

Abstract
Herein, we present an efficient and mild methodology for the synthesis of aromatic phosphonate esters in good to excellent yields using 10H-phenothiazine, an inexpensive commodity chemical, as a photoredox catalyst. The reaction exhibits wide functional group compatibility enabling the transformation in the presence of ketone, amide, ester, amine, and alcohol moieties. Importantly, the reaction proceeds using a green solvent mixture primarily composed of water, thus lowering the environmental footprint of this transformation compared to current methods. The transformation also proceeds under atmospheric conditions, which further differentiates it from current methods that require inert atmosphere. Mechanistic work using fluorescence quenching experiments and radical trapping approaches support the proposed mechanism.
Keywords: Radical phosphonation, Photoredox, Atmospheric conditions, Aqueous solvent, Ammonium salts, Inexpensive photocatalyst
Graphical Abstract
INTRODUCTION
The aromatic phosphonate ester motif is found in a wide variety of pharmaceuticals, photoelectric materials, agrochemical compounds, and in ligands for metal catalysis.1–5 For these reasons, the synthesis of C–P bonds has attracted significant interest in the past decade.6–10 Traditional ways to access aryl phosphonates requires the activation of P–H bonds in reagents such as diethyl phosphite (H(O)P(OEt)2) using transition-metal catalysis (Scheme 1A). Coupling partners for these reactions have been widely explored and include aryl halides, aryl triflates, and aryl boronic acids.11–14 However, the expensive and sometimes toxic nature of the catalysts and reagents required for these transformations, alongside substrate scope limitations because of harsh reaction conditions, have encouraged the development of milder methodologies. In the past decade, photoredox-catalyzed methods to generate aryl phosphonates has gained traction as an attractive alternative to traditional methods because of their milder conditions and broader functional group tolerance (Scheme 1B).15–22 Pioneering work by Toste achieved phosphonation of aryldiazonium salts using H(O)P(OEt)2 in the presence of a gold(I) catalyst and a Ru photocatalyst.15 Later work by König achieved these transformations using a Ru photocatalyst and a rhodamine dye.16,17 More recently, the combination of photoredox catalysis and electrochemistry enabled Wickens to achieve the phosphonation of aryl chlorides.18
Scheme 1.
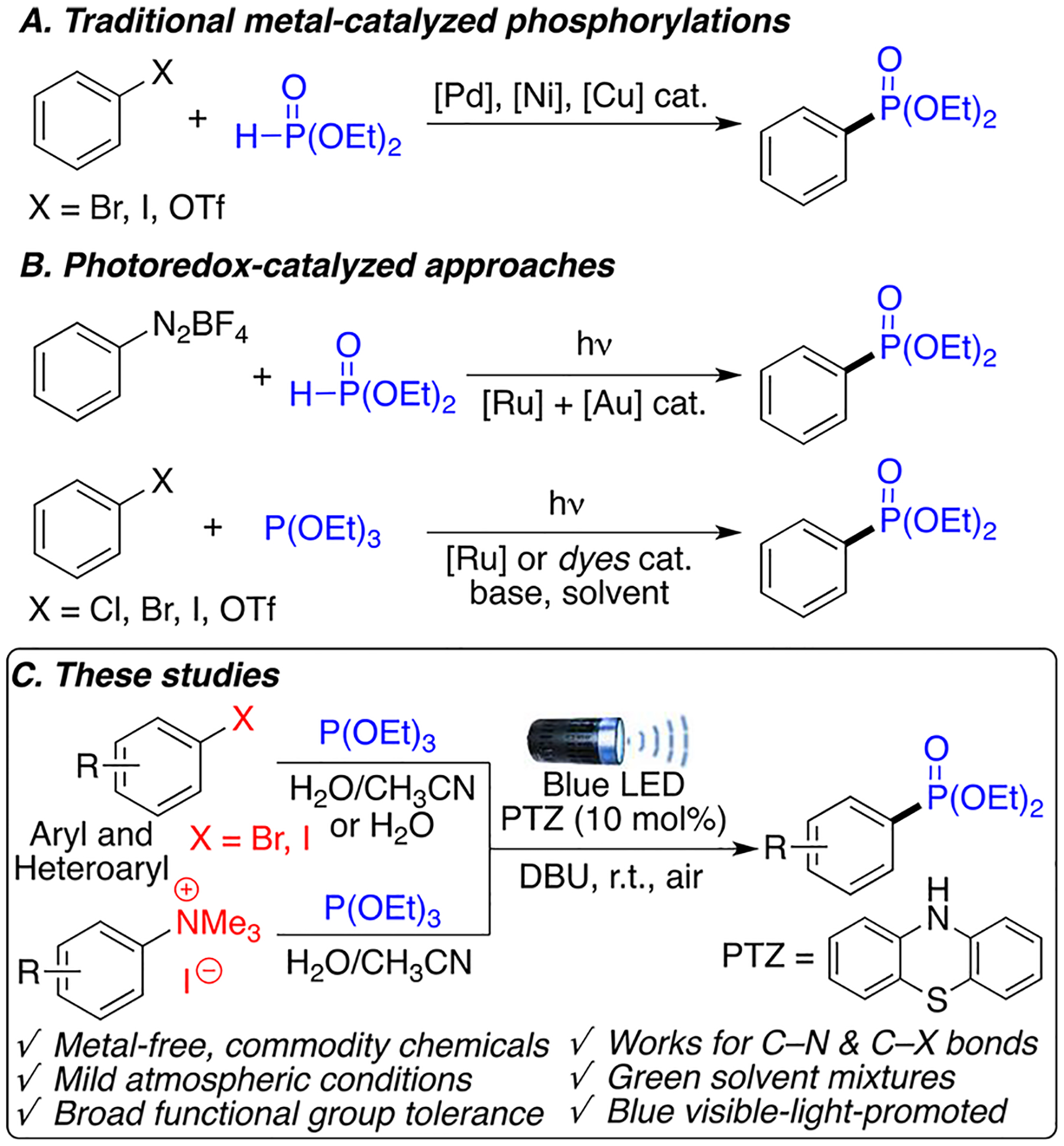
Current and Proposed Approaches to Aryl C–P Bond Formation: (A) Traditional Metal-Catalyzed Phosphorylations, (B) Photoredox-Catalyzed Approaches, and (C) These Studies
Despite all the progress achieved by these methods, their environmental footprint is far from desirable. The uses of rare metals, problematic solvents, highly toxic organic dyes,23 or anhydrous and inert atmosphere remain challenges to the green synthesis of these valuable and industrially relevant compounds. Importantly, improving the sustainability of these transformations has to be achieved without compromising substrate scope and functional group compatibility.
