

Abstract
Accumulation of cytoplasmic inclusions of TAR-DNA binding protein 43 (TDP-43) is seen in both neurons and glia in a range of neurodegenerative disorders, including amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD) and Alzheimer’s disease (AD). Disease progression involves non-cell autonomous interactions among multiple cell types, including neurons, microglia and astrocytes. We investigated the effects in Drosophila of inducible, glial cell type-specific TDP-43 overexpression, a model that causes TDP-43 protein pathology including loss of nuclear TDP-43 and accumulation of cytoplasmic inclusions. We report that TDP-43 pathology in Drosophila is sufficient to cause progressive loss of each of the 5 glial sub-types. But the effects on organismal survival were most pronounced when TDP-43 pathology was induced in the perineural glia (PNG) or astrocytes. In the case of PNG, this effect is not attributable to loss of the glial population, because ablation of these glia by expression of pro-apoptotic reaper expression has relatively little impact on survival. To uncover underlying mechanisms, we used cell-type-specific nuclear RNA sequencing to characterize the transcriptional changes induced by pathological TDP-43 expression. We identified numerous glial cell-type specific transcriptional changes. Notably, SF2/SRSF1 levels were found to be decreased in both PNG and in astrocytes. We found that further knockdown of SF2/SRSF1 in either PNG or astrocytes lessens the detrimental effects of TDP-43 pathology on lifespan, but extends survival of the glial cells. Thus TDP-43 pathology in astrocytes or PNG causes systemic effects that shorten lifespan and SF2/SRSF1 knockdown rescues the loss of these glia, and also reduces their systemic toxicity to the organism.
Introduction
Pathological cytoplasmic inclusions of TDP-43 are seen in post-mortem brain tissues from >90% of patients with ALS, ~45% of FTD cases, and nearly 50% of AD cases1–8. TDP-43 is an RNA and DNA binding protein that is normally found in the nucleus of healthy cells, where it performs a variety of cellular functions, including regulation of splicing, transcription, and silencing of retrotransposons (RTEs) and endogenous retroviruses (ERVs)5,9–14. Although a few percent of familial ALS cases are caused by amino acid substitutions in the TDP-43 protein coding region, the vast majority of cases involve inclusions with TDP-43 protein of normal amino acid sequence3,15. The upstream mechanisms that trigger TDP-43 aggregation are poorly understood, but TDP-43 protein levels are under tight regulatory control, including via an autoregulatory mechanism in which nuclear TDP-43 binds to its own transcript to inhibit protein expression16.
Under pathological conditions, TDP-43 is lost from the nucleus and becomes sequestered into cytoplasmic inclusions, which removes this autoregulatory mechanism16–18. This may further increase cytoplasmic concentrations of the protein. The most commonly used animal models rely on overexpression of transgenic TDP-43 to drive aggregation19–22. Such over-expression largely sidesteps the role of upstream disease initiators. Although it is an imperfect representation of the spontaneous appearance of pathology in sporadic disease, overexpression can drive robust accumulation of hyperphosphorylated, insoluble cytoplasmic inclusions and loss of nuclear localization3,5,23.
The loss of nuclear function is in fact a shared feature of over-expression and knock out models, but the over-expression approach also has the potential to model impacts from presence of cytoplasmic inclusions24. Over-expression models of TDP-43 pathology have revealed myriad cellular roles for TDP-43 as well as downstream effects of pathological TDP-43 that are found to be relevant to disease5,24–28 The majority of animal model studies have focused on impacts of TDP-43 pathology in neurons. In this study, we investigated the mechanisms at play in glia, including specialized glial cell types that form the Drosophila blood brain barrier (BBB). There are several motivations for these experiments. First, pathological TDP-43 in patients is observed both in neurons and in glial cells2,29–32 and there are established roles in disease progression of both astrocytes and microglia33–37. For example, primary astrocytes derived from post-mortem sporadic ALS patients are toxic to motor neurons as revealed either with co-culture or application of astrocyte conditioned media38,39.
Such astrocyte toxicity has also been observed in in vivo rodent and fly models40–43. Activated microglia also may stimulate astrocytes to adopt a pro-inflammatory state that drives non-cell-autonomous toxicity37,44. In addition to these glial impacts, there are clear effects of blood brain barrier dysfunction45–47. Findings in a Drosophila TDP-43 model are convergent with this literature, implicating a variety of non-neuronal cells in toxicity to neurons42,43. We have previously demonstrated that initiating TDP-43 protein pathology by overexpression in Drosophila glial cells is sufficient to cause severe motor deficits, drive TDP-43 pathology in nearby neurons, cause neuronal cell death, and greatly reduce lifespan23,42,43.
Drosophila have five major glial cell types in the adult central brain, whose functional roles and anatomical features bear many similarities to non-neuronal cells (including glia) in the mammalian brain48–50. The perineurial glia (PNG) and subperineurial glia (SPG) form an epithelial like BBB on the brain surface, held together by cell-cell junctions51. Although they have a glial origin, the SPG undergo a mesenchymal-to-epithelial transition48. In addition to these specialized BBB forming glial cells, the cortex glia (CG), astrocyte-like glia (ALG), and ensheathing glia (EG) provide many of the functional characteristics of mammalian astrocytes and microglia48,50,52,53. We previously reported that TDP-43 pathology in astrocytes, PNG and SPG each contribute non-cell autonomous effects to nearby neurons42,43.
Here, we used cell-type-specific nuclear RNA-seq both to better characterize the baseline transcriptional profiles of the 5 major glial cell types and to investigate mechanisms underlying glial cell type specific impacts of TDP-43 pathology. We identified numerous glial cell-type-specific transcriptional changes in response to induced TDP-43 expression. Notably, these included SF2/SRSF1, a known suppressor of C9orf72 ALS/FTD models54 that we also recently identified in a screen for suppression of TDP-43 toxicity in motor neurons55. We observe that in response to TDP-43 pathology, SF2/SRSF1 is down-regulated in both the PNG and SPG specialized BBB glia. We demonstrate among the 5 glial cell types, TDP-43 toxicity in PNG is a primary driver of organismal toxicity. And we demonstrate that the systemic effects on the animal’s survival from TDP-43 pathology in PNG can be ameliorated by PNG specific knockdown of SF2/SRSF1.
Results
Organismal impacts from glial cell-type specific TDP-43 pathology reveals PNG as a primary driver of systemic toxicity
We have previously demonstrated that in Drosophila, pan-glial over-expression of TDP-43 causes loss of nuclear localization and accumulation of cytoplasmic hyperphosphorylated TDP-4323. Such pan-glial TDP-43 toxicity reduces lifespan and causes non-cell autonomous toxicity to neurons that appears to involve some combination of astrocytes, PNG and SPG23,42. Importantly, the toxicity to neurons from glial-specific TDP-43 over-expression in a humanized Drosophila model also triggers pathological cytoplasmic accumulation of TDP-43 in nearby neurons where TDP-43 is not over-expressed43. We sought to distinguish the contribution of TDP-43 proteinopathy in each glial cell type to the animal’s survival. Using cell-type specific Gal4 lines (methods) and the temperature sensitive Gal80 (Gal-80ts) approach, we induced post development over-expression of TDP-43 in each of the 5 individual glial cell types. This temperature switch expression system allowed us to avoid effects on development from over-expressing TDP-43 during glial specification, and also permitted us to determine the time-course of lethality after induction on the first day of adult life (Fig 1A). In order to monitor effects on survival of each glial cell type, we co-expressed a nuclear-envelope tagged fluorescent reporter (5X-UAS-unc84–2X-GFP). We then monitored the effects over time of inducing glial-cell type specific TDP-43 over-expression on both organismal and cell type survival. We found induced TDP-43 expression in each of the five glial cell types was sufficient to significantly reduce lifespan (Figs 1B-F) though we note this reduction is quite modest in size for the cases of the EG and CG and is intermediate in magnitude for the case of SPG. In contrast, we see more substantial reduction in lifespan for PNG (median survival reduced from 42 for control to 18 days) and ALG (median survival reduced from 36 vs. 28 days).
Figure 1. Effect on lifespan of post-developmental TDP-43 overexpression in individual glial cell types of the Drosophila CNS.

(A) To induce post development expression of both hTDP-43 and 5XUAS-Unc84–2XGFP, flies were reared at 21°C and shifted to 29°C upon eclosion (day 0). (B-F) Lifespan analyses with expression in each individual glial cell type, compared to a control without TDP-43; p<0.0001. (G) Lifespan analysis for pan-glial TDP-43 expression compared to a control without TDP-43; p<0.0001. Gal4 lines and full genotypes used for each cell type are listed in methods.
