

Abstract
Glioblastoma (GBM) is characterized by extensive cellular and genetic heterogeneity. Its initial presentation as primary disease (pGBM) has been subject to exhaustive molecular and cellular profiling. By contrast, our understanding of how GBM evolves to evade the selective pressure of therapy is starkly limited. The proteomic landscape of recurrent GBM (rGBM), which is refractory to most treatments used for pGBM, are poorly known. We therefore quantified the transcriptome and proteome of 134 patient-derived pGBM and rGBM samples, including 40 matched pGBM-rGBM pairs. GBM subtypes transition from pGBM to rGBM towards a preferentially mesenchymal state at recurrence, consistent with the increasingly invasive nature of rGBM. We identified immune regulatory/suppressive genes as important drivers of rGBM and in particular 2–5-oligoadenylate synthase 2 (OAS2) as an essential gene in recurrent disease. Our data identifies a new class of therapeutic targets that emerge from the adaptive response of pGBM to therapy, emerging specifically in recurrent disease and may provide new therapeutic opportunities absent at pGBM diagnosis.
Keywords: Proteomics, Glioblastoma, Immunosuppression, OAS2.
Introduction
Glioblastoma (GBM) is the most commonly diagnosed primary malignant brain tumor in adults and constitutes ~60% of all neuroepithelial tumors [25]. Despite aggressive multimodal treatment with surgical resection followed by radiotherapy and chemotherapy, GBM remains incurable. Almost all patients experience relapse 7–9 months post-diagnosis and median survival has remained around 16 months for the past decade [40].
Cellular and molecular characterization of treatment-naïve primary GBM (pGBM) has revealed extensive inter- and intra-tumoral heterogeneity caused by multiple types of molecular dysregulation [19, 20, 30, 39]. Recent studies have suggested that GBM evolves significantly in response to therapy [26, 34]. For example, an evolutionary analysis of 21 paired primary and locally-recurrent GBM specimens found that overall mutation burden was not elevated in rGBM, but additional driver mutations were frequently acquired [22]. New driver mutations were not only acquired in rGBM, but pGBM clones harbouring specific drivers were preferentially lost. For example, focal EGFR amplifications were detected in ~95% of pGBM but these either reverted or the underlying clone went extinct in 27% of rGBM [22]. It is thus clear that GBM evolves significantly in response to therapy, making it refractory to first-line therapies, but the key signaling pathways mediating these changes remain unclear.
To fill this gap in our understanding of rGBM, we characterize how GBMs evolved and adapted their signaling in response to conventional therapy with the longitudinal proteomic analysis of 134 tissue samples from patients with recurrent GBM. Using this resource, we identified new therapeutic targets specifically activated at GBM recurrence, many of which regulate processes related to maintenance of an immunosuppressive niche. These data imply that GBM treatment resistance may evolve not only due to escape of cancer stem cell populations, but also an altered tumor immune microenvironment (TIME) in response to selective pressures of therapy.
Material and Methods
Patient samples and clinical data
Human GBM patient samples and fetal brain samples (normal brain derived cell lines) were collected from the Hamilton Health Sciences (Juravinski Cancer Centre and Hamilton General Hospital) from consenting patients as approved by the Hamilton Health Sciences (HHS)/McMaster Health Sciences research ethics board (REB #07–366 and REB# 08–005) and at University Health Network (REB #19–6350). Electronic health record software including Citrix, Meditech and MOSAIQ databases were used to search the Hamilton Health Sciences records (2001–2016) to collect primary-recurrent GBM matched-pair formalin-fixed paraffin-embedded (FFPE) samples and patient’s clinicopathological information.
Sample preparation for tissue microarray construction, proteomics and NanoString analysis
The hematoxylin and eosin (H&E)-stained slides associated with each FFPE block were used to mark the area of interest (tumor tissue and normal tissue adjacent to the tumor) by neuropathologist Dr. Cynthia Hawkins, University of Toronto. The marked FFPE blocks were then used for constructing tissue microarrays (TMA) and collecting tissue punches for proteomics and NanoString analysis. Briefly, three tissue cores from each donor block were acquired in circular spots form (1 mm in diameter) using a tissue microarray automated machine (3DHISTECH TMA Master, Quorum Technologies) and were placed in an empty paraffin block. In addition to sampling for constructing TMA blocks, 3 – 4 more tissue cores (1.5 mm in diameter) from each block were collected for proteomics and NanoString analysis. This allowed us to collect a total number of 143 samples: 45 pGBM-rGBM matched pairs, 20 normal tissue adjacent to the tumor (NAT), 9 unmatched pGBM and 22 unmatched rGBM. All samples were used for constructing a TMA.
Proteomic profiling was performed on 134 of these 143 samples (17 NATs, 40 pGBM-rGBM pairs (n= 84, three patients (GBM09, GBM18, GBM24) had two pGBM samples and one patient (GBM21) had two rGBM samples), 9 unmatched pGBMs and 24 unmatched rGBMs obtained from 22 patients). Moreover, 22 of the matched pGBM-rGBM pairs were used for NanoString analysis.
FFPE tissue sample preparation for mass spectrometry (MS) analysis
GBM FFPE tissue cores (1 mm) were deparaffinized twice using 500 μL of xylene (Sigma, Cat# 534056) with continuous end-to-end rotation for 5 minutes at room temperature. The samples were centrifuged at 14,000-g for 5 minutes and the supernatant was discarded. The tissue cores were rehydrated using sequential stepwise gradient treatment with 100%, 90%, 70% and 50% ethanol followed with water as the final step, with 5 minutes of end-to-end rotation for each step. The rehydrated tissue cores were lysed in 100 μL of 50% (v/v) 2,2,2-Trifluoroethanol (Sigma, Cat# 96924) with 300 mM Tris (pH 8) and sonicated using 5 cycles of pulse sonication 10 seconds each. The protein lysates were heated at 95°C for 2 hours for decrosslinking of the proteins. The fresh-frozen GBM tissue (1.5–2mg) samples were pulverized using Covaris cryoPREP Pulverizer and lysed in 500 μL of 50% (v/v) 2,2,2-Trifluoroethanol with 100 mM ammonium bicarbonate (pH 8). Two pmol of Suc2 (yeast invertase, Sigma, Cat# I4504) was added as digestion control in all samples. The disulphide bonds were reduced using 5 mM dithiothreitol for 30 minutes at 60 °C, the reduced disulphide bridges were alkylated with 25 mM iodoacetamide for 30 minutes at room temperature in the dark. The samples were diluted 1:5 with 100 mM ammonium bicarbonate (pH 8.0) and 2 mM CaCl2 was added. The proteins were digested overnight with 2 μg of trypsin/Lys-C enzyme mix (Promega, Cat# V5072) at 37 °C. The reaction was quenched with addition of formic acid and the peptides were desalted by C18-based solid phase extraction, then lyophilized in a SpeedVac vacuum concentrator. The peptides were solubilized in mass spectrometer-grade 0.1% formic acid in water and the peptide concentration determined with NanoDrop Lite (at 280 nm) [29, 38].
MS sample processing and data analysis
Prior to data acquisition, synthetic iRT peptides (Biognosys, Cat#Ki-3002) were spiked into each sample at a ratio of 1:10. LC-MS/MS data was acquired using an Easy nLC 1000 (Thermo) nano-flow liquid chromatography system with a 50 cm EasySpray ES803 column (Thermo) coupled to a Orbitrap Fusion tandem mass spectrometer (Thermo). Peptides were separated by reverse phase chromatography using a 4-hour non-linear chromatographic gradient of 4–48% buffer B (0.1% FA in ACN) at a flow rate of 250 nL/minute. Mass spectrometry data was acquired in positive-ion data-dependent mode. MS1 data was acquired at a resolution of 240,000 in the orbitrap with maximum injection time (maxIT) of 1000 ms and 40s dynamic exclusion, while MS2 data was acquired in the ion trap at ‘Normal’ scan rate, maxIT of 10 ms. HCD fragmentation was done at a normalized collision energy of 31%. The raw files were searched in MaxQuant [7] (version 1.6.2.3) using a UniProt protein sequence database containing human protein sequences from Uniprot (complete human proteome; Released 2019–09) merged with, Suc2 (yeast) protein sequences from Uniprot, and iRT synthetic peptide sequences (Biognosis). Searches were performed with a maximum of two missed cleavages, and carbamidomethylation of cysteine as a fixed modification. The oxidation at methionine, acetylation (N-term), arginine and lysine methylation was set as variable modifications. The FDR for the target-decoy search was set to 1% for protein and peptide level. Intensity-based absolute quantification (iBAQ), label-free quantitation (LFQ), and match between runs (matching and alignment time windows set as 2 and 20 minutes, respectively) were enabled. The proteinGroups.txt file was used for subsequent analysis. Proteins matching decoy and contaminant sequences were removed, and proteins identified with two or more unique peptides were carried forward. LFQ intensities were used for protein quantitation [6]. For proteins with missing LFQ values, median-adjusted iBAQ values were used as replacement [48]. The missing data was imputed using the normal distribution where the missing values were imputed from the lower half of the Gaussian distribution (width = 0.3, downshift = 1.8). Differentially expressed proteins among the GBM pairs were identified using paired sample Mann-Whitney U-test with multiple test correction using FDR. Pathway enrichment analysis was performed using Gene Set Enrichment Analysis (GSEA) [28, 41]. Pre-rank mode was used to perform GSEA using hallmark gene set from molecular signature database (MSigDB) [23, 24, 41] with the following parameters, number of permutations = 1000, where FDR < 0.25 was considered significant.
