

Abstract
The Staphylococcus xylosus gene hprK, encoding HPr kinase (HPrK), has been isolated from a genomic library. The HPrK enzyme, purified as a His6 fusion protein, phosphorylated HPr, the phosphocarrier protein of the bacterial phosphotransferase system, at a serine residue in an ATP-dependent manner, and it also catalyzed the reverse reaction. Therefore, the enzyme constitutes a bifunctional HPr kinase/phosphatase. Insertional inactivation of the gene in the genome of S. xylosus resulted in the concomitant loss of both HPr kinase and His serine-phosphorylated-HPr phosphatase activities in cell extracts, strongly indicating that the HPrK enzyme is also responsible for both reactions in vivo. HPrK deficiency had a profound pleiotropic effect on the physiology of S. xylosus. The hprK mutant strain showed a severe growth defect in complex medium upon addition of glucose. Glucose uptake in glucose-grown cells was strongly enhanced compared with the wild type. Carbon catabolite repression of three tested enzyme activities by glucose, sucrose, and fructose was abolished. These results clearly demonstrate the prominent role of HPr kinase in global control to adjust catabolic capacities of S. xylosus according to the availability of preferred carbon sources.
Carbon catabolite repression (CR) is a global regulatory mechanism by which microorganisms adjust gene expression and enzyme activities to ensure preferential use of readily metabolizable carbon sources. Signal transduction leading to CR appears to be quite distinct in different groups of bacteria (42, 44). In enteric bacteria, the phosphorylation state of the glucose-specific enzyme IIA of the phosphoenolpyruvate:carbohydrate phosphotransferase system (PTS) triggers CR (37). Another PTS component, the phosphocarrier protein HPr, is the central signaling protein in AT-rich gram-positive bacteria (40), whereas a glucose kinase is essential for CR in Streptomyces coelicolor and perhaps in other GC-rich gram-positive organisms (1, 29).
In AT-rich gram-positive bacteria, the activity of the ATP-dependent HPr kinase is crucial for mediating CR. The enzyme, originally detected in Streptococcus pyogenes (11), phosphorylates a serine residue at position 46 in HPr. One of the regulatory consequences of the appearance of His serine-phosphorylated HPr (P-Ser-HPr) is the activation of the catabolite control protein A (CcpA), the central transcriptional regulator of CR (22, 23). Additionally, P-Ser-HPr appears to be involved in various forms of inducer control (45). In Bacillus subtilis, Crh, a protein similar to HPr, is also phosphorylated by HPr kinase and also participates in CR (17, 18, 53). Recent experiments with purified HPr kinases from Enterococcus faecalis (27) and Streptococcus salivarius (5) questioned the long-accepted view that HPr kinases are activated by metabolites such as fructose-1,6-diphosphate (FDP) (40). The enzymes from these organisms could not be stimulated by glycolytic intermediates, whereas purified B. subtilis HPr kinase required FDP for full activity (19, 39). Meanwhile, it was also recognized that the HPr kinases from E. faecalis and B. subtilis efficiently catalyze the dephosphorylation of P-Ser-HPr, thus constituting P-Ser-HPr phosphatases as well (27). The availability of a number of microbial genome sequences revealed HPr kinase genes in bacterial species where they had not been anticipated (The Institute of Genomic Research [TIGR] microbial database [http://www.tigr.org]). The presence of such a gene in Treponema pallidum is especially striking, as this organism does not possess a complete PTS (16). It appears, therefore, that the regulatory role(s) of HPr kinases in bacterial physiology may be quite diverse. It will be interesting to analyze HPr kinase function in a variety of organisms, to define common principles as well as differences.
So far, only the B. subtilis HPr kinase gene has been inactivated, resulting in a pleiotropic loss of CR (19, 32, 39). In this communication, we report the identification of the HPr kinase gene of Staphylococcus xylosus, a nonpathogenic AT-rich gram-positive bacterium, and the consequences of its insertional inactivation. We also demonstrate that this gene, hprK, encodes the P-Ser-HPr phosphatase.
MATERIALS AND METHODS
Bacterial strains and plasmid vectors.
S. xylosus C2a served as the wild-type strain in this study, and all mutants are derived from C2a. S. xylosus C2a has been obtained by curing S. xylosus DSM 20267 (47) of the endogenous plasmid pSX267 (21). The ccpA insertion mutant TX154 has been described previously (12). Cloning in Escherichia coli was carried out in DH5α [Φ80dlacZΔM15 Δ(lacZYA-argF) recA1 endA1 hsdR17 supE44 thi-1 gyrA96 relA1 deoR]. Plasmid pQE9 (Qiagen) was used to overexpress His6-fused HPr kinase (His6-HPrK) enzyme in E. coli M15 (lac ara gal mtl) harboring the LacI expression plasmid pRep4. Plasmid pGEM-T (Promega) served to clone PCR-generated fragments. Plasmids pUC18 (51) and pBluescript KS+ (Stratagene) were used as general cloning vectors and for sequencing. The shuttle vector pRB473 (7) served to complement the hprK mutation with cloned hprK in S. xylosus. Allelic replacements were achieved with the temperature-sensitive shuttle pBT2 and the ermB fragment from Tn551 cloned in pEC5 (6).
Growth media, DNA manipulations, and transformation.
DNA manipulations, plasmid DNA isolation, Southern blot analysis, transformation of E. coli, and preparation of media and agar plates for bacterial growth were performed according to standard procedures (46). Plasmid DNA was introduced into S. xylosus by electroporation using glycine-treated electrocompetent cells (6). PCR was carried out with Taq polymerase (Boehringer Mannheim GmbH), Vent polymerase (New England Biolabs), or the Expand long template system (Boehringer). S. xylosus was grown in B medium consisting of 1% peptone, 0.5% yeast extract, 0.5% NaCl, and 0.1% K2HPO4. To test for catabolite repression, sugars were added to a final concentration of 25 mM. Utilization of carbohydrates was monitored on fermentation test agar plates (34) containing 0.5% sugar.
Primers used for PCR and primer extension.
To clone an internal hprK fragment, the following degenerate primers containing inosine at variable positions were used (the positions refer to the nucleotide sequence AJ243915): Kin1 (5′-GICCIGGIITIGAIATGGCIGG; 2226 to 2246) and Kin4 (5′-GTITCITCITTIAIICCIACICIITC; 2847 to 2871). The primers Kin11 (5′-GCCAGATGAAGAACGTAAAGGACGC; 2320 to 2344) and Kin12 (5′-CTGTTAGTATCGATCCAGCGCC; 2768 to 2789) were used to detect the hprK gene in the amplified S. xylosus library. To complement the hprK mutation, the gene was amplified with primers Kin17 (5′-TATGGATCCTGAAGGTGGCGATGGTGG; 1699 to 1718) and Kin18 (5′-ATAGGATCCGTACCATCGGACTGAAATCGG; 3122 to 3144) to yield plasmid pKIN7. The His6-HPrK enzyme expression plasmid pKIN9 was constructed using primers Kin15 (5′-AACCTGCAGCCAATGTTAACTACAAAAAG; 2126 to 2145) and Kin16 (5′-TCTAAGCTTATTTCTCCTCACCATTATTAC; 3051 to 3075). The hprK deletion plasmid pKIN5 was constructed with primers Kin13 (CCTGTTGCAGTGAAGTGCCGC; 2520 to 2540) and Kin14 (CGGCGTAGGTGTACTAATAACTGGTG; 2557 to 2582).
To map the transcription start site of hprK, primer Kin20 (5′-CCCCGCCATTTCCAATCC; 2231 to 2248) with an IRD700 infrared dye at the 5′ end was used.
Isolation of the hprK gene and construction of hprK plasmids.
