

Abstract
Modulation of corticostriatal plasticity alters the information flow throughout basal ganglia circuits and represents a fundamental mechanism for motor learning, action selection, and reward. Synaptic plasticity in the striatal direct- and indirect-pathway spiny projection neurons (dSPNs and iSPNs) is regulated by two distinct networks of GPCR signaling cascades. While it is well-known that dopamine D2 and adenosine A2a receptors bi-directionally regulate iSPN plasticity, it remains unclear how D1 signaling modulation of synaptic plasticity is counteracted by dSPN-specific Gi signaling. Here, we show that striatal dynorphin selectively suppresses long-term potentiation (LTP) through Kappa Opioid Receptor (KOR) signaling in dSPNs. Both KOR antagonism and conditional deletion of dynorphin in dSPNs enhance LTP counterbalancing with different levels of D1 receptor activation. Behaviorally, mice lacking dynorphin in D1 neurons show comparable motor behavior and reward-based learning, but enhanced flexibility during reversal learning. These findings support a model in which D1R and KOR signaling bidirectionally modulate synaptic plasticity and behavior in the direct pathway.
INTRODUCTION
The striatum is the gateway into the basal ganglia and receives convergent glutamatergic inputs from the cortex and thalamus. Activity-dependent excitatory synaptic plasticity in the striatum alters the information transfer through the basal ganglia and is crucial for fine motor control, motor learning, habit formation, and action selection (1–3). Maladaptive and/or loss of synaptic plasticity are thought to mediate motor symptoms seen in movement disorders, such as Parkinson’s disease and L-DOPA-induced dyskinesia (4–7). In the striatum, more than 90% of neurons are spiny projection neurons (SPNs), which form two parallel pathways with distinct axonal projections and expression of neuropeptides and neuromodulatory receptors (8). In particular, dynorphin and dopamine D1 receptor are exclusively expressed in direct-pathway SPNs (dSPNs), whereas enkephalin, dopamine D2 receptor, and adenosine A2a receptor are exclusively expressed in indirect-pathway SPNs (iSPNs) (9, 10). This pathway-specific expression of neuromodulatory receptors is optimally positioned to regulate excitatory synaptic plasticity (11). For example, in iSPNs, activation of the Gs-coupled adenosine A2a receptor promotes long-term potentiation (LTP) expression, and activation of the Gi-coupled D2 receptor suppresses LTP expression (4, 12–14). Conversely, what remains unclear is whether there is a dSPN-specific counterpart of D2R that suppresses LTP expression. One candidate is the Gi-coupled M4 muscarinic receptor (M4R), which has been shown to successfully shunt LTP in dSPNs (15). The effect of M4R is assumed to be low in iSPNs because it is preferentially expressed on dSPNs (9, 16). However, there is still substantial M4R expression in iSPNs and its effect on LTP in the indirect pathway has not been examined. In addition, M4R is expressed at corticostriatal axonal terminals and on striatal cholinergic interneurons (17, 18), which makes M4R not suitable to modulate plasticity exclusively in the direct pathway.
Another pathway-specific candidate is the Gi-coupled Kappa Opioid Receptor (KOR). KOR is also preferentially expressed in dSPNs (19, 20), and importantly, the endogenous ligand, dynorphin (Dyn), is exclusively expressed in dSPNs (21). Even though the distinct expression pattern of Dyn in the dSPNs is well-characterized, the endogenous function of Dyn in the striatal direct pathway is still not well understood. Previous studies have largely focused on its acute regulation of synaptic transmission and long-term depression. For example, activation of KOR by Dyn can suppress dopamine (DA) release in the striatum (22, 23) and the effect is short-term and can be reversed after washout (24). In addition, acute KOR activation can also suppress glutamatergic synaptic transmission (25, 26) or prolonged activation of KOR causes long-term depression (LTD) (27) through pre-synaptic mechanisms.
Here, we used a combination of pharmacological manipulation of KOR and a Dyn conditional knockout (cKO) mouse to probe the endogenous function of Dyn in D1 neurons. In particular, we focus on Dyn/KOR modulation of excitatory spike-timing-dependent LTP (STDP-LTP). We demonstrate that Dyn can suppress STDP-LTP selectively in dSPNs. By contrast, M4R activation suppresses STDP-LTP in both dSPNs and iSPNs, showing that M4R-mediated suppression of LTP is not pathway-specific. Furthermore, we characterized the interaction of Dyn/KOR with D1R signaling in dSPNs and found that Dyn/KOR counteracts to varying levels the effects of D1R-modulation of STDP-LTP. Finally, we found mice lacking Dyn in D1 neurons (D1-Dyn cKO mice) showed enhanced performance in re-learning a reversed reward contingency. These mice exhibited comparable motor behavior, working memory, anxiety level, and initial reward learning suggesting that Dyn/KOR plasticity plays a role in reversal learning. Taken together, our studies provide novel mechanistic insights into dichotomous regulation of striatal direct-pathway synaptic plasticity by Dyn/KOR signaling and demonstrate a link between endogenous function of striatal Dyn, striatal direct pathway synaptic plasticity and behavioral cognitive flexibility.
MATERIALS AND METHODS
Experimental animals
Mice were housed under a 12 h light/dark cycle and water and food were available ad libitum. Animal experiments were conducted following protocols approved by Administrative Panel on Laboratory Animal Care at Stanford University. pDynfl/fl mice were generated by Stanford Transgenic, Knockout and Tumor model Center (TKTC). Double or triple transgenic mice were generated by crossing pDynfl/fl with D1-cre mice (MMRRC stock # 017264-UCD) (28), and Rosa-CAG-LSL-ChR2(H134R)-eYFP mice (Ai32, purchased from JAX #012569). For electrophysiological studies, D1-cre;pDyn+/+;Ai32 were used as wildtype (WT) control mice to compare with the D1-Dyn cKO (D1-cre;pDynfl/fl;Ai32) mice. The expression of ChR2 allowed us to optically identify dSPN during the recording. Neurons with SPN intrinsic properties and failed to respond to blue light stimulation (450nm, 5–10 mW) were identified as iSPNs. For electrophysiology, both male and female (4–16 weeks) mice were used. For behavioral tests, only D1-cre;pDyn+/+ (WT control) and D1-cre;pDynfl/fl(D1-Dyn cKO) without Ai32 allele were used. Both male and female adult (8–16 weeks) mice were used for behavioral tests. We used the same cohort of mice for the open field test, Y maze, three-chamber social interaction test, and elevated plus maze test; one cohort for 4-choice behavior, and another cohort for spatial reversal learning. During 4-choice behavior and spatial reversal-learning task, mice started to be food-restricted 3 days before the tasks and monitored daily to maintain at 90% body weight.
Reagents
VU10010, (−)-U50488, (±)-U50488, Colchicine, norbinaltorphimine, SCH23390, SKF81297, and Picrotoxin were purchased from Tocris. Gramicidin A and biocytin were from Sigma-Aldrich. Alexa Fluor 594 was purchased from Invitrogen. Tetrodotoxin citrate was purchased from A.G.Scientific.
Generation of pDyn cKO mice
The murine prodynorphin gene (pDyn) contains 4 exons. A BAC clone containing exon 4 genomic sequence was used to generate the loxP-flanked pDyn targeting construct. A PCR amplicon containing the loxP-NEO/Kan-loxP cassette from the pSV-cre plasmid flanked by 70 bp of homologous sequence at both ends, which matched sites in the third intron of pDyn, was generated using the following primers: DYN-LOXP-F1, 5’ TGTAAGCGCAGGGCAATCGGAAGATCTTGCTTCTACTTTTGGCCTTGCCACCCGTGTCTCTGCGCCTTTGGCAGCCCAATTCCGATCATATTCAATAACC 3’ and DYN-LOXP-R1, 5’ CTGAAAGGATGCAAGAGTTTTGTCATGTGAGAAATGAGAAATTAGACTTACAGAGCCTAGGCAGCTATCCCGCTCTAGAACTAGTGGATCCCCTCGAGGGACC 3’. The PCR amplicon was transformed into electrocompetent EL350 cells that had been previously transformed with the aforementioned pDyn BAC, followed by arabinose-induced expression of cre recombinase to remove LoxP-NEO/Kan-LoxP selective cassette (29). The modified pDyn BAC containing a loxP site in the third intron was then transformed into EL250 cells. A second PCR amplicon containing the FRT-NEO/Kan-FRT-loxP flanked by 70 bp of homologous sequence at both ends, which matched sites in the 3’-end downstream genomic region of pDyn, was generated using the following primers: DYN-LOXP-F2, 5’ ACATGTATAGCTGTATTGGAGGATTGAACCTAGTTCCTCATGCATGATAGGCAAACACTCTACCACCAAGC-CGACGGTATCGATAAGCTTGATATCGAATTCC 3’ and DYN-LOXP-R2, 5’ ACACTTGCTTTCTTGCTTTCTGTTATTCCACACTGTATTTTCTAATATGACTACAGGGGCTGGAGCTATG-GCTCTAGAACTAGTGGATCCACCTAATAACTTC 3’.
The PCR amplicon was then transformed into EL250 cells containing the modified pDyn BAC. A targeting construct was derived from this modified BAC spanning the region 5 kb upstream of the loxP site inserted into intron 3 and 2 kb downstream of the loxP site inserted into 3’-end genomic region. The resulting targeting construct was then electroporated into mouse embryonic stem (ES) cells (W4/129S6, Taconic, NY). Correctly targeted ES clones were identified and injected into blastocysts resulting in several 100% male chimeras. Chimeras were bred to mice bearing a flp-recombinase transgene (ROSA-Flp, JAX #003946) to remove the neomycin selection marker, generating mice with one allele of pDyn flanked by loxP sites at exon 4 (pDynfl/+). The resulted pDynfl/+ offspring were then backcrossed with C57BL/6 mice for seven generations. The following primers were used for genotyping PCR setups: pDyn-F: 5’ CATGCCTGATGAAGGTCGGTAG 3’; pDyn-R: 5’ CATCTTCCAAGTCATCCTTGCC3’; and pDyn-D: 5’ GCAGTACCTCTCTCTGTCCTC3’, with amplified PCR products of 315 bp for wildtype alleles, 420 bp for loxP-flanked alleles, and 552 bp for cre-deleted alleles. We then crossed pDynfl/+ with D1-cre mice (MMRRC stock # 017264-UCD) (28) to obtain D1-cre;pDynfl/+ mice. For experiments, dynorphin conditional knockout mice (D1-cre;pDynfl/fl) and littermate controls (D1-cre;pDyn+/+) were obtained from breeding pairs of D1-cre;pDynfl/+ × pDynfl/+.
Immunohistochemistry
To stain for dynorphin, mice were intracerebroventricularly injected with the axonal transport blocker colchicine (75 μg/kg body weight) 48 hours before procedures to reveal dynorphin immunoreactivity within neuronal cell bodies (30). Mice were anesthetized with isoflurane and intracardially perfused with ice-cold 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS). Brains were dissected, post-fixed 24 h at 4 °C, and transferred to a 30% sucrose/PBS solution for at least 24 h at 4 °C, then cut into 30-μm sections for immunostaining. Free floating sections were blocked for 1 h in PBS containing 0.1% Triton X-100 (G-Biosciences) and 5% normal goat serum (Cell Signaling). Sections were then incubated with rabbit anti-Dyn antisera (1:1000, Cat# T4268, Peninsula Labs) overnight at 4 °C in a blocking solution. Sections were washed three times in PBS, followed by incubation for 2 h at room temperature in Alexa488 goat anti-rabbit IgG (1:1000, Cat# A11008, Invitrogen), and nuclear dye DAPI (1:500, Neurotrace, Invitrogen). Sections were washed three times in PBS and mounted on glass slides with Hard set Vectashield (Vector Labs) for microscopy.
To confirm the presence of enkephalin in the recorded cells during the LTP experiment with acute brain slices (300 μm), the cells were loaded with 0.2% biocytin after recording, and the slices were post-fixed in 4% PFA overnight at 4 °C. After fixation, slices were washed and blocked in PBS containing 0.5% Triton X-100 and 5% normal goat serum. Slices were then incubated at 4 °C for 48 h with rabbit anti-Met-Enkephalin antibody (1:100, # 20065, Immunostar), and overnight with Alexa594 goat anti-rabbit IgG (1:1000, Cat# A11012, Invitrogen), Streptavidin-Cy5 (1:1000, Cat# SA1011, Invitrogen) to reveal biocytin and DAPI. Slices were then washed three times in PBS and mounted.
