

Abstract
Catalase from the facultatively psychrophilic bacterium Vibrio rumoiensis S-1T, which was isolated from an environment exposed to H2O2 and exhibited high catalase activity, was purified and characterized, and its localization in the cell was determined. Its molecular mass was 230 kDa, and the molecule consisted of four identical subunits. The enzyme, which was not apparently reduced by dithionite, showed a Soret peak at 406 nm in a resting state. The catalytic activity was 527,500 U · mg of protein−1 under standard reaction conditions at 40°C, 1.5 and 4.3 times faster, respectively, than those of the Micrococcus luteus and bovine catalases examined under the same reaction conditions, and showed a broad optimum pH range (pH 6 to 10). The catalase from strain S-1T is located not only in the cytoplasmic space but also in the periplasmic space. There is little difference in the activation energy for the activity between strain S-1T catalase and M. luteus and bovine liver catalases. The thermoinstability of the activity of the former catalase were significantly higher than those of the latter catalases. The thermoinstability suggests that the catalase from strain S-1T should be categorized as a psychrophilic enzyme. Although the catalase from strain S-1T is classified as a mammal type catalase, it exhibits the unique enzymatic properties of high intensity of enzymatic activity and thermoinstability. The results obtained suggest that these unique properties of the enzyme are in accordance with the environmental conditions under which the microorganism lives.
Aerobic organisms possess specific enzymes to eliminate hydrogen peroxide (H2O2), which is produced as a by-product of oxygen metabolism and is toxic to cells. Among these enzymes, catalase is well known to eliminate H2O2. On the other hand, even under anaerobic conditions, catalase is considered necessary to certain parasitic microorganisms for protection against H2O2 produced by host organisms (36). The relationship between either parasitic or symbiotic microorganisms and hosts, producing catalase and H2O2, respectively, has been reported in several cases (21, 36, 40). There are also several studies on catalases from agents that cause human disease in relation to protection against the oxidative bursts of macrophages (1, 2, 3). Furthermore, the gene regulatory system for the response of bacteria to oxidative stress has been extensively studied in enteric bacteria (10).
There have been many reports of microorganisms that are able to grow in extreme environments, such as extreme temperatures, high pressure in the deep sea, high salinity, alkaline and acidic conditions, and high concentrations of chemicals such as organic solvents (30). Apparently, these microorganisms have acquired the ability to survive under these extreme environmental pressures through long-term evolutionary processes, and they possess specific mechanisms for survival in such environments. Among such adaptational processes, organic molecules, such as enzymes that sustain their metabolisms, might have been affected by environmental pressures and induced to change via evolutionary processes. Recently, we began to conduct studies in order to understand how a bacterium adapts to an oxidative environment and why such an adaptable bacterium exists in certain environments. A facultatively psychrophilic bacterium exhibiting high catalase activity was isolated from a drain pool of a herring egg processing plant that uses H2O2 as a bleaching agent (43). The psychrophilic isolate, strain S-1T, was identified as a new species, Vibrio rumoiensis, based on its taxonomic characteristics (44). This bacterium is regarded as resistant to hyperoxidative conditions. Although individual cells of strain S-1T do not exhibit strong resistance to H2O2, a certain number of cells together do exhibit strong H2O2 resistance. The fragility of the cell structure facilitates the rapid release of catalase from the cells and therefore protects the group by reducing the amount of H2O2 which exists around the cells (20). Moreover, the catalase activity in cell extracts of strain S-1T was 1 or 2 orders of magnitude higher than those of Escherichia coli and Bacillus subtilis (43, 44). This system may work well to enable the survival of this microorganism.
In the present study, in order to understand the molecular features of catalase from the facultatively psychrophilic bacterium V. rumoiensis S-1T that enable it to survive under high concentrations of H2O2, we purified catalase from the strain, characterized it, and determined its localization in the cells. The results demonstrated that this catalase had higher activity than any previously reported catalases and that it exhibited the characteristics of a psychrophilic enzyme. As far as we know, this catalase is the first psychrophilic heme-containing enzyme reported. It is also an unusual psychrophilic enzyme in that it exhibited higher activity than its mesophilic or thermophilic counterparts.
MATERIALS AND METHODS
Bacterial strain.
The strain that we examined was V. rumoiensis strain S-1T, which exhibits high catalase activity. The organism was cultivated aerobically up to the late-logarithmic-growth phase at 27°C in PYS-2 medium (pH 7.5) containing (per liter of deionized water) 8.0 g of polypeptone (Nihon Pharmaceutical, Tokyo, Japan), 3.0 g of yeast extract (Kyokuto, Tokyo, Japan), and 5.0 g of NaCl. The organism was cultured in 20 liters of the above medium using a 30-liter stainless-steel fermentor with an agitation speed of 200 rpm · min−1. The cells were harvested by centrifugation at 10,000 × g for 20 min at 4°C, and the collected cells were stored at −85°C until use. The freezing process did not reduce the catalase activity of the cells.
Physical and chemical measurements.
