

Abstract
To compare the immunopathology of immune checkpoint inhibitor‐induced myasthenia gravis (ICI‐MG) and idiopathic MG, we profiled the respective AChR autoantibody pathogenic properties. Of three ICI‐MG patients with AChR autoantibodies, only one showed complement activation and modulation/blocking potency, resembling idiopathic MG. In contrast, AChR autoantibody‐mediated effector functions were not detected in the other two patients, questioning the role of their AChR autoantibodies as key mediators of pathology. The contrasting properties of AChR autoantibodies in these cases challenge the accuracy of serological testing in establishing definite ICI‐MG diagnoses and underscore the importance of a thorough clinical assessment when evaluating ICI‐related adverse events.
Introduction
Myasthenia gravis (MG) is a rare autoimmune disease of the neuromuscular junction (NMJ). Autoantibodies that bind to the acetylcholine receptor (AChR) are detected in ~85% of MG patients. When MG is clinically suspected, autoantibody detection by radioimmunoassay (RIA) or cell‐based assay (CBA) establishes a definitive diagnosis. 1 Mechanistically, AChR autoantibodies are key mediators in the manifestation of MG, because IgG binding to AChR results in detrimental effects on the NMJ and failure of neuromuscular transmission. The pathogenic potential of AChR autoantibodies derives from their capacity to induce antigenic modulation (internalization), complement fixation, and receptor blocking. 2 Although no unequivocal trigger has yet been identified in the pathogenesis of the disease, a growing number of MG cases has been causally associated with the administration of immune checkpoint inhibitors (ICIs). 3 In clinical practice, ICIs have provided lasting remission for patients with metastatic and earlier‐stage cancers. 4 , 5
Notwithstanding these beneficial effects, ICI administration can cause off‐target toxicities termed immune‐related adverse events (irAEs), potentially affecting any organ. 6 ICI‐induced MG (ICI‐MG), albeit rare, stands out from other irAEs because of its high mortality rate of nearly 30%. 7 This poor outcome has been attributed to a frequent inflammatory overlap involving the skeletal and/or cardiac muscle, which is hardly ever found in idiopathic MG. Furthermore, AChR autoantibodies are frequently detected in ICI‐MG patients but, unlike idiopathic MG, the precise mechanisms through which they induce pathology are currently unknown. 7 , 8 Elucidating this immunological aspect may provide deeper insights into disease pathogenesis and ultimately lead to better treatment strategies. In this study, we leveraged a suite of experimental assays to measure autoantibody binding capacity, effector functions, and epitope specificity in ICI‐MG patients. Our results call into question the pathogenic role of AChR autoantibodies in patients diagnosed with ICI‐MG and suggest that a humoral and/or cellular factor—other than AChR autoantibodies—may be the true mechanistic driver of disease symptoms and outcome.
Methods
Study approval
This study was approved by the Institutional Review Boards of Yale University, Duke University, and the University of North Carolina at Chapel Hill. Patients or their legally authorized representatives consented to participate in the study.
Patient selection
In this multicenter retrospective case series study, three academic MG biorepositories were sourced to identify serum or plasma samples collected between 2016 and 2020 from patients diagnosed with MG following ICI treatment. MG diagnosis was based on a compatible clinical phenotype and at least one of the following: (1) a positive AChR or muscle‐specific tyrosine kinase (MuSK) autoantibody result by radioimmunoassay (RIA); (2) decremental response on repetitive nerve stimulation (RNS) or increased jitter on single‐fiber electromyography (SFEMG); and (3) clinical response to cholinesterase inhibitors.