Proteomic subtype identification
Consensus clustering of the proteomics data was performed using the R package ConsensusClusterPlus v1.52.0 [47] using proteins (n = 1,595) detected in all matched primary and recurrence pairs (n = 84 samples; 40 pairs; three patients had two pGBM samples, and one patient had two rGBM samples) with protein clusters (k) varying from 2 to 20. Hierarchical clustering was performed using Euclidean distance and Ward linkage, with 80% gene resampling and 80% item resampling with 1,000 iterations. Clustering of samples and proteins was performed separately. The optimal cluster number (k = 5, for both sample and protein clustering) was chosen based on the delta area, which is the relative change in the area under the CDF curve comparing k and k-1.
Comparison to CPTAC proteomics
For the Clinical Proteomic Tumor Analysis Consortium GBM (CPTAC-GBM) data, the processed TMT protein abundance table and its associated clinical data were downloaded from the CPTAC data portal (https://cptac-data-portal.georgetown.edu/study-summary/S057). The experimental procedure and quantification were described in the original report [45]. To eliminate samples that were potentially mislabelled or swapped, Pearson’s correlation was calculated between all samples and samples with a correlation above 90% were removed (n = 2). Proteins missing in more than 90% of the samples were excluded from the analysis. We performed a Student’s t-test for each protein comparing tumor and NAT. The Benjamini-Hochberg test was performed to correct for multiple-testing. Spearman’s correlation was calculated by comparing the log2 fold change between NAT and pGBM samples in CPTAC and the Hamilton Health Sciences (HHS). Venn diagrams of the overlapping proteins were created using the R package VennDiagram [4] (v1.6.20). Centroids were created in the HHS dataset using the median abundance per protein in each of the five proteomic subtypes. CPTAC proteomic profiles were correlated to each centroid and samples were classified based on the highest positive Spearman’s correlation.
Comparison of pGBM proteomics with TCGA RNA abundance
Level 3 TCGA RNA-Sequencing data was downloaded from the Genomic Data Commons Data Portal and only primary samples were retained. The median abundance per gene was used to calculate the Spearman’s correlation between TCGA RNA and pGBM and rGBM abundance separately.
NanoString analysis
RNA was extracted from FFPE tissue cores using the Qiagen Allprep DNA/RNA FFPE kit per manufacturer’s protocol. 300 ng of total RNA of each sample were analyzed by NanoString gene expression assay using a custom codeset profiling four housekeeping genes and 30 classifier genes (one probe per gene) corresponding to the classical (CL), mesenchymal (MES), proneural (PN), and neural (NL) subtypes. Raw counts were background subtracted then normalized using the geometric mean of the samples analyzed using NanoString nSolver (nSolver Analysis Software: https://hdmzstaging.nanostring.com/products/analysis-software/nsolver). After removal of outlier probe values, data from 44 samples from 22 patients was classified using non-negative matrix factorization (NMF) into up to four groups. NMF was run with 200 iterations at ranks k = 2 to k = 7, approximating at each rank (1) metagenes representing the expression pattern of discriminatory genes, and (2) the weights of each metagene per sample. Rank k = 4 was the most parsimonious solution yielding clusters with a high cophenetic correlation coefficient, and separation of subtyping genes among metagenes. Metagene 2 includes both PN and NL classifier genes and was thereafter used to represent both groups. Metagene 3 corresponds to the CL subtype. The MES classifier genes were split between metagenes 1 and 4, potentially indicating two MES subtypes. Samples were assigned to subtypes via hard clustering using the maximum weights of each metagene. To account for the presence of multiple metagene contributions in each sample, weights were converted to percentages. Using these values, significant changes in subtype composition between paired primary-recurrence samples were identified using a paired t-test and corrected using the Benjamini-Hochberg procedure.
Cell culture
The cells used in this study were from patient-derived GBM cell lines. To isolate and propagate Brain Tumor Initiating Cells (BTICs), human brain tumor tissues were processed upon surgical resection according to the previously described protocol [36, 37, 44]. Briefly, tumor specimens were dissociated in enzymatic solution consisting of PBS (ThermoFisher, Cat#10010049) and 0.2 Wünsch unit/mL Liberase Blendzyme 3 (Millipore Sigma, Cat#5401119001) and incubated on a shaker at 37 °C for 15 minutes. The dissociated tissue was then filtered through a 70 μm cell strainer (Falcon, Cat#08–771-2) and collected by centrifugation at 1200 rpm for 5 minutes. Ammonium chloride solution (STEMCELL Technologies, Cat#07850) was used for lysing the red blood cells. BTICs were cultured in NeuroCult complete (NCC) media, a chemically defined serum-free neural stem cell medium (STEMCELL Technologies, Cat#05751), complemented with human recombinant epidermal growth factor (hrEGF) (20ng/mL: STEMCELL Technologies, Cat#78006), basic fibroblast growth factor (bFGF) (10ng/mL; STEMCELL Technologies Cat#78006), heparin (2 mg/mL 0.2% Heparin Sodium Salt in PBS; STEMCELL technologies, Cat#07980), antibiotic-antimycotic (1X; Wisent, Cat# 450–115-EL). GBM BTICs were plated on ultra-low attachment plates (Corning, Cat#431110), cultured as neurospheres and propagated by minimally-culturing (< 20 passages) human GBM samples and plating them on polyornithine-laminin coated plates for adherent growth. After enough expansion, adherent cells were replated in low-binding plates and cultured as tumorspheres. These cells were maintained as spheres upon serial passaging in vitro and retained their self-renewal potential and were capable of in vivo tumor formation. Normal brain cells used in this paper included Neural Stem Cells (NSCs) and Normal Human Astrocytes (NHAs). NSCs were isolated and propagated from fetal brain samples (approved by the Hamilton Health Sciences/McMaster Health Sciences) and NHAs were purchased from Lonza.
Cloning of OAS2 knockout lentivectors and generation of lentiviruses
Guide RNAs (gRNAs) targeting AAVS1 (5’-GGGGCCACTAGGGACAGGAT-3’) and OAS2 (A: 5’-TATGGCCACTCCCTGCACCA-3’, B: 5’-AGGGCATACGGAGACAGCGA-3’, C: 5’-ACTGGCATTTGTCTTATCCA-3’) were obtained from TKOv3 [16] and cloned into a single-gRNA lentiCRISPRv2 construct (Addgene 52961). Sequences were verified using Sanger sequencing and each construct was packaged independently into lentivirus using second-generation packaging constructs. Briefly, 16 hours prior to transfection, HEK293T cells were seeded into tissue-culture treated T75 cm2 flasks at a density of 10 million cells per flask using high-glucose DMEM with 2 mM L-glutamine and 1 mM sodium pyruvate (ThermoFisher, Cat#: 11995065), supplemented with 1% non-essential amino acid solution (ThermoFisher, Cat#: 11140050) and 10% fetal bovine serum (Gibco, Cat#: 12483020). The following day, the HEK293T media was replaced with viral harvesting media which is HEK culture media that is supplemented with 10 mM HEPES (ThermoFisher, Cat#: 15630080) and 1 mM sodium butyrate (Sigma-Aldrich, Cat#: 303410). Next, 15 μg of transfer plasmid (lentiCRISPRv2, AAVS1, OAS2-Knockout (KO) 1/A, OAS2-KO2/B and OAS2-KO3/C), 7.2 μg of psPAX2 (Addgene), and 4.8 μg of pMD2.G (Addgene) were mixed with polyethylenimin (PEI; Sigma-Aldrich, Cat#: 408719) at a 1:3 ratio (m:v) in 1.3 mL of Opti-MEM. After complexing for 15 minutes at room temperature, the PEI/DNA mixture was transferred to the HEK293T-containing flasks in dropwise fashion. Viral supernatants were collected 24 and 48 hours after transfection and then concentrated using ultracentrifugation (25,000 RPM for 2 hours at 4 °C) before being snap frozen and stored at −80 °C.
Lentiviral transduction of GBM cells
One million tumor or normal cells (BT972, BT241, BT618 or BT935, or human astrocytes) were plated in cell-repellent dishes (Greiner Bio, Cat#662970) and infected with lentivirus containing single-gRNA lentiCRISPRv2 constructs targeting AAVS1 or OAS2 (three gRNAs). Twenty-four hours post-infection, virus-containing media was replaced with fresh NCC media containing puromycin (1–2 μg/mL) (ThermoFisher, Cat#A1113803) for 48–72 hours.
Cell proliferation assay
Upon confirmation of OAS2 knockout by Western blotting analysis, OAS2 KO and AAVS1 transduced cells were dissociated, and 1,000 single cells were plated in 180 μL NCC per well in pentaplicate in a 96-well plate (Greiner Bio, Cat#655970) and incubated for three days. 20 μLs of Presto Blue (ThermoFisher, Cat#A13262), a fluorescent cell viability (metabolism) indicator, was added to each well two hours prior to the readout time point. FLUOstar Omega Fluorescence 556 Microplate reader (BMG LABTECH) was used to measure the fluorescence signal at excitation and emission wavelengths of 544 nm and 590 nm, respectively. Readings were analyzed using Omega analysis software (version: 5.11).