All the sections were imaged using confocal microscopy (Leica TCS SPE confocal microscope) with consistent settings. Cells were quantified in ImageJ.
Electrophysiology
1. Slice preparation
Brain slices (300 μm) were obtained using standard techniques (31). Briefly, animals were anesthetized with isoflurane and decapitated. The brain was exposed and chilled with ice-cold artificial CSF (ACSF) containing 125 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 1.25 mM NaH2PO4, 1 mM MgCl2, 25 mM NaHCO3, and 15 mM D-glucose. ACSF was saturated with 95% O2 and 5% CO2. Osmolarity was adjusted to 300–305 mOsm. Oblique horizontal brain slices containing the dorsal striatum were prepared with a vibrating microtome (Leica VT1200 S, Germany) and left to recover in ACSF for 30 min at 34°C and then at room temperature for an additional 30 min before recording. For pharmacological treatment with norBNI (1 μM), (−)-U50488 (1 μM), SCH23390 (3 μM), SKF81297 (3 μM), and VU10010 (5 μM), the ACSF for recovering also contains the corresponding drugs. After the recovery period, slices were moved to a submerged recording chamber perfused with ACSF at a rate of 2–3 ml/min at 30–31°C, and brain slices were recorded within 5 hours after recovery.
2. Fast-scan cyclic voltammetry
Brain slices from D1-cre;pDyn+/+ and D1-cre;pDynfl/fl mice were used for fast-scan cyclic voltammetry recordings to measure the extracellular dopamine release. Recordings were performed using carbon-fiber microelectrodes (7 μm diameter carbon fiber extending 50–100 μm beyond the tapered glass seal). Cyclic voltammograms were measured with a triangular potential waveform (−0.4 to +1.3 V versus Ag/AgCl reference electrode, 400 V/s scan rate, 8.5 ms waveform width) applied at 100 ms intervals. The carbon fiber microelectrode was held at −0.4 V between scans. Cyclic voltammograms were background-subtracted by averaging 10 background scans. Dopaminergic axon terminals were stimulated locally (100–200 μm from carbon fiber) with a concentric bipolar electrode (FHC) with two single stimulations (100 μA intensity, 0.2 ms length) separated by 15s, or a theta burst train that was used in the LTP induction protocol (Figure 1A), which consists of five bursts repeated at 0.1 Hz, with each burst composed of three stimulations at 50 Hz (100 μA intensity, 0.2 ms length). Recordings were obtained using TarHeel CV system. Changes in dopamine concentration by electrode stimulation were quantified by plotting the peak oxidation current of the voltammogram over time. Carbon-fiber microelectrode was calibrated at the end of each day of experiments to convert oxidation current to dopamine concentration using 10 μM dopamine in ACSF.
Figure 1. Pathway-specific modulation of striatal STDP-LTP by KOR.
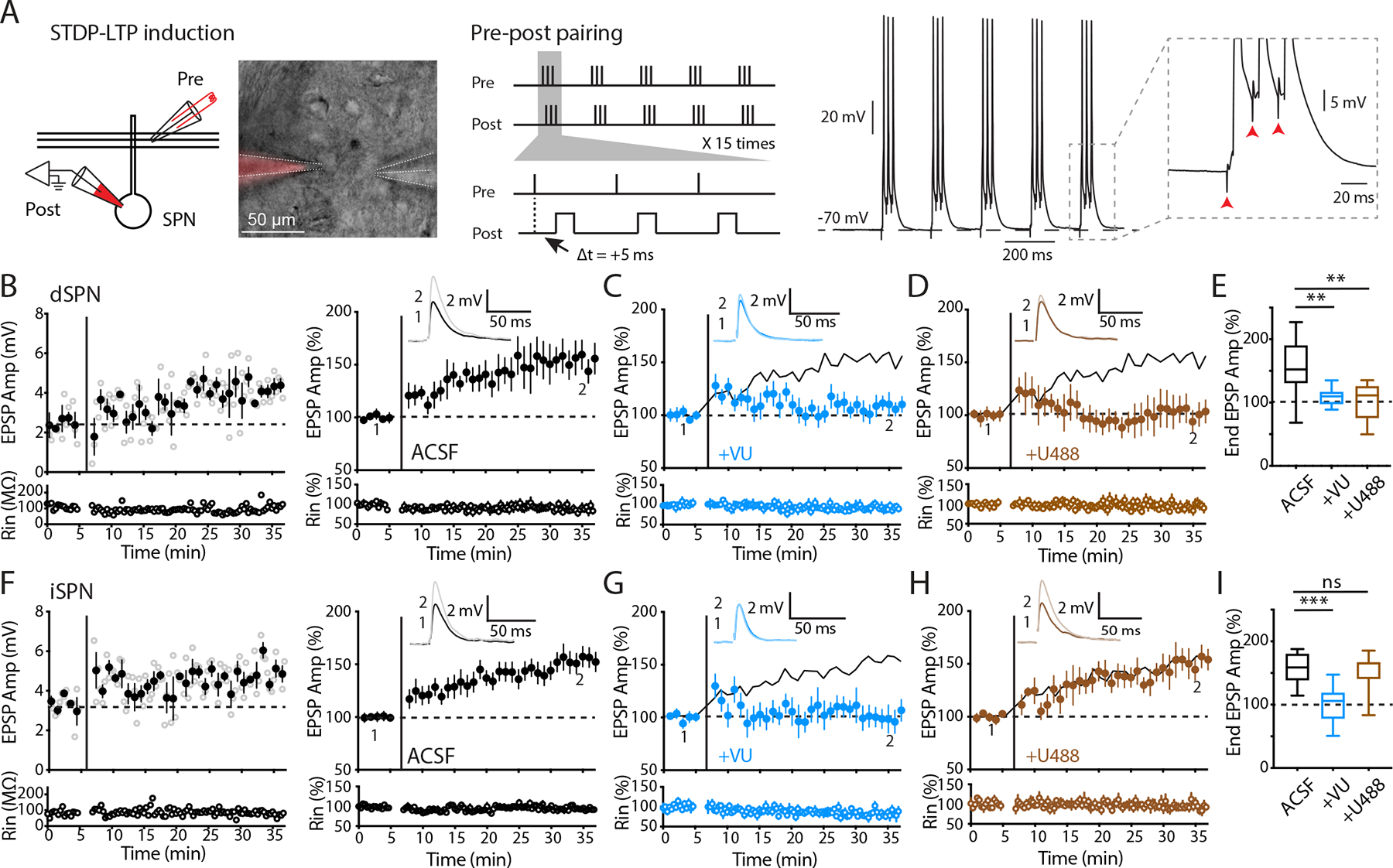
A Left: schematic and image depicting perforated patch recording from a postsynaptic SPN with presynaptic stimulation using a two-barrel theta glass. Middle: the theta-burst spike-timing-dependent plasticity (STDP) pairing protocol for LTP induction. Right: a representative trace showing action potentials and EPSPs recorded during STDP-LTP induction. B, F Left: example experiments of dSPN (B) and iSPN (F) recordings from slices obtained from WT mice. Open circles show EPSP amplitudes (Amp) and input resistance (Rin) throughout the recording. Filled circles are averages of 3 trials (± SEM). STDP-LTP induction is indicated by the vertical bar. The dashed line is the average baseline EPSP amplitude before induction. Right: summary plots of the normalized EPSP amplitudes and Rin as a function of time. Inset: representative average EPSP traces of the baseline (1) and the last 5 min (2). C, G Summary plots of recordings obtained from WT mice with administration of M4R PAM (VU10010). VU10010 suppressed LTP in both dSPNs (C) and iSPNs (G). Solid lines are control LTP for reference. D, H Summary plots of recordings obtained from WT mice with administration of KOR agonist (U50488). U50488 suppressed LTP in dSPNs (D) but not iSPNs (H). Solid lines are control LTP for reference. E, I Box-plot summaries showing the normalized EPSP amplitudes from the last 5 min of recording (end EPSP). E dSPN LTP was suppressed by VU10010 and U50488. Control (ACSF), n = 12/8, 153.04% ± 12.64; +VU, n = 10/4, 109.37% ± 4.42; +U488, n = 8/4, 102.09% ± 10.59. Control vs +VU, p = 0.0034; control vs +U488, p = 0.0055, Mann-Whitney. I iSPN LTP was suppressed by VU10010 but not U50488. Control (ACSF), n = 14/8, 153.69% ± 7.71; +VU, n = 9/4, 99.98% ± 9.77; +U488, n = 9/5, 151.99% ± 9.79. Control vs +VU, p = 0.0005; control vs +U488, p = 0.98, Mann-Whitney. Data are presented as mean ± SEM. ** p < 0.01, ns: not significant.
3. miniature EPSC recording
Brain slices from D1-cre;pDyn+/+;Ai32 and D1-cre;pDynfl/fl;Ai32 mice were used to record miniature EPSCs (mEPSCs). SPNs were visualized under infrared illumination using an Olympus BX51WI microscope equipped with DIC, a water-immersion objective (40× NA 0.8), and a CMOS camera (Hamamatsu Photonics). Whole-cell voltage-clamp recording was performed with borosilicate glass microelectrodes (3–5 MΩ) filled with a Cs+-based low Cl− internal solution (126 mM CsMeSO3, 8 mM NaCl, 10 mM HEPES, 2.9 mM QX-314, 8 mM Na2-Phosphocreatine, 0.3 mM GTP-Na, 4 mM ATP-Mg, 0.1 mM CaCl2, 1 mM EGTA; pH 7.2–7.3; osmolarity 285–290 mOsm). The access resistance was < 25MΩ (no compensation), and the data were discarded if the access resistance changed more than 25% during recording. The presence of ChR2 current evoked by a 450 nm laser pulse (0.5 ms, OptoEngine, USA) was used to distinguish dSPNs and iSPNs. For recording of miniature EPSC (mEPSC), TTX (1 μM) was included, and neurons were held at a membrane potential of the Cl− reversal potential (−70 mV, liquid junction potential not corrected). Recordings were obtained with a Multiclamp 700B amplifier (Molecular Devices) using the WinWCP software (University of Strathclyde, UK). Signals were filtered at 2 kHz, digitized at 10 kHz (NI PCIe-6259, National Instruments), and analyzed offline using Clampfit 10.0 (Molecular Devices) and Mini Analysis Program (Synaptosoft).
4. Perforated patch and LTP induction
Brain slices from D1-cre;pDyn+/+;Ai32 and D1-cre;pDynfl/fl;Ai32 were used to record LTP. The perforated patch and LTP induction protocol was adapted from a previous study (12). Electrical access was achieved through the perforated-patch method using Gramicidin A (Sigma). Perforated patch was performed with a borosilicate glass microelectrode (3–3.5 MΩ), front-filled with 1 μl K+-based internal solution (135 mM KMeSO3, 8.1 mM KCl, 10 mM HEPES, 8 mM Na2-Phosphocreatine, 0.3 mM GTP-Na, 4 mM ATP-Mg, 0.1 mM CaCl2, 1 mM EGTA; pH 7.2–7.3; osmolarity 285–290 mOsm), and back-filled with 10 μl Gramicidin A-containing internal solution. The Gramicidin A-containing internal solution was made fresh before use: a stock solution of Gramicidin A (20 mg/mL) in dimethyl sulfoxide (DMSO) was prepared and diluted in the K+-based internal solution yielding a final concentration of 200 μg/mL. The solution was thoroughly mixed by vortexing and then sonicated for 5 min and filtered with a centrifuge tube filter (0.22 μm, Spin-X, Costar). The fluorescent dye Alexa594 (10 μM) was also added in the internal solution for visualizing the integrity of the perforated patch configuration throughout the recording. After the microelectrode formed a giga seal with the cell membrane, access resistance was continuously monitored during perforation by applying a −5 mV pulse from a holding potential of −70 mV, under the voltage-clamp mode. A stable perforated patch normally formed within 30–60 minutes, and the access resistance stays around 30–50 MΩ without further decreasing. Subsequently, the presence of ChR2 current evoked by a 450 nm laser pulse (0.5 ms, OptoEngine, USA) was used to distinguish dSPNs from iSPNs. Then the recording was switched to current-clamp mode, and serial resistance was compensated with amplifier bridge balance. The data were excluded if the input resistance (Rin) changed more than 25% over the course of the experiment. Presynaptic inputs, recorded as EPSPs, were evoked by focal extracellular stimulation with a small theta glass electrode positioned 50–100 μm from the recorded cell body. Stimulation intensity (0.2 ms, 5–30 μA) was adjusted to evoke stable EPSPs with an amplitude of around 2–5 mV. EPSPs were evoked every 20 s for 5 minutes as the baseline. Then LTP was induced using the theta-burst spike-timing-dependent plasticity (STDP) pairing protocol. The protocol consisted of 15 trains of five bursts repeated at 0.1 Hz, with each burst composed of three postsynaptic spikings preceded (+5 ms) with three presynaptic inputs (EPSPs) at 50 Hz (Figure 1A). The postsynaptic spikes were evoked by direct somatic current injection (5 ms, 1–1.5 nA). During the induction, the postsynaptic cell membrane potential was depolarized to −70 mV. After the STDP induction, EPSPs were recorded for another 30 minutes to monitor the change of amplitudes. GABAA were blocked by the bath application of 100 μM picrotoxin throughout the recording. For recordings with norBNI (1 μM), (−)-U50488 (1 μM), SCH23390 (3 μM), or SKF81297 (3 μM), or VU10010 (5 μM), slices were incubated with these drugs during slice recovery (one hour) as well as throughout the recording, which was to reduce the acute effect of a receptor agonist or antagonist treatment.
