TIARA-PD-1, see Methods).
Given that the features defining TIARA-PD-1 were computed based upon anti-PD-1 response in our aggregate cohort of baseline, cutaneous melanoma tumors, it was unsurprising that TIARA-PD-1 score was significantly increased among biopsies of patients with CR/PR to anti-PD-1 compared to PD (Figure 5D). Consistent with our previous analysis, this difference was greater in magnitude when including prior anti-CTLA-4 treatment as a covariate in biopsies of patients with anti-PD-1 response, but not among nonresponsive tumors. We also computed TIDE, estimating T cell dysfunction and exclusion, and IMPRES, a predictor of ICI response in melanoma that includes 15 pairwise transcriptomics relations between immune checkpoint genes.13,15 Both TIDE and IMPRES support interactions with prior anti-CTLA-4 experience and response to anti-PD1 (Figure S4C). However, while TIARA-PD1 variance is associated with CR/PR in both the naive and anti-CTLA-4 experienced groups, TIDE was only able to stratify anti-PD-1 response in the anti-CTLA-4-experienced setting (Figure S4D). Importantly, computation of TIDE scores required manual supervision of whether the patients had experienced prior immunotherapy or not to identify these interactions (see Methods).
We sought to validate whether TIARA-PD-1 was associated with clinical response in melanoma tumors in other ICI contexts by quantifying immune cell-type signatures and TIARA-PD-1 across both baseline and on-treatment RNAseq samples from tumors treated with other ICI regimens associated with this study (Figure S5A–B). Consistent with our anti-PD-1 results in baseline, cutaneous melanoma, clinical response (CR/PR) to ICI in other treatment regimens tended to have larger TIARA-PD-1 scores (Figure S5C). Additionally, median TIARA-PD-1 was higher in on-treatment samples, compared to baseline, in both the anti-PD-1 and sequential anti-CTLA-4-to-anti-PD-1 treatment regimens, suggesting increased global immune infiltration on-therapy with ICI. We also performed an independent validation of TIARA-PD-1 in a small clinical trial in which patients with advanced melanoma were treated with either anti-CTLA-4 monotherapy or anti-CTLA-4 plus anti-PD-1 combination therapy.31 Tumors with either PR or stable disease (SD) in this cohort did show a trend of increased TIARA-PD-1 over the course of therapy, comparing lesion-paired on-treatment samples (N=10) with baseline samples (N=10) (Figure S5D–E). When we evaluated TIARA-PD-1 in melanoma subtypes other than cutaneous melanoma, TIARA-PD-1 was associated with anti-PD-1 response in mucosal melanoma, but not in tumors with acral, uveal, or unknown site of origin (Figure S5F). Notably, these comparisons were less powered than the analysis in cutaneous melanomas.
To better understand the associations between the tumor microenvironment and potential tumor-intrinsic processes, we performed differential expression analysis in baseline, cutaneous melanoma tumors treated with anti-PD-1 based upon TIARA-PD-1 score as an outcome variable. Gene expression analysis identified 3,961 significantly different genes (FDR<=0.1, Figure 5E). Genes contributing to the immune signatures used to compute TIARA-PD-1 were excluded from this analysis. Unsurprisingly, increased TIARA-PD-1 was positively associated with LCK, immunoglobulins, XCL1, and other cytokines (Figure 5E), and GSEA on ranked genes revealed enrichment for Hallmark gene sets associated with T-cell function, interferon signaling, and TNFA signaling via NFKB across all clinical subsets (Figure 5F, Table S5). Conversely, TIARA-PD-1 was negatively associated with myosin and actin family genes, with enrichment of genes associated with myogenesis, epithelial to mesenchymal transition, glycolysis, MYC targets, and hypoxia (Figure 5E–F).
TIARA-PD-1 was informed by the cell-types enriched in anti-PD-1 responding biopsies, and our prior analysis showed that this signal was greater in anti-CTLA-4-experienced tumors. Differential gene expression analysis of each clinical subset showed the greatest number of significant genes associated with TIARA-PD-1 among anti-CTLA-4-naive responders, followed by anti-PD-1 nonresponders, anti-CTLA-4-experienced non-responders, then anti-CTLA-4-experienced responders (Figure 5G). The low number of significant genes associated with TIARA-PD1 in the anti-CTLA-4 experienced subsets may be a result of the smaller sample size, as seen in our previous transcriptomic analysis. When GSEA was performed within each clinical subset, many gene sets were consistently enriched in the same direction, particularly those that were negatively associated with TIARA-PD-1 (Figure 5H, Table S6). Gene sets that had opposing directions in enrichment scores were only significant in one or two clinical subsets. In particular, MYC and E2F targets showed diverging associations with TIARA-PD-1 in anti-PD-1-responsive tumors, with positive enrichment in anti-CTLA-4-experienced tumors and negative enrichment in anti-CTLA-4-naive tumors. MYC targets were also negatively enriched in anti-CTLA-4-experienced, anti-PD-1 non-responding tumors (Figure 5H). Collectively, these results suggest differential roles in these processes in either coordinating with or facilitating antitumor immune activity responsible for anti-PD-1 response.
Mutation subtype may be associated with immune cell infiltration and anti-PD1 response in an anti-CTLA-4-experienced dependent manner
We sought to further understand whether the patterns with respect to prior anti-CTLA-4 and anti-PD1 response observed in baseline cutaneous melanoma samples were associated with known canonical genetic drivers of melanoma (BRAF hotspot, NRAS hotspot, NF1-mutant, or triple-wildtype [TWT]). Driver mutations detected in WES data were annotated based upon the detection of hotspot mutations in BRAF (V600) or NRAS (G12, G13, Q61), mutations in NF1, or the lack of these mutations (TWT). Clinical benefit to anti-PD1 or prior anti-CTLA4 experience was not associated with any genetic subtype (two-sided Fisher’s Exact Test, p=0.53). We performed differential expression analysis comparing anti-PD-1 response (CR/PR versus PD) for anti-CTLA-4-naive and anti-CTLA-4-experienced tumors within each genetic subtype. There were no significant genes identified in anti-CTLA-4-naive tumors. In the anti-CTLA-4-experienced group, IGHM was the only significant gene positively associated with CR/PR in tumors with BRAF hotspot mutations (log2-fold change=3.56, padj=0.099, Table S7). Notably, sample sizes were small after stratification by anti-PD-1 response, anti-CTLA-4 experience, and mutation subtype.
Regardless of genetic subtype, anti-CTLA-4-experienced patients that responded to anti-PD1 tended to have greater global immune infiltration than those without benefit (Figure S6A). There was no significant association between immune cell signatures across mutant subtypes or within clinical groups, stratified by mutant subtypes. TIARA-PD1 analysis of immune signatures was consistent across genetic subtypes, with highest TIARA-PD1 in anti-CTLA-4-experienced, anti-PD-1 responders (Figure S6B–C), with one exception. Anti-CTLA-4-naive and anti-CTLA-4-experienced tumors with BRAF V600 mutations that responded to anti-PD-1 had equally high TIARA-PD1 scores. The inverse relationship between TIDE and TIARA-PD1 was strongest in BRAF V600 and TWT tumors, and IMPRES did not stratify differences across clinical groups within any of the genetic subtypes (Figure S6C). Together, the trends supported by TIDE and TIARA-PD-1 suggest that there may be differences in T-cell infiltration and dysfunction across genetic subtypes, which could further implicate the concurrent or sequential targeting of these alterations by MAPK inhibitors in modulating the tumor microenvironment and response to anti-PD-1 therapy.32 It is important to note that these patients were not annotated for their prior treatment with MAPK inhibitors.