Secondary sphere formation assay
After confirming OAS2 knockout by western blotting, tumorspheres were dissociated using enzymatic digestion solution containing 10 μL Liberase Blendzyme3 (0.2 Wunsch unit/mL) plus 10 μL of DNase in 1 mL PBS for 5 minutes at 37°C. Single cell GBM BTICs were plated at 200 cells per well in 200 μL of NCC media in a 96-well plate (Greiner Bio, Cat#655970). Cultures were left untouched at 37°C, 5% CO2. The number of secondary spheres per well was counted every day from day three to seven and used to estimate the mean number of spheres per 2,000 cells.
In Vitro Limiting dilution assay
GBM BTICs (BT972) were seeded in a 96-well plate at different cell density (0, 5, 10, 20, 40, or 80 cells) per well with 24 replicates for each concentration. After two weeks, the number of tumor-spheres formed in each well was recorded and data were further analyzed using the software available at http://bioinf.wehi.edu.au/software/elda/.
Immunohistochemistry (IHC)
OAS2, CD3 and CD163 IHC was performed on TMA consisting of patient’s GBM samples on the Leica Bond RX (Leica Biosystems). IBA1 IHC was performed on Patient Derived Xenograft (PDX) brain tissues coming from BT972 GBM AAVS1 and OAS2 KO engrafted mice. Antigen retrieval was performed in Epitope Retrieval Buffer (ER2) (Leica, Cat#AR9640-Leica) for 20 minutes at 100°C. Antibodies were diluted in Powervision IHC Super Blocker (Leica, Cat#PV6122) and stained for 15 minutes as follows: rabbit monoclonal CD3 1:150 (abcam, Cat#ab16669), rabbit monoclonal CD163 1:1000 (abcam, Cat#182422) and mouse monoclonal OAS2 1:800 (Origene, Cat#CF802824). IBA1 antibody (abcam, Cat#ab178846) was diluted in TBS buffer with 1% BSA (1:300) and stained overnight at 4°C. For the mouse antibodies, a post primary antibody contained in the detection kit was applied before a polymer/HRP reagent. For rabbit antibodies, only the polymer reagent was applied. Both the post primary and polymer reagents were incubated for 8 minutes each. Slides were treated to a peroxidase block, developed with DAB and counterstained with hematoxylin all contained in the Leica Bond Polymer Refine Detection Kit (Leica, Cat#DS9800). Slides were then coverslipped with Permount. The digitization of the immunohistochemically stained TMA histology slides was performed using the Olympus® VS120 Slide Scanner. The cellular data was acquired through the HALO® Image Analysis Platform by Indica Labs. The quantitative tissue analysis was performed using HALO® Multiplex IHC module in combination with HALO® TMA module. This technology allowed us to detect and quantify the total number of OAS2+, CD3+ and CD163+ cells present in each tissue core of the TMA as well as the percentage of those cells that contain sufficient chromogenic IHC stain to be considered positive for the protein of interest.
Western blotting
Total protein was isolated from GBM BTICs and brain normal cells (Neural Stem Cells and Normal Human Astrocytes) with 1X RIPA buffer. Denatured proteins resolved on a 4–15% Mini-PROTEAN® TGX Stain-Free™ precast polyacrylamide gel, 1.5 mm, 10-well (Bio-Rad Cat#4568084) using 10x Tris/Glycine/SDS running buffer (Bio-Rad, Cat#1610732). Thereafter, resolved proteins were transferred onto polyvinylidene difluoride (PVDF) membranes, and membranes blocked in ODYSSEY buffer (LI-COR, Cat# 927–60001) diluted in TBS (1:1) for 30 minutes at room temperature. Following blocking, the membranes were incubated with primary antibodies (1:300 Mouse anti human monoclonal OAS2, [Origene, Cat#CF802824]) or GAPDH (1:2000 Mouse anti human monoclonal antibody, [abcam, Cat#ab8245]) as a loading control at 4°C overnight. Following primary antibody overnight incubation, membranes were thoroughly washed in 1X TBS-T for 3 × 5 minutes before subsequent incubation with HRP-conjugated secondary antibody (Goat Anti-Mouse IgG (H + L)-HRP Conjugate [Bio-Rad, Cat#1706516]) for one hour at room temperature. Band visualization was performed using Clarity™ Western ECL Substrate, (Bio-Rad, Cat#1705060). Data acquisition and protein detection was done using Chemidoc. Immunoblots were quantified and normalized to the loading control using ImageJ (1.52K) software.
Quantitative polymerase chain reaction (qPCR)
Total RNA was isolated from cells (BT972) using the Norgen RNA extraction kit and reverse transcribed into cDNA with the qScript cDNA SuperMix (Quanta Biosciences) and the C1000 Thermo Cycler (Bio-Rad). Real time PCR (qPCR) was performed in the Cfx96 (Bio-Rad) system with SYBR Green (Bio-Rad). Expression values were normalized to GAPDH. Gene specific primers included:
RNase L forward (5’-AAGAAGCACTTGGGTTTGGTGCAG-3’),
RNase L reverse (5’-TCCGCCTCGCTGTCATAACAAGAT-3’),
RIG-1 forward (5’-AGTGAGCATGCACGAATGAA-3’),
RIG-1 reverse (5’-GGGATCCCTGGAAACACTTT-3’),
TGFB1 forward (5’-GTACCTGAACCCGTGTTGCT-3’),
TGFB1 reverse (5’-GTATCGCCAGGAATTGTTGC-3’),
IL4 forward (5’-ACAGCAGTTCCACAGGCACAAG-3’),
IL4 reverse (5’-CGTACTCTGGTTGGCTTCCTTCAC-3’),
GAPDH forward (5’-AAGGTGAAGGTCGGAGTCAAC-3’), and
GAPDH reverse (5’-GGGGTCATTGATGGCAACAATA-3’).
In vivo experiments: intracranial injections, histological analysis for tumor size measurement and survival studies
All animal studies were conducted according to McMaster University Animal Research Ethics Board approved protocols. Intracranial transplantation of GBM BTICs was performed as previously described [35]. 100,000 BT972 AAVS1 or 100,000 BT972 OAS2 KO cells were injected into the right frontal lobes of 6− to 8-week old immunocompromised NSG mice. Briefly, mice were anaesthetized using Isoflurane gas (5% induction, 2.5% maintenance). A 1.5 cm vertical midline incision was made on top of the skull using a 15-blade scalpel and a small burr hole was then generated 2–3 mm anterior to the coronal suture, 3 mm lateral to midline by a drill held perpendicular to the skull. Tumor cells which were suspended in 10 μL PBS, were injected into the frontal lobe using a Hamilton syringe (Hamilton, Cat#7635–01) while it was inserted through the burr hole to a 5 mm depth. The incision was closed using interrupted stitches and sutures (Ethicon, Cat#J493G) and were sealed with a tissue adhesive (3M Vetabond, Cat#70200742529). The tumor formation and progression were tracked by MRI imaging. All mice were sacrificed at endpoint, brains were collected, formalin-fixed, and paraffin-embedded for H&E staining to assess the tumor burden. Images were captured using an Aperio Slide Scanner and analyzed using ImageScope v11.1.2.760 (Aperio) and imageJ (1.52K) software. The number of days of survival were also recorded for survival analysis.
scRNA sequencing analyses
Data Source.
Single cell RNA seq data from Neftel et al. (2019) [30] from (GEO; accession number GSE131928); and Ochocka et al. (2021) [31] from GEO (accession number GSE136001).
Data preprocessing.
scRNA-seq data sets were normalized, scaled, dimensionally-reduced and visualized on a UMAP using the Seurat (v 4.0.5) workflow implemented in scPipeline [27]. In brief, count matrices were loaded into a Seurat object and normalized using NormalizeData (…, normalization.method = ‘LogNormalize’, scale.factor = 10000). Variable features were identified using FindVariableFeatures (…, selection.method = ‘mvp’, mean.cutoff = c(0.1,8), dispersion.cutoff = c(1,Inf)) and then data were scaled using ScaleData(). Principal component analysis and UMAP embedding was performed using RunPCA() and RunUMAP(…, dims = 1:30), respectively. Metadata and cell type markers from original publications were used to annotate cell types.
GBM subtype classification.
To assign GBM subtypes to scRNA-seq data from Neftel et al. (2019), the AddModuleScore () function in Seurat was used to compute gene signature scores for each gene panel, and for each tumor cell, the signature with the highest score was taken as the subtype. The Neftel subtypes included MES1 (mesenchymal type 1), MES2 (mesenchymal type 2), NPC1 (neural projenitor type 1), NPC2 (neural progenitor type 2), OPC (oligodendrocyte progenitor cells), and AC (astrocyte-like).
Transcriptome analysis.
To visualize the expression of OAS2 in public scRNAseq data, cell-level expression was projected onto a UMAP using the FeaturePlot() function (Seurat) [14]. The mean normalized expression and expression fraction for each cell type was computed and visualized using barplots and dotplots overlaid on a common axis [expression.Plot(…), scMiko R package, v0.1.0]. Cell types were arranged based on hierarchical clustering performed on normalized expression and expression fraction values. Cell-type specific differences in OAS2 expression between primary and recurrent samples were then determined using student’s t-test.