In a subset of the recordings in which the recorded cells were post-hoc immunostained for enkephalin, 0.2% biocytin was included, while Alexa594 was excluded in the internal solution. The perforated patch configuration was maintained during the induction and recording of LTP. After recording, the perforated patch was ruptured to the whole-cell mode by applying rapid negative pressure, and the biocytin was allowed to dialyze the cell for at least 15 min. The slice was then fixed in PFA, followed by the immunostaining procedures described above.
Behavioral assays
1. Open field test
Mice were individually placed in the center of illuminated white plastic boxes (40 × 40 × 40 cm) and allowed to roam freely for 60 min. Locomotion was video recorded and analyzed using Viewer III tracking system (Biobserve). Total distance traveled and time spent in the 20 × 20 cm center of the square were quantified. The total distance was used to evaluate locomotor activity and the time spent in center area was used to estimate the anxiety level in an open environment. The total distance was quantified over each 5-min bin and analyzed with repeated measurement two-way ANOVA. The time spent in the center were analyzed with Student’s t-test.
2. Balance beam test
The animal’s ability to navigate across a beam was tested using two wooden beams (cylindrical beam 95 cm in length, 1 cm and 2 cm in diameter, respectively). The beams were fixed with ring stands on both ends 50 cm above the ground with one end leading into a small hide box. During the trials, the 2 cm beam was first used. Mice were placed at the start line near one end of the beam, facing the hide box, which was 80 cm away to the other end. Mice were left to walk across the beam to the box. After reaching the hide box, mice were let to stay there for 1 min and placed at the start line again for a total of three trials. Repeat the trials with 1 cm beam. The speed, number of foot slips for each mouse were scored and averaged across 3 trials for each beam. Data were analyzed with Student’s t-test.
3. Spontaneous Y maze
The Y-maze contained three gray arms (each arm: 40 × 10 × 17 cm) placed at 120° angles. Mice were placed in the distal end of one arm and allowed to freely explore the maze for 10 min under video monitoring. One entry was counted when the whole body including tail of a mouse entered an arm. An alternation was defined when the mouse visited all three different arms in three consecutive entries. The alternation percentage was calculated as [number of alternations/total number of entries] × 100. Y maze is based on the innate tendency of rodents to explore novel environment; thus, a mouse would try to make more spontaneous alternations in the Y maze, which depends on its spatial working memory. The alternation percentage was used to evaluate the spatial working memory of mice. Data were analyzed with Student’s t-test.
4. Elevated plus maze
The maze was elevated 40 cm above the floor and consisted of two open arms without walls and two closed arms with 10-cm tall walls. Each arm was 30 × 8 cm, connected by an 8 × 8 cm center area. Mice were habituated in the testing room 1 hour before testing. Mice were placed in the center zone of the EPM facing one of the open arms and were allowed to explore freely for 10 minutes. Locomotion was video recorded and analyzed using Viewer III tracking system (Biobserve). Total distance traveled, time and distance spent in the closed and open arms were quantified. Data were analyzed with Student’s t-test.
5. Three-chamber sociability test
A transparent three-chamber apparatus (60 × 30 × 30 cm per chamber) was used for sociability tests. One day before testing, the mice were first habituated in the entire apparatus for 20 minutes. During testing, a stranger C57BL/6J mouse confined in an upside-down wired-mesh cup was placed in one chamber (social chamber), and an identical but empty cup was placed in the right chamber (non-social chamber). The mice were placed in the center chamber and left to freely explore the three chambers for 30 min. The activity was recorded with Viewer III tracking system, and time spent in each chamber was quantified. Data was analyzed with Student’s t-test.
6. Spatial reversal learning task
The training task was adapted and modified from previous studies (32, 33). The training was conducted daily. All mice underwent one training session per day throughout the whole task. The mice started to be food restricted 3 days before the training and monitored to maintain around 90% body weight.
6.1. Operant apparatus
Each operant chamber was measured 21.6 × 17.8 × 12.7 cm (model # ENV-307W; Med Associates) and was housed within a sound and light attenuating cubicle (ENV-022MD; Med Associates). The back and ceiling and a forward-opening door were transparent plexiglass, and the floor was stainless steel grid. A pellet dispenser delivering a reward pellet (20 mg, #F0071; BioServ) singly into a food magazine was located at one side wall, and the nose-poke ports were situated left and right of the food magazine. Each nose-poke port had a white lamp in the recess. The nose pokes and pellet retrievals were detected via infra-red beam. A house light and tone generator were located on the other side wall. A house light was provided during the training and task sessions. All elements were removed and cleaned with 70% ethanol after each session. Four such operant chambers were run in parallel, with two chambers for males and females, respectively. Each mouse was assigned to a specific chamber. The four chambers were controlled from one PC using Med-PC V interface (Med Associates). A customized program scripted in Trans V was run to automatically control and record all experimental events and outputs. Only one session was conducted for each mouse per day.
6.2. Habituation and shaping
Mice were taken to the chambers for habituation for 3 days. The house light was lighted, and food magazine was loaded with pellets, and mice were placed in the operant chambers individually to explore freely for 10 minutes. This was to habituate the mice to the chamber and to retrieving pellets out of the food magazine.
During shaping, the session started with turning on the house light, and a pellet was dispensed along with a 2-s tone (CS, conditioned stimulus) every 45s. Total 39 pellets were dispensed in the 30-minute session. This step was to condition the mice to associate reward pellets with the tone, which facilitated later trainings. The criteria for each mouse to proceed to the next stage was retrieving at least 30 pellets for two consecutive sessions.
6.3. Pre-training/nose-poke learning
For all subsequent training stages, some common procedures were kept consistent as below:
During each trial, nose-poke port lights were illuminated to signal the beginning of a trial.
Immediately after each correct nose poke, the 2-s CS tone was presented, the nose-poke port lights were shut off, and a reward pellet was delivered. Before the mouse retrieved the pellet from the magazine, further nose pokes had no consequence. After the mouse retrieved the pellet, an inter-trial-interval (ITI) of 2.5s was initiated, during which the nose-poke port lights remained off, and nose pokes had no consequence. The onset of the next trial was signaled by switching on nose-poke port.
After mice poking into the wrong port, a timeout punishment of ITI of 5.0 s was initiated, during which the nose-poke port lights were switched off and further nosepokes had no consequences. After 5 s, the nose-poke port lights were switched on automatically to signal the new trial.
The pre-training was to get mice familiar with the operant task of nose poking to get reward. Training took place on each port separately with FR1 schedule. Only one of nose poke ports was presented and illuminated (so there was no wrong poke). Crushed pellets were initially placed in the port to encourage the mice to poke. One poke would trigger the CS tone and pellet reward delivery. The criterion was to reach 30 pellets in two consecutive sessions. Each session lasts up to 30 minutes or when the mice reached the criteria earlier. If the criterion was not achieved, the same training was repeated the next day. After mice reached criteria for one port, the same training took place in the other port. The order of left and right ports was counterbalanced across subjects.
6.4. Acquisition of spatial discrimination
After the mice learned the operant task of nose poking into one port, they were now presented with two illuminated ports, however, with one port as correct and the other as wrong. The assignment of left or right port as active was counterbalanced across subjects. As described above, in each trial, the correct poke led to reward delivery, while the wrong poke led to 5 s timeout punishment. The criteria to complete the acquisition phase was to have 9 correct responses in 10 consecutive trials in a session. Each session lasted up to 30 minutes or when the mice reached the criteria earlier. The main variables analyzed were the total number of trials and errors (failed trials) to reach criteria in the Acquisition stage. Some mice took more than one session to reach the criteria, and in this case, the variables described above from all sessions were combined. To proceed to the next stage, mice needed to complete another “retention” session, to reinforce the acquisition of spatial discrimination.
6.5. Reversal learning
The reversal learning task examined differences in behavioral flexibility. For experiments examining the effects of U50488, (±)-U50488 (5 mg/kg, i.p., dissolved in saline as 0.5 mg/mL) or saline were injected, and the mice were placed in home cages for 30 min before the reversal learning task. During the task, correct and wrong nose poke ports were switched, while the other procedures remained the same with the discrimination phase. The learning criteria were the same as the discrimination phase, with 9 correct responses out of 10 consecutive trials. The total numbers of trials and errors before reaching the criteria were measured. The errors were refined to “regressive” and “perseverative” errors. A regressive error was defined as an error following a successful trial. A perseverative error was an error following a previous error.
6.6. Progressive ratio schedule task
After the mice completed reversal learning task, they underwent progressive ratio schedule task once to assess the differences in incentive-motivation between control and pDyn cKO mice. Only one nose-poke port was deployed and illuminated. The reinforcement was a 2-s CS tone and a pellet, the same as above, but the nose-poke port light remained on. The number of nose pokes required to get a reinforcement reward was based on exponential progression derived from the formula (5 × e0.2n) −5, where n was the position in the ratio sequence (34). The session lasted up to 60 minutes or after 5 min elapsed without any responses. The nosepoke rate over the whole session and reward number achieved were analyzed.
7. The 4-choice odor discrimination and reversal task
The task followed the protocols from a previous study (35). The task was conducted daily for three days. Mice were food deprived over 3 days before the task to 90% of their original body weight and fed to maintain 90% body weight during training.
7.1. Apparatus
The 4-choice arena was a square clear acrylic box (12″ × 12″ × 9″). The arena was partially divided into 4 quadrants by four 3” wide inner walls. A white ceramic bowl (2.88” in diameter, 1.75” deep) was placed in each quadrant to hold scented wood shavings to present the odors, and a Honey Nut Cheerio piece (General Mills, Minneapolis, MN) buried in the shavings as reward. A removable 6” diameter cylinder was used to confine the mice in the center between trials.
7.2. Habituation
The first day of training was a habituation phase that allowed mice to become familiar with the arena and bowls containing Cheerio. A bowl containing 10 mg cheerio (no shavings) was placed in each quadrant. A mouse was placed in the cylinder. The cylinder was then lifted to let the mouse explore freely and eat from the bowls for 10 minutes. After 10 minutes, the mouse was returned to the start cylinder and bowls were re-baited. This procedure was repeated twice for a total habituation time of 30 min. The maze was wiped with 70% ethanol between animals.
7.3. Shaping
The second day of training was a shaping phase to train mice to dig wood shavings for cheerio pieces. Each mouse underwent 12 training trials. In each trial, one of the four quadrants was set up with a bowl containing 10 mg cheerio buried with wood shavings. The quadrant holding the bowl was rotated over trials (SE to NW to SW to NE). And the wood shaving amounts were increased over trials: the first two with no shavings, the next two with a thin layer, then two with a quarter full, two with half full, and four trials with the bowl full of shavings burying the cheerio. Trials were not timed.
