

Abstract
T cell differentiation requires Notch1 signaling. Here we show that an enhancer upstream of Notch1 active in double-negative (DN) mouse thymocytes is responsible for raising Notch1 signaling intra-thymically. This enhancer is required to expand multipotent progenitors intra-thymically while delaying early differentiation until lineage restrictions are established. Early thymic progenitors lacking the enhancer show accelerated differentiation through the DN stages and increased frequency of B-, ILC-, and NK-cell differentiation. Transcription regulators for T cell lineage restriction and commitment are expressed normally, but ILC- and NK-cell gene expression persists after T cell lineage commitment and TCRβ V-DJ recombination, Cd3 expression and β-selection are impaired. This Notch1 enhancer is inactive in double-positive (DP) thymocytes. Its aberrant reactivation at this stage in Ikaros mutants is required for leukemogenesis. Thus, the DN-specific Notch1 enhancer harnesses the regulatory architecture of DN and DP thymocytes to achieve carefully orchestrated changes in Notch1 signaling required for early lineage restrictions and normal T cell differentiation.
Introduction
Hematopoietic stem cells (HSCs) in the bone marrow (BM) rely on Notch signaling to generate thymus seeding progenitors (TSPs)1, 2, 3. Upon entry into the thymus, Notch1 receptors on TSPs engage with ligand delta-like 4 (DLL4) expressed on thymic epithelial cells (TECs) to induce and sustain differentiation into T cell-related cell fates4, 5. Deletion of Notch1 in hematopoietic progenitors, or DLL4 in TECs blocks TSP differentiation and increases the number of thymic B cell precursors and dendritic cells6, 7, 8, 9, 10. Deletion of RBPjk, a DNA binding protein that associates with the released intracellular form of Notch (ICN) and forms a nuclear complex that induces Notch signaling-dependent gene expression causes a similar phenotype, indicating that Notch1 signaling promotes T cell differentiation in part by repressing differentiation into other related lineages3, 11. On the other hand, ectopic expression of ICN1 in BM progenitors, a product of active Notch1 signaling, or ectopic expression of DLL4 on BM stroma causes extrathymic T cell differentiation12, 13, 14. These studies show that increasing Notch signaling in multipotent progenitors an event that normally happens in the thymus is important for T cell differentiation.
Intra-thymic Notch1 signaling in TSPs initiates a cascade of transcriptional events that mediates restriction into early thymic progenitors (ETPs) and further differentiation into double negative 2a (DN2a) thymocyte precursors where commitment to the T cell lineage occurs15. The combined activities of Notch1 signaling, TCF1, Runx1 and GATA3 are responsible for induction of Bcl11b, a transcription factor implicated in T cell lineage commitmen16. Bcl11b upregulates expression of T cell differentiation genes and downregulates multipotency factors, thereby achieving T cell lineage commitment at the DN2b stage15, 17. Bcl11b supports T cell precursor survival and β-selection at the DN3 stage by facilitating Runx1 recruitment and V-DJ rearrangement at the T cell receptor β locus and by inducing expression of genes related to the T cell receptor signaling pathway17, 18. Notch1 signaling also supports proliferation and survival during β-selection19, 20, 21, 22, 23, 24. Whether Notch1 operates upstream of Bcl11b or in concert with Bcl11b during early T cell differentiation remains unclear.
After completing β-selection, thymocytes transition to the double positive (DP) stage and downregulate Notch1 expression and signaling25, 26. Aberrant reactivation of Notch1 signaling during this transition causes development of thymic T cell leukemias27, 28, 29, 30. Thus, both early activation and later repression of Notch1 signaling are critical events for T cell differentiation and homeostasis.
Here, we show that the level of Notch1 signaling required for intra-thymic T cell differentiation is supported by a DN-specific enhancer located upstream of the Notch1 locus. Using genetic models of inactivation of the DN-specific Notch1 enhancer combined with single cell analysis of ETPs and lineage-committed thymic progenitors we provide new insight into how Notch1 signaling controls progression of TSPs through a multipotent thymic progenitor stage and supports T cell lineage restriction, commitment and β-selection.
Results
A DN-specific regulatory region resides upstream of Notch1
Notch1 mRNA is first detected within the HSC compartment. It is upregulated in ETPs, peaks in DN3 thymocyte precursors, and is attenuated at the pre-DP and DP stages (Fig. 1a, b)25, 26, 31. In addition to an increase in the canonical Notch1 transcript, three transcripts that map upstream of the Notch1 promoter were also detected from ETP through DN3, correlating with induction of Notch signaling at these developmental stages (Fig. 1a, c). One of these transcripts, referred to as DN-specific 1 (DNS1) was transcribed in the same direction as Notch1 and contained two exons that overlapped with the previously reported alternative Notch1 exons 1a and 1b in leukemic cells (Fig. 1d)27, 32. However, splicing of DNS1 to the Notch1 canonical exons was a very infrequent event in normal DN thymocytes (Fig. 1d, e). The two other transcripts, DNS2 and DNS3, were transcribed in the opposite direction of Notch1 with no obvious open reading frame (Fig. 1d).
Fig. 1. A 5’ regulatory region controls Notch1 expression in T cell progenitors.
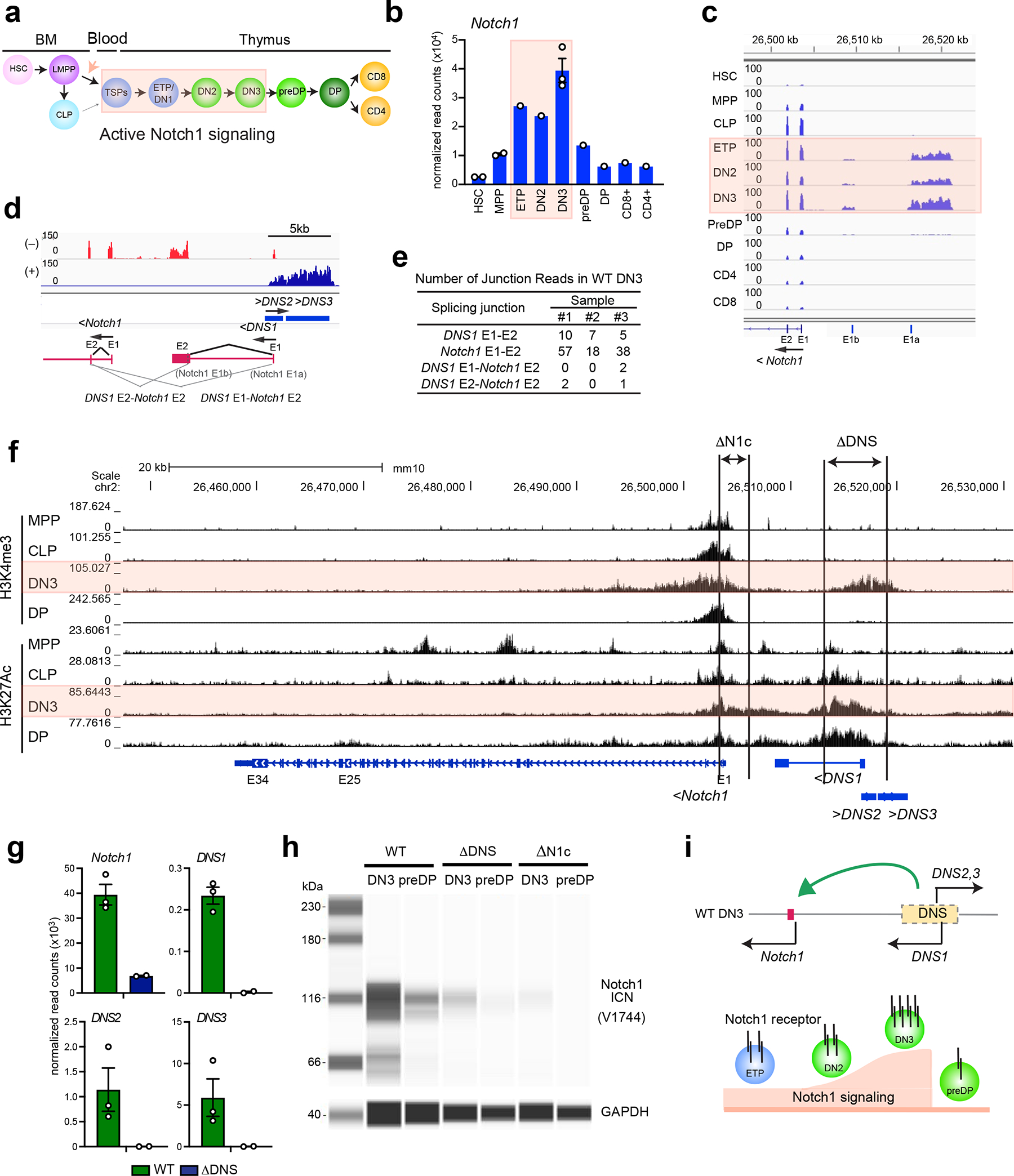
(a) Schematic of T cell development with active Notch1 signaling highlighted in pink. HSC: hematopoietic stem cell, LMPP: lympho-myeloid-primed progenitor, CLP: common lymphoid progenitor, TSP: thymus seeding progenitor, ETP: early thymic progenitor, DN: CD8−CD4− double negative cells, DP: CD4+CD8+ double positive cells, CD4:CD4+ single positive cells, CD8: CD8+ single positive cells (b) Expression of Notch1 mRNA during T cell development with data presented as mean values +/− SEM when replicates are available. (c-d) Genome browser tracks of normalized and strand-specific RNAseq reads at the 5’ region of Notch1 are shown for each developmental stage and in DN3. Bi-directionally transcribed DNS RNAs are schematically representated. Exons and splicing events are shown by a closed square and angled line respectively. (e) Number of RNA-seq reads that span splicing junctions are shown for three independent WT DN3 data sets. (f) Genome browser tracks of ChIP-seq for histone modifications (H3K4me3; promoter mark, H3K27Ac; active enhancer and promoter mark) at Notch1 at different stages of differentiation are shown. Targeting of DNS and Notch1 promoter and exon1 are depicted as ΔDNS and ΔN1c. (g-h) Expression of Notch1, DNS1, DNS2 and DNS3 RNAs in DN3 (mean +/− SEM) and of ICN1 protein is shown in DN3 and preDP. (i) A model of regulation of Notch1 transcription and signaling by the DNS region.
Data shown in (b, c, d, e, and g) were generated from one (ETP, DN2, preDP, DP, CD8+ and CD4+), two (ΔDNS DN3) and three (WT DN3) independent experimental groups with pooled samples from at least two biological replicates. Two replicates of HSC, MPP, and CLP were downloaded from publicly available data (GEO: GSE77695). Data shown in (f), H3K4me3 ChIPs in DN3 and DP and H3K27Ac ChIP in DP were generated from one experimental group with pooled samples from at least two biological replicates. H3K4me3 and H3K27Ac ChIPs in MPP, CLP, and DN3 were deduced from published data sets (GEO: GSE60103, GSE73143, GSE115742). Data in (h) were representative of two independent experiments with pooled samples from three biological replicates.
Islands of H3K4me3 revealed the presence of two active promoters; the Notch1 canonical promoter detected from the early stages of hematopoiesis and through T cell differentiation, and a second in the region that expresses the DNS transcripts and detected only at the DN stages (Fig. 1f). H3K27Ac, a mark of transcriptionally active enhancers and promoters, was highly enriched at the Notch1 canonical promoter and over the DNS region (Fig. 1f). In contrast to the canonical Notch1 promoter where the H3K27Ac and H3K4me3 peaks overlapped, at the DNS region these peaks were well separated suggesting the presence of distinct enhancer and promoter elements (Fig. 1f).
Intra-thymic Notch1 signaling requires the DNS region
The role of the DNS region in regulating Notch1 expression was tested by conditionally deleting an encompassing 4.6 kb region (ΔDNSCD2cre) that progressively acquires permissive histone modifications through the DN stages (Fig. 1f and Extended Data Fig. 1a). A previously described Notch1 conditional null model, caused by deletion of the Notch1 canonical promoter and exon 1 was tested in parallel (Fig. 1f, ΔN1cCD2cre)6. As previously reported during normal T cell development, the ΔN1c model is effective as a Notch1 null6, 19. However, under conditions of leukemic transformation, alternative Notch1 promoters are induced that support Notch1 expression and signaling from the conditionally deleted allele27, 29.
Expression of the canonical Notch1 and DNS1-3 RNAs was examined at the DN3 stage of T cell differentiation in both Notch1 deletion mouse models. The expected complete loss of DNS1-3 expression was accompanied by a marked reduction in Notch1 mRNA in ΔDNSCD2cre compared to WT DN3 cells (Fig. 1g and Extended Data Fig. 1b). In Notch1 null (ΔN1cCD2cre) DN3 cells, no expression of Notch1 exon1 was detected, while the remaining exons (exons 2–34) were transcribed at a lower level (Extended Data Fig. 1b, c). However, expression of DNS1-3 and the splicing frequency of DNS1 exons to the Notch1 canonical exons was increased in ΔN1cCD2cre relative to WT DN3 cells (Extended Data Fig. 1c, d, e).
We next evaluated whether these transcriptional changes affected Notch1 signaling by testing levels of the intracellular domain of Notch1 (ICN1) that is released upon receptor-ligand engagement and serves as a co-activator for RBPjk upon entry into the nucleus33. ICN1 was highly expressed in WT DN3 and reduced in pre-DP cells, consistent with the reported up- and down-regulation of Notch signaling at these stages of differentiation (Fig. 1h). A dramatic reduction in levels of ICN1 was seen in the ΔDNSCD2cre DN3 and preDP cells, although compared to the Notch1 null some signal was detected (Fig. 1h). Differential gene expression analysis in WT, ΔDNSCD2cre, and ΔN1cCD2cre DN3 cells further supported these observations. ΔDNSCD2cre mutant showed gene expression changes that were similar albeit milder compared to ΔN1cCD2cre DN3 (Extended Data Fig. 1f, g). No new genes were deregulated in ΔDNSCD2cre vs. WT compared to ΔN1cCD2cre vs. WT, indicating that the DNS region and its associated transcripts did not contribute to gene expression outside the Notch1 locus in DN3 cells. Thus our studies show that during early T cell development both induction of Notch1 expression and Notch signaling relies on the DNS region (Fig. 1i).
The DNS region prevents intra-thymic B cell differentiation
Given the dramatic decrease in Notch1 signaling caused by deletion of the DNS region, we evaluated whether and how this mutation affected T cell differentiation. The DNS region was conditionally inactivated using the interferon-inducible Mx-Cre that deletes targeted alleles in all hematopoietic cells including early BM progenitors (Fig. 2a, ΔDNSMxcre) and mice were analyzed 30–35 days after the first injection of pIpC (Fig. 2a). Notch1 null mutant (ΔN1cMxcre) mice were analyzed as controls (Fig. 2a). Thymocyte cellularity and the absolute number of cells at major stages of T cell differentiation were 4-fold reduced in ΔDNSMxcre compared to WT (Fig. 2b, c, d). A greater reduction (64 fold) in total thymocytes and their subsets was detected in ΔN1cMxcre (Fig. 2b, c, d). Pre-committed thymic progenitors residing in the lineage negative (Lin−) DN compartment were further evaluated. The absolute number of ETPs (CD44+cKit+CD25−), the earliest T cell progenitors, and their DN2 progeny (CD44+cKit+CD25+) were significantly decreased in both mutants compared to WT, with reduction being smaller in ΔDNSMxcre compared to ΔN1cMxcre (Fig. 2e, f).
Fig. 2. The DNS region is required for T lineage restriction.