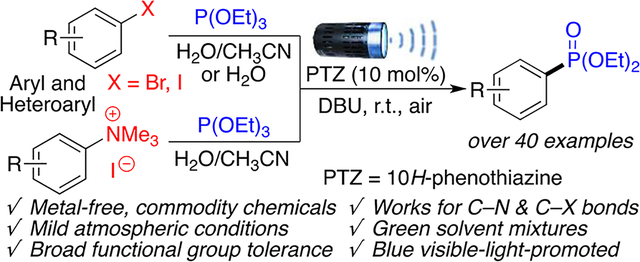
Our interest in the development of metal-free green synthetic methodologies,24–26 recently led us to explore new approaches for the radical activation of aryl halides.25 Here, we report the first metal-free photoredox-catalyzed phosphonation of aryl trimethylammonium salts and aryl halides (iodides and bromides) using affordable phosphite reagents in green aqueous solvent mixtures. This reaction uses 10H-phenothiazine (PTZ), a common and inexpensive commodity chemical, as a photoredox catalyst. In addition to lowering the environmental footprint provided by the possible use of water as a solvent, this transformation proceeds unaffected under atmospheric conditions without the need for degassing or distilling of the solvents, or placing the reaction under inert atmosphere. These mild reaction conditions also provide broad functional group tolerance in the presence of reactive ketone, amide, ester, amine, and alcohol moieties.
RESULTS AND DISCUSSION
Our initial studies aimed at the phosphonation of 4-iodoanisole (1a) using triethyl phosphite (2a) as our phosphorus containing reagent (Table 1). To achieve our objectives of performing this transformation under the most environmentally friendly and cost-effective conditions possible, we focused our attention to the screening of bases, solvents, and atmospheric conditions using PTZ as our photocatalyst (< $90/kg).27
Table 1.
Optimization of the Reaction Conditionsa
|
|||||
|---|---|---|---|---|---|
| entry | base (equiv) | 2a (equiv) | solvent | 3 yield (%)b | 1a recovered (%)b |
| 1 | DBU (3.0) | 3.0 | CH3CN | 89 (A) | - |
| 2 | Et3N (3.0) | 3.0 | CH3CN | 10 (A) | 90 |
| 3 | DIPEA (3.0) | 3.0 | CH3CN | 12 (A) | 87 |
| 5 | K2CO3 (3.0) | 3.0 | CH3CN | 34 (B) | 60 |
| 6 | LiOtBu (3.0) | 3.0 | CH3CN | 58 (B) | 39 |
| 7 | - | 3.0 | CH3CN | 9 (B) | 90 |
| 8c | DBU (3.0) | 3.0 | CH3CN | 6 (B) | 83 |
| 9d | DBU (3.0) | 3.0 | CH3CN | - (B) | 97 |
| 10e | DBU (3.0) | 3.0 | CH3CN | 85 (B) | trace |
| 11 | DBU (2.0) | 3.0 | CH3CN | 92 (B) | trace |
| 12 | DBU (3.0) | 2.0 | CH3CN | 66 (B) | 23 |
| 13e | DBU (2.0) | 3.0 | CH3CN/H2O (1:3) | 90 (B) | 4 |
| 14e | DBU (2.0) | 3.0 | CH3CN/H2O (1:9) | 87 (B) | 6 |
| 15e | DBU (2.0) | 3.0 | H2O | 72 (B) | 15 |
Reaction conditions: 1a (0.2 mmol), 2a (0.6 mmol), DBU (0.6 mmol), solvent (1 mL), room temperature around the reaction flask is 35 °C (heating caused by the 456 (A) or 427 nm (B) LED lamp), under argon, 24 h.
Yields are based on 1a, determined by 1H NMR using dibromomethane as an internal standard.
The reaction was performed in the absence of PTZ.
The reaction was performed in the dark, covered by aluminum foil.
The reaction was performed in air.
Gratifyingly, the reaction proceeded in excellent yield (89%) in acetonitrile using DBU (1,8-diazabicyclo(5.4.0)undec-7-ene) as a base (entry 1). Four other organic and inorganic bases were screened providing moderate yields for the desired product and poor conversions of the starting material 1a (Table 1, entries 2–6). The main byproduct of these reactions is the proto-dehalogenation of the aryl halide 1a. In the absence of base (entry 7) or photocatalyst (entry 8), the yields decreased dramatically (<10%), with an almost complete recovery of the starting material. Similarly, no desired product was obtained when the reaction was performed in the dark (entry 9). Importantly, the transformation did not seem to be negatively affected by the use of atmospheric conditions (entry 10). While reducing the amount of DBU to 2.0 equiv improved the yield (entry 11), reducing the amount of phosphite 2a had a deleterious effect on yield (entry 12). The reaction was also minimally affected by the addition of water (entries 13–14). Still, the use of water as the sole solvent (entry 15) afforded the desired product in good yield with minimal decomposition of 1a (72% product yield and 15% recovered starting material). However, the used of other solvents and bases resulted in lower yields (see Supporting Information for full table of optimization, S6).