An internal hprK fragment was amplified with degenerate primers Kin1 and Kin4 from chromosomal S. xylosus DNA, subcloned to pGEM-T, and sequenced. From this sequence, specific primers Kin11/Kin12 were designed and used to screen a plasmid library containing chromosomal S. xylosus DNA (7). The library had been constructed with partially Sau3AI-restricted fragments and pBR322 (48) and was stored as pools of plasmid DNA isolated from 50 colonies. A 0.5-kb PCR fragment was detected in two plasmid mixtures within the first 20 pools that were tested. E. coli DH5α was transformed with aliquots of these mixtures, and the transformants were tested by PCR for the presence of the hprK-containing plasmid. Two colonies harboring an identical plasmid were identified in one of the transformations. The plasmid, which turned out to contain the hprK gene, was designated pKIN6.
To overexpress hprK in E. coli and for complementation experiments in S. xylosus, a PCR fragment generated with primer pair Kin17-Kin18 and Vent polymerase was cloned into pBluescript KS+ (pKIN7) and the shuttle vector pRB473 (pKIN11). The fragment starts within the 3′ end of uvrA (Fig. 1) and contains the putative transcriptional terminator of uvrA, the uvrA-hprK intergenic region, hprK, and the first 60 bp of the following lgt gene (Fig. 1).
FIG. 1.

Genetic organization of the hprK region of S. xylosus. The sequenced region around the HPr kinase gene is shown. The thick line represents the DNA that was obtained from a genomic S. xylosus library. DNA indicated by the thin line was obtained by PCR. Sizes and orientations of the genes were deduced from the nucleotide sequence. The site of insertion and the orientation of the erythromycin resistance gene ermB in the HPr kinase mutants S. xylosus TX66 and TX67 are indicated. The nucleotide sequence of the hprK promoter region is shown below the genetic organization. Numbering refers to the complete DNA sequence (accession no. AJ243915). The transcriptional start site is marked by an arrow.
Inactivation of the hprK gene in the genome of S. xylosus.
To inactivate the hprK kinase gene in the chromosome of S. xylosus, a 5.4-kb fragment was produced by inverse high-fidelity long-range PCR using ligated HindIII-restricted chromosomal DNA as the template and the primers Kin13 and Kin14. The fragment was cleaved with HindIII and BamHI, and the resulting fragments of 2.9 and 2.5 kb were subcloned to pBluescript to yield pKIN2 and pKIN3. From pKIN2, the 2.9-kb HindIII-BamHI fragment was recovered, while the 2.5-kb fragment was removed from pKIN3 as a KpnI-BamHI fragment. Subsequently, both fragments were combined with plasmid pBT2 restricted with KpnI and HindIII. The resulting plasmid, pKIN5, contained most of the hprK region as depicted in Fig. 1, with a short (20-bp) deletion in the middle of the hprK gene and a newly created unique BamHI restriction site. In this site, a BamHI-BclI fragment from pEC5 (6) was inserted in both orientations harboring the erythromycin resistance gene (ermB) of Tn551. The resulting plasmids, pKIN5E1 and pKIN5E2, were introduced into S. xylosus, and insertional inactivation of hprK was achieved as described elsewhere (6). The resulting hprK mutant strains, TX66 (hprK::ermB) and TX67 (hprK::ermB), harbored ermB in the same orientation as hprK (TX66) or opposite to hprK (TX67). The genomic organization of the hprK region in both strains was confirmed by PCR analysis.
Purification of His6-HPrK enzyme.
To purify HPr kinase with a His tag extension at the amino terminus, a PCR fragment was produced with Vent polymerase using primers Kin15 and Kin16 and cloned as a PstI-HindIII fragment into the expression plasmid pQE9 to yield pKIN9. By this cloning, a fusion gene was created expressing an HPr kinase protein with a 17-amino-acid extension. The deduced amino acid sequence of the N terminus of that fusion protein is MRGSHHHHHHGSVDLQPMLTTK (boldface characters indicate the authentic N terminus of HPrK).
After transformation of E. coli M15(pREP-4) with pKIN9, the strain was grown at 37°C in 100 ml of 2xTY medium (46) to an optical density at 578 nm (OD578) of 0.6. Expression of the fusion gene was induced by 1 mM isopropyl-β-d-thiogalactoside. After 4 h, the cells were collected by centrifugation and broken by glass beads in 8 ml of a 50 mM Tris (pH 7.5)–50 mM NaCl buffer as described elsewhere (50). Then 1 ml of Ni-nitriloacetic acid (NTA) agarose (Qiagen) was added to the lysate and incubated for 1 h at room temperature with stirring. A column was packed with this material and washed with 20 ml of buffer A (50 mM Tris [pH 7.5], 200 mM NaCl) and with 20 ml of buffer B (50 mM Tris [pH 7.5], 200 mM NaCl, 20 mM imidazole). The bound fusion protein was eluted with buffer E (50 mM Tris [pH 7.5], 50 mM NaCl, 250 mM imidazole). Fractions containing the fusion protein were combined and desalted through 10 ml of Sephadex G-25 (PD-10; Amersham Pharmacia) columns, resulting in about 2 mg of His6-HPrK enzyme in 50 mM Tris (pH 7.5)–50 mM NaCl. The enzyme was stored at 4°C and found to be active for several weeks.
HPr kinase assay.
As the substrate for HPr kinase, we used a mutant form of HPr from Staphylococcus aureus where the histidine at position 15 was replaced by alanine (27). By this mutation, phosphoenolpyruvate-dependent, enzyme I-mediated phosphorylation at His-15 is prevented. Purified His-15-Ala-HPr was kindly provided by W. Hengstenberg, Bochum, Germany. HPr kinase assays were done according to the procedure described for E. faecalis HPr kinase (27). The reactions were carried out in 50 mM Tris (pH 7.5)–50 mM NaCl–10 mM MgCl2–5 mM ATP with up to 5 μg of His-15-Ala-HPr and various amounts of purified His6-HPrK enzyme or bacterial whole cell lysates. Stimulation of HPr kinase activity by FDP was tested by adding FDP to a final concentration of 2 mM. The assay mixture was incubated at 37°C for 20 min, heated for 5 min at 70°C, and loaded onto nondenaturing 15% polyacrylamide gels that were prepared according to Laemmli (30) without the addition of sodium dodecyl sulfate (SDS). HPr and P-Ser-HPr are easily distinguished on Coomassie blue-stained gels, as phosphorylated HPr runs considerably faster than HPr.
Purification of P-Ser-HPr.
To purify P-Ser-HPr, 20 μl of the Ni-NTA agarose column material with bound His6-HPrK enzyme was incubated with 30 μg of His-15-Ala-HPr as described for the HPr kinase assay. The phosphorylation reaction was monitored on nondenaturing gels. After completion of the reaction, Ni-NTA agarose was removed by centrifugation and the supernatant was further purified by a second centrifugation through a Ni-NTA agarose spin column (Qiagen). Subsequently, remaining ATP was removed by gel filtration through a Sephadex G-25 column (PD-10; Amersham Pharmacia), yielding P-Ser-HPr in 50 mM Tris (pH 7.5)–50 mM NaCl.
P-Ser-HPr phosphatase assay.
Phosphatase activity was tested with purified P-Ser-HPr, purified His6-HPr kinase, or cell extracts in a buffer containing 50 mM Tris (pH 7.5), 50 mM NaCl, 10 mM MgCl2, and 1 mM NaH2PO4. The assay mixture was incubated at 37°C for 20 min and analyzed by nondenaturing polyacrylamide gel electrophoresis (PAGE) as described for the HPr kinase assay above.
Determination of catabolic enzyme activities in cell extracts.
For determination of catabolic enzymatic activities in S. xylosus, cells were grown in B medium to an OD578 of 1. The culture was then split; one half was supplemented with 25 mM appropriate carbon source(s), whereas the other half grew without addition. After 1 h, the OD was determined and the cells were harvested by centrifugation. Crude extracts were prepared by disrupting the cells with glass beads (51), and β-galactosidase, β-glucuronidase, and α-glucosidase activities were determined as described previously (12). Protein concentrations were determined by the method of Bradford (4).