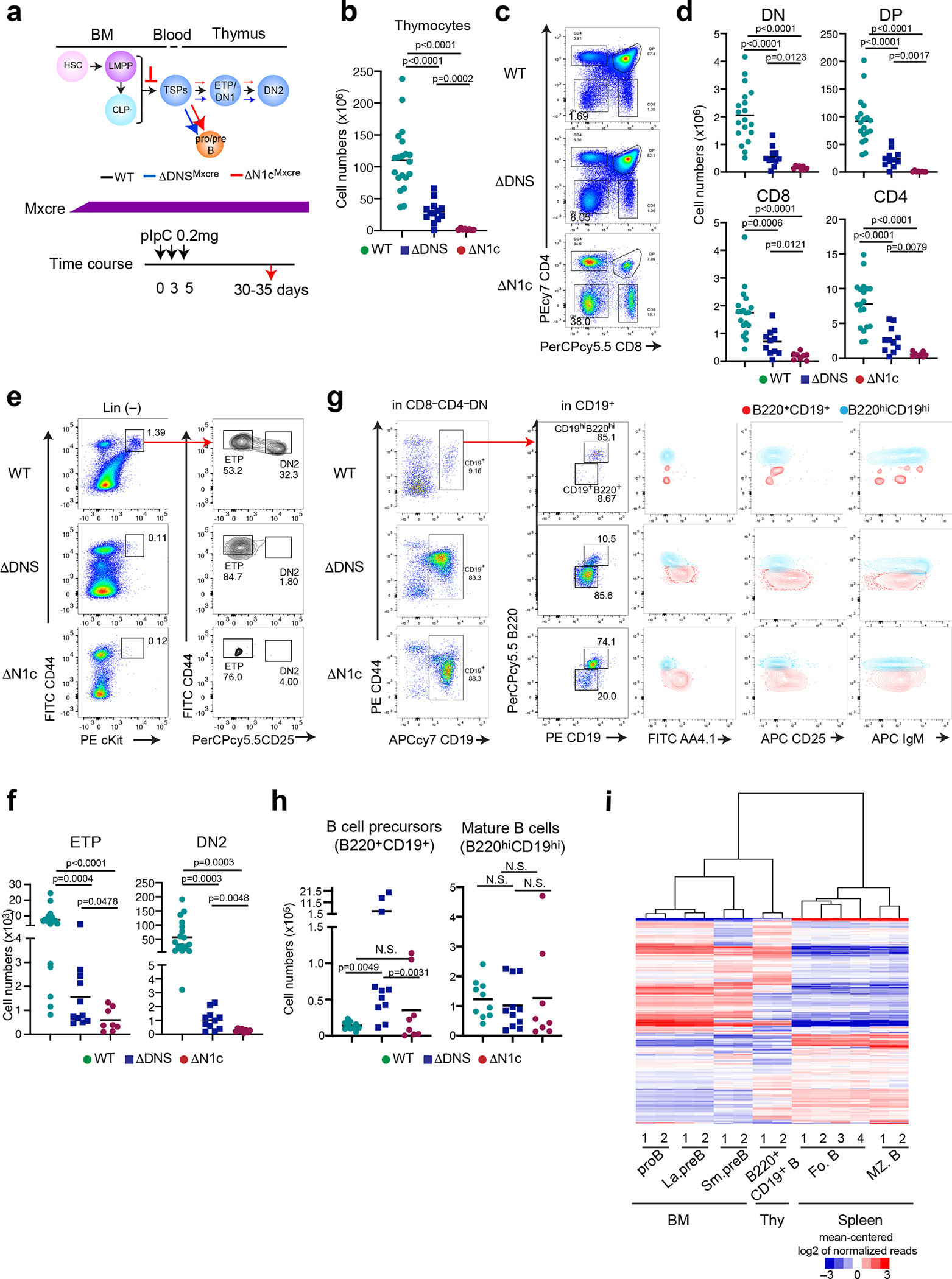
(a) Inducible deletion of DNS or Notch1 by Mx-cre and effects on T cell differentiation are shown. Mx-cre was induced by pIpC administered to 1.5 month-old mice every three days and analyzed 30–35 days later. (b) Thymocyte cellularity is shown for WT, ΔDNSMxcre, and ΔN1cMxcre mice. Statistical analysis was performed by one-way ANOVA and a multiple comparison test. Significance of the differences between samples is provided. (c-d) Analysis of T cell progenitors from WT, ΔDNSMxcre, and ΔN1cMxcre thymus was performed as in (b). (e, f) Analysis of ETP and DN2 progenitors identified after lineage depletion by expression of CD44, cKit, and CD25 is shown as in (b). (g) Analysis of B cells was performed in the CD8−CD4− compartment of WT, ΔDNSMxcre, and ΔN1cMxcre mice. (h) Absolute numbers for B cell progenitors (B220+CD19+) and mature B cells (B220hiCD19hi) in WT, ΔDNS Mxcre, and ΔN1cMxcre thymi are provided with statistical analysis of differences between conditions preformed by one-way ANOVA after exlusion of outliers. N.S., Not signifcant (i) Hierarchical clustering of genes expressed in B cell progenitors isolated from WT BM, mature B cells from WT spleen, and B220+CD19+ B cells from DNS mutant thymus. proB; pro B cells, La.preB; large pre B cells, Sm.preB; small pre B cells, CD19+B; B220+CD19+ B cells, Fo.B; Follicular B cells, MZ.B; marginal zone B cells.
Data shown in (b) were generated from 7 independent experiments with WT N=20, ΔDNSMxcre N=13, and ΔN1cMxcre N=8 mice, in (d) from 6 independent experiments with WT N=19, ΔDNSMxcre N=11, and ΔN1cMxcre N=8 mice, in (f) from 6 independent experiments with WT N=19, ΔDNSMxcre N=11, and ΔN1cMxcre N=8, in (h) from of 5 independent experiments with WT N=10, ΔDNSMxcre N=11, and ΔN1cMxcre N=8. Representative FACS profiles from respective studies are shown in (c), (e) and (g). Data shown in (i) were from two independent experimental groups from WT proB, large preB and small preB in the BM and DNS mutant B220+CD19+ B in the thymus. Data for the four replicates of follicular B and two replicates of marginal zone B were obtained from the ImmGen consortium (GSE122597).
In the WT thymus, a small number of B cells are identified within the DN population (Fig. 2g). The majority (85%) are mature recirculating B cells (CD19hiB220hiAA4.1−IgM+) with a small number (9%) of B cell precursors also detected (Fig. 2g). Surprisingly, the increase in thymic B cell precursors (B220+CD19+ AA4.1+CD25−/+IgM−) caused by reduction of Notch signaling in the ΔDNSMxcre thymus was greater than in the ΔN1cMxcre, although the latter showed greater reduction in thymocyte progenitors and precursors (Fig. 2g, h)6, 7, 8. Thymic B cell precursors (CD19+B220+AA4.1+CD25−/+IgM−) were increased by reduction in Notch1 signaling. They were classified as “BM-like pro/pre-B” cells because their gene expression profile was more similar to that of BM B cell precursors than mature B cell subsets from the spleen (Fig. 2i).
Thus, deletion of the DNS region caused a smaller reduction in ETPs and their T cell progeny but a greater increase in thymic pro/pre-B cell-like precursors compared to the Notch1 null mutant (Fig. 2a). A recent study has shown that Notch signaling induced within the lympho-myeloid-primed progenitors (LMPPs)34 in the BM is important for the generation and migration of TSPs to the thymus3. Complete loss of Notch signaling from these multipotent progenitors accounts for a more severe reduction in ETPs and progeny. On the other hand, loss of the DNS region, not active in BM HSCs and LMPPs, is unlikely to impact the number of TSPs, but alters their intra-thymic differentiation and accounts for a less severe reduction in T cell progenitors and greater increase in B cell precursors compared to the Notch1 null mutation.
A DNS region enhancer guides cell fate decisions
The 4.6 kb DNS region contains a bidirectional promoter and a putative enhancer that may contribute to Notch1 expression. To evaluate their functional contributions, a 600 bp region that lies 4 kb away from the bidirectional DNS promoter and contains evolutionarily conserved transcription factor binding sites in a nucleosome-free region was deleted in the mouse germ line (Fig. 3a and Extended Data Fig. 2a). These DNS enhancer homozygote mutants (ΔDNSe) were born with the expected Mendelian ratio and developed without obvious health issues although Notch1 mRNA expression in DN3 thymocytes was greatly reduced (Fig. 3b). DNS1-3 transcripts, abundantly expressed in WT DN3 cells, were not detectable in ΔDNSe thymocytes (Fig. 3c). Reanalysis of Hi-C data from DN3 thymocytes35 using 10kb bins highlighted statistically significant physical interactions36 preferentially bringing together the DNS enhancer in close proximity to Notch1 promoter (Fig. 3d and Extended Data Fig. 2b). The small genomic distance between DNS enhancer and promoter did not allow for a statistical assessment of their specific looping. However, these two elements and Notch1 promoter were all encompassed within a significant interaction of size 20kb as well as several other spanning interactions up to 150kb in size suggesting all three are within a strongly interacting domain or a sub-domain that facilitates regulation of gene expression by the DNS enhancer (Fig. 3d). Taken together these studies demonstrate that this 600bp region serves as a transcriptional enhancer acting on both the Notch1 and DNS promoters (Fig. 3e).
Fig. 3. Notch1 expression depends on a conserved enhancer.
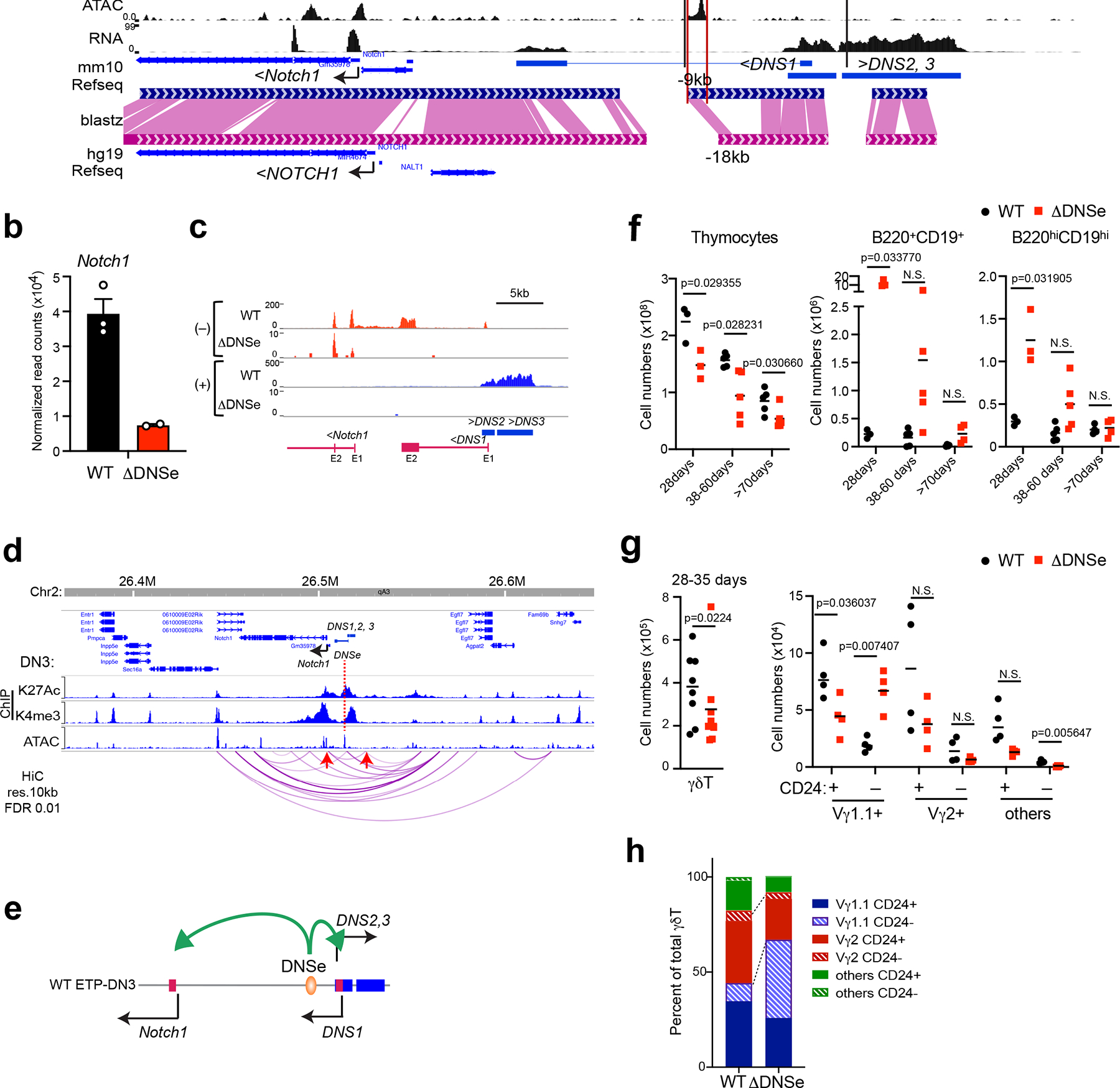
(a) The 600bp region targeting DNSe for deletion as well as histone modifications (ChIPseq), chromatin accessibility (ATAC-seq), and gene expression (RNAseq) are shown in DN3 thymocytes. Sequence conservation between mouse (mm10) and human (hg19) are depicted by dark blue (mouse) and magenta pink (human) with conserved regions highlighted in pink. (b-c) Expression of Notch1, DNS1, DNS2, and DNS3 are shown in WT and ΔDNSe DN3 thymocytes (mean +/− SEM). (d) Statistically significant (FDR <0.01) long distance interactions at Notch1 locus identified by HiC in DN3 thymocytes are shown together with histone modifications and chromatin accessibility. Red arrows highlight a significant interaction between Notch1 promoter and 20kb upstream region. (e) A model of regulation of Notch1 and DNS1-3 transcripts by the DNSe in DN thymocytes. (f) Absolute numbers of total thymocytes, B220+CD19+ B cell precursors and B220hiCD19hi mature B cells from WT and ΔDNSe thymi are provided at different ages with unpaired t-test (two-tailed) used for statistical analysis. N.S. not significant (g) Absolute numbers of total γδT cells and immature (CD24+) or mature (CD24−) γδT cell subsets (Vγ1.1, Vγ2, and others) in the thymus of 28–35 days old WT and ΔDNSe mice are shown with statistical analysis performed as in (f). N.S. not significant (h) Proportional distribution of each γδT cell subset is shown.
ATACseq data shown in (a and d) for DN3 thymocytes was obtained from ImmGen (GSE100738) with other datasets described in Fig. 1. Data shown in (b-c) for ΔDNSe were generated from two independent experiments with pooled samples from at least two biological replicates. Data for WT DN3 is also used in Fig. 1g. HiC analysis of published data (GSE79422) is shown in (d). Data in (f) were generated from 4 independent experiments with mice at different age groups (WT; N=3, N=5, N=5 at 28, 38–60, >70 days, respectively, ΔDNSe; N=3, N=5, N=6 or 4 at 28, 38–60, >70 days, respectively), in (g and h) were generated from 3 independent experiments with WT; N=8 and ΔDNSe; N=8.
Compared to littermate controls, thymic cellularity and the absolute number of αβT cell subsets (DN1-DN4, DP, CD8+SP and CD4+SP) in ΔDNSe mice were reduced through all the age groups tested (Fig. 3f and Extended Data Fig. 3a, b). A similar increase in intra-thymic B cell precursors was seen in ΔDNSe and ΔDNSMxcre mice (Fig. 3f, and Extended Data Fig. 3a). At 28 days, thymic B cell precursors (B220+CD19+) showed a 52 fold increase in absolute number in the ΔDNSe compared to WT thymus (Fig. 3f). Although the difference decreased with age, a 10-fold increase in B cell precursors was still detected at later time points (Fig. 3f). In contrast, the smaller increase (4-fold) in mature B cells (B220hiCD19hi) dissipated with age to control levels (Fig. 3f).
The absolute number of total γδ T cells, derived from committed T cell precursors (i.e., DN2 and DN3), was also decreased in ΔDNSe compared to WT with the proportional distribution skewed towards the Vγ1.1+ subset and the absolute number of Vγ1.1+CD24− mature γδ T cells increased (Fig. 3g, h and Extended Data Fig. 3c).
Taken together our studies show that the ability of the DNS region to control cell fate decisions in early thymic progenitors relies on an evolutionally conserved enhancer (hereinafter referred to as DNS enhancer/DNSe). Deletion of the DNSe did not alter the lineage choice between αβT and γδT cells but affected the Vγ T cell selection and maturation. The DNS enhancer also restricted intra-thymic differentiation into the B cell lineage.
The DNSe controls establishment of multipotent ETPs
We next tested whether loss of the DNS enhancer affected the composition and lineage differentiation properties of ETPs37, which were reduced by 40% in ΔDNSe compared to WT (Fig. 4a–c).
Fig. 4. ETP multipotency requires the Notch1 DNS enhancer.