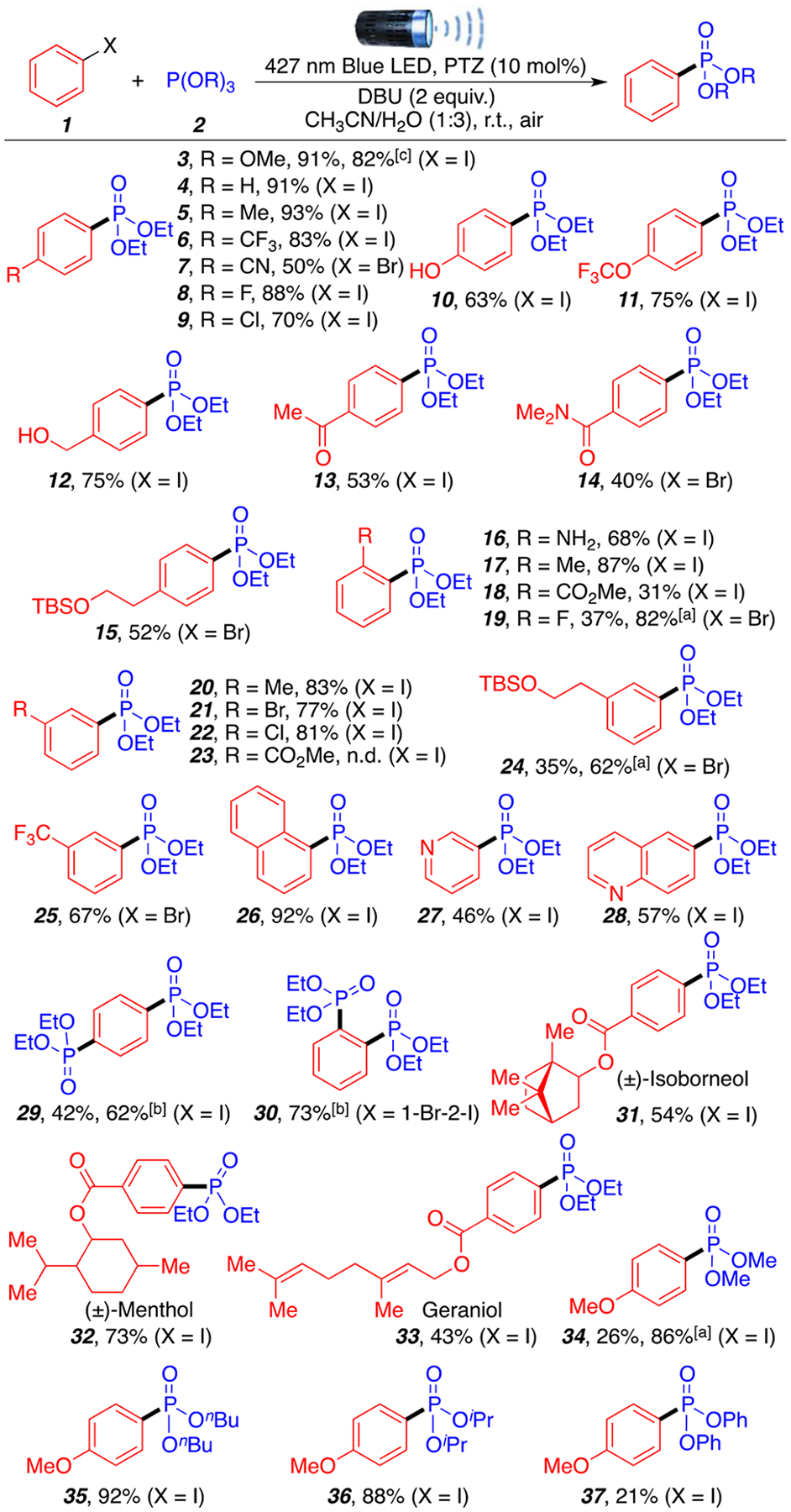
With a wide range of possible reaction conditions available, we chose to explore the substrate scope using the aqueous mixture of CH3CN/H2O (1:3) as it provides a good overview on how efficient the transformation can be, and allows for future possible substrate optimization in acetonitrile or water as sole solvents. A variety of aryl iodides and bromides (1), as well as different phosphite reagents (2) were investigated (Scheme 2). Various aryl iodides and bromides were first reacted with triethyl phosphite (2a) as the coupling partner. The reaction proceeds smoothly in the presence of electrondonating groups (3–5, 91–93%) and electron-withdrawing groups (6–9, 50–88%). A large-scale (5 mmol, 1.17 g) reaction was also performed under optimal reaction conditions and afforded product 3 in 82% yield (see SI, S5 for large scale procedure). Acidic and protic functional groups such as phenol and alcohol (10, 12) were well tolerated (63–75%). Ketone (13) and amide (14) moieties afforded the product in moderate yield (40–53%). Of interest to synthetic chemist, silyl protected alcohols (15, 24) afforded the desired phosphonated products in good yields (52–62%). It is important to highlight that all the yields can be further enhanced by improving the solubility of the starting substrates through the use of acetonitrile as main solvent. Sterics at both ortho (16–19) and meta (20–25) positions were well tolerated (31–87%) even in the presence of aniline group; the only exception was substrate 23. 2-Iodonaphthalene afforded the desired product is excellent yield (26, 92%), and the method tolerated heteroaromatic pyridine (27) and quinoline (28) moieties often found in bioactive compounds (46–57%). The reaction of 1,4-diiodobenzene and 1-bromo-2-iodobenzene afforded the disubstituted products 29 and 30 in moderate to good yield (62–73%). Moreover, various medicinal and natural product frameworks containing esters were investigated (31–33) and generated the desired phosphonated products in moderate to good yield (43–73%), further showing functional group compatibility and the possibility for late-stage functionalization of complex molecules. It is important to highlight that the reactivity of the aryl halide substrates increases as the halide becomes a better leaving group (ArI > ArBr > ArCl ≫ ArF), with aryl chlorides providing only trace amounts of products and aryl fluorides remaining completely unreactive (see SI for additional substrates and experiments; S23). Finally, different trialkyl and triaryl phosphite reagents were tested (34–37) affording the desired products in low to excellent yields (21–92%).
