

Abstract
The aryl hydrocarbon receptor (AHR) is a ligand-dependent transcription factor that binds DNA and regulates genes in response to halogenated and polycyclic aromatic hydrocarbons. AHR also regulates the development and function of the liver and the immune system. In the canonical pathway, AHR binds a consensus DNA sequence, termed the xenobiotic response element (XRE), recruits protein coregulators, and regulates target gene expression. Emerging evidence suggests that AHR may regulate gene expression via an additional pathway, by binding to a non-consensus DNA sequence termed the non-consensus XRE (NC-XRE). The prevalence of NC-XRE motifs in the genome is not known. Studies using chromatin immunoprecipitation and reporter genes provide indirect evidence of AHR-NC-XRE interactions, but direct evidence for an AHR-NCXRE interaction that regulates transcription in a natural genomic context is lacking. Here, we analyzed AHR binding to NC-XRE DNA on a genome-wide scale in mouse liver. We integrated ChIP-seq and RNA-seq data and identified putative AHR target genes with NC-XRE motifs in regulatory regions. We also performed functional genomics at a single locus, the mouse Serpine1 gene. Deleting NC-XRE motifs from the Serpine1 promoter reduced the upregulation of Serpine1 by TCDD, an AHR ligand. We conclude that AHR upregulates Serpine1 via NC-XRE DNA. NC-XRE motifs are prevalent throughout regions of the genome where AHR binds. Taken together, our results suggest that AHR regulates genes via NC-XRE motifs. Our results will also improve our ability to identify AHR target genes and their physiologic relevance.
INTRODUCTION
The toxic effects of secondhand cigarette smoke, fossil fuels, and byproducts of industrial combustion are caused by halogenated and polycyclic aromatic hydrocarbons such as TCDD. Understanding the signaling pathways by which such compounds cause toxicity is crucial for predicting tolerable exposure levels and for reversing the effects of adverse exposure. Aromatic hydrocarbons activate aryl hydrocarbon receptors (AHR), ligand-dependent transcription factors that recruit protein coregulators to DNA and directly regulate gene expression (Denison et al., 2011). AHR forms a dimer with aryl hydrocarbon receptor nuclear translocator protein (ARNT1, also known as HIF1B) to regulate transcription (Hankinson, 2005; Reyes et al., 1992). The AHR-ARNT1 complex binds a consensus DNA sequence (GCGTG) termed the xenobiotic response element (XRE) (Denison et al., 1988; Yao and Denison, 1992).
Recent evidence complicates these findings. AHR may regulate gene expression by binding to non-consensus XRE sequences. A chromatin immunoprecipitation study found that half of TCDD-AHR complexes in mouse liver were bound to DNA lacking a consensus XRE (Dere et al., 2011). Exploring the genome more closely, Huang and colleagues identified a non-consensus DNA sequence to which AHR binds (Huang and Elferink, 2012). Chromatin immunoprecipitation, gel shift and reporter assays support the hypothesis that AHR binds a non-consensus XRE DNA sequence (nucleotides GGGA, referred to as NC-XRE motif) (Huang and Elferink, 2012; Jackson et al., 2014; Joshi et al., 2015; Wilson et al., 2013). Such studies were limited because they did not analyze AHR-NCXRE interactions on a genome-wide scale. Additionally, chromatin immunoprecipitation identifies where AHR binds to DNA does not reveal whether transcription actually occurs. Gel shift and reporter gene assays sometimes fail to reflect what occurs in a natural genomic context. Direct evidence for an AHR-NCXRE interaction that regulates transcription in a natural genomic context is lacking.
Here, we sought to analyze AHR binding to non-canonical DNA elements on a genome-wide scale and, in the case of a single gene, directly test whether NC-XRE motifs are required for AHR-dependent target gene expression.
RESULTS
Frequency of NC-XRE motifs
To determine the distribution of AHR binding to DNA following TCDD exposure, we analyzed published AHR ChIP-seq data from livers of C57BL/6 male and female mice (28 days old) following 2 hour exposure to 30 μg/kg TCDD (GEO accession #GSE97634, GSE97636) (Fader et al., 2017; Fader et al., 2019). This data was previously assayed for AHR binding to consensus XRE DNA [GCGTG] but was not interrogated for AHR binding to non-consensus DNA such as NC-XRE [GGGA] or RelBAHRE [GGGTGCAT]. We found that, on average, 30% of peaks (AHR binding to DNA) contained a NC-XRE motif without a nearby XRE. 9% of peaks contained an XRE motif without a nearby NC-XRE, while 51% of peaks contained an XRE and NC-XRE (Fig 1A, C). Fewer than 1% of peaks contained RelBAHRE.
Figure 1. Frequency of NC-XRE motifs in DNA bound by AHR.

(A) Following TCDD exposure, we analyzed mouse liver AHR ChIP-seq peaks for the presence of known AHR motifs: canonical xenobiotic response element (XRE), non-canonical xenobiotic response element (NC-XRE) or RelB AHR response element (RelBAHRE). Pie-chart shows the percent of AHR binding sites (AHR ChIP peaks) containing one or more of the motifs indicated. A majority of AHR peaks contain canonical and non-canonical xenobiotic response element sequences (XRE+NC-XRE). About a quarter of AHR peaks contain NC-XRE motifs alone. (B) Percent of AHR peaks containing XRE or NC-XRE sequences that are associated with promoters, enhancers or indicated genomic feature. A higher ratio of XRE+NC-XRE sites overlap with promoters compared to NC-XRE sites alone. (C) Venn diagram showing number of AHR peaks containing XRE, NCXRE, RelBAHRE alone or in combination. No AHR peaks contain RelBAHRE alone, two AHR peaks contain RelBAHRE + XRE. (D) Likelihood of finding XRE, NC-XRE or RelBAHRE motifs in AHR ChIP peaks versus in a random region of DNA. Table shows number of AHR peaks containing at least one XRE, NC-XRE or RelBAHRE motif. P value derived from Fisher’s exact test.