7.4. Acquisition of odor discrimination
The third day consisted of an odor discrimination phase and a reversal phase. In the odor discrimination phase, mice had to discriminate four odors and learn which odor was associated with cheerio reward. Wood shavings scented with odors (O1-O4: anise, clove, litsea, and thyme, respectively) were placed in separate bowls. Only O1 was rewarded with 10 mg cheerio buried. To avoid spatial association, the four odors were randomly placed in different quadrants and an odor was never in the same location two trials in a row. The mouse was first confined in the center cylinder, and the trial began when the cylinder was lifted. The mouse could freely explore the quadrants until it began digging in one of the bowls, which was immediately followed by the lowering of the cylinder to prevent multiple digging choices. The digging was defined as consistent moving shavings with both front paws, but not as sniffing or chewing shavings. When the mouse began digging in an incorrect odor, the trial was terminated, and the mouse was returned to the center cylinder; if the mouse began digging in the correct odor, it was allowed to retrieve the reward before returned to the center cylinder. If the mouse didn’t make a digging choice within 3 minutes, the trial was terminated and recorded as an omission. Analysis of the latency to dig did not include omissions. Between each trial, the odors were rearranged and re-baited if necessary. The criterion for this phase was 8 correct digging responses out of 10 consecutive trials, including the omissions.
7.5. Reversal learning
Once criterion was met for discrimination, the reversal phase was initiated immediately within the same session. In this phase, O2 (clove) became the rewarded odor and O4 (thyme) was switched out for a novel odor O5 (eucalyptus). The criterion to complete this phase was the same as discrimination: 8 correct digging responses out of 10 consecutive trials. To quantify the reversal learning phenotype, their number of trials and errors before reaching criteria was analyzed. Specific errors were refined as follows (36): regressive – errors made after the first correct trial; perseverative – errors made before the first correct trial. To analyze their motivational aspects during the reversal learning, the time latency from trial starts to digging was analyzed.
Statistical analyses
Statistical analyses were performed using GraphPad Prism (GraphPad Software) and MATLAB (Mathworks). Behavioral performances were analyzed with Student’s unpaired t-test and repeated measurement 2-way ANOVA. The cumulative plot of the percentage of mice reaching the criteria were compared with the Kolmogorov-Smirnov test. Averaged mEPSC amplitudes and frequencies, spine densities, and evoked [DA]o were analyzed with the Mann-Whitney Rank Sum test, and cumulative distribution of mEPSC amplitudes and inter-event intervals were analyzed with the Kolmogorov-Smirnov test. The EPSP amplitudes at the baseline and the end of recordings in LTP experiments were compared with Paired Wilcoxon signed-rank test to determine if LTP or LTD were generated, and the normalized End EPSP amplitudes were compared between conditions using the Mann-Whitney Rank Sum test. All data were represented as mean ± SEM. Within the figures: * p < 0.05, ** p < 0.01, *** p < 0.001 and ns = not significant, p > 0.05.
RESULTS
To investigate pathway specific regulation of plasticity by neuromodulatory receptors, we utilized a spike-timing-dependent plasticity (STDP) protocol combined with perforated-membrane patch clamp recording to induce LTP in the dorsal striatum,(12). The induction protocol comprised of a series of 50 Hz bursts of “pre-post” pairing, in which three presynaptic inputs preceded three postsynaptic spikes by 5 ms (Figure 1A). Presynaptic inputs were stimulated with a small theta glass pipette close to the recorded SPN (50–100 μm), and postsynaptic spiking was induced by current injection through the perforated patch. Before induction, we recorded theta glass-evoked excitatory postsynaptic potentials (EPSPs) to obtain a stable 5-minute baseline, and after STDP-LTP induction, we continued to record the evoked EPSPs for at least 30 minutes to monitor changes in EPSP amplitudes. To distinguish dSPNs and iSPNs, we used D1-cre;Ai32 mice, in which Channelrhodopsin-2 (ChR2) is specifically expressed in D1 neurons, enabling the identification of dSPNs and iSPNs based on the presence or absence of the optically evoked response, respectively (please see Methods for details). In both dSPNs and iSPNs, this STDP protocol reliably induced LTP (Figure 1B,F).
KOR suppresses LTP specifically in dSPNs
To investigate whether the activation of Gi-coupled M4R can differentially regulate LTP in a pathway-specific manner, we incubated the brain slices obtained from wildtype (WT) mice with the M4R positive allosteric modulator (PAM) VU10010 (VU, 5 μM), which enhances the response to endogenous acetylcholine. Consistent with the previous finding (15), the M4R PAM VU10010 blunted LTP expression in dSPNs (Figure 1C,E). It has been generally assumed that the effect of M4R is small in iSPNs because M4R is preferentially expressed in dSPNs (9, 16). On the contrary, we observed a similar level of suppression of LTP in iSPNs by M4R PAM VU10010 (Figure 1G,I). This result indicates that M4R activation broadly regulates LTP in both dSPNs and iSPNs.
Because of the unique expression pattern of Dyn (21) and preferential expression of KOR in the dSPNs (19, 20), we next tested the effect of KOR activation on LTP in both dSPNs and iSPNs in WT mice. We used the same recording condition and replaced M4R PAM with KOR agonist U50488 (U488, 1 μM). We observed that U488 abolished LTP expression in dSPNs (Figure 1D,E), while LTP in iSPNs remained intact (Figure 1H,I), indicating that KOR signaling regulates striatal LTP in a pathway-specific manner.
D1-pDyn cKO enhances LTP specifically in dSPNs
One caveat associated with KOR agonists is that KOR activation can directly affect the basal glutamatergic and dopaminergic synaptic transmission (23, 25, 27). To confirm the pathway-specific regulation of LTP by endogenous KOR signaling in the striatum, we next used genetic tools to selectively remove the endogenous KOR ligand, Dyn, by generating a striatum direct-pathway-specific Dyn conditional knockout mice.
The dynorphin family consists of several variants derived from the preprodynorphin (pDyn) gene (37), all of which share the amino acid sequence encoded in the exon 4, conferring them the ligand binding specificity to KORs (38). Therefore, we generated pDyn-floxed (pDynfl/fl) mice by flanking exon 4 of the pDyn gene with loxP sites (Figure 2A). To knock out Dyn in the striatum, we crossed the pDynfl/fl mice with D1-cre mice, which express cre recombinase in D1 dopamine receptor (D1R)-containing cells. While D1R is also expressed at low levels in several other brain areas (28), dynorphin and D1R co-expression occurs mainly in the striatal dSPNs. Therefore, D1-cre;pDynfl/fl mice primarily lack dynorphin in the striatal direct pathway (Figure 2B).
Figure 2. Direct pathway-specific deletion of pDyn.

A Gene targeting strategy scheme for pDyn conditional knockout (cKO) mice, including the diagrams of wildtype pDyn allele, targeted allele, loxP-flanked allele, and cre-deleted allele. In the targeted allele, pDyn Exon 4 and a neo cassette are flanked by loxP sites. The neo cassette is removed from the targeted allele by FLP recombinase. pDyn exon 4 is then deleted in D1 cells by breeding to a D1-cre mouse line. The primers used for genotyping are also indicated as arrows. B Immunohistochemistry of dynorphin in striatum and motor cortex (M1). Top: sample images of a coronal section of cortex (left), striatum (middle) and substantia nigra (right), stained for dynorphin (green) and DAPI (blue). Bottom: quantification of co-labelling of dynorphin with DAPI in the cortex (WT, n = 3500 cells / 4 mice, 31.6%; D1-pDyn cKO, n = 3500 cells / 4 mice, 33%) and striatum (WT, n = 4002 cells / 4 mice, 52.9%; D1-pDyn cKO, n = 3996 cells / 4 mice, 2.9%).
To assess the deletion efficiency in the striatum, we performed immunostaining with anti-Dyn antisera. We quantified the percentage of DAPI labeled neurons that were dynorphin positive in the dorsal striatum. In control striatal sections, approximately half of the neurons were dynorphin positive (WT control: 52.9%; Figure 2B) consistent with previous reports (8). By contrast, Dyn expression in the striatum of D1-cre;pDynfl/fl mice was almost fully abolished in the striatum (D1-pDyn cKO: 2.9%; Figure 2B). We also immunostained and imaged the substantia nigra region, where the axonal projections of dSPNs terminate, and again observed a diminished Dyn signal in cKO slices. Dyn expression in the motor cortex (including all layers) was also quantified, which on the other hand was not affected in the D1-cre;pDynfl/fl mice (WT: 31.6%, D1-pDyn cKO: 33%; Figure 2B), confirming that deletion of Dyn in D1-cre;pDynfl/fl mice was indeed specific to the striatum.
We recorded STDP-LTP in dSPN from the D1-cre;pDynfl/fl mice. Strikingly, we found that LTP in dSPNs was significantly enhanced in the Dyn cKO mice (Figure 3A,B). Pharmacologically blocking KOR with the antagonist norbinaltorphimine (norBNI, 1 μM) in the D1-cre;pDyn+/+ mice resulted in a similar enhancement in LTP in dSPNs (Figure 3C,D). These results are congruent with the effects we observed of KOR agonism (Figure 1D,E). In addition, KOR antagonism did not have any effects on STDP-LTP in iSPNs confirming the pathway specificity of the effect. Together, these data show that LTP is bi-directionally regulated by KOR signaling: Dyn/KOR signaling suppresses LTP expression in dSPNs and antagonizing KOR or genetically removing its ligand Dyn enhances LTP.
Figure 3. D1-pDyn cKO enhanced LTP in dSPNs, but not iSPNs.

A Left: an example experiment of dSPN recordings from slices from D1-pDyn cKO mice (D1-cre;pDynfl/fl). Right: summary plot showing an enhanced LTP in D1-pDyn cKO compared with WT (D1-cre;pDyn+/+) group. The solid line shows the control LTP from Figure 1B for reference. Notably, the controls were interleaved with various treatment conditions. B Box-plot summary showing enhanced dSPN LTP by D1-pDyn cKO. WT, n = 12/8, 153.04% ± 12.64; D1-pDyn cKO, n = 10/7, 220.86% ± 23.28. p = 0.0206, Mann-Whitney. C Left: an example experiment of dSPN recordings from WT mice (D1-cre;pDyn+/+) with administration of KOR antagonist (+nBNI). Right: summary plot showing that norBNI significantly enhanced the LTP to a similar level as cKO. The solid black and red lines are LTP recorded from WT and D1-pDyn cKO mice, respectively, for reference. D Box-plot summary showing enhanced dSPN LTP by norBNI. Control (ACSF), n = 12/8, 153.04% ± 12.64; +nBNI, n = 8/3, 212.98% ± 23.66. control vs +nBNI, p = 0.0473, Mann-Whitney. E Left: an example experiment of iSPN recordings from D1-pDyn cKO slices. Right: summary plot showing similar LTP levels in control and D1-pDyn cKO. The solid line is the control LTP from Figure 1F for reference. F Box-plot summary showing similar iSPN LTP between control and D1-pDyn cKO. WT, n = 14/8, 153.69% ± 7.71; D1-pDyn cKO, n = 14/9, 154.61% ± 8.96. WT vs D1-pDyn cKO, p = 0.95, Mann-Whitney. Data are presented as mean ± SEM. * p < 0.05, ns: not significant.
In addition, we recorded LTP in iSPNs from the D1-cre;pDynfl/fl mice. LTP in iSPNs of D1-pDyn cKO mice were comparable to WT control mice (Figure 3E,F), confirming that suppression of LTP by dyn/KOR signaling is pathway specific. Notably, in a subset of the recordings, we included 0.2% biocytin in the internal solution and performed post hoc immunostaining for enkephalin (Figure S1) to verify the identity of iSPNs. These verified iSPNs showed similar levels of LTP between control and D1-pDyn cKO groups.