Spectrophotometric measurements were performed with a Hitachi (Tokyo, Japan) U-3210 spectrophotometer using a 1-cm-light-path cuvette. The molecular masses of the subunits were determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on a 15% (wt/vol) acrylamide gel according to the method of Laemmli (25). The prestained molecular weight standard for SDS-PAGE was purchased from Bio-Rad (Hercules, Calif.). The molecular mass of the native enzyme was determined by gel filtration using two 7.8- by 300-mm Protein PAK 300 columns (Nihon Waters, Tokyo, Japan) equilibrated with 0.1 M potassium phosphate buffer (pH 7.0). The high-performance liquid chromatography (HPLC) system consisted of a solvent delivery pump (model L-7100; Hitachi) and a spectrophotometric detector (model L-7400; Hitachi) set at 280 nm. For molecular mass standards, the following proteins were used: thyroglobulin (669 kDa), apoferritin (443 kDa), β-amylase (200 kDa), alcohol dehydrogenase (150 kDa), bovine serum albumin (66.2 kDa), and carbonic anhydrase (29 kDa). The amino acid composition of catalase was analyzed with a Hitachi L-8500A automated amino acid analyzer after the sample had been hydrolyzed with 6 N HCl for 24 h at 105°C in an evacuated sealed tube. The protoheme content was determined by the pyridine ferrohemochrome method: 10% pyridine, 0.2 N NaOH, and a small amount of Na2S2O4 were added to the catalase solution, the absorbance at 557 nm was measured, and the heme content was calculated on the basis of the extinction coefficient (ɛ) of 34.4 mM−1 cm−1 for pyridine ferroprotohemochrome (31). The protein content was determined by the method of Lowry et al. (28) with bovine serum albumin as the standard.
References for amino acid composition.
As references for the amino acid composition of strain S-1T, the deduced amino acid compositions of the following catalases (with GenBank gene sequence accession numbers in parentheses) were used: Vibrio fischeri KatA (AF011784), Micrococcus luteus CatA (P29422), and Haemophilus influenzae HktE (U02682).
Catalase activity assay conditions.
Catalase activity was measured spectrophotometrically by monitoring the decrease in A240 resulting from the elimination of H2O2, using a Hitachi U-3210 spectrophotometer. The ɛ for H2O2 at 240 nm was 43.6 M−1 cm−1 (17). The standard reaction mixture for the assay contained 50 mM potassium phosphate buffer (pH 7.0), 30 mM H2O2, and 3 μl of catalase-containing solution for a total volume of 1.0 ml. The reaction was run at 20°C unless otherwise stated, and only the initial linear rate was used to estimate the catalase activity. The amount of enzyme activity that decomposed 1 μmol of H2O2 per min was defined as 1 U of activity. Results shown are averages and standard deviations from experiments performed at least eight times using at least two independent samples. Statistical analysis was conducted with Student's t test, and a P value of 0.05 was considered significant.
Purification of catalase from V. rumoiensis S-1T.
Frozen cells (approximately 80 g of wet cells) were suspended in 100 ml of 10 mM Tris-HCl buffer at pH 8.0, containing 1 mM disodium EDTA and 10 μM phenylmethylsulfonyl fluoride (PMSF) (buffer A). The suspension was gently stirred for 30 min at 30°C after the addition of DNase (1.5 μg/ml) and was then homogenized with a Teflon homogenizer. The suspension was passed through a French pressure cell (SLM-AMINCO Instruments, Inc., Rochester, N.Y.) at 18,000 lb/in2 and centrifuged at 15,000 × g for 20 min to remove unbroken cells. The resulting supernatant was recentrifuged at 105,000 × g for 1 h to obtain the soluble fraction. The soluble fraction was diluted with buffer A and subjected to the first chromatography on a Q-Sepharose Fast Flow column (2.8 by 18.5 cm) which had been equilibrated with buffer A. The enzyme was eluted with a linear gradient of NaCl (0.075 to 0.225 M) produced from 600 ml of buffer A. The eluates that contained the catalase were combined and diluted with 10 mM Tris-HCl buffer at pH 8.0 (buffer B) and then subjected to a second chromatography on a Q-Sepharose Fast Flow column (2.8 by 11 cm) which had been equilibrated with buffer B. The adsorbed enzyme was eluted with a linear gradient of NaCl (0 to 0.4 M) produced from 800 ml of buffer B. The eluate was concentrated by Centriflo CF25 Membrane Cones (Amicon, Beverly, Mass.) and then subjected to gel filtration with a Sephacryl S-300 column (2.6 by 89 cm) which had been equilibrated with buffer B containing 0.25 M NaCl. The eluted catalase fractions were combined and dialyzed against 50 mM Tris-HCl at pH 8.0 for 6 h and used as the purified enzyme preparation. A summary of the purification of catalase from V. rumoiensis S-1T is shown in Table 1.
TABLE 1.
Purification of V. rumoiensis S-1T catalase
| Step | Protein (mg · ml−1) | Total protein (mg) | Total activity (U) | Sp act (U · mg of protein−1) | Yield (%) | Purification (fold) |
|---|---|---|---|---|---|---|
| Crude extract | 12.3 | 3,190 | 23,200,000 | 7,300 | 100 | 1.0 |
| 1st Q-Sepharose fast flow | 1.88 | 84.8 | 10,400,000 | 123,000 | 45 | 16.8 |
| 2nd Q-Sepharose fast flow | 0.780 | 23.4 | 7,350,000 | 314,000 | 32 | 43.0 |
| Sephacryl S-300 | 0.534 | 11.8 | 4,660,000 | 395,000 | 20 | 54.1 |
Fractionation of bacteria.