We wondered whether XRE and NC-XRE peaks were distributed equally among promoter and enhancer DNA. We observed more peaks at promoter regions (within 3 kb of transcription start sites) containing XRE motifs alone or XRE+NC-XRE motifs together compared to promoter regions containing NC-XRE alone (Fig 1B). This suggests that AHR binding to XRE is stronger in promoter regions, whereas AHR binding to NC-XRE is stronger in enhancer regions.
XRE, NC-XRE and RelBAHRE motifs are different lengths, which affects the frequency with which they are found in the genome. We assessed the likelihood of finding XRE, NC-XRE or RelBAHRE motifs in AHR ChIP peaks versus in a random region of DNA. NC-XRE and XRE motifs are statistically significantly enriched in AHR ChIP peaks, whereas RelBAHRE motifs are not (Fig 1D). Because of the low number of RelBAHRE in AHR peaks, we focus on the XRE and NC-XRE motifs in this study.
Repeated NC-XRE motifs associated with AHR target genes
The preceding analysis examined presence of NC-XRE motifs in all regions where AHR was bound to DNA. However, transcription factors like AHR may bind DNA without affecting gene expression. Therefore, we sought to identify AHR ChIP-seq peaks associated with AHR target genes. To identify AHR target genes, we integrated the AHR ChIP-seq data with published RNA-seq data from mouse liver (Fader et al., 2017; Nault et al., 2015; Nault et al., 2018). To maximize identification of AHR target genes, we included RNA-seq data from mouse livers treated with a variety of doses of TCDD for various durations of exposure (Table 1). We define AHR target genes as differentially expressed genes (RNA-seq, fold difference in TCDD vs vehicle exceeding 1.5x to cover both up- and downregulation, FDR<5%) containing AHR binding site (ChIP peak) within 10 kb of a gene body. The 10 kb parameter allows us to include promoter and gene proximal regulatory DNA peaks, important because our analysis of ChIP-seq data suggested that AHR binds NC-XRE at enhancers. Of the differentially expressed genes in TCDD vs vehicle, 520 had AHR binding peaks at XRE, NC-XRE or XRE+NC-XRE motifs within 10 kb of a gene body. 28 of these genes had AHR binding peaks at NC-XRE but not at XRE or XRE+NC-XRE motifs.
Table 1.
Datasets analyzed in this manuscript
| GEO accession | PMID | Assay | Sex | Conditions |
|---|---|---|---|---|
| GSE97634 | 28213091, 31015483 | ChIP-seq | male C57BL/6 liver | exposed to 30 ug/kg TCDD via oral gavage for 2 hours, n=5 mice |
| GSE97636 | 26582802 | ChIP-seq | female C57BL/6 liver | exposed to 30 ug/kg TCDD via oral gavage for 2 hours, n=5 mice |
| GSE62902 | 25958198 | RNA-seq | female C57BL/6 liver | exposed to 0.01, 0.03, 0.1, 0.3, 1, 3, 10, or 30 ug/kg TCDD or sesame oil vehicle via oral gavage, every 4 days for 28 days |
| GSE87519 | 28213091 | RNA-seq | male C57BL/6 liver | exposed to 0.01, 0.03, 0.1, 0.3, 1, 3, 10, or 30 ug/kg TCDD or sesame oil vehicle via oral gavage, every 4 days for 28 days. At 7, 15, and 23d after the first dose, mice (vehicle and 30 μg/kg TCDD groups) were fasted for 6 hours (access to water but not food). At 22d after the first dose, oral glucose tolerance tests (OGTT) were performed (vehicle and 30 μg/kg TCDD groups; fasted for 6h). Briefly, at time 0 minutes (min) animals were orally gavaged with 2 g/kg glucose in a 25% solution and tail blood glucose was measured at 0, 5, 15, 25, 30, 60, and 120 min. At 26d after the first dose, mice (vehicle and 30 μg/kg TCDD groups) were transferred to Innocages lacking bedding for 2h, with access to water but not food. |
| GSE109863 | 29752288 | RNA-seq | male C57BL/6 liver | On postnatal day 28 mice were orally gavaged with sesame oil vehicle or 30 μg/kg TCDD for 2, 4, 8, 12, 24, 72, and 168 hours |
The preceding analysis is for any single instance of XRE or NC-XRE DNA sequence. For some transcription factors, repeated DNA binding sites produce a more robust response (Klein-Hitpass et al., 1988). We sought to examine the association between repeated runs of XRE or NCXRE motifs and target gene expression. Fig 2A shows the relative frequency of AHR target genes with AHR peaks containing between 3 and 10 NC-XRE motifs separated by up to 50 basepairs (bp), compared to target genes found near similar patterns of XRE peaks. 24% of AHR peaks contain 2 or more NC-XRE motifs separated by 25 bp or less (Fig 2B). AHR peaks containing 5 NC-XRE motifs separated by 25 bp or less were found proximal to 82 AHR target genes. Compared to repeated XRE motifs, runs of 5 or more NC-XREs are enriched at AHR binding sites proximal to transcription start sites (H3K4me, H3K27ac, Fig 2C). This suggests that NCXREs may be more potent when they occur in tandem repeats. The top differentially regulated genes with 5 or more NCXRE motifs are shown in Fig 2D. As validation, the known AHR target gene Serpine1 (aka Pai1) contains 5 NCXRE motifs in the putative promoter, consistent with results that AHR upregulates Serpine1 via NC-XRE DNA (Huang and Elferink, 2012). AHR binding was also detected at NC-XRE DNA near genes like Nqo1, a canonical AHR target gene thought to be regulated by XRE motifs (Yeager et al., 2009). It is possible that NC-XRE DNA also contributes to the regulation of NQO1 expression.
Figure 2. Repeated NC-XRE motifs associated with AHR target genes.