The effects on lifespan with induced TDP-43 pathology in PNG (Fig.1B) were particularly severe, and approached the magnitude of effect on lifespan seen with pan-glial hTDP-43 expression (Fig. 1G). Thus, inducing TDP-43 pathology within each of the 5 glial cell types does impact survival of the animals, but the magnitude of effect on lifespan from TDP-43 pathology varies greatly between glial cell types, with the largest effect being from PNG.
Similar intracellular toxicity of induced TDP-43 across glial cell types.
The observed differences in magnitude of effects on lifespan seen above could result from (1) differential intracellular sensitivity of a given cell type to TDP-43 toxicity, (2) differences in severity of effects on brain homeostasis from loss of each glial cell type, or (3) from cell-type specificity in non-cell autonomous toxic effects from TDP-43 proteinopathy42,43. We first tested whether there were differences in sensitivity to the toxicity of induced TDP-43 proteinopathy within each glial cell type. This could derive from either biological differences in the resistance of each cell type, from differences in expression levels within that cell type, or from a combination of the two. To distinguish these possibilities, we took advantage of the co-induced 5X-UAS-unc84–2X-GFP fluorescent reporter, to monitor cell number over a time-course after induction of TDP-43. With each genotype, we dissected brains at 2, 5, and 10-days post-induction of both TDP-43 and used confocal microscopy to quantify the numbers of nuclei labeled with GFP.
With each cell type, we observed a progressive loss in the numbers of labeled nuclei over time. With PNG, ALG, EG and CG, we see a similar time-course of cell loss (Fig.2). With each of those four glial cell types, there is a near complete loss of labeled nuclei by 10 days post induction. With SPG, the rate of loss appears to be somewhat slower (Fig. 2C,D). Despite the fact that PNG, ALG, EG and CG appear to be lost at similar rates, TDP-43 over-expression in PNG and ALG caused a palpably more severe shortening in lifespan compared to the other cell types, with the PNG expression causing the most severe effect (Fig.1). The SPG exhibit an intermediate severity of lifespan impact and a slower loss of cell number, making comparisons more difficult. But comparing PNG with EG and CG, there is a starkly higher organismal impact that cannot be explained by differences in rate of loss of these glial subtypes.
Figure 2. TDP-43 overexpression leads to age-dependent loss of each glial cell type.
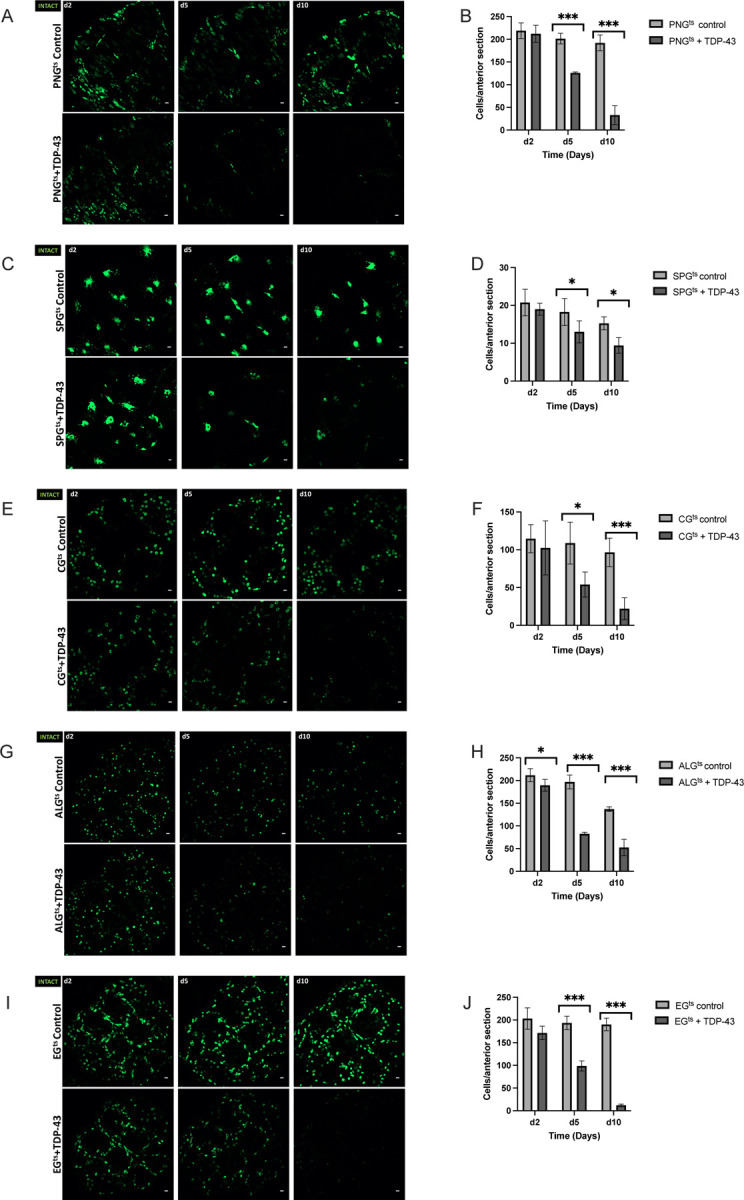
Representative confocal images of equivalent anterior section of the Drosophila brain on days 2,5, and 10 post eclosion for flies control (top) vs flies that express TDP-43 in PNGts (A), SPGts (C), CG (E) ALG (G), EG (I). Quantification of the number of each glial cell type in an equivalent anterior section was determined by counting nuclei labeled by Unc84–2X-GFP, which was co-expressed (B,D,F,H,J). Gal4 lines and full genotypes used for each cell type are listed in methods. Scale bar = 10μm. * p<0.5, ***p<0.001
Lifespan deficit observed with PNG cell-type-specific TDP-43 induction is not recapitulated by PNG ablation.
Given the severe impact on lifespan that we observe with PNG specific TDP-43 induction, we wondered if this reflected loss of an essential function that is specific to this specialized group of BBB glial cells. To test this idea, we compared the effects on lifespan of inducing expression in PNG of TDP-43 vs the pro-apoptotic gene reaper (rpr). In order to quantify effects on PNG cell loss, we co-expressed a UAS-nuclear-GFP. In each case, we used the same Gal-80ts approach to induce post development expression of GFP and either TDP-43 or rpr. As above, animals were reared at 21°C and then shifted to 29°C after eclosion (Day 0) in order to induce expression. We then quantified the effects on lifespan and PNG cell loss (Fig.3).
Figure 3. Lifespan deficit observed with TDP-43 expression in PNG is not recapitulated by PNG ablation.

(A) confocal images of an anterior section of the Drosophila brain on days 2,5, and 10 for flies expressing of PNGts + TDP-43 (top) or PNGts + rpr (bottom). (B) quantification of PNG cells in equivalent anterior sections was performed on days 2,5, and 10 post induction by counting the number of nuclei labeled with GFP, which was co-expressed using the UAS-WM reporter (Methods). (C) Lifespan analysis of flies expressing rpr (PNGts + rpr) or TDP-43 (PNGts + TDP-43). Gal4 lines and full genotypes are listed in methods. Scale bar = 10μm. * p<0.5, **p<0.01, ***p<0.001
As with induction of TDP-43 pathology, expression of rpr causes progressive loss of the PNG cell population. This occurs with similar kinetics across both groups (Figs. 3A,B). Despite the indistinguishable effects on PNG cell survival, we find that lifespan of the animals after inducing TDP-43 pathology in PNG is greatly reduced compared to lifespan after induction of rpr (Fig.3C, p<0.0001). Thus, the lifespan deficit resulting from TDP-43 pathology in PNG cannot be explained merely by a loss of this population of glial cells. Instead, this supports the interpretation that TDP-43 pathology in PNG causes systemic effects that reduce survival42 of the animals.