The experimental procedure for fractionation of bacterial cells was a modification of the method of Klotz and Hutcheson (23). Cells were incubated in a 500-ml flask containing 250 ml of PYS-2 broth medium, which was set on a reciprocal shaker (130 rpm · min−1) at 27°C for 21 h. The cells were harvested by centrifugation at 4,100 × g for 15 min. Because the cells were too vulnerable to wash, they were directly suspended in buffer I (10 mM Tris-HCl–30 mM MgCl2 [pH 7.3]), and 15 μl of chloroform was added. The suspension was then immediately centrifuged at 4,100 × g for 15 min at 4°C. The resulting supernatant was passed through a sterile 0.20-μm-pore-size Dismic-25 filter (Toyo Roshi, Tokyo, Japan) for sterilization. The fraction obtained was used as the periplasmic fraction. The pelleted cells were suspended in buffer I, passed through a French pressure cell (SLM-AMINCO Instruments) at 18,000 lb/in2, and then centrifuged at 105,000 × g for 20 min. The fraction obtained was used as the cytoplasmic fraction. The activity of malate dehydrogenase, a cytoplasmic enzyme, was determined by measuring the increase in A340 in an assay solution containing 10 μl of a fraction, 2.7 mM β-NAD+, and 18 mM l-malic acid in 50 mM Tris-HCl buffer (pH 8.0) in a final total volume of 1.0 ml. The reaction was run at 25°C. The amount of enzymatic activity that decomposed 1 μmol of malate per min was defined as 1 U of activity. The activity of alkaline phosphatase, a periplasmic enzyme, was determined spectrophotometrically by monitoring the release of para-nitrophenyl phosphate (PNPP) at A405 in an assay solution containing an appropriate amount of a fraction and 380 μmol of PNPP in 200 mM sodium glycine buffer (pH 10.3) in a final total volume of 1 ml. The reaction was run at 30°C. The amount of enzyme activity that decomposed 1 μmol of PNPP per min was defined as 1 U of activity.
Western blot analysis.
Antibodies for V. rumoiensis S-1T catalase were raised by the injection of rabbits four times with approximately 1 mg of total protein. The enzyme for the antigen was emulsified in 0.25 ml of Freund's complete adjuvant (FCA) and was used for the first subcutaneous injection into the rabbit. Instead of FCA, Freund's incomplete adjuvant (FIC) was used for the second through fourth injections. The existence of catalase in fractionated cells of V. rumoiensis S-1T was determined by Western blotting (immunoblotting). One-dimensional SDS-PAGE was performed using a 15.0% separating gel. The SDS-PAGE gel was blotted onto a polyvinylidene difluoride (PVDF) membrane at room temperature as described by Towbin et al. (39). As a secondary antibody, a goat anti-rabbit immunoglobulin G (heavy and light chains) [IgG(H+L)] (human IgG adsorbed)-horseradish peroxidase (HRP) conjugate (Bio-Rad) was used. The antigen-antibody complex was detected using an HRP conjugate substrate kit (Bio-Rad).
Chemicals.
Q-Sepharose Fast Flow and Sephacryl S-300 were purchased from Pharmacia (Uppsala, Sweden), DNase and 3-amino-1,2,4,-triazole from Sigma (St. Louis, Mo.), PMSF and β-NAD+ from Wako Pure Chemical Industries (Osaka, Japan), PNPP from ICN Biomedicals, Inc. (Aurora, Ohio), and molecular weight standards for SDS-PAGE from Bio-Rad. A G. P. sensor to detect the presence of glycoprotein on the purified-catalase-loaded SDS-PAGE gel blotted onto a PVDF membrane was purchased from Honen (Tokyo, Japan). Catalases from bovine liver and Micrococcus luteus (lysodeikticus) were purchased from Sigma and Nagase (Tokyo, Japan), respectively. We used the bovine liver catalase as purchased, without further purification. Catalase from M. luteus was purified by gel filtration with a Sephacryl S-300 column and used as purified enzyme. All other chemicals were of the highest grade commercially available.
RESULTS
Purification of V. rumoiensis S-1T catalase.
The purification steps for catalase from cell extracts are summarized in Table 1. The catalase was purified by two-step anion-exchange chromatography and one-step gel filtration. The procedure demonstrated approximately 55-fold purification with a 20% yield. The purified catalase showed a high final specific activity of approximately 400,000 U/mg of protein. Although it is considered that the microorganism did not produce extracellular proteinase according to the taxonomic characteristics (44), the addition of PMSF improved the retention of activity. This was probably due to production of intracellular proteinase by the organism. There was only one peak of catalase activity in the first Q-Sepharose elution, suggesting that the microorganism possessed only one kind of catalase. The purified enzyme preparation showed a single protein band in SDS-PAGE. When the purified-catalase-loaded SDS-PAGE gel was blotted onto a PVDF membrane, staining revealed that the catalase was a glycoprotein (data not shown).
Molecular weight and isoelectric point.
The molecular weight of a subunit of the catalase was estimated to be 57.3 kDa by SDS-PAGE (Fig. 1). The native molecular weight of the enzyme was estimated to be 230 kDa by gel filtration using a Protein PAK 300. These results suggested that the purified catalase was composed of four identical subunits.
FIG. 1.
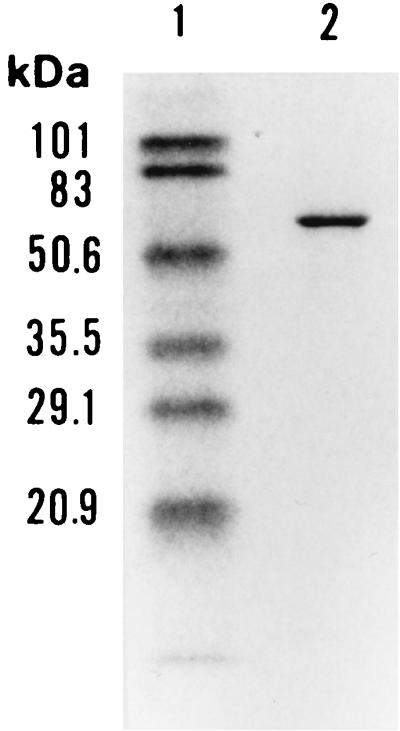
SDS-PAGE of V. rumoiensis S-1T catalase (lane 2). The marker proteins (lane 1) are commercially obtained prestained standards as described in Materials and Methods: phosphorylase B (101 kDa), bovine serum albumin (83 kDa), ovalbumin (50.6 kDa), carbonic anhydrase (35.5 kDa), soybean trypsin inhibitor (29.1 kDa), and lysozyme (20.9 kDa).