Scheme 2.
Aryl Halides Substrate Scope and Functional Group Compatibilitya
aReaction conditions: aryl halides 1 (0.2 mmol, 1 equiv), trialkyl phosphites 2 (0.6 mmol, 3 equiv), DBU (0.4 mmol, 2 equiv), PTZ (0.02 mmol, 10 mol %), CH3CN/H2O (0.25/0.75 mL), room temperature around reaction flask is 35 °C (heating caused by the 427 nm LED lamp), air, 24 h. All yields are isolated. [a] CH3CN/H2O (0.9/0.1 mL). [b] Trialkyl phosphites 2 (1.2 mmol, 6 equiv). [c] Gram scale reaction: 1 (5 mmol), 2 (10 mmol), DBU (10 mmol), PTZ (0.5 mmol), CH3CN/H2O (12/4 mL), 48 h.
Subsequently, we decided to apply this procedure to the phosphonation of aryl trimethylammonium salts. To this day, there are no examples of photoredox-catalyzed phosphonations of aryl ammonium salts. Current C–N phosphonation examples are limited to the use of aryl diazonium salts and hydrazine derivatives using transition-metal catalyzed approaches.15,28–31 To our delight, the reaction successfully proceeds in comparable yields as with aryl halides, although a change in solvent mixture and light source was required. Indeed, quaternary arylammonium salts requires the use of CH3CN as the major solvent and a 390 nm purple light source (Scheme 3).
Scheme 3.
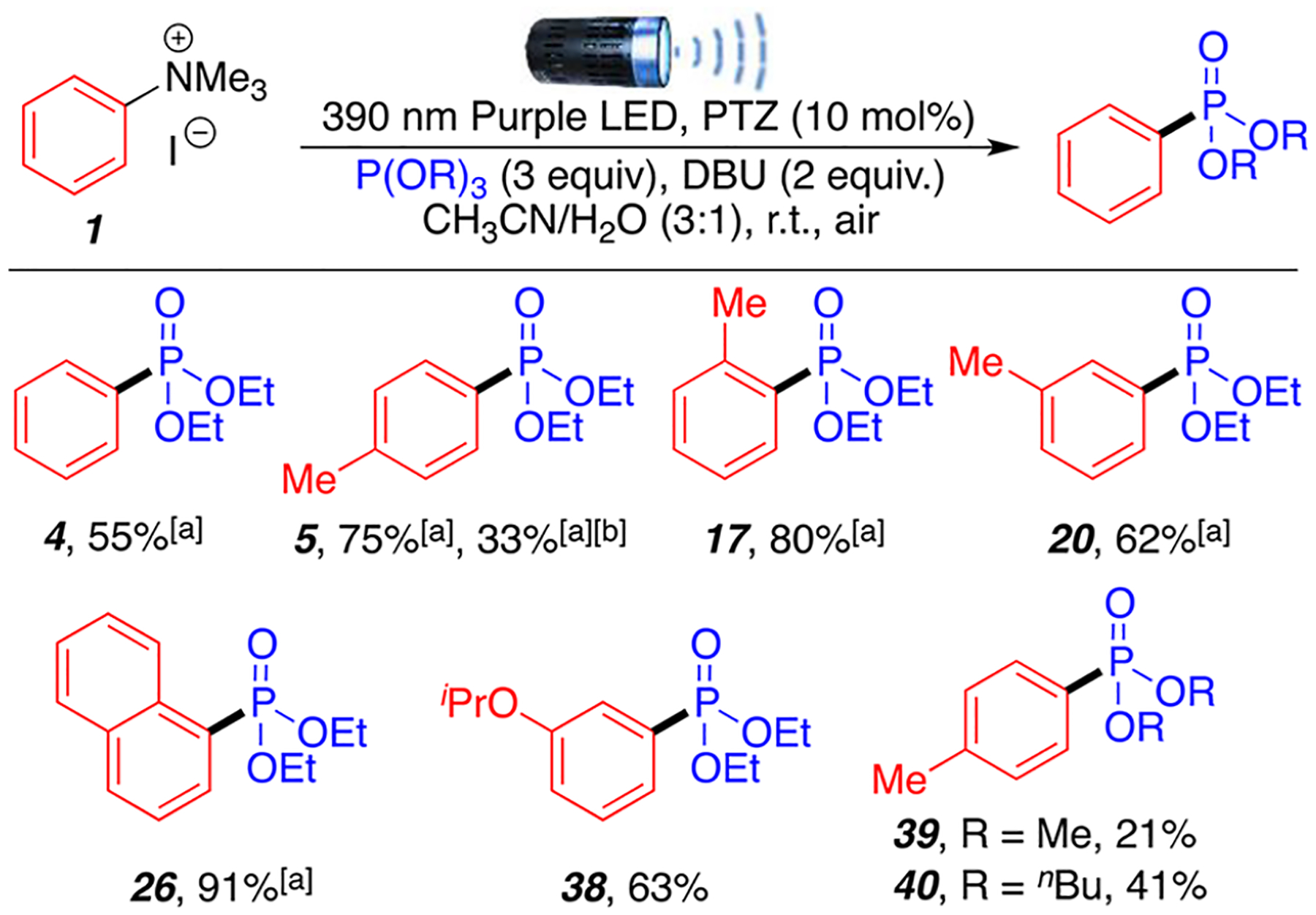
Aryl Trimethylammonium Salt Substrate Scopea
aReaction conditions: aryl ammonium salt 1 (0.2 mmol, 1 equiv), trialkyl phosphites 2 (0.6 mmol, 3 equiv), DBU (0.4 mmol, 2 equiv), PTZ (0.02 mmol, 10 mol %), CH3CN/H2O (0.75/0.25 mL), room temperature around flask is 35 °C (heating caused by the 390 nm LED lamp), air, 24 h. All yields are isolated unless noted otherwise. [a] Yields are based on 1, determined by 1H-NMR using dibromomethane as internal standard. [b] CH3CN/H2O (0.25/0.75 mL) with 427 nm LED lamp.