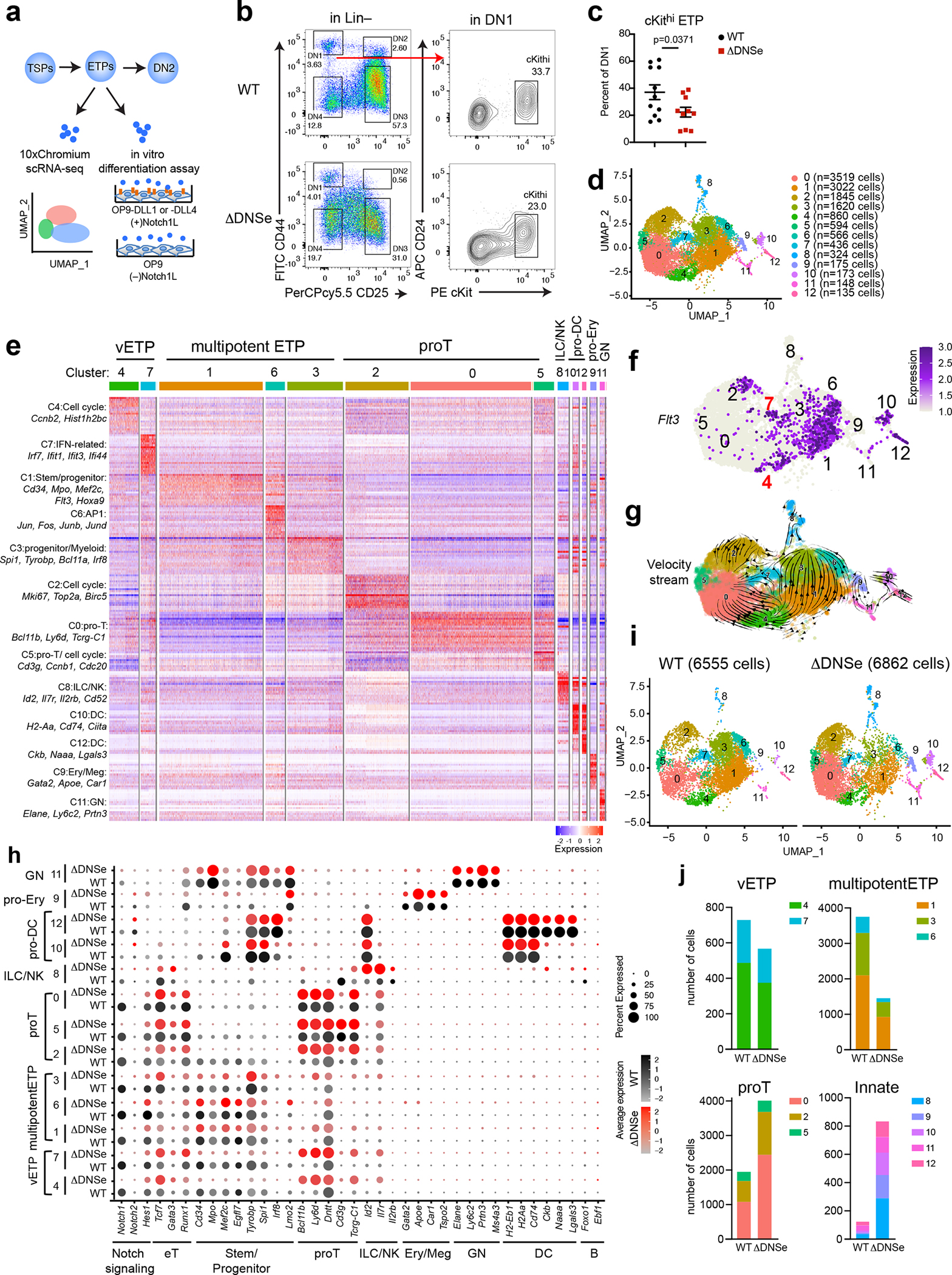
(a) Experimental strategy to test heterogeneity and lineage differentiation potential of ETPs using scRNA-seq and in vitro differentiation assays. (b) ETP FACS analysis is shown in WT and ΔDNSe mice (28–35 days old). ETPs (ckithi) were identified by differential expression of cKit, and CD24 in Lin(−)CD44+CD25−DN1 cells. (c) Relative numbers of cKithi ETPs in WT and ΔDNSe DN1 cells are shown. Average and SEM are provided for each population with unpaired t-test (two-tailed) used for statistical analysis. (d) Integrated analysis and UMAP visualization of WT ETPs (6555 cells) and ΔDNSe ETPs (6862 cells), is shown in 13 colored sub-clusters. Absolute cell number per cluster is shown in parenthesis. (e) Heatmap of the top 20 enriched genes in each sub-cluster ordered by approximate developmental progression based on gene expression and connectivity in the UMAP display. (f) Flt3 expression is shown in the UMAP defined clusters. (g) RNA velocity trajectories are shown in the UMAP space for the combined WT and ΔDNSe ETP single cell data sets. The vector calculation was performed without the 1248 cell-cycle-associated genes that belonged to either GO term: GO:0022402 or Reactome Pathway ID: R-MMU-1640170. (h) Expression of genes that define ETP subsets and downstream stages of T cell differentiation as well as genes representative of innate lymphoid/natural killer (ILC/NK), granulocyte (GN), erythroid/megakaryocyte (Ery/Meg), dendritic cell (DC), and B cell differentiation are shown separately for WT and ΔDNSe. Color intensity is proportional to the average gene expression across cells in respective clusters. The size of circles is proportional to the percent of cells expressing the indicated genes. (i) Independent visualization of WT and ΔDNSe ETPs by UMAP reveals a differential impact of the DNSe mutation on ETP subclusters 0–12. (j) The effect of the DNSe mutation on ETP subcluster composition is shown by bar graph format.
Data shown in (b) were representative FACS profiles from 9 independent experiments with WT (N=11) and ΔDNSe (N=10) samples pooled from at least 3 biological replicates as in (c).
Changes in ETPs were evaluated by single-cell RNA-seq (scRNA-seq). ETPs clustered into 13 subpopulations in two independent experiments (Fig. 4d and Extended Data Fig. 4). Previously established gene expression profiles from HSC, early lineage progenitors and thymic DC were used to annotate the 13 clusters (Extended Data Fig. 5a and Supplementary Table 1). Based on wide-spread expression of genes linked to hematopoietic progenitors three clusters (1, 3 and 6), were identified as multipotent progenitors (multipotentETP) (Fig. 4d, e and Extended Data Fig. 5a). The smaller cluster 4, expressed Flt3 and progenitor genes and was tentatively identified as the most primitive of ETPs (very Early Thymic Progenitors/vETPs) based on similarity to a previously reported ETP subtype (Fig. 4d–f and Extended Data Fig. 5b, c)5, 38, 39. Cluster 7 was included in this vETP designation (Fig. 4d–f and Extended Data Fig. 5b, c). Three clusters expressed early T cell markers and were identified as “pro-T cells” (proT), with clusters 0 and 2 classified as quiescent and cycling respectively and cluster 5, expressing CD3 complex genes, as more advanced in T cell differentiation (Fig. 4d, e, and Extended Data Fig. 5a). Smaller groups of cells, clusters 8–12, were identified as innate lineage progenitors (Fig. 4d, e and Extended Data Fig. 5a). In this analysis, it is noteworthy that early progenitor populations lie in the center of the gene expression space with more committed proT clusters on one side and the expanded progenitor pool on the other (Fig. 4d, e). This suggests that early ETPs choose between two developmental paths, either a direct transition towards proT cells or expansion as less committed progenitors. Velocity analysis used to infer developmental trajectories supported this conclusion and further predicted that these expanded progenitors can then give rise to proT cells as well as progenitors biased towards other lineages (Fig. 4g and Extended Data Fig. 6). In the transition from vETP to multipotentETP, “pluripotent progenitor” gene expression increases with augmented expression of progenitor, proT, and innate cell genes driving the velocity trajectories. The transition from vETP to proT is associated with increased expression of T cell lineage genes.
Notch1 was widely expressed in the primitive (4,7), progenitor (1,3,6) and pro-T (0,2,5) cell clusters (e.g., 0–7) in the WT but not in the ΔDNSe (Fig. 4h). Although the overall pattern of WT vs. ΔDNSe ETP clusters was similar important differences were noted. The multipotentETP clusters 1,3 and 6 were reduced by 2–4-fold in ΔDNSe relative to WT (Fig. 4i, j). In contrast, the proT ETP clusters (e.g., 0 and 2) were increased by 2-fold (Fig. 4i, j). Furthermore, pro-T cell marker genes were expressed at higher levels in the ΔDNSe proT cells (Fig. 4h). The reciprocal effects on pro-T and multipotentETP clusters was also observed in the vETP clusters 4 and 7 (Fig. 4h–i). In WT, cells in these clusters were distributed equally along the gene expression space, but in the ΔDNSe they exhibited a clear shift to the proT side of the spectrum (Fig. 4i). Velocity analysis in the ΔDNSe supported this observation. In the absence of the augmented Notch1 expression in the ΔDNSe, the choice between a direct proT path vs. expansion as multipotentETP appears to be strongly biased towards the proT cell path (Extended Data Fig. 6a). Cells making this transition exhibit the augmented expression of ETP thymus-seeding and T cell lineage genes. Cells taking the path from vETP to multipotentETP in the DNSe mutant show an increased contribution of innate lineage genes to the velocity analysis (Extended Data Fig. 6b). Velocity analysis also linked the ILC/NK cluster 8 to the multipotent ETP clusters, confirming their derivation from the multipotent ETP progenitor pool and revealing an increased transition to cluster 8 in ΔDNSe (Extended Data Fig. 6a). Notably, these more committed clusters that express low levels of Notch1 in WT showed a 6–8 fold increase among ΔDNSe ETPs compared to WT ETPs, larger than the 2–4 fold increase observed among the proT clusters that express Notch1 in wild type (Fig. 4h–j).
Our studies reveal that levels of Notch1 expression and signaling achieved by the DNS enhancer help to establish and maintain the majority of ETPs in an uncommitted progenitor state while ΔDNSe ETPs are more likely to directly transition to a pro-T cell state. An increase in ETPs with an innate cell progenitor phenotype, normally a very small minority among WT ETPs, is also observed in ΔDNSe.
T cell lineage restriction and β-selection depend on the DNSe
The differentiation potential of ΔDNSe ETPs was compared to WT by co-culture with OP9 stroma cells expressing either DLL4, a ligand for Notch1, or DLL1, a ligand for all Notch receptors (Fig. 4a and Fig. 5). ETPs from both genotypes differentiated predominantly into T cell precursors (Thy1hiCD25+) (Fig. 5a). ΔDNSe ETPs generated a ~10 fold higher number of ILC (CD44+Thy1hiICOS+) and NK (CD44+NK1.1+Thy1+/−) cell precursors, consistent with the dramatic increase in ETPs with ILC/NK progenitor signatures seen by scRNAseq (Fig. 5a, Fig. 4h–j cluster 8). The frequency of ETP differentiation into the T, NK and ILC lineages was quantified by a limiting dilution assay on OP9-DLL1 (Fig. 5b, Supplementary Table 2). A 21.3-fold increase in the frequency of NK cells (WT: 1 in 1335.3, ΔDNSe: 1 in 62.6), and a 3.2-fold increase in ILC precursors (WT: 1 in 132.8, ΔDNSe: 1 in 40.3) were detected in ΔDNSe compared to WT ETPs co-cultures (Fig. 5b). In contrast, a 1.8-fold reduction in T cell precursors (WT: 1 in 4.17, ΔDNSe: 1 in 7.53) was seen (Fig. 5b). Increase in B cell differentiation frequency was also detected with ΔDNSe ETPs (Fig. 5c, d, and Supplementary Table 2).
Fig. 5. Increase in innate lymphoid potential and developmental arrest of ΔDNSe DN3.
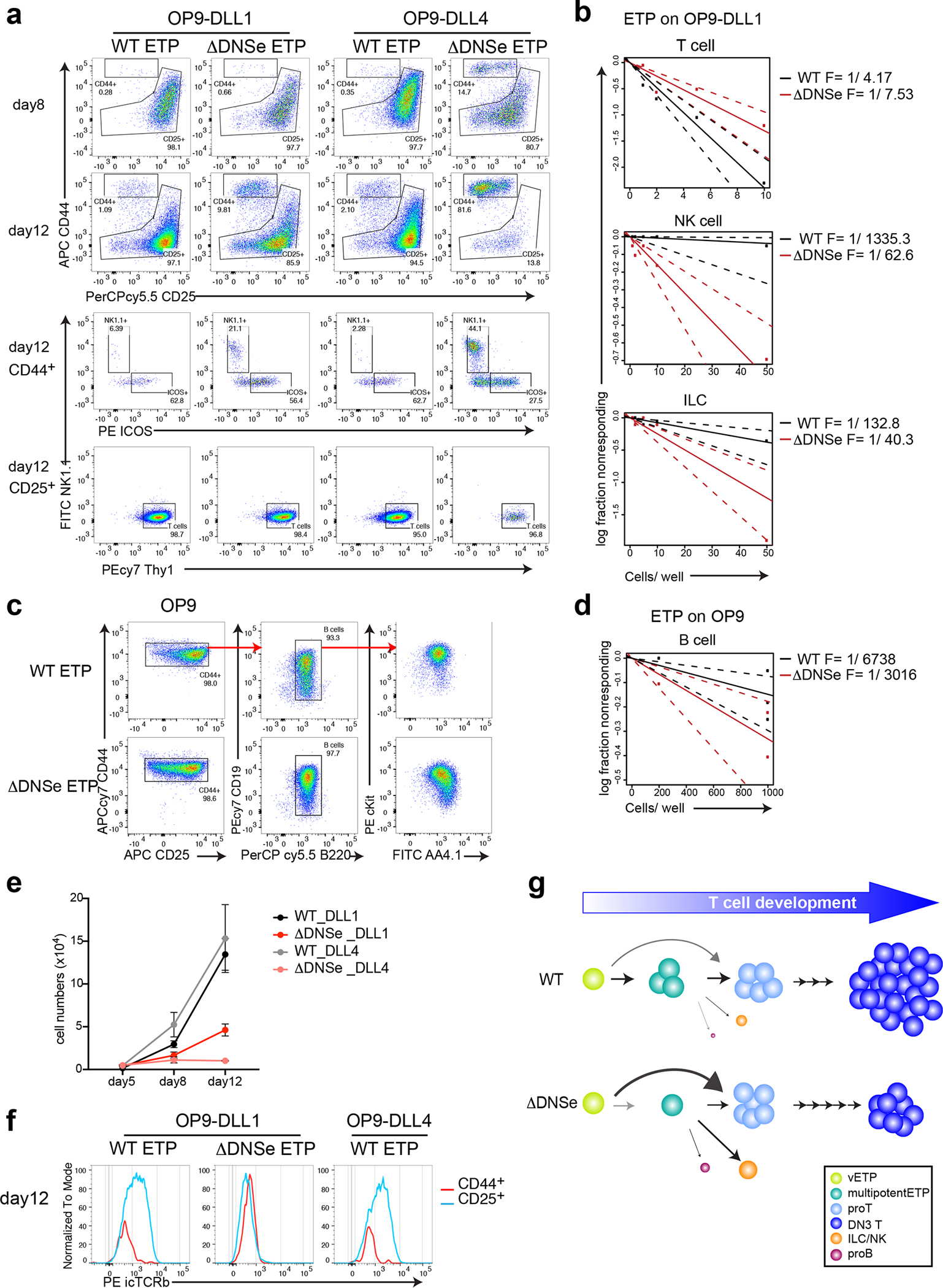
(a, b, e, f) Lineage potential of ETPs after co-culture on OP9-DLL1 or OP9-DLL4 stroma under T cell differentiation conditions (2ng/ml of IL7 and 5ng/ml of Flt3L). (a) FACS profiles of ETPs are shown after 8–12 days in culture. Cells were tested for expression of T (Thy1+CD44−CD25lo/+), NK (Thy+/− NK1.1+CD44+CD25−/lo), or ILC (Thy1+ICOS+CD44+CD25−/lo) cell lineage differentiation markers. (b) T, NK, and ILC cell lineage differentiation frequencies of WT and ΔDNSe ETPs are shown. (c) FACS profiles of ETPs after 13 days in culture on OP9 stroma for WT or 8 days for ΔDNSe are shown. Expanded cells expressed markers of early B cell differentiation (B220+CD19+AA4.1+cKit+). (d) B cell lineage frequency of WT and ΔDNSe ETPs is shown. Dashed lines in (b) and (d) indicate the 95% confidence interval for the regression line. (e) Absolute number of hematopoietic cells at different time points of culture under T cell differentiation condition are shown (mean +/− SEM). (f) Recombination at the Trb locus was evaluated by expression of intracellular TCRβ (icTCRβ) after 12 days in culture. (g) A model on ETP differentiation by Notch1 signaling. High levels of Notch1 signaling (WT) supported by the DNS enhancer promote the transition of very early ETPs (Flt3+) through a multipotent to a pro-T cell state prior to lineage restriction. Deletion of the DNS enhancer and reduction in Notch1 signaling (ΔDNSe) reduces the number of multipotentETPs and increases precocious ETP differentiation into T and ILC/NK cell lineages.