The purified catalase was electrophoresed on a polyacrylamide gel with a pH gradient of 3 to 10, and the isoelectric point was determined to be 6.5.
Spectroscopic properties of the catalase.
The absorption spectrum of catalase purified as described above exhibited a Soret band at 406 nm and an additional minor peak at 630 nm (data not shown). Treatment of the enzyme with sodium dithionite did not alter the spectral shape (data not shown). The pyridine ferrohemochrome of the enzyme showed an absorption spectrum typical of the hemochrome of protoheme IX; peaks appeared at 419, 525, and 557 nm (data not shown). From the spectrum of its pyridine ferrohemochrome, the protoheme content in the enzyme was estimated to be 3.0 molecules per tetrameric molecule. The A406/A280 ratio of 0.93 is slightly higher than those for other purified bacterial catalases (0.82) (22).
Effect of pH on catalase activity and pH stability of the catalase.
The activity versus pH profiles for the catalase activity of purified V. rumoiensis S-1T catalase were studied in a pH range of 3.0 to 10.0 (data not shown). A broad optimum pH range was observed from pH 6.0 to 10.0. Residual activity was observed from pH 4.0 to 5.0, while the activity was completely eliminated below pH 3.0. When the enzyme was incubated in a buffer solution (pH range, 3.0 to 10.0) at 30°C for 30 min, the enzyme was stable in the pH range from 6.0 to 10.0 and more than 80% of the activity remained at pH 5.0. Enzymatic activity was completely eliminated at pH 3.0.
Temperature dependence and thermal stability.
Catalase activity was assayed at various temperatures using the enzyme purified from V. rumoiensis S-1T and was compared with those from bovine liver and M. luteus (Fig. 2). Although compared with that of most enzymes, the temperature dependence of catalase activity was not great, the optimum temperature for the enzymatic activity in strain S-1T was approximately 40°C and those in M. luteus and bovine liver were approximately 40°C and 40 to 60°C, respectively (Fig. 2A). The temperature-dependent activities of V. rumoiensis S-1T catalase fluctuated more than those of the M. luteus and bovine liver catalases; a slight temperature dependency was exhibited by the two reference catalases we examined. The catalytic activity of strain S-1T was 527,500 U · mg of protein−1 at 40°C, 1.5 and 4.3 times faster than those of M. luteus catalase and bovine liver catalase, respectively, under the same reaction conditions. There are no significant differences in activation energy among the three enzymes (0.8 to 1.1 kcal/mol). The stability of S-1T catalase activity was examined by incubation of the purified enzyme at a predetermined temperature for 15 min (Fig. 3). Even when S-1T was incubated at 35 to 40°C, its catalase activity was slightly suppressed, while the activities of M. luteus and bovine liver catalases were stable at the temperature ranges of 30 to 55 and 30 to 45°C, respectively. The S-1T catalase was completely suppressed by incubation at 60°C, while the M. luteus and bovine liver catalases were suppressed at 70 and 65°C, respectively. These results suggested that the heat stability of the activity of the S-1T catalase was lower than those of the M. luteus and bovine liver catalases.
FIG. 2.

Effect of temperature on the catalases purified from V. rumoiensis S-1T, M. luteus, and bovine liver. (A) The catalase activity was assayed as described in Materials and Methods at the temperatures indicated. (B) The logarithm of the specific activity (V) (units per milligram of protein) was plotted against the reciprocal of absolute temperature (T). Values shown are activation energies calculated from the linear part of the plot. Symbols: ○, V. rumoiensis S-1T; □, M. luteus; ▵, bovine liver.
FIG. 3.

Effect of temperature on stability of catalases purified from V. rumoiensis S-1T, M. luteus, and bovine liver. Enzymes were incubated for 15 min at the indicated temperatures prior to the initiation of the reaction. Catalase activity was assayed at 20°C as described in Materials and Methods. Symbols: ○, V. rumoiensis S-1T; □, M. luteus; ▵, bovine liver.
Catalytic properties of the catalase.
The catalytic activity of the purified catalase from strain S-1T was found to be inhibited 56% after incubation for 50 min with 20 mM 3-amino-1,2,4-triazole. Treatment with 0.01 mM KCN or 0.1 mM NaN3 for 2 min inhibited enzyme activity by 73 or 97%, respectively. The purified catalase from strain S-1T was more unstable with H2O2 as a substrate than catalases from M. luteus and bovine liver. When the substrate was at concentrations higher than 70 mM H2O2, the catalase began to be inactivated, whereas catalases from M. luteus and bovine liver were inactivated at concentrations higher than 80 mM H2O2 (Fig. 4). The velocity of the catalytic activity of purified S-1T catalase was higher than that exhibited by the catalases from M. luteus and bovine liver, although catalase itself is known to exhibit very high activity compared to those of most known enzymes.
FIG. 4.

Effect of H2O2 concentration on the catalases purified from V. rumoiensis S-1T (○), M. luteus (□), and bovine liver (▵). The enzyme activity was assayed at 20°C as described in Materials and Methods.
Hydrophobic properties and effect of H2O2.
It has been known that exposing a cell extract to a mixture of ethanol and chloroform results in the denaturation of many coexisting proteins and that this can take place without affecting the catalase (33). The cell extract from strain S-1T was vortexed for 10 min at room temperature with reagents at a cell extract/95% ethanol/chloroform ratio of 10:5:3 (vol/vol/vol). The catalase activity of S-1T was 100% recovered with the denaturation of coexisting proteins. The effect of H2O2 on catalase activity was estimated using purified S-1T catalase. One milliliter of enzyme preparation at a concentration of 0.2 mg/ml was dialyzed against 2 mM H2O2 in 33 mM sodium phosphate (pH 6.8)–10 mM EDTA at 30°C for 10 to 60 min and compared with the control, which was dialyzed against the buffer without H2O2. The dialysis of the S-1T catalase against H2O2 exhibited no inactivation during this incubation period.