To explore the mechanism for this transformation, we carried out a series of radical trapping and fluorescence quenching experiments (Scheme 4). The addition of four different radical trapping reagents (TEMPO, 1,1-DPE, 1,4-DNB, and BHT) hampered the reactions and aryl radical intermediates were successfully trapped and observed via GC-MS (Scheme 4A). Also, the complete quenching of the reaction by 1,4-DNB strongly suggests the formation of a radical anion intermediate.
Scheme 4.
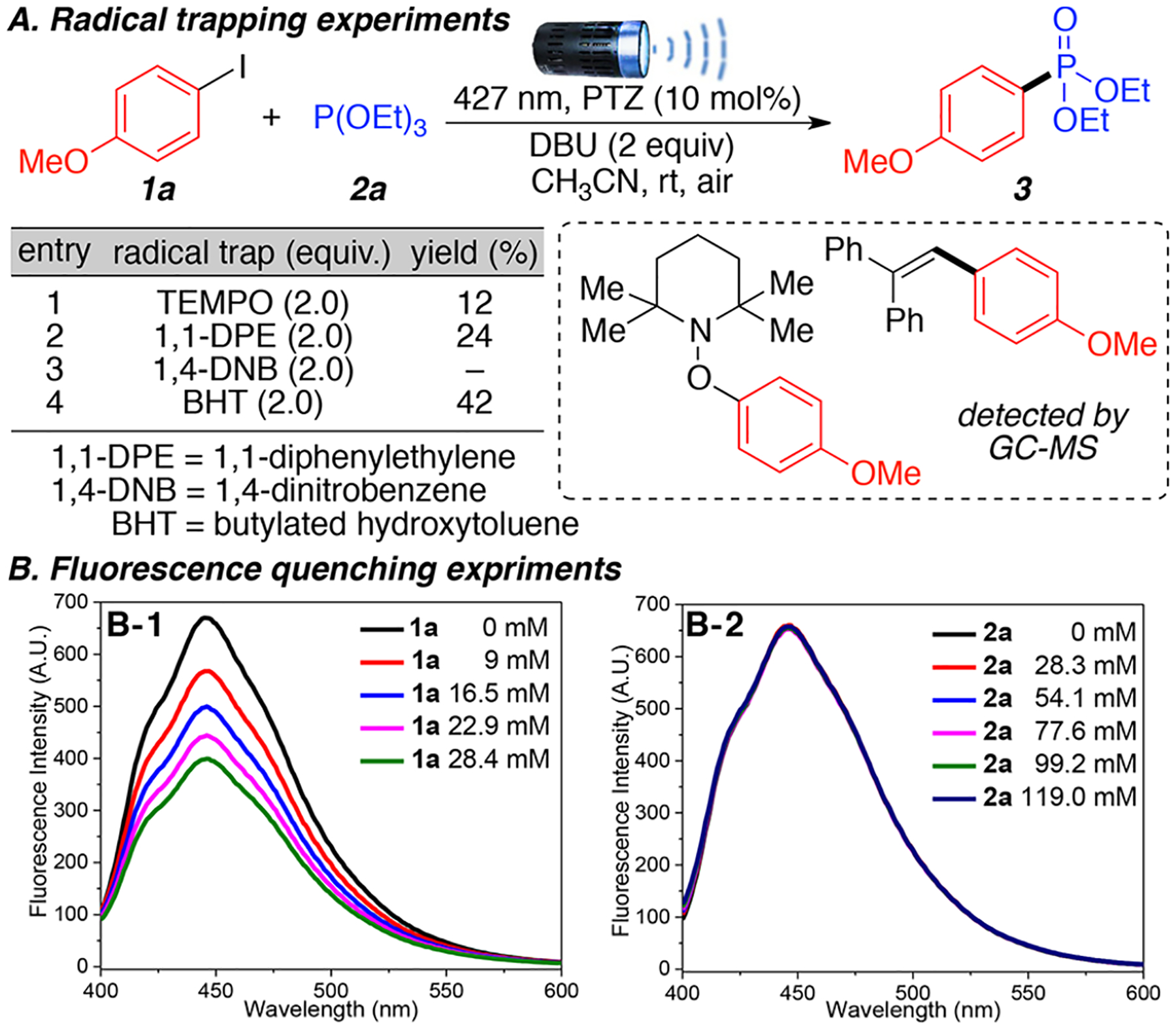
Mechanistic Experimentsa
a(A) Radical trapping experiments were performed aryl halides 1 (0.2 mmol, 1 equiv), trialkyl phosphites 2 (0.6 mmol, 3 equiv), DBU (0.4 mmol, 2 equiv), PTZ (0.02 mmol, 10 mol %), and radical trapping agents ((0.4 mmol, 2 equiv), in CH3CN (1 mL) using 427 nm LED. (B-1) Addition of aryl halide 1a to a mixture of PTZ + DBU. (B-2) Addition of phosphite 2a to a mixture of PTZ + DBU.