Glial-cell type specific RNA-sequencing reveals differential effects of TDP-43
In order to investigate glial cell-type specific impacts, we profiled both baseline expression in each glial cell type, and differential expression in response to TDP-43 induction. To accomplish this, we used RNA sequencing of nuclei purified from individual glial cell types using tandem affinity purification of INTACT nuclei (TAPIN)56,57. Here too, we used the Gal80ts approach to induce the UAS-unc84–2X-GFP nuclear tag either alone or with TDP-43. The 5X-UAS-unc84–2X-GFP reporter is tethered to the inner nuclear membrane, but has the GFP molecule on the outer surface of the nucleus. This provides the means for affinity purification of labeled nuclei from heterogeneous tissue (Fig S1, Table S1). In this TAPIN method, GFP-expressing nuclei are purified via two rounds of affinity purification, and then nuclear RNA is extracted and used to produce barcoded Illumina sequencing libraries.
Using this method, we generated nuclear RNA sequencing libraries from each of the 5 cell types and compared expression 2 days after induction of either the GFP tag alone (baseline), or the GFP tag plus TDP-43 (N=3 libraries per group). Because each of the cell types are comprised of different numbers of cells per brain (Table S1), we adjusted the number of brains to yield approximately similar numbers of cells per library. Of these 30 samples, 28 yielded high read depth and passed quality control metrics (Table 1, methods). One CG baseline library and one ALG baseline library were removed from analysis due to low sequencing depth (<5 million reads for one CG sample) and poor correlation with other samples (for one ALG sample). Among the remaining 28 libraries the total number of reads ranged from ~62.5 million to ~103.5 million, with an average of 85% of reads mapped to the reference genome (Table 1). This corresponded to detection of ~15,000 features [i.e. genes, transposable elements (TEs), structural RNAs] on average.
Table 1. Nuclear RNA-seq from each of five glial cell types, with and without TDP-43 induction.
TAPIN RNA-seq library metrics at day 2 across each glial cell type with and without TDP-43.
| Library Metric | PNGts Control (n=3) | PNGts + TDP-43 (n=3) | SPGts Control (n=3) | SPGts + TDP-43 (n=3) | CGts Control (n=2) | CGts + TDP-43 (n=3) | ALGts Control (n=2) | ALGts + TDP-43 (n=3) | EGts Control (n=3) | EGts + TDP-43 (n=3) |
|---|---|---|---|---|---|---|---|---|---|---|
| Total # of reads | 70,421,315 | 62,494,049 | 81,368,497 | 77,664,506 | 64,108,924 | 103,515,309 | 48,552,166 | 62,506,607 | 71,281,357 | 74,018,004 |
| % mapped (ave.) | 86.7 | 85.9 | 83.4 | 84.5 | 84.8 | 84.7 | 82.3 | 84.2 | 85.2 | 85.0 |
| % uniquely aligned (ave.) | 53.3 | 58.2 | 52.7 | 54.7 | 48.8 | 64.3 | 65.4 | 70.0 | 58.7 | 65.9 |
| Features detected | 15,315 | 15,009 | 14,301 | 14,354 | 14,954 | |||||
| # of upreg. features (p<.05) | 1,289 | 1,012 | 2,931 | 1,483 | 2,498 | |||||
| # of downreg. features (p<.05) | 748 | 782 | 2,316 | 1,025 | 1,772 | |||||
When comparing the baseline expression profiles across each of the five glial cell types, several things are of note. First, we found that the CG and EG show higher correlation in gene expression with each other than with the other cell types (Fig. S2A,B). Similarly, the PNG and SPG, which constitute the BBB, are more similar to each other in gene expression profiles than they are with the other cell types (Fig. S2B,C). Within each cell type, we also found that induction of TDP-43 over-expression resulted in differential expression in numerous genes, including ones that were cell-type specific. Overall, differential expression analysis with DESeq2 revealed upregulation of ~1,000 to ~3,000 features and downregulation of ~750 to ~2,300 features across the various cell types. The majority of the differentially expressed features were cellular genes (Fig. 4A,C,E,G,I, Table S2), although we also detected many changes in expression of TEs, including RTEs and ERVs as expected (Fig.4.B,D,F,H,J; Table S3)23,42,43,58–68.
Figure 4. Differentially expressed genes and TEs in response to TDP-43 overexpression in each glial cell type.
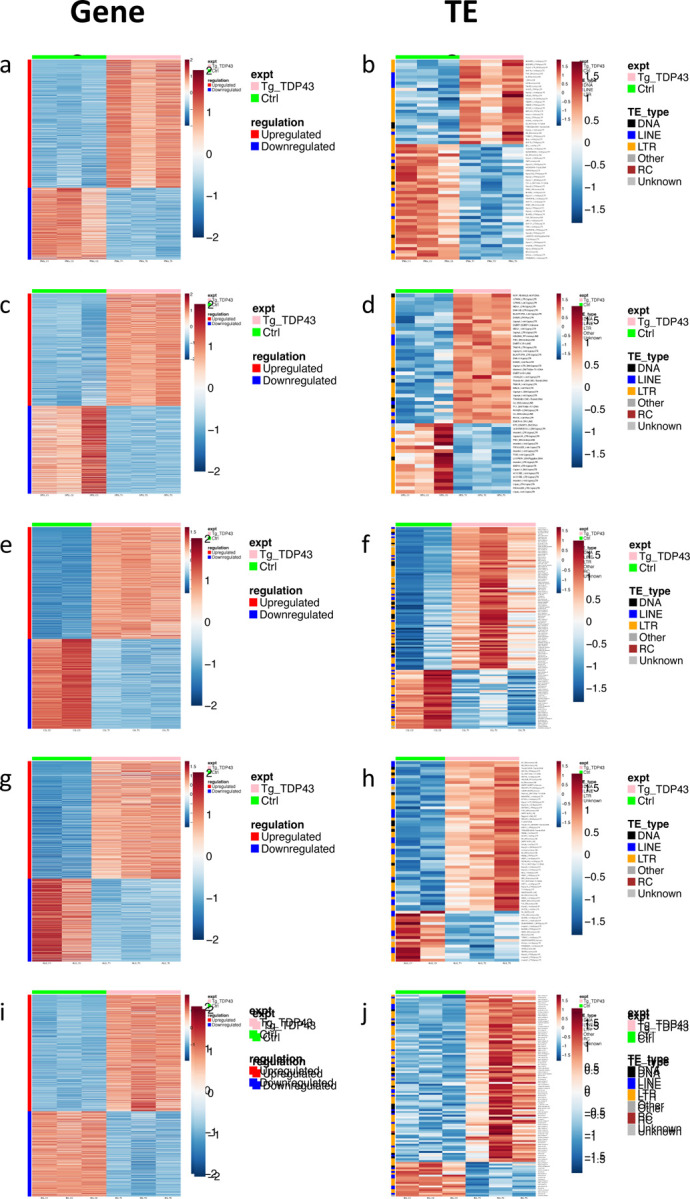
Significantly (p<.05, see methods) upregulated (blue) and downregulated (red) genes (A,C,E,G,I) and TEs (B,D,F,H,J) are shown for TDP43 expressing (Green bar, left) vs control (Pink bar, right) for each cell type. Genes or TEs that are significantly (p<.05) upregulated (blue) and downregulated (red) are shown for PNG (A, B), SPG (C,D), CG (E,F), ALG (G,H) or and EG (I,J). Full genotypes and Gal4 lines are listed in methods.
The transcriptional responses to TDP-43 pathology within each glial cell type included significant differential expression of many genes from cellular pathways that previously have been implicated in TDP-43 related neurodegeneration. These included genes involved in neuroinflammation68,69, DNA damage/repair23,42,70, chromatin organization55,71 and nucleocytoplasmic transport55,72–74, and many transposable elements14,23,42,43,58,59,61,68,75,76, each of which have been extensively implicated in TDP-43 related neurodegeneration (Table S3-S7). Although GO analyses do not identify enrichment for effects within these pathways, this may reflect differences in cell-type-specific transcriptomes and/or the fact that we profiled an early timepoint (Day 2 post TDP43 induction), in order to identify early effects. By contrast, expression profiling from human brain samples are at a disease endpoint. Given how soon after induction our expression profiling was done, it was of note that we identified many statistically significant changes in expression of TEs (including RTEs and ERVs), which was true across cell types. Indeed, we found a total of 299 upregulated TEs and 131 downregulated TEs across all five cell types (Fig.4B,D,F,H,J; Table S3). This is convergent with an emerging literature demonstrating that RTEs/ERVs become expressed in response to TDP-43 proteinopathy and may contribute to ALS progression23,42,43,58,59,61–63,65,66,68. In this fly model, we have previously demonstrated that RTEs/ERVs play a key role in mediating the toxicity of TDP-43 in glia, as well as in the intercellular toxicity to neurons23,42,43.