Data shown in (a, f) are representative FACS profiles from 3 independent experiments with more than 3 technical replicates per genotype per experiment, in (b) are combined data from 2 independent experiments. Data shown in (c) are representative FACS profiles from 3 independent experiments, in (d) are combined data from 3 experiments. Data shown in (e) are representative of 3 independent experiments. Technical replicates (3 wells/each genotype) were analyzed at each time point.
We next tested whether the loss of DNSe affected progression through the DN stages of T cell differentiation. After eight days of culture on either OP9-DLL1 or OP9-DLL4, WT ETPs differentiated into DN2 and DN3 thymocytes (Fig. 5a). In the same time frame, ΔDNSe ETPs showed accelerated differentiation to the DN3 stage (Fig. 5a). By day 12, ΔDNSe DN3 cells were greatly reduced in OP9-DLL4 cultures, indicating that Notch1 signaling was also required for DN3 precursor survival (Fig. 5a, e). However, this was not seen when ΔDNSe DN3 cells were cultured on OP9-DLL1, indicating that signaling through other Notch receptors could support survival40. Despite their ability to survive, ΔDNSe DN3 cells did not rearrange or express intracellular TCRβ, a hallmark of successful rearrangement at the Trb locus that was observed in WT cultures (Fig. 5a, e, f).
Thus, loss of the DNS enhancer augments the frequency of ETP differentiation into innate cell lineages such as NK and ILC and affects T cell precursor differentiation and survival (Fig. 5g). Although T cell precursors with reduced levels of Notch1 signaling (i.e., ΔDNSe) reach the DN3 stage, they fail to survive and undergo TCRβ rearrangement and β-selection. Upon loss of Notch1 signaling, other Notch family members can rescue thymocyte precursor survival but fail to support β-selection.
Repression of extra-lineage transcripts in committed proTs
To gain insight into how the DNSe affected DN3 differentiation and β-selection, we examined the transcription profiles of WT and ΔDNSe DN3 cells generated on OP9-DLL1 by bulk mRNA analysis. Consistent with their cell surface phenotypes, both WT and ΔDNSe DN3 cells generated in culture were transcriptionally similar to in vivo generated DN3 thymocytes and distinct from earlier T cell progenitors (ETP or DN2) or progenitors of innate lymphoid cells obtained from the BM (ILC2 or NK) (Extended Data Fig. 7a). Comparative analysis of WT vs ΔDNSe DN3 cells also identified differentially expressed genes (DEGs) as potential targets of Notch1 signaling mediated by the DNSe (Fig. 6a, Up: 1003 genes, Down: 898 genes). Known Notch signaling gene targets such as Dtx1, Hes1, Notch3, Nrp1, Hivep3, Myc, and IL2Ra were downregulated (Fig. 6b, Supplementary Table 3). Gene set enrichment analysis (GSEA) confirmed that genes activated by Notch signaling and its downstream target Myc were downregulated in ΔDNSe DN3 cells (Extended Data Fig. 7b).
Fig. 6. Promiscuous expression of innate lymphoid genes in ΔDNSe DN3 cells.
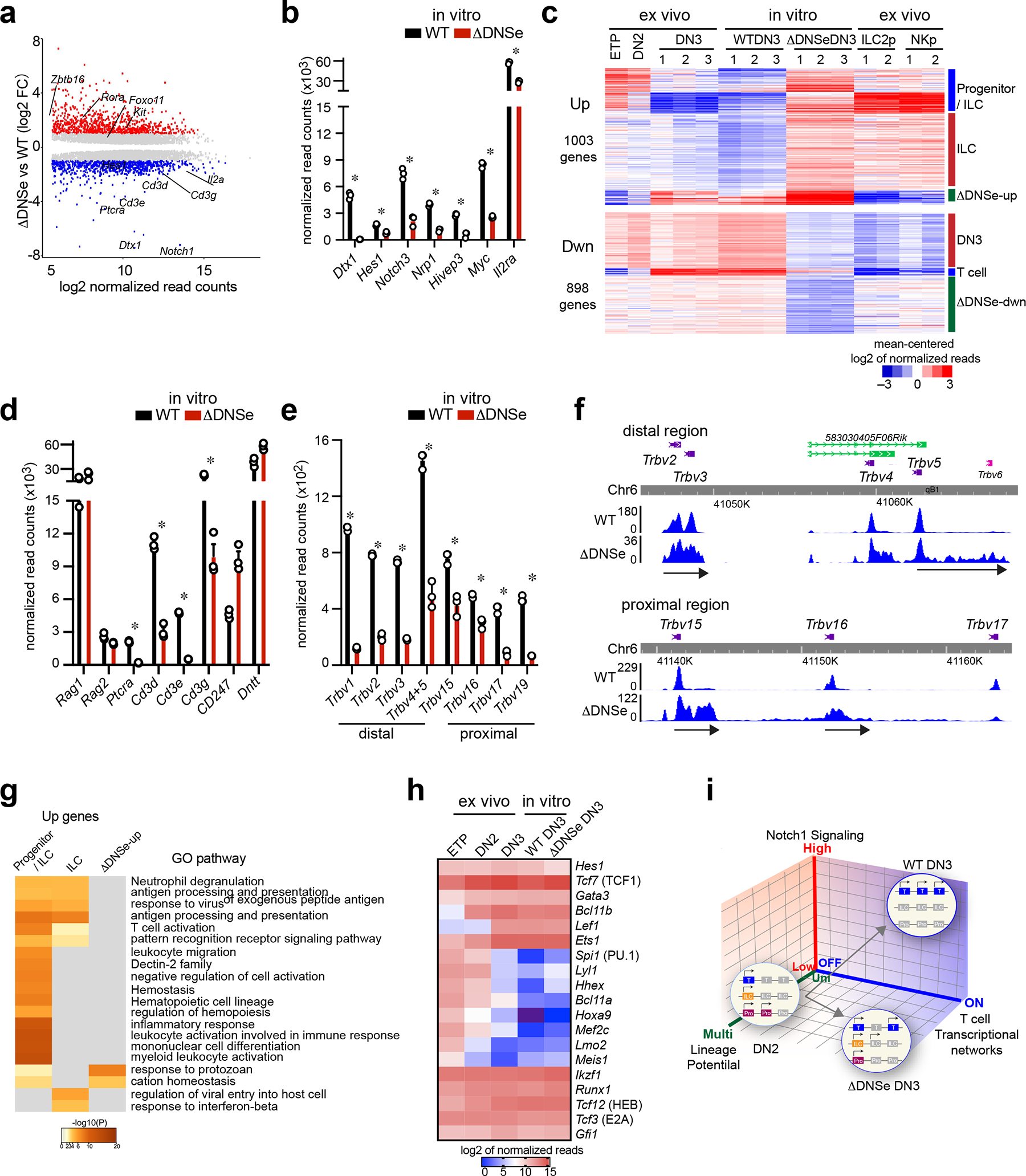
(a) Differentially expressed genes (mean normalized read counts>50, adj. p <0.05) are shown for cultured ΔDNSe vs WT DN3. Up- (log2FC>1, adj. p <0.05, 1003 genes) and Down- (log2FC <−1, adj. p <0.05, 898 genes) regulated genes are highlighted in red and blue, respectively. (b) Expression of Notch1 downstream target genes in cultured DN3 cells is shown (mean+/−sem). * ΔDNSe vs WT DN3: log2FC <−1 or >1, adj. p <0.05 (c) Heatmap of K-means clustering for Up- and Down-regulated genes in cultured ΔDNSe vs WT DN3 is provided. Deregulated genes were co-clustered with expression data from ex vivo WT T cell progenitors (ETP, DN2 DN3 cells) and WT innate lymphoid progenitors (ILC2p and NKp). (d) Expression of Rag1, Rag2, Dntt, Ptcra, and Cd3 genes in cultured DN3 cells is shown (mean+/−sem). * ΔDNSe DN3 vs WT DN3: log2FC <−1 or >1, adj. p <0.05 (e and f) Expression of Trbv genes is shown by bar graph (mean+/−sem, * adj. p <0.01) (e) and RNAseq read distribution is visualized by genome browser (f). (g) Heatmap based on significance of functional pathway enrichment (−log10(pval)) is shown for up-regulated genes classified into three signatures as in (c). (h) Heatmap of gene expression for key transcription factors in T cell development is provided. Analysis of combined datasets from ex vivo WT DN3 and cultured DN3 from WT and ΔDNSe is shown from three independent samples. (i) A model on how strength of Notch1 signaling controls induction of T cell transcriptional networks and lineage restriction is provided. Genes associated with the T cell lineage, early progenitors, or innate lymphocytes are depicted by blue, purple, and orange boxes, respectively. Silenced genes are shown in gray.
Data for cultured DN3 cells were generated from three independent experimental groups for each genotype. Data for ex vivo WT ETP and DN2 were generated from one experimental group and for ex vivo WT DN3 from three independent experimental groups as in Fig. 1. Data for the two replicates of ILC2p and NKp were obtained from published datasets (GEO: GSE77695).
Lineage affiliation of DEGs was tested by co-clustering with gene expression data sets from ETP, DN2, ILC2 and NK cell progenitors (Fig. 6c). Genes down-regulated in ΔDNSe mutant thymocytes fell into three categories; one that is specifically and strongly induced in DN3, a second that is expressed in DN3 and downregulated in innate lymphoid cell precursors, and a third that is only downregulated in ΔDNSe mutant DN3, respectively designated as DN3, T cell, and ΔDNSe-down signatures (Fig. 6c and Supplementary Table 3,4). Prominent among genes in the T cell signature were those encoding proteins of the pre-TCR complex (Fig. 6d, Supplementary Table 3). Expression of Ptcra and Cd3d, Cd3e, Cd3g of the CD3 complex that reside within the same genomic locus and are co-regulated41, 42 were significantly reduced in the mutant, whereas Cd247 (Cd3ζ) that lies in a different chromosome was increased (Fig. 6d). Analysis of Trb locus transcripts revealed that transcription at Vb region promoters (both distal and proximal) was active in ΔDNSe DN3 cells, albeit at lower levels compared to WT (Fig. 6e, Extended Data Fig. 7c). Whereas in WT DN3 cells the Vb region Trb transcripts corresponded precisely to the encoding exon, in ΔDNSe DN3 cells an extension into the 3’ intragenic region was seen (Fig. 6f). This indicated expression of sterile Vb transcripts and a pre-V-DJ recombination status at the TCR locus. In contrast, expression of Rag1 and Rag2 or Dntt, key regulators of V-DJ recombination were not affected (Fig. 6d). Thus, reduction in Ptcra, Cd3d, Cd3e, and Cd3g and lack in Trb V-DJ recombination are responsible for lack of a pre-TCR complex in the ΔDNSe DN3 cells. In line with reduction in the pre-TCR complex, downregulated genes in the ΔDNSe DN3 and especially in the ΔDNSe-down signature were involved in cell proliferation that are normally induced by a combination of Notch1 and pre-TCR signaling (Fig. 6c, Extended Data Fig. 7d, Supplementary Table 4).
Genes up-regulated in ΔDNSe DN3 cells fell into three categories; one expressed in earlier T cell progenitors and ILCs and normally downregulated in DN3 (progenitor/ILC signature), a second that is specifically induced in innate lymphoid progenitors (ILC/NK signature), and a third that is normally induced in DN3 and was further upregulated in mutant cells (ΔDNSe-up signature) (Fig. 6c, Supplementary Table 5). Genes in all three clusters were associated with pathways that support innate immune responses and effector cell functions, such as antigen processing and presentation, leukocyte differentiation, pattern recognition receptor signaling as well as response to virus and protozoa (Fig. 6g, Supplementary Table 5). Expression of the kit receptor that supports survival and expansion of early progenitors was upregulated in mutant DN3 cells (Fig. 6a). Genes that belonged to the progenitor/ILC signature and transcription regulators normally expressed in innate lymphoid progenitors such as Rora, Foxo1, and Zbtb16 (PLZF) were also upregulated (Fig. 6a and Extended Data Fig. 7e).
Consistent with the scRNAseq of ETPs, Tcf7, Lef1, Gata3, Ets1 and Bcl11b, key transcriptional regulators that work in concert to establish expression of genes that define T cell identity, such as Cd3d, Cd3e, Cd3g, Dntt, Rag1 and Rag2, were normally induced in ΔDNSe DN3 cells (Fig. 6h)15. Additionally, Spi1 (PU.1), Lmo2, and Mef2c, key regulators of transcription in earlier multipotent progenitors, were attenuated normally in the DNS enhancer mutant DN3 cells (Fig. 6h)15. Expression of Id2, a critical transcription factor for innate lymphoid cell development that is directly repressed by Bcl11b in committed T cell precursors, was normally expressed (Extended Data Fig. 7e, f)17, 43, 44, 45, 46. However, other genes repressed by Bcl11b such as Cd7, Itgae, itgb7, Klf2, Cd163l1 and Cited417 were upregulated indicating a co-dependence on Notch1 signaling for repression (Extended Data Fig. 7f).
Thus committed T cell precursors rely on the DNSe to provide Notch1 signaling that supports T cell differentiation transcriptional programs while restricting innate cell related transcription (Fig. 6i). Although transcription factors that play a key role in establishing T cell identity are induced in ΔDNSe T cell precursors, these cells appear to retain a lineage-diverse (ILC and NK) transcriptional profile that may prevent Trb rearrangement and β-selection, hallmarks of early T cell differentiation. Alternatively, nuclear effectors of Notch1 signaling may directly participate in these T cell-specific molecular processes.
Lineage-specific transcription factors regulate the DNSe
Analysis of publicly available ChIPseq data sets revealed that transcription factors involved in establishing T cell identity such as Ikaros, E2A, HEB, PU.1, TCF1, Runx1, and Bcl11b bound at the DNSe during T cell development (Fig. 7a). De novo motif discovery confirmed that Ikaros, PU.1, Runx1 and TCF1 target through their own DNA binding activity (Fig. 7b). The active form of Notch1 (ICN1) also bound the DNSe in DP leukemic cells supporting a positive feed-back loop in Notch1 regulation (Fig. 7a)27.
Fig. 7. Regulation of DNSe by lineage-specific transcription factors.