Statistical models to predict response to anti-PD-1 in cutaneous melanoma are improved by including prior anti-CTLA-4 experience
Differential gene expression analyses yielded consistent patterns across clinical subsets defined by prior anti-CTLA-4 experience and anti-PD-1 response; however, we aimed to identify which features of the defined genomic, transcriptomic, or clinical features would best predict anti-PD-1 response in cutaneous melanoma. Utilizing a lasso-regularized logistic regression (LRLR) for variable selection,33–35 we compared anti-PD-1 responsive and nonresponsive tumors with both WES and RNAseq data from baseline tumors (N=90). Features included statistically significant results across both data types from the previous analyses, as well as published features (Table S3, Figure S7A). Features selected by the model that positively predicted antiPD-1 response included PIK3C2G mutations, B cell signatures, TIARA-PD-1, while features selected to predict anti-PD-1 nonresponse included expression of AREG or MAPK10 (Figure 6A, Figure S7B). Some features were only selected based upon the status of prior anti-CTLA-4 therapy, further demonstrating the interaction between prior anti-CTLA-4 therapy with gene expression features in predicting anti-PD-1 response, such as the Hallmark genes downregulated by KRAS activation as a predictor of anti-PD-1 nonresponse in anti-CTLA-4-naive tumors.
Figure 6. Statistical models are improved by including prior anti-CTLA-4 experience to predict response to anti-PD-1 in cutaneous melanoma.
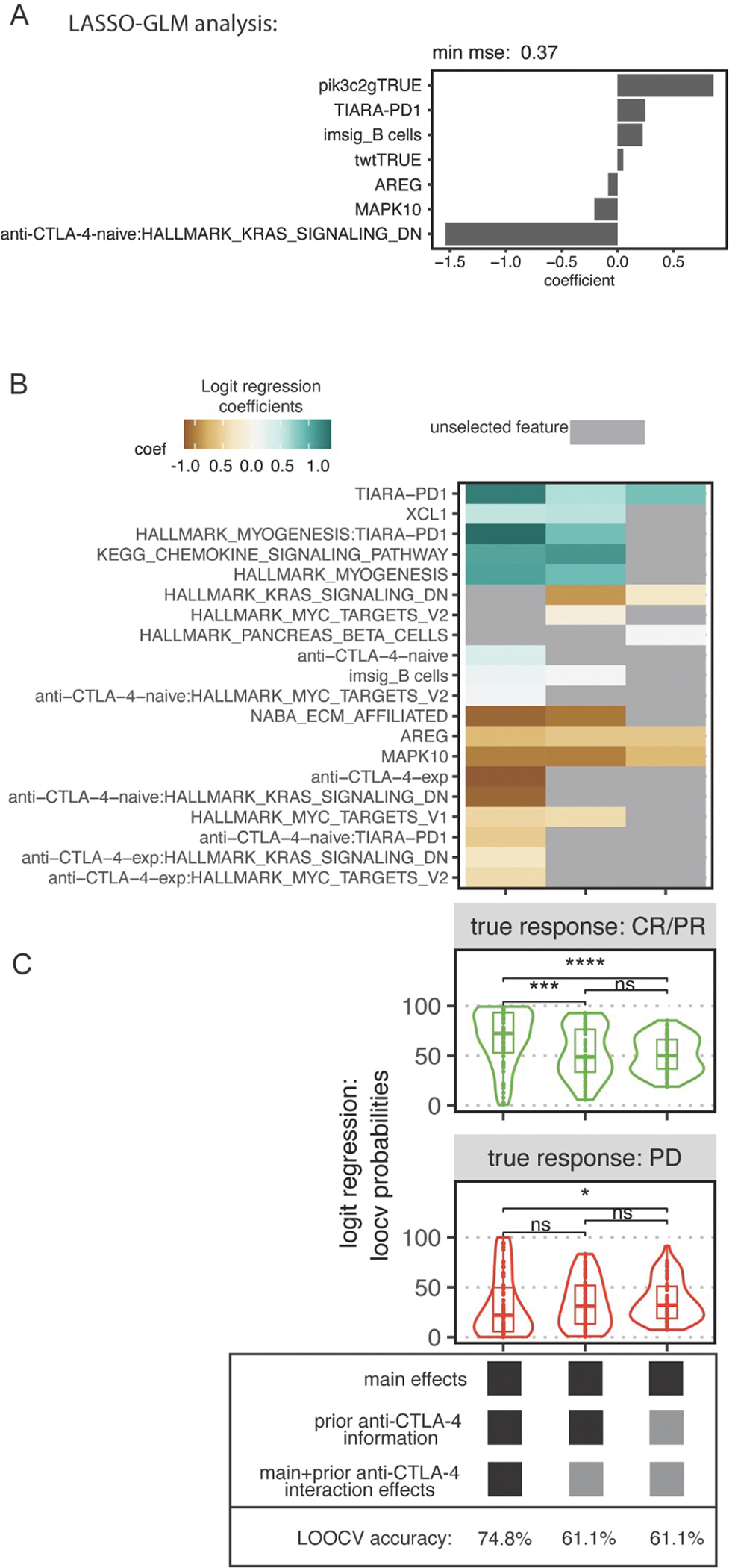
(A) Feature selection using cross-validated lasso-regularized logistic regression for 90 paired WES and RNAseq baseline cutaneous melanoma tumors treated with anti-PD-1, with or without prior anti-CTLA-4, comparing CR/PR to PD. (B) Heatmap of selected features for 3 fitted models. Color indicates magnitude and direction of the standardized coefficient for that feature. Gray indicates that the feature was not selected. (C) Predicted probabilities from leave-one-out cross-validation (LOOCV) logistic regression comparing CR/PR vs PD using all selected features from (B) in each respective model. The y-axis indicates probability of response. The top and bottom facets indicate the clinically annotated true response to anti-PD-1. LOOCV accuracy is at the bottom. See Figure S7.
Due to the lack of genomic correlates in the integrated analysis and to increase statistical power, we attempted three additional approaches for all anti-PD-1-treated cutaneous melanoma tumors with RNAseq data available (N=131), regardless of matched WES. We performed LRLR with two distinct models: Model A was a complete model that integrates statistically significant factors related to response to anti-PD-1 regardless of association with anti-CTLA-4 experience, and Model B only considered statistically significant factors without interaction with anti-CTLA-4 experience (Figure 6B–C, Figure S7C). Model A was subsequently divided into two approaches, utilizing either LRLR-selected features in a complete model containing interaction effects with anti-CTLA-4 experience (A1) or excluding those interaction effects (A2).
Features selected by these models, consisting of only transcriptomic features, were consistent with the first approach, which integrated both genomic and transcriptomic features (Figure 6A). However, there were additional features selected, including genes related to the extracellular matrix across all samples and the interaction effects in association with genes regulated by KRAS activation or MYC, myogenesis, and TIARA-PD-1 (Figure 6B–C). Exclusion of prior anti-CTLA-4 interactions in Models A2 and B selected similar features as Model A1, but inclusion of the interactions improved the LOOCV predicted probabilities and accuracy of the complete model (Figure 6B–C). Model A1 showed significant improvement in predicting anti-PD-1 response over Models A2 (Wilcoxon rank sum test, p=8.1e-05) and B (Wilcoxon rank sum test, p=4.8e-07). This was shown by predicting the greatest probability of response in anti-PD-1 responding biopsies, and the lowest probability of response in anti-PD-1 non-responding biopsies (Figure 6C). Furthermore, Model A1 had the highest overall accuracy in predicting anti-PD-1 response (74.8%), followed by A2 (65.6%) and B (62.6%) (Figure 6C). A random forest classifier selected features similar to our LRLR results, with an accuracy of 66.7% (Figure S7D–E). Collectively, this analysis suggests that annotation of anti-CTLA-4 experience is highly impactful in the context of studying anti-PD-1 response in cutaneous melanoma, and its inclusion may significantly refine the discovery of predictors or mechanisms responsible for anti-PD-1 response.