Knockdown of SF2/SRSF1 in astrocytes or PNG ameliorates the toxicity of TDP-43.
The differential expression analysis identified numerous cell type specific responses to TDP-43, as well as responses that were found to be in common across several cell types (Fig.S2C). Among those that were cell type specific, we made particular note of the differential expression of SF2/SRSF1 (Table S8) because we and others have recently identified knock-down of this gene as a potent suppressor of TDP-43 and C9orf72 pathology54,55,77. We previously reported that knockdown of SF2/SRSF1 in animals that overexpress TDP-43 in a subset of motor neurons is sufficient to largely prevent the age-dependent decline in escape locomotion behavior55. We also reported that SF2/SRSF1 knock down in glia was sufficient to extend lifespan of animals in which we had induced glial TDP-43 pathology. In that study, we did not examine cell type specificity, but instead used pan-glial manipulations that included all five of the major glial cell types. Our differential expression analysis identified SF2/SRSF1 as being down regulated in response to TDP-43 expression in PNG (−1.31 log2 fold change, p<0.0001) and SPG (−0.85 log2 fold change, p<.001), but not in CG, ALG, or EG (Table S8).
The fact that SF2/SRSF1 levels are reduced in SPG and PNG, but not other glial cell types, is intriguing given that pan-glial knockdown of SF2/SRSF1 can ameliorate the lifespan effects of pan-glial TDP-43 pathology (Fig. 5A)55. We observe a substantial impact on lifespan from pathological TDP-43 induction in PNG and ALG (Fig.1), but only see down regulation of SF2/SRSF1 in PNG (Table S8). We therefore wondered whether cell-type specific knockdown of SF2/SRSF1 would impact the lifespan and cell survival from TDP-43 induction in PNG vs Astrocytes. We used the PNG or Astrocyte Gal4 lines, together with the Gal80ts method, to induce TDP-43 together with either a control shRNA against mCherry or the shRNA that targets the SF2/SRSF1 gene55. We used the same Gal80ts induction conditions described in previous experiments to turn on expression after development (reared at 21°C, shifted to 29°C on adult day 0). We found that SF2/SRSF1 knockdown significantly suppressed the lifespan defects caused by TDP-43 induction in either PNG or ALG (Fig. 5B,C). Thus, despite the fact that TDP-43 induction itself causes down-regulation of SF2/SRSF1 in PNG, further knockdown in PNG has a substantial protective effect that extends organismal survival. And although TDP-43 induction in astrocytes does not on its own cause a decrease in SF2/SRSF1 expression, knock-down in astrocytes of SF2/SRSF1 by RNAi does ameliorate the effects on lifespan from astrocytic induction of TDP-43 pathology.
Figure 5. SF2/SRSF1 knockdown prevents glial cell loss and extends lifespan after induced TDP-43 overexpression.
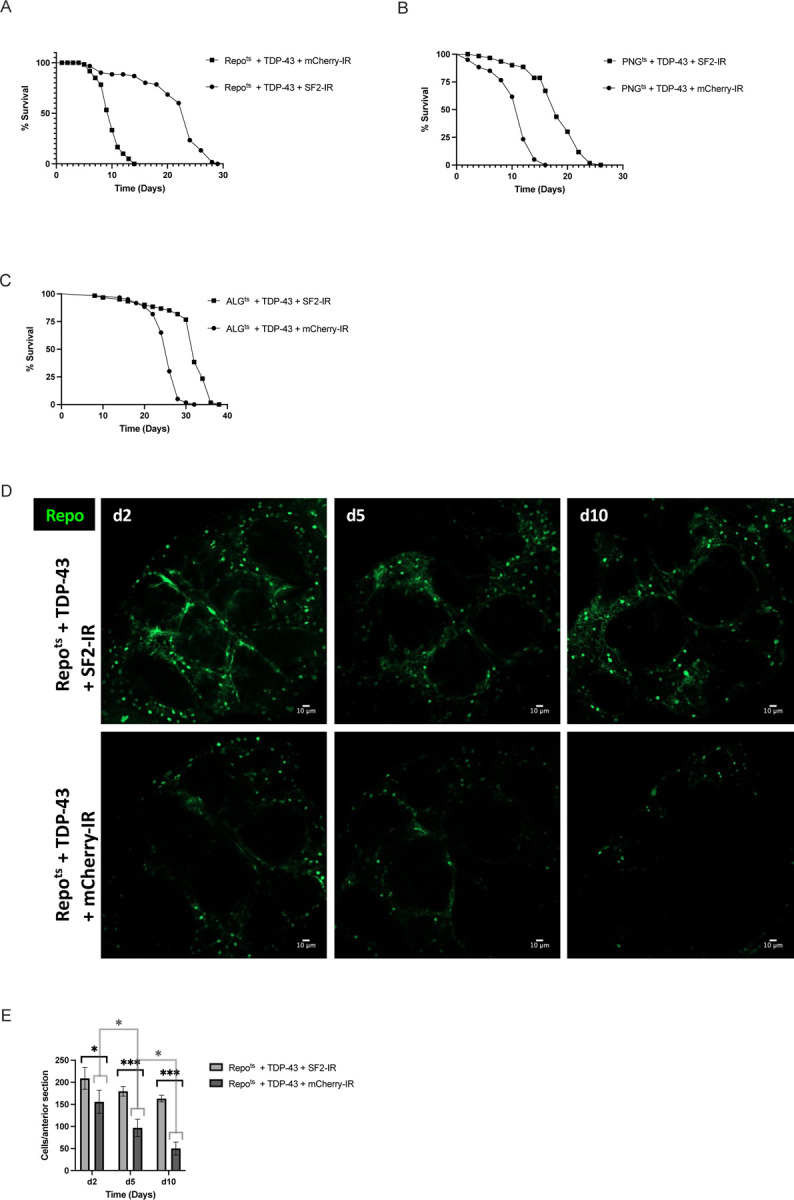
(A-C) Lifespan analysis of (A) Repots + TDP-43 + SF2-IR vs Repots + TDP-43 + mCherry-IR, (B) PNGts + TDP-43 + SF2-IR vs PNGts + TDP-43 + mCherry-IR and (C) ALGts + TDP-43 + SF2-IR vs ALGts + TDP-43 + mCherry-IR. (D) confocal images of an anterior section of the Drosophila brain on days 2,5, and 10 for flies expressing of Repots + TDP-43 + SF2-IR (top) or Repots + TDP-43 + mCherry-IR (bottom). (E) quantification of Repo+ glial cells on days 2,5, and 10 per anterior brain section. Full genotypes and Gal4 lines used are listed in methods. Scale bar = 10μm. * p<0.5, ***p<0.001
Inducing TDP-43 in PNG causes loss of most of this population of specialized BBB glia within about 10 days (Fig. 2), but the effects on lifespan (Fig. 1) are not explained by loss of essential functions of PNG because ablating these cells with rpr expression (Fig. 2) does not recapitulate the severe lifespan defect seen with TDP-43 induction. This is consistent with the interpretation that in the presence of TDP-43 pathology, these glia become neurotoxic before they die. Indeed, we previously demonstrated that TDP-43 induction in either SPG, PNG or astrocytes results in non-cell autonomous toxicity that causes the appearance of TDP-43 pathology and DNA damage in neurons, leading to neuronal cell death42,43. And in that context, blocking apoptosis of glia was sufficient to prevent glial loss, but this increased toxicity to neurons42. In light of these findings, two possibilities could explain the amelioration of the systemic effects on lifespan from knocking down SF2/SRSF1 in PNG or astrocytes. First, it is possible that knock down of SF2/SRSF1 in glia prevents release of a toxic factor that kills neurons. Alternatively, knocking down SF2/SRSF1 may cause glia to die more quickly, thereby preventing their release of a toxic substance. But this second hypothesis can be ruled out because we find that knockdown of SF2/SRSF1 actually delays the loss of glia in response to TDP-43 induction (Figs. 5D,E). Taken together, these findings support the hypothesis that the protective effects from SF2/SRSF1 are mediated by both an amelioration of the lethality of TDP-43 pathology to these glial cell types, as well as a reduction in the non-cell autonomous toxicity of the glia to surrounding neurons.