Then, fluorescence quenching experiments (Scheme 4B, see Supporting Information for full experiments and Stern–Volmer plots; S21) show that the addition of aryl halide 1a quenches fluorescence of the excited state of the photocatalyst (Scheme 4B-1), while addition of phosphite 2a does not (Scheme 4B-2). This indicates that the excited state of the catalyst initially reacts with the aryl halides 1a via a single electron transfer. It is important to highlight that, under our reaction conditions, we did not observe a significant difference in quenching by 1a of a solution of PTZ alone and a solution of PTZ that contained DBU (see Supporting Information). These results differs from previous observations by the Larionov group,32 which observed enhanced quenching when a mixture of PTZ + Cs2CO3 was used. These results suggest that, under our reaction conditions, PTZ alone is capable of reducing aryl iodides and that DBU only marginally increases PTZ’s reactivity. DBU is, however, an essential base in the final steps of the reaction mechanism.
On the basis of the above obtained mechanistic results and previous literature,32 a plausible reaction mechanism is proposed (Scheme 5). First, photoexcited PTZ reduces aryl halide via a single electron transfer to generate aryl radical anion A. Loss of halogen produces aryl radical that then couples with triethyl phosphite and forms phosphoranyl radical B. Facile oxidation of B ()33 by PTZ•+ radical cation () produces phosphonium species C and regenerates the active photocatalyst. Further reaction of phosphonium intermediate C with DBU affords the desired final product. While other groups have proposed that the base behaves as a terminal oxidant to close the catalytic cycle,34,35 we do not believe that to be the case in this reaction. Indeed, DBU has an oxidation potential of , which makes the redox reaction with PTZ•+ () not spontaneous.10
Scheme 5.
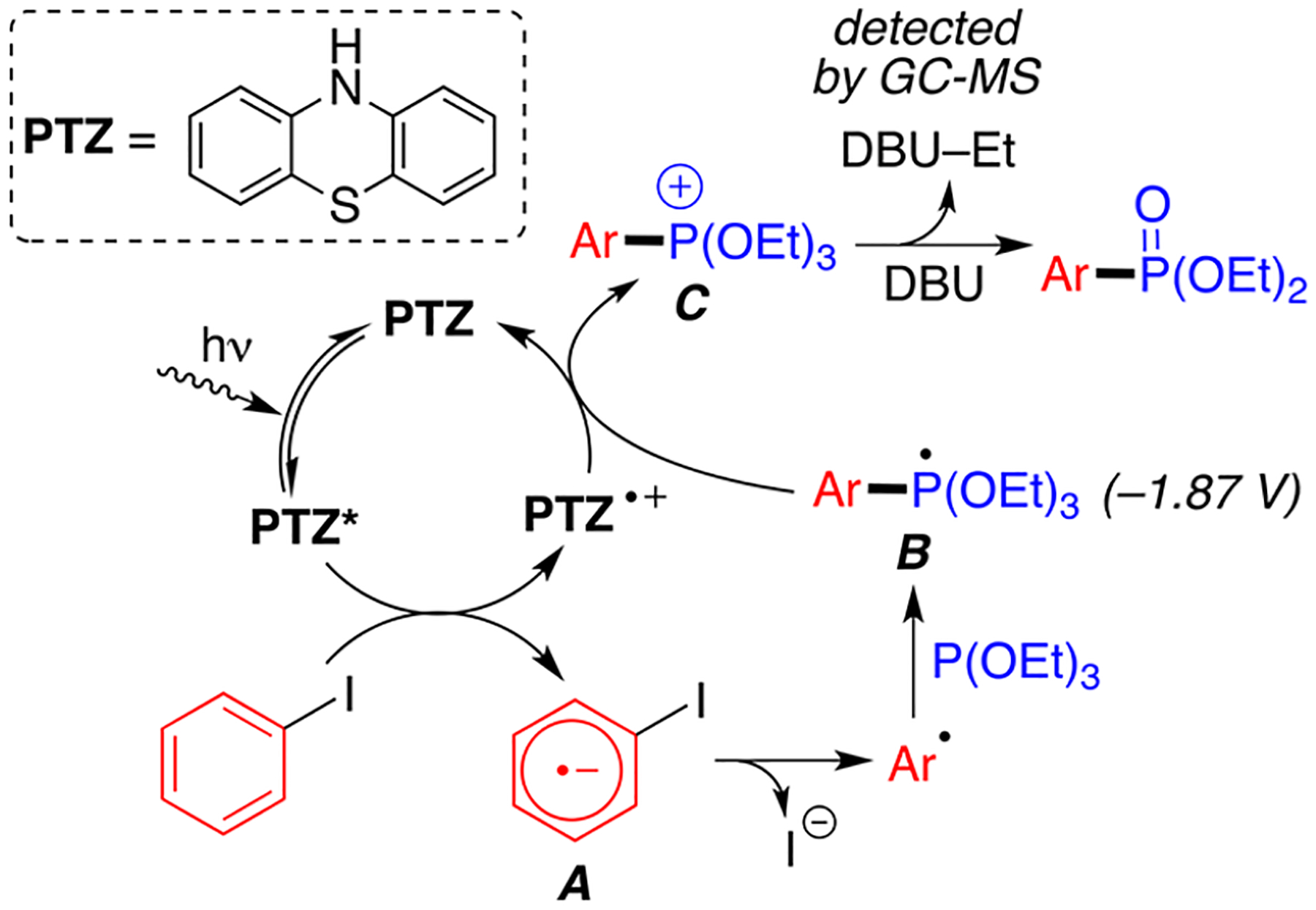
Proposed Reaction Mechanism
CONCLUSION
In summary, we have developed a simple and efficient photoredox cross-coupling reaction between aryl halides or aryl ammonium salts with trialkyl phosphites to form aromatic phosphonates. This transformation is the first to achieve the photoredox phosphonation of aryl ammonium salts. Additionally, this transformation is the first example of a photoredox-catalyzed phosphonation reaction that can be performed under atmospheric conditions in a wide range of aqueous solvent mixtures and in the absence of transition-metals. The method exhibits broad tolerance of functional groups enabling the late stage phosphonation of complex compounds in the presence of ester, ketone, amide, amine, alcohol, and silyl functional groups.