(A) Relative frequency of putative AHR target genes within 10kb of AHR ChIP peaks containing 3–10 NC-XRE motifs with 1–50 basepair (bp) spacers, compared to AHR peaks containing 3–10 XRE motifs with similar spacing. AHR ChIP peaks containing 5 NC-XRE motifs separated by 25 basepairs or less were found within 10 kb of 82 target genes. There are 4.9 fold more AHR target genes associated with AHR ChIP peaks containing 5 NCXRE motifs vs 5 XRE motifs separated by 25 bp or less. (B) Frequency of repeated runs of NCXRE motifs in AHR ChIP peaks. In 24% of AHR ChIP-seq peaks there are 2 or more NCXRE motifs separated by 25 basepairs or less. (C) Binding strength of H3K4me3 and H3K27ac at AHR peaks containing 5 or more NC-XRE or XRE motifs separated by 25 basepairs or less. Runs of NCXRE are enriched at AHR binding sites proximal to transcription start sites, as marked by H3K4me or H3K27ac ChIP peaks. (D) Examples of differentially expressed AHR target genes (fold change TCDD vs vehicle, mouse liver) containing AHR ChIP peaks with 5 or more NC-XRE motifs separated by 25 basepairs or less. We define AHR target genes as differentially expressed genes (RNA-seq, fold difference in TCDD vs vehicle > 1.5x, FDR<5%) containing AHR ChIP peak within 10 kb of a gene body.
Motifs flanking XRE or NC-XRE sequence
Transcription factors bind regulatory DNA in a complex composed of multiple different transcription factors. For example, AHR is associated with the transcription factor estrogen receptor alpha at regulatory DNA (Beischlag and Perdew, 2005; Klinge et al., 2000; Madak-Erdogan and Katzenellenbogen, 2012). Another transcription factor, ARNT, is thought to be an obligate co-regulator of AHR (Lo and Matthews, 2012; Reyes et al., 1992). Previous studies have focused on transcription factor binding at XRE motifs. We identified known transcription factor motifs flanking NC-XRE motifs. We focused on AHR ChIP peaks containing at least 2 NC-XRE motifs within 25 basepairs of each other. The top 12 most frequently occurring known transcription factor motifs are shown in Figure 3. We found several motifs enriched in peaks containing both XRE and NC-XRE motifs. As expected, the ARNT motif was enriched in both, supporting the previously known association with XRE motifs and suggesting that the AHR-ARNT complex could be associated with NC-XRE motifs. Several motifs were enriched flanking NC-XRE but not XRE, such as androgen receptor binding site and SMAD3 site. This suggests that there may be crosstalk between AHR and other signaling pathways, such as TGF-beta, at NC-XRE motifs.
Figure 3. Transcription factor motifs flanking XRE or NC-XRE motifs in AHR peaks.

Top 12 most frequently occuring known transcription factor motifs in the 50 basepairs flanking repeated NCXRE and XRE motifs (2 or more motifs within 25 basepairs of each other) in genomic regions where AHR binds. Highlighted motifs are enriched flanking NCXRE but not XRE. Significance defined as p<10−11
Testing NC-XRE function in a natural genomic context
We mined published RNA-seq and ChIP-seq data and identified candidate genes upregulated by TCDD in mouse liver, where such genes contain an AHR ChIP peak containing XRE or NCXRE (or both) within 10 kb of the gene body. We exposed mouse Hepa1–6 liver cells to 10 nM TCDD for 2 hours and assayed expression of 8 candidate genes identified from our analysis of mouse liver. Three genes were upregulated more than 2-fold compared to vehicle in Hepa1–6 cells (Figure 4A). For one of these genes, Serpine1, ChIP studies in mouse liver identified an AHR binding site ~150 bp upstream of the transcription start site, which contains a run of 5 NC-XRE motifs (Fig 4B) (Huang and Elferink, 2012). Indirect evidence suggests that AHR upregulates Serpine1 expression via NC-XRE motifs, but this has never been tested directly.
Figure 4. Mutations in NCXRE DNA in Serpine1 promoter reduced TCDD-dependent expression of Serpine1.
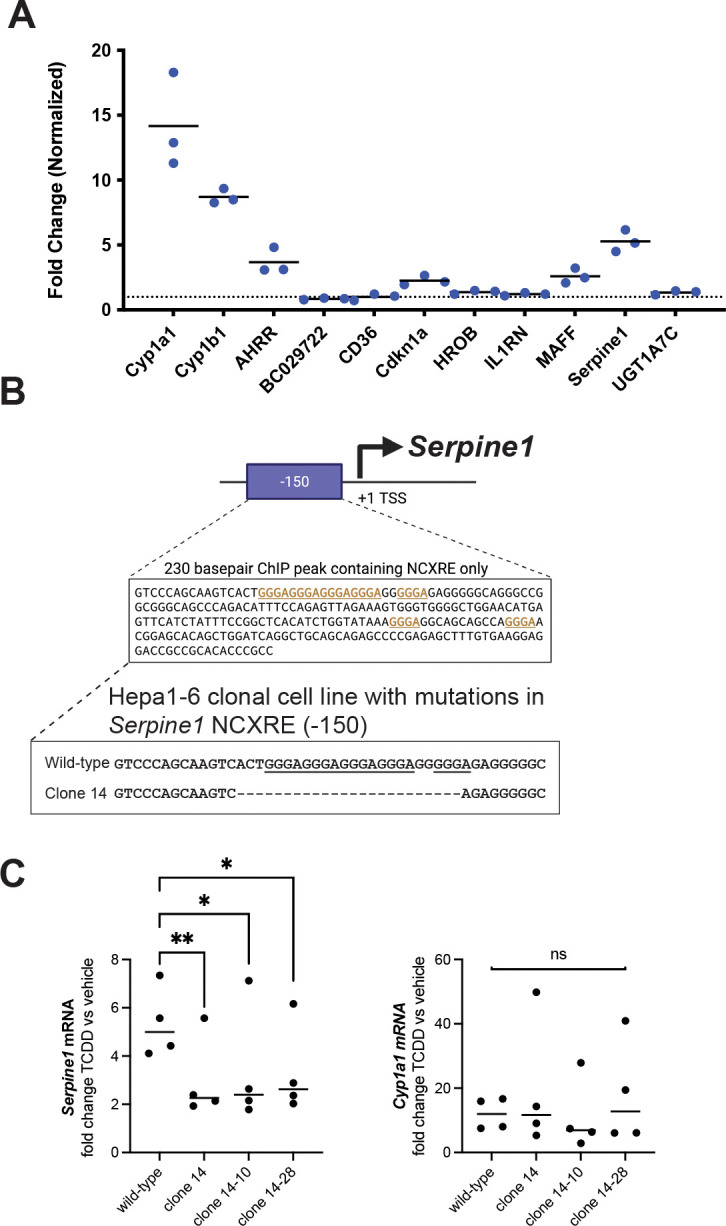