Discussion
Correlates of ICI response have been studied using genomic, transcriptomic, and pathologic approaches in a global attempt to stratify patient populations, through the analysis of individual and aggregate clinical cohorts.7,12 Gaining access to such datasets is a challenge in itself, and pooling larger cohorts requires additional methodology and expertise, both scientifically and clinically. Here, we harmonized the molecular and clinical annotation of seven clinical cohorts of patients with melanoma treated with ICIs, including two previously unpublished datasets. The integration of these studies allowed us to leverage the increased statistical power of an aggregate resource to more rigorously address the evaluation of anti-PD-1 response in cutaneous melanoma. We found that both genomic and transcriptomic features predictive of anti-PD-1 response were modified by prior anti-CTLA-4 administration, highlighting key biological differences across baseline melanoma tumors studied across retrospective immunotherapy trials.
One of the primary benefits of harmonizing molecular datasets is the increased statistical power for more rigorous identification of biological features of patient subgroups. Other studies have attempted to increase statistical power by combining various clinical datasets, including those spanning different tumor types or treatment modalities. Alternatively, we focused on the utility of deeper clinical annotation, particularly with regards to melanoma subtype, treatment regimen, and treatment history. While the Liu et al. cohort previously reported some correlates of anti-PD-1 response, controlling for melanoma subtype and anti-CTLA-4 experience, some were not reported as significant due to multiple testing correction. However, the rigorous harmonization of cohorts, as well as the addition of the Checkmate 064 and 067 datasets, made it possible to detect signals associated with anti-PD-1 response, such as PIK3C2G missense mutations. This study highlights correlative findings that may support or drive experimental studies moving forward, so that the mechanistic understanding of these correlations can be properly implemented clinically. Importantly, we found that the refined stratification of cutaneous melanoma tumors by both anti-CTLA-4 experience and anti-PD-1 response led to distinct sets of findings regarding TMB, inflammatory gene expression patterns, and potential tumor-intrinsic gene expression patterns. The clinical datasets interrogated in this study comprised samples spanning melanoma subtypes; however, our assessment of TMB, confirming known differences in histologically defined subtypes, demonstrated the need for histopathologic annotation of samples for more rigorous analysis of molecular and clinical correlates. Collectively, this emphasizes why these efforts in data curation and harmonization are critical to clarify our understanding of predictive biomarkers for ICI treatment strategies, and may also explain the lack of clarity, inconsistency, or lack of statistical signal in previous reports across these settings.
It is important to consider the relevance of our findings from these retrospective melanoma datasets in the context of the current treatment paradigm. Given the establishment of anti-PD-1 alone or in combination with anti-CTLA-4, as well as the recent approval of combined anti-PD-1/anti-LAG-3 therapy in the first-line setting for advanced melanoma, patients do not receive single agent anti-CTLA-4 as frontline therapy. Our analysis shows far fewer differences between anti-PD-1 responsive and nonresponsive tumors in the anti-CTLA-4-naive setting, which may highlight the need for refinement of predictive biomarkers to anti-PD-1, specifically in this setting. The known mechanism of PD-1 blockade involves the recognition of tumor cells by CD8 T cells, and the detection of infiltrating T cells in baseline samples has been a strong predictor of clinical benefit following anti-PD-1 therapy.11 Anti-CTLA-4 stratified analysis of bulk RNAseq tumor infiltration signatures revealed that prior CTLA-4 blockade was associated with significantly higher global immune infiltration, with enrichment for CD8+ T cell, B cell, TLS, and cDC1 signatures, which have all been previously implicated in anti-PD-1 responsiveness 9,11. These results agree with pathology analysis data supporting that CTLA-4 blockade therapy increases intratumor T cell infiltrates regardless of clinical response to therapy.29,36 There was also a lower enrichment of cell-types commonly associated with immune suppression, such as neutrophil and Treg cell signatures, among patients with clinical benefit to anti-PD1. Furthermore, there were much lower signals associated with immune cell populations in anti-PD-1 nonresponsive tumors, regardless of anti-CTLA-4 experience.
We established TIARA-PD-1 as a summary metric of immune signatures differentially associated with response to anti-PD1 in cutaneous melanoma, and found diverse transcriptional signatures as a function of immune infiltration, based upon anti-PD-1 response and prior anti-CTLA-4 experience. Specifically, increase in TIARA-PD1 was strongly associated with a decrease in extracellular matrix, matrisome-associated proteins, glycoproteins, and myogenesis as well as glycolysis and hypoxia signatures. These signatures were consistent and more striking in anti-CTLA-4 experienced patients, while non-responders to anti-PD1 did not demonstrate a significant negative enrichment of many ECM and hypoxia signatures despite an increase in TIARA-PD1. This is consistent with reports recognizing the ECM as a crucial microenvironmental component affecting the immune response in tumors, as well as the role of a hypoxic TME in inducing immune suppression and resistance.37–40 In addition, TIARA-PD-1 was more similar across anti-CTLA-4-experienced and -naive tumors specifically in the BRAF V600-mutant tumors. These results warrant further study of combinatorial treatment strategies to enhance immunotherapy and potentiate cancer immunity for tumors resistant to immunotherapy despite strong immune infiltration. This should include both immunotherapeutic strategies and targeted therapies, given the potential synergy in combining or sequencing MAPK inhibitor therapies in the context of ICIs.41,42
Early ICI genomics studies reported the association between clinical response and TMB, 17,25,26,43 which has subsequently been used to positively identify ICI responders across tumor types.7,12 Generally, it is thought that the presence of more mutations increases the chances of neoantigens arising that can be targeted by the immune system. However, in cancers that commonly have high TMB such as melanoma, it can be difficult to identify specific recurrently mutated genes associated with response, since there are many mutation events to consider when performing statistical tests. Our approach, assessing genes with corresponding mutation types in a larger dataset, enabled us to identify the positive association between PIK3C2G missense mutations and ICI response. However, since PIK3C2G is infrequently expressed overall in melanoma, the association between genomic and gene expression profiles remains unclear.12 This is further supported by the lack of association between TMB and TIARA-PD-1, an aggregate metric of immune cell populations strongly associated with predicted anti-PD-1 response in cutaneous melanoma. Together, these findings suggest that defining the immunogenicity of tumors may require more specific or alternative genomic metrics, in conjunction with assessment of immune recognition at the tumor site.
The reported, harmonized dataset presented in this study sets a foundation for investigating the heterogeneity of clinical responses to ICIs in melanoma. These harmonized molecular and clinical annotation efforts enabled focus on specific questions related to anti-PD-1 response in cutaneous melanoma, with the appropriate account of critical clinical and technical batch effects. These systematic approaches are necessary to advance the rigorous and complex analyses required to optimize ICI treatment strategies.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Katie Campbell (katiecampbell@mednet.ucla.edu).
Materials Availability
There were no new physical or biological materials generated with this study.