Measurement of glucose uptake.
Uptake of glucose in S. xylosus was measured with whole cells grown in B medium or B medium supplemented with 25 mM glucose as described above. Staphylococcal cells were harvested, washed in transport buffer (0.1 M morpholinepropanesulfonic acid, 0.5 mM MgSO4, 10 mM NaCl [pH 7.0]), and resuspended in the same buffer to a final OD578 of 3.0. After addition of 200 μM [14C]glucose (6.2 mCi/mmol) to 1 ml of prewarmed (30°C) cells, 0.15-ml samples were taken at intervals, collected on membrane filters with a pore size of 0.45 μm, and washed with 5 ml of transport buffer. Filters were dried at 80°C for 1 h. The radioactivity was determined by liquid scintillation counting. Uptake rates are expressed in nanomoles of glucose per minute per milligram of cellular protein. The amount of protein was determined by the method of Bradford (4).
Determination of methylglyoxal accumulation in the medium.
Methylglyoxal production and release from the cells was measured by reaction of the growth medium with 2,4-dinitrophenylhydrazine (9, 25). The cells were removed by centrifugation and various volumes of the medium (20 to 320 μl) were incubated with 120 μl of 2,4-dinitrophenylhydrazine (10 mg/ml in 2 M HCl) for 15 min at 30°C. After addition of 560 μl of 10% (wt/vol) NaOH and incubation at room temperature for 10 min, the OD550 was determined. An absorbance of 16.4 corresponds to 1 μmol of methylglyoxal that was present in the reaction mix (9).
RNA preparation and primer extension analysis.
Preparation of RNA and primer extension reactions were done as described previously (3). The primer used in these experiments contained an infrared dye IRD700 at the 5′ end. Reverse transcripts were run on 8% polyacrylamide-urea gels and detected by a Li-Cor DNA sequencer. The file containing the image of that gel was imported into Photoshop and printed on glossy paper.
Nucleotide sequence accession number.
The DNA sequence reported here is available from the EMBL, GenBank, and DDBJ databases under accession no. AJ243915.
RESULTS
Nucleotide sequence of the S. xylosus hprK region.
The HPr kinase gene (hprK) of S. xylosus was identified in a plasmid library as outlined in Materials and Methods, and the nucleotide sequence of the cloned S. xylosus DNA was determined. In addition, an adjacent genomic fragment that was cloned during hprK inactivation was also sequenced. The nucleotide sequence of 6,196 bp harbors four complete and two truncated open reading frames on the same DNA strand (Fig. 1). The second gene in the sequenced region (Fig. 1) encodes HPr kinase, a protein of 314 amino acids with a molecular mass of 35.3 kDa. Identity with other HPr kinases ranges from 86% with the enzyme from S. aureus (TIGR microbial database) to 32% with the kinases from Mycoplasma pneumoniae (24) and Mycoplasma genitalium (15). HPr kinase from B. subtilis and S. xylosus share 62% identical amino acids. The proposed signature sequence for HPr kinases (39) is present at positions 202 through 211 in the S. xylosus HPr kinase, and an ATP/GTP-binding motif is found at positions 158 to 165.
Apart from hprK, the function of three gene products in this region can be predicted based on protein similarity comparisons. First, ′uvrA, which is truncated at the 5′ end, specifies subunit A of (A)BC excinuclease, an enzyme complex needed for nucleotide excision repair (36). Second, lgt, the gene following hprK, encodes prolipoprotein diacylglyceryltransferase, an enzyme involved in lipid modification of lipoproteins (38). Third, the last gene in the sequence, trxB′, which lacks the 3′ end, encodes thioredoxin reductase, a general stress protein required to maintain the thioredoxins in the reduced state (2). The remaining genes, yvoF and yvcD, encode counterparts of putative proteins without assigned function from B. subtilis (28). Both genes were designated according to the B. subtilis homologs.
The genetic organization of the hprK region of S. xylosus (Fig. 1) is identical to that in S. aureus (TIGR microbial database) and partially conserved in other bacteria. The HPr kinase gene is followed by lgt in the sequenced mycoplasmas (15, 24) as well as in AT-rich gram-positive bacteria analyzed so far (5, 19, 27, 39). In contrast, the gene at the corresponding place in T. pallidum encodes the general PTS protein HPr (16). The gene yvoF, located immediately downstream of lgt in S. xylosus (Fig. 1), is found two positions further downstream in B. subtilis (19, 39). Notably, yvoE (hprP), located in the vicinity of hprK and originally described to encode P-Ser-HPr phosphatase in B. subtilis (19), is missing in the hprK region of S. xylosus.
HPr kinase and P-Ser-HPr phosphatase activity of the His6-HPrK enzyme.
To design a strategy for HPr kinase purification, we tested whether active HPr kinase could be obtained by expressing hprK in E. coli. Three hprK plasmids were used for this purpose. Plasmid pKIN6 contained the chromosomal S. xylosus DNA fragment from the library (Fig. 1), pKIN7 harbored a PCR-generated fragment consisting of hprK with its own promoter, and pKIN9 contained a His-tagged hprK fusion gene expressed from a strong E. coli phage T5 promoter (8). Cell extracts of the three strains and of a control without cloned hprK were prepared, and high levels of HPr kinase activity were detected in the samples from hprK-containing strains (data not shown). The highest activity was found to be mediated by pKIN9. In addition, SDS-PAGE revealed a prominent band at about 36 kDa in the pKIN9-harboring strain that was not detected in the other cell extracts (data not shown). Therefore, the enzyme with the N-terminal extension including six histidines was produced in high quantity and was apparently active. The recombinant His6-HPrK enzyme was subsequently purified as described in Materials and Methods and tested for HPr kinase activity.
As shown in Fig. 2 (lane 2), the purified enzyme efficiently converts HPr from S. aureus to a faster-migrating form. The conversion was dependent on the addition of ATP and did not occur when a mutant HPr containing an alanine instead of serine at position 46 was used (data not shown). Therefore, the protein exhibits HPr(Ser) kinase activity.
FIG. 2.

HPr kinase and P-Ser-HPr phosphatase activity of purified His6-HPrK enzyme. As substrates, a mutant form of S. aureus HPr, His-15-Ala-HPr, and the P-Ser derivative thereof were applied. HPr and phosphorylated HPr were separated on 15% nondenaturing polyacrylamide gels and visualized by Coomassie blue staining. Lane 1 and 4 (controls), 2 μg of HPr and P-Ser-HPr, respectively; lane 2, 2 μg of HPr and 0.5 μg of His6-HPrK enzyme, incubated in the presence of 5 mM ATP–50 mM Tris (pH 7.5)–50 mM NaCl–10 mM MgCl2 for 20 min at 37°C; lane 3, 2 μg of P-Ser-HPr and 0.5 μg of His6-HPrK enzyme, incubated in the presence of 1 mM NaH2PO4 instead of ATP–50 mM Tris (pH 7.5)–50 mM NaCl–10 mM MgCl2 for 20 min at 37°C. The positions of HPr, P-Ser-HPr, and His6-HPrK on the gel are indicated.
Assays of stimulation of HPr kinase activity by FDP yielded inconclusive results (data not shown). Stimulation was detectable only with low amounts (less than 0.01 μg) of the His6-HPrK enzyme. Under these conditions, no HPr kinase activity was visible without FDP. When more enzyme (0.05 μg to 0.1 μg) was used, HPr kinase activity became apparent without FDP, but it was not further enhanced by FDP addition. In these assays, about 70% of HPr was left unphosphorylated, which should have been enough substrate to detect higher kinase activity. Adding 0.5 μg or more of His6-HPrK led to the complete phosphorylation of HPr, as shown in Fig. 2. The same results were obtained with reduced ATP concentrations (2 instead of 5 mM).