Serological assays
All samples were tested on live clustered AChR, MuSK, and low‐density lipoprotein receptor‐related protein 4 (LRP4) CBAs using flow cytometry, as previously described. 9 For the LRP4 CBA, human embryonic kidney (HEK) 293T cells were transfected with human LRP4 plasmid (a generous gift from Dr. Stephan Kröger). 10 Experimental CBA controls included AChR‐ and MuSK‐specific human recombinant monoclonal autoantibodies (637 mAb and MuSK‐1A, respectively), 11 , 12 as well as an anti‐LRP4 recombinant mouse monoclonal antibody (Addgene plasmid #177513). AChR autoantibody‐mediated complement fixation was evaluated with a CBA as we have previously described. 13 Autoantibody‐mediated modulation and blocking of AChR were measured jointly on a human cell line. Briefly, CN21 cells derived from the TE671 rhabdomyosarcoma cell line were incubated at 37°C for 18 h in the presence of heat‐inactivated serum (or plasma) samples. Surface AChR expression was labeled with Alexa Fluor 647‐conjugated ‐bungarotoxin, and changes from baseline (untreated condition) were assessed by flow cytometry. Epitope mapping was performed using a Jurkat cell line genetically engineered to express the ectodomains of single AChR subunits. To test for the presence of AChR autoantibodies targeting the main immunogenic region (MIR) located at the extracellular end of subunits, competitive binding between serum (or plasma) samples and fluorescently labeled MIR‐specific 637 mAb was assessed on a live clustered AChR CBA. 14 For each assay evaluating autoantibody effector functions, sera from patients with an MG diagnosis following other cancer immunotherapies (OCI‐MG), ICI‐naïve AChR autoantibody‐positive (AChR+) MG patients, and healthy subjects were included as controls.
Results
Five ICI‐MG patients met the inclusion criteria. The median (range) age at symptom onset and sampling was 71 (36–86) years, and 4 (80%) were male. Table 1 summarizes patients' demographics, clinical features, laboratory findings, and experimental results. The control group included 32 patients with idiopathic MG and two OCI‐MG patients (OCI‐MG#1, on imatinib for leukemia, and OCI‐MG#2, on interferon alfa‐2A for metastatic melanoma). At diagnostic workup, 3 out of 5 ICI‐MG patients (60%) and 1 out of 2 OCI‐MG patients (50%) tested positive for AChR autoantibodies by RIA. Electrodiagnostic findings in the ICI‐MG cohort are summarized in Table 1. To improve autoantibody testing sensitivity, we performed a serological screening using live CBAs (Fig. 1; Table 1). RIA results were confirmed by a clustered AChR CBA, but neither AChR nor MuSK autoantibodies were detected in the sera of RIA‐based seronegative patients. Neither ICI‐MG nor OCI‐MG patients tested positive for LRP4 autoantibodies (Fig. S1). Within the AChR+ ICI‐MG subgroup, only one patient (ICI‐MG#3) showed complement fixation and modulation/blocking activity when these autoantibody effector functions were assessed in vitro, mirroring the case of OCI‐MG#1 (Fig. 2A–C; Table 1). For ICI‐MG#3 (and OCI‐MG#1, data not shown), epitope mapping demonstrated a polyclonal autoimmune response directed to multiple AChR subunits (Fig. 2D; Table 1), including the subunit. The patient's plasma and MIR‐specific 637 mAb showed competitive binding to AChR, suggesting the MIR as one of the target epitopes of ICI‐MG#3's autoantibody repertoire. (Fig. S2). Conversely, the two remaining AChR+ ICI‐MG patients (ICI‐MG#1 and ICI‐MG#2) showed neither autoantibody effector functions in vitro nor reactivities to single AChR subunits.
Table 1.
Demographics, clinical features, laboratory findings, and experimental data of ICI‐MG patients.