In vitro and in vivo statistical analyses
All experiments were performed in duplicates or triplicates. Applicable data were analyzed and represented using GraphPad Prism 6 software or within the R statistical environment (v3.6.0). Data are presented as means ± S.E.M. Unpaired Student’s t-tests were used for statistical analysis of two groups and one-way ANNOVA analysis of variance with Tukey/Newman–Keuls test was used for statistical analysis of more than two groups with a p-value < 0.05 deemed as statistically significant. Visualization in R was performed using the BPG package [33] (v6.0.2) and ggplot2 (3.2.1) [46] .
Data availability
All mass spectrometry raw data has been deposited to the Mass Spectrometry Interactive Virtual Environment (MassIVE) with the following MassiVE ID: MSV000087947 and FTP link: ftp://massive.ucsd.edu/MSV000087947/. NanoString data is available on GEO with accession ID: GSE177549.
Results
Primary and recurrent GBM have distinct transcriptomic and protein landscapes
To identify potential neoplastic drivers generated throughout the course of therapy, we interrogated a cohort of 143 samples with either proteomics (n=134) or NanoString transcriptomics (n=44) using FFPE punches. In addition, three cores from each block were used to construct a tissue microarray (TMA). Detailed clinico-pathological information was collected for each patient (Fig. 1a; Table 1; Supplementary Table 1).
Fig. 1: pGBM and rGBM have distinct genomic and protein landscapes.
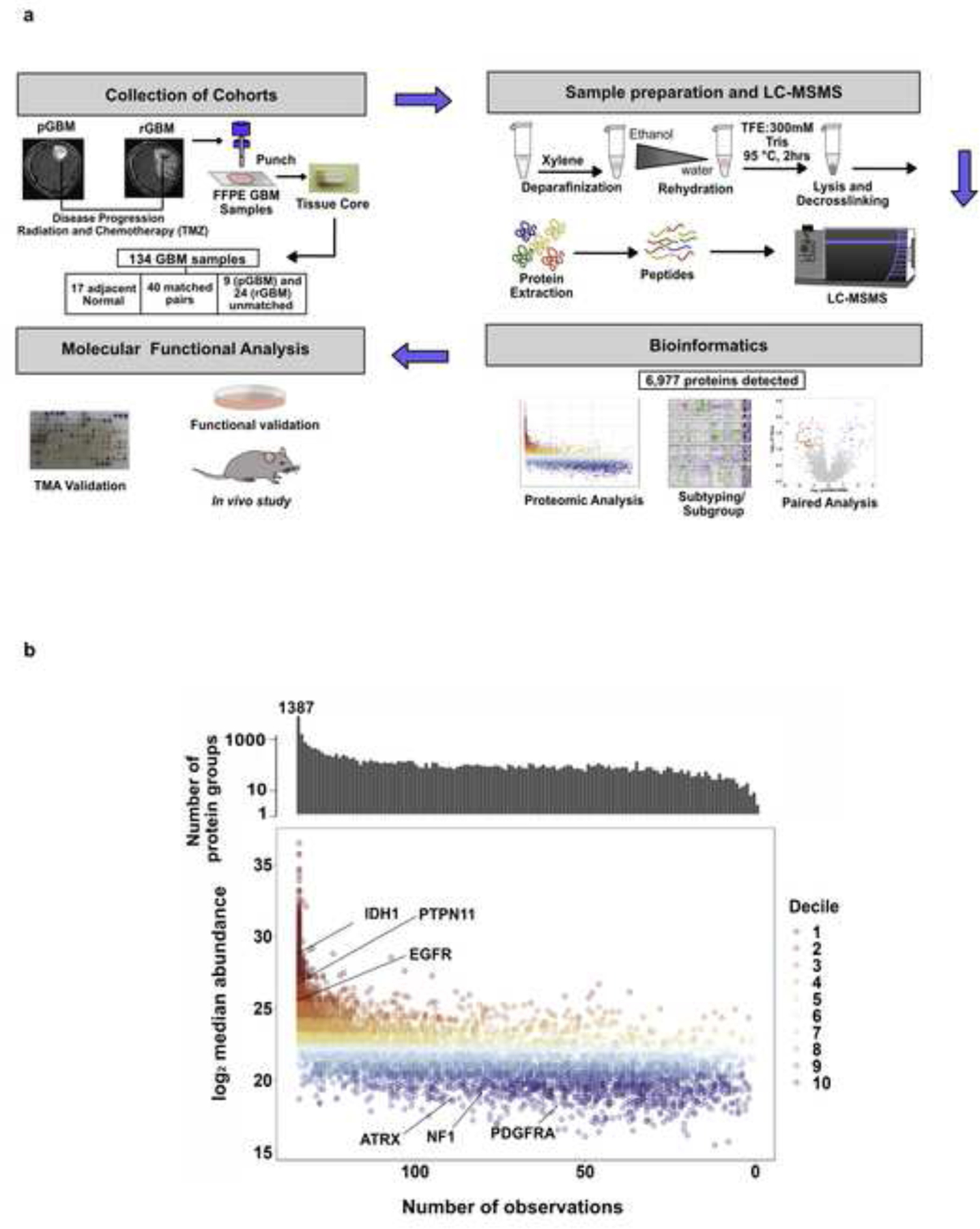
a, Schematic representation of sample collection and preparation, MS proteomics, target identification and functional analysis workflow. b, Distribution of protein quantitation measured as median intensity by the number of samples they are detected in. Bar plot on top represents the total counts of proteins quantified by the number of samples they are present in. Missing values were omitted when calculating the median.
Table 1:
Patient demographics indicating clinico-pathological features of the cohort
| Cohort | Primary | Recurrent | Matched |
|---|---|---|---|
| Sex | |||
| Male Female |
4 5 |
16 6 |
31 9 |
| Age at Treatment | |||
| Mean Median |
61.0 61.4 |
56.5 55.8 |
58.1 56.5 |
| Time to recurrence (Months) | |||
| Mean Median |
10.8 9.5 |
8.3 8.1 |
9.2 8.4 |
| Time to death (Months) | |||
| Mean Median |
23 19.2 |
14.2 14.1 |
14.9 14.6 |
| Total (Patients) | 9 | 22 | 40 |
Proteomic profiling was performed on 134 of these 143 samples (17 NAT, 40 pGBM-rGBM pairs (n= 84, three patients (GBM09, GBM18, GBM24) had two pGBM samples and one patient (GBM21) had two rGBM samples), 9 unmatched pGBMs and 24 unmatched rGBMs obtained from 22 patients using a previously established mass spectrometry workflow [38] (Fig. 1a). To evaluate our proteomics workflow 11 iRT peptides were added to each sample as method controls and elution profiles of these peptides showed consistent chromatographic performance (Supplementary Figure 1a). In order to ensure consistent performance of the instrument, HeLa cell lysate controls were run after every ten biological samples, and these were highly reproducible (R2 = 0.937; Supplementary Figure 1b–c). Furthermore, as an additional quality control, one biological sample from the cohort was run after every twenty samples (injection replicate, n = 5) and these technical replicates also showed high correlation (R2 = 0.938) (Supplementary Figure 1c–d). Using label free quantification, 6,977 distinct protein groups were detected, of which 2,515 were detected in at least 95% of samples (Fig. 1b, Supplementary Table 2). The well-known GBM markers IDH1, EGFR and PTPN11 were detected in all samples (Fig. 1b). The pGBM proteome (n = 51) was positively correlated to the pGBM transcriptome characterized by TCGA [2] (n = 166; Spearman’s ρ = 0.58, P < 2.2 × 10−16; Supplementary Figure 2a). There was a similarly high correlation between the mean rGBM protein abundance and mean pGBM RNA abundance (ρ = 0.56, P < 2.2 × 10−16; Supplementary Figure 2b).
To identify the proteomic subtypes of GBM across disease states, we performed consensus clustering on the 40 matched pGBM-rGBM pairs (n = 84) using the 1,595 proteins detected in all matched pairs. We identified five sample subtypes (S1 through S5) and five protein subtypes (P1 through P5) (Fig. 2a, Supplementary Figures 2c–f). Sample subtypes differed by age at diagnosis (P = 0.02; one-way ANOVA), with the median age in S5 (63.6 years) being older and S1 or S2 being the youngest (54.9 years). Sample subtypes were independent of sex (P = 0.21), treatment group (P = 0.153; Pearson’s Χ2 test, Supplementary Figure 2g) and disease state (pGBM vs. rGBM; P = 0.66; Pearson’s Χ2 test, Supplementary Figure 2h). If rGBM and pGBM proteomic profiles were alike, we would expect matched pGBM-rGBM tumors to be classified in the same sample subtype, but this did not occur. Subtype concordance was only 22.5%, with 9/40 pGBM and rGBM pairs classified into the same subtype (P = 9.0 × 10–4; proportion test; Fig. 2b). Interestingly, of the three patients with more than one pGBM sample, two were classified into different subtypes. These results may reflect widespread rewiring of signaling pathways during response to therapy or heterogeneity across the tissue. pGBM tumors assigned in the S2 subtype preferentially reclassified to the S1 subtype at recurrence (83%; 5/6). However, presence of normal brain constituents or regional sampling bias as contributors to subtype switching cannot be excluded.
Fig. 2: Proteomic subtypes of GBM across disease states.

a, Proteomic subtypes were identified in 40 matched pGBM-rGBM pairs. Clinical covariates indicating tumour classification, age at treatment (years), transcriptomic subtype, sex and pathway enrichment for proteomic subtypes are shown. b, pGBM and rGBM samples from the same patient classify as different proteomic (n = 40; replicate pGBM or rGBM samples were removed for simplicity) and c, transcriptomic subtypes (n = 22).