In the course of this work, it was noticed that the HPr kinases from E. faecalis and B. subtilis also exhibit P-Ser-HPr phosphatase activity (27). The His6-HPrK was therefore tested for dephosphorylation of P-Ser-HPr. To that end, the enzyme was incubated with purified P-Ser-HPr without ATP but instead with 1 mM potassium phosphate. Under these conditions, P-Ser-HPr was completely dephosphorylated (Fig. 2, lane 3). Therefore, the S. xylosus HPrK enzyme shows both HPr kinase and P-Ser-HPr phosphatase activities. Subsequent experiments indicated that His6-HPrK did not require phosphate to exhibit phosphatase activity (data not shown).
HPr kinase and P-Ser-HPr phosphatase activities in S. xylosus wild-type and hprK mutant strains.
After demonstration of the in vitro activities of HPrK, our major interest was to examine the regulatory role of HPrK in vivo by comparing wild-type S. xylosus with an HPrK-deficient strain. For that purpose, hprK was inactivated by a small deletion and by insertion of the ermB resistance gene. To account for possible polar effects, ermB was integrated in two orientations allowing transcriptional readthrough from ermB into the downstream genes (14) when hprK and ermB have the same orientation (Fig. 1). For the initial characterization of the two resulting strains, S. xylosus TX66 and TX67, cell extracts were prepared and tested for HPr kinase activity. As shown in Fig. 3A, no phosphorylation occurred in extracts of the hprK mutant strains, while wild-type extracts converted most of the HPr to the faster-migrating P-Ser-HPr. Incubation of the reactions for up to 4 h resulted in completion of the reaction with the wild-type lysate but did not reveal any phosphorylation with the hprK mutant extracts (data not shown). Therefore, the HPrK enzyme appears to be the only one in S. xylosus exhibiting HPr(Ser) kinase activity.
FIG. 3.

HPr kinase and P-Ser-HPr phosphatase activity in cell extracts of S. xylosus wild-type and hprK mutant strains. (A) HPr kinase activity in cell extracts of S. xylosus wild-type C2a and the hprK mutant strains TX66 and TX67. His-15-Ala HPr from S. aureus served as the substrate. HPr forms were separated together with whole cell extracts on 15% nondenaturing polyacrylamide gels and visualized by Coomassie blue staining. The assays were carried out with 15 μg of cell extracts, 3 μg of HPr, and 5 mM ATP in 50 mM Tris (pH 7.5)–50 mM NaCl–10 mM MgCl2 for 20 min at 37°C. Lanes 1 (3 μg of HPr) and 5 (15 μg of wild-type [C2a] extract, no HPr), controls; lane 2, S. xylosus wild-type (C2a) extract; lane 3, TX66 (hprK::ermB) extract; lane 4, TX67 (hprK::ermB) extract. The positions of HPr and P-Ser-HPr are indicated. (B) P-Ser-HPr phosphatase activity in cell extracts of S. xylosus wild-type C2a and the hprK mutant strain TX66. The P-Ser derivative of S. aureus His-15-Ala-HPr served as the substrate. HPr forms were separated together with whole cell extracts on 15% nondenaturing polyacrylamide gels and visualized by Coomassie blue staining. The assays were carried out with 40 μg of cell extracts, 4 μg of P-Ser-HPr, and 1 mM NaH2PO4, in 50 mM Tris (pH 7.5)–50 mM NaCl–10 mM MgCl2 for 20 min at 37°C. Lane 1 (1 μg of HPr), lane 4 (S. xylosus wild-type extract, no P-Ser-HPr), and lane 5 (1 μg of P-Ser-HPr), controls; lane 2, S. xylosus wild-type (C2a) extract; lane 3, TX66 (hprK::ermB) extract. The positions of HPr and P-Ser-HPr are indicated.
When FDP stimulation of HPr kinase activity was analyzed, the same phenomenon as with the purified His-tagged enzyme was observed. Higher activity in the presence of FDP was observed only with low amounts of cell extract. When more cellular protein was applied, HPr kinase activity became independent of FDP addition, although the reaction was not completed, leaving about 60% HPr in the assay (data not shown).
Since purified His6-HprK exhibited kinase as well as phosphatase activity, hprK inactivation should result in the concomitant loss of both activities. Therefore, P-Ser-HPr phosphatase activity was compared in wild-type and TX66 (hprK::ermB) extracts. As shown in Fig. 3B, no phosphatase was detected in the hprK mutant strain TX66. Incubation for several hours or incubation with different buffers did not reveal any conversion of P-Ser-HPr to HPr (data not shown). Therefore, HPrK seems to be the only enzyme able to dephosphorylate P-Ser-HPr in S. xylosus.
Growth of the hprK mutant strains in the presence of glucose.
To further characterize the consequences of hprK inactivation, utilization of several sugars, glucose, sucrose, fructose, and lactose, was tested by monitoring acid production on fermentation test plates (34). Surprisingly, the mutant strains TX66 and TX67 did not grow on glucose-containing plates even after several days. The mutants grew with the other tested sugars, but they formed smaller colonies and produced more acid on lactose- and sucrose-containing plates than the wild type.
As shown in Fig. 4, the glucose-mediated growth defect was also detected in liquid culture. The hprK mutant strains TX66 and TX67 were grown in complex B medium to an OD578 of 0.25. When glucose was added to a final concentration of 25 mM, a severe growth retardation occurred (Fig. 4), whereas growth of the wild-type strain C2a was accelerated (Fig. 4). The adverse effect of glucose was concentration dependent. While the hprK mutant strain TX66 grew within 6 h to an OD578 of 1.05 with 5 mM glucose in the medium, it reached only an OD578 of 0.5 with 50 mM glucose. Addition of 25 mM glucose resulted in an intermediate OD578 of 0.8 (Fig. 4). Without extra glucose, only a slight reduction of the growth rate was observed for the hprK mutants (Fig. 4).
FIG. 4.

Growth of S. xylosus wild-type C2a and the hprK mutant strains TX66 and TX67 in the absence and presence of glucose. The strains were grown in complex B medium. Glucose to a final concentration of 25 mM was added, when the strains were grown to an OD578 of 0.25.
Since the genetic organization of the hprK region suggested cotranscription of lgt, yvoF, and yvcD with hprK, integration of ermB into hprK may exert polar effects on these genes, depending on the orientation of the resistance marker (Fig. 1). Therefore, it was necessary to determine that the glucose sensitivity is due only to hprK inactivation. To that end, pKIN11, a shuttle plasmid harboring the hprK gene with its own promoter, was introduced into the hprK mutants TX66 and TX67. Growth experiments showed that the strains lost their glucose sensitivity (data not shown). Therefore, this phenotype is indeed caused by hprK inactivation and is fully reversed by cloned hprK. In the course of these complementation experiments, we noticed that the hprK mutant strains were about 10-fold less transformable than the wild type.
Glucose uptake in the hprK mutant strain TX66.
To explain the glucose-mediated growth retardation of the HPr kinase-deficient strains, we reasoned that enhanced glucose uptake and subsequent accumulation of intermediates could be the cause. Therefore, glucose uptake was studied. Since the two mutants were phenotypically identical, only one, the hprK mutant TX66, was taken for these measurements and for the subsequent experiments. S. xylosus TX66 was grown in B medium to an OD578 of 1. The culture was split, and glucose (final concentration, 25 mM) was added to one half. After 1 h of further incubation, the cells were harvested. Under these conditions, growth retardation in the presence of glucose was not as strong as in the experiments shown in Fig. 4 but was clearly detectable (Table 1). Glucose uptake in S. xylosus TX66 was strongly enhanced when the cells were grown with glucose (Fig. 5B). In contrast, glucose transport was slightly repressed in the wild type (Fig. 5A). Since the hprK mutation should greatly diminish, and perhaps even abolish, CcpA activity, it was of interest to determine glucose uptake in the ccpA mutant strain TX154 (12). As shown in Fig. 5C, glucose transport is enhanced in TX154 by glucose in the medium, showing that CcpA, directly or indirectly, controls genes involved in glucose transport. However, the extent of glucose induction in TX154 was clearly lower than in the hprK mutant strain TX66. Therefore, HPr kinase exerts a major regulatory effect on glucose uptake in S. xylosus, but only part of the control depends on CcpA.