| ICI‐MG#1 | ICI‐MG#2 | ICI‐MG#3 | ICI‐MG#4 | ICI‐MG#5 | |
|---|---|---|---|---|---|
| Sex | Male | Male | Male | Male | Female |
| Age at symptom onset and sampling | 71 | 69 | 36 | 86 | 75 |
| Cancer | Renal cell carcinoma |
Prostate cancer |
Thymoma |
Prostate cancer |
Renal cell carcinoma |
| ICI type | Anti‐PD1/CTLA4 | Anti‐PD1/CTLA4 | Anti‐PD1 | Anti‐PD1 | Anti‐PD1/CTLA4 |
| ICI cycles before MG onset (time from first administration to symptoms) | 6 (4 months) | 1 (2 days) | 4 (2 years) | 3 (1.5 months) | 3 (1.5 months) |
| MGFA class at sampling | I | IIIB | IIIB | IIIB | IVA |
| Clinical and research laboratory findings | |||||
| AChR IgG RIA (nmol/L) 1 | 15.3 | 0.68 | 15.8 | Negative | Negative |
| MuSK IgG RIA (nmol/L) 1 | Not performed | Not performed | Not performed | Negative | Negative |
| AChR CBA (IgG titer) 2 | Positive (1:540) | Positive (1:180) | Positive (1:1620) | Negative | Negative |
| MuSK/LRP4 CBAs | Negative | Negative | Negative | Negative | Negative |
| Peak CK level (U/L) 3 since MG onset | Not available | 4063 U/L | 85 U/L 4 | 966 U/L | 37 U/L |
| AChR autoantibody effector functions and epitope specificity | |||||
| Complement fixation | No | No | Yes | NA | NA |
| Modulation/blocking | No | No | Yes | NA | NA |
| AChR single subunit reactivity | No | No | , | NA | NA |
| Electrodiagnostic findings | |||||
| Decrement on RNS | No | No | Not performed 4 | Yes | Yes |
| Abnormal SFEMG | Not performed | Mildly abnormal | Yes 4 | Yes | Not performed |
| Overlap syndromes | |||||
| Myopathy 5 | Insufficient data | Yes | No 4 | Yes | No |
| Myocarditis 6 | No | Yes | No | No 7 | No |
| Concomitant MG treatments at sampling | |||||
| Pyridostigmine | No | Yes | No | Yes | No |
| Immunosuppressants/IVIG/PLEX | No | Yes | Yes | Yes | Yes |
| Outcome; time from MG onset | No follow‐up | Death; 1 month | No follow‐up | Death; 2 months | Death; 2 weeks |
AChR, acetylcholine receptor; CBA, cell‐based assay; CK, creatine kinase; CTLA4, cytotoxic lymphocyte antigen‐4; ICI, immune checkpoint inhibitor; IVIG, intravenous immunoglobulin; LRP4, low‐density lipoprotein receptor‐related protein 4; MG, myasthenia gravis; MGFA, Myasthenia Gravis Foundation of America; MuSK, muscle‐specific tyrosine kinase; NA, not applicable; PD‐1, programmed cell death protein 1; PLEX, plasma exchange; RIA, radioimmunoassay; RNS, repetitive nerve stimulation; SFEMG, single‐fiber electromyography.
Autoantibody testing by radioimmunoassay; normal range <0.02 nmol/L.
IgG serum titer was measured through six threefold serial dilutions, ranging from 1:20 to 1:14.580.
Creatine kinase reference range: 30–220 U/L.
Two years prior to the detection of AChR autoantibodies and MG symptom onset, the patient developed non‐fluctuating bulbar and proximal limb weakness following the first ICI administration and was diagnosed with ICI‐related myositis (CK value: 2753 U/L; RNS and SFEMG tests were normal; AChR and MuSK autoantibodies were negative by RIA).
Diagnosis based on elevated creatine kinase levels and electrodiagnostic findings of myopathic motor unit potentials.
Diagnosis based on clinical, laboratory, and imaging findings.
Diagnosis of congestive heart failure.
Figure 1.