Pathway enrichment analysis showed that each protein subtype (P1 - P5) was enriched in different biological pathways, as shown in Fig. 2a. To investigate the phenomenon of subtype transition in matched pGBM and rGBM at the transcriptome level, we defined RNA subtypes for 22 paired pGBM and rGBM samples using NanoString data (Supplementary Figure 2i, Supplementary Table 3). The transcriptome and proteome were well-correlated in the 44 matched samples (median Spearman’s ρ = 0.41; Supplementary Figure 2j). As in the proteome, the majority of rGBM samples transitioned to different transcriptional subtypes than their pGBM counterparts (13/22 paired samples; Fig. 2c). Transcriptomic and proteomic subtypes were associated with one another (P = 0.03; Pearson’s Χ2 test). For example, 10/14 tumors with the proneural/neural transcriptomic signature were classified into the proteomic sample subtype S3, while tumors with the mesenchymal transcriptomic subtype were preferentially classified into the S1, S2 and S4 proteomic subtypes (Supplementary Figure 2k).
To validate the proteomics subtypes, we created a centroid-based single-sample subtype-classifier. This classifier was used to subtype a set of 99 publicly available pGBM proteomes (CPTAC) [45]. (Supplementary Figure 2l). While the methods used to quantify proteins differed between the two studies (TMT vs. LFQ), there was a high overlap in the proteins observed (HHS = 91.5%, 6,384/6,977; CPTAC = 58.2%, 6,384/10,970; Supplementary Figure 2m). S1 and S3 were present in similar proportions between the two datasets, while a higher proportion of samples were classified as S2 in the current dataset (HHS) and a higher proportion of samples were classified as S4 and S5 in the CPTAC dataset (Supplementary Figure 2n; P = 1.27 × 10–3; Pearson’s Χ2 test). Although these two proteomics datasets were created using different technologies (TMT vs. LC-MS/MS) the same proteomic patterns are reproducibly detected.
Adjacent normal tissue has a distinct proteome from pGBM
Normal adjacent tissue (NAT) is commonly used as a control in many cancer studies, however, on a molecular level they may not be entirely normal. Nevertheless, NAT presents an intermediate state between healthy and tumor state. In our study we used NAT as a normal control compared to pGBM and rGBM. Hierarchical clustering of NAT, pGBM and rGBM samples using proteins detected in all samples showed clear separation of NAT from the tumor samples (Supplementary Fig. 3a). Comparison of the proteomics profile between NAT (n = 17) and pGBM (n = 50) samples identified 2,902 proteins significantly differentially abundant (Q < 0.1, |log2FC| > 1; Supplementary Fig. 3b, Supplementary Table 4). Gene set enrichment analysis revealed that neuron activity related pathways were upregulated in NAT, whereas mRNA processing, DNA repair and immune related pathways were enriched in pGBM (Supplementary Figure 3c, Supplementary Table 4). A similar analysis comparing normal brain tissue and pGBM samples in the CPTAC cohort, which included proteomics from 10 normal samples from the reference GTEx dataset [45] showed similar effect sizes in the overlapping detected proteins (ρ = 0.79, P < 2.2 × 10−16; Supplementary Figure 3d).
Proteomic profiling of pGBM and rGBM matched samples reveal rGBM is associated with immunosuppression
Next, we asked what signaling pathways were changed during the evolutionary response of pGBMs to the selective pressure of treatment. We performed differential abundance analysis with 40 pGBM-rGBM matched pairs and identified 165 proteins that differed significantly (Q < 0.1, |log2FC| > 1, Mann-Whitney U-test; Fig. 3a, Supplementary Table 4). The rGBM proteome was enriched in proteins associated with interferon alpha and gamma (broad inflammatory response), epithelial to mesenchymal transition, TNF-a signaling, fatty acid metabolism, hypoxia and oxidative phosphorylation pathways (Fig. 3b, Supplementary Table 5).
Fig. 3: Differential expression analysis between pGBM-rGBM matched pairs introduces OAS2, an essential gene for rGBM with significant upregulation at the recurrent stage.
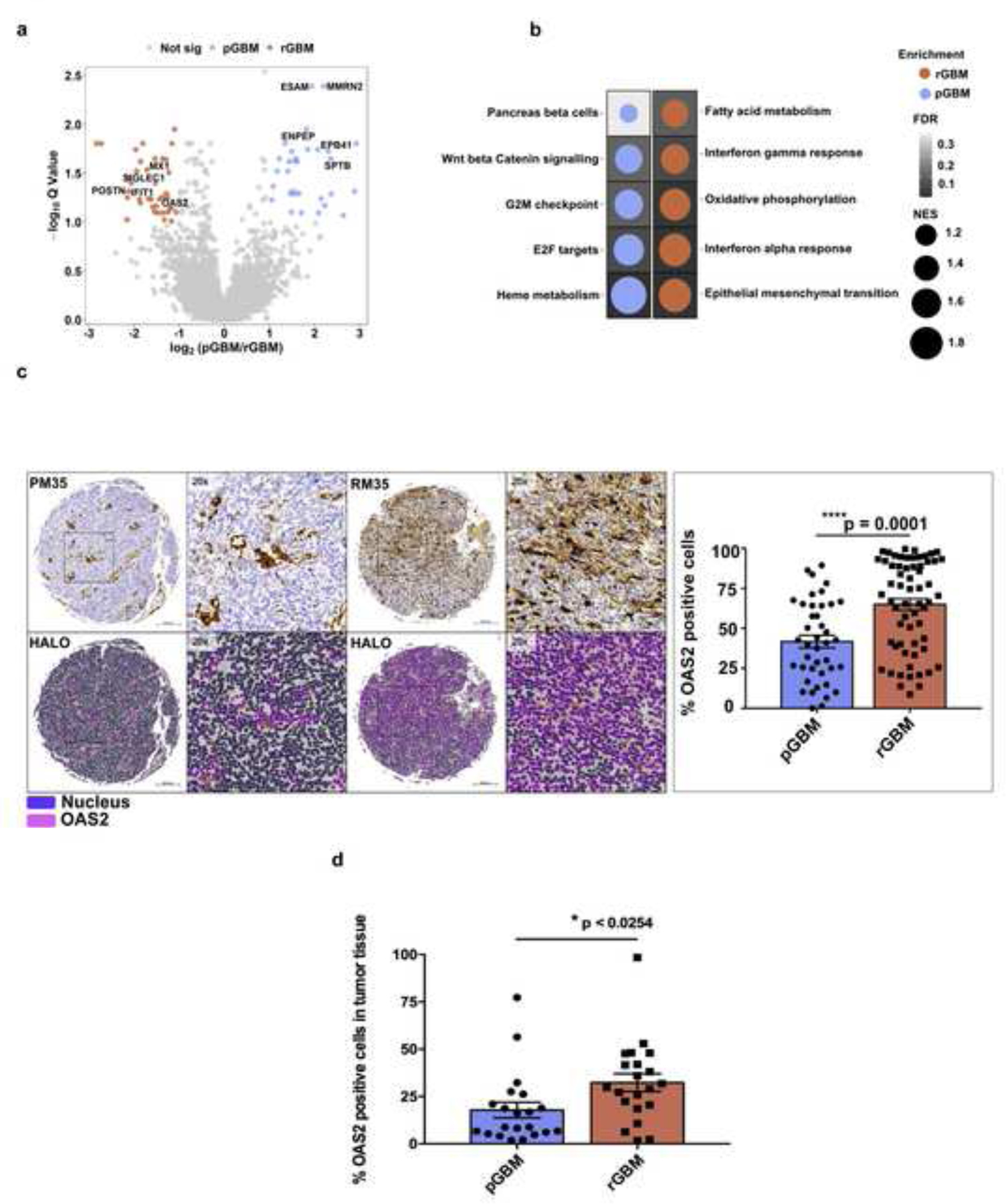
a, Volcano plot depicting differential abundance analysis between pGBM and rGBM in matched sample pairs. Proteins significantly enriched in rGBM patients are indicated in red, while proteins enriched in pGBM are indicated in blue (Q value < 0.1; log2 fold change > 1). b, Gene set enrichment analysis (GSEA) showing the top significantly enriched pathways in pGBM vs. rGBM. The dot plot shows the normalized enrichment scores (NES) with the background color gradient representing the Q values. c, Immunohistochemical analysis of OAS2 on TMA consisting of both matched and unmatched pGBM-rGBM samples indicated a significantly higher level of OAS2 at the recurrent stage. (The representative image shows pGBM (PM35)-rGBM (RM35) matched samples) (Scale bar: 200 um, P value: *** 0.0002, **** < 0.0001). d, Overexpression of OAS2 in rGBM was confirmed by immunohistochemical analysis on a TMA construct consisting of an independent cohort of 20 pGBM-rGBM matched samples.
To identify the top potentially targetable proteins enriched in rGBM, we prioritized these 165 candidates based on three measures: the magnitude of differential abundance between primary and recurrent GBMs while requiring the protein to be highly upregulated at the recurrence, low abundance in brain normal tissue (NAT) and a known role in immunosuppression. Following target validation using immunohistochemical analysis on TMA as well as several functional assays, 2–5-oligoadenylate synthetase 2 (OAS2) was identified as the top candidate (Supplementary Figure 4a). OAS2 plays a crucial role in regulating immunosuppression and has been previously implicated in solid cancers including oral and breast cancer [8, 18]. In addition, Periostin (POSTN), previously described in the literature [52] as a tumor associated macrophage (TAM) marker of rGBM, was also enriched in the rGBM cohort, further validating the observation of upregulated immune-related proteins in rGBMs.