(A) Hepa1–6 cells were exposed to 10 nM TCDD or vehicle for 2 hours, followed by RNA extraction and qPCR for the indicated genes. Gene expression normalized to vehicle, 18S rRNA used as reference gene. Each circle represents a different biological replicate, horizontal black lines indicate average expression. Dotted line indicates 1x fold change. Cyp1a1, Cyp1b1 and Ahrr were used as positive controls since they are known targets of TCDD-AHR. Cdkn1a, Maff and Serpine1 exhibit more than 2x increase in expression following TCDD exposure. (B) Schematic of mouse Serpine1 gene showing AHR ChIP peak upstream of transcription start site (TSS). An AHR peak 150 basepairs (bp) upstream of TSS contains NCXRE and no XRE sequences. We transfected Hepa1–6 cells with Cas9 plus guide RNA to generate mutations in these NCXRE motifs, picked single cells and derived a clonal population (clone 14) that completely lacks the NCXRE motifs. We then picked single cells from clone 14 and derived additional clonal populations, designated clone 14–10 and 14–28, with indentical mutations (deletion of the entire NCXRE motifs). (C) Hepa1–6 wild-type or mutant cells were exposed to 10 nM TCDD or vehicle for 2 hours, followed by RNA extraction and qPCR for Serpine1 or Cyp1a1. Mutant cell lines lacking the entire NCXRE exhibit reduced increase in Serpine1 following TCDD exposure compared to wild-type cells. Mutant cells showed similar increase in Cyp1a1 following TCDD exposure as wild-type cells. One-tailed paired t test, * p < 0.05, ** p < 0.01, ns not significant (p > 0.05).
We deleted the 5x NC-XRE motif in the Serpine1 promoter, generated clonal mutant cell lines, and directly tested whether the 5x NC-XRE motif contributes to TCDD-dependent Serpine1 expression. We transfected Hepa1–6 cells with Cas9 protein together with a guide RNA targeting this NCXRE sequence. We picked single cells and established a clonal cell line (clone 14) containing mutations in the target NCXRE (Fig 4B). Bulk sequencing of clone 14 cells revealed a heterogenous population with 57% of the DNA sequence containing a 24bp deletion of the 5x NC-XRE, 14% had a 2 bp deletion that does not disrupt the NC-XRE motifs, and 28% of the DNA was wild-type. We then derived additional single cell colonies from the original clone 14, denoted as clone 14–10 and clone 14–28. Bulk DNA sequencing of each of these clones revealed no wild-type DNA. 95% of the DNA contained the expected 24bp deletion encompassing the 5x NC-XRE, while 5% had the same 2bp deletion that does not disrupt the NC-XRE motifs.
We exposed mutant and wild-type cells to TCDD or vehicle for 2 hours and assayed Serpine1 expression by RT-qPCR. In all clones, deletion of the NCXRE reduced upregulation of Serpine1 in response to TCDD (Fig 4C). To test whether AHR function was normal, we also examined expression of Cyp1a1, a prototypical AHR target gene thought to be regulated by XRE motifs. All cell lines exhibited increased Cyp1a1 expression following TCDD exposure, and there was no difference in mean Cyp1a1 expression levels between wild-type and any mutant cell line. Our results suggest that AHR transcription factor function is normal in NC-XRE clone 14 mutant cell lines. We conclude that AHR upregulates Serpine1 via this NCXRE sequence 150 bp upstream of the transcription start site.
DISCUSSION
We performed a genome-wide assessment of NC-XRE motifs and find they are prevalent in regulatory regions associated with AHR target genes. We also provide functional evidence that NC-XRE motifs in the Serpine1 promoter are necessary for full upregulation of Serpine1 in response to TCDD.
There are few examples where a transcription factor binding site DNA was mutated in an endogenous genomic context, allowing investigators to test whether a specific DNA sequence is required for transcription factor activity. Carleton and colleagues used catalytically inactive Cas9 to identify and block enhancer DNA bound by estrogen receptor alpha (Carleton et al., 2017). Other labs have deleted hundreds or thousands of basepairs in enhancer DNA to interrogate enhancer function (reviewed in (Lopes et al., 2016)). These studies are fruitful but lack the resolution to identify specific DNA sequence to which transcription factors bind. Here we used targeted, precise mutation to demonstrate the necessity of repeated NC-XRE sequence to regulate an AHR target gene.
However, we have only a basic understanding of the physiologic relevance of AHR signaling at NC-XRE motifs. Only two target genes (Serpine1 and Cdkn1a) are known to be directly regulated by AHR at NC-XRE DNA, and these interactions were explored in only one cell type, hepatocytes, in adulthood (Huang and Elferink, 2012; Jackson et al., 2014). The degree to which other AHR target genes are regulated by non-consensus DNA sequence, in additional tissues and cell types and at different stages of organismal development, is not known.