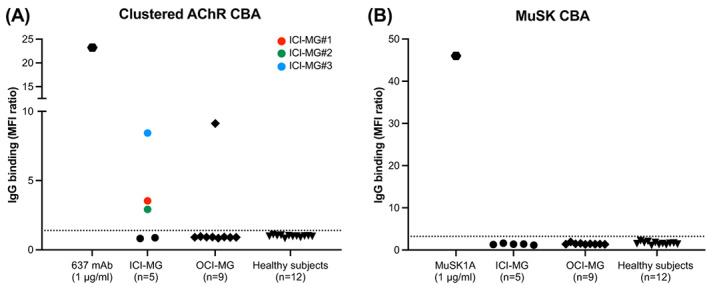
Clustered AChR and MuSK cell‐based assay results. Scatter plots of clustered AChR (A) and MuSK (B) cell‐based assay screening results using flow cytometry. Serum or plasma samples from ICI‐MG patients (n = 5), 2 OCI‐MG patients (n = 9 longitudinal samples), and healthy subjects (n = 12 samples) were tested at 1:20 dilution. Each symbol is the mean of duplicate experiments. Alexa Fluor 647‐conjugated anti‐human Fcγ was used as a secondary antibody to detect IgG binding to the AChR‐ or MuSK‐expressing cells. For every sample tested, the median Alexa Fluor 647 fluorescence intensity (MFI) was measured in two cell populations: GFP‐positive (transfected) and GFP‐negative (untransfected) HEK 293T cells. The ratio between the two MFIs was calculated (MFI ratio) and plotted on the Y‐axis. The positivity cutoff (dotted line) was set at the mean MFI ratio of healthy subjects plus 5 standard deviations. In the scatter plot of the clustered AChR CBA, three ICI‐MG patients are above the positivity cutoff (ICI‐MG#1, #2, #3, shown as a red, green, and blue dot symbols respectively).
Figure 2.
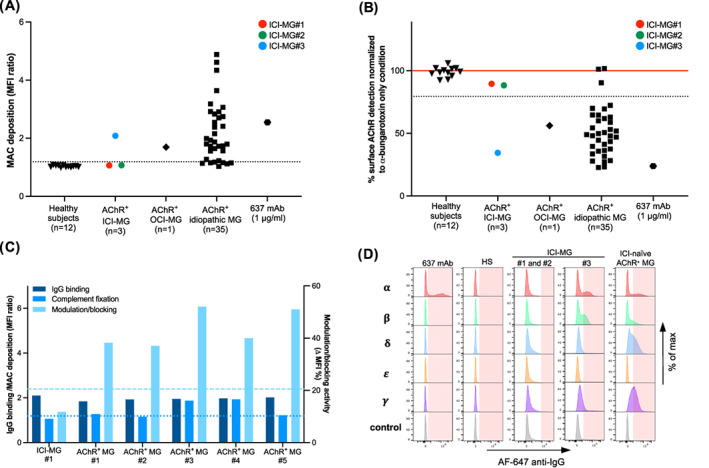
Evaluation of autoantibody effector functions and epitope specificity in AChR+ ICI‐MG patients. (A) Flow cytometric measurement of AChR autoantibody‐mediated complement fixation. Serum or plasma samples from AChR+ ICI‐MG patients (n = 3 samples), AChR+ OCI‐MG patient (n = 1 sample), 32 idiopathic AChR+ MG patients (n = 35 samples), and healthy subjects (n = 12 samples) were included. Each symbol is the mean of experimental triplicates. Membrane attack complex (MAC) deposition was detected using a C9 neoantigen‐specific antibody and a secondary fluorophore‐labeled antibody. For every sample tested, the median fluorescence intensity (MFI) was measured in two cell populations: GFP‐positive (transfected) and GFP‐negative (untrasfected) HEK 293T cells. The MFI ratio, which expresses the magnitude of MAC deposition, is plotted on the Y‐axis. The cutoff for positivity (dotted line) was set at the mean MFI ratio of healthy subjects plus 5 standard deviations. (B) Flow cytometric measurement of combined autoantibody‐mediated modulation and blocking activity. Comparison of antigenic modulation/blocking activity using serum or plasma samples from AChR+ ICI‐MG patients (n = 3 samples), AChR+ OCI‐MG patient (n = 1 sample), 32 idiopathic AChR+ MG patients (n = 35 samples), and healthy subjects (n = 12 samples). The red line corresponds to the baseline signal of Alexa Fluor 647‐conjugated ‐bungarotoxin labeling untreated cells (no incubation with serum). Patients' MFI values were normalized to the baseline signal. Each symbol is the mean of experimental triplicates. The cutoff for positivity (dotted line) was set at the mean MFI ratio of healthy subjects minus 5 standard deviations. (C) Comparison of IgG binding strength and effector functions in a subgroup of patients including one AChR+ ICI‐MG patient (ICI‐MG#1) and five ICI‐naïve AChR+ MG patients (AChR+ MG#1 to #5). The left Y‐axis shows IgG binding strength and MAC deposition, while the right Y‐axis indicates the magnitude of modulation/blocking activity (MFI %: baseline MFI– MFI of each sample %). The bar graph shows patients with similar IgG binding strength on the CBA, but a variable degree of autoantibody effector functions ranging from high to undetectable values. The positivity cutoffs for IgG binding and complement fixation coincide and are shown by a single dotted line. The positivity cutoff for