Supplementary Material
Funding
This publication was made possible with support from the National Institute of Dental & Craniofacial Research grant number 5R21DE029156-02. This research was supported in part by Lilly Endowment, Inc. through its support for the Indiana University Pervasive Technology Institute.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssuschemeng.1c07394.
1H and 13C{1H} NMR spectra for all phosphonated products, GC–MS spectra, and substrate scope and its limitations (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acssuschemeng.1c07394
The authors declare no competing financial interest.
Contributor Information
Lei Pan, Department of Chemistry and Chemical Biology, Indiana University-Purdue University Indianapolis, Indianapolis, Indiana 46202, United States.
Alexandra S. Kelley, Department of Chemistry and Chemical Biology, Indiana University-Purdue University Indianapolis, Indianapolis, Indiana 46202, United States
Maria Victoria Cooke, Department of Chemistry and Chemical Biology, Indiana University-Purdue University Indianapolis, Indianapolis, Indiana 46202, United States.
Macy M. Deckert, Department of Chemistry and Chemical Biology, Indiana University-Purdue University Indianapolis, Indianapolis, Indiana 46202, United States
Sébastien Laulhé, Department of Chemistry and Chemical Biology, Indiana University-Purdue University Indianapolis, Indianapolis, Indiana 46202, United States.
REFERENCES
- (1).Moonen K; Laureyn I; Stevens CV Synthetic Methods for Azaheterocyclic Phosphonates and Their Biological Activity. Chem. Rev 2004, 104, 6177–6215. [DOI] [PubMed] [Google Scholar]
- (2).Jiang W; Allan G; Fiordeliso JJ; Linton O; Tannenbaum P; Xu J; Zhu P; Gunnet J; Demarest K; Lundeen S; Sui Z New Progesterone Receptor Antagonists: Phosphorus-Containing 11 β-Aryl-Substituted Steroids. Bioorg. Med. Chem 2006, 14, 6726–6732. [DOI] [PubMed] [Google Scholar]
- (3).Lassaux P; Hamel M; Gulea M; Delbruck H; Mercuri PS; Horsfall L; Dehareng D; Kupper M; Frere JM; Hoffmann K; Galleni M; Bebrone C Mercapto Phosphonate Compounds as Broad-Spectrum Inhibitors of the Metallo-Beta-Lactamases. J. Med. Chem 2010, 53, 4862–4876. [DOI] [PubMed] [Google Scholar]
- (4).Van derJeught S; Stevens CV Direct Phosphonylation of Aromatic Azaheterocycles. Chem. Rev 2009, 109, 2672–2702. [DOI] [PubMed] [Google Scholar]
- (5).Erion MD; van Poelje PD; Dang Q; Kasibhatla SR; Potter SC; Reddy MR; Reddy KR; Jiang T; Lipscomb WN MB06322 (CS-917): A Potent and Selective Inhibitor of Fructose 1,6-Bisphosphatase for Controlling Gluconeogenesis in Type 2 Diabetes. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 7970–7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Baillie C; Xiao J Catalytic Synthesis of Phosphines and Related Compounds. Curr. Org. Chem 2003, 7, 477–514. [Google Scholar]
- (7).Tappe FMJ; Trepohl VT; Oestreich M Transition-Metal-Catalyzed C-P Cross-Coupling Reactions. Synthesis. 2010, 2010, 3037–3061. [Google Scholar]
- (8).Demmer CS; Krogsgaard-Larsen N; Bunch L Review on Modern Advances of Chemical Methods for the Introduction of a Phosphonic Acid Group. Chem. Rev 2011, 111, 7981–8006. [DOI] [PubMed] [Google Scholar]
- (9).Queffelec C; Petit M; Janvier P; Knight DA; Bujoli B Surface Modification Using Phosphonic Acids and Esters. Chem. Rev 2012, 112, 3777–3807. [DOI] [PubMed] [Google Scholar]
- (10).Montchamp J-L Phosphinate Chemistry in the 21st Century: A Viable Alternative to the Use of Phosphorus Trichloride in Organophosphorus Synthesis. Acc. Chem. Res 2014, 47, 77–87. [DOI] [PubMed] [Google Scholar]
- (11).Liu C; Szostak M Decarbonylative Phosphorylation of Amides by Palladium and Nickel Catalysis: The Hirao Cross-Coupling of Amide Derivatives. Angew. Chem., Int. Ed 2017, 56, 12718–12722. [DOI] [PubMed] [Google Scholar]
- (12).Zhuang R; Xu J; Cai Z; Tang G; Fang M; Zhao Y Copper-Catalyzed C-P Bond Construction via Direct Coupling of Phenylboronic Acids with H-Phosphonate Diesters. Org. Lett 2011, 13, 2110–2113. [DOI] [PubMed] [Google Scholar]
- (13).Bai Y; Liu N; Wang S; Wang S; Ning S; Shi L; Cui L; Zhang Z; Xiang J Nickel-Catalyzed Electrochemical Phosphorylation of Aryl Bromides. Org. Lett 2019, 21, 6835–6838. [DOI] [PubMed] [Google Scholar]
- (14).Kalek M; Jezowska M; Stawinski J Preparation of Arylphosphonates by Palladium (0)-Catalyzed Cross-Coupling in the Presence of Acetate Additives: Synthetic and Mechanistic Studies. Adv. Synth. Catal 2009, 351, 3207–3216. [Google Scholar]