Amino acid composition.
The amino acid composition of S-1T catalase, a group III catalase (data not shown), was compared with those of three kinds of catalases of different origins: V. fischeri, a mesophile that belongs to the same genus as strain S-1T and possesses a group III catalase (40); M. luteus, a mesophile that produces a well-known, highly active catalase, whose group, however, is unknown; and H. influenzae, a mesophile that is parasitic to humans and possesses a group III catalase (3) (Table 2). The amino acid composition of S-1T catalase was similar to those of the catalases from other sources, as shown in Table 2. However, although the content of proline (13) was higher in S-1T catalase than in other catalases, the amino acid composition of S-1T catalase showed properties of a cold-active enzyme, such as a low isoleucine content and a low ratio of arginine content to arginine-plus-lysine content (30, 32).
TABLE 2.
Comparison of the amino acid composition of catalase from V. rumoiensis S-1T with catalases from V. fischeri, M. luteus, and H. influenzaea
| Amino acid | Concn (mol %)
|
|||
|---|---|---|---|---|
| V. rumoiensis | V. fischeri | M. luteus | H. influenzae | |
| Asx | 12.5 | 13.1 | 12.7 | 12.8 |
| Thr | 5.4 | 5.0 | 6.8 | 5.3 |
| Ser | 4.0 | 4.2 | 6.0 | 4.5 |
| Glx | 11.7 | 11.2 | 10.3 | 10.0 |
| Pro | 6.5 | 5.8 | 5.2 | 6.1 |
| Gly | 8.4 | 6.9 | 8.9 | 6.9 |
| Ala | 9.5 | 8.7 | 7.9 | 10.0 |
| Cys | NDb | 0.6 | 0.0 | 0.9 |
| Val | 5.5 | 5.4 | 7.7 | 4.9 |
| Met | 1.9 | 1.9 | 1.6 | 2.2 |
| Ile | 2.8 | 3.7 | 3.0 | 3.2 |
| Leu | 6.8 | 7.3 | 6.0 | 7.1 |
| Tyr | 3.5 | 2.7 | 3.2 | 3.2 |
| Phe | 5.7 | 6.6 | 5.4 | 6.3 |
| Lys | 5.5 | 2.7 | 3.2 | 4.1 |
| His | 3.8 | 4.2 | 3.4 | 3.0 |
| Arg | 6.5 | 7.1 | 6.4 | 7.3 |
| Trp | ND | 1.9 | 2.2 | 2.2 |
| Arg/Arg + Lys | 54.2 | 72.4 | 66.7 | 64.0 |
The amino acid composition of V. rumoiensis was determined using an amino acid analyzer as described in Materials and Methods. Other amino acid compositions were deduced from gene sequences obtained from the gene database.
ND, not determined.
Localization of catalase in the cell.
It is known that E. coli has two kinds of catalases, hydroperoxidase I (HPI) (7) and hydroperoxidase II (HPII) (8). HPI bifunctional catalase levels increase in response to the presence of ascorbate or H2O2, and the catalase is associated with plasma membranes, whereas HPII monofunctional catalase levels do not respond to ascorbate or H2O2, and the catalase is localized in the cytoplasm (15, 26). To determine the cellular localization of the catalase in strain S-1T, the activities of catalase, malate dehydrogenase (a cytoplasmic enzyme), and alkaline phosphatase activities (a periplasmic enzyme) were assayed using cytoplasmic and periplasmic extracts from strain S-1T. Malate dehydrogenase activity was detected only in the cytoplasmic fraction (1.40 U · mg of protein−1). The malate dehydrogenase activity was not inactivated by the concentration of chloroform used. On the other hand, alkaline phosphatase activity was detected only in the concentrated periplasmic fraction (2,301 U · mg of protein−1). We estimated protein content in culture supernatant and in periplasmic and cytoplasmic fractions. The protein content of the cytoplasmic fraction was 1.3 mg · ml−1, whereas those of the culture supernatant and the periplasmic fraction were too low (less than 1 μg · ml−1) to estimate the content. The ratio of catalase activity in the culture supernatant/periplasm/cytoplasm was 1:7:202. By the results described above, the specific activities of the periplasmic and cytoplasmic fractions will be obviously different. This is evidence that the fractionation in this experiment was successful. The catalase activity was detected in both the cytoplasmic and periplasmic fractions. As far as we know, strain S-1T has only one kind of catalase in the cell, according to the results of activity staining after cell extract-loaded native gel electrophoresis and fractionation of the cell extract by anion-exchange chromatography and gel filtration. Therefore, it is considered that the same molecule of catalase is contained in both the cytoplasmic and periplasmic fractions. This was confirmed by the results of Western blot analysis on a PVDF membrane blotted with both the cytoplasmic and periplasmic fractions, separated by SDS-PAGE (Fig. 5). Although in the case of E. coli, HPI and HPII localized in the periplasm and cytoplasm, respectively, the sole catalase of strain S-1T was contained in the periplasm as well as in the cytoplasm, like Pseudomonas aeruginosa KatB (4) and Pseudomonas syringae CatF (24).
FIG. 5.

SDS-PAGE (lanes 1 through 4) and Western blot analysis (lanes 5 through 8) of fractionated V. rumoiensis S-1T cells. Lanes 1 and 5, cytoplasmic fraction; lanes 2 and 6, concentrated periplasmic fraction; lanes 3 and 7, purified catalase; lanes 4 and 8, marker proteins as used in Fig. 1. Malate dehydrogenase activity was used as a marker of the cytoplasmic fraction.