ARNT1 is considered an obligate binding partner of AHR (Reyes et al., 1992), but these studies focused on AHR-ARNT1 interactions at XRE DNA. Whether ARNT1 binds AHR at NC-XRE DNA is not known. Studies suggest that under certain conditions AHR can act independently of ARNT1 to upregulate gene expression. In mouse liver, AHR was bound to NC-XRE in the promoter of the Serpine1 gene following TCDD exposure, but no ARNT1 was detected (Huang and Elferink, 2012). In a rat hepatoma cell line containing a Serpine1:luciferase reporter construct, TCDD increased luciferase levels but knockdown of ARNT1 had no effect (Huang and Elferink, 2012). These results suggest that AHR may bind NC-XRE in the absence of ARNT1.
A limitation of the previous studies is that none considered ARNT2. ARNT2 was shown to interact with AHR and XRE DNA in vitro (Hirose et al., 1996; Rowatt et al., 2003; Tanguay et al., 2000), but to our knowledge there is absence of evidence whether ARNT2 interacts with AHR in vivo. DNA binding, coregulator recruitment and target gene expression could be different between AHR-ARNT1 and AHR-ARNT2 transcriptional complexes. One possibility is that ARNT1 preferentially associates with AHR-XRE complexes, while ARNT2 preferentially associates with AHR-NC-XRE complexes.
We appreciate that DNA regulatory elements may be incompletely conserved between species. Showing that AHR binds NCXRE DNA to regulate gene expression in any species is important foundational knowledge. While the specific number or configuration of NCXRE sequence at a given promoter or enhancer may differ between strains of animals or between species, it is extremely unlikely that AHR regulates gene expression by binding to NCXRE in mice but not in humans. Considering that AHR binds identical XRE DNA elements in zebrafish, mice and humans (Andreasen et al., 2002; Denison et al., 1988; Tanguay et al., 1999; Yao and Denison, 1992), we argue that the same is likely true for NCXRE.
METHODS
Re-analysis of published murine liver ChIP-Seq datasets
ChIP-Seq datasets of AHR in murine livers after 2hrs of TCDD treatment in male (GSE97634) and female (GSE97636) mice were downloaded from NCBI Gene Expression Omnibus (GEO) (Fader et al., 2017; Fader et al., 2019; Nault et al., 2016). Data were trimmed using trim_galore, mapped to the mouse genome build UCSC mm10 using bowtie2, and peaks were called using MACS2 using the provided controls (Langmead and Salzberg, 2012; Zhang et al., 2008). Peaks were merged using bedtoools across both sexes (Quinlan and Hall, 2010). Each peak was searched for AHR motifs using an in-house script. To test the likelihood of finding AHR motifs anywhere in the genome, random DNA peaks were generated matching the chromosome and size distribution as previously described (Coarfa et al., 2020), then searched for AHR motifs. Enrichment of AHR motifs compared to the random controls was assessed using a Fisher’s exact test, with significance achieved at p-value<0.05. Venn diagrams of AHR motifs in the merged AHR peaks were derived using bedtools. Genomic distribution of AHR peaks over genomic elements was inferred using bedtools and visualized using GraphPad Prism.
Runs of AHR motifs, eg sequences of successive motifs within a maximum distance from each other, were determined using an in-house Python script. Flanking sequences were determined using the bedtools software. Enriched motifs within the flanking sequences were determined using the HOMER software (Heinz et al., 2010).
Additional ChIP-Seq datasets of histone modifications in the mouse liver were downloaded from NCBI GEO using Encode mouse liver from 8 week old mice: GSM1000140, GSM769014 (Yue et al., 2014). Data were trimmed using trim_galore, mapped to the mouse genome build UCSC mm10 using bowtie2 (Langmead and Salzberg, 2012), and ChIP-Seq signal distribution over genomic regions was computed using the HOMER software.
Re-analysis of published murine liver AhR RNA-Seq datasets and integration with ChIP-seq datasets
Several RNA-seq datasets of mouse liver after treatment with TCDD at different doses and for multiple timepoints were downloaded from NCBI GEO: GSE109863, GSE62902, and GSE87519 (Fader et al., 2017; Nault et al., 2015; Nault et al., 2018)(Table 1). Data was trimmed using trim_galore, then mapped onto the mouse genome build UCSC mm10 using STAR (Dobin et al., 2013), and gene expression was quantified using feature_counts (Liao et al., 2014). Differentially expressed genes between TCDD and vehicle treatment were inferred using the EdgeR and RUVr R packages (Risso et al., 2014; Robinson et al., 2010), with significance achieved at fold change exceeding 1.5x and FDR-adjusted p-value<0.05. Genomic locations (represented as bed files) and gene signatures were integrated using bedtools. To capture both promoter and proximal enhancer effects, peaks were considered as associated with genes if the gene body was within 10,000 basepairs of a peak.
Cell culture
The mouse hepatoma cell line Hepa 1–6 was obtained from ATCC (Manassas, VA) (Darlington et al., 1980). Hepa 1–6 cells cells were cultured in 10 cm plates in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 1% (v/v) Penicillin/Streptomycin and 10% (v/v) FBS in a humidified incubator (5% CO2, 37°C).
CRISPR mutagenesis of Hepa 1–6 cells
We used CHOPCHOP (Labun et al., 2019) to design guide RNAs targeting the 5x NC-XRE motif region 150bp upstream of transcription start in the mouse Serpine1 gene (target sequence CAGCAAGTCACTGGGAGGGAGGG, PAM motif underlined). Synthetic single guide RNA—modified with 2’-O-methyl at 3 first and last bases, and 3’-phosphorothioate bonds between first 3 and last 2 bases—and purified SpCas9–2NLS protein was purchased from Synthego (Redwood City, CA). We mixed guide RNA and Cas9 (1.3:1 ratio) to form ribonucleoproteins according to Synthego’s recommended protocol. Guide RNA and Cas9 ribonucleoprotein complexes were transfected into Hepa1–6 cells using Lipofectamine™ CRISPRMAX™ Cas9 Transfection reagent (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s protocol. 100,000 Hepa-1–6 cells were mixed with the ribonucleoprotein-transfection solution and split into two wells for genomic analysis and clonal expansion. Cells were incubated in a humidified incubator (5% CO2, 37 °C) for 24 hours. We then changed the media and allowed the cells to incubate for another 3 days before genotyping or deriving single-cell colonies for clonal expansion.