No change on basal dopaminergic and glutamatergic transmission in D1-cre;pDynfl/fl mice
Previous studies have shown that acute activation of dyn/KOR signaling can suppress the dopamine (DA) (22, 23, 39) and glutamatergic release (25, 27, 40, 41). To test whether dopaminergic and glutamatergic signaling are affected in D1-pDyn cKO mice, we measured basal dopaminergic and glutamatergic transmission. We first used fast scan cyclic voltammetry (FSCV) to assess dopaminergic transmission in the dorsal striatum. We used a local bipolar electrode to evoke DA release with a single stimulation or a theta burst train that is identical as that of the LTP induction protocol, and measured the evoked local extracellular DA concentration ([DA]o). We did not find any differences in evoked [DA]o between WT and D1-pDyn cKO mice (Figure S2A–C,E). We also did not observe any differences in the paired-pulse ratio (PPR) indicative of the release probability (Figure S2D). We next recorded the spontaneous miniature excitatory postsynaptic currents (mEPSCs) (Figure S3A,B) to assess basal glutamatergic transmission. We did not find any significant differences in mESPC mean amplitude or frequency in either SPN types between WT control and D1-pDyn cKO mice (Figure S3C,D). Furthermore, using 2-photon imaging in acute brain slices in which SPNs were filled with the red fluorophore Alexa594 through the whole-cell recording pipette, we examined the density of spiny protrusions along SPN dendrites, where most glutamatergic synapses are formed on SPNs. We found that there was no significant differences in spine density in either dSPNs or iSPNs between WT and D1-pDyn cKO mice (Figure S3E,F). These results indicated that deletion of striatal Dyn did not affect the basal levels of dopaminergic or glutamatergic transmission in the dorsal striatum. Our results thus provide evidence that the enhanced LTP expression in dSPNs in D1-pDyn cKO mice is not due to an altered baseline DA or glutamatergic release, but rather suggests a direct effect of autonomous activation of KOR signaling in dSPNs.
Interaction between Dyn/KOR and D1R signaling on LTP induction
Opposing signaling cascades from different G-protein coupled receptor (GPCR) subunits exert a push and pull that control striatal synaptic plasticity (9). In dSPNs, D1Rs are crucial for LTP induction (12). D1Rs activation promotes LTP induction while D1Rs blockade leads to LTD even with STDP-LTP induction protocols (12). In our STDP experiment described above, D1Rs are activated by dopamine released from en passant dopaminergic fibers stimulated during STDP induction. However, dopamine levels might be considerably different in physiological conditions. For example, midbrain DA neurons exhibit phasic bursts of firing in response to unexpected rewards and decreased firing rate when an expected reward is omitted (42, 43), which might result in transient high-level activation or suppression of D1R signaling in dSPNs. Therefore, we investigate how D1R signaling interacts with Dyn/KOR signaling to modulate LTP expression at different D1R activation levels. We treated the striatal slices with the D1R full agonist SKF81297 (SKF, 3 μM) or antagonist SCH23390 (SCH, 3 μM) to examine whether dSPN LTP is modulated by KOR signaling when D1R is fully activated or blocked, respectively. In the presence of the D1R agonist, SKF81297, we observed that WT control expressed enhanced levels of LTP (Figure 4A), consistent with previous findings (12, 44); while D1-pDyn cKO dSPNs showed similar magnitudes of LTP (Figure 4B), suggesting that high levels of D1R activation occlude the LTP enhancement by Dyn deletion. Next, we examine LTP induction and expression when both D1R and KOR are activated by the administration of both agonists SKF 81297 and U50488. Interestingly, U50488 failed to suppress LTP in the presence of the D1R agonist (Figure 4C), suggesting D1R outweighs KOR signaling. Notably, all groups showed comparable high levels of LTP expression regardless of whether KOR is activated or blocked (Figure 4D). Together, these results suggest that D1R signaling dominates the KOR pathway when both are activated in tandem (Figure 4E).
Figure 4. D1R signaling modulated the effect of dyn/KOR on dSPN LTP.

A Summary plot showing enhanced LTP in the presence of D1 agonist (SKF81297) recorded from WT (D1-cre;pDyn+/+)mice. The solid line shows LTP recorded without SKF81297 for reference (same as Figure 1B). B Summary plot showing enhanced LTP in the presence of D1 agonist (SKF81297) recorded from D1-pDyn cKO (D1-cre;pDynfl/fl)mice. SKF81297 did not further enhance LTP in D1-pDyn cKO mice. The solid line shows LTP recorded from D1-pDyn cKO mice without SKF81297 for reference (Same as Figure 3A). C Summary plot showing enhanced LTP in the presence of D1 agonist (SKF81297) and KOR antagonist (U50488) recorded from WT (D1-cre;pDyn+/+) mice. In the presence of SKF81297, U50488 failed to block the LTP. The solid line shows recording in the presence of +U50488 alone for reference (Same as Figure 1D). D Box-plot summary showing LTP levels in the presence of SKF81297. WT+SKF, n = 7/3, 206.76% ± 24.5; cKO+SKF, n = 7/3, 211.83% ± 19.76; WT+U488+SKF, n = 7/2, 210.82% ± 29.05. WT+SKF vs cKO+SKF, p = 0.805; WT+SKF vs WT+U488+SKF, p = 0.71; cKO+SKF vs WT+U488+SKF, p = 0.99, Mann-Whitney. E The schematic depicting the signaling pathway of D1R and KOR activation. D1R activation enhanced dSPN LTP expression regardless of whether Dyn/KOR pathway is activated. F Summary plot showing the same STDP induction protocol resulted in LTD in the presence of D1R antagonist (SCH23390) recorded from WT (D1-cre;pDyn+/+) mice. The solid line shows control LTP for reference. G Summary plot showing LTP is dampened to WT control LTP level in the presence of D1 antagonist (SCH23390) recorded from D1-pDyn cKO (D1-cre;pDynfl/fl) mice. The solid red line shows LTP in cKO mice and solid black line shows WT control LTP for reference. H Summary plot showing LTP recorded from WT (D1-cre;pDyn+/+) mice in the presence of both D1 antagonist (SCH23390) and KOR antagonist (norBNI). When D1R is blocked, blocking KOR resulted in normal LTP. The solid orange line shows WT+norBNI LTP and solid black line shows control LTP for reference. I Box-plot summary showing LTP levels in the presence of D1R antagonist (SCH23390). Loss of KOR function rescued LTP in the presence of D1R antagonist. WT+SCH, n = 9/4, 59.95% ± 11.72; cKO+SCH, n = 9/3, 156.01% ± 19.31; WT+norBNI+SCH, n = 9/4, 154.68% ± 14.14. WT+SCH vs cKO+SCH, p = 0.0008; WT+SCH vs WT+norBNI+SCH, p = 0.0005; cKO+SCH vs WT+norBNI+SCH, p = 0.99, Mann-Whitney. J The schematic depicting the signaling pathway of D1R and KOR blockade. Blockade of D1R suppressed dSPN LTP, deletion or blockade of KOR rescued LTP in the presence of D1R antagonist. Data are presented as mean ± SEM. ns: not significant, p > 0.05, *** p < 0.001.
Conversely, in the presence of the D1R antagonist SCH23390 (3 μM), dSPNs from WT D1-cre;pDyn+/+ mice expressed long-term depression (LTD) even though an LTP induction protocol was used (Figure 4F) and is consistent with previous findings (12). Interestingly, D1R antagonist SCH23390 reduced but did not fully block the LTP in dSPNs from D1-cre;pDynfl/fl mice (Figure 4G). Consistently, when we blocked KOR with norBNI in dSPNs from WT D1-cre;pDyn+/+ mice, LTP was reduced by SCH23390 compared to LTP in the presence of norBNI alone (Figure 4H), but again LTP was not fully blocked by D1 antagonist SCH23390. Notably, unlike the results above with the D1R agonist, where decreasing or increasing dyn/KOR does not alter the predominant effect of D1R activation, here, LTP induction and expression are comparable to WT control levels when Dyn is conditionally deleted from dSPNs or KOR is blocked (Figure 4G,H,I).
In summary, these results showed that signaling of endogenous striatal dynorphin interacts closely with dopamine D1R signaling to modulate striatal plasticity in direct pathway. Dyn/KOR plays a more prominent role in suppressing LTP in the absence or at lower levels of D1R activation, and its effect may be dampened during higher levels of D1R activation (Figure 4J).
Enhanced instrumental learning in the reversal phase
Having established the role of striatal dynorphin in regulating synaptic plasticity in direct pathway, we next ask, what is the behavioral relevance? One of the essential functions of the dorsal striatum is to facilitate reward-based instrumental learning, in which mice acquire the association between behavioral actions and reward outcomes (45). The dorsal striatum has been shown to encode action-outcome (A-O) contingency, action sequence and habit formation (1, 2, 46). Given that LTP is commonly correlated with learning and memory and an essential role of basal ganglia is motor control (47–49), we test whether mice lacking Dyn in D1 neurons would have any behavioral deficits using a battery of behavioral tests, including motor control, anxiety, working memory and instrumental learning performance.
The Dyn cKO and their WT control littermates performed equally in a variety of behavioral assays, including the open field task for testing locomotion and anxiety (Figure 5A), balance beam for fine movement control (Figure 5B), Y maze for working memory (Figure 5C), and elevated plus maze for anxiety (Figure 5D). There were no statistically significant differences between WT control (D1-cre;pDyn+/+) and D1-pDyn cKO (D1-cre;pDynfl/fl) mice in these tasks suggesting that deletion of Dyn in dSPNs does not cause any gross behavioral deficits.
Figure 5. Behavioral assessment of pDyn WT and cKO mice.

A Open field test. Left, distance travelled over 60 minutes (WT D1-cre;pDyn+/+, n = 25; D1-pDyn cKO D1-cre;pDynfl/fl, n = 19; genotype: F1,42 = 0.2985, p = 0.59, 2-way RM ANOVA). Right, time spent in the center (WT, 5.54 ± 0.89; D1-pDyn cKO, 5.89 ± 0.85, p = 0.78, t test). B Balance beam test with 2 or 1 cm-diameter suspended beam. Upper, speed on 2 cm (WT, n = 7, 26.39 ± 4.93; D1-pDyn cKO, n = 10, 21.40 ± 1.8; p = 0.37, t-test) and 1 cm beam (WT, 16.56 ± 2.42; D1-pDyn cKO, 13.58 ± 2.34, p = 0.39, t-test). Lower, foot slips on 2 cm (WT, 0.47 ± 0.27; D1-pDyn cKO 0.19 ± 0.13, p = 0.38, t-test) and 1 cm beam (WT, 0.96 ± 0.25; D1-pDyn cKO, 0.63 ± 0.19, p = 0.31, t-test). C Y-maze task. Left, arm entries (WT, n = 13, 34.08 ± 3.47; D1-pDyn cKO, n = 16, 34.88 ± 2.72, p = 0.89, t-test). Right, spontaneous alternations % (WT, 67.46 ± 2.38; D1-pDyn cKO, 64.81 ± 1.53, p = 0.34, t-test). D Elevated plus maze test. Upper, tracklength in closed (WT, n = 25, 34.09 ± 5.03; D1-pDyn cKO, n = 19, 32.19 ± 5.56, p = 0.8, t-test) and open arms (WT, 3.45 ± 0.76; D1-pDyn cKO, 2.97 ± 0.74, p = 0.65, t-test). Lower, duration in closed (WT, 239.36 ± 7.19; D1-pDyn cKO, 235.87 ± 7.59, p = 0.74, t-test) and open arms (WT, 24.46 ± 4.17; D1-pDyn cKO, 20.23 ± 3.45, p = 0.44, t-test). E Experimental scheme and behavior apparatus of 2-choice spatial discrimination reversal learning task. F It took similar session numbers for both WT and cKO mice to learn nose-poking in pre-training stage. 1st hole: WT, n = 17, 3.41 ± 0.32; D1-pDyn cKO, n = 13, 3.15 ± 0.24. 2nd hole: WT, 2.41 ± 0.15; D1-pDyn cKO 2.62 ± 0.24, p = 0.48, t test. G Left: trial numbers the mice took to reach criteria in the discrimination and reversal phases. Both WT and D1-pDyn cKO mice needed comparable numbers of trials to reach the criteria during the discrimination phase (WT, 40.9 ± 6.06; D1-pDyn cKO 48.8 ± 9.89, p = 0.51, t test), while D1-pDyn cKO group needed significantly fewer trials to reach the criteria during the reversal phase (WT, 48.17 ± 6.56; D1-pDyn cKO, 27 ± 2.7, p = 0.0071, t test). Right: cumulative plots showing the percentage of mice that reached criteria over training trials. The curves during the discrimination phase were not significantly different (p = 0.84, Kolmogorov-Smirnov test), while D1-pDyn cKO reached the criteria significantly faster during the reversal phase (## p = 0.0019, Kolmogorov-Smirnov test). H During the reversal phase, D1-pDyn cKO made significantly fewer total errors (WT, 22.7 ± 3.67; D1-pDyn cKO, 12.3 ± 1.72, p = 0.018, t-test) and specifically fewer regressive errors (WT, 9.24 ± 1.72; D1-pDyn cKO, 3.38 ± 0.79, p = 0.0054, t-test), but comparable numbers of perseverative errors (WT, 12.71 ± 2.67; D1-pDyn cKO, 7.92 ± 1.39, p = 0.15, t test). I In the progressive ratio task, there is no difference between WT and D1-pDyn cKO groups in the cumulative plot of nose poke numbers (F1,23 = 0.00058, p = 0.94, 2-way RM ANOVA). And the number of rewards achieved in the session was comparable (control 15.54 ± 0.55, cKO 15.17 ± 0.89, p = 0.72, t test). Data are presented as mean ± SEM. Individual data points of male and female mice are shown by the empty and filled circles, respectively. * p < 0.05, ** p < 0.01, ns: not significant.