modulation/blocking activity is shown by a dashed line. (D) Representative flow cytometric histograms of AChR subunit reactivities using Jurkat cell lines expressing the ectodomains of adult () and fetal ( AChR subunits. IgG binding to singly expressed subunits was detected using Alexa Fluor 647‐conjugated anti‐human Fcγ secondary antibody on flow cytometry. For each sample, the positivity threshold gate (light red shaded area) was set based on the negative control (Jurkat cells expressing no AChR subunit). From left to right: (1) 637 mAb with known epitope‐specificity: ‐binder; (2) lack of reactivity to any subunit (examples include one representative healthy subject, ICI‐MG#1, and ICI‐MG#2); and (3) reactivity to multiple subunits (ICI‐MG#3: reactivities to ; one representative ICI‐naïve AChR+ MG patient: reactivities to , and ).
Discussion
The immunological features of ICI‐MG remain largely unknown. To address this absence, we performed a serological assessment of five symptomatic patients diagnosed with ICI‐MG using highly sensitive, quantitative assays. We confirmed AChR autoantibody seropositivity in three patients, while two were triple‐seronegative. Within the AChR+ ICI‐MG subgroup, autoantibody‐mediated complement fixation and modulation/blocking potency were detected in only one patient (ICI‐MG#3). Mirroring the case of idiopathic MG patients, epitope mapping of ICI‐MG#3 showed targeting of multiple AChR subunits, including the MIR on the subunits. Notably, this patient, the youngest in the ICI‐MG cohort, first presented with ICI‐related myositis, and MG was diagnosed after 2 years from ICI initiation (Table 1). Because the patient had thymic cancer, the possibility of thymoma‐associated disease merits acknowledgment. Despite no conclusive link between ICI and MG, this case nonetheless underscores the importance of extended clinical surveillance for potential late‐onset irAEs.
Conversely, the other two AChR+ ICI‐MG patients (ICI‐MG#1 and ICI‐MG#2) showed CBA‐confirmed IgG binding to extracellular AChR epitopes but undetectable autoantibody effector functions. RNS testing did not reveal a functional NMJ defect in either patient. Based on laboratory and electromyographic findings, a diagnosis of concurrent myositis was made for ICI‐MG#2. In this patient, SFEMG performed on a clinically weak facial muscle was only mildly abnormal, suggesting that the facial weakness owed primarily to the concomitant myopathic process (Table 1).
Taken together, such findings cast doubt on the direct involvement of AChR autoantibodies in the muscle pathology of ICI‐MG#1 and ICI‐MG#2 and corroborate the hypothesis that, in specific contexts, AChR autoantibodies are not the key mediators of disease but rather represent a bystander autoimmune epiphenomenon. 15 This hypothesis is further substantiated by the two‐day lag between ICI initiation and symptom onset in ICI‐MG#2, which suggests that AChR autoantibodies predated ICI administration and argues against a de novo humoral response to AChR. Overall, experimental results and available clinical data make it tempting to reassess the clinical manifestations of ICI‐MG#1 and ICI‐MG#2 as oculobulbar presentations of ICI‐associated myopathy and challenge the diagnostic role of AChR autoantibodies in post‐ICI muscle weakness. 16 These observations do not preclude the existence of true overlap syndromes characterized by a detrimental autoimmune response affecting both the NMJ and skeletal muscle fibers. Such cases implicate diagnostic and therapeutic pitfalls that warrant future research.