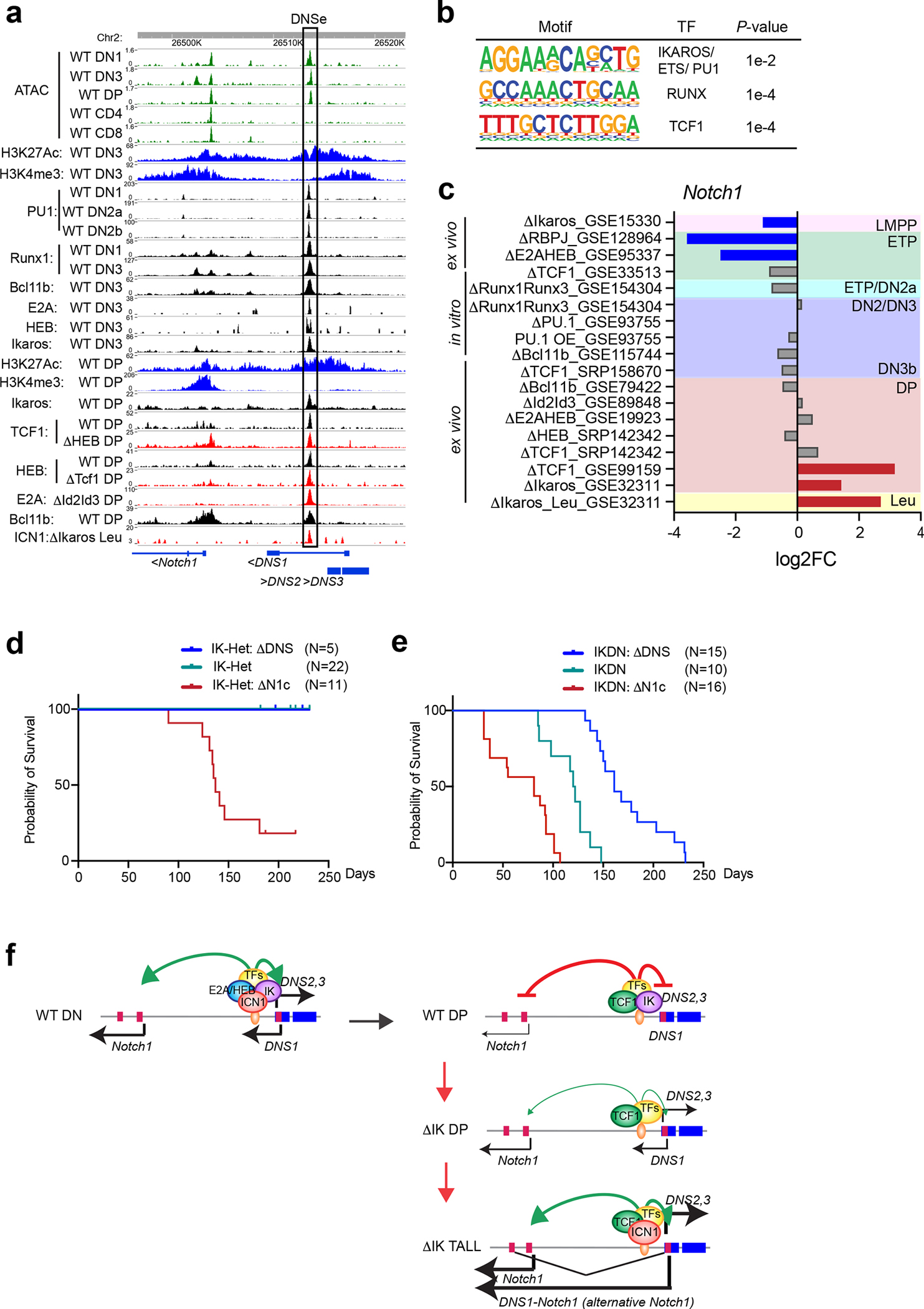
(a) Chromatin accessibility (ATAC-seq), histone modifications and transcription factor enrichment (ChIP-seq) at the DNS enhancer are shown for the DN and DP stages of WT and TF knock-out (Δ) thymocytes. (b) De novo transcription factor-binding motifs are shown at the 0.6kb DNSe region. (c) Changes in Notch1 expression during T cell differentiation are shown in multipotent progenitors and thymocytes from single or double knock out (Δ), or after overexpression (OE) of developmentally relevant transcription factors. Up-regulation (log2FC>1, FDR<0.05) and down-regulation (log2FC <−1, FDR<0.05) of Notch1 relative to WT or control are highlighted in red or blue, respectively with non-significant changes shown in gray. (d and e) Kaplan-Meier survival curves for mice with mutations in Notch1, DNS, and Ikzf1 (Ikaros) are shown. (d) All IK-Het mice (N=22) and IK-Het:ΔDNSCD2cre (N=5) remained healthy during the observation period. 9/11 IK-Het:ΔN1cCD2cre mice died of leukemia. The survival curve for IK-Het is not visible due to overlap with that of the IK-Het:ΔDNSCD2cre. (e) Median survival of IKDNCD2cre mice (N=10) was 121 days, whereas that of IKDN:ΔN1cCD2cre mice (N=16) was shortened to 81 days. In contrast, the onset of leukemia was delayed in IKDN:ΔDNSCD2cre mice (N=15), with median survival of 161 days. (f) A model of regulation of the DNS enhancer by Ikaros and other developmentally relevant transcription factors.
Data shown in (a), H3K4me3 and H3K27Ac ChIPs in DN3 and DP thymoyctes were generated in this study as in Fig. 1. The rest of the data set, i.e., ATAC, ChIP, and RNA expression, are from published datasets listed in Supplementary Table 6. Data shown in (d) were generated from IK-Het mice (N=22), IK-Het:ΔDNSCD2cre (N=5), and IK-Het:ΔN1cCD2cre (N=11), and in (e) were generated from IKDNCD2cre mice (N=10), IKDN:ΔDNSCD2cre (N=15), and IKDN:ΔN1cCD2cre (N=16).
Publicly available gene expression data sets were used to address the effect of inactivating these factors in progenitors prior to and after commitment into the T cell lineage on Notch1 expression. At the LMPP, the earliest lymphoid progenitor, priming of Notch1 expression depended on Ikaros (Fig. 7c)47. Further increase in Notch1 expression at the onset of T cell differentiation in the ETP required RBPjk, E2A and HEB (Fig. 7c)3, 48. DP thymocytes represent a stage of T cell differentiation when Notch1 signaling is normally terminated and its persistence can cause leukemic transformation (Fig. 7c)27. Loss of either Ikaros or TCF1 caused aberrant Notch1 expression in DP (Fig. 7c). This was further elevated upon leukemic transformation of Ikaros mutant DPs consistent with ICN binding at the DNSe and a positive feed-forward loop of Notch signaling on Notch1 expression (Fig. 7c)27.
Thus, the Notch1 locus is primed for expression by Ikaros at the earliest stage of lymphopoiesis and is also repressed by Ikaros at the DP stage of differentiation. Increase of Notch1 expression to levels required for T cell progenitor and early T cell precursor differentiation (i.e., at the DN stages) is supported by E2A, HEB, and the nuclear effectors of Notch1 signaling working through the DNSe.
DNSe repression in DP thymocytes prevents leukemogenesis
To test the hypothesis that Ikaros suppresses Notch1 expression and signaling after β-selection by repressing the DNSe, we crossed ΔDNSCD2cre or ΔN1cCD2Cre to two different Ikaros inactivation mouse models; one heterozygous for an Ikaros null mutation (Ikzf1+/−: IK-Het) that displays a very mild leukemogenic phenotype and a second that is heterozygous for a dominant negative Ikaros mutation (IkE5 fl/+; CD2cre: IKDNCD2cre) with a severe leukemogenic phenotype27, 49. As shown in our past studies, the Notch1 null genotype that lacks the canonical Notch1 promoter and exon 1 augments the mild IK-Het leukemogenic phenotype (Fig. 7d, IK-Het:ΔN1cCD2cre)27. In these mice, leukemias develop within 5 months at high penetrance due to activation of the DNS promoter and, in the absence of the canonical Notch1 promoter, induction of DNS1-originating Notch1 transcripts (Fig. 7d)27. In contrast, mice with combined mutations in ΔDNS and IK-Het (IK-Het:ΔDNSCD2cre) did not develop leukemia by the end of the experimental period (250 days) (Fig. 7d). In the more severe loss-of- Ikaros leukemogenesis model, IKDNCD2cre, mice developed leukemia rapidly with a median survival of 121 days (Fig. 7e). The combined IKDN:ΔN1cCD2cre mutations accelerated the disease process and reduced the median survival to 81 days (Fig. 7e). In sharp contrast, disease development was decelerated in the IKDN:ΔDNSCD2cre mice with the median survival increasing to 161 days (Fig. 7e).
The DNS deletion used in this study includes both the DNSe and the alternative Notch1 promoter and the attenuating effects on Ikaros loss-induced leukemogenesis could be caused by ablation of either of these elements. However, the DNSe is directly bound by Ikaros and is also required for activation of the Notch1 canonical promoter, while the DNS promoter plays no detectable role in Notch1 expression when the canonical promoter is present. We therefore conclude that Ikaros directly represses the activity of the DNSe after β-selection and that re-activation of this regulatory unit is in part responsible for initiation of loss-of-Ikaros mediated leukemogenesis through induction of Notch1 transcripts (Fig. 7f).
Discussion
Induction of Notch1 signaling in the hematopoietic system activates cellular pathways that support multipotent progenitor migration to the thymus1, 2, 3. Upon entry into the thymus, re-induction or increase in Notch1 signaling triggers T cell differentiation while erasing the potential for multi-lineage differentiation4, 5. Our genetic studies on an enhancer that lies upstream of Notch1 reveal an unexpected role for Notch1 signaling and provide important revisions for the molecular process that controls early T cell development.
Among bone marrow hematopoietic progenitors, Notch1 is first induced in the LMPP34, and expression increases during early stages of T cell differentiation in the thymus. A set of noncoding RNAs (DNS1-3) transcribed from a bidirectional promoter that lies upstream of the Notch1 promoter were specifically expressed at the early DN stages of thymocyte differentiation. Evaluation of this upstream genomic region (DNS region) for chromatin accessibility, chromatin interaction, transcription factor enrichment and histone modifications identified a 600bp putative enhancer. An engineered deletion spanning the DNS RNAs, their promoter, and this putative enhancer revealed a requirement in this region for normal T cell development in the thymus. A deletion of the enhancer region that left the promoter and DNS transcribed regions intact confirmed that this was a DN stage-specific enhancer that increases Notch1 expression from the canonical promoter and is also required for expression of the DNS RNAs. The absolute dependence of the DNS1-3 transcripts on DNSe precluded further conclusion on their role in the mouse models we generated.
The DNSe features a high density of transcription factor binding motifs conserved between mouse and human. Binding of transcription factors involved in T cell development is observed in this region regardless of the stage of differentiation. Ikaros, PU.1, Runx1, Bcl11b, E2A and HEB are bound in DN thymocytes when the enhancer is active, while Ikaros, HEB, TCF1, and Bcl11b are bound in DP thymocytes when the enhancer is inactive27, 35. Transcriptome meta-analysis provided further insight into stage-dependent regulation of Notch1 by lineage specific transcription factors. Ikaros is required for priming of Notch1 in the LMPPs in the BM, whereas RBPjk and E2A/HEB are required to elevate Notch1 expression at the onset of T cell differentiation in the thymus3, 47, 48. Later in T cell differentiation, Notch1 signaling and expression must be attenuated to prevent leukemogenesis with Ikaros27 and TCF1 involved in the repression process. The active form of Notch1 (ICN1) is bound at this enhancer in leukemic thymocytes that also express aberrant levels of Notch127, indicating that positive feedback of Notch1 signaling on Notch1 expression is effected through this enhancer. It also highlights the importance of repression mechanisms operating through this regulatory region in preventing leukemogenesis (Fig. 7f).
Deletion of the DNS enhancer caused an accumulation of B cell precursors within the thymus, normally a rare population. The increase, although more profound in young was also seen in old mice. Transcriptional profiling identified these cells as thymic pro/pre-B cells indicating that they were produced intra-thymically, possibly by disruption in lineage restriction of early thymic progenitors such as the ETP. Increased expression of Ly6d in the vETP cluster (Fig. 4h) may presage the enhanced capacity for B cell differentiation in the ETP pool50. In contrast to the milder effect on ETP cellularity seen with the DNSe mutation compared to the Notch1 null mutation, a more profound B cell precursor accumulation was observed. Loss in Notch1 expression in BM progenitors is expected to affect their migration into the thymus and therefore to impact the number of the earliest thymic immigrants and their progeny, whereas the DNSe mutation effects are expected to manifest within established thymic progenitors.
Single cell RNA-based velocity analysis confirmed that a previously reported developmental flux from a Flt3hi vETP subset to a Flt3lo multipotentETP subset with increased expression of progenitor markers is the dominant path in WT (Fig. 4f). However, it also revealed a more direct conversion into proT-ETPs as an alternative path, albeit a more minor one among WT ETPs. In the absence of the DNSe, vETPs showed increased expression of early T cell markers and both velocity analysis and relative cell numbers suggested that more direct differentiation into proT-ETPs becomes the dominant pathway. An increase in the expression of T cell differentiation markers was also seen among ΔDNSe ETP proT cells. The premature activation of T cell differentiation genes and expansion of ETPs with a proT cell transcriptome in vivo is consistent with the accelerated differentiation of ΔDNSe ETPs to the DN3 stage relative to WT.
ETPs normally exhibit a latent differentiation potential towards innate cell lineages. Loss in the DNSe increased ETP differentiation into innate cell lineages. The effect was revealed both by an increase in the frequency of ETP differentiation into ILC/NK cells in vitro and by an increase in ETPs with a transcriptional profile of ILC and NK progenitors in vivo. Thus, by preserving ETP multipotency, the Notch1 DNSe not only ensures the timely restriction into the T cell lineage but also prevents differentiation into innate cell lineages.
Despite lower levels of Notch signaling, ΔDNSe thymocyte precursors reached the DN3 stage and exhibited normal expression of genes encoding transcription factors that establish T cell identity, such as TCF1, GATA3, Runx1, and Bcl11b. Thus, the level of Notch1 signaling provided by the Notch1 DNS enhancer is not required for induction of T cell commitment factors. However, transcription programs associated with innate developmental pathways were aberrantly expressed at the DN3 stage even in the presence of T cell commitment factors. Since these results are based on bulk RNA analysis of DN3 cells we cannot exclude the possibility that expression of innate cell markers represents a distinct subset of cells with a DN3 cell phenotype. Nonetheless, nuclear effectors of Notch1 signaling may repress innate cell fates both prior to and after T cell commitment.
Induction of the Cd3 genes and rearrangement of the Trb locus, defining events of T cell differentiation at the DN3 stage, are regulated by T cell commitment factors including Bcl11b17, 18. V-DJ recombination did not occur in the DNSe mutant DN3 cells despite normal transcript levels of Bcl11b, other cooperating transcription factors (e.g., Runx1 and Tcf3) and the Rag1 and Rag2 genes, and expression of Vb germline transcripts at the Trb locus. The failure to concomitantly repress genes associated with innate cell fates and resulting conflict between multi-lineage transcriptional cascades may explain the impaired ability to establish pre-TCR signaling and promote T cell lineage progression. Alternatively, lack of TCR rearrangement may reflect a direct role for Notch1 in the process. This failure to survive at the DN3 stage and progress to DP stages may underly the commonly held belief that Notch1 activity promotes T cell differentiation in early thymocytes. Our detailed analysis of the DNSe mutant mice revealed that this later positive role for augmented Notch signaling in promoting T cell differentiation is preceded by an earlier role in ETPS delaying early progress towards T cell differentiation and promoting expansion of less committed progenitors.
Notch1 expression and signaling are rapidly inactivated after β-selection25, 26 and aberrant re-activation of Notch1 in DP thymocytes causes leukemic transformation27, 28, 29, 30. Ikaros, a key regulator of T cell homeostasis, binds to the DNS enhancer in DP thymocytes and represses transcription at the Notch1 locus27. Thymocytes with Ikaros inactivating mutations have a strong leukemogenic potential. Counterintuitively, leukemogenesis in Ikaros mutant thymocytes is augmented by the Notch1 null mutation. However, this mutation leaves the DNS enhancer intact and results in expression of a constitutively active form of Notch derived from alternative promoters27. Notch1 ICN binds to the enhancer, creating a positive feedback loop that amplifies Notch signaling in these mutants. Consistent with these observations, the DNSe mutation suppresses T cell leukemogenesis caused by Ikaros mutations. Thus, Ikaros repression of Notch1 activity at the DP stage is mediated through the DNSe and averts leukemogenesis.
In summary, we have shown that a DN-specific Notch1 enhancer is responsible for raising Notch1 expression to levels that establish a multipotent progenitor pool in early thymic progenitors and that delays rather than accelerates T cell differentiation at this stage. We propose that this harbor for multipotentETP progenitors set up by Notch1 signaling is an important stage in T cell differentiation that allows for the generation of an epigenetic landscape that supports appropriate changes in transcriptional programs required for reliable T cell differentiation. Either as a consequence of the failure to correctly set the epigenetic landscape or a more direct requirement for Notch1 activity, cells lacking the DNSe fail to undergo critical steps in T cell differentiation as they rapidly transition through the DN stages and most fail to progress past DN3. DNSe-driven Notch1 activity becomes a liability at the DP stage and it is actively repressed to prevent leukemogenesis. The Notch1 DNSe harnesses the regulatory architecture of DN and DP cells to achieve a carefully orchestrated burst of Notch1 activity to guide a critical period of T cell development.
Methods
Mice
The DNSLoxpF/LoxpF (designated as DNSfl/fl) mouse line was generated by inserting loxP sites flanking the 4.6 kb region consisting of DNS promoter and transcription factor binding enhancer element by standard gene targeting. The DNSe−/− and DNSe fl/fl mouse lines were generated by CRISPR-Cas9-mediated gene editing. Briefly, guide RNAs were designed using the Alt-R CRISPR HDR Design tool (https://www.idtdna.com/pages/tools/alt-r-crispr-hdr-design-tool). The synthesized CRISPR RNAs (crRNAs), trans-activating crRNA (tracrRNA), donor vector consisting of 600bp DNSe fragment flanked by loxP sites and 1kb outside arms on each side, and Cas9 protein were microinjected into fertilized eggs obtained from C57BL/6 mice. All mutant lines were established from founder mice carrying loxP insertions or deletion by mating with C57BL/6 syngeneic mice. Ikzf1+/− (IK-Het) and IKE5fl/+ (IKDN-Het) lines were generated in the Georgopoulos laboratory49, 51. Notch1 fl/fl (N1c fl/fl), CD2-Cre and Mx-Cre lines were obtained from Drs. F. Radtke, D. Kioussis and K. Rajewsky, respectively6, 52, 53. All mice were bred and maintained under pathogen-free conditions, a 12h/12h light dark cycle, at 19–22°C and 40–70% humidity in the animal facility at Massachusetts General Hospital, Bldg. 149–8. All animal experiments were done according to protocols approved by the Subcommittee on Research Animal Care at Massachusetts General Hospital (Charlestown, MA) and in accordance with the guidelines set forth by the National Institutes of Health.