DISCUSSION
Most bacterial catalases are characterized as one of two types: typical catalases, such as mammal type catalases, and bifunctional catalase-peroxidases. The mammal type catalases are commonly isolated from animals, plants, fungi, and bacteria, and their molecular features are similar to each other: they are composed of four subunits of equal size containing 2.5 to 4 protohemes IX per tetramer, with a molecular mass range of 225 to 270 kDa. They exhibit a broad optimum pH range of 5 to 10, are resistant to treatment with organic solvents, are glycoproteins, and are inhibited by 3-amino-1,2,4-triazole (6, 8, 22, 33, 38). The catalase-peroxidases, which are isolated from bacteria and fungi, have several properties that distinguish them from the typical catalases: they are reduced by dithionite, they are not glycoproteins, their activity is pH dependent, and they are more sensitive to heat, organic solvents, and H2O2 than the typical catalases, but they are insensitive to 3-amino-1,2,4-triazole (5, 11, 18, 29, 33, 42). Table 3 compares the characteristics of strain S-1T catalase with those of the Rhodobacter capsulatus bifunctional catalase-peroxidase (18) as well as those of a typical monofunctional catalase, eukaryotic catalase (9, 37). Although there are differences in the heme content, the intensity of the activity, and the isoelectric point, the S-1T catalase is classified as a monofunctional mammal type catalase.
TABLE 3.
Comparison of the enzymatic properties of V. rumoiensis S-1T catalase with those of eukaryotic catalase and R. capsulatus catalase-peroxidase
| Characteristic | Eukaryotic catalasea | V. rumoiensis S-1T catalaseb | R. capsulatus catalase-peroxidasec |
|---|---|---|---|
| Mr | 240,000 | 230,000 | 236,000 |
| No. of subunits | 4 | 4 | 4 |
| Heme/tetramer | 4 | 2.9 | 2.5–4 |
| Soret peak (nm) | 405 | 406 | 403 |
| Specific activity (U · mg of protein−1) | 97,000 | 396,900 | 7,800 |
| CN− and N3− inhibition | + | + | + |
| Aminotriazole inhibition | + | + | − |
| Optimum pH range | Broad | Broad | Narrow |
| Temperature stability | Unstable | Unstable | Unstable |
| H2O2 stability | Stable | Stable | Unstable |
| Organic solvent stability | Stable | Stable | Unstable |
| Na2S2O4 reduction | − | − | + |
| Peroxidase activity | − | − | + |
| Glycoprotein | + | + | − |
| Isoelectric point | 5.5 | 6.5 | 4.5 |
To date, there have been several reports of unique catalases from halophiles (5, 6, 12), thermophiles (27, 41), and alkaliphiles (16, 42). However, there have been no reports of psychrophilic catalases isolated from psychrophilic microorganisms. We attempted to isolate and characterize the first example of a psychrophilic catalase, which is a psychrophilic heme protein. It is commonly considered that the cold-adapted enzyme exhibits a shift in optimum activity toward low temperatures, a low activation energy, and a weak thermal stability. Gerday et al. (13) proposed that a psychrophilic enzyme is characterized by a higher specific activity over a temperature range roughly covering 0 to 30°C than its mesophilic counterpart and by a relative instability. On the other hand, Gerike et al. (14) characterized a psychrophilic citrate synthase from an antarctic bacterium, and it exhibited a lower specific activity over a temperature range of 5 to 30°C than the mesophilic counterpart. Based on the results, these authors considered that comparison factors such as optimum temperature, thermostability, and specific activity are not necessarily good indicators of the psychrophilic fitness of an enzyme; the crucial question is whether the activity at low temperatures is sufficient to permit cell growth and function. In the case of S-1T catalase, although the extent of the activity was higher than that of its mesophilic counterpart, there are no significant differences in optimum temperature and activation energy. From these results, it is considered that the extent of activity at a low temperature (e.g., 0 to 30°C), the optimum temperature, and the activation energy are not always good indicators of psychrophilic enzymes, because these factors might change depending on the absolute extent of the enzymatic activity and how much activity is necessary to sustain the metabolism and function for survival and for its own enzymatic properties. On the other hand, S-1T catalase exhibited a relatively higher thermoinstability than its mesophilic counterparts. As far as we know, there are no reports of psychrophilic enzymes and proteins that exhibit higher thermostability than their mesophilic counterparts. From molecular evolution and diversity points of view, thermoinstability may be important in order to sustain the metabolism of organisms growing in cold temperatures because thermoinstability may be in accordance with low-temperature-specific protein turnover. A low-temperature-specific proteolytic system has been described for the psychrophilic bacterium Arthrobacter globiformis SI55 (34, 35). On the basis of our experimental results and previously reported examples of psychrophilic enzymes, it is considered that thermoinstability is one of the most fundamental features of the psychrophilic enzyme.
In the present study, we purified and characterized a catalase which exhibited extensive activity and thermoinstability from a facultatively psychrophilic microorganism living under conditions of exposure to H2O2. It is suggested that these unique properties of this characterized catalase are in accordance with two independent environmental pressures on the microorganism: cold and an oxidative environment. In general, the extents of activity of psychrophilic enzymes were lower than those of mesophilic counterpart enzymes in comparisons of individual enzymes under the optimum condition. The oxidative environmental stress in addition to the cold selective pressure might have produced this unique psychrophilic enzyme that exhibited higher activity than its mesophilic or thermophilic counterpart enzymes.
ACKNOWLEDGMENT
This work was supported by the Special Coordination Fund for Promoting Science and Technology of the Science and Technology Agency of the Japanese Government.