Immunologically, the reason why AChR autoantibodies bind to their target but do not exert apparent effector functions in vitro remains enigmatic. Corroborating our earlier findings, 13 this study further confirmed that AChR+ MG patients may exhibit heterogeneity in the magnitude of autoantibody effector functions despite sharing similar IgG binding capacity on CBA (Fig. 2C). Several factors including IgG titer and subclasses, affinity maturation, epitope specificity, post‐translational modifications, and treatment status may play a role in conferring pathogenic potential (or lack thereof) to AChR autoantibodies. Recently, synergy among autoantibodies of different epitope specificities has been proposed as a further mechanism that promotes autoantibody binding and enhances effector functions. 17 , 18 Whether this represents an absolute requirement for autoantibody pathogenicity in MG awaits confirmation. Interestingly, both ICI‐MG#1 and ICI‐MG#2 samples showed IgG binding to AChR in its native conformation but no reactivity to individual AChR subunits (Fig. 2D), potentially indicating an autoimmune response to epitopes derived from the interaction of adjacent subunits.
This study has several limitations, including its retrospective design, the limited number of patients due to the rarity of ICI‐MG, and the absence of biospecimens for histopathology evaluation. In addition, we cannot exclude that the lack of measurable effector functions and subunit specificities in some patients was due to suboptimal assay sensitivity. In this respect, we recognize that our experimental assays lack the molecular complexity of the NMJ (which is a highly specialized synapse), and co‐expression of additional NMJ proteins—other than the autoantibody target—may be required to effectively trigger autoantibody effector functions, as well as to measure them more accurately. Of note, we also identified a subset of ICI‐naïve AChR+ MG patients harboring autoantibodies that did not demonstrate pathogenic capacity in our in vitro assays, and fitting with this observation, these patients had no clinical evidence of muscle weakness at sampling. Conversely, in the AChR+ ICI‐MG subgroup, all patients were overtly symptomatic. Prospective investigations on larger ICI‐MG cohorts, along with direct evaluation of IgG‐mediated effects on muscle endplates, are warranted to confirm these findings. Furthermore, given the identification of triple‐seronegative ICI‐MG cases (ICI‐MG#4 and ICI‐MG#5), future studies will also be necessary to elucidate the target and mechanism(s) of muscle damage in such patients.
In sum, our study shows that patients diagnosed with ICI‐MG may harbor AChR autoantibodies with molecular properties similar to those found in idiopathic MG. In a subset of ICI‐treated patients, however, AChR autoantibodies lack pathogenic effector functions in vitro, suggesting that other humoral and/or cellular factors, including those not related to established MG pathology, may have a more prominent role in their disease. Translated into clinical practice, these findings challenge the accuracy of serological testing alone in establishing a definite ICI‐MG diagnosis and corroborate the need for more in‐depth ancillary investigations when evaluating muscle‐related irAEs. 19
Authors Contributions
Concept and design: Kevin C. O'Connor and Gianvito Masi. Acquisition, analysis, or interpretation of data: All authors. Initial drafting of the manuscript: Gianvito Masi. Critical revision of the manuscript for important intellectual content and final approval: All authors. Supervision: Kevin C. O'Connor and Gianvito Masi.