- (15).He Y; Wu H; Toste FD A Dual Catalytic Strategy for Carbon-Phosphorus Cross-Coupling via Gold and Photoredox Catalysis. Chem. Sci 2015, 6, 1194–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Shaikh RS; Düsel SJS; König B Visible-Light Photo-Arbuzov Reaction of Aryl Bromides and Trialkyl Phosphites Yielding Aryl Phosphonates. ACS Catal. 2016, 6, 8410–8414. [Google Scholar]
- (17).Ghosh I; Shaikh RS; König B Sensitization-Initiated Electron Transfer for Photoredox Catalysis. Angew. Chem., Int. Ed 2017, 56, 8544–8549. [DOI] [PubMed] [Google Scholar]
- (18).Neumeier M; Sampedro D; Majek M; de la Pena O’Shea VA; Jacobi von Wangelin A; Perez-Ruiz R Dichromatic Photocatalytic Substitutions of Aryl Halides with a Small Organic Dye. Chem.— Eur. J 2018, 24, 105–108. [DOI] [PubMed] [Google Scholar]
- (19).Zeng H; Dou Q; Li C-J Photoinduced Transition-Metal-Free Cross-Coupling of Aryl Halides with H-Phosphonates. Org. Lett 2019, 21, 1301–1305. [DOI] [PubMed] [Google Scholar]
- (20).Cowper NGW; Chernowsky CP; Williams OP; Wickens ZK Potent Reductants via Electron-Primed Photoredox Catalysis: Unlocking Aryl Chlorides for Radical Coupling. J. Am. Chem. Soc 2020, 142, 2093–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Sobhani S; Vahidi Z; Zeraatkar Z; Khodadadi S A Pd Complex of a NNN Pincer Ligand Supported on γ-Fe2O3@SiO2 as the First Magnetically Recoverable Heterogeneous Catalyst for C-P Bond Forming Reactions. RSC Adv. 2015, 5, 36552–36559. [Google Scholar]
- (22).Sobhani S; Ramezani Z Synthesis of Arylphosphonates Catalyzed by Pd-imino-Py-γ-Fe2O3 as a New Magnetically Recyclable Heterogeneous Catalyst in Pure Water Without Requiring Any Additive. RSC Adv. 2016, 6, 29237–29244. [Google Scholar]
- (23). Two organic dyes have been previously reported: (i) Rhodamine 6G (ref 16) is acutely toxic (inhibits mitochondrial metabolic activity), and it is very toxic to aquatic life with long lasting effects. (ii) 9,10-Dicyanoanthracene (ref 18) is also acutely toxic.
- (24).Pan L; Elmasry J; Osccorima T; Cooke MV; Laulhé S Photochemical Regioselective C(sp3)-H Amination of Amides Using N-haloimides. Org. Lett 2021, 23, 3389–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Pan L; Cooke MV; Spencer A; Laulhé S Dimsyl Anion Enables Visible-Light-Promoted Charge Transfer in Cross-Coupling Reactions of Aryl Halides. Adv. Synth. Catal 2021, DOI: 10.1002/adsc.202101052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Niu B; Blackburn BG; Sachidanandan K; Cooke MV; Laulhé S Metal-free visible-light-promoted C(sp3)-H functionalization of aliphatic cyclic ethers using trace O2. Green Chem. 2021, 23, 9454–9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Sigma-Aldrich catalogue no. 88580: $89.90/kg (checked on 10/2021)
- (28).Yang B; Wang Z-X Ni-Catalyzed C-P Coupling of Aryl, Benzyl, or Allyl Ammonium Salts with P(O)H Compounds. J. Org. Chem 2019, 84, 1500–1509. [DOI] [PubMed] [Google Scholar]
- (29).Chen SY; Zeng RS; Zou JP; Asekun OT Copper-Catalyzed Coupling Reaction of Arylhydrazines and Trialkylphosphites. J. Org. Chem 2014, 79, 1449–1453. [DOI] [PubMed] [Google Scholar]
- (30).Xu W; Hu G; Xu P; Gao Y; Yin Y; Zhao Y Palladium-Catalyzed C-P Cross-Coupling of Arylhydrazines with H-Phosphonates via C-N Bond Cleavage. Adv. Synth. Catal 2014, 356, 2948–2954. [Google Scholar]
- (31).Berrino R; Cacchi S; Fabrizi G; Goggiamani A; Stabile P Arenediazonium Tetrafluoroborates in Palladium-Catalyzed C-P Bond-Forming Reactions. Synthesis of Arylphosphonates, -Phosphine Oxides, and -Phosphines. Org. Biomol. Chem 2010, 8, 4518–4520. [DOI] [PubMed] [Google Scholar]
- (32).Jin S; Dang HT; Haug GC; He R; Nguyen VD; Nguyen VT; Arman HD; Schanze KS; Larionov OV Visible Light Induced Borylation of C-O, C-N, and C-X Bonds. J. Am. Chem. Soc 2020, 142, 1603–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33). Calculated redox potentials, see Supporting Information for details.
- (34).Gazi S; Ananthakrishnan R Metal-Free-Photocatalytic Reduction of 4-Nitrophenol by Resin-Supported Dye under the Visible Irradiation. Appl. Catal. B Environ 2011, 105, 317–325. [Google Scholar]
- (35).Tahara K; Hisaeda Y Eco-Friendly Molecular Transformations Catalyzed by a Vitamin B12 Derivative with a Visible-Light-Driven System. Green Chem. 2011, 13, 558–561. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.