REFERENCES
- 1.Archibald F S, Duong M-N. Superoxide dismutase and oxygen toxicity defenses in the genus Neisseria. Infect Immun. 1986;51:631–641. doi: 10.1128/iai.51.2.631-641.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bishai W R, Howard N S, Winkelstein J A, Smith H O. Characterization and virulence analysis of catalase mutants of Haemophilus influenzae. Infect Immun. 1994;62:4855–4860. doi: 10.1128/iai.62.11.4855-4860.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bishai W, Smith H O, Barcak G J. A peroxide/ascorbate-inducible catalase from Haemophilus influenzae is homologous to the Escherichia coli katE gene product. J Bacteriol. 1994;176:2914–2921. doi: 10.1128/jb.176.10.2914-2921.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown S M, Howell M L, Vasil M L, Anderson A J, Hassett D J. Cloning and characterization of the katB gene of Pseudomonas aeruginosa encoding a hydrogen peroxide-inducible catalase: purification of KatB, cellular localization, and demonstration that it is essential for optimal resistance to hydrogen peroxide. J Bacteriol. 1995;177:6536–6544. doi: 10.1128/jb.177.22.6536-6544.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown-Peterson N J, Salin M L. Purification of a catalase-peroxidase from Halobacterium halobium: characterization of some unique properties of the halophilic enzyme. J Bacteriol. 1993;175:4197–4202. doi: 10.1128/jb.175.13.4197-4202.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown-Peterson N J, Salin M L. Purification and characterization of mesohalic catalase from the halophilic bacterium Halobacterium halobium. J Bacteriol. 1995;177:378–384. doi: 10.1128/jb.177.2.378-384.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Claiborne A, Fridovich I. Purification of the o-dianisidine peroxidase from Escherichia coli B. J Biol Chem. 1979;254:4245–4252. [PubMed] [Google Scholar]
- 8.Claiborne A, Malinowski D P, Fridovich I. Purification and characterization of hydroperoxidase II of Escherichia coli B. J Biol Chem. 1979;254:11664–11668. [PubMed] [Google Scholar]
- 9.Deisseroth A, Dounce A L. Catalase: physical and chemical properties, mechanism of catalysis and physiological role. Physiol Rev. 1970;50:319–375. doi: 10.1152/physrev.1970.50.3.319. [DOI] [PubMed] [Google Scholar]
- 10.Farr S B, Kogoma T. Oxidative stress response in Escherichia coli and Salmonella typhimurium. Microbiol Rev. 1991;55:561–585. doi: 10.1128/mr.55.4.561-585.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fraaije M W, Roubroeks H P, Hagen W R, Van Berkel W J H. Purification and characterization of an intracellular catalase-peroxidase from Penicillium simplicissimum. Eur J Biochem. 1996;235:192–198. doi: 10.1111/j.1432-1033.1996.00192.x. [DOI] [PubMed] [Google Scholar]
- 12.Fukumori Y, Fujiwara T, Okada-Takahashi Y, Mukohata Y, Yamanaka T. Purification and properties of a peroxidase from Halobacterium halobium L-33. J Biochem. 1985;98:1055–1061. doi: 10.1093/oxfordjournals.jbchem.a135352. [DOI] [PubMed] [Google Scholar]
- 13.Gerday C, Aittaleb M, Arpigny J L, Baise E, Chessa J-P, Garsoux G, Petrescu I, Feller G. Psychrophilic enzyme: a thermodynamic challenge. Biochim Biophys Acta. 1997;1342:119–131. doi: 10.1016/s0167-4838(97)00093-9. [DOI] [PubMed] [Google Scholar]
- 14.Gerike U, Danson M J, Russel N J, Hough D W. Sequencing and expression of the gene encoding a cold-active citrate synthase from an Antarctic bacterium, strain DS2-3R. Eur J Biochem. 1997;248:49–57. doi: 10.1111/j.1432-1033.1997.00049.x. [DOI] [PubMed] [Google Scholar]
- 15.Heimberger A, Eisenstark A. Compartmentalization of catalase in Escherichia coli. Biochem Biophys Res Commun. 1988;154:392–397. doi: 10.1016/0006-291x(88)90698-5. [DOI] [PubMed] [Google Scholar]
- 16.Hick D B. Purification of three catalase isozymes from facultatively alkaliphilic Bacillus firmus OF4. Biochim Biophys Acta. 1995;1299:347–355. doi: 10.1016/0005-2728(95)00016-c. [DOI] [PubMed] [Google Scholar]
- 17.Hildebrandt A G, Roots I. Reduced nicotinamide adenine dinucleotide phosphate (NADPH)-dependent formation and breakdown of hydrogen peroxide during mixed function oxidation reaction in liver microsomes. Arch Biochem Biophys. 1975;171:385–397. doi: 10.1016/0003-9861(75)90047-8. [DOI] [PubMed] [Google Scholar]
- 18.Hochman A, Goldberg I. Purification and characterization of a catalase-peroxidase from the photosynthetic bacterium Rhodopseudomonas capsulata. J Biol Chem. 1991;262:6871–6876. [PubMed] [Google Scholar]
- 19.Horikoshi K, Grant W D. Superbugs; microorganisms in extreme environments. Tokyo, Japan: Japan Scientific Societies Press; 1991. [Google Scholar]
- 20.Ichise N, Morita N, Hoshino T, Kawasaki K, Yumoto I, Okuyama H. A mechanism of resistance to hydrogen peroxide in Vibrio rumoiensis S-1. Appl Environ Microbiol. 1999;65:73–79. doi: 10.1128/aem.65.1.73-79.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katsuwon J, Anderson A J. Characterization of catalase activities in root colonizing isolates of Pseudomonas putida. Can J Microbiol. 1992;38:1026–1032. [Google Scholar]