Interferon induced gene targeting
DNS fl/fl; Mx-Cre and N1c fl/fl; Mx-Cre and their littermate controls (WT; DNS fl/+; Mx-Cre, DNS fl/+, DNS fl/fl, N1c fl/+; Mx-Cre, N1c fl/+, and N1c fl/fl ) were subjected to intraperitoneal injection with 0.2mg of polyinosinic:polycytidylic acid (pIpC) at days 0, 3, and 5 and analyzed 30–35 days after first injection. At the time of analysis mice were 2.5–3 months of age.
Antibodies
A complete list of antibodies for lineage depletion, flow cytometry, immunoblotting, and ChIP-seq studies is provided in Supplementary Table 7.
Flow cytometry and cell sorting
Thymocytes were isolated by mechanical disruption. For analyzing DN compartment, total thymocytes were incubated with antibodies against lineage markers such as Ter119, CD11b, Ly6G/Gr1, IgM, CD19, CD4, CD8α, TCRβ, TCRγδ, NK1.1 and CD49b and lineage positive cells were removed by BioMag goat anti-rat IgG (QIAGEN) or streptavidin conjugated microbeads (Miltenyi Biotec). Total thymocytes or cells after depletion were labeled with fluorochrome-conjugated antibodies against T-, B-, NK-, ILC- cell markers for phenotypic analysis. Flow cytometric analysis was performed using a two-laser FACSCanto™ (BD Biosciences) or a three-laser FACS Aria (BD Biosciences). Cell sorting was performed using a three-laser FACS Fortessa (BD Biosciences) or a three-laser SONY SH800 Cell sorter (SONY Biotechnology). The resulting files were uploaded to FlowJo v10 (BD Biosciences) for further analysis.
Preparation for bulk RNA-seq and ChIP-seq libraries
Total RNAs from FACS sorted DN3 cells and thymic B cells were purified using the Direczol RNA extraction kit (Zymo) and cDNA libraries were constructed for sequencing. The Truseq stranded RNA sample prep kit (Illumina) was used for total RNAs greater than 50ng. The NEB Next Single Cell/Low Input RNA Library Prep Kit for Illumina (NEB) was used for total RNAs less than 5ng.
ChIP for histone modifications was performed from sorted DN3 and DP cells as described previously54. DNA recovered from ChIP was used to generate libraries for sequencing. Briefly, DNA was end-repaired, end-adenylated, and ligated with Illumina TruSeq-indexed adaptors. The ligated DNA was purified with AMPure XP beads (Beckman Coulter) and amplified with KAPA HiFi DNA polymerase (KAPA Biosystems). After amplification, the library DNA was separated on a 2% agarose gel, and DNA fragments in the 200- to 500-bp range were purified with a gel DNA recovery kit (Zymo Research).
Sequencing and data analysis for bulk RNAseq and ChIPseq
The purified DNA libraries was multiplexed and sequenced using Illumina Hiseq2000 or NextSeq 550. Read alignment was performed on the mm10 assembly of the mouse genome with the STAR genome alignment algorithm55. Read normalization and differential gene expression were performed using the HOMER v4.1.156 scripts with implementation of DESeq2 v1.36.0 or EdgeR v3.38.4 through R57 58. For data visualization, RPKM (reads per kilobase per million mapped reads) normalized reads were derived using DeepTools v3.3.159 and generated coverage tracks were uploaded onto the UCSD genome browser (https://genome.ucsc.edu/) or the WashU genome browser (https://epigenomegateway.wustl.edu/). Heat maps of hierarchical clustering or K-mean clustering of normalized read counts were generated with Cluster 3.0 (open source software by Michael Eisen) and visualized with Java TreeView (open source software by Alok J. Saldanha). MA plots were generated with ggplot2 v3.3.5 package and bar graphs were generated with Prism 9 software. The metascape, a free gene annotation and analysis resource, was used for Gene Ontology (GO) analysis (https://metascape.org). Gene set enrichment analysis (GSEA) was performed with GSEA 4.2.2 60, 61. HOMER de novo motif discovery algorithm was used for motif discovery analysis of TFs56. NGS datasets used in this study are listed in Supplementary Table 6.
Capillary Immunoblotting analyses
Capillary Western analyses were performed on a Wes blot system (ProteinSimple) according to the manufacturer’s instructions. Briefly, sorted DN3 and preDP cells were lysed in RIPA buffer. The cell lysates were diluted, combined with fluorescent master mix and heated for 5 min at 95°C. The prepared samples, blocking reagent, primary antibodies (1:50 dilution for ICN1 and 1:200 dilution for GAPDH), secondary antibodies, and chemiluminescent substrate were dipensed into designated wells in the assay plate. The electrophoresis and immunodetection steps were carried out in the fully automated capillary system. Data were analyzed using Compass software v4.0.0 (ProteinSimple).
HiC data analysis
Sequencing reads were aligned on the mm10 assembly and processed into normalized contact maps using HiC-Pro v2.862. HiC contact maps were analyzed at 10kb resolution. Statistically significant interactions were called using FitHiC v2.0.736 with default parameters and an FDR threshold of 0.01 for each sample. The datasets used in this study are listed in Supplementary Table 6.
Single cell RNA-seq
Sorted ETPs were counted and measured for viability using the Cellometer Auto 2000 (Nexcelom Bioscience). The sample was then washed, resuspended in DMEM or HBSS supplemented with 10% FCS, and loaded into each 10x Chromium v3.1 lane, and subsequent preparation was conducted following the instruction manual of Chromium Single Cell 3’ v3.1 library kit (10x Genomics). The cDNA library and final library after indexing were evaluated with Agilent 4200 TapeStation (Agilent) for quality control. After evaluating concentrations of the final libraries by Qubit fluorometer (Thermo Fisher Scientific) and Kapa DNA Quantification reagents (Kapa Biosystems), they were multiplexed and sequenced using Illumina NextSeq 550. The reads were mapped onto the mm10 assembly using standard Cell Ranger pipeline (10x Genomics). We followed the Seurat v4.1.1 package integrated analysis and comparative analysis workflows to cluster cells and to perform differential gene expression analyses 63. Briefly, genes expressed in less than 10 cells, cells expressed with less than 1000 genes, cells expressed with more than 4500 genes (potential doublets), cells with more than 10% mitochondrial genes were excluded for downstream analysis in each dataset. Cell cycle score for each cell was calculated by CellCycleScoring function from Seurat using mouse cell cycle genes. SCTransform function was invoked to normalize the dataset, regress out mitochondrial (percent.MT) and cell cycle (S and G2M) contents and identify variable genes. We further excluded cells expressing Hemoglobin beta (Hb-bt >2) from the analysis. The QC filter resulted 13,417 cells in replicates1 (WTETPs: 6,555 cells, ΔDNSe ETPs: 6,862 cells) and 6,293 cells in replicates2 (WTETPs: 3,341 cells, ΔDNSe ETPs: 2,952 cells). For RNA velocity analysis, the Velocyto v0.17.17 package 64 was used to obtain the spliced and un-spliced count matrices from the bam files that were processed with Cell Ranger (10x Genomics). The RNA velocities were then estimated in the stochastic mode of scVelo v0.2.4 package 65 with default parameters, based on normalized counts and UMAP coordinates obtained from Seurat 63. The datasets used in this study are listed in Supplementary Table 6.
Cell culture
OP9-GFP, OP9-DLL1, OP9-DLL4 cells were maintained and co-cultured as previously described47, 66, 67. Briefly, 200–1000 sorted ETPs were co-cultured with OP9, OP9-DLL4, or OP9-DLL1 stroma in the presence of indicated amounts of IL-7 and Flt3L. Equal number of WT and ΔDNSe cells were plated and harvested for counts and phenotypic analysis at indicated time points.
Limiting dilution analysis
For T, NK and ILC differentiation assay, WT and ΔDNSe ETPs were directly sorted on the OP9-DLL1 stroma in a 96 well plate (50, 30, 10, 5, 2, to 1 per well) and were cultured for 14–16 days in the presence of 5ng/ml of IL7 and Flt3L. For B cell differentiation assay, sorted ETPs were plated at 200 or 1000 cells on OP9 stroma in a 96 well and cultured for 8–16 days in the presence of 5ng/ml of IL7 and Flt3L. Wells with >30 hematopoietic cells were scored as positive and their lineage identities were determined by expression of cell surface markers. The mean frequency of T, ILC, NK and B cells was calculated using ELDA software (https://bioinf.wehi.edu.au/software/elda/)68.
RNA microarray data analysis
GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/) was used for the analysis.
Statistical analysis
Except for NGS-data, statistical tests were performed with Prism 9 software (GraphPad). No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those reported in previous publications 66,27. Randomization was not applied because all samples are genetically unique in this study. All available samples from male and female mice were analyzed, and no gender differences were observed. Data collection and analysis were not performed blindly. Data were presumed to be normally distributed. Unpaired Welch’s t-test (two-tailed) was performed for two groups comparison. One-way analysis of variance (ANOVA) was performed and multiple comparisons were corrected using Dunnett’s post hoc test or Benjamini, Krieger and Yekutieli’s two-stage post hoc test for three groups comparison. p values of < 0.05 were considered statistically significant.
Extended Data
Extended Data Fig. 1. Generation of mice with deletion of the DNS region and effects of the DNS and Notch1 deletions on gene expression in DN3 thymocytes.
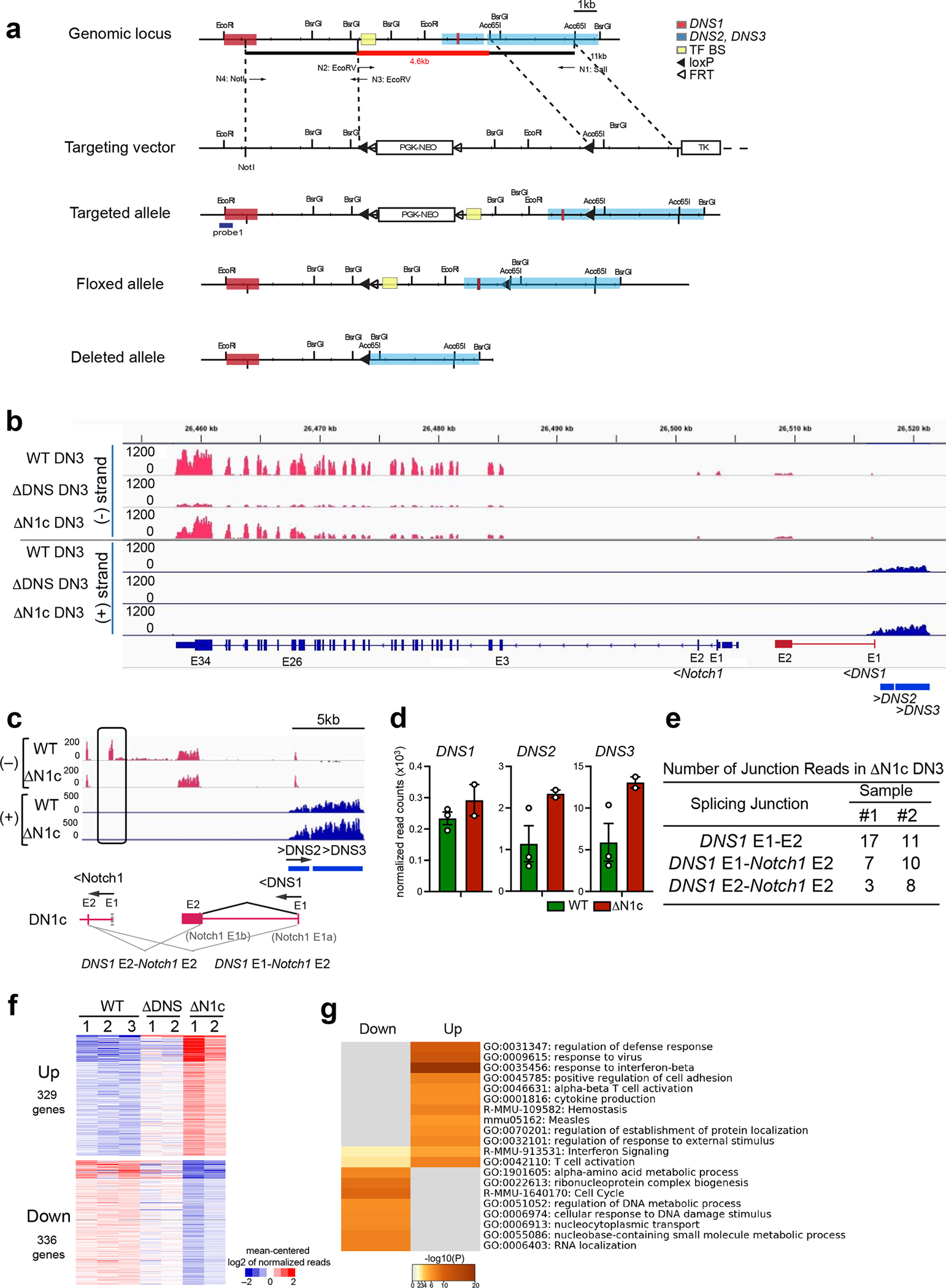
(a) Strategy to generate a conditional deletion of the DNS region. A 4.6 kb region containing the DNS promoter and a region enriched for transcription factor binding sites (TF BS) were flanked by loxP sites (black arrowhead). The Neo marker was flanked by Frt sites (white arrowhead). Flp recombinase excises the Neo gene from the targeting construct and generates the DNS floxed allele. CD2-Cre or Mx-Cre recombinase were used to induce deletion of the DNS region in T cell progenitors or in all hematopoietic cells and progenitors, respectively. DNS1 exons (red square) and DNS2 and DNS3 exons (blue square) and TF BS (yellow square) are shown. (b, c) Genome browser tracks of normalized RNAseq reads at Notch1 ((−) strand), DNS1 ((−) strand), and DNS2 and DNS3 ((+) strand) genes in WT, ΔDNSCD2cre, and ΔN1cCD2cre DN3 thymocytes are shown. Complete loss of Notch1 exon1 (E1) was detected in ΔN1cCD2cre DN3 (b, c). Splicing events in ΔN1cCD2cre are drawn by angled lines (c). (d) Expression of DNS1-3 RNAs in DN3 is shown (mean +/− SEM). Data shown for WT DN3 is also used in Fig. 1g. (e) Number of RNA-seq reads that span splicing junctions are shown for two independent ΔN1cCD2cre DN3 data sets. (f) K-means clustering of genes significantly up-or down- regulated in ΔN1cCD2cre vs WT (log2FC>0 or <0, adj.p <0.05) in WT, ΔDNSCD2cre, and ΔN1cCD2cre DN3 cells is shown. A similar trend of up- and down-regulated genes was seen with ΔDNSCD2cre and ΔN1cCD2cre DN3 cells relative to WT DN3 cells although gene expression changes observed in ΔDNSCD2cre were smaller than those seen in ΔN1cCD2cre DN3. (g) GO Pathway analyses of up- or down-regulated genes in ΔN1cCD2cre vs WT.
Data shown were generated from two (ΔDNSCD2cre DN3 and ΔN1cCD2cre DN3) and three (WT DN3) independent experimental groups with pooled samples from at least two biological replicates.
Extended Data Fig. 2. Generation of mice with deletion of the DNS enhancer and interaction of the DNS enhancer with the canonical Notch1 and DNS promoters.
(a) Strategy to generate the DNS enhancer mutant allele. A 600 bp region containing the conserved TF BS were deleted by CRISPR-Cas9-mediated gene editing. DNS1 (red square) and DNS2 and 3 (blue square), and TF BS (yellow square) are shown by respectively colored squares. (b) Statistically significant (FDR <0.01) long distance interactions at Notch1 locus identified by HiC in DN2 thymocytes are shown together with histone modifications and chromatin accessibility.
HiC data shown in (b) was downloaded publicly available data (GSE90958).
Extended Data Fig. 3. Characterization of ΔDNSe mutant thymus.
(a) FACS profiles of total thymocytes with markers for, DN, DP, and SP stages of abT and B cells are shown. (b) Absolute cell numbers for abT cell subsets in the WT and ΔDNSe thymi at different ages are shown. Unpaired t-test (two-tailed) was used for statistical analysis. N.S., not significant. (c) FACS profiles of γδT cells are shown.