We then utilized instrumental reversal learning task to assess the animal’s performance during initial learning and the animal’s flexibility to adapt to a new action-reward contingency during the reversal learning phase (50). We first employed a spatial reversal learning task using operant chambers with two nose-poking holes (Figure 5E; see Methods). In this task, mice needed to poke one of the two holes to receive a food reward. Mice were pre-trained to proficiently poke into both holes and to retrieve rewards from the food magazine. We found that it took both WT control (D1-cre;pDyn+/+) and D1-pDyn cKO (D1-cre;pDynfl/fl) mice similar numbers of training sessions during the nosepoke pre-training phase (Figure 5F), suggesting that D1-pDyn cKO did not affect general action sequence learning. Next, during the spatial discrimination phase, one of the two holes was associated with reward, while the other with 5-second timeout punishment. We found that the trial numbers to reach criteria (9 correct responses out of 10 trials) during the discrimination phase was similar for both groups (Figure 5G). These results suggest D1-pDyn cKO did not change the initial acquisition of action-reward association. In the subsequent reversal phase, the correct and wrong nosepoke holes were reversed. The mice initially poked the previously rewarded hole and were punished with a timeout, and gradually switched to the updated correct hole. Strikingly, the trial number to reach criteria during reversal was significantly reduced in the D1-pDyn cKO group (Figure 5G). Consistently, D1-pDyn cKO mice made significantly fewer errors (failed trials in which mice poked into the previously rewarded but currently unrewarded hole) during the reversal phase (Figure 5H). We further classified the errors into perseverative errors (an error following a previous error) and regressive errors (an error following a correct trial), which are used to measure the sensitivity to negative feedback and their flexibility in adapting to the new reward contingency, respectively (50). We found that regressive errors were significantly reduced in the D1-pDyn cKO mice, while perseverative errors were comparable (Figure 5H). These results suggested that loss of dynorphin elevated the animal’s flexibility in adapting to new action-reward contingencies during the reversal phase.
To test if activating KOR could reverse the enhanced reversed learning phenotype seen in the D1-pDyn cKO mice, we treated a separate cohort of cKO mice with either the KOR agonist, U50488 (U488, 5 mg/kg, i.p.), or saline before the reversal session (Figure S4A). We found that U488 treated D1-pDyn cKO mice spent more trials to reach the criteria (Figure S4B) and made more errors than saline-treated D1-pDyn cKO mice (Figure S4C). These results further strengthened the conclusion that the lack of Dyn/KOR signaling is responsible for the enhanced reversal learning in cKO mice. Notably, U488 increased the perseverative errors significantly while not affecting regressive errors, suggesting that activation of KOR may prevent the enhancement of reversal learning in D1-pDyn cKO mice by lowering their sensitivity to negative feedback.
Since previous studies showed that Dyn/KOR might modulate the perception of reward value (51–54), we next examined if the enhanced performance during reversal learning was due to a change in motivation to work for the reward. We conducted a progressive ratio task with the same mice. Nosepoke rates over the entire session were indistinguishable between D1-pDyn cKO (D1-cre;pDynfl/fl) mice and control (D1-cre;pDyn+/+) mice groups (Figure 5I), suggesting that loss of striatal Dyn did not change the motivation or the perceived value of the food reward.
We further confirmed this unique behavioral phenotype with a different task - 4-choice odor discrimination reversal learning task (35). A new cohort of mice was trained to dig for food reward (habituation and shaping phases) and then trained to associate a specific odor with the food reward (discrimination phase) (Figure 6A; see Methods). After mice reached the discrimination criteria (8 correct responses out of 10 trials), the food reward was paired with one of the other odors (reversal phase). Consistent with our results in the spatial reversal paradigm, we found that while the trial numbers to reach criteria in the discrimination phase were similar, the trial numbers in the reversal phase were significantly reduced in D1-pDyn cKO (D1-cre;pDynfl/fl) mice (Figure 6B). We again quantified the errors during the reversal phase and found that cKO mice made fewer total errors, with a specific decrease in the number of regressive errors but not perseverative errors (Figure 6C), suggesting that D1-pDyn cKO (D1-cre;pDynfl/fl) mice have a faster adaptation of the new action-reward contingency. To assess reward motivation in this task, we measured the time latency from trial starts to digging during the reversal phase and found no significant difference (Figure 6D).
Figure 6. Enhanced reversal learning in pDyn cKO mice in 4-choice odor discrimination task.

A Schematic of the experiment. The shavings were unscented in the shaping phase and scented with four of the five odors (O1–O5) during the discrimination and reversal phases. B Left: trial numbers the mice took to reach criteria in the discrimination (Disc) and reversal (Rev) phases. There is no significant difference in the number of trials for WT and D1-pDyn cKO groups to reach the criteria during the discrimination (WT, n = 22, 24.32 ± 1.79; D1-pDyn cKO, n = 21, 20.95 ± 1.09, p = 0.12, t-test), while D1-pDyn cKO mice need significantly fewer trials to reach criteria during the reversal phase (WT, 28.73 ± 1.54; D1-pDyn cKO, 20.67 ± 1.82, p = 0.0016, t test). Right: cumulative curves of the percentage of mice that reached criteria over training trials. The plots are not significantly different during the discrimination phase (p = 0.87, Kolmogorov-Smirnov test), and D1-pDyn cKO group increases significantly faster than WT during the reversal phase (## p = 0.0021, Kolmogorov-Smirnov test). C During the reversal phase, D1-pDyn cKO mice made significantly fewer total errors (WT, 14.46 ± 1.17; D1-pDyn cKO, 9.05 ± 1.48, p = 0.006, t test) and significantly fewer regressive errors (WT, 6.32 ± 0.66; D1-pDyn cKO, 3.38 ± 0.72, p = 0.0044, t test), but similar numbers of perseverative errors (WT, 4.09 ± 0.78; D1-pDyn cKO, 3.14 ± 1.01, p = 0.24, t test). D Cumulative plot of the time latencies from trial starts to digging over the first 10 trials in the reversal phase. 2-way RM ANOVA revealed no effect by genotype (F1,39 = 1.453, p = 0.24). Data are presented as mean ± SEM. Individual data points of male and female mice are shown by the empty and filled circles, respectively. p > 0.05, ** p < 0.01, ns: not significant.
In summary, D1-pDyn cKO (D1-cre;pDynfl/fl) mice and control (D1-cre;pDyn+/+) mice demonstrated similar performance during the initial acquisition of action-reward contingency in both discrimination reversal learning behavioral paradigms, however, the D1-pDyn cKO mice showed increased flexibility in adapting new reward contingency during the reversal phase.
DISCUSSION
Neuropeptides, such as Dyn, are often associated with a specific cell population within a given region. In the striatum, Dyn is almost exclusively expressed in the direct pathway, and frequently used as a marker for dSPNs (55), however, surprisingly, its endogenous functions remain unclear. Because of its unique expression pattern – the ligand, Dyn, is exclusively expressed in the dSPNs, and the receptor, KOR, is also highly differentially expressed in dSPNs – Dyn/KOR has been hypothesized to dampen dSPN output, functioning similarly to an autoreceptor (56). In the present study, we probed the functional role of Dyn using pharmacology and pathway-specific deletion of Dyn, we found an unexpected role of Dyn/KOR signaling for the regulation of striatal synaptic plasticity and behavior. We find that Dyn/KOR signaling bidirectionally regulates striatal plasticity in a pathway-specific manner, together with dopamine D1R signaling, to fine tune dSPN LTP expression. We further revealed an interesting behavioral phenotype in mice lacking Dyn in dSPNs using two different discrimination reversal learning paradigms, suggesting a crucial role of D1 and Dyn-expressing neurons in cognitive flexibility.
Dyn/KOR signaling regulates LTP in dSPNs
Striatal SPNs receive convergent glutamatergic inputs from various areas of the cerebral cortex and the thalamus, as well as the midbrain dopaminergic inputs (8). Previous studies have shown that Dyn can activate presynaptic KOR to inhibit glutamatergic and DA release through a Gi/o-coupled signaling cascade (22–25, 27). Studies from other brain regions also suggest that Dyn/KOR regulates synaptic plasticity, for example, studies in the hippocampus have shown that Dyn released from dentate granule cells blocks LTP of excitatory inputs (57–60). Similarly, in the VTA, KOR activation blocks LTP of inhibitory inputs onto dopaminergic cells (61, 62).
Here, we show Dyn/KOR signaling bi-directionally regulates LTP expression in dSPNs using both genetic and pharmacological tools. A previous study reported that acutely evoked release of Dyn suppresses LTP in dorsal striatum indirectly via reducing striatal dopamine levels (63). It is certainly true that acute KOR activation alters DA levels in the ex vivo slice. However, we demonstrated that D1-pDyn cKO did not affect dopaminergic basal transmission, therefore, altered DA release is unlikely to be responsible for bi-directional regulation of dSPN LTP. Rather, KOR signaling directly participates in LTP expression in dSPNs. It is possible that dSPN LTP is suppressed by Dyn that is acutely released during STDP protocol or by the ambient dynorphin tone in the striatum. Notably, KORs are expressed in both pre- and postsynaptic sites (64). In addition to its prominent presynaptic actions on glutamatergic and dopaminergic inputs to SPNs (56), postsynaptic KOR activation can also lead to AMPARs endocytosis via protein kinase A (PKA)/calcineurin-dependent mechanisms in interneurons (26). Because LTP expression in dSPNs requires postsynaptic activation of D1R, which subsequently upregulates the adenylate cyclase (AC)/protein kinase A (PKA) cascade pathway (4), and KOR can downregulate AC/PKA signaling through Gi/o-coupled mechanism (65, 66), it is likely that Dyn/KOR signaling directly interacts with D1R signaling to bi-directionally regulate LTP expression in dSPNs through a postsynaptic mechanism.
Interaction between Dyn/KOR and dopaminergic D1R signaling
Previous studies established a model in which different GPCR cascades modulate synaptic plasticity in SPNs (4). In particular, D1R activation is required to enable Hebbian LTP induction in dSPNs (44), and deactivating D1R blocks LTP and shift the plasticity polarity to LTD (12). Our current study adds an additional player - Dyn/KOR signaling - to the model. Gs-coupled D1R signaling and Gi/o-coupled KOR signaling pathways converge and regulate cAMP/PKA levels in opposing directions in the postsynaptic cell, which has been shown to be critical in mediating the direction of plasticity (67). Similar mechanisms involving bi-directional regulation of postsynaptic cAMP/PKA were also demonstrated in iSPNs (68).
Importantly, we functionally characterized that the relative strength of D1R and KOR is subtly different. We show that when D1R is fully activated LTP expression is enhanced regardless of whether Dyn/KOR signaling is activated or blocked, supporting the model that D1R activation is permissive for LTP expression, and high levels of D1R activation can saturate LTP expression machinery, thus occluding the effect of pDyn cKO or KOR blockade. Conversely, Dyn/KOR signaling may exert a more prominent effect under lower levels of D1R activation (i.e., the basal slice condition or with D1R blockade). Under the basal slice conditions without pharmacological D1R manipulations, blocking KOR and pDyn cKO both enhanced LTP expression, and KOR activation abolished LTP expression. Remarkably, blocking KOR or deleting Dyn both were able to rescue LTP expression even when D1R is fully blocked. In psychomotor disorders, such as Parkinson’s disease, dopamine depletion leads to a lack of LTP in dSPNs (4, 7) and a disrupted balance of the striatal network. In addition to dopamine replacement therapy to restore dopamine receptor signaling, KOR antagonism may hold potential to rescue striatal plasticity and alleviate striatal circuit dysfunctions.