Discussion
We use glial cell-type-specific TDP-43 overexpression in Drosophila, which triggers pathological cytoplasmic inclusions23,42,43, to identify the relative contribution of each cell type to systemic effects on organismal lifespan, cell autonomous effects on glial cell loss, and transcriptional changes within each cell type. The PNG, which constitute a specialized glial cell type that contributes to BBB function48–50, emerged as a primary driver of the effects of TDP-43 induction on lifespan despite similar kinetics of TDP-43 induced cellular loss of each of the glial cell types. It is unlikely that this dramatic effect on lifespan from PNG driven TDP-43 pathology could be explained by loss of essential functions from these specialized BBB cells because ablation of PNG by induced expression of the pro-apoptotic rpr gene, does not phenocopy the shortened lifespan seen with TDP-43 expression despite a similar rate of loss of the PNG cell population.
The PNG and SPG, the ‘surface glia’, are thought to function analogously to the vertebrate BBB. PNG, which comprise the outer most layer of surface glia, far outnumber SPG (~2246 PNG vs ~300 SPG per brain)48, and lie directly in contact with the hemolymph (analog of blood). Together the PNG and SPG isolate the CNS from hemolymph components by blocking paracellular diffusion48,51,78,79. Although BBB function is vital for many aspects of normal physiology79,80, we demonstrate that loss of these BBB glia is not sufficient to explain the lifespan defects seen when TDP-43 pathology is driven in PNG. Our data argue rather that induced expression of TDP-43 in these glia triggers systemic effects with more severe impact on survival than is seen with ablation of the cell type. This is consistent with the hypothesis that PNG are induced to release toxic factors that contribute to systemic effects that cause shortened lifespan. And our findings also indicate that SF2/SRSF1 plays a key role in mediating the cell autonomous toxicity from TDP-43 over-expression to PNG and astrocytes, but also the systemic toxicity to the animal from such cell-type specific expression.
A sizeable literature supports the idea that release of factors from disease-activated glial cells is sufficient to promote toxicity to other cells including neurons38,40,44,81–91. This phenomenology is seen across a variety of models, including vertebrate systems and ALS patient-derived glial cells81–84. Given the fact that the PNG provide the outermost layer of the BBB, they are in direct contact with the hemolymph. Thus, the systemic effects of induced TDP-43 within the PNG could involve release of toxic factors into hemolymph. Dysfunction of the BBB from loss of PNG and increased permeability could also synergize with other systemic effects driven by TDP-43 proteinopathy. BBB defects have been noted in patients across a range of neurodegenerative diseases including ALS, FTD and AD. On the other hand, PNG are also in close contact with the underlying SPG, which in turn are in contact with neuronal somata that lie near the surface of the brain.
Our prior work has in fact demonstrated that TDP-43 proteinopathy in SPG is sufficient to trigger loss of nearby neurons, an effect that is mediated by the gypsy-ERV (also known as mdg4, and we will use that name to avoid repeating an ethnic slur) as a pathogenic factor in glial cells expressing TDP-4323,42,43. For example, TDP-43 over-expression in PNG, SPG and astrocytes is sufficient to drive expression of mdg-4, and mdg-4 mediates much of the toxicity both within those glial cell types, and to surrounding neurons42,43. Importantly, expression of the mdg4-ERV or the human ERV, HERV-K (HML-2) are sufficient to trigger TDP-43 proteinopathy43, and TDP-43 proteinopathy is sufficient to trigger RTE and ERV expression14,23,42,43,58,59,61,68,75,76. Thus TDP-43 pathology and ERV expression constitute a positive feedback loop, that also causes non-cell autonomous toxicity to neurons43.
Cell type expression profiling provides some insight into the functional differences between glial cell types in Drosophila, and also to the non-cell autonomous effects of TDP-43. Although several prior publications have reported baseline expression profiling of several glial-cell-types during larval development, and adult stages in Drosophila92–95, this is the first direct comparison of RNA expression profiles across all five major glial subtypes in the adult, using a single platform. This dataset supports the idea that CG and EG are functionally most similar, and that PNG and SPG also are more similar to each other than to the other glial cell types (Fig. S2; Table S2). We anticipate this dataset will serve as a valuable resource to investigate functional properties of each of these glial cell types in Drosophila. Differential gene expression, in response to induction of TDP-43 over-expression within each glial cell type, identifies a number of genes and signaling pathways that have previously been implicated in neurodegeneration, with some core similarities across cell types, but also many cell type specific effects (Fig. 4, Tables S2-7). This included numerous differentially expressed TEs (Fig.4 and Table S3), consistent with previous reports14,23,42,43,58,59,61,68,75,76. Notably, the SF2/SRSF1 gene exhibits decreased expression in PNG and SPG, but not other glial cell types (Table S8).
Several previous reports identified SF2/SRSF1 as a suppressor of both TDP-43 and C9orf72 pathology54,55,77. As we previously reported, knockdown of SF2/SRSF1 in animals that overexpress TDP-43 in either motor neurons or all glia is sufficient to largely prevent the systemic effects on locomotion or lifespan, respectively55. But findings reported here indicate that the effects of SF2/SRSF1 knockdown impact systemic, non-cell autonomous effects of TDP-43. Knockdown of SF2/SRSF1 in either PNG or ALG partially rescued effects of TDP-43 pathology on lifespan (Fig.5), but importantly, this also extended cellular survival of the glial cells. This is in contrast with the effects of expressing caspase-3 inhibitors in glial cells that over-express TDP-4342. Like knockdown of SF2/SRSF1, inhibition of apoptotic cell death prevents glial cell death, but this exacerbates neuronal cell death and further shortens lifespan42, the opposite sign of effect seen with SF2/SRSF1 knockdown (Fig. 5). We therefore hypothesize that SF2 decreases both the cell autonomous and non-autonomous toxicity of TDP-43 over-expression, either by interacting directly with TDP-43 or through some other mechanism.
Methods
Drosophila strains
Flies were reared at 21°C and maintained on standard propionic acid food for all experiments in this study. A list of transgenic lines used in this study can be found in the table below. All were backcrossed to Canton-S derivative w1118 (isoCJ1), our laboratory wild-type strain, for at least five generations. Female flies were utilized throughout the study.
| Drosophila Strains |
|---|
| Canton-S derivative w1118 (isoCJ1) |
| R85G01-Gal4 (PNG) |
| R54C07-Gal4 (SPG) |
| R54H02-Gal4 (CG) |
| R86E01-Gal4 (ALG) |
| R56F03-Gal4 (EG) |
| tub-Gal80ts (2nd chromosome) |
| 5X-UAS-unc84–2X-GFP (INTACT) |
| UAS-hTDP-43 (3rd chromosome) |
| UAS-hTDP-43 (2nd chromosome) |
| UAS-rpr |
| UAS-WM |
| UAS-SF2-IR |
| UAS-mcherry-IR |
| Repo-Gal4 |
| UAS-nlsGFP |
| tub-Gal80ts (3rd chromosome) |
Lifespan analysis
All flies were reared at 21°C and shifted to 29°C upon eclosion, in order to relieve the Gal80ts repression, allowing Gal4 mediated expression. Each lifespan experiments included 20 female flies per vial of a single genotype, with at least 60 flies total per genotype in each experiment. Flies were counted and flipped onto fresh food every other day. The Log-Rank (Mantel-Cox) test and the Gehan-Breslow-Wilcoxon test were used to compare the survival curves.