Data and Code Availability
Raw sequencing data is available through the Sequence Read Archive accession identifier PRJNA923698. Processed data, including annotated variants and gene expression values, are available at https://github.com/ParkerICI/MORRISON-1-public. Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
DNA and RNA sequencing data were downloaded for previously reported datasets.6,8,22–24 The clinical results of CheckMate 064 and CheckMate 067 were previously described.20,21
For consideration in our study, samples had to have been collected from patients with a melanoma diagnosis, with melanoma subtype (cutaneous, acral, mucosal, uveal, unknown), and complete annotation for the following demographics: evaluable response by RECIST 1.1 criteria44, patient gender and age, treatment regimen, and whether the patient previously received anti-CTLA-4 therapy. Throughout the study, samples from responsive (RECIST 1.1 categories of CR or PR) tumors were grouped together and compared to samples from tumors with progressive disease (RECIST 1.1 PD) following anti-PD-1 therapy, in cutaneous melanoma tumors. Samples from tumors with stable disease (SD) following anti-PD-1 therapy were excluded from categorical comparisons of response. Samples denoted “anti-CTLA-4-naive” corresponded to tumors from patients that were not treated with anti-CTLA-4 prior to sample collection while “anti-CTLA-4-experienced” corresponds to tumors from patients previously treated with anti-CTLA-4 therapy.
METHOD DETAILS
WES analysis
Methods for whole exome sequencing (WES) for the CheckMate 038, Gide, et al., Hugo, et al., Liu, et al., and Van Allen, et al. were previously described.6,8,22–24 For CheckMate 064 and CheckMate 067, WES was performed as previously described, with one modification.22 CheckMate 067 WES libraries were generated using the Agilent SureSelect All Exon V5 capture reagent.
Whole exome sequencing data was preprocessed (Figure S1) according to GATK best practices,45 and aligned to the human reference genome (GRCh38) by BWA-MEM v0.7.15.46 Somatic variants were detected by comparing tumor WES with patient-matched normal WES by Mutect2,47 Varscan2,48 Strelka,49 and SomaticSniper.50 Single nucleotide variants (SNVs) were further annotated using a deep learning model, DeepSVR,51 for to further filter false positive variants.52 Multi-nucleotide variants (MNVs) were identified by extracting the read names associated with neighboring SNV calls and re-annotating them as MNVs if at least three shared read names correspond to all neighboring SNVs (Supplementary Figure S1) (https://github.com/kcampbel/public/scripts/mnvDetection.py).
SNVs and indels were subsequently filtered to those called by a minimum of two out of four variant callers, and SNVs corresponding to MNVs were removed. Filtered SNVs and Indels were annotated by Ensembl Variant Effect Predictor (VEP), using the Ensembl annotation database v94 53. TMB was quantified by normalizing the number of nonsilent mutations by the total number of bases sequenced and tumor purity, as previously described.54 Nonsilent mutations included any variants with ‘HIGH’ or ‘MODERATE’ impacts on proteins, determined by Ensembl VEP.
Sequenza was used to detect copy number alterations (CNAs) and regions of loss-of-heterozygosity (LOH) and quantify tumor cellularity.55 Tumor-in-normal contamination was quantified using GATK4 CalculateContamination.56 Tumor WES samples with cellularity less than or equal to 0.1, average depth of sequencing in targeted regions less than or equal to 100X, or estimated contamination of at least 0.03 were removed from additional analysis (Figure S3). We estimated that with a targeted 100X coverage by WES, in samples with more than 3% of reads associated with cross-sample contamination, false-positive variants due to this type of contamination would be detected due to having more than 1–3 reads of sequencing read support. Notably, 20 out of 23 processed tumor WES samples from the Hugo et al. cohort failed the contamination thresholds; due to the high rate of contamination in this dataset, this cohort was entirely removed for downstream analysis. Tumor-matched RNAseq samples were also removed, if WES did not meet the defined criteria.
RNAseq analysis
Methods for transcriptome sequencing (RNAseq) for the CheckMate 038, Gide, et al., Hugo, et al., Liu, et al., and Van Allen, et al. were previously described.6,8,22–24 For CheckMate 064 and CheckMate 067, RNAseq was performed as previously described, with one modification.22 CheckMate 064 RNAseq libraries were prepared using the Illumina Stranded mRNA sequencing kit, and CheckMate 067 RNAseq libraries were prepared using the Illumina TruSeq RNA Exome kit.
RNAseq data was aligned to the human reference genome (GRCh38) using HISAT257 (Supplementary Figure S1). Gene expression was quantified by HTseq-counts58 and Stringtie.59 Differential gene expression analysis was performed using edgeR60 with cohort included as a covariate to account for batch effects due to differences in library prep methods. Genes were filtered by expression prior to statistical analysis using the edgeR::filterByExpr function and the Benjamini-Hochberg (BH) procedure was used for multiple hypothesis testing. Statistical significance by edgeR glmQLF test (edgeR: quasi-likelihood negative binomial generalized log-linear model) was determined at an adjusted pvalue cutoff <= 0.1 after multiple hypothesis correction. Gene set enrichment analysis (GSEA) was performed with genes ranked according to differential expression, and run using clusterProfiler in R on Hallmark and KEGG pathways from the Molecular Signatures Database (MSigDB) and statistics were evaluated using clusterProfiler::GSEA Kolmogorov Smirnov (K-S) test.61,62 Prior to running GSEA and computing statistical significance, MSigDB gene sets of interest for the analysis were manually curated from different collections (Tables S1–S2). Pathways that reached statistical significance in one of the sub-analyses were visualized to reduce the final number of pathways included in visualizations. Statistically significant GSEA results were evaluated after an adjusted p-value cutoff of 0.10 using the BH procedure. BH-correction was stringently performed across all comparisons and all subsets in aggregate. Gene Ontology Enrichment Analysis (GOEA) was performed using XGR on differentially expressed genes (positively or negatively) from each comparison group.63 Significance was determined using fisher’s exact test in XGR::xEnricherGenes. Similar to GSEA, BH-correction was stringently performed across all comparisons and all subsets in aggregate at an adjusted p-value cutoff of 0.10.
The R package Combat-Seq (https://github.com/zhangyuqing/ComBat-seq) was used for batch-effect correction of raw sequencing counts prior to gene signature analysis.64 Combat-Seq was not used prior to differential gene expression analysis in edgeR. Combat-Seq transformed counts were normalized to total mapped reads and log-scaled prior to analysis.
Principal Component Analysis was performed on the cohort-corrected log cpm gene matrix. Genes were mean-centered and scaled before transformation by PCA.
Single-sample scores for pathways or immune cell-type signatures were generated using a custom signature scoring method, similar to those used in published RNAseq analysis.65 The score is calculated using the average expression level computed for a single-sample based on an input gene list. Using the Combat-Seq-corrected, log-CPM gene expression table, the average expression of each gene was calculated across all samples, and genes were ranked from highest to lowest and divided into 30 bins of equal size to generate a control feature set. The control gene-sets for each gene in the gene-set were generated by randomly selecting 100 genes from the same expression bin in the control feature set for that gene. The final sample score was computed by subtracting the control gene-set from the sample’s gene-set score.