Dynorphin from D1 neurons impedes cognitive flexibility during learning
Dyn in the central nervous system has long been recognized as a major mediator of dysphoria, anxiety, and depression during stress and drug withdrawal (69–71). Constitutive knockout of dynorphin reduces anxiety and depression levels (38), and evoked dynorphin release in vivo by optogenetic activation of dSPNs leads to aversion (72). The D1-pDyn cKO mice showed normal anxiety levels, suggesting the reduced anxiety phenotype may be mediated by other brain regions (71).
By contrast, studies examining the role of Dyn in learning and memory have generated conflicting results (73–76). Such discrepancy may result from the widespread expression of Dyn and KOR, and the functional complexity of Dyn/KOR signaling across various brain regions. Therefore, using more targeted cell-type specific manipulations of Dyn can shed light on its role within a specific node of a brain circuit. Here, we identified enhanced cognitive flexibility during reversal learning as the phenotype of D1-pDyn cKO mice.
Though somewhat unexpected, our finding is consistent with previous studies that reported the neurotoxic lesions of the dorsal striatum impaired the performance in various reversal learning tasks (32, 77) as well as a study showing disrupting dorsal striatal dSPN LTP by reducing NMDA receptor signaling decreased learning flexibility (78). In addition, treatment with both D1 (79) and D2 (80) agonists have been shown to impair reversal learning indicating that proper balance between dSPNs and iSPNs is critical. Accordingly, the dSPN-specific suppression of LTP by Dyn/KOR signaling may lead to the net effect of decreasing the excitability of dSPNs relative to iSPNs. It should be noted that using our genetic intersectional strategy, conditional knockout of Dyn also takes places in D1 neurons in the brain outside of the dorsal striatum. Indeed, Dyn expression overlaps with D1 receptor expression in the nucleus accumbens (81). In addition, Dyn and D1 receptors are also expressed in the central amygdala (82), the paraventricular hypothalamus (83), and the insular cortex (84), but much less overlapped in defined cell types. Our manipulation cannot exclude the contribute of these brain regions to the behavioral phenotype observed here.
Reversal learning has also been shown to be impaired in conditions where dopamine signaling is disrupted, such as Parkinson’s disease (85) and addiction (86). Additionally, 6-OHDA lesions of the dorsal medial striatum impairs reversal learning performance (85). It is interesting to examine then how D1 and Dyn/KOR signaling may work synergistically to mediate reversal learning. Phasic dopamine transients in response to reward-prediction error act as the teaching signaling during the reward learning (42, 43), and D1R activation is highly dependent on the phasic dopamine release due to its relatively low ligand-binding affinity (87). Given that the suppression of LTP by Dyn/KOR is masked by high D1R activation and revealed under low D1R activation (Figure 4), we speculate that the dopamine levels are different during the initial learning phase and the reversal phase, and the relatively lower dopamine levels during the reversal phase permitted the enhancement of LTP caused by Dyn deletion and hence enhanced the performance in Dyn cKO mice. However, we acknowledge that the behavioral effects might involve mechanisms other than LTP, and regions beyond dorsal striatum. In summary, our study not only reasserted the role of striatum in reversal learning, but also provided mechanistic insights linking Dyn/KOR and D1 signaling, striatal plasticity, and flexible reversal-learning behavior.
In conclusion, our results imply that Dyn release, putatively in the striatum, impedes cognitive flexibility. This trait seems disadvantageous, however, it may enhance the performance in the case of probabilistic reversal learning task, where there are infrequent but “misleading” punishment (probabilistic error) at the correct stimulus, which resembles the natural changing environment. Excessive flexibility leads to frequent shifting from the just-punished but correct stimulus and may impair the overall performance (88, 89). Therefore, dynorphin may contribute to maintaining the correct response in a probabilistic environment by strategically reducing flexibility. In addition, ethological studies showed a potential trade-off between the ability to rapidly acquire new memories and the ability to retain old ones (90, 91). For instance, many food-caching species that displayed long-lasting spatial memory performed significantly worse than their closely related non-caching species during reversal learning. Therefore, dynorphin may contribute to long-term memory retention by reducing cognitive flexibility.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Eddy Albarran, Dr. Richard Roth for advice on the manuscript, Drs. Yu Liu and Xiaobai Ren for technical assistance, and the members of Ding lab for valuable discussions. This work was funded by NINDS/NIH NS091144 (to J.B.D.), NS107315, DK108797, DK046200 (to D.K.) the GG gift fund (to J.B.D.), the Stanford Bio-X Bowes Graduate Student Fellowship (to R.Y.), and Stanford Neuroscience Institute Interdisciplinary Scholar Awards (to R.R.L.)
Footnotes
CONFLICT OF INTEREST
The authors declare no competing interests.
REFERENCES
- 1.Graybiel AM, Grafton ST. The striatum: where skills and habits meet. Cold Spring Harb Perspect Biol. 2015;7(8):a021691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gremel CM, Costa RM. Orbitofrontal and striatal circuits dynamically encode the shift between goal-directed and habitual actions. Nat Commun. 2013;4:2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8(11):844–58. [DOI] [PubMed] [Google Scholar]
- 4.Surmeier DJ, Graves SM, Shen W. Dopaminergic modulation of striatal networks in health and Parkinson’s disease. Curr Opin Neurobiol. 2014;29:109–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giordano N, Iemolo A, Mancini M, Cacace F, De Risi M, Latagliata EC, et al. Motor learning and metaplasticity in striatal neurons: relevance for Parkinson’s disease. Brain. 2018;141(2):505–20. [DOI] [PubMed] [Google Scholar]
- 6.Girasole AE, Lum MY, Nathaniel D, Bair-Marshall CJ, Guenthner CJ, Luo L, et al. A Subpopulation of Striatal Neurons Mediates Levodopa-Induced Dyskinesia. Neuron. 2018;97(4):787–95 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pisani A, Centonze D, Bernardi G, Calabresi P. Striatal synaptic plasticity: implications for motor learning and Parkinson’s disease. Mov Disord. 2005;20(4):395–402. [DOI] [PubMed] [Google Scholar]
- 8.Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhai S, Shen W, Graves SM, Surmeier DJ. Dopaminergic modulation of striatal function and Parkinson’s disease. J Neural Transm (Vienna). 2019;126(4):411–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calabresi P, Picconi B, Tozzi A, Ghiglieri V, Di Filippo M. Direct and indirect pathways of basal ganglia: a critical reappraisal. Nat Neurosci. 2014;17(8):1022–30. [DOI] [PubMed] [Google Scholar]
- 11.Gerfen CR, Young WS 3rd. Distribution of striatonigral and striatopallidal peptidergic neurons in both patch and matrix compartments: an in situ hybridization histochemistry and fluorescent retrograde tracing study. Brain Res. 1988;460(1):161–7. [DOI] [PubMed] [Google Scholar]
- 12.Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321(5890):848–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Higley MJ, Sabatini BL. Competitive regulation of synaptic Ca2+ influx by D2 dopamine and A2A adenosine receptors. Nat Neurosci. 2010;13(8):958–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kozorovitskiy Y, Peixoto R, Wang W, Saunders A, Sabatini BL. Neuromodulation of excitatory synaptogenesis in striatal development. Elife. 2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen W, Plotkin JL, Francardo V, Ko WK, Xie Z, Li Q, et al. M4 Muscarinic Receptor Signaling Ameliorates Striatal Plasticity Deficits in Models of L-DOPA-Induced Dyskinesia. Neuron. 2015;88(4):762–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan Z, Flores-Hernandez J, Surmeier DJ. Coordinated expression of muscarinic receptor messenger RNAs in striatal medium spiny neurons. Neuroscience. 2001;103(4):1017–24. [DOI] [PubMed] [Google Scholar]
- 17.Bernard V, Normand E, Bloch B. Phenotypical characterization of the rat striatal neurons expressing muscarinic receptor genes. J Neurosci. 1992;12(9):3591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hersch SM, Gutekunst CA, Rees HD, Heilman CJ, Levey AI. Distribution of m1-m4 muscarinic receptor proteins in the rat striatum: light and electron microscopic immunocytochemistry using subtype-specific antibodies. J Neurosci. 1994;14(5 Pt 2):3351–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tejeda HA, Wu J, Kornspun AR, Pignatelli M, Kashtelyan V, Krashes MJ, et al. Pathway- and Cell-Specific Kappa-Opioid Receptor Modulation of Excitation-Inhibition Balance Differentially Gates D1 and D2 Accumbens Neuron Activity. Neuron. 2017;93(1):147–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oude Ophuis RJ, Boender AJ, van Rozen AJ, Adan RA. Cannabinoid, melanocortin and opioid receptor expression on DRD1 and DRD2 subpopulations in rat striatum. Front Neuroanat. 2014;8:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reiner A, Anderson KD. The patterns of neurotransmitter and neuropeptide co-occurrence among striatal projection neurons: conclusions based on recent findings. Brain Res Brain Res Rev. 1990;15(3):251–65. [DOI] [PubMed] [Google Scholar]
- 22.Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A. 1988;85(14):5274–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spanagel R, Herz A, Shippenberg TS. Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway. Proc Natl Acad Sci U S A. 1992;89(6):2046–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ehrich JM, Messinger DI, Knakal CR, Kuhar JR, Schattauer SS, Bruchas MR, et al. Kappa Opioid Receptor-Induced Aversion Requires p38 MAPK Activation in VTA Dopamine Neurons. J Neurosci. 2015;35(37):12917–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mu P, Neumann PA, Panksepp J, Schluter OM, Dong Y. Exposure to cocaine alters dynorphin-mediated regulation of excitatory synaptic transmission in nucleus accumbens neurons. Biol Psychiatry. 2011;69(3):228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coleman BC, Manz KM, Grueter BA. Kappa opioid receptor modulation of excitatory drive onto nucleus accumbens fast-spiking interneurons. Neuropsychopharmacology. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Atwood BK, Kupferschmidt DA, Lovinger DM. Opioids induce dissociable forms of long-term depression of excitatory inputs to the dorsal striatum. Nat Neurosci. 2014;17(4):540–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gong S, Doughty M, Harbaugh CR, Cummins A, Hatten ME, Heintz N, et al. Targeting Cre recombinase to specific neuron populations with bacterial artificial chromosome constructs. J Neurosci. 2007;27(37):9817–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee EC, Yu D, Martinez de Velasco J, Tessarollo L, Swing DA, Court DL, et al. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics. 2001;73(1):56–65. [DOI] [PubMed] [Google Scholar]
- 30.Li Y, van den Pol AN. Differential target-dependent actions of coexpressed inhibitory dynorphin and excitatory hypocretin/orexin neuropeptides. J Neurosci. 2006;26(50):13037–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu YW, Kim JI, Tawfik VL, Lalchandani RR, Scherrer G, Ding JB. Input- and cell-type-specific endocannabinoid-dependent LTD in the striatum. Cell Rep. 2015;10(1):75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Castane A, Theobald DE, Robbins TW. Selective lesions of the dorsomedial striatum impair serial spatial reversal learning in rats. Behav Brain Res. 2010;210(1):74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ineichen C, Sigrist H, Spinelli S, Lesch KP, Sautter E, Seifritz E, et al. Establishing a probabilistic reversal learning test in mice: evidence for the processes mediating reward-stay and punishment-shift behaviour and for their modulation by serotonin. Neuropharmacology. 2012;63(6):1012–21. [DOI] [PubMed] [Google Scholar]
- 34.Richardson NR, Roberts DC. Progressive ratio schedules in drug self-administration studies in rats: a method to evaluate reinforcing efficacy. J Neurosci Methods. 1996;66(1):1–11. [DOI] [PubMed] [Google Scholar]