To derive colonies from single cells, we took 10 cells / mL suspension and aliquoted 100 μl into each well of the 96-well plate. The plates were checked daily and wells with one cell were marked. Cells were monitored and allowed to grow for 1–2 weeks before transfer to a 6 well plate for genotyping and further use.
Genotyping cell lines
Genomic DNA was extracted from cells using phenol-chloroform extraction. Cells were lysed with DNA lysis buffer (10 mM Tris, 200 mM NaCl, 5 mM EDTA, 1% SDS, 0.4 mg/ml proteinase K). Equal volume of phenol:choloroform:isoamyl alcohol was added for DNA extraction into upper phase followed by ethanol-sodium acetate precipitation of DNA. We used PCR to amplify the region flanking the NC-XRE motifs using primers 5’- AAGCCAGGCCAACTTTTCCT and 5’- CGGTCCTCCTTCACAAAGCT. Amplicons were then ligated into pCR4-TOPO vector using a TOPO TA cloning kit (catalogue 450030, ThermoFisherScientific, Waltham, MA) according to the manufacturer’s protocol. Plasmids were transfected into competent cells, plated and grown overnight. 10–20 bacterial colonies per plate were picked for miniprep DNA extraction and Sanger sequencing using M13 forward primer. DNA sequence was aligned to wild-type using MacVector software (MacVector Inc., Apex, NC). If 19 out of 20 colonies contained the same mutant DNA sequence, and 1 colony contained wild-type DNA, we would conclude that 95% of that cell line contained mutant DNA, and 5% wild-type. Clone 14 was selected based on the presence of a 24 bp deletion that encompassed the entire 5x NC-XRE motif (see Figure 4).
Quantitative reverse-transcription PCR
70–80% confluent Hepa-1–6 wild-type and mutant cells were exposed to media containing 10 nM TCDD or DMSO (vehicle) for 2 hours. To extract RNA, cells were lysed in TRIzol followed by RNA extraction according to the manufacturer’s protocol using Direct-zol RNA Miniprep Kit, including the on-column DNase digestion (catalogue 11–331, Zymo Research, Irvine, CA). RNA concentration was measured using a NanoDrop 2000 Spectrophotometer (ThermoFisherScientific, Waltham, MA). Purity of the RNA (A260/A280) was ≥ 2 with the yield of 60 ng/μl or higher. We converted 1000 ng of the RNA into cDNA using iScript Reverse Transcription Supermix for RT-qPCR (Biorad, Hercules, CA; cat no 1708841) according to the manufacturer’s protocol (total volume 20 μl, incubated for 5 min at 25°C, followed by incubation at 46°C for 20 min, then 95°C for 1 min). For the qPCR reaction, 4 μl of 1:10 dilution of cDNA was mixed with SsoAdvanced Universal SYBR Green Supermix (cat no 1725271, Bio-Rad, Hercules, CA) and 500 nM each primer up to a total volume of 10 μl and amplified in a CFX96 Real-Time System (Bio-Rad) with the following program: 95C for 30 sec, then 95C for 10 sec and 60C for 30 sec for 40 cycles. Primer pairs were either designed using MacVector software or were previously published. Ct values were calculated using CFX Maestro software version 4.1.2433.1219 (Bio-Rad). Target gene expression was compared to reference gene expression using the 2−ΔΔCt method. The results shown were the average of three or more independent experiments. Statistical analysis was performed using Prism version 9.4.2 (GraphPad Software, Boston, MA).
Primer sequences:
CYP1B1 gene (Liu et al., 2015)
Cyp1b1 FP: 5’-CCAGATCCCGCTGCTCTACA-3’
Cyp1b1 RP: 5’-TGGACTGTCTGCACTAAGGCTG-3’
AHRR (Aryl Hydrocarbon Receptor Repressor) gene (Bernshausen et al., 2006)
AHRR FP: 5’-GTTGGATCCTGTAGGGAGCA-3’
AHRR RP: 5’-AGTCCAGAGGCTCACGCTTA-3’
Mmp24os1 (BC029722) gene
BC029722 FP: 5’-CGCTTTCTAATCGCCTGCAC-3’
BC029722 RP: 5’-TGAGGAGATAAAAGCCAGGCC-3’
Cd36 gene (Niu et al., 2018)
CD36 FP: 5’-TTGTGGAGCTCAAAGACCTG-3’
CD36 RP: 5’-TGCAAGAAGCGGGATGTAGTC-3’
P21Cip1 gene (Jackson et al., 2014)
Cdkn1a FP: 5’-TGTCTTGCACTCTGGTGTCTGAG-3’
Cdkn1a RP: 5’-CAATCTGCGCTTGGAGTGATAG-3’
HROB gene
HROB FP: 5’-AAGAGGAGCTCTCAGAGGCA-3’
HROB RP: 5’-ATGGTGGATGCCCTGTCTTG-3’
IL1RN Gene (Isoda et al., 2005)
IL1RN FP: 5′-CTTTACCTTCATCCGCTCTGAGA-3′
IL1RN RP: 5′-TCTAGTGTTGTGCAGAGGAACCA-3′
MAF BZIP Transcription Factor F gene
MAFF FP: 5’ ATGGCTGTGGATCCCTTATCT-3’
MAFF RP: 5’ CATCAGCGCTTCATCCGACA-3’
UGT1A7C gene
UGT1A7C FP: 5’-GTCATCCAAAGACTCGGGCA-3’
UGT1A7C RP: 5’-GGGCATCATCACCATCGGAA-3’
18s rRNA (Turner et al., 2018)
m18s-FP: 5’-GTAACCCGTTGAACCCCATT-3’
m18s-RP: 5’-CCATCCAATCGGTAGTAGCG-3’
CYP1A1 gene (Jackson et al., 2014)
CYP1A1-FP: 5’-GCCTAACTCTTCCCTGGATGC-3’
CYP1A1-RP: 5’- GACATCACAGACAGCCTCATTGA -3’
Serpine1 gene (aka PAI-1) (Eren et al., 2014)
Serpine1-FP: 5′-ACGCCTGGTGCTGGTGAATGC-3′
Serpine1-RP: 5′-ACGGTGCTGCCATCAGACTTGTG-3′
ACKNOWLEDGMENTS
TDP, SLG and CC were partially supported by The Cancer Prevention Institute of Texas (CPRIT) [RP210227, RP200504], NIH/NCI P30 shared resource grant [CA125123], NIH/NIEHS center grants [P30 ES030285] and [P42 ES027725], and NIH/NIMHD [P50MD015496]. DAG supported by NIEHS R01 ES026337.