Genotypes used in Figures 1–5
| Figure | Drosophila Strains |
|---|---|
| Figs. 1B; 2A,B; 4A,B | Tubulin-Gal80 ts /5XUAS-Unc84–2XGFP; UAS-TDP-43/ R85G01-Gal4 |
| Tubulin-Gal80 ts /5XUAS-Unc84–2XGFP; R85G01-Gal4/+ | |
| Figs. 1C; 2C,D; 4C,D | Tubulin-Gal80 ts /5XUAS-Unc84–2XGFP; UAS-TDP-43/ R54C07-Gal4 |
| Tubulin-Gal80 ts /5XUAS-Unc84–2XGFP; R54C07-Gal4/+ | |
| Figs. 1D; 2E,F; 4E,F | Tubulin-Gal80 ts /5XUAS-Unc84–2XGFP; UAS-TDP-43/ R54H02-Gal4 |
| Tubulin-Gal80 ts /5XUAS-Unc84–2XGFP; R54H02-Gal4/+ | |
| Figs. 1E; 2G,H; 4G,H | Tubulin-Gal80 ts /5XUAS-Unc84–2XGFP; UAS-TDP-43/ R86E01-Gal4 |
| Tubulin-Gal80 ts /5XUAS-Unc84–2XGFP; R86E01-Gal4/+ | |
| Figs. 1F; 2I,J; 4I,J | Tubulin-Gal80 ts /5XUAS-Unc84–2XGFP; UAS-TDP-43/ R56F03-Gal4 |
| Tubulin-Gal80 ts /5XUAS-Unc84–2XGFP; R56F03-Gal4/+ | |
| Fig. 1G | Tubulin-Gal80 ts /5XUAS-Unc84–2XGFP; UAS-TDP-43/ Repo-Gal4 |
| Fig. 3A-C | Tubulin-Gal80 ts /UAS-TDP-43; UAS-WM/ R85G01-Gal4 |
| Tubulin-Gal80 ts /UAS-rpr; UAS-WM/ R85G01-Gal4 | |
| Fig. 5A,D,E | Tubulin-Gal80 ts / UAS-SF2-IR; UAS-TDP-43/ Repo-Gal4 |
| Tubulin-Gal80 ts / UAS-mcherry-IR; UAS-TDP-43/ Repo-Gal4 | |
| Fig. 5B | Tubulin-Gal80 ts / UAS-SF2-IR; UAS-TDP-43/ R85G01-Gal4 |
| Tubulin-Gal80 ts / UAS-mcherry-IR; UAS-TDP-43/ R85G01-Gal4 | |
| Fig. 5C | Tubulin-Gal80 ts / UAS-SF2-IR; UAS-TDP-43/ R86E01-Gal4 |
| Tubulin-Gal80 ts / UAS-mcherry-IR; UAS-TDP-43/ R86E01-Gal4 |
Immunohistochemistry
Brains from adult drosophila were dissected in phosphate-buffered-saline (PBS) on days 2, 5, and 10. After dissection flies were transferred to 4% paraformaldehyde (PFA) solution (1XPBS, 4% PFA, and 0.2% Triton-X-100) and fixed for one hour in a vacuum to remove air from trachea. Brains were then washed 3 times in 1XPBST for 10 minutes each prior to either (1) being mounted directly for imaging; or (2) transferred to a blocking solution (10% normal goat serum in 1X PBST; ThermoFisher: 31872) overnight at 4°C on a nutator in preparation for staining. After blocking brains were washed once in 1X PBST for 10 minutes and transferred to primary antibodies against Repo (1:10, Developmental Studies Hybridoma Bank 8D12) and Elav (1:10, Developmental Studies Hybridoma Bank 7E8A10) in 1XPBST and incubated overnight at 4°C on a nutator. Following primary antibody incubation brains were washed 4 times for 15 minutes each in a 3% salt solution (in 1X PBST) and transferred to secondary antibodies (Abcam: ab175673; ThermoFisher: A-21236) in 1XPBST and incubated overnight at 4°C on a nutator. After secondary incubation brains were washed 4 times for 15 minutes each in a 3% salt solution (in 1X PBST) and mounted in FocusClear (CelExplorer).
Confocal imaging and quantification
Brains were imaged within 24 hours of mounting using a Zeiss LSM 800 confocal microscope at 20X. All images were processed by Zeiss Zen software package. A single standard anterior section, defined by anatomy, was chosen for analysis for all cell quantification. Images were analyzed using FIJI (ImageJ). Mixed-effect analysis and two-way ANOVA were used to compare cell counts.
TAPIN
Methods are described in detail in prior studies56,57. Briefly, 2 day old female flies were anesthetized with CO2 and flash frozen in liquid nitrogen before being stored at −80°C until an appropriate number of flies were collected for each group (>1,000,000 cells for all groups except SPG, for which cell number was >300,000). Fly heads were separated, homogenized, and incubated with an antibody against GFP (ThermoFisher: G10362) before being bound to protein A beads, cleaved, and rebound to protein G beads. Twice purified TAPIN-tagged nuclei were then burst and nuclear RNA was collected and stored at −80°C.
Library preparation and sequencing
TAPIN-generated nuclear RNA was treated with DNase and purified by the Arcturus PicoPure system. Purified RNA was amplified to cDNA via Nugen Ovation v2 system (Nugen: 7102–32) and was then fragmented with a Covaris s-series to 200bp. Fragmented cDNA was repaired and underwent second strand synthesis before being ligated to barcoded linkers (Nugen: 0319–32, 0320–32). Libraries were quantified via KAPA qPCR before being sequenced on Illumina NextSeq to 76bp read length.
Computational analysis of RNA-seq libraries
Reads were aligned to the dm6 genome using STAR v2.7.6a96, allowing for 4% mismatch and up to 100 alignments per read to ensure capture of young transposon sequences. Abundance of gene (UCSC curated Refseq, March 2018) and transposon (UCSC RepeatMasker) sequences was calculated with TEtranscripts v2.2.197. Differential analysis was performed using DESeq2 v1.28.198 using the DESeq normalization strategy and negative binomial modeling. B-H corrected FDR P-value threshold of p< 0.05 was used to determine significance. For correlation plots, Pearson correlation was calculated using normalized count values from DESeq2. For heatmap visualization, counts were normalized using a variance stabilizing transformation in DESeq2
Statistical analysis
With the exception of the TAPINseq expression analysis, all statistical analyses were performed in GraphPad Prism v9.2. Specific statistical analyses are listed under each methods description and include mixed-effect analysis, two-way ANOVA, Log-rank (Mantel-Cox) test, and Gehan-Breslow-Wilcoxon test.
Supplementary Material
Acknowledgements:
We are grateful to Roger Sher, Lillian Talbot, Shreevidya Korada, Isabel Hubbard, Enas Gad El-Karim, Richard Keegan, Jorge Azpurua, Meng-Fu Shih, and Yung-Heng Chang for many helpful discussions. We also thank Yung-Heng Chang for comments on the manuscript, and Gilbert L Henry for technical help and advice with the nuclear sequencing methods. This work was supported by NIA awards RF1AG057338, R01AG078788, and RF1AG076493 to JD.