Tumor Immunogenicity Associated with Response to Anti-PD-1 (TIARA-PD-1)
TIARA-PD-1 was established to aggregate the immune cell gene expression signatures associated with anti-PD-1 response into a single metric. TIARA-PD-1 was generated using the baseline RNAseq data derived from cutaneous melanoma tumors that either responded (CR/PR) or did not respond (PD) following anti-PD-1 monotherapy. First, the mean score was calculated for each immune cell-type signature across anti-PD-1 responsive tumors and across anti-PD-1 nonresponsive tumors, and the difference was taken between the two means. Immune cell-types were separated into anti-PD-1 responder-up or -down, based upon whether the immune cell-type was higher in samples from patients with response or no response to anti-PD-1, respectively. Next, within each sample, the averages were taken across immune cell-type signatures in each of the responder-up and -down categories. TIARA-PD-1 is the resulting difference between the aggregate responder-up and responder-down groups, a calculation that can be applied to any binary comparison group.66 Based upon this calculation, TIARA-PD-1 is increased in magnitude due to more positive responder-up or more negative responder-down aggregate scores. We evaluated this calculation using the mean, median, or sum of immune cell-type signatures, and these all exhibited similar results (data not shown).
EPIC, TIDE, and IMPRES
EPIC to estimate immune, stromal, and other cell fraction estimates from the bulk RNAseq data was computed using the batch-effect corrected gene expression matrix and the EPIC R package. TIDE was computed using the web-based application provided by http://tide.dfci.harvard.edu/.15 The batch-effect corrected gene expression matrix was normalized using the all sample average as the normalization control, as instructed by the developers. The normalized matrix was then split into anti-CTLA-4 naive and anti-CTLA-4 experienced files and inputted into the TIDE web application separately, and we selected the “Previous immunotherapy” option for the file containing the anti-CTLA-4-experienced samples. TIDE did not identify differences in an anti-CTLA-4 dependent manner without this “Previous immunotherapy” option. IMPRES was calculated using the batch-effect corrected gene expression matrix with 14 of the 15 pairwise transcriptomic relations described in the author’s methods.13 CD27/PD1 is 1 of the transcriptomic pairs and CD27 was filtered out during the batch-effect correction step as it did not meet the minimum count and sample abundance. Using the unfiltered, uncorrected gene expression matrix with all 15 pairs yielded the same results (results not shown).
Model generation and evaluation
Lasso was used in R67 to identify markers associated with response33–35. To construct a lasso model, we used the glmnet34 implementation of lasso in R, including a built-in cross-validation function to tune the L1 regularization parameter λ. We used the minimum mean-cross validated error. To compare probability of response across the main effect, prior anti-CTLA-4 informed, and interaction effect informed models, we used each model’s selected features and assessed performance using LOOCV. Linear discriminant analysis (LDA) was used for supervised dimensionality reduction68,69 to assess gene importance associated with groups defined by anti-PD-1 response and anti-CTLA-4 experience. The Combat-Seq corrected, log-CPM table was filtered to include only differentially significant gene results from prior analysis comparing anti-PD-1 response in the context of anti-CTLA-4 experience across all samples. This filtered table was an input for LDA to find genes that maximally separated samples based on both anti-CTLA-4 experience and anti-PD-1 response.
QUANTIFICATION AND STATISTICAL ANALYSIS
All analyses and statistics were performed using R (v 4.2.0).67 TMB was compared across clinically defined groups using Kruskal-Wallis (across group) and Wilcoxon (pairwise) tests. Recurrently mutated genes in clinical groups were identified by applying logistic regression to the genes and mutation types across clinical subsets, controlling for tumor purity, TMB, melanoma subtype based on site of origin (cutaneous, mucosal, acral, uveal), and anti-CTLA-4 experience. Significant gene-mutation pairs were selected for by p-value cutoffs of 0.05 and Benjamini-Hochberg adjusted p-value cutoffs of 0.20. Significant differences in gene expression, gene sets, and scores were selected by Benjamini-Hochberg adjusted p-value cutoffs of 0.10 across clinically defined groups. Correlations were reported by the Spearman rank correlation and reported with Benjamini-Hochberg adjusted p-values.
Supplementary Material
Supplementary Table S1. Gene set enrichment analysis by anti-CTLA-4 experience. Related to Figure 3.
Gene set enrichment analysis corresponding to comparison of anti-CTLA-4-experienced and anti-CTLA-4-naive tumors.
Supplementary Table S2. Gene ontology pathways for gene set enrichment analysis. Related to Figure 3.
List of GO:BP pathways from MSIGDB used in GSEA for analysis of significantly different genes.
Supplementary Table S3. Gene set enrichment analysis by anti-PD-1 response. Related to Figure 4.
Gene set enrichment analysis corresponding to comparison of anti-PD-1 response in cutaneous melanoma tumors.
Supplementary Table S4. Gene sets used in rank-based gene set enrichment analysis and features used in statistical analysis. Related to Figures 3, 4, 5, and 6.
Manually curated list of HALLMARK and KEGG pathways from MSIGDB used in rank-based GSEA for analysis of prior anti-CTLA-4 experience or clinical response to anti-PD1. List of HALLMARK and NABA pathways from MSIGDB used in rank-based Gene Set Enrichment Analysis. List of Genomic, gene, and gene set features used for statistical models.
Supplementary Table S5. Gene set enrichment analysis across TIARA-PD-1. Related to Figure 5.
Gene set enrichment analysis for processes associated with TIARA-PD1.
Supplementary Table S6. Gene sets associated with TIARA-PD-1 within clinical groups. Related to Figure 5.
Gene set enrichment analysis for processes associated with TIARA-PD1 within clinical groups.
Supplementary Table S7. Differential gene expression analysis within genetic subtypes. Related to Figure 5.
Differential gene expression analysis results within melanoma genetic subtypes..
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Tumor biopsies and peripheral blood collected from melanoma patients (CheckMate 064) | Weber, et al. (PMID: 27269740) | https://clinicaltrials.gov/ct2/show/NCT01783938 |
| Tumor biopsies and peripheral blood collected from melanoma patients (CheckMate 067) | Wolchok, et al. (PMID: 28889792) | https://clinicaltrials.gov/ct2/show/NCT01844505 |
| Critical commercial assays | ||
| Agilent SureSelect All Exome V2 | Agilent | https://www.agilent.com/en/product/next-generation-sequencing/hybridization-based-next-generation-sequencing-ngs/exome-probes/sureselect-clinical-research-exome-v2-232867#productdetails |
| Agilent SureSelect All Exome V5 | Agilent | https://www.agilent.com/en/product/next-generation-sequencing/hybridization-based-next-generation-sequencing-ngs/exome-probes/sureselect-human-all-exon-232866 |
| Illumina Stranded mRNA | Illumina | https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/stranded-mrna-prep.html |
| Illumina TruSeq RNA Exome | Illumina | https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/truseq-rna-access.html |
| Deposited data | ||
| Processed data | This paper | https://github.com/ParkerICI/MORRISON-1-public |
| CheckMate 064 and 067 WES data | This paper | BioProject: PRJNA923698 |
| CheckMate 064 and 067 RNAseq data | This paper | BioProject: PRJNA923698 |