Conflicts of Interest
Dr. Gianvito Masi, Minh C. Pham, and Tabitha Karats report no disclosures. Dr. Sangwook Oh has received patent licensing payments from Cabaletta Bio. Dr. Aimee S. Payne has received equity, research support, patent licensing and other payments from Cabaletta Bio; patent licensing payments from Novartis; and consultant fees from Janssen. Dr. Richard J. Nowak has received research support from the NIH, Genentech, Alexion Pharmaceuticals, argenx, Annexon Biosciences, UCB Ra Pharmaceuticals, Myasthenia Gravis Foundation of America, Momenta, Immunovant, Grifols, and Viela Bio, now (Horizon Therapeutics). RJN has served as consultant/ advisor for Alexion Pharmaceuticals, argenx, Cabaletta Bio, CSL Behring, Grifols, Ra Pharmaceuticals, now a part of UCB Pharma, Immunovant, Momenta, and Viela Bio, now a part of Horizon Therapeutics. Dr. James F. Howard Jr. has received research support from Alexion Pharmaceuticals, argenx BVBA, the Myasthenia Gravis Foundation of America, the Muscular Dystrophy Association, the National Institutes of Health (including the National Institute of Neurological Disorders and Stroke and the National Institute of Arthritis and Musculoskeletal and Skin Diseases), PCORI, Ra Pharmaceuticals (now UCB) and Takeda Pharmaceuticals; Honoraria from Alexion Pharmaceuticals, argenx BVBA, Immunovant Inc., Ra Pharmaceuticals (now UCB), Regeneron Pharmaceuticals, Sanofi US and Viela Bio Inc. and non‐financial support from Alexion Pharmaceuticals, argenx BVBA, Ra Pharmaceuticals (now UCB) and Toleranzia AB. Dr. Jeffrey T. Guptill has received consulting fees/honoraria from Immunovant, Alexion, Apellis, Momenta, Ra Pharma, Cabaletta, Regeneron, argenx, Janssen, UCB, and Toleranzia. JTG receives industry grant support from UCB pharma for a fellowship training grant; is a site investigator for Alexion, Janssen, UCB Pharma, argenx, Takeda and grant/research support from: NIH (NIAID, NINDS, NIMH), MGFA, CDC. Dr. Vern C. Juel has received research support from the MGFA and NIH RDN for Myasthenia Gravis. He has served as consultant/advisor for Accordant Health Services, Alexion and Immunovant. VCJ is a site principal investigator for Alexion, Janssen, and argenx. Dr. Kevin C. O'Connor has received research support from Ra Pharma, now (UCB Pharma), Alexion, now (AstraZeneca), Viela Bio, now (Horizon Therapeutics), and argenx. KCO is a consultant and equity shareholder of Cabaletta Bio. KCO has served as a consultant/advisor for Alexion Pharmaceuticals, now (AstraZeneca), and Roche.
Role of the Funder/Sponsor
The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Supporting information
Figure S1.
Acknowledgements
Dr. Gianvito Masi is an MGNet Scholar Awardee. The MGNet is a member of the Rare Disease Clinical Research Network Consortium (RDCRN) NIH U54 NS115054. All RDCRN consortia are supported by the network's Data Management and Coordinating Center (DMCC) (U2CTR002818). Funding support for the DMCC is provided by the National Center for Advancing Translational Sciences (NCATS) and the National Institute of Neurological Disorders and Stroke (NINDS). Dr. Kevin C. O'Connor is supported by the National Institute of Allergy and Infectious Diseases of the NIH under award numbers R01‐AI114780 and R21‐AI164590; through an award provided through the Rare Diseases Clinical Research Consortia of the NIH and MGNet (award number U54‐NS115054); by a High Impact Clinical Research and Scientific Pilot Project award and Seronegative MG award both from the Myasthenia Gravis Foundation of American (MGFA).
Funding Statement
This work was funded by MGNet grants GR116693 and U54‐NS115054; National Institute of Allergy and Infectious Diseases grants R01‐AI114780 and R21‐AI164590.