- 35.Johnson C, Wilbrecht L. Juvenile mice show greater flexibility in multiple choice reversal learning than adults. Dev Cogn Neurosci. 2011;1(4):540–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo SX, Timbang L, Kim JI, Shang Y, Sandoval K, Tang AA, et al. TGF-beta Signaling in Dopaminergic Neurons Regulates Dendritic Growth, Excitatory-Inhibitory Synaptic Balance, and Reversal Learning. Cell Rep. 2016;17(12):3233–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Healy DJ, Meador-Woodruff JH. Prodynorphin-derived peptide expression in primate cortex and striatum. Neuropeptides. 1994;27(5):277–84. [DOI] [PubMed] [Google Scholar]
- 38.Schwarzer C 30 years of dynorphins--new insights on their functions in neuropsychiatric diseases. Pharmacol Ther. 2009;123(3):353–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schlosser B, Kudernatsch MB, Sutor B, ten Bruggencate G. Delta, mu and kappa opioid receptor agonists inhibit dopamine overflow in rat neostriatal slices. Neurosci Lett. 1995;191(1–2):126–30. [DOI] [PubMed] [Google Scholar]
- 40.Hjelmstad GO, Fields HL. Kappa opioid receptor inhibition of glutamatergic transmission in the nucleus accumbens shell. J Neurophysiol. 2001;85(3):1153–8. [DOI] [PubMed] [Google Scholar]
- 41.Hjelmstad GO, Fields HL. Kappa opioid receptor activation in the nucleus accumbens inhibits glutamate and GABA release through different mechanisms. J Neurophysiol. 2003;89(5):2389–95. [DOI] [PubMed] [Google Scholar]
- 42.Watabe-Uchida M, Eshel N, Uchida N. Neural Circuitry of Reward Prediction Error. Annu Rev Neurosci. 2017;40:373–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science. 1997;275(5306):1593–9. [DOI] [PubMed] [Google Scholar]
- 44.Centonze D, Grande C, Saulle E, Martin AB, Gubellini P, Pavon N, et al. Distinct roles of D1 and D5 dopamine receptors in motor activity and striatal synaptic plasticity. J Neurosci. 2003;23(24):8506–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yin HH, Ostlund SB, Balleine BW. Reward-guided learning beyond dopamine in the nucleus accumbens: the integrative functions of cortico-basal ganglia networks. Eur J Neurosci. 2008;28(8):1437–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Balleine BW, O’Doherty JP. Human and rodent homologies in action control: corticostriatal determinants of goal-directed and habitual action. Neuropsychopharmacology. 2010;35(1):48–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Albarran E, Raissi A, Jaidar O, Shatz CJ, Ding JB. Enhancing motor learning by increasing the stability of newly formed dendritic spines in the motor cortex. Neuron. 2021;109(20):3298–311 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clem RL, Celikel T, Barth AL. Ongoing in vivo experience triggers synaptic metaplasticity in the neocortex. Science. 2008;319(5859):101–4. [DOI] [PubMed] [Google Scholar]
- 49.Rioult-Pedotti MS, Friedman D, Donoghue JP. Learning-induced LTP in neocortex. Science. 2000;290(5491):533–6. [DOI] [PubMed] [Google Scholar]
- 50.Izquierdo A, Brigman JL, Radke AK, Rudebeck PH, Holmes A. The neural basis of reversal learning: An updated perspective. Neuroscience. 2017;345:12–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carlezon WA Jr., Beguin C, DiNieri JA, Baumann MH, Richards MR, Todtenkopf MS, et al. Depressive-like effects of the kappa-opioid receptor agonist salvinorin A on behavior and neurochemistry in rats. J Pharmacol Exp Ther. 2006;316(1):440–7. [DOI] [PubMed] [Google Scholar]
- 52.Negus SS. Effects of the kappa opioid agonist U50,488 and the kappa opioid antagonist nor-binaltorphimine on choice between cocaine and food in rhesus monkeys. Psychopharmacology (Berl). 2004;176(2):204–13. [DOI] [PubMed] [Google Scholar]
- 53.Schindler AG, Li S, Chavkin C. Behavioral stress may increase the rewarding valence of cocaine-associated cues through a dynorphin/kappa-opioid receptor-mediated mechanism without affecting associative learning or memory retrieval mechanisms. Neuropsychopharmacology. 2010;35(9):1932–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Todtenkopf MS, Marcus JF, Portoghese PS, Carlezon WA Jr. Effects of kappa-opioid receptor ligands on intracranial self-stimulation in rats. Psychopharmacology (Berl). 2004;172(4):463–70. [DOI] [PubMed] [Google Scholar]
- 55.Edwards NJ, Tejeda HA, Pignatelli M, Zhang S, McDevitt RA, Wu J, et al. Circuit specificity in the inhibitory architecture of the VTA regulates cocaine-induced behavior. Nat Neurosci. 2017;20(3):438–48. [DOI] [PubMed] [Google Scholar]
- 56.Nestler EJ. Historical review: Molecular and cellular mechanisms of opiate and cocaine addiction. Trends Pharmacol Sci. 2004;25(4):210–8. [DOI] [PubMed] [Google Scholar]
- 57.Drake CT, Terman GW, Simmons ML, Milner TA, Kunkel DD, Schwartzkroin PA, et al. Dynorphin opioids present in dentate granule cells may function as retrograde inhibitory neurotransmitters. J Neurosci. 1994;14(6):3736–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Terman GW, Wagner JJ, Chavkin C. Kappa opioids inhibit induction of long-term potentiation in the dentate gyrus of the guinea pig hippocampus. J Neurosci. 1994;14(8):4740–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wagner JJ, Terman GW, Chavkin C. Endogenous dynorphins inhibit excitatory neurotransmission and block LTP induction in the hippocampus. Nature. 1993;363(6428):451–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weisskopf MG, Zalutsky RA, Nicoll RA. The opioid peptide dynorphin mediates heterosynaptic depression of hippocampal mossy fibre synapses and modulates long-term potentiation. Nature. 1993;365(6442):188. [DOI] [PubMed] [Google Scholar]
- 61.Polter AM, Bishop RA, Briand LA, Graziane NM, Pierce RC, Kauer JA. Poststress block of kappa opioid receptors rescues long-term potentiation of inhibitory synapses and prevents reinstatement of cocaine seeking. Biol Psychiatry. 2014;76(10):785–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Graziane NM, Polter AM, Briand LA, Pierce RC, Kauer JA. Kappa opioid receptors regulate stress-induced cocaine seeking and synaptic plasticity. Neuron. 2013;77(5):942–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hawes SL, Salinas AG, Lovinger DM, Blackwell KT. Long-term plasticity of corticostriatal synapses is modulated by pathway-specific co-release of opioids through kappa-opioid receptors. J Physiol. 2017;595(16):5637–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Svingos AL, Chavkin C, Colago EE, Pickel VM. Major coexpression of kappa-opioid receptors and the dopamine transporter in nucleus accumbens axonal profiles. Synapse. 2001;42(3):185–92. [DOI] [PubMed] [Google Scholar]
- 65.Gentleman S, Parenti M, Neff NH, Pert CB. Inhibition of dopamine-activated adenylate cyclase and dopamine binding by opiate receptors in rat striatum. Cell Mol Neurobiol. 1983;3(1):17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bruchas MR, Chavkin C. Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology (Berl). 2010;210(2):137–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Augustin SM, Beeler JA, McGehee DS, Zhuang X. Cyclic AMP and afferent activity govern bidirectional synaptic plasticity in striatopallidal neurons. J Neurosci. 2014;34(19):6692–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lerner TN, Kreitzer AC. RGS4 Is Required for Dopaminergic Control of Striatal LTD and Susceptibility to Parkinsonian Motor Deficits. Neuron. 2012;73(2):347–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Van’t Veer A, Carlezon WA Jr. Role of kappa-opioid receptors in stress and anxiety-related behavior. Psychopharmacology (Berl). 2013;229(3):435–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C. The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J Neurosci. 2008;28(2):407–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Crowley NA, Bloodgood DW, Hardaway JA, Kendra AM, McCall JG, Al-Hasani R, et al. Dynorphin Controls the Gain of an Amygdalar Anxiety Circuit. Cell Reports. 2016;14(12):2774–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Soares-Cunha C, de Vasconcelos NAP, Coimbra B, Domingues AV, Silva JM, Loureiro-Campos E, et al. Nucleus accumbens medium spiny neurons subtypes signal both reward and aversion. Mol Psychiatry. 2020;25(12):3241–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Carey AN, Lyons AM, Shay CF, Dunton O, McLaughlin JP. Endogenous kappa opioid activation mediates stress-induced deficits in learning and memory. J Neurosci. 2009;29(13):4293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hiramatsu M, Hoshino T. Involvement of kappa-opioid receptors and sigma receptors in memory function demonstrated using an antisense strategy. Brain Res. 2004;1030(2):247–55. [DOI] [PubMed] [Google Scholar]
- 75.Kuzmin A, Madjid N, Terenius L, Ogren SO, Bakalkin G. Big dynorphin, a prodynorphin-derived peptide produces NMDA receptor-mediated effects on memory, anxiolytic-like and locomotor behavior in mice. Neuropsychopharmacology. 2006;31(9):1928–37. [DOI] [PubMed] [Google Scholar]
- 76.Ukai M, Itoh J, Kobayashi T, Shinkai N, Kameyama T. Effects of the kappa-opioid dynorphin A(1–13) on learning and memory in mice. Behav Brain Res. 1997;83(1–2):169–72. [DOI] [PubMed] [Google Scholar]
- 77.Braun S, Hauber W. The dorsomedial striatum mediates flexible choice behavior in spatial tasks. Behav Brain Res. 2011;220(2):288–93. [DOI] [PubMed] [Google Scholar]
- 78.Darvas M, Palmiter RD. Specific contributions of N-methyl-D-aspartate receptors in the dorsal striatum to cognitive flexibility. Neuroscience. 2015;284:934–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Izquierdo A, Wiedholz LM, Millstein RA, Yang RJ, Bussey TJ, Saksida LM, et al. Genetic and dopaminergic modulation of reversal learning in a touchscreen-based operant procedure for mice. Behav Brain Res. 2006;171(2):181–8. [DOI] [PubMed] [Google Scholar]
- 80.Boulougouris V, Castane A, Robbins TW. Dopamine D2/D3 receptor agonist quinpirole impairs spatial reversal learning in rats: investigation of D3 receptor involvement in persistent behavior. Psychopharmacology (Berl). 2009;202(4):611–20. [DOI] [PubMed] [Google Scholar]
- 81.Al-Hasani R, McCall JG, Shin G, Gomez AM, Schmitz GP, Bernardi JM, et al. Distinct Subpopulations of Nucleus Accumbens Dynorphin Neurons Drive Aversion and Reward. Neuron. 2015;87(5):1063–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim J, Zhang X, Muralidhar S, LeBlanc SA, Tonegawa S. Basolateral to Central Amygdala Neural Circuits for Appetitive Behaviors. Neuron. 2017;93(6):1464–79 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li MM, Madara JC, Steger JS, Krashes MJ, Balthasar N, Campbell JN, et al. The Paraventricular Hypothalamus Regulates Satiety and Prevents Obesity via Two Genetically Distinct Circuits. Neuron. 2019;102(3):653–67 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pina MM, Pati D, Hwa LS, Wu SY, Mahoney AA, Omenyi CG, et al. The kappa opioid receptor modulates GABA neuron excitability and synaptic transmission in midbrainprojections from the insular cortex. Neuropharmacology. 2020;165:107831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Grospe GM, Baker PM, Ragozzino ME. Cognitive Flexibility Deficits Following 6-OHDA Lesions of the Rat Dorsomedial Striatum. Neuroscience. 2018;374:80–90. [DOI] [PubMed] [Google Scholar]
- 86.Izquierdo A, Jentsch JD. Reversal learning as a measure of impulsive and compulsive behavior in addictions. Psychopharmacology (Berl). 2012;219(2):607–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Richfield EK, Penney JB, Young AB. Anatomical and affinity state comparisons between dopamine D1 and D2 receptors in the rat central nervous system. Neuroscience. 1989;30(3):767–77. [DOI] [PubMed] [Google Scholar]
- 88.Evers EA, Cools R, Clark L, van der Veen FM, Jolles J, Sahakian BJ, et al. Serotonergic modulation of prefrontal cortex during negative feedback in probabilistic reversal learning. Neuropsychopharmacology. 2005;30(6):1138–47. [DOI] [PubMed] [Google Scholar]
- 89.Murphy FC, Michael A, Robbins TW, Sahakian BJ. Neuropsychological impairment in patients with major depressive disorder: the effects of feedback on task performance. Psychol Med. 2003;33(3):455–67. [DOI] [PubMed] [Google Scholar]
- 90.Lea SEG, Chow PKY, Leaver LA, McLaren IPL. Behavioral flexibility: A review, a model, and some exploratory tests. Learn Behav. 2020;48(1):173–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tello-Ramos MC, Branch CL, Kozlovsky DY, Pitera AM, Pravosudov VV. Spatial memory and cognitive flexibility trade-offs: to be or not to be flexible, that is the question. Anim Behav. 2019;147:129–36. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.