To further confirm the upregulation of OAS2 in rGBM, we performed whole cell proteomic analysis on an independent cohort of fresh frozen pGBM (n= 8) and rGBM (n= 6) samples. This validation analysis showed enrichment of OAS2 in the rGBM samples (Supplementary Figure 4b) compared to the pGBM samples in both the datasets, further corroborating our results.
To further characterize OAS2 across a broader cohort of GBM patients, we assessed its abundance through immunohistochemical analysis in pGBM-rGBM intact tissues. We observed a significant upregulation in OAS2 expression on tumor-intact tissues, particularly in endothelial cells and foamy macrophages, as assessed by immunohistochemical analysis on 45 pGBM-rGBM matched pairs, nine unmatched pGBM and 24 rGBM unmatched tissues (Fig. 3c and Supplementary Figure 4c), as well as in an independent cohort of 20 pGBM-rGBM matched pairs (Fig. 3d). Moreover, there was a significant increase in OAS2 protein abundance in rGBM compared to pGBM in purely enriched patient-derived BTICs while no to very low levels were observed in normal brain cells (NSCs and NHAs) (Fig. 4a). The role of OAS2 in GBM pathogenesis has not yet been investigated, although we have demonstrated its expression both within rGBM cells (specifically BTICs) and in cells comprising the rGBM tumor immune microenvironment. Therefore, our next step was focused on further validating the role of OAS2 in rGBM tumorigenesis.
Fig. 4: OAS2, an essential gene for rGBM, has significant upregulation at the recurrent stage.
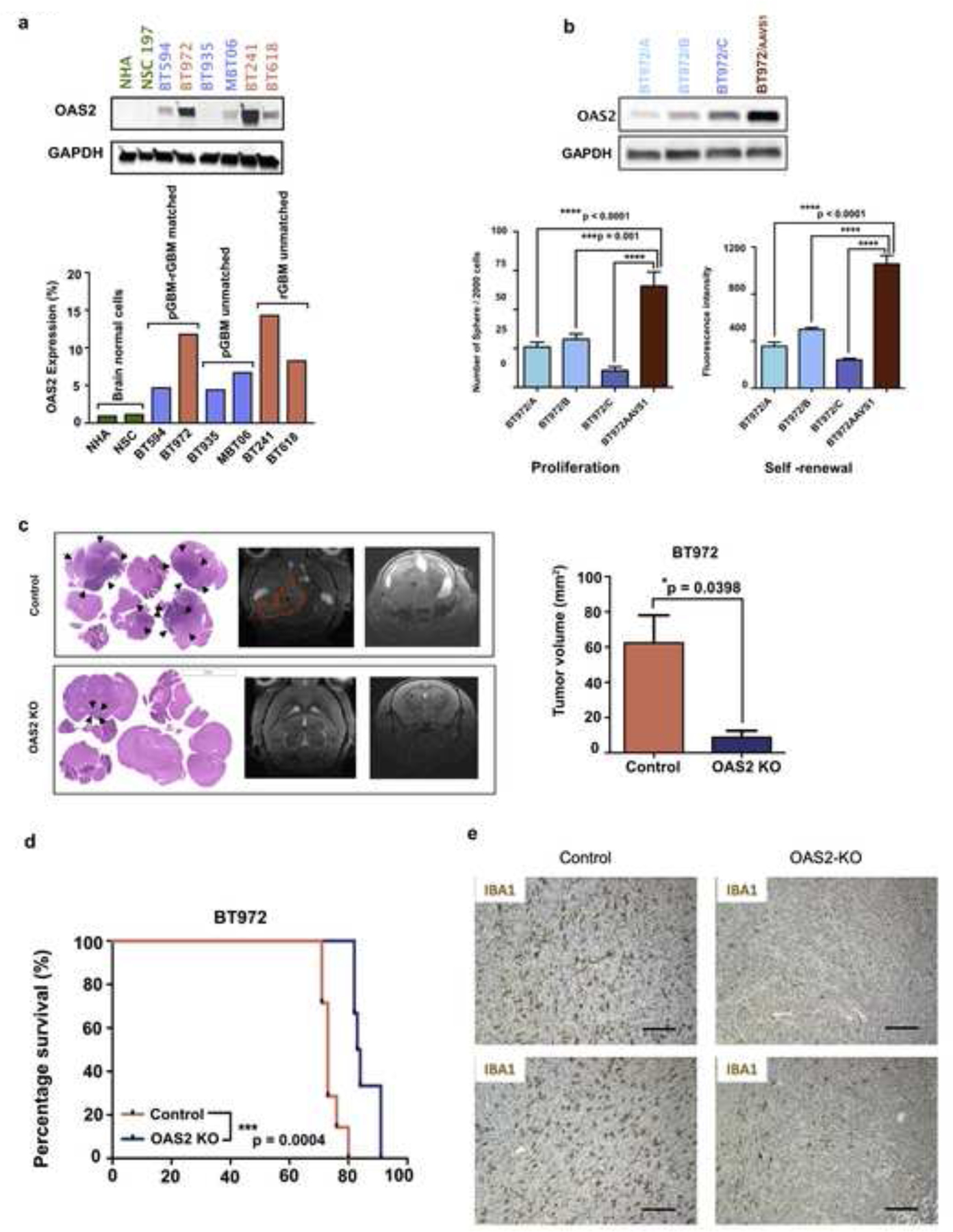
a, Western blotting analysis on the protein lysate of GBM BTIC (pGBM-rGBM matched and unmatched samples) confirmed a two-fold increase in OAS2 expression. b, BT972, an OAS2 high expressing recurrent GBM BTIC line, was used for functional analysis. OAS2 was knocked out using CRISPR knockout gene editing. Following confirmation of gene knockout (construct A, B and C) by western blotting, the effect of OAS2 on self-renewal and proliferation capacity of OAS2 KO BT972 vs OAS2 WT BT972 (BT972 AAVS1) were measured using secondary sphere formation assay and PrestoBlue proliferation assay, respectively (P value: *** p < 0.001, **** p < 0.0001) (One way ANOVA). c-d, OAS2 KO BT972 and BT972 control (BT972 AAVS1) (100,000) were intracranially implanted into the right frontal lobe of NSG mice (n=6). The tumor size was tracked weekly using MRI imaging. e, IHC analysis of IBA1 on GBM xenografts from OAS2 KO or WT GBM cell engrafted mice revealed higher levels of IBA1 expression in OAS2 WT vs OAS2 KO engrafted brains. Scale bars represent 100 μm.
OAS2 is an essential driver of treatment-resistant cell populations in rGBM
To specifically investigate the effect of OAS2 on GBM BTIC stem-like properties including self-renewal and proliferative capacity, secondary sphere formation, limiting dilution assay and proliferation assays were performed. Upon knocking out (KO) OAS2 in OAS2-high rGBM lines, we observed a marked reduction in clonogenicity, and proliferative capacity of OAS2 KO cells (constructs A, B and C) compared to OAS2 control cells (AAVS1) as measured by secondary sphere formation as well as limiting dilution assay and proliferation assays, respectively (Fig. 4b, Supplementary Figure 4d and 4e). This indicated that OAS2 has the ability to drive stem-like properties in GBM BTICs which in turn fuels cancer progression. Moreover, we showed that depletion of OAS2 only affects the cell proliferation in rGBM and has no effect on normal human astrocytes and pGBM suggesting that OAS2 is an essential driver for rGBM. (Supplementary Figure 4f). To further validate the role of OAS2 in GBM progression, the effect of OAS2 depletion on tumor progression was investigated. For this purpose, an OAS2 high expressing rGBM line, BT972, was tested at the in vivo level. In detail, BT972 OAS2 KO and BT972 OAS2 control cells (AAVS1) were intracranially engrafted into the right frontal region of the mouse brain and tumor growth was tracked using weekly MRI imaging until mice reached endpoint (Fig. 4c and Supplementary Figure 4g). Mice engrafted with OAS2 KO BT972 had significant reduction in tumor burden as shown by histological analysis and MRI imaging (Fig. 4c) as well as an increased survival advantage (Fig. 4d) when compared to their control counterparts (engrafted mice with BT972 AAVS1). This provides another layer of confirmation for the key role of OAS2 in rGBM tumor progression. Moreover, using published scRNA-seq data from GBM patients and syngeneic murine models (GL261 glioma) [27, 30, 31] we demonstrate that OAS2 is preferentially expressed in infiltrating myeloid cells (macrophages/monocytes) (Supplementary Figure 5a and 5b), which promote an immunosuppressive environment in GBM [10]. Relative to GBM-associated immune infiltrates, OAS2 expression in GBM tumor cells was low but detectable. Within the malignant populations observed in the Neftel cohorts [30], the highest OAS2 expression was consistently observed in MES1 (mesenchymal type 1) and AC (astrocyte)-like GBM subtypes indicating OAS2 expression is associated with the GBM subtypes which have shown the worst prognosis (Supplementary Figure 5a and 5b). Together, these scRNA-seq analyses demonstrated the OAS2 expression is associated with immunosuppression and poor prognosis [12, 32]. In addition, through immunohistochemical analysis of IBA1 on PDX tissues derived from BT972 GBM AAVS1 and OAS2 KO engrafted mice, we indicated significantly higher level of TAMs infiltration in the brain tissues of BT972 GBM AAVS1 cells compared to OAS2 KO engrafted mouse brain. As TAMs are major immunosuppressive cells in GBM [12, 13] this data reveals the key role of OAS2 in the recruitment of TAMs to the tumor tissue and consequently generating an immunosuppressive microenvironment (Fig. 4e). This was further confirmed by assessing the effect of OAS2 on immunosuppressive cytokines including TGFβ and IL4. We observed that OAS2 disruption largely decreases TGFβ and IL4 gene expression suggesting that OAS2 depletion in GBM cells impairs the tumor supportive cytokines. Together, these data revealed that OAS2 enhances TAMs infiltration in GBM and regulates the release of protumorigenic cytokines such as TGFβ and IL4, suggesting that OAS2 plays an essential role for suppressing TIME in GBM (Supplementary Figure 6a). We also observed that disruption of OAS2 affects the OAS2 downstream signaling pathway by significantly decreasing RNase L and RIG-1 gene expression suggesting that OAS2 signaling pathway is involved in recurrent GBM [9] (Supplementary Figure 6b). Moreover, we indicated that TAMs with M2-like phenotype were more abundant at recurrence, as supported by CD3 and CD163 immunohistochemistry on TMAs (Supplementary Figure 6c) confirming significant amount of immunosuppression at the recurrent stage of the disease.