TABLE 1.
Catabolic enzyme activities in the S. xylosus wild-type C2a and in the hprK mutant strain TX66
| Enzyme | Growth conditionsa | C2a (wild type)
|
TX66 (hprK::ermB)
|
||
|---|---|---|---|---|---|
| Growth rateb | Enzyme activityc | Growth rate | Enzyme activity | ||
| β-Galactosidase | B + lactose | 1.1 | 106 | 1.0 | 142 |
| B + lactose + glucose | 1.1 | 7 | 0.5 | 103 | |
| B + lactose + sucrose | 1.1 | 42 | 0.6 | 155 | |
| B + lactose + fructose | 1.1 | 41 | 1.0 | 149 | |
| β-Glucuronidase | B | 1.1 | 26 | 1.0 | 38 |
| B + glucose | 1.3 | 6 | 0.6 | 44 | |
| B + sucrose | 1.2 | 8 | 1.0 | 34 | |
| B + fructose | 1.1 | 9 | 1.0 | 34 | |
| α-Glucosidase | B | 1.1 | 13 | 1.0 | 14 |
| B + glucose | 1.3 | 3 | 0.6 | 14 | |
| B + sucrose | 1.2 | 7 | 1.0 | 12 | |
| B + fructose | 1.1 | 8 | 1.0 | 13 | |
Cells were grown in complex B medium (B) to an OD578 of 1; 25 mM carbohydrate was added as indicated. After 1 h of further growth, the OD578 was determined, and the cells were harvested and disrupted with glass beads. Extracts prepared from 40 ml of cells were used for determination of the enzyme activities.
For the last hour of each culture, expressed in doublings per hour.
Determined using p-nitrophenyl-β-d-galactopyranoside (β-galactosidase), p-nitrophenyl-β-d-glucuronide (β-glucuronidase), and p-nitrophenyl-α-d-glucopyranoside (α-glucosidase) as substrates. Values of at least three independent experiments are given in nanomoles of nitrophenol released per minute per milligram of protein. Standard deviations were in the range of ±14%.
FIG. 5.

Glucose uptake in S. xylosus wild-type C2a, in the hprK mutant TX66, and in the ccpA mutant TX154. (A) Glucose uptake in S. xylosus C2a. The cells were grown in B medium. Glucose was added to a final concentration of 25 mM, when the strains were grown to an OD578 of 1. Glucose uptake was determined after 1 h of further growth, using 200 μM [14C]glucose (6.2 mCi/mmol). The values represent measurements of two cultures. Standard deviations were in the range of ±14%. (B) Glucose uptake in the hprK mutant TX66. The cells were grown in B medium. Glucose was added to a final concentration of 25 mM, when the strains were grown to an OD578 of 1. Glucose uptake was determined after 1 h of further growth, using 200 μM [14C]glucose (6.2 mCi/mmol). The values represent measurements of three cultures. Standard deviations were in the range of ±22%. (C) Glucose uptake in the ccpA mutant TX154. The cells were grown in B medium. Glucose was added to a final concentration of 25 mM, when the strains were grown to an OD578 of 1. Glucose uptake was determined after 1 h of further growth, using 200 μM [14C]glucose (6.2 mCi/mmol). The values represent measurements of three cultures. Standard deviations were in the range of ±16%.
The same results have been obtained using the nonmetabolizable 2-deoxyglucose as substrate (data not shown). Since the activity of the recently described PTS-independent glucose uptake protein GlcU is not detectable with 2-deoxyglucose (14), the observed alterations in glucose uptake should reflect PTS-mediated glucose transport.
Methylglyoxal production in the hprK mutant strain TX66.
In E. coli, excessive carbon intake may lead to methylglyoxal production and excretion, when the carbon source can act as a direct supply of the glycolytic intermediate dihydroxyacetone phosphate (13, 25). From dihydroxyacetone phosphate, the toxic electrophile methylglyoxal is produced by methylglyoxal synthase (49), resulting in growth arrest and cell death. Since the hprK mutant TX66 showed enhanced glucose uptake, methylglyoxal accumulation in the medium was measured and compared with that of the wild type. Under the growth conditions described above to measure glucose transport, 0.12 mM methylglyoxal was found in the medium of the glucose-containing culture of the hprK mutant TX66. No methylglyoxal was excreted from the wild type under these growth conditions. When glycerol was added as carbon source, 0.29 mM methylglyoxal was detected in the TX66 culture, and even 0.08 mM was found in the wild-type culture.
Carbon catabolite repression in the hprK mutant strain TX66.
Inactivation of the HPr kinase gene should result in a severe reduction of CR, perhaps even in a complete loss of regulation. Therefore, we measured three catabolic enzyme activities that were shown in previous studies to be subject to CR (12, 14, 50). Two of these activities, α-glucosidase and β-glucuronidase, are expressed constitutively in B medium, whereas efficient β-galactosidase expression requires induction by lactose (3). The cells were grown under the same conditions as described above for the determination of glucose transport. As observed in previous experiments, glucose in the medium impaired growth of the hprK mutant strain (Table 1). When glucose and lactose were added to test repression of β-galactosidase activity, the adverse effect was most pronounced. Concomitant addition of sucrose and lactose also resulted in retardation, although sucrose alone had no effect (Table 1). On the other hand, fructose did not alter the growth behavior of the hprK mutant.
Repression of α-glucosidase and β-glucuronidase activities by the three tested sugars is completely lost in the hprK mutant strain TX66 (Table 1). Repression of β-galactosidase activity by sucrose and fructose was also abolished, but the 15-fold glucose-mediated repression, found in the wild type, was diminished to a 1.5-fold reduction. This slight effect may well be caused by the reduced growth rate of the mutant under these conditions. Therefore, it appears that CR is virtually lost upon hprK inactivation.
Transcriptional start point of hprK.
To determine the transcriptional start point of hprK, RNA was isolated from wild-type cells grown in B medium with (25 mM) or without glucose. Primer extension reactions were carried out with an hprK-specific primer and separated on denaturing gels (Fig. 6). The start site of the hprK promoter was localized 43 bp in front of the hprK start codon (Fig. 1). Comparison of the amount of reverse transcript obtained with RNA from cells grown in B medium with or without glucose revealed no significant difference (Fig. 6). Thus, transcription of the HPr kinase gene is not altered by the presence of glucose in the culture medium. Constitutive expression of hprK would support the concept that modulation of HPr kinase activity is important for CR rather than control of hprK expression.
FIG. 6.
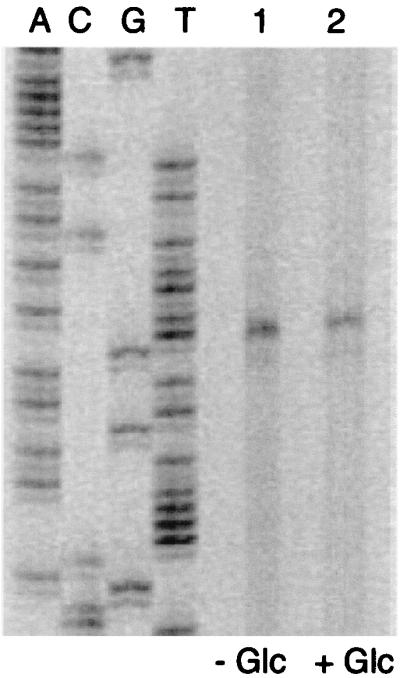
Primer extension analysis of hprK transcription. RNA was prepared from S. xylosus C2a grown without and with glucose. Primer extension products were separated along with DNA sequencing reactions on an 8% polyacrylamide-urea gel. Lane 1, primer extension reaction with 15 μg of RNA from cells grown without glucose; lane 2, primer extension reaction with 15 μg of RNA from cells grown with 25 mM glucose.