- 22.Kim H, Lee J S, Hah Y C, Roe J H. Characterization of the major catalase from Streptomyces coelicolor ATCC 10147. Microbiology. 1994;140:3391–3397. doi: 10.1099/13500872-140-12-3391. [DOI] [PubMed] [Google Scholar]
- 23.Klotz M G, Hutcheson S W. Multiple periplasmic catalases in phytopathogenic strains of Pseudomonas syringae. Appl Environ Microbiol. 1992;58:2468–2473. doi: 10.1128/aem.58.8.2468-2473.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klotz M G, Kim Y C, Katsuwon J, Anderson A J. Cloning, characterization, and phenotypic expression in Escherichia coli of catF, which encodes the catalytic subunit of catalase isozyme CatF of Pseudomonas syringae. Appl Environ Biotechnol. 1995;43:656–666. doi: 10.1007/BF00164770. [DOI] [PubMed] [Google Scholar]
- 25.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;277:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 26.Loewen P C, Switala J, Triggs-Raine B L. Catalase HPI and HPII in Escherichia coli are induced independently. Arch Biochem Biophys. 1985;243:144–149. doi: 10.1016/0003-9861(85)90782-9. [DOI] [PubMed] [Google Scholar]
- 27.Loprasert S, Negoro S, Okada H. Thermostable peroxidase from Bacillus stearothermophilus. J Gen Microbiol. 1988;134:1971–1976. doi: 10.1099/00221287-134-7-1971. [DOI] [PubMed] [Google Scholar]
- 28.Lowry O H, Rosebrough N J, Farr A L, Randall R J. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 29.Maricinkeviciene J A, Magliozzo R S, Blancard J S. Purification and characterization of Mycobacterium smegmatis catalase-peroxidase involved in isoniazid activation. J Biol Chem. 1995;270:22290–22295. doi: 10.1074/jbc.270.38.22290. [DOI] [PubMed] [Google Scholar]
- 30.Menéndez-Arias L, Argos P. Engineering protein thermal stability: sequence statistics point to residue substitutions in alpha helices. J Mol Biol. 1989;206:397–406. doi: 10.1016/0022-2836(89)90488-9. [DOI] [PubMed] [Google Scholar]
- 31.Morrison M, Connelly J, Petix J, Stotz E. Properties and structural consideration of hemin a. J Biol Chem. 1960;235:1202–1205. [PubMed] [Google Scholar]
- 32.Muir J M, Russel R J M, Hough D W, Danson M J. Citrate synthase from the hyperthermophilic Archaeon, Pyrococcus furiosus. Protein Eng. 1995;8:583–592. doi: 10.1093/protein/8.6.583. [DOI] [PubMed] [Google Scholar]
- 33.Nadler V, Goldberg I, Hochman A. Comparative study of bacterial catalase. Biochim Biophys Acta. 1986;882:234–241. [Google Scholar]
- 34.Potier P, Drevet P, Gounot A M, Hipkiss A R. ATP-dependent and -independent protein degradation in extracts of psychrotrophic bacterium Arthrobacter sp. SI55. J Gen Microbiol. 1987;133:2797–2806. [Google Scholar]
- 35.Potier P, Drevet P, Gounot A M, Hipkiss A R. Protein turnover in a psychrotrophic bacterium: proteolytic activity in extracts of cells grown at different temperatures. FEMS Microbiol Lett. 1987;44:267–271. [Google Scholar]
- 36.Rocha E R, Selby T, Coleman J P, Smith C J. Oxidative stress response in an anaerobe, Bacteroides fragilis: a role for catalase in protection against hydrogen peroxide. J Bacteriol. 1996;178:6895–6903. doi: 10.1128/jb.178.23.6895-6903.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schonbaum G R, Chance B. Catalase. In: Boyer P D, editor. The enzymes. Vol. 13. New York, N.Y: Academic Press; 1976. pp. 363–408. [Google Scholar]
- 38.Terzenbach D P, Blaut M. Purification and characterization of a catalase from the nonsulfur phototrophic bacterium Rhodobacter sphaeroides ATH 2.4.1 and its role in the oxidative stress response. Arch Microbiol. 1998;169:503–508. doi: 10.1007/s002030050603. [DOI] [PubMed] [Google Scholar]
- 39.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Visick K L, Ruby E G. The periplasmic, group III catalase of Vibrio fischeri is required for normal symbiotic competence and is induced both by oxidative stress and by approach to stationary phase. J Bacteriol. 1998;180:2087–2092. doi: 10.1128/jb.180.8.2087-2092.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang H, Tokushige Y, Shinoyama H, Fujii T, Urakami T. Purification and characterization of thermostable catalase from broth of Thermoascus auranticus. J Ferment Bioeng. 1998;85:169–173. [Google Scholar]
- 42.Yumoto I, Fukumori Y, Yamanaka T. Purification and characterization of catalase from a facultative alkalophilic Bacillus. J Biochem. 1990;108:583–587. doi: 10.1093/oxfordjournals.jbchem.a123246. [DOI] [PubMed] [Google Scholar]
- 43.Yumoto I, Yamazaki K, Kawasaki K, Ichise N, Morita N, Hoshino T, Okuyama H. Isolation of Vibrio sp. S-1 exhibiting extraordinarily high catalase activity. J Ferment Bioeng. 1998;85:113–116. [Google Scholar]
- 44.Yumoto I, Iwata H, Sawabe T, Ueno K, Ichise N, Matsuyama H, Okuyama H, Kawasaki K. Characterization of a facultatively psychrophilic bacterium, Vibrio rumoiensis sp. nov., that exhibits high catalase activity. Appl Environ Microbiol. 1999;65:67–72. doi: 10.1128/aem.65.1.67-72.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]