REFERENCES
- Andreasen E. A., Hahn M. E., Heideman W., Peterson R. E. and Tanguay R. L. (2002). The zebrafish (Danio rerio) aryl hydrocarbon receptor type 1 is a novel vertebrate receptor. Mol. Pharmacol. 62, 234–249. [DOI] [PubMed] [Google Scholar]
- Beischlag T. V. and Perdew G. H. (2005). ER alpha-AHR-ARNT protein-protein interactions mediate estradiol-dependent transrepression of dioxin-inducible gene transcription. J. Biol. Chem. 280, 21607–21611. [DOI] [PubMed] [Google Scholar]
- Bernshausen T., Jux B., Esser C., Abel J. and Fritsche E. (2006). Tissue distribution and function of the Aryl hydrocarbon receptor repressor (AhRR) in C57BL/6 and Aryl hydrocarbon receptor deficient mice. Arch. Toxicol. 80, 206–211. [DOI] [PubMed] [Google Scholar]
- Carleton J. B., Berrett K. C. and Gertz J. (2017). Multiplex Enhancer Interference Reveals Collaborative Control of Gene Regulation by Estrogen Receptor α-Bound Enhancers. Cell Syst. 5, 333–344.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coarfa C., Grimm S. L., Katz T., Zhang Y., Jangid R. K., Walker C. L., Moorthy B. and Lingappan K. (2020). Epigenetic response to hyperoxia in the neonatal lung is sexually dimorphic. Redox Biol. 37, 101718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darlington G. J., Bernhard H. P., Miller R. A. and Ruddle F. H. (1980). Expression of liver phenotypes in cultured mouse hepatoma cells. J Natl Cancer Inst 64, 809–819. [PubMed] [Google Scholar]
- Denison M. S., Fisher J. M. and Whitlock J. P. (1988). Inducible, receptor-dependent protein-DNA interactions at a dioxin-responsive transcriptional enhancer. Proc Natl Acad Sci USA 85, 2528–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denison M. S., Soshilov A. A., He G., DeGroot D. E. and Zhao B. (2011). Exactly the same but different: promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol. Sci. 124, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dere E., Lo R., Celius T., Matthews J. and Zacharewski T. R. (2011). Integration of genome-wide computation DRE search, AhR ChIP-chip and gene expression analyses of TCDD-elicited responses in the mouse liver. BMC Genomics 12, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M. and Gingeras T. R. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren M., Boe A. E., Murphy S. B., Place A. T., Nagpal V., Morales-Nebreda L., Urich D., Quaggin S. E., Budinger G. R. S., Mutlu G. M., et al. (2014). PAI-1-regulated extracellular proteolysis governs senescence and survival in Klotho mice. Proc Natl Acad Sci USA 111, 7090–7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fader K. A., Nault R., Kirby M. P., Markous G., Matthews J. and Zacharewski T. R. (2017). Convergence of hepcidin deficiency, systemic iron overloading, heme accumulation, and REV-ERBα/β activation in aryl hydrocarbon receptor-elicited hepatotoxicity. Toxicol. Appl. Pharmacol. 321, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fader K. A., Nault R., Doskey C. M., Fling R. R. and Zacharewski T. R. (2019). 2,3,7,8-Tetrachlorodibenzo-p-dioxin abolishes circadian regulation of hepatic metabolic activity in mice. Sci. Rep. 9, 6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hankinson O. (2005). Role of coactivators in transcriptional activation by the aryl hydrocarbon receptor. Arch. Biochem. Biophys. 433, 379–386. [DOI] [PubMed] [Google Scholar]
- Heinz S., Benner C., Spann N., Bertolino E., Lin Y. C., Laslo P., Cheng J. X., Murre C., Singh H. and Glass C. K. (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose K., Morita M., Ema M., Mimura J., Hamada H., Fujii H., Saijo Y., Gotoh O., Sogawa K. and Fujii-Kuriyama Y. (1996). cDNA cloning and tissue-specific expression of a novel basic helix-loop-helix/PAS factor (Arnt2) with close sequence similarity to the aryl hydrocarbon receptor nuclear translocator (Arnt). Mol. Cell. Biol. 16, 1706–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G. and Elferink C. J. (2012). A novel nonconsensus xenobiotic response element capable of mediating aryl hydrocarbon receptor-dependent gene expression. Mol. Pharmacol. 81, 338–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isoda K., Sawada S., Ayaori M., Matsuki T., Horai R., Kagata Y., Miyazaki K., Kusuhara M., Okazaki M., Matsubara O., et al. (2005). Deficiency of interleukin-1 receptor antagonist deteriorates fatty liver and cholesterol metabolism in hypercholesterolemic mice. J. Biol. Chem. 280, 7002–7009. [DOI] [PubMed] [Google Scholar]
- Jackson D. P., Li H., Mitchell K. A., Joshi A. D. and Elferink C. J. (2014). Ah receptor-mediated suppression of liver regeneration through NC-XRE-driven p21Cip1 expression. Mol. Pharmacol. 85, 533–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi A. D., Mustafa M. G., Lichti C. F. and Elferink C. J. (2015). Homocitrullination is a novel histone H1 epigenetic mark dependent on aryl hydrocarbon receptor recruitment of carbamoyl phosphate synthase 1. J. Biol. Chem. 290, 27767–27778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein-Hitpass L., Kaling M. and Ryffel G. U. (1988). Synergism of closely adjacent estrogen-responsive elements increases their regulatory potential. J. Mol. Biol. 201, 537–544. [DOI] [PubMed] [Google Scholar]