| CheckMate 038 WES and RNAseq data | Riaz, et al. (PMID: 29033130) | SRA: SRP094781; SRA: SRP095809 |
| Gide, et al. RNAseq dataa | Gide, et al. (PMID: 30753825) | ENA: PRJEB23709 |
| Hugo, et al. WES and RNAseq | Hugo, et al. (PMID: 26997480) | SRA: SRP067938; GEO: GSE78220 |
| Liu, et al. WES and RNAseq | Liu, et al. (PMID: 31792460) | dbGaP: phs000452.v3.p1 |
| Van Allen, et al. RNAseq | Van Allen, et al. (PMID: 26359337) | dbGaP: phs000452.v2.p1 |
| Software and algorithms | ||
| GATK4 | Van der Auwera, et al. | https://github.com/broadinstitute/gatk/releases |
| BWA-MEM v0.7.15 | Li (PMID: 19451168) | https://bio-bwa.sourceforge.net/bwa.shtml |
| Mutect2 | Benjamin, et al. (https://doi.org/10.1101/861054) | https://gatk.broadinstitute.org/hc/en-us/articles/360036713131-Mutect2 |
| Varscan2 | Koboldt, et al. (PMID: 22300766) | https://varscan.sourceforge.net/ |
| Strelka | Saunders, et al. (PMID: 22581179) | https://github.com/Illumina/strelka |
| SomaticSniper | Larson, et al. (PMID: 22155872) | https://gmt.genome.wustl.edu/packages/somatic-sniper/ |
| DeepSVR | Ainscough, et al. (PMID: 30397337) | https://github.com/griffithlab/DeepSVR |
| MNVdetect | This paper | https://github.com/kcampbel/public/scripts/mnvDetection.py |
| Ensembl-VEP | McLaren, et al. (PMID: 27268795) | https://uswest.ensembl.org/info/docs/tools/vep/index.html |
| Sequenza | Favero, et al. (PMID: 25319062) | https://cran.r-project.org/web/packages/sequenza/vignettes/sequenza.html |
| HISAT2 | Kim, et al. (PMID: 31375807) | https://daehwankimlab.github.io/hisat2/ |
| HTseq-counts | Anders, et al. (PMID: 25260700) | https://htseq.readthedocs.io/en/release_0.11.1/count.html |
| Stringtie | Pertea, et al. (PMID: 25690850) | https://ccb.jhu.edu/software/stringtie/ |
| edgeR | Robinson, et al. (PMID: 19910308) | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| MSigDB | Liberzon, et al (PMID: 26771021) | https://www.gsea-msigdb.org/gsea/msigdb/ |
| clusterProfiler | Yu, et al. (PMID: 22455463) | https://github.com/YuLab-SMU/clusterProfiler |
| XGR | Fang, et al. (PMID: 27964755) | https://xgr.r-forge.r-project.org/ |
| ComBat-seq) | Zhang, et al. (PMID: 33015620) | https://github.com/zhangyuqing/ComBat-seq |
| TIDE | Jiang, et al. (PMID: 30127393) | http://tide.dfci.harvard.edu |
| Glmnet | Friedman, et al. (PMID: 20808728) | https://glmnet.stanford.edu/articles/glmnet.html |
Highlights.
Harmonization of molecular and clinical annotation clarifies anti-PD-1 response patterns
Prior anti-CTLA-4 treatment modifies predictors of anti-PD-1 response in melanoma
Immune cell signature differences are enhanced in tumors with prior anti-CTLA-4
Acknowledgements
We would like to acknowledge Drs. Tuba Gide and James Wilcott for their support in data annotation. KMC is supported by the Cancer Research Institute Irvington Postdoctoral Fellowship Program, the V Foundation Gil Nickel Melanoma Research Fellowship, and the Parker Institute for Cancer Immunotherapy (PICI) and V Foundation Bridge Fellows Program. JSW is supported by the NYU Langone Medical Center Cancer Center Support Grant, NIH grants R01 CA 231295-03, R01 CA 227505-03, R01 CA 244936-02, R01 CA 243486-02, NYU Melanoma SPORE, the Mark Foundation, and the Melanoma Research Alliance. JDW is supported by the NIH/NCI Cancer Center Support Grant P30 CA008748, the PICI, the Ludwig Collaborative and Swim Across America Laboratory and Department of Medicine at Memorial Sloan Kettering Cancer Center, and Weill Cornell Medicine. AR is supported by NIH grants R35 CA197633 and P01 CA167575, the PICI, the Ressler Family Fund, and the support from Ken and Donna Schultz, and of Thomas Stutz.
Footnotes
Declaration of interests
KMC is a shareholder in Geneoscopy LLC and received consulting fees from PACT Pharma, Tango Therapeutics, Geneoscopy LLC, and Flagship Labs 81 LLC. MA received consulting fees from PICI. JSW received consulting fees from Merck, Genentech, AstraZeneca, GlaxoSmithKline, Novartis, Nektar, Celldex, Incyte, Biond, ImCheck, Sellas, Evaxion, and EMD Serono, is or has been a member of the scientific advisory board for Bristol Myers Squibb (BMS), CytoMx, Incyte, ImCheck, Biond, Sellas, Instil Bio, OncoC4 and Neximmune, and holds equity in Biond, Evaxion, Instil Bio, and Neximmune. JSW has been named on patents filed by Moffitt Cancer Center (ipilimumab biomarker and TIL growth method), and Biosedix (PD-1). NYU has received research support from BMS, Merck, GlaxoSmithKline, Moderna, Pfizer, Novartis, and AstraZeneca. JDW received consulting fees from Apricity, CellCarta, Ascentage Pharma, AstraZeneca, Astellas, Bicara Therapeutics, Boehringer Ingelheim, BMS, Dragonfly, Georgiamune, Imvaq, Larkspur, Maverick Therapeutics, Psioxus, Recepta, Tizona, and Sellas, has received research/grant support from BMS and Sephora, and has equity in Apricity, Arsenal IO, Ascentage, Beigene, Imvaq, Linnaeus, Georgiamune, Maverick, Tizona Pharmaceuticals, and Trieza. JL has worked in a consulting/advisory role for iOnctura, Apple Tree, Merck, BMS, Eisai, Debipharm, and Incyte, received honoraria from AstraZeneca, BMS, Eisai, EUSA Pharma, GlaxoSmithKline, Incyte, Ipsen, Merck, touchEXPERTS, Royal College of Physicians, Cambridge Healthcare Research, Royal College of General Practitioners, VJOncology, Agence Unik, Merck Sharp & Dohme, Novartis, Aptitude, Pierre Fabre, Pfizer, Roche, Seagen, Inselgruppe, eCancer, Ultimovacs, Calithera, and Goldman Sachs, and received research/grant support from Achilles Therapeutics, BMS, Immunocore, Aveo, Pharmacyclics, MSD, Nektar Therapeutics, Covance, Novartis, Pfizer, and Roche. FSH received research support from the NCI of the NIH, BMS, Novartis, and Genentech; royalties or licenses from BMS and Novartis; consulting fees from BMS, EMD Serono, Surface, Sanofi, Genentech, Gossamer, Trillium, Immunocore, Merck, Novartis, Compass Therapeutics, Pieris, Bioentre, Iovance, Catalym, and Amgen; patents for methods for treating MHC class I polypeptide-related sequence A disorders (#20100111973; pending, with royalties paid), tumour antigens and uses thereof (#7250291; issued), angiopoiten-2 biomarkers predictive of anti-immune checkpoint response (#20170248603; pending), compositions and methods for the identification, assessment, prevention, and treatment of melanoma using PD-L1 isoforms (#20160340407; pending), therapeutic peptides (#20160046716; #20140004112; #20170022275; #20170008962; all pending. #9402905; issued), methods of using pembrolizumab and trebananib (pending), vaccine compositions and methods for restoring NKG2D pathway function against cancers (#10279021; issued), antibodies that bind to MHC class I polypeptide-related sequence A (#10106611; issued), and anti-galectin antibody biomarkers predictive of anti-immune checkpoint and anti-angiogenesis responses (#20170343552; pending); data safety monitoring board and advisory board participation for Aduro and Checkpoint Therapeutics; scientific advisory board leadership for Bicara and Apricity; and stock options in Checkpoint Therapeutics, Pionyr, Apricity, and Bicara. SB, LS, DT, and TT are employees of BMS. SB, LS, DT, and TT are employees of BMS. DKW is an employee and equity holder at Santa Ana Bio; holds equity in Immunai, and has received consulting fees from Rubius Therapeutics and DeepMind. AR has received honoraria from consulting with Amgen, BMS, Chugai, Genentech, Merck, Novartis, Roche, and Sanofi, is or has been a member of the scientific advisory board and holds stock in Advaxis, Arcus Biosciences, Bioncotech Therapeutics, Compugen, CytomX, Five Prim, FLX-Bio, ImaginAb, Isoplexis, Kite-Gilead, Lutris Pharma, Merus, PACT Pharma, Rgenix, and Tango Therapeutics, and has received research funding from Agilent Technologies and BMS through Stand Up to Cancer.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. (2010). Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, et al. (2015). Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 372, 2521–2532. [DOI] [PubMed] [Google Scholar]