References
- 1.Forman M. S., Trojanowski J. Q. & Lee V. M. TDP-43: a novel neurodegenerative proteinopathy. Curr Opin Neurobiol 17, 548–555, doi: 10.1016/j.conb.2007.08.005 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neumann M. et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133, doi: 10.1126/science.1134108 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Ling S. C., Polymenidou M. & Cleveland D. W. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438, doi: 10.1016/j.neuron.2013.07.033 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saberi S., Stauffer J. E., Schulte D. J. & Ravits J. Neuropathology of Amyotrophic Lateral Sclerosis and Its Variants. Neurol Clin 33, 855–876, doi: 10.1016/j.ncl.2015.07.012 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prasad A., Bharathi V., Sivalingam V., Girdhar A. & Patel B. K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front Mol Neurosci 12, 25, doi: 10.3389/fnmol.2019.00025 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neumann M., Kwong L. K., Sampathu D. M., Trojanowski J. Q. & Lee V. M. TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: protein misfolding diseases without amyloidosis. Arch Neurol 64, 1388–1394, doi: 10.1001/archneur.64.10.1388 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Amador-Ortiz C. et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol 61, 435–445, doi: 10.1002/ana.21154 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uryu K. et al. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J Neuropathol Exp Neurol 67, 555–564, doi: 10.1097/NEN.0b013e31817713b5 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Copley K. E. & Shorter J. Repetitive elements in aging and neurodegeneration. Trends Genet, doi: 10.1016/j.tig.2023.02.008 (2023). [DOI] [PMC free article] [PubMed]
- 10.Dubnau J. The Retrotransposon storm and the dangers of a Collyer’s genome. Curr Opin Genet Dev 49, 95–105, doi: 10.1016/j.gde.2018.04.004 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Donde A. et al. Splicing repression is a major function of TDP-43 in motor neurons. Acta Neuropathol 138, 813–826, doi: 10.1007/s00401-019-02042-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bjork R. T., Mortimore N. P., Loganathan S. & Zarnescu D. C. Dysregulation of Translation in TDP-43 Proteinopathies: Deficits in the RNA Supply Chain and Local Protein Production. Front Neurosci 16, 840357, doi: 10.3389/fnins.2022.840357 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buratti E. & Baralle F. E. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci 13, 867–878, doi: 10.2741/2727 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Liu E. Y. et al. Loss of Nuclear TDP-43 Is Associated with Decondensation of LINE Retrotransposons. Cell Rep 27, 1409–1421 e1406, doi: 10.1016/j.celrep.2019.04.003 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sreedharan J. et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672, doi: 10.1126/science.1154584 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ayala Y. M. et al. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J 30, 277–288, doi: 10.1038/emboj.2010.310 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koyama A. et al. Increased cytoplasmic TARDBP mRNA in affected spinal motor neurons in ALS caused by abnormal autoregulation of TDP-43. Nucleic Acids Res 44, 5820–5836, doi: 10.1093/nar/gkw499 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sugai A. et al. Non-genetically modified models exhibit TARDBP mRNA increase due to perturbed TDP-43 autoregulation. Neurobiol Dis 130, 104534, doi: 10.1016/j.nbd.2019.104534 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Ash P. E. et al. Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum Mol Genet 19, 3206–3218, doi: 10.1093/hmg/ddq230 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liachko N. F., Guthrie C. R. & Kraemer B. C. Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J Neurosci 30, 16208–16219, doi: 10.1523/JNEUROSCI.2911-10.2010 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wils H. et al. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A 107, 3858–3863, doi: 10.1073/pnas.0912417107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu Y. F. et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci 30, 10851–10859, doi: 10.1523/JNEUROSCI.1630-10.2010 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krug L. et al. Retrotransposon activation contributes to neurodegeneration in a Drosophila TDP-43 model of ALS. PLoS Genet 13, e1006635, doi: 10.1371/journal.pgen.1006635 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wegorzewska I. & Baloh R. H. TDP-43-based animal models of neurodegeneration: new insights into ALS pathology and pathophysiology. Neurodegener Dis 8, 262–274, doi: 10.1159/000321547 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alrafiah A. R. From Mouse Models to Human Disease: An Approach for Amyotrophic Lateral Sclerosis. In Vivo 32, 983–998, doi: 10.21873/invivo.11339 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Halliday G. et al. Mechanisms of disease in frontotemporal lobar degeneration: gain of function versus loss of function effects. Acta Neuropathol 124, 373–382, doi: 10.1007/s00401-012-1030-4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gendron T. F., Rademakers R. & Petrucelli L. TARDBP mutation analysis in TDP-43 proteinopathies and deciphering the toxicity of mutant TDP-43. J Alzheimers Dis 33 Suppl 1, S35–45, doi: 10.3233/JAD-2012-129036 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gendron T. F. & Petrucelli L. Rodent models of TDP-43 proteinopathy: investigating the mechanisms of TDP-43-mediated neurodegeneration. J Mol Neurosci 45, 486–499, doi: 10.1007/s12031-011-9610-7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arai T. et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351, 602–611, doi: 10.1016/j.bbrc.2006.10.093 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Igaz L. M. et al. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am J Pathol 173, 182–194, doi: 10.2353/ajpath.2008.080003 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang H. et al. TDP-43-immunoreactive neuronal and glial inclusions in the neostriatum in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol 115, 115–122, doi: 10.1007/s00401-007-0285-7 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Hasegawa M. et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol 64, 60–70, doi: 10.1002/ana.21425 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamashita H., Komine O., Fujimori-Tonou N. & Yamanaka K. Comprehensive expression analysis with cell-type-specific transcriptome in ALS-linked mutant SOD1 mice: Revisiting the active role of glial cells in disease. Front Cell Neurosci 16, 1045647, doi: 10.3389/fncel.2022.1045647 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Naito M. et al. Pathological features of glial cells and motor neurons in the anterior horn of the spinal cord in sporadic ALS using ADAR2 conditional knockout mice. J Neurol Sci 444, 120520, doi: 10.1016/j.jns.2022.120520 (2023). [DOI] [PubMed] [Google Scholar]
- 35.Vahsen B. F. et al. Human iPSC co-culture model to investigate the interaction between microglia and motor neurons. Sci Rep 12, 12606, doi: 10.1038/s41598-022-16896-8 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szebenyi K. et al. Human ALS/FTD brain organoid slice cultures display distinct early astrocyte and targetable neuronal pathology. Nat Neurosci 24, 1542–1554, doi: 10.1038/s41593-021-00923-4 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guttenplan K. A. et al. Knockout of reactive astrocyte activating factors slows disease progression in an ALS mouse model. Nat Commun 11, 3753, doi: 10.1038/s41467-020-17514-9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haidet-Phillips A. M. et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol 29, 824–828, doi: 10.1038/nbt.1957 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Re D. B. et al. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 81, 1001–1008, doi: 10.1016/j.neuron.2014.01.011 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tong J. et al. Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. EMBO J 32, 1917–1926, doi: 10.1038/emboj.2013.122 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ditsworth D. et al. Mutant TDP-43 within motor neurons drives disease onset but not progression in amyotrophic lateral sclerosis. Acta Neuropathol 133, 907–922, doi: 10.1007/s00401-017-1698-6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang Y. H. & Dubnau J. The Gypsy Endogenous Retrovirus Drives Non-Cell-Autonomous Propagation in a Drosophila TDP-43 Model of Neurodegeneration. Curr Biol 29, 3135–3152 e3134, doi: 10.1016/j.cub.2019.07.071 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang Y.-H. & Dubnau J. Endogenous retroviruses and TDP-43 proteinopathy form a sustaining feedback driving intercellular spread of Drosophila neurodegeneration. Nature Communications 14, 966, doi: 10.1038/s41467-023-36649-z (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liddelow S. A. et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487, doi: 10.1038/nature21029 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miyazaki K. et al. Disruption of neurovascular unit prior to motor neuron degeneration in amyotrophic lateral sclerosis. J Neurosci Res 89, 718–728, doi: 10.1002/jnr.22594 (2011). [DOI] [PubMed] [Google Scholar]