- Klinge C. M., Kaur K. and Swanson H. I. (2000). The aryl hydrocarbon receptor interacts with estrogen receptor alpha and orphan receptors COUP-TFI and ERRalpha1. Arch. Biochem. Biophys. 373, 163–174. [DOI] [PubMed] [Google Scholar]
- Labun K., Montague T. G., Krause M., Torres Cleuren Y. N., Tjeldnes H. and Valen E. (2019). CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 47, W171–W174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B. and Salzberg S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G. K. and Shi W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu X., Huang T., Li L., Tang Y., Tian Y., Wang S. and Fan C. (2015). CYP1B1 deficiency ameliorates obesity and glucose intolerance induced by high fat diet in adult C57BL/6J mice. Am. J. Transl. Res. 7, 761–771. [PMC free article] [PubMed] [Google Scholar]
- Lopes R., Korkmaz G. and Agami R. (2016). Applying CRISPR-Cas9 tools to identify and characterize transcriptional enhancers. Nat. Rev. Mol. Cell Biol. 17, 597–604. [DOI] [PubMed] [Google Scholar]
- Lo R. and Matthews J. (2012). High-resolution genome-wide mapping of AHR and ARNT binding sites by ChIP-Seq. Toxicol. Sci. 130, 349–361. [DOI] [PubMed] [Google Scholar]
- Madak-Erdogan Z. and Katzenellenbogen B. S. (2012). Aryl hydrocarbon receptor modulation of estrogen receptor α-mediated gene regulation by a multimeric chromatin complex involving the two receptors and the coregulator RIP140. Toxicol. Sci. 125, 401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nault R., Fader K. A. and Zacharewski T. (2015). RNA-Seq versus oligonucleotide array assessment of dose-dependent TCDD-elicited hepatic gene expression in mice. BMC Genomics 16, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nault R., Fader K. A., Kirby M. P., Ahmed S., Matthews J., Jones A. D., Lunt S. Y. and Zacharewski T. R. (2016). Pyruvate Kinase Isoform Switching and Hepatic Metabolic Reprogramming by the Environmental Contaminant 2,3,7,8-Tetrachlorodibenzo-p-Dioxin. Toxicol. Sci. 149, 358–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nault R., Doskey C. M., Fader K. A., Rockwell C. E. and Zacharewski T. (2018). Comparison of Hepatic NRF2 and Aryl Hydrocarbon Receptor Binding in 2,3,7,8-Tetrachlorodibenzo-p-dioxin-Treated Mice Demonstrates NRF2-Independent PKM2 Induction. Mol. Pharmacol. 94, 876–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu B., He K., Li P., Gong J., Zhu X., Ye S., Ou Z. and Ren G. (2018). SIRT1 upregulation protects against liver injury induced by a HFD through inhibiting CD36 and the NF-κB pathway in mouse kupffer cells. Mol. Med. Report. 18, 1609–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A. R. and Hall I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes H., Reisz-Porszasz S. and Hankinson O. (1992). Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science 256, 1193–1195. [DOI] [PubMed] [Google Scholar]
- Risso D., Ngai J., Speed T. P. and Dudoit S. (2014). Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 32, 896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M. D., McCarthy D. J. and Smyth G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowatt A. J., DePowell J. J. and Powell W. H. (2003). ARNT gene multiplicity in amphibians: characterization of ARNT2 from the frog Xenopus laevis. J. Exp. Zool. B Mol. Dev. Evol. 300, 48–57. [DOI] [PubMed] [Google Scholar]
- Tanguay R. L., Abnet C. C., Heideman W. and Peterson R. E. (1999). Cloning and characterization of the zebrafish (Danio rerio) aryl hydrocarbon receptor. Biochim. Biophys. Acta 1444, 35–48. [DOI] [PubMed] [Google Scholar]
- Tanguay R. L., Andreasen E., Heideman W. and Peterson R. E. (2000). Identification and expression of alternatively spliced aryl hydrocarbon nuclear translocator 2 (ARNT2) cDNAs from zebrafish with distinct functions. Biochim. Biophys. Acta 1494, 117–128. [DOI] [PubMed] [Google Scholar]
- Turner N., Lim X. Y., Toop H. D., Osborne B., Brandon A. E., Taylor E. N., Fiveash C. E., Govindaraju H., Teo J. D., McEwen H. P., et al. (2018). A selective inhibitor of ceramide synthase 1 reveals a novel role in fat metabolism. Nat. Commun. 9, 3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson S. R., Joshi A. D. and Elferink C. J. (2013). The tumor suppressor Kruppel-like factor 6 is a novel aryl hydrocarbon receptor DNA binding partner. J. Pharmacol. Exp. Ther. 345, 419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao E. F. and Denison M. S. (1992). DNA sequence determinants for binding of transformed Ah receptor to a dioxin-responsive enhancer. Biochemistry 31, 5060–5067. [DOI] [PubMed] [Google Scholar]
- Yeager R. L., Reisman S. A., Aleksunes L. M. and Klaassen C. D. (2009). Introducing the “TCDD-inducible AhR-Nrf2 gene battery”. Toxicol. Sci. 111, 238–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue F., Cheng Y., Breschi A., Vierstra J., Wu W., Ryba T., Sandstrom R., Ma Z., Davis C., Pope B. D., et al. (2014). A comparative encyclopedia of DNA elements in the mouse genome. Nature 515, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]