- 3.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob J-J, Rutkowski P, Lao CD, Cowey CL, Schadendorf D, Wagstaff J, Dummer R, et al. (2019). Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 381, 1535–1546. [DOI] [PubMed] [Google Scholar]
- 4.Center for Drug Evaluation, and Research FDA grants accelerated approval to dostarlimab-gxly for dMMR advanced solid tumors. U.S. Food and Drug Administration. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-dostarlimab-gxly-dmmr-advanced-solid-tumors. [Google Scholar]
- 5.Marcus L, Fashoyin-Aje LA, Donoghue M, Yuan M, Rodriguez L, Gallagher PS, Philip R, Ghosh S, Theoret MR, Beaver JA, et al. (2021). FDA Approval Summary: Pembrolizumab for the Treatment of Tumor Mutational Burden-High Solid Tumors. Clin. Cancer Res. 27, 4685–4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM, et al. (2015). Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Litchfield K, Reading JL, Puttick C, Thakkar K, Abbosh C, Bentham R, Watkins TBK, Rosenthal R, Biswas D, Rowan A, et al. (2021). Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell 184, 596–614.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu D, Schilling B, Liu D, Sucker A, Livingstone E, Jerby-Arnon L, Zimmer L, Gutzmer R, Satzger I, Loquai C, et al. (2019). Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med. 25, 1916–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, Yizhak K, Sade-Feldman M, Blando J, Han G, et al. (2020). B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, et al. (2017). IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 127, 2930–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, Sher X, Liu XQ, Lu H, Nebozhyn M, et al. (2018). Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 362. 10.1126/science.aar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Auslander N, Zhang G, Lee JS, Frederick DT, Miao B, Moll T, Tian T, Wei Z, Madan S, Sullivan RJ, et al. (2018). Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat. Med. 24, 1545–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Damotte D, Warren S, Arrondeau J, Boudou-Rouquette P, Mansuet-Lupo A, Biton J, Ouakrim H, Alifano M, Gervais C, Bellesoeur A, et al. (2019). The tumor inflammation signature (TIS) is associated with anti-PD-1 treatment benefit in the CERTIM pan-cancer cohort. J. Transl. Med. 17, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B, et al. (2018). Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 24, 1550–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu S, Stein JE, Rimm DL, Wang DW, Bell JM, Johnson DB, Sosman JA, Schalper KA, Anders RA, Wang H, et al. (2019). Comparison of Biomarker Modalities for Predicting Response to PD-1/PD-L1 Checkpoint Blockade: A Systematic Review and Meta-analysis. JAMA Oncol 5, 1195–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Samstein RM, Lee C-H, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, Barron DA, Zehir A, Jordan EJ, Omuro A, et al. (2019). Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 10.1038/s41588-018-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rose AAN, Armstrong SM, Hogg D, Butler MO, Saibil SD, Arteaga DP, Pimentel Muniz T, Kelly D, Ghazarian D, King I, et al. (2021). Biologic subtypes of melanoma predict survival benefit of combination anti-PD1+anti-CTLA4 immune checkpoint inhibitors versus anti-PD1 monotherapy. J Immunother Cancer 9. 10.1136/jitc-2020-001642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang M, Banik I, Shain AH, Yeh I, and Bastian BC (2022). Integrated genomic analyses of acral and mucosal melanomas nominate novel driver genes. Genome Med. 14, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weber JS, Gibney G, Sullivan RJ, Sosman JA, Slingluff CL Jr, Lawrence DP, Logan TF, Schuchter LM, Nair S, Fecher L, et al. (2016). Sequential administration of nivolumab and ipilimumab with a planned switch in patients with advanced melanoma (CheckMate 064): an open-label, randomised, phase 2 trial. Lancet Oncol. 17, 943–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob J-J, Cowey CL, Lao CD, Wagstaff J, Schadendorf D, Ferrucci PF, et al. (2017). Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 377, 1345–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, Hodi FS, Martín-Algarra S, Mandal R, Sharfman WH, et al. (2017). Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 171, 934–949.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. (2016). Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 165, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gide TN, Quek C, Menzies AM, Tasker AT, Shang P, Holst J, Madore J, Lim SY, Velickovic R, Wongchenko M, et al. (2019). Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 35, 238–255.e6. [DOI] [PubMed] [Google Scholar]
- 25.Wu Y, Xu J, Du C, Wu Y, Xia D, Lv W, and Hu J (2019). The Predictive Value of Tumor Mutation Burden on Efficacy of Immune Checkpoint Inhibitors in Cancers: A Systematic Review and Meta-Analysis. Front. Oncol. 9, 1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wood MA, Weeder BR, David JK, Nellore A, and Thompson RF (2020). Burden of tumor mutations, neoepitopes, and other variants are weak predictors of cancer immunotherapy response and overall survival. Genome Med. 12, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schou Nørøxe D, Flynn A, Westmose Yde C, Østrup O, Cilius Nielsen F, Skjøth-Rasmussen J, Brennum J, Hamerlik P, Weischenfeldt J, Skovgaard Poulsen H, et al. (2022). Tumor mutational burden and purity adjustment before and after treatment with temozolomide in 27 paired samples of glioblastoma: a prospective study. Mol. Oncol. 16, 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang N-AAS, Andrews MC, Sharma P, Wang J, Wargo JA, Pe’er D, et al. (2017). Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 170, 1120–1133.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang RR, Jalil J, Economou JS, Chmielowski B, Koya RC, Mok S, Sazegar H, Seja E, Villanueva A, Gomez-Navarro J, et al. (2011). CTLA4 blockade induces frequent tumor infiltration by activated lymphocytes regardless of clinical responses in humans. Clin. Cancer Res. 17, 4101–4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Friedman CF, Spencer C, Cabanski CR, Panageas KS, Wells DK, Ribas A, Tawbi H, Tsai K, Postow M, Shoushtari A, et al. (2022). Ipilimumab alone or in combination with nivolumab in patients with advanced melanoma who have progressed or relapsed on PD-1 blockade: clinical outcomes and translational biomarker analyses. J Immunother Cancer 10. 10.1136/jitc-2021-003853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dixon-Douglas JR, Patel RP, Somasundram PM, and McArthur GA (2022). Triplet Therapy in Melanoma - Combined BRAF/MEK Inhibitors and Anti-PD-(L)1 Antibodies. Curr. Oncol. Rep. 24, 1071–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tibshirani R (1996). Regression Shrinkage and Selection via the Lasso. J. R. Stat. Soc. Series B Stat. Methodol. 58, 267–288. [Google Scholar]
- 34.Friedman J, Hastie T, and Tibshirani R (2010). Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 33, 1–22. [PMC free article] [PubMed] [Google Scholar]
- 35.James G, Witten D, Hastie T, and Tibshirani R (2013). An Introduction to Statistical Learning: with Applications in R (Springer Texts in Statistics) 1st ed. (Springer; ). [Google Scholar]