DISCUSSION
The gene encoding HPr kinase from S. xylosus has been identified. The HPrK protein phosphorylates HPr at a serine residue in an ATP-dependent manner, but it also hydrolyzes P-Ser-HPr. Thus, the S. xylosus enzyme constitutes HPr kinase as well as P-Ser-HPr phosphatase, consistent with the findings for HPrK from E. faecalis and B. subtilis (27). In B. subtilis, a second enzyme, product of the yvoE gene, which is located in the putative hprK operon, was found to dephosphorylate P-Ser-HPr (19), albeit at a lower rate than the HPrK enzyme (27). In extracts from HPrK-deficient S. xylosus cells (TX66), no P-Ser-HPr phosphatase activity could be detected. In addition, a yvoE homolog is not present in the vicinity of hprK. Thus, removal of the phosphate group from P-Ser-HPr in S. xylosus appears to depend on the bifunctional HPrK enzyme.
A key issue in understanding CR in gram-positive bacteria is the conditions activating HPr kinase activity according to the availability of carbon sources. Conflicting results have been reported regarding the stimulatory action of FDP. While the kinase activity of the enzyme from B. subtilis was stimulated by FDP (19, 39), the activity of the enzymes from E. faecalis and S. salivarius were not affected (5, 27). In our hands, kinase activity of HPrK was enhanced with FDP only at low concentrations of the enzyme. Identification of the phosphatase activity of the enzyme clearly complicates the interpretation of the kinase experiments and hinders comparisons. Different purification and assay conditions may have resulted in enzymes with higher specific kinase than phosphatase activity, and vice versa. Clearly, more work is required to elucidate the mechanism of HPrK activation.
At present there is no information on the amount of HPrK enzyme present in bacterial cells. According to our primer extension experiments (Fig. 6), the S. xylosus hprK promoter is relatively weak, suggesting that the kinase experiments using low levels of the enzyme may reflect the in vivo situation. Therefore, we still favor the idea that FDP, perhaps together with high ATP levels, shifts HPrK activity toward the kinase function and thereby triggers CR in gram-positive bacteria. However, the complex control of the activities of kinases and phosphatases in the B. subtilis phosphorelay by various proteins (35) demonstrates that alternative ways to adjust HPrK kinase and phosphatase activities are conceivable.
Inactivation of the HPr kinase gene led to a rather surprising phenotype, glucose sensitivity. Glucose in the medium severely impaired growth of the hprK mutant strain in a concentration-dependent manner. As S. xylosus can also grow at the expense of amino acids as carbon source in complex B medium, the presence of 25 mM glucose probably constitutes a situation of excess carbohydrate. Concomitantly with retardation of growth, glucose uptake was strongly enhanced. It appeared to be inducible by glucose in the medium. Since the initial uptake was relatively fast in the mutant, the results presented in Fig. 5B may even underestimate stimulation, which is at least four- to fivefold compared with the wild type. This enhanced glucose influx may eventually result in growth inhibition, if incoming glucose exceeds metabolic capacities of the cell. Under these circumstances, deleterious levels of metabolites, e.g., glucose-6-phosphate, may be accumulated and arrest growth. Enhanced methylglyoxal production in the hprK mutant TX66 does not appear to be the cause for growth retardation. Even higher levels of methylglyoxal were detected with glycerol, but no growth inhibition occurred. Methylglyoxal production apparently indicates unbalanced metabolism, substantiating the role of HPr kinase in cell physiology.
In contrast to the hprK mutant, glucose uptake was not inducible in the wild type when glucose was present in the medium (Fig. 5A). Unexpectedly, prevention of induction was due to CcpA (Fig. 5C), a function not previously attributed to this regulator (12, 22). However, glucose transport in the ccpA mutant did not reach the levels observed in the hprK mutant, and the ccpA mutant did not show glucose sensitivity (12). Apparently, HPr kinase still acts negatively on glucose transport in the absence of a functional CcpA.
A conceivable target for this negative regulation would be the HPr-mediated phosphotransfer activity of the PTS (43, 52). It has been shown in vitro that P-Ser-HPr is a poor substrate for enzyme I (10). Consequently, ATP-dependent phosphorylation of HPr at Ser-46 should lower PEP-dependent phosphate transfer to the PTS permeases, resulting in reduced sugar transport and phosphorylation. In addition, expression of PTS permease genes, which are controlled by antitermination, would be diminished, provided the antiterminators require PTS-dependent phosphorylation to be active (31). Experiments are under way to isolate the general PTS genes as well as the glucose-specific PTS permease gene(s) to examine their regulation.
In contrast to S. xylosus, glucose-related growth retardation in HPr kinase-deficient B. subtilis mutants have not been reported (19, 39), while a ptsH1 crh double mutant, preventing phosphorylation of HPr and Crh by HPr kinase, exhibited growth defects (54). In these studies, differently composed minimal media were used, indicating that not only the sugar content of the medium may influence the phenotypic consequences of HPr kinase deficiency. It will be important to develop a suitable minimal medium for S. xylosus and examine growth of the HPr kinase mutant under various conditions.
Another apparent difference between the S. xylosus and B. subtilis HPr kinase-deficient strains concerns the uptake of glucose. While the B. subtilis mutant transports slightly less glucose than the wild type (39), glucose uptake is strongly enhanced in the S. xylosus mutant. In addition, our results differ from earlier studies in S. aureus, where no influence of Ser-HPr phosphorylation on glucose uptake was detected (41). In these experiments, HPr mutants of B. subtilis have been tested in the heterologous host, which may explain the discrepancy. These conflicting findings reflect either differences in the experimental design, e.g., growth conditions, or indeed variations in the physiological role of HPr kinase in these bacteria. In any case, it will be interesting to construct and analyze HPr kinase mutants in a wider range of organisms.
Inactivation of the hprK gene in S. xylosus resulted in a complete relief of three catabolic enzyme activities from CR, which is in accordance with the B. subtilis HPr kinase mutant (19, 39). In previous studies, repression of two of these activities, α-glucosidase and β-glucuronidase, was lost upon ccpA inactivation, while repression of the third, β-galactosidase, was only diminished (12). The failure of CcpA to control α-glucosidase and β-glucuronidase expression in the hprK mutant strongly suggests that P-Ser-HPr is absolutely required for CcpA activity in S. xylosus. This finding does not necessarily rule out the participation of CcpA corepressors other than P-Ser-HPr (20, 26, 33), as CcpA may be able to bind more than one effector (26).
Since CcpA-independent CR of β-galactosidase expression is also abolished in the hprK mutant, additional mechanisms of CR also rely on HPr kinase. As the nature of the CcpA-independent control of β-galactosidase expression is not yet clear, it remains to be determined which regulatory process is affected by HPr kinase.
In conclusion, inactivation of the HPr kinase gene of S. xylosus led to a profound perturbation of physiology, enhanced glucose uptake, glucose sensitivity, and loss of CR. For future work, it will be important to define exactly how HPr kinase/phosphatase senses the metabolic state of the cell and how this information is transmitted to control metabolic fluxes.
ACKNOWLEDGMENTS
We thank W. Hengstenberg for the generous gift of His-15-Ala HPr from S. aureus. We also thank W. Hillen, F. Titgemeyer, J. Deutscher, and W. Hengstenberg for communicating results prior to publication, and we thank F. Götz, in whose laboratory this work was carried out, for his support.