DISCUSSION
The field of GBM research currently lacks a quantitative, comparative analysis of the proteomes of primary and recurrent GBMs [3, 38]. To date, the vast majority of molecular datasets of patient GBMs comprise primary tumors. Recently, large consortia have significantly advanced the field of proteogenomics, including in brain malignancies [43, 49, 53], but focused almost exclusively on primary treatment-naïve disease. Surprisingly, while the GBM genome and transcriptome have been well elucidated in primary tumors, the GBM proteome and its relation to up-stream genomic alterations are poorly documented. Previous studies have shown that tumor cells are exposed to IFNγ [17, 42] produced by the TME, and that tumor associated-macrophages (TAMs) might be the source of IFNγ production, leading to immune evasion [11]. We identified numerous proteins related to IFNγ signalling that were increased in abundance in rGBM relative to pGBM, including OAS1, OAS2, MX1 and IFIT1. OAS2 is a member of the template-independent nucleotidyltransferase protein family and is an interferon (IFN)-induced antiviral enzyme involved in the antiviral innate immune response [5]. OAS2 has been reported as a prognostic biomarker in breast cancer [51] and causes immunosuppression in oral cancers by down regulating CD3-ζ chain expression through induction of caspase-3 activation. The upregulation of OAS2 in rGBM was further confirmed in an independent cohort of fresh frozen GBM samples. Further, we showed with in vivo and in vitro CRISPR knockouts that OAS2 is essential for GBM progression through an as-yet unknown mechanism. However, we acknowledge the shortcomings of the use of immunocompromised models to further evaluate the immunomodulatory role of OAS2 in the tumor microenvironment.
Consistent with this broad proteomic change in IFNγ signalling, our data revealed a dominant immunosuppressive phenotype for rGBM, with elevated Siglec-1 (CD169) abundance occurring during tumor evolution under therapeutic pressure. CD169 is a sialic acid receptor expressed on a specific type of macrophages. In triple-negative breast cancer CD169+ macrophages support tumor growth and metastasis by causing immune escape, negatively affecting CD8+ T cell accumulation in tumors and causing the JAK2/STAT3 signaling pathway to be activated upon exposure to tumor cells [18]. TAMs with M2-like phenotype were more abundant at recurrence, as supported by CD3 and CD163 immunohistochemistry on TMAs. Moreover, OAS2 expression was positively correlated with higher levels of TAM infiltration into tumor tissue as well as increased gene expression of immunosuppressive cytokines including TGFβ and IL4. These data provide another layer of confirmation for the crucial role of macrophages in GBM progression and immunosuppression which is associated with poor patient prognosis [50]. This finding is aligned with recent studies reporting the significant role of macrophages in inducing GBM immunosuppression, seen predominantly in GBMs of the mesenchymal subtype as the most aggressive subtype for GBM [15, 21]. This was also further confirmed by published scRNA-seq data on glioblastoma patient samples as well as syngeneic glioma mouse model which also revealed the high level of expression of OAS2 on myeloid cells as immunosuppressive cell population in GBM as well as tumor cells with mesenchymal and classical subtypes resulting in therapy resistance and as a result which results in patient’s poor survival [1, 27, 30, 31].
This work describes the first proteomic landscape analysis of matched pGBM-rGBM, which could promote the development of new, selective targeted therapies and immunotherapies. This proteomic characterization of rGBM could begin to instruct novel and rational combinatorial poly-therapeutic approaches to provide more effective and personalized treatments for therapy-resistant rGBM. However, to further understand the role of OAS2 in GBM progression, related signaling pathways and mechanistic studies need to be explored in the future. This study provides clear rationale for increased research and clinical profiling of rGBM, including by non-invasive assays measuring circulating tumor DNA, RNA and protein.
Supplementary Material
Acknowledgements
This work was partially supported by a CIHR Project Grant (PJT154357) to TK, the NIH/NCI under awards P30CA016042, U24CA248265 and P50CA211015 to PCB and TFRI 1065. This research was funded in part by the Ontario Ministry of Health and Long-Term Care. SKS and TK were supported through the Canadian Research Chair program. We thank Dr. Kaiyun Yang for her contribution in generating patient’s information.
Footnotes
Ethics declarations
The authors have declared that no conflict of interest exists.
References
- 1.Behnan J, Finocchiaro G, Hanna G (2019) The landscape of the mesenchymal signature in brain tumours. Brain 142: 847–866 Doi 10.1093/brain/awz044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SHet al (2013) The somatic genomic landscape of glioblastoma. Cell 155: 462–477 Doi 10.1016/j.cell.2013.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Casey AE, Sinha A, Singhania R, Livingstone J, Waterhouse P, Tharmapalan P, Cruickshank J, Shehata M, Drysdale E, Fang Het al (2018) Mammary molecular portraits reveal lineage-specific features and progenitor cell vulnerabilities. J Cell Biol 217: 2951–2974 Doi 10.1083/jcb.201804042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen H, Boutros PC (2011) VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics 12: 35 Doi 10.1186/1471-2105-12-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi UY, Kang JS, Hwang YS, Kim YJ (2015) Oligoadenylate synthase-like (OASL) proteins: dual functions and associations with diseases. Exp Mol Med 47: e144 Doi 10.1038/emm.2014.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M (2014) Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13: 2513–2526 Doi 10.1074/mcp.M113.031591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26: 1367–1372 Doi 10.1038/nbt.1511 [DOI] [PubMed] [Google Scholar]
- 8.Dar AA, Pradhan TN, Kulkarni DP, Shah SU, Rao KV, Chaukar DA, D’Cruz AK, Chiplunkar SV (2016) Extracellular 2’5’-oligoadenylate synthetase 2 mediates T-cell receptor CD3-zeta chain down-regulation via caspase-3 activation in oral cancer. Immunology 147: 251–264 Doi 10.1111/imm.12560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drappier M, Michiels T (2015) Inhibition of the OAS/RNase L pathway by viruses. Curr Opin Virol 15: 19–26 Doi 10.1016/j.coviro.2015.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Exley MA, Garcia S, Zellander A, Zilberberg J, Andrews DW (2022) Challenges and Opportunities for Immunotherapeutic Intervention against Myeloid Immunosuppression in Glioblastoma. J Clin Med 11: Doi 10.3390/jcm11041069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gangoso E, Southgate B, Bradley L, Rus S, Galvez-Cancino F, McGivern N, Guc E, Kapourani CA, Byron A, Ferguson KMet al (2021) Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell 184: 2454–2470 e2426 Doi 10.1016/j.cell.2021.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goswami S, Walle T, Cornish AE, Basu S, Anandhan S, Fernandez I, Vence L, Blando J, Zhao H, Yadav SSet al (2020) Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat Med 26: 39–46 Doi 10.1038/s41591-019-0694-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guerriero JL (2018) Macrophages: The Road Less Traveled, Changing Anticancer Therapy. Trends Mol Med 24: 472–489 Doi 10.1016/j.molmed.2018.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hao Y, Hao S, Andersen-Nissen E, Mauck WM, 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager Met al (2021) Integrated analysis of multimodal single-cell data. Cell 184: 3573–3587.e3529 Doi 10.1016/j.cell.2021.04.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hara T, Chanoch-Myers R, Mathewson ND, Myskiw C, Atta L, Bussema L, Eichhorn SW, Greenwald AC, Kinker GS, Rodman Cet al (2021) Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma. Cancer Cell 39: 779–792 e711 Doi 10.1016/j.ccell.2021.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hart T, Tong AHY, Chan K, Van Leeuwen J, Seetharaman A, Aregger M, Chandrashekhar M, Hustedt N, Seth S, Noonan Aet al (2017) Evaluation and Design of Genome-Wide CRISPR/SpCas9 Knockout Screens. G3 (Bethesda) 7: 2719–2727 Doi 10.1534/g3.117.041277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoekstra ME, Bornes L, Dijkgraaf FE, Philips D, Pardieck IN, Toebes M, Thommen DS, van Rheenen J, Schumacher TNM (2020) Long-distance modulation of bystander tumor cells by CD8(+) T cell-secreted IFNgamma. Nat Cancer 1: 291–301 Doi 10.1038/s43018-020-0036-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jing W, Guo X, Wang G, Bi Y, Han L, Zhu Q, Qiu C, Tanaka M, Zhao Y (2020) Breast cancer cells promote CD169(+) macrophage-associated immunosuppression through JAK2-mediated PD-L1 upregulation on macrophages. Int Immunopharmacol 78: 106012 Doi 10.1016/j.intimp.2019.106012 [DOI] [PubMed] [Google Scholar]