- 46.Garbuzova-Davis S. et al. Impaired blood-brain/spinal cord barrier in ALS patients. Brain Res 1469, 114–128, doi: 10.1016/j.brainres.2012.05.056 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Winkler E. A. et al. Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc Natl Acad Sci U S A 111, E1035–1042, doi: 10.1073/pnas.1401595111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kremer M. C., Jung C., Batelli S., Rubin G. M. & Gaul U. The glia of the adult Drosophila nervous system. Glia 65, 606–638, doi: 10.1002/glia.23115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stork T., Bernardos R. & Freeman M. R. Analysis of glial cell development and function in Drosophila. Cold Spring Harb Protoc 2012, 1–17, doi: 10.1101/pdb.top067587 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Freeman M. R. & Doherty J. Glial cell biology in Drosophila and vertebrates. Trends Neurosci 29, 82–90, doi: 10.1016/j.tins.2005.12.002 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Limmer S., Weiler A., Volkenhoff A., Babatz F. & Klambt C. The Drosophila blood-brain barrier: development and function of a glial endothelium. Front Neurosci 8, 365, doi: 10.3389/fnins.2014.00365 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yildirim K., Petri J., Kottmeier R. & Klambt C. Drosophila glia: Few cell types and many conserved functions. Glia 67, 5–26, doi: 10.1002/glia.23459 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Kim T., Song B. & Lee I. S. Drosophila Glia: Models for Human Neurodevelopmental and Neurodegenerative Disorders. Int J Mol Sci 21, doi: 10.3390/ijms21144859 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hautbergue G. M. et al. SRSF1-dependent nuclear export inhibition of C9ORF72 repeat transcripts prevents neurodegeneration and associated motor deficits. Nat Commun 8, 16063, doi: 10.1038/ncomms16063 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Azpurua J., El-Karim E. G., Tranquille M. & Dubnau J. A behavioral screen for mediators of age-dependent TDP-43 neurodegeneration identifies SF2/SRSF1 among a group of potent suppressors in both neurons and glia. PLoS Genet 17, e1009882, doi: 10.1371/journal.pgen.1009882 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Henry G. L., Davis F. P., Picard S. & Eddy S. R. Cell type-specific genomics of Drosophila neurons. Nucleic Acids Res 40, 9691–9704, doi: 10.1093/nar/gks671 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davis F. P. et al. A genetic, genomic, and computational resource for exploring neural circuit function. Elife 9, doi: 10.7554/eLife.50901 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li W., Jin Y., Prazak L., Hammell M. & Dubnau J. Transposable elements in TDP-43-mediated neurodegenerative disorders. PLoS One 7, e44099, doi: 10.1371/journal.pone.0044099 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li W. et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci Transl Med 7, 307ra153, doi: 10.1126/scitranslmed.aac8201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li W. et al. Activation of transposable elements during aging and neuronal decline in Drosophila. Nat Neurosci 16, 529–531, doi: 10.1038/nn.3368 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Douville R., Liu J., Rothstein J. & Nath A. Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann Neurol 69, 141–151, doi: 10.1002/ana.22149 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Douville R. N. & Nath A. Human Endogenous Retrovirus-K and TDP-43 Expression Bridges ALS and HIV Neuropathology. Front Microbiol 8, 1986, doi: 10.3389/fmicb.2017.01986 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Manghera M., Ferguson-Parry J. & Douville R. N. TDP-43 regulates endogenous retrovirus-K viral protein accumulation. Neurobiol Dis 94, 226–236, doi: 10.1016/j.nbd.2016.06.017 (2016). [DOI] [PubMed] [Google Scholar]
- 64.Nadeau M. J., Manghera M. & Douville R. N. Inside the Envelope: Endogenous Retrovirus-K Env as a Biomarker and Therapeutic Target. Front Microbiol 6, 1244, doi: 10.3389/fmicb.2015.01244 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alfahad T., Levey A. & Nath A. Detection Of Increased Transcripts Of Human Endogenous Retrovirus In Plasma Of Patients With Frontotemporal Dementia Associated Amyotrophic Lateral Sclerosis (S40.004). Neurology 82, S40.004 (2014). [Google Scholar]
- 66.Tam O. H., Ostrow L. W. & Gale Hammell M. Diseases of the nERVous system: retrotransposon activity in neurodegenerative disease. Mob DNA 10, 32, doi: 10.1186/s13100-019-0176-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jonsson M. E., Garza R., Johansson P. A. & Jakobsson J. Transposable Elements: A Common Feature of Neurodevelopmental and Neurodegenerative Disorders. Trends Genet 36, 610–623, doi: 10.1016/j.tig.2020.05.004 (2020). [DOI] [PubMed] [Google Scholar]
- 68.Tam O. H. et al. Postmortem Cortex Samples Identify Distinct Molecular Subtypes of ALS: Retrotransposon Activation, Oxidative Stress, and Activated Glia. Cell Rep 29, 1164–1177 e1165, doi: 10.1016/j.celrep.2019.09.066 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhan L., Xie Q. & Tibbetts R. S. Opposing roles of p38 and JNK in a Drosophila model of TDP-43 proteinopathy reveal oxidative stress and innate immunity as pathogenic components of neurodegeneration. Hum Mol Genet 24, 757–772, doi: 10.1093/hmg/ddu493 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guerrero E. N. et al. Amyotrophic lateral sclerosis-associated TDP-43 mutation Q331K prevents nuclear translocation of XRCC4-DNA ligase 4 complex and is linked to genome damage-mediated neuronal apoptosis. Hum Mol Genet 28, 2459–2476, doi: 10.1093/hmg/ddz062 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Berson A. et al. TDP-43 Promotes Neurodegeneration by Impairing Chromatin Remodeling. Curr Biol 27, 3579–3590 e3576, doi: 10.1016/j.cub.2017.10.024 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jovicic A. et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci 18, 1226–1229, doi: 10.1038/nn.4085 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Boeynaems S. et al. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci Rep 6, 20877, doi: 10.1038/srep20877 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Freibaum B. D. et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525, 129–133, doi: 10.1038/nature14974 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Prudencio M. et al. Repetitive element transcripts are elevated in the brain of C9orf72 ALS/FTLD patients. Hum Mol Genet 26, 3421–3431, doi: 10.1093/hmg/ddx233 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Romano G., Klima R. & Feiguin F. TDP-43 prevents retrotransposon activation in the Drosophila motor system through regulation of Dicer-2 activity. BMC Biol 18, 82, doi: 10.1186/s12915-020-00816-1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Castelli L. M. et al. SRSF1-dependent inhibition of C9ORF72-repeat RNA nuclear export: genome-wide mechanisms for neuroprotection in amyotrophic lateral sclerosis. Mol Neurodegener 16, 53, doi: 10.1186/s13024-021-00475-y (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schwabe T., Li X. & Gaul U. Dynamic analysis of the mesenchymal-epithelial transition of blood-brain barrier forming glia in Drosophila. Biol Open 6, 232–243, doi: 10.1242/bio.020669 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hindle S. J. & Bainton R. J. Barrier mechanisms in the Drosophila blood-brain barrier. Front Neurosci 8, 414, doi: 10.3389/fnins.2014.00414 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhao Z., Nelson A. R., Betsholtz C. & Zlokovic B. V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 163, 1064–1078, doi: 10.1016/j.cell.2015.10.067 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Philips T. & Rothstein J. D. Glial cells in amyotrophic lateral sclerosis. Exp Neurol 262 Pt B, 111–120, doi: 10.1016/j.expneurol.2014.05.015 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Valori C. F., Brambilla L., Martorana F. & Rossi D. The multifaceted role of glial cells in amyotrophic lateral sclerosis. Cell Mol Life Sci 71, 287–297, doi: 10.1007/s00018-013-1429-7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vahsen B. F. et al. Non-neuronal cells in amyotrophic lateral sclerosis - from pathogenesis to biomarkers. Nat Rev Neurol 17, 333–348, doi: 10.1038/s41582-021-00487-8 (2021). [DOI] [PubMed] [Google Scholar]
- 84.Van Harten A. C. M., Phatnani H. & Przedborski S. Non-cell-autonomous pathogenic mechanisms in amyotrophic lateral sclerosis. Trends Neurosci 44, 658–668, doi: 10.1016/j.tins.2021.04.008 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mitra J. et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc Natl Acad Sci U S A 116, 4696–4705, doi: 10.1073/pnas.1818415116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kia A., McAvoy K., Krishnamurthy K., Trotti D. & Pasinelli P. Astrocytes expressing ALS-linked mutant FUS induce motor neuron death through release of tumor necrosis factor-alpha. Glia 66, 1016–1033, doi: 10.1002/glia.23298 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ferraiuolo L. et al. Oligodendrocytes contribute to motor neuron death in ALS via SOD1-dependent mechanism. Proc Natl Acad Sci U S A 113, E6496–E6505, doi: 10.1073/pnas.1607496113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Endo F. et al. Astrocyte-derived TGF-beta1 accelerates disease progression in ALS mice by interfering with the neuroprotective functions of microglia and T cells. Cell Rep 11, 592–604, doi: 10.1016/j.celrep.2015.03.053 (2015). [DOI] [PubMed] [Google Scholar]
- 89.Liao B., Zhao W., Beers D. R., Henkel J. S. & Appel S. H. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp Neurol 237, 147–152, doi: 10.1016/j.expneurol.2012.06.011 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xiao Q. et al. Mutant SOD1(G93A) microglia are more neurotoxic relative to wild-type microglia. J Neurochem 102, 2008–2019, doi: 10.1111/j.1471-4159.2007.04677.x (2007). [DOI] [PubMed] [Google Scholar]
- 91.Nagai M. et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci 10, 615–622, doi: 10.1038/nn1876 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.DeSalvo M. K. et al. The Drosophila surface glia transcriptome: evolutionary conserved blood-brain barrier processes. Front Neurosci 8, 346, doi: 10.3389/fnins.2014.00346 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lago-Baldaia I. et al. A Drosophila glial cell atlas reveals that transcriptionally defined cell types can be morphologically diverse. bioRxiv, 2022.2008.2001.502305, doi: 10.1101/2022.08.01.502305 (2022). [DOI]
- 94.Huang Y., Ng F. S. & Jackson F. R. Comparison of larval and adult Drosophila astrocytes reveals stage-specific gene expression profiles. G3 (Bethesda) 5, 551–558, doi: 10.1534/g3.114.016162 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ng F. S. et al. TRAP-seq Profiling and RNAi-Based Genetic Screens Identify Conserved Glial Genes Required for Adult Drosophila Behavior. Front Mol Neurosci 9, 146, doi: 10.3389/fnmol.2016.00146 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dobin A. & Gingeras T. R. Optimizing RNA-Seq Mapping with STAR. Methods Mol Biol 1415, 245–262, doi: 10.1007/978-1-4939-3572-7_13 (2016). [DOI] [PubMed] [Google Scholar]
- 97.Jin Y., Tam O. H., Paniagua E. & Hammell M. TEtranscripts: a package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics 31, 3593–3599, doi: 10.1093/bioinformatics/btv422 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Love M. I., Huber W. & Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550, doi: 10.1186/s13059-014-0550-8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.