The work was supported by the Deutsche Forschungsgemeinschaft by an individual grant (BR947/3-1) and within the priority program, Molecular Analysis of Regulatory Networks in Bacteria (BR947/4-1).
REFERENCES
- 1.Angell S, Lewis C G, Buttner M J, Bibb M J. Glucose repression in Streptomyces coelicolor A3(2): a likely regulatory role for glucose kinase. Mol Gen Genet. 1994;244:134–143. doi: 10.1007/BF00283514. [DOI] [PubMed] [Google Scholar]
- 2.Aslund F, Beckwith J. The thioredoxin superfamily: redundancy, specificity, and gray-area genomics. J Bacteriol. 1999;181:1375–1379. doi: 10.1128/jb.181.5.1375-1379.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bassias J, Brückner R. Regulation of lactose utilization genes in Staphylococcus xylosus. J Bacteriol. 1998;180:2273–2279. doi: 10.1128/jb.180.9.2273-2279.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:246–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 5.Brochu D, Vadeboncoeur C. The HPr(Ser) kinase of Streptococcus salivarius: purification, properties, and cloning of the hprK gene. J Bacteriol. 1999;181:709–717. doi: 10.1128/jb.181.3.709-717.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brückner R. Gene replacement in Staphylococcus carnosus and Staphylococcus xylosus. FEMS Microbiol Lett. 1997;151:1–8. doi: 10.1111/j.1574-6968.1997.tb10387.x. [DOI] [PubMed] [Google Scholar]
- 7.Brückner R, Wagner E, Götz F. Characterization of a sucrase gene from Staphylococcus xylosus. J Bacteriol. 1993;175:851–857. doi: 10.1128/jb.175.3.851-857.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bujard H, Gentz R, Lanzer M, Stüber D, Müller D, Ibrahimi M, Häuptle M T, Dobberstein B. A T5 promoter based transcription-translation system for the analysis of proteins in vivo and in vitro. Methods Enzymol. 1987;155:416–433. doi: 10.1016/0076-6879(87)55028-5. [DOI] [PubMed] [Google Scholar]
- 9.Cooper R A. Methylglyoxal synthase. Methods Enzymol. 1975;41:502–508. doi: 10.1016/s0076-6879(75)41106-5. [DOI] [PubMed] [Google Scholar]
- 10.Deutscher J, Kessler U, Hengstenberg W. Streptococcal phosphoenolpyruvate:sugar phosphotransferase system: purification and characterization of a phosphoprotein phosphatase which hydrolyzes the phosphoryl bond in seryl-phosphorylated histidine-containing protein. J Bacteriol. 1985;163:1203–1209. doi: 10.1128/jb.163.3.1203-1209.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deutscher J, Saier M H., Jr ATP-dependent protein kinase-catalyzed phosphorylation of a seryl residue in HPr, a phosphate carrier protein of the phosphotransferase system in Streptococcus pyogenes. Proc Natl Acad Sci USA. 1983;80:6790–6794. doi: 10.1073/pnas.80.22.6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Egeter O, Brückner R. Catabolite repression mediated by the catabolite control protein CcpA in Staphylococcus xylosus. Mol Microbiol. 1996;21:739–749. doi: 10.1046/j.1365-2958.1996.301398.x. [DOI] [PubMed] [Google Scholar]
- 13.Ferguson G P, Tötemeyer S, MacLean M J, Booth I R. Methylglyoxal production in bacteria: suicide or survival? Arch Microbiol. 1998;170:209–219. doi: 10.1007/s002030050635. [DOI] [PubMed] [Google Scholar]
- 14.Fiegler H, Bassias J, Jankovic I, Brückner R. Identification of a gene in Staphylococcus xylosus encoding a novel glucose uptake protein. J Bacteriol. 1999;181:4929–4936. doi: 10.1128/jb.181.16.4929-4936.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fraser C M, Gocayne J D, White O, Adams M D, Clayton R A, Fleischmann R D, Bult C J, Kerlavage A R, Sutton G, Kelley J M, et al. The minimal gene complement of Mycoplasma genitalium. Science. 1995;270:397–403. doi: 10.1126/science.270.5235.397. [DOI] [PubMed] [Google Scholar]
- 16.Fraser C M, Norris S J, Weinstock G M, White O, Sutton G G, Dodson R, Gwinn M, Hickey E K, Clayton R, Ketchum K A, Sodergren E, Hardham J M, McLeod M P, Salzberg S, Peterson J, Khalak H, Richardson D, Howell J K, Chidambaram M, Utterback T, McDonald L, Artiach P, Bowman C, Cotton M D, Venter J C, et al. Complete genome sequence of Treponema pallidum, the syphilis spirochete. Science. 1998;281:375–388. doi: 10.1126/science.281.5375.375. [DOI] [PubMed] [Google Scholar]
- 17.Galinier A, Deutscher J, Martin-Verstraete I. Phosphorylation of either Crh or HPr mediates binding of CcpA to the Bacillus subtilis xyn cre and catabolite repression of the xyn operon. J Mol Biol. 1999;286:307–314. doi: 10.1006/jmbi.1998.2492. [DOI] [PubMed] [Google Scholar]
- 18.Galinier A, Haiech J, Kilhoffer M C, Jaquinod M, Stülke J, Deutscher J, Martin-Verstraete I. The Bacillus subtilis crh gene encodes a HPr-like protein involved in carbon catabolite repression. Proc Natl Acad Sci USA. 1997;94:8439–8444. doi: 10.1073/pnas.94.16.8439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galinier A, Kravanja M, Engelmann R, Hengstenberg W, Kilhoffer M C, Deutscher J, Haiech J. New protein kinase and protein phosphatase families mediate signal transduction in bacterial catabolite repression. Proc Natl Acad Sci USA. 1998;95:1823–1828. doi: 10.1073/pnas.95.4.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gösseringer R, Küster E, Galinier A, Deutscher J, Hillen W. Cooperative and non-cooperative DNA binding modes of catabolite control protein CcpA from Bacillus megaterium result from sensing two different signals. J Mol Biol. 1997;266:665–676. doi: 10.1006/jmbi.1996.0820. [DOI] [PubMed] [Google Scholar]
- 21.Götz F, Zabielski J, Philipson L, Lindberg M. DNA homology between the arsenate resistance plasmid pSX267 from Staphylococcus xylosus and the penicillinase plasmid pI258 from Staphylococcus aureus. Plasmid. 1983;9:126–137. doi: 10.1016/0147-619x(83)90015-x. [DOI] [PubMed] [Google Scholar]
- 22.Henkin T M. The role of CcpA transcriptional regulator in carbon metabolism in Bacillus subtilis. FEMS Microbiol Lett. 1996;135:9–15. doi: 10.1111/j.1574-6968.1996.tb07959.x. [DOI] [PubMed] [Google Scholar]
- 23.Henkin T M, Grundy F J, Nicholson W L, Chambliss G H. Catabolite repression of alpha-amylase gene expression in Bacillus subtilis involves a trans-acting gene product homologous to the Escherichia coli lacl and galR repressors. Mol Microbiol. 1991;5:575–584. doi: 10.1111/j.1365-2958.1991.tb00728.x. [DOI] [PubMed] [Google Scholar]
- 24.Himmelreich R, Hilbert H, Plagens H, Pirkl E, Li B C, Herrmann R. Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res. 1996;24:4420–4449. doi: 10.1093/nar/24.22.4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kadner R J, Murphy G P, Stephens C M. Two mechanisms for growth inhibition by elevated transport of sugar phosphates in Escherichia coli. J Gen Microbiol. 1992;138:2007–2014. doi: 10.1099/00221287-138-10-2007. [DOI] [PubMed] [Google Scholar]