References
- 1. Punga AR, Maddison P, Heckmann JM, Guptill JT, Evoli A. Epidemiology, diagnostics, and biomarkers of autoimmune neuromuscular junction disorders. Lancet Neurol. 2022;21(2):176‐188. [DOI] [PubMed] [Google Scholar]
- 2. Huijbers MG, Marx A, Plomp JJ, le Panse R, Phillips WD. Advances in the understanding of disease mechanisms of autoimmune neuromuscular junction disorders. Lancet Neurol. 2022;21(2):163‐175. [DOI] [PubMed] [Google Scholar]
- 3. Dubey D, David WS, Reynolds KL, et al. Severe neurological toxicity of immune checkpoint inhibitors: growing Spectrum. Ann Neurol. 2020;87(5):659‐669. [DOI] [PubMed] [Google Scholar]
- 4. Larkin J, Chiarion‐Sileni V, Gonzalez R, et al. Five‐year survival with combined Nivolumab and Ipilimumab in advanced melanoma. N Engl J Med. 2019;381(16):1535‐1546. [DOI] [PubMed] [Google Scholar]
- 5. Puri S, Shafique M, Gray JE. Immune checkpoint inhibitors in early‐stage and locally advanced non‐small cell lung cancer. Curr Treat Options Oncol. 2018;19(8):39. [DOI] [PubMed] [Google Scholar]
- 6. Postow MA, Sidlow R, Hellmann MD. Immune‐related adverse events associated with immune checkpoint blockade. N Engl J Med. 2018;378(2):158‐168. [DOI] [PubMed] [Google Scholar]
- 7. Marini A, Bernardini A, Gigli GL, et al. Neurologic adverse events of immune checkpoint inhibitors: a systematic review. Neurology. 2021;96(16):754‐766. [DOI] [PubMed] [Google Scholar]
- 8. Safa H, Johnson DH, Trinh VA, et al. Immune checkpoint inhibitor related myasthenia gravis: single center experience and systematic review of the literature. J Immunother Cancer. 2019;7(1):319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Masi G, Li Y, Karatz T, et al. The clinical need for clustered AChR cell‐based assay testing of seronegative MG. J Neuroimmunol. 2022;367:577850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pevzner A, Schoser B, Peters K, et al. Anti‐LRP4 autoantibodies in AChR‐ and MuSK‐antibody‐negative myasthenia gravis. J Neurol. 2012;259(3):427‐435. [DOI] [PubMed] [Google Scholar]
- 11. Graus YF, de Baets MH, van Breda Vriesman PJ, Burton DR. Anti‐acetylcholine receptor fab fragments isolated from thymus‐derived phage display libraries from myasthenia gravis patients reflect predominant specificities in serum and block the action of pathogenic serum antibodies. Immunol Lett. 1997;57(1–3):59‐62. [DOI] [PubMed] [Google Scholar]
- 12. Takata K, Stathopoulos P, Cao M, et al. Characterization of pathogenic monoclonal autoantibodies derived from muscle‐specific kinase myasthenia gravis patients. JCI Insight. 2019;4(12):e127167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Obaid AH, Zografou C, Vadysirisack DD, et al. Heterogeneity of acetylcholine receptor autoantibody‐mediated complement activity in patients with myasthenia gravis. Neurol Neuroimmunol Neuroinflamm. 2022;9(4):e1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luo J, Taylor P, Losen M, de Baets MH, Shelton GD, Lindstrom J. Main immunogenic region structure promotes binding of conformation‐dependent myasthenia gravis autoantibodies, nicotinic acetylcholine receptor conformation maturation, and agonist sensitivity. J Neurosci. 2009;29(44):13898‐13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mammen AL, Rajan A, Pak K, et al. Pre‐existing antiacetylcholine receptor autoantibodies and B cell lymphopaenia are associated with the development of myositis in patients with thymoma treated with avelumab, an immune checkpoint inhibitor targeting programmed death‐ligand 1. Ann Rheum Dis. 2019;78(1):150‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shelly S, Triplett JD, Pinto MV, et al. Immune checkpoint inhibitor‐associated myopathy: a clinicoseropathologically distinct myopathy. Brain Commun. 2020;2(2):fcaa181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kreye J, Wright SK, van Casteren A, et al. Encephalitis patient‐derived monoclonal GABAA receptor antibodies cause epileptic seizures. J Exp Med. 2021;218(11):e20210012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rose N, Holdermann S, Callegari I, et al. Receptor clustering and pathogenic complement activation in myasthenia gravis depend on synergy between antibodies with multiple subunit specificities. Acta Neuropathol. 2022;144(5):1005‐1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guidon AC, Burton LB, Chwalisz BK, et al. Consensus disease definitions for neurologic immune‐related adverse events of immune checkpoint inhibitors. J Immunother Cancer. 2021;9(7):e002890. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.