Data shown in in (a) were representative FACS profiles from one experiment with 28-day-old WT (N=3) and ΔDNSe (N=3) mice, in (b) were generated from 4 independent experiments with mice at different age groups (WT N=13, ΔDNSe N=12), in (c) were representative FACS profiles from 3 independent experiments with WT (N=8) and ΔDNSe (N=8) mice.
Extended Data Fig. 4. scRNAseq analysis with replicate 2 samples.
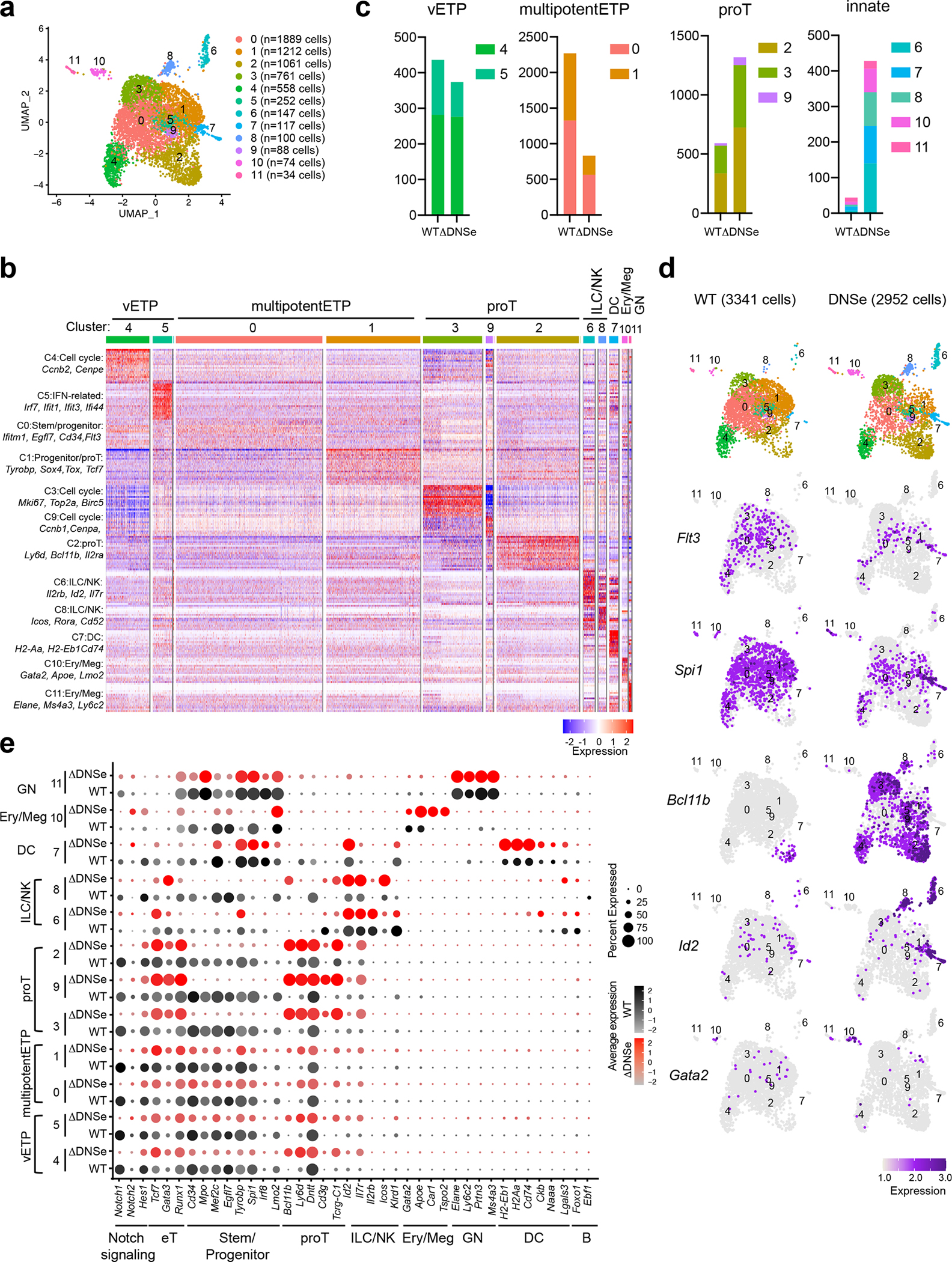
(a) Integrated analysis and UMAP visualization of WT ETPs (3341 cells) and ΔDNSe ETPs (2952 cells), is shown in 12 colored sub-clusters. Absolute cell number in each cluster is shown in parenthesis. (b) Heatmap of the top 20 enriched genes in each sub-cluster ordered by approximate developmental progression based on gene expression and connectivity in the UMAP display is shown. (c) ETP subcluster composition in WT and ΔDNSe ETPs are shown by bar graph. (d) Independent visualization of WT and ΔDNSe ETP subclusters and expression of Flt3 (a vETP marker), Spi1 (a marker for multipotentETP and innate cells), Bcl11b (a marker for proT), Id2 (a marker for ILC/NK), and Gata2 (a marker for Ery/Meg) in the UMAP defined clusters are shown. (e) Expression of genes that define ETP subsets and downstream stages of T cell differentiation as well as genes representative of ILC/NK, GN, Ery/Meg, DC, and B cell differentiation are shown. Color intensity is proportional to the average gene expression across cells in the indicated clusters. The size of circles is proportional to the percent of cells expressing the indicated genes.
Extended Data Fig. 5. Deducing lineage affiliation of cells in ETP sub-clusters (replicate 1).
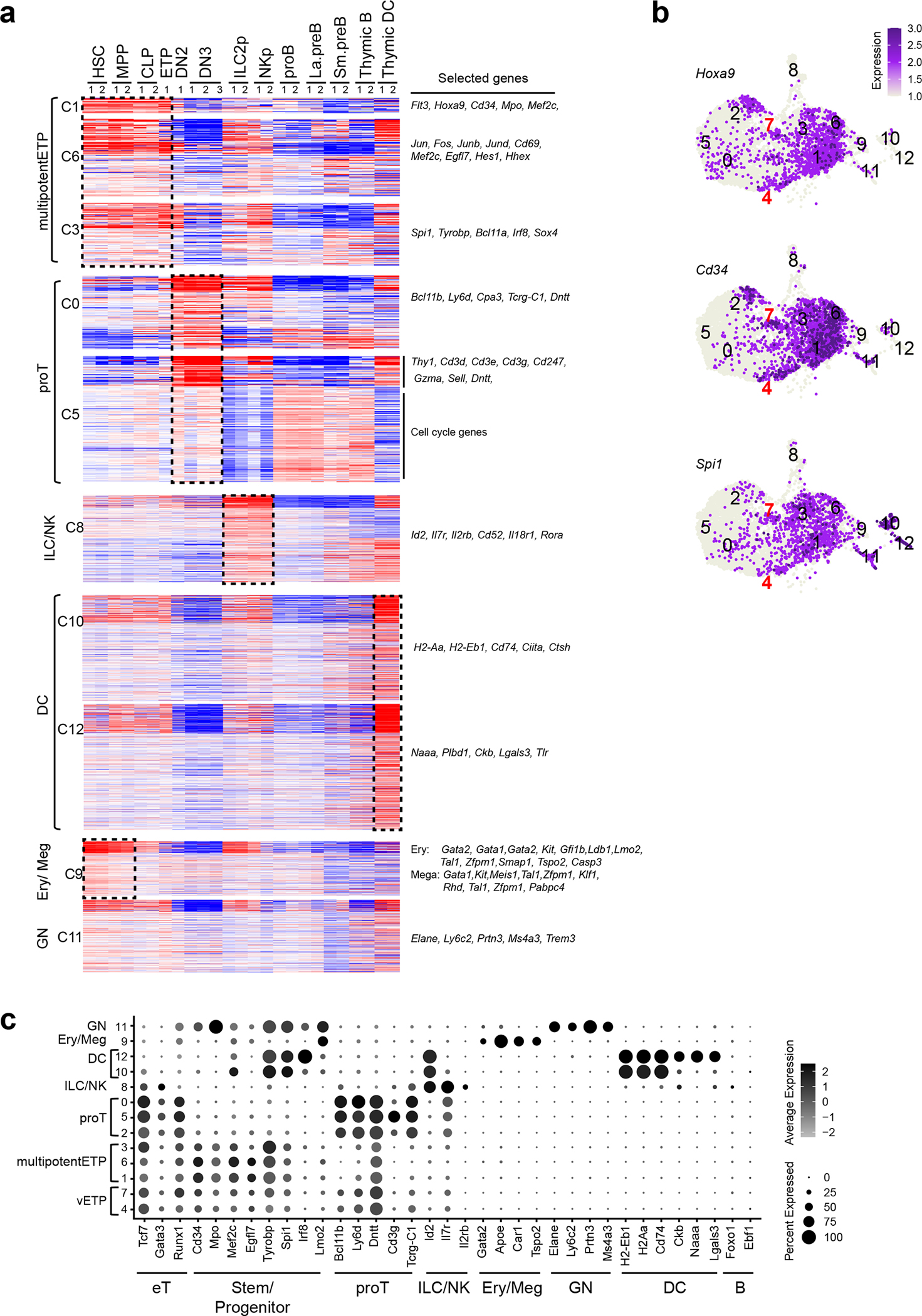
(a) Previously established gene expression data sets from HSC, early lineage progenitors and thymic DC were used to define the cell type affiliation of ETP cluster-defining gene markers. The list of markers is summarized in Supplementary Table 1. (b) Expression of Hoxa9, Cd34, and Spi1 in the UMAP defined clusters are shown. (c) Expression of genes that define ETP subsets and downstream stages of T cell differentiation as well as genes representative of ILC/NK, GN, Ery/Meg, DC, and B cell differentiation are shown. Color intensity is proportional to the average gene expression across cells in the indicated clusters. The size of circles is proportional to the percent of cells expressing the indicated genes.
Two replicates of HSC, MPP, CLP, ILC2p, NKp, and thymic DC were obtained from published datasets (GEO: GSE77695, GSE183056). ETP, DN2, DN3, proB, La.preB, Sm.preB, and thymic B data sets were generated in this study (as described in Fig. 1 and Fig. 2).
Extended Data Fig. 6. Analysis of RNA velocity (replicate 1).
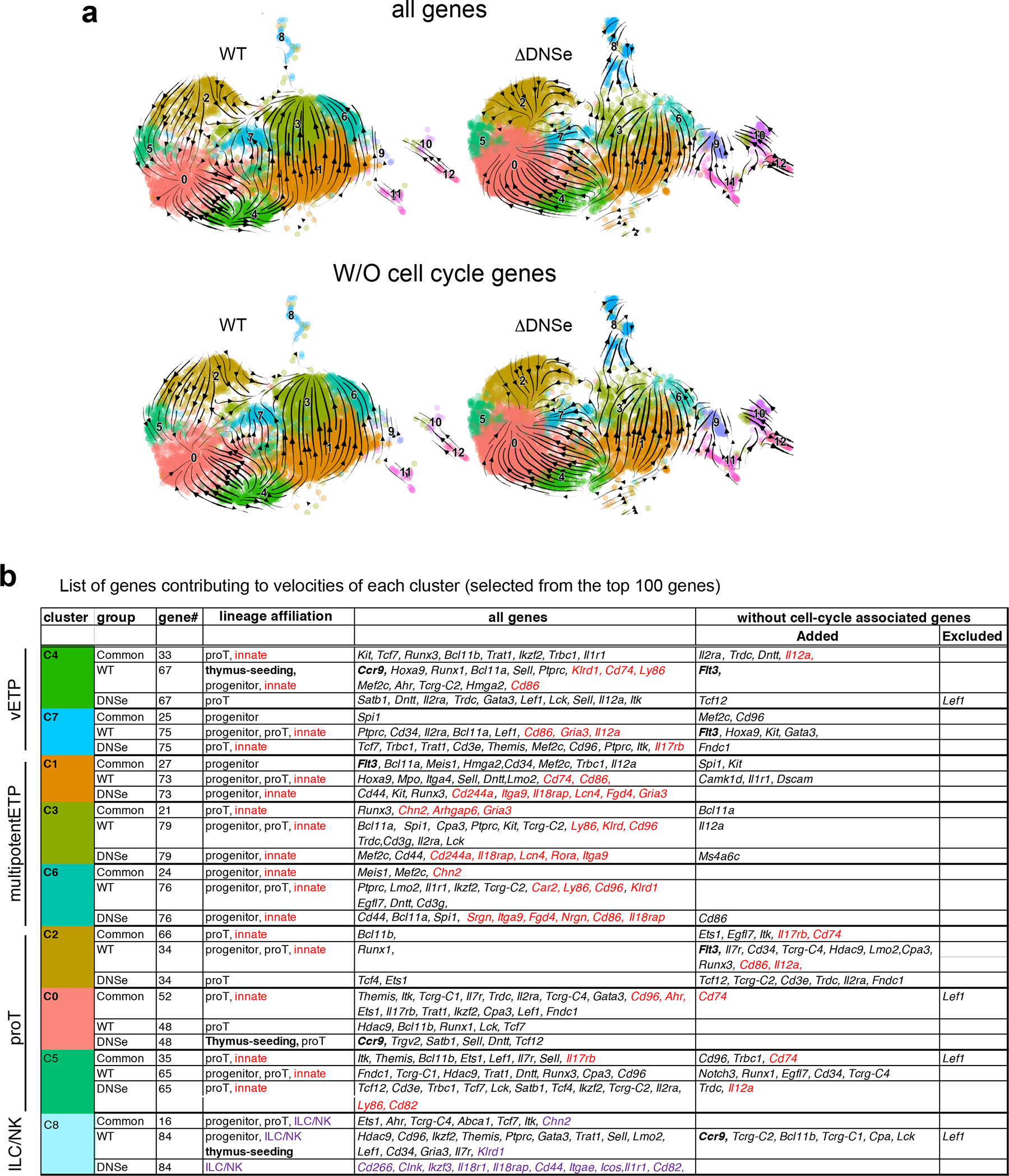
(a) Velocity streams of scRNA analysis for WT and ΔDNSe ETPs are shown separately. Stream arrows represent relative transition probabilities based on un-spliced over spliced transcript ratio calculations. The vector calculation was performed with (top) or without (bottom) 1248 cell-cycle-associated genes that belonged to either GO term: GO:0022402 or Reactome Pathway ID: R-MMU-1640170. (b) Selected genes contributing to velocities of each cluster is shown. Genes were classified as 1) common to WT and ΔDNSe, 2) only in WT, 3) only in ΔDNSe. Genes added to or excluded from the list after exclusion of cell-cycle associated genes are also shown.
Extended Data Fig. 7. Gene expression profiling for cultured WT and ΔDNSe DN3 cells.