- 26.Kim J H, Voskuil M I, Chambliss G H. NADP, corepressor for the Bacillus catabolite control protein ccpA. Proc Natl Acad Sci USA. 1998;95:9590–9595. doi: 10.1073/pnas.95.16.9590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kravanja M, Engelmann R, Dossonnet V, Blüggel M, Meyer H E, Frank R, Galinier A, Deitscher J, Schnell N, Hengstenberg W. The hprK gene of Enterococcus faecalis encodes a novel bifunctional enzyme: the HPr kinase/phosphatase. Mol Microbiol. 1999;31:59–66. doi: 10.1046/j.1365-2958.1999.01146.x. [DOI] [PubMed] [Google Scholar]
- 28.Kunst F, Ogasawara N, Moszer I, Albertini A M, Alloni G, Azevedo V, Bertero M G, Bessieres P, Bolotin A, Borchert S, Borriss R, Boursier L, Brans A, Braun M, Brignell S C, Bron S, Brouillet S, Bruschi C V, Caldwell B, Capuano V, Carter N M, Choi S K, Codani J J, Connerton I F, Danchin A, et al. The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature. 1997;390:249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
- 29.Kwakman J H J M, Postma P W. Glucose kinase has a regulatory role in carbon catabolite repression in Streptomyces coelicolor. J Bacteriol. 1994;176:2694–2698. doi: 10.1128/jb.176.9.2694-2698.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 31.Lindner C, Galinier A, Hecker M, Deutscher J. Regulation of the activity of the Bacillus subtilis antiterminator LicT by multiple PEP-dependent, enzyme I- and HPr-catalysed phosphorylation. Mol Microbiol. 1999;31:995–1006. doi: 10.1046/j.1365-2958.1999.01262.x. [DOI] [PubMed] [Google Scholar]
- 32.Martin-Verstraete I, Deutscher J, Galinier A. Phosphorylation of HPr and Crh by HprK, early steps in the catabolite repression signalling pathway for the Bacillus subtilis levanase operon. J Bacteriol. 1999;181:2966–2969. doi: 10.1128/jb.181.9.2966-2969.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miwa Y, Nagura K, Eguchi S, Fukuda H, Deutscher J, Fujita Y. Catabolite repression of the Bacillus subtilis gnt operon exerted by two catabolite-responsive elements. Mol Microbiol. 1997;23:1203–1213. doi: 10.1046/j.1365-2958.1997.2921662.x. [DOI] [PubMed] [Google Scholar]
- 34.Morse M L, Alire M L. An agar medium indicating acid production. J Bacteriol. 1958;76:270–271. doi: 10.1128/jb.76.3.270-271.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perego M. Kinase-phosphatase competition regulates Bacillus subtilis development. Trends Microbiol. 1998;6:366–370. doi: 10.1016/s0966-842x(98)01350-x. [DOI] [PubMed] [Google Scholar]
- 36.Petit C, Sancar A. Nucleotide excision repair: from E. coli to man. Biochimie. 1999;81:15–25. doi: 10.1016/s0300-9084(99)80034-0. [DOI] [PubMed] [Google Scholar]
- 37.Postma P W, Lengeler J W, Jacobson G R. Phosphoenolpyruvate: carbohydrate phosphotransferase systems of bacteria. Microbiol Rev. 1993;6:543–594. doi: 10.1128/mr.57.3.543-594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qi H Y, Sankaran K, Gan K, Wu H C. Structure-function relationship of bacterial prolipoprotein diacylglyceryl transferase: functionally significant conserved regions. J Bacteriol. 1995;177:6820–6824. doi: 10.1128/jb.177.23.6820-6824.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reizer J, Hoischen C, Titgemeyer F, Rivolta C, Rabus R, Stülke J, Karamata D, Saier M H, Jr, Hillen W. A novel protein kinase that controls carbon catabolite repression in bacteria. Mol Microbiol. 1998;27:1157–1169. doi: 10.1046/j.1365-2958.1998.00747.x. [DOI] [PubMed] [Google Scholar]
- 40.Reizer J, Romano A H, Deutscher J. The role of phosphorylation of HPr, a phosphocarrier protein of the phosphotransferase system, in the regulation of carbon metabolism in gram-positive bacteria. J Cell Biochem. 1993;51:19–24. doi: 10.1002/jcb.240510105. [DOI] [PubMed] [Google Scholar]
- 41.Reizer J, Sutrina S L, Saier M H, Stewart G C, Peterkofsky A, Reddy P. Mechanistic and physiological consequences of HPr(ser) phosphorylation on the activities of the phosphoenolpyruvate:sugar phosphotransferase system in gram-positive bacteria: studies with site-specific mutants of HPr. EMBO J. 1989;8:2111–2120. doi: 10.1002/j.1460-2075.1989.tb03620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saier M H., Jr Cyclic AMP-independent catabolite repression in bacteria. FEMS Microbiol Lett. 1996;138:97–103. doi: 10.1111/j.1574-6968.1996.tb08141.x. [DOI] [PubMed] [Google Scholar]
- 43.Saier M H, Jr, Chauvaux S, Cook G M, Deutscher J, Paulsen I T, Reizer J, Ye J J. Catabolite repression and inducer control in Gram-positive bacteria. Microbiology. 1996;142:217–230. doi: 10.1099/13500872-142-2-217. [DOI] [PubMed] [Google Scholar]
- 44.Saier M H, Jr, Chauvaux S, Deutscher J, Reizer J, Ye J J. Protein phosphorylation and regulation of carbon metabolism in gram-negative versus gram-positive bacteria. Trends Biochem Sci. 1995;20:267–271. doi: 10.1016/s0968-0004(00)89041-6. [DOI] [PubMed] [Google Scholar]
- 45.Saier M H, Jr, Crasnier M. Inducer exclusion and the regulation of sugar transport. Res Microbiol. 1996;147:482–489. doi: 10.1016/s0923-2508(96)90150-3. [DOI] [PubMed] [Google Scholar]
- 46.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 47.Schleifer K H, Kloos W E. Isolation and characterization from human skin. I. Amended descriptions of Staphylococcus epidermidis and Staphylococcus saprophyticus and description of three new species: Staphylococcus cohnii, Staphylococcus haemolyticus and Staphylococcus xylosus. J Syst Bacteriol. 1975;174:3042–3048. [Google Scholar]
- 48.Sutcliffe J G. Complete sequence of Escherichia coli plasmid pBR322. Cold Spring Harbor Symp Quant Biol. 1979;43:77–90. doi: 10.1101/sqb.1979.043.01.013. [DOI] [PubMed] [Google Scholar]
- 49.Tötemeyer S, Booth N A, Nichols W W, Dunbar B, Booth I R. From famine to feast: the role of methylglyoxal production in Escherichia coli. Mol Microbiol. 1998;27:553–562. doi: 10.1046/j.1365-2958.1998.00700.x. [DOI] [PubMed] [Google Scholar]
- 50.Wagner E, Marcandier S, Egeter O, Deutscher J, Götz F, Brückner R. Glucose kinase-dependent catabolite repression in Staphylococcus xylosus. J Bacteriol. 1995;177:6144–6152. doi: 10.1128/jb.177.21.6144-6152.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yanisch-Perron P, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 1985;33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- 52.Ye J J, Reizer J, Cui X, Saier M H., Jr Inhibition of the phosphoenolpyruvate:lactose phosphotransferase system and activation of a cytoplasmic sugar-phosphate phosphatase in Lactococcus lactis by ATP-dependent metabolite-activated phosphorylation of serine 46 in the phosphocarrier protein HPr. J Biol Chem. 1994;269:11837–11844. [PubMed] [Google Scholar]
- 53.Zalieckas J M, Wray L V, Jr, Fisher S H. trans-acting factors affecting carbon catabolite repression of the hut operon in Bacillus subtilis. J Bacteriol. 1999;181:2883–2888. doi: 10.1128/jb.181.9.2883-2888.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]