- 36.Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, Allison JP, Ribas A, and Sharma P (2019). Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3+ Regulatory T Cells (Tregs) in Human Cancers. Clin. Cancer Res. 25, 1233–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nicolas-Boluda A, Vaquero J, Vimeux L, Guilbert T, Barrin S, Kantari-Mimoun C, Ponzo M, Renault G, Deptula P, Pogoda K, et al. (2021). Tumor stiffening reversion through collagen crosslinking inhibition improves T cell migration and anti-PD-1 treatment. Elife 10. 10.7554/eLife.58688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zandberg DP, Menk AV, Velez M, Normolle D, DePeaux K, Liu A, Ferris RL, and Delgoffe GM (2021). Tumor hypoxia is associated with resistance to PD-1 blockade in squamous cell carcinoma of the head and neck. J Immunother Cancer 9. 10.1136/jitc-2020-002088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang Y, Zhang H, Wang J, Liu Y, Luo T, and Hua H (2022). Targeting extracellular matrix stiffness and mechanotransducers to improve cancer therapy. J. Hematol. Oncol. 15, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuczek DE, Larsen AMH, Thorseth M-L, Carretta M, Kalvisa A, Siersbæk MS, Simões AMC, Roslind A, Engelholm LH, Noessner E, et al. (2019). Collagen density regulates the activity of tumor-infiltrating T cells. J Immunother Cancer 7, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Subbiah V, Baik C, and Kirkwood JM (2020). Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer Res. 6, 797–810. [DOI] [PubMed] [Google Scholar]
- 42.Atkins MB, Lee SJ, Chmielowski B, Ribas A, Tarhini AA, Truong T-G, Davar D, O’Rourke MA, Curti BD, Brell JM, et al. (2021). DREAMseq (Doublet, Randomized Evaluation in Advanced Melanoma Sequencing): A phase III trial—ECOG-ACRIN EA6134. J. Clin. Orthod. 39, 356154–356154. [Google Scholar]
- 43.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, et al. (2014). Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 371, 2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwartz LH, Litière S, de Vries E, Ford R, Gwyther S, Mandrekar S, Shankar L, Bogaerts J, Chen A, Dancey J, et al. (2016). RECIST 1.1-Update and clarification: From the RECIST committee. Eur. J. Cancer 62, 132–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. (2013). From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li H (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv [q-bio.GN]. [Google Scholar]
- 47.Benjamin D, Sato T, Cibulskis K, Getz G, Stewart C, and Lichtenstein L (2019). Calling Somatic SNVs and Indels with Mutect2. bioRxiv, 861054. 10.1101/861054. [DOI] [Google Scholar]
- 48.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, and Cheetham RK (2012). Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817. [DOI] [PubMed] [Google Scholar]
- 50.Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, Ley TJ, Mardis ER, Wilson RK, and Ding L (2012). SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics 28, 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ainscough BJ, Barnell EK, Ronning P, Campbell KM, Wagner AH, Fehniger TA, Dunn GP, Uppaluri R, Govindan R, Rohan TE, et al. (2018). A deep learning approach to automate refinement of somatic variant calling from cancer sequencing data. Nat. Genet. 50, 1735–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barnell EK, Ronning P, Campbell KM, Krysiak K, Ainscough BJ, Sheta LM, Pema SP, Schmidt AD, Richters M, Cotto KC, et al. (2019). Standard operating procedure for somatic variant refinement of sequencing data with paired tumor and normal samples. Genet. Med. 21, 972–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, and Cunningham F (2016). The Ensembl Variant Effect Predictor. Genome Biol. 17, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anagnostou V, Niknafs N, Marrone K, Bruhm DC, White JR, Naidoo J, Hummelink K, Monkhorst K, Lalezari F, Lanis M, et al. (2020). Multimodal genomic features predict outcome of immune checkpoint blockade in non-small-cell lung cancer. Nat Cancer 1, 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Favero F, Joshi T, Marquard AM, Birkbak NJ, Krzystanek M, Li Q, Szallasi Z, and Eklund AC (2015). Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann. Oncol. 26, 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ulintz PJ, Wu W, and Gates CM (2019). Bioinformatics Analysis of Whole Exome Sequencing Data. Methods Mol. Biol. 1881, 277–318. [DOI] [PubMed] [Google Scholar]
- 57.Kim D, Paggi JM, Park C, Bennett C, and Salzberg SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, and Salzberg SL (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu G, Wang L-G, Han Y, and He Q-Y (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fang H, Knezevic B, Burnham KL, and Knight JC (2016). XGR software for enhanced interpretation of genomic summary data, illustrated by application to immunological traits. Genome Med. 8, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang Y, Parmigiani G, and Johnson WE (2020). ComBat-seq: batch effect adjustment for RNA-seq count data. NAR Genom Bioinform 2, lqaa078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haynes WA, Vallania F, Liu C, Bongen E, Tomczak A, Andres-Terrè M, Lofgren S, Tam A, Deisseroth CA, Li MD, et al. (2017). EMPOWERING MULTI-COHORT GENE EXPRESSION ANALYSIS TO INCREASE REPRODUCIBILITY. Pac. Symp. Biocomput. 22, 144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Team, R.C., and Others (2013). R: A language and environment for statistical computing.
- 68.Venables WN, and Ripley BD Modern Applied Statistics with S (Springer; New York: ). [Google Scholar]
- 69.Amouzgar M, Glass DR, Baskar R, Averbukh I, Kimmey SC, Tsai AG, Hartmann FJ, and Bendall SC (2022). Supervised dimensionality reduction for exploration of single-cell data by HSS-LDA. Patterns (N Y) 3, 100536. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. Gene set enrichment analysis by anti-CTLA-4 experience. Related to Figure 3.
Gene set enrichment analysis corresponding to comparison of anti-CTLA-4-experienced and anti-CTLA-4-naive tumors.
Supplementary Table S2. Gene ontology pathways for gene set enrichment analysis. Related to Figure 3.
List of GO:BP pathways from MSIGDB used in GSEA for analysis of significantly different genes.
Supplementary Table S3. Gene set enrichment analysis by anti-PD-1 response. Related to Figure 4.
Gene set enrichment analysis corresponding to comparison of anti-PD-1 response in cutaneous melanoma tumors.
Supplementary Table S4. Gene sets used in rank-based gene set enrichment analysis and features used in statistical analysis. Related to Figures 3, 4, 5, and 6.
Manually curated list of HALLMARK and KEGG pathways from MSIGDB used in rank-based GSEA for analysis of prior anti-CTLA-4 experience or clinical response to anti-PD1. List of HALLMARK and NABA pathways from MSIGDB used in rank-based Gene Set Enrichment Analysis. List of Genomic, gene, and gene set features used for statistical models.
Supplementary Table S5. Gene set enrichment analysis across TIARA-PD-1. Related to Figure 5.
Gene set enrichment analysis for processes associated with TIARA-PD1.
Supplementary Table S6. Gene sets associated with TIARA-PD-1 within clinical groups. Related to Figure 5.
Gene set enrichment analysis for processes associated with TIARA-PD1 within clinical groups.
Supplementary Table S7. Differential gene expression analysis within genetic subtypes. Related to Figure 5.
Differential gene expression analysis results within melanoma genetic subtypes..
Data Availability Statement
Raw sequencing data is available through the Sequence Read Archive accession identifier PRJNA923698. Processed data, including annotated variants and gene expression values, are available at https://github.com/ParkerICI/MORRISON-1-public. Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.