- 19.Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, Fouse SD, Yamamoto S, Ueda H, Tatsuno Ket al (2014) Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343: 189–193 Doi 10.1126/science.1239947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim H, Zheng S, Amini SS, Virk SM, Mikkelsen T, Brat DJ, Grimsby J, Sougnez C, Muller F, Hu Jet al (2015) Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res 25: 316–327 Doi 10.1101/gr.180612.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim Y, Varn FS, Park SH, Yoon BW, Park HR, Lee C, Verhaak RGW, Paek SH (2021) Perspective of mesenchymal transformation in glioblastoma. Acta Neuropathol Commun 9: 50 Doi 10.1186/s40478-021-01151-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Korber V, Yang J, Barah P, Wu Y, Stichel D, Gu Z, Fletcher MNC, Jones D, Hentschel B, Lamszus Ket al (2019) Evolutionary Trajectories of IDH(WT) Glioblastomas Reveal a Common Path of Early Tumorigenesis Instigated Years ahead of Initial Diagnosis. Cancer Cell 35: 692–704 e612 Doi 10.1016/j.ccell.2019.02.007 [DOI] [PubMed] [Google Scholar]
- 23.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P (2015) The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1: 417–425 Doi 10.1016/j.cels.2015.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP (2011) Molecular signatures database (MSigDB) 3.0. Bioinformatics 27: 1739–1740 Doi 10.1093/bioinformatics/btr260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW (2016) The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131: 803–820 Doi 10.1007/s00401-016-1545-1 [DOI] [PubMed] [Google Scholar]
- 26.Meyer M, Reimand J, Lan X, Head R, Zhu X, Kushida M, Bayani J, Pressey JC, Lionel AC, Clarke IDet al (2015) Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc Natl Acad Sci U S A 112: 851–856 Doi 10.1073/pnas.1320611111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mikolajewicz N, Brown KR, Moffat J, Han H (2022) Multi-level cellular and functional annotation of single-cell transcriptomes. bioRxiv: 2022.2003.2013.484162 Doi 10.1101/2022.03.13.484162 [DOI] [PMC free article] [PubMed]
- 28.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila Eet al (2003) PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34: 267–273 Doi 10.1038/ng1180 [DOI] [PubMed] [Google Scholar]
- 29.Nassiri F, Liu J, Patil V, Mamatjan Y, Wang JZ, Hugh-White R, Macklin AM, Khan S, Singh O, Karimi Set al (2021) A clinically applicable integrative molecular classification of meningiomas. Nature 597: 119–125 Doi 10.1038/s41586-021-03850-3 [DOI] [PubMed] [Google Scholar]
- 30.Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, Richman AR, Silverbush D, Shaw ML, Hebert CMet al (2019) An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 178: 835–849 e821 Doi 10.1016/j.cell.2019.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ochocka N, Segit P, Walentynowicz KA, Wojnicki K, Cyranowski S, Swatler J, Mieczkowski J, Kaminska B (2021) Single-cell RNA sequencing reveals functional heterogeneity of glioma-associated brain macrophages. Nat Commun 12: 1151 Doi 10.1038/s41467-021-21407-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ott M, Tomaszowski KH, Marisetty A, Kong LY, Wei J, Duna M, Blumberg K, Ji X, Jacobs C, Fuller GNet al (2020) Profiling of patients with glioma reveals the dominant immunosuppressive axis is refractory to immune function restoration. JCI Insight 5: Doi 10.1172/jci.insight.134386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.P’ng C, Green J, Chong LC, Waggott D, Prokopec SD, Shamsi M, Nguyen F, Mak DYF, Lam F, Albuquerque MAet al (2019) BPG: Seamless, automated and interactive visualization of scientific data. BMC Bioinformatics 20: 42 Doi 10.1186/s12859-019-2610-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RLet al (2014) Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344: 1396–1401 Doi 10.1126/science.1254257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qazi M, Mann A, van Ommeren R, Venugopal C, McFarlane N, Vora P, Singh SK (2014) Generation of murine xenograft models of brain tumors from primary human tissue for in vivo analysis of the brain tumor-initiating cell. Methods Mol Biol 1210: 37–49 Doi 10.1007/978-1-4939-1435-7_4 [DOI] [PubMed] [Google Scholar]
- 36.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB (2003) Identification of a cancer stem cell in human brain tumors. Cancer Res 63: 5821–5828 [PubMed] [Google Scholar]
- 37.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB (2004) Identification of human brain tumour initiating cells. Nature 432: 396–401 Doi 10.1038/nature03128 [DOI] [PubMed] [Google Scholar]
- 38.Sinha A, Huang V, Livingstone J, Wang J, Fox NS, Kurganovs N, Ignatchenko V, Fritsch K, Donmez N, Heisler LEet al (2019) The Proteogenomic Landscape of Curable Prostate Cancer. Cancer Cell 35: 414–427 e416 Doi 10.1016/j.ccell.2019.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, Curtis C, Watts C, Tavare S (2013) Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U S A 110: 4009–4014 Doi 10.1073/pnas.1219747110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, Toms S, Idbaih A, Ahluwalia MS, Fink Ket al (2017) Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. Jama 318: 2306–2316 Doi 10.1001/jama.2017.18718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ESet al (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102: 15545–15550 Doi 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thibaut R, Bost P, Milo I, Cazaux M, Lemaitre F, Garcia Z, Amit I, Breart B, Cornuot C, Schwikowski Bet al (2020) Bystander IFN-gamma activity promotes widespread and sustained cytokine signaling altering the tumor microenvironment. Nat Cancer 1: 302–314 Doi 10.1038/s43018-020-0038-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vasaikar S, Huang C, Wang X, Petyuk VA, Savage SR, Wen B, Dou Y, Zhang Y, Shi Z, Arshad OAet al (2019) Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 177: 1035–1049 e1019 Doi 10.1016/j.cell.2019.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Venugopal C, McFarlane NM, Nolte S, Manoranjan B, Singh SK (2012) Processing of primary brain tumor tissue for stem cell assays and flow sorting. J Vis Exp: Doi 10.3791/4111 [DOI] [PMC free article] [PubMed]
- 45.Wang LB, Karpova A, Gritsenko MA, Kyle JE, Cao S, Li Y, Rykunov D, Colaprico A, Rothstein JH, Hong Ret al (2021) Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 39: 509–528 e520 Doi 10.1016/j.ccell.2021.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wickham H (2016) Data analysis. ggplot2 Springer, City, pp 189–201 [Google Scholar]
- 47.Wilkerson MD, Hayes DN (2010) ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 26: 1572–1573 Doi 10.1093/bioinformatics/btq170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wojtowicz EE, Lechman ER, Hermans KG, Schoof EM, Wienholds E, Isserlin R, van Veelen PA, Broekhuis MJ, Janssen GM, Trotman-Grant Aet al (2016) Ectopic miR-125a Expression Induces Long-Term Repopulating Stem Cell Capacity in Mouse and Human Hematopoietic Progenitors. Cell Stem Cell 19: 383–396 Doi 10.1016/j.stem.2016.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang M, Vesterlund M, Siavelis I, Moura-Castro LH, Castor A, Fioretos T, Jafari R, Lilljebjorn H, Odom DT, Olsson Let al (2019) Proteogenomics and Hi-C reveal transcriptional dysregulation in high hyperdiploid childhood acute lymphoblastic leukemia. Nat Commun 10: 1519 Doi 10.1038/s41467-019-09469-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zeiner PS, Preusse C, Golebiewska A, Zinke J, Iriondo A, Muller A, Kaoma T, Filipski K, Müller-Eschner M, Bernatz Set al (2019) Distribution and prognostic impact of microglia/macrophage subpopulations in gliomas. Brain Pathol 29: 513–529 Doi 10.1111/bpa.12690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y, Yu C (2020) Prognostic characterization of OAS1/OAS2/OAS3/OASL in breast cancer. BMC Cancer 20: 575 Doi 10.1186/s12885-020-07034-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, Wu L, Sloan AE, McLendon RE, Li Xet al (2015) Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol 17: 170–182 Doi 10.1038/ncb3090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu Y, Orre LM, Johansson HJ, Huss M, Boekel J, Vesterlund M, Fernandez-Woodbridge A, Branca RMM, Lehtio J (2018) Discovery of coding regions in the human genome by integrated proteogenomics analysis workflow. Nat Commun 9: 903 Doi 10.1038/s41467-018-03311-y [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All mass spectrometry raw data has been deposited to the Mass Spectrometry Interactive Virtual Environment (MassIVE) with the following MassiVE ID: MSV000087947 and FTP link: ftp://massive.ucsd.edu/MSV000087947/. NanoString data is available on GEO with accession ID: GSE177549.