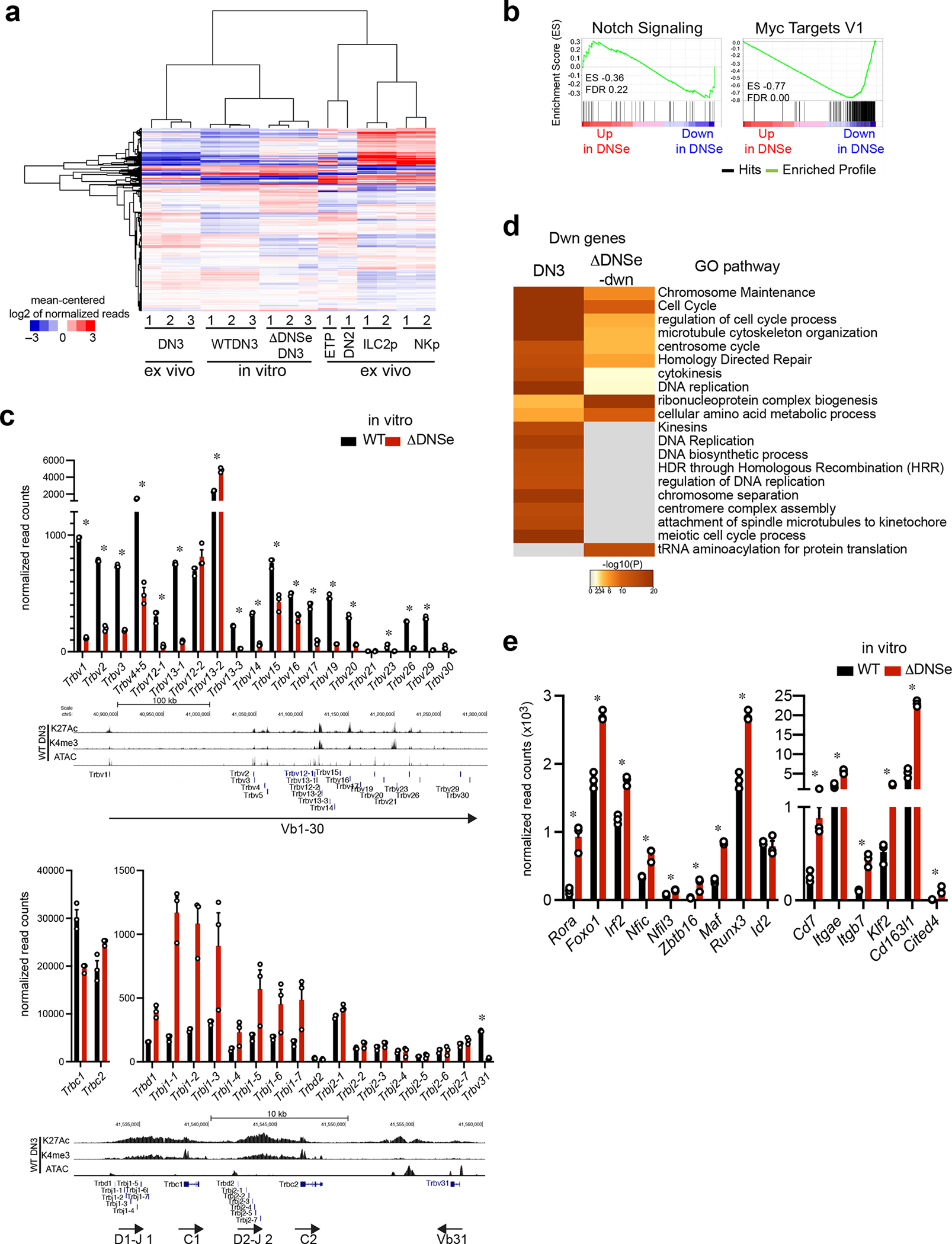
(a) Hierarchical clustering of genes expressed in cultured DN3, ex vivo T cell progenitors and innate lymphoid progenitors is shown. Genes with > 50 reads in at least one data set were clustered. (b) Enrichment of ΔDNSe deregulated genes in the Notch Signaling (Hallmark Notch Signaling) and Myc (Hallmark Myc target V1) pathways are shown by GSEA. Genes ranked from most upregulated (left end) to most downregulated (right end) in ΔDNSe relative to WT DN3 cells are plotted on the X axis. Enrichment profiles of genes are plotted on Y axis and calculated enrichment score (ES) and FDR are shown. (c) The full expression panel for Trbv, Trbc, Trbd and Trbj genes in cultured DN3 cells as determined by RNAseq is shown (mean +/− SEM. * adj. p <0.01 Expression data for Trbv genes is also shown in Fig. 6e. Reduction in expression of genes encoding Vβ region (Trbv) were detected in ΔDNSe relative to WT DN3 with the exception of Trbv12-2 and Trbv13-2. Expression of genes encoding Cβ, Dβ and Jβ regions (Trbc, Trbd, and Trbj) were similar or even higher in ΔDNSe DN3, suggesting that ΔDNSe DN3 have already undergone D-J rearrangement. (d) Heatmaps based on significance of functional pathway enrichment (−log10(pval)) are shown for down-regulated genes classified as a DN3 and ΔDNSe-dwn signatures as shown in Fig. 6c. (e) Expression of genes related to innate lymphoid lineages in cultured DN3 cells as determined by RNAseq is shown (mean +/− SEM). * adj. p <0.01
Data for cultured DN3 cells were generated from three independent experimental groups for each genotype. Data for ex vivo ETP, DN2 and DN3 were generated in this study as in Fig. 1. Data for ILC2p and NKp were obtained from published datasets (GEO: GSE77695) as in Fig. 6 and Extended Data Fig. 5.
Supplementary Material
Acknowledgements
We thank Dr Howard Petrie at the Scripps Research Institute for providing us sorted ex vivo T cell progenitors. We thank Drs Kristin White, Jin Mo Park, and Fotini Gounari for critical review of the manuscript, and Eleanor Wu and Robert Czyzewski for mouse husbandry. Research was supported by NIH R01HL140622 and R01CA158006 to K.G and NIH R21AR074748 to B.A.M, and and NIH R35GM128938 to F.A. Gene targeting was performed at CBRC and at the targeting core at University of Chicago. Cell sorting was performed at the Flow and Mass Cytometry Core/MGH and at CBRC. High-throughput RNA sequencing was performed at the Bauer Center for Genomic research at Harvard University and at CBRC.
Footnotes
Competing interests
The authors declare no competing interests.
Data availability
All data that support the findings of this study are available from the corresponding authors upon request. ChIP-seq and RNA-seq datasets generated during this study have been deposited in the Gene Expression Omnibus (GEO) under accession code GSE186764 and GSE211079. The publicly available NGS datasets used during this study can be found in the GEO or PRJNA; GSE60103, GSE73143, GSE61149, GSE115742, GSE33679, GSE32311, GSE77695, GSE183056, GSE109125, GSE15330, GSE128964, GSE95337, GSE33513, GSE154304, GSE93755, GSE115744, PRJNA487507, GSE79422, GSE89848, GSE19923, PRJNA451505, GSE99159, GSE100738, GSE31233, GSE103953, GSE93572, GSE30518, GSE162292, GSE89847, GSE90958, GSE79422 and are listed in Supplementary Table 6. Source data were provided with this paper.
References
- 1.Yu VW et al. Specific bone cells produce DLL4 to generate thymus-seeding progenitors from bone marrow. J Exp Med 212, 759–774 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tikhonova AN et al. The bone marrow microenvironment at single-cell resolution. Nature 569, 222–228 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen ELY, Thompson PK & Zuniga-Pflucker JC RBPJ-dependent Notch signaling initiates the T cell program in a subset of thymus-seeding progenitors. Nat Immunol 20, 1456–1468 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tan JB, Visan I, Yuan JS & Guidos CJ Requirement for Notch1 signals at sequential early stages of intrathymic T cell development. Nat Immunol 6, 671–679 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Sambandam A et al. Notch signaling controls the generation and differentiation of early T lineage progenitors. Nat Immunol 6, 663–670 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Radtke F et al. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity 10, 547–558 (1999). [DOI] [PubMed] [Google Scholar]
- 7.Wilson A, MacDonald HR & Radtke F Notch 1-deficient common lymphoid precursors adopt a B cell fate in the thymus. J Exp Med 194, 1003–1012 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feyerabend TB et al. Deletion of Notch1 converts pro-T cells to dendritic cells and promotes thymic B cells by cell-extrinsic and cell-intrinsic mechanisms. Immunity 30, 67–79 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Hozumi K et al. Delta-like 4 is indispensable in thymic environment specific for T cell development. J Exp Med 205, 2507–2513 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koch U et al. Delta-like 4 is the essential, nonredundant ligand for Notch1 during thymic T cell lineage commitment. J Exp Med 205, 2515–2523 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han H et al. Inducible gene knockout of transcription factor recombination signal binding protein-J reveals its essential role in T versus B lineage decision. Int Immunol 14, 637–645 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Pui JC et al. Notch1 expression in early lymphopoiesis influences B versus T lineage determination. Immunity 11, 299–308 (1999). [DOI] [PubMed] [Google Scholar]
- 13.Dorsch M et al. Ectopic expression of Delta4 impairs hematopoietic development and leads to lymphoproliferative disease. Blood 100, 2046–2055 (2002). [PubMed] [Google Scholar]
- 14.Yan XQ et al. A novel Notch ligand, Dll4, induces T-cell leukemia/lymphoma when overexpressed in mice by retroviral-mediated gene transfer. Blood 98, 3793–3799 (2001). [DOI] [PubMed] [Google Scholar]
- 15.Yui MA & Rothenberg EV Developmental gene networks: a triathlon on the course to T cell identity. Nat Rev Immunol 14, 529–545 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rothenberg EV, Ungerback J & Champhekar A Forging T-Lymphocyte Identity: Intersecting Networks of Transcriptional Control. Adv Immunol 129, 109–174 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hosokawa H et al. Bcl11b sets pro-T cell fate by site-specific cofactor recruitment and by repressing Id2 and Zbtb16. Nat Immunol 19, 1427–1440 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wakabayashi Y et al. Bcl11b is required for differentiation and survival of alphabeta T lymphocytes. Nat Immunol 4, 533–539 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Wolfer A, Wilson A, Nemir M, MacDonald HR & Radtke F Inactivation of Notch1 impairs VDJbeta rearrangement and allows pre-TCR-independent survival of early alpha beta Lineage Thymocytes. Immunity 16, 869–879 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Tanigaki K et al. Regulation of alphabeta/gammadelta T cell lineage commitment and peripheral T cell responses by Notch/RBP-J signaling. Immunity 20, 611–622 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Schmitt TM, Ciofani M, Petrie HT & Zuniga-Pflucker JC Maintenance of T cell specification and differentiation requires recurrent notch receptor-ligand interactions. J Exp Med 200, 469–479 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ciofani M et al. Obligatory role for cooperative signaling by pre-TCR and Notch during thymocyte differentiation. J Immunol 172, 5230–5239 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Ciofani M & Zuniga-Pflucker JC Notch promotes survival of pre-T cells at the beta-selection checkpoint by regulating cellular metabolism. Nat Immunol 6, 881–888 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Maillard I et al. The requirement for Notch signaling at the beta-selection checkpoint in vivo is absolute and independent of the pre-T cell receptor. J Exp Med 203, 2239–2245 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taghon T, Yui MA, Pant R, Diamond RA & Rothenberg EV Developmental and molecular characterization of emerging beta- and gammadelta-selected pre-T cells in the adult mouse thymus. Immunity 24, 53–64 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Yashiro-Ohtani Y et al. Pre-TCR signaling inactivates Notch1 transcription by antagonizing E2A. Genes Dev 23, 1665–1676 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gomez-del Arco P et al. Alternative promoter usage at the Notch1 locus supports ligand-independent signaling in T cell development and leukemogenesis. Immunity 33, 685–698 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ashworth TD et al. Deletion-based mechanisms of Notch1 activation in T-ALL: key roles for RAG recombinase and a conserved internal translational start site in Notch1. Blood 116, 5455–5464 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jeannet R et al. Oncogenic activation of the Notch1 gene by deletion of its promoter in Ikaros-deficient T-ALL. Blood 116, 5443–5454 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X, Gounari F, Protopopov A, Khazaie K & von Boehmer H Oncogenesis of T-ALL and nonmalignant consequences of overexpressing intracellular NOTCH1. J Exp Med 205, 2851–2861 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mingueneau M et al. The transcriptional landscape of alphabeta T cell differentiation. Nat Immunol 14, 619–632 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsuji H, Ishii-Ohba H, Ukai H, Katsube T & Ogiu T Radiation-induced deletions in the 5’ end region of Notch1 lead to the formation of truncated proteins and are involved in the development of mouse thymic lymphomas. Carcinogenesis 24, 1257–1268 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Mumm JS & Kopan R Notch signaling: from the outside in. Dev Biol 228, 151–165 (2000). [DOI] [PubMed] [Google Scholar]
- 34.Yoshida T, Ng SY & Georgopoulos K Awakening lineage potential by Ikaros-mediated transcriptional priming. Curr Opin Immunol 22, 154–160 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu G et al. Transformation of Accessible Chromatin and 3D Nucleome Underlies Lineage Commitment of Early T Cells. Immunity 48, 227–242 e228 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaul A, Bhattacharyya S & Ay F Identifying statistically significant chromatin contacts from Hi-C data with FitHiC2. Nat Protoc 15, 991–1012 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Porritt HE et al. Heterogeneity among DN1 prothymocytes reveals multiple progenitors with different capacities to generate T cell and non-T cell lineages. Immunity 20, 735–745 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Zhou W et al. Single-Cell Analysis Reveals Regulatory Gene Expression Dynamics Leading to Lineage Commitment in Early T Cell Development. Cell Syst 9, 321–337 e329 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramond C et al. Two waves of distinct hematopoietic progenitor cells colonize the fetal thymus. Nat Immunol 15, 27–35 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Suliman S et al. Notch3 is dispensable for thymocyte beta-selection and Notch1-induced T cell leukemogenesis. PLoS One 6, e24937 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Georgopoulos K, van den Elsen P, Bier E, Maxam A & Terhorst C A T cell-specific enhancer is located in a DNase I-hypersensitive area at the 3’ end of the CD3-delta gene. EMBO J 7, 2401–2407 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clevers H et al. Characterization and expression of the murine CD3-epsilon gene. Proc Natl Acad Sci U S A 85, 8623–8627 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Satoh-Takayama N et al. IL-7 and IL-15 independently program the differentiation of intestinal CD3-NKp46+ cell subsets from Id2-dependent precursors. J Exp Med 207, 273–280 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yokota Y et al. Development of peripheral lymphoid organs and natural killer cells depends on the helix-loop-helix inhibitor Id2. Nature 397, 702–706 (1999). [DOI] [PubMed] [Google Scholar]
- 45.Zook EC et al. Transcription factor ID2 prevents E proteins from enforcing a naive T lymphocyte gene program during NK cell development. Sci Immunol 3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seillet C et al. Deciphering the Innate Lymphoid Cell Transcriptional Program. Cell Rep 17, 436–447 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Ng SY, Yoshida T, Zhang J & Georgopoulos K Genome-wide lineage-specific transcriptional networks underscore Ikaros-dependent lymphoid priming in hematopoietic stem cells. Immunity 30, 493–507 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miyazaki M et al. The E-Id Protein Axis Specifies Adaptive Lymphoid Cell Identity and Suppresses Thymic Innate Lymphoid Cell Development. Immunity 46, 818–834 e814 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Joshi I et al. Loss of Ikaros DNA-binding function confers integrin-dependent survival on pre-B cells and progression to acute lymphoblastic leukemia. Nat Immunol 15, 294–304 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Inlay MA et al. Ly6d marks the earliest stage of B-cell specification and identifies the branchpoint between B-cell and T-cell development. Genes Dev 23, 2376–2381 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only references
- 51.Georgopoulos K et al. The Ikaros gene is required for the development of all lymphoid lineages. Cell 79, 143–156 (1994). [DOI] [PubMed] [Google Scholar]
- 52.de Boer J et al. Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur J Immunol 33, 314–325 (2003). [DOI] [PubMed] [Google Scholar]
- 53.Kuhn R, Schwenk F, Aguet M & Rajewsky K Inducible gene targeting in mice. Science 269, 1427–1429 (1995). [DOI] [PubMed] [Google Scholar]
- 54.Hu Y et al. Superenhancer reprogramming drives a B-cell-epithelial transition and high-risk leukemia. Genes Dev 30, 1971–1990 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robinson MD, McCarthy DJ & Smyth GK edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramirez F, Dundar F, Diehl S, Gruning BA & Manke T deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42, W187–191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mootha VK et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34, 267–273 (2003). [DOI] [PubMed] [Google Scholar]
- 62.Servant N et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol 16, 259 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hao Y et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.La Manno G et al. RNA velocity of single cells. Nature 560, 494–498 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bergen V, Lange M, Peidli S, Wolf FA & Theis FJ Generalizing RNA velocity to transient cell states through dynamical modeling. Nat Biotechnol 38, 1408–1414 (2020). [DOI] [PubMed] [Google Scholar]
- 66.Yoshida T, Ng SY, Zuniga-Pflucker JC & Georgopoulos K Early hematopoietic lineage restrictions directed by Ikaros. Nat Immunol 7, 382–391 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mohtashami M, Zarin P & Zuniga-Pflucker JC Induction of T Cell Development In Vitro by Delta-Like (Dll)-Expressing Stromal Cells. Methods Mol Biol 1323, 159–167 (2016). [DOI] [PubMed] [Google Scholar]
- 68.Hu Y & Smyth GK ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 347, 70–78 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data that support the findings of this study are available from the corresponding authors upon request. ChIP-seq and RNA-seq datasets generated during this study have been deposited in the Gene Expression Omnibus (GEO) under accession code GSE186764 and GSE211079. The publicly available NGS datasets used during this study can be found in the GEO or PRJNA; GSE60103, GSE73143, GSE61149, GSE115742, GSE33679, GSE32311, GSE77695, GSE183056, GSE109125, GSE15330, GSE128964, GSE95337, GSE33513, GSE154304, GSE93755, GSE115744, PRJNA487507, GSE79422, GSE89848, GSE19923, PRJNA451505, GSE99159, GSE100738, GSE31233, GSE103953, GSE93572, GSE30518, GSE162292, GSE89847, GSE90958, GSE79422 and are listed in Supplementary Table 6. Source data were provided with this paper.