

Summary
Extracellular vesicles (EVs) are nanovesicles released by all eukaryotic cells. This work reports the first nanoscale fluorescent visualization of tumor-originating vesicles bearing an angiogenic microRNA (miR)-126 cargo. In a validated experimental model of lethal murine vascular neoplasm, tumor-originating EV delivered its miR-126 cargo to tumor-associated macrophages (TAMs). Such delivery resulted in an angiogenic (LYVE+) change of state in TAM that supported tumor formation. Study of the trafficking of tumor-originating fluorescently tagged EV revealed colocalization with TAM demonstrating uptake by these cells. Ex vivo treatment of macrophages with tumor-derived EVs led to gain of tumorigenicity in these isolated cells. Single-cell RNA sequencing of macrophages revealed that EV-borne miR-126 characterized the angiogenic change of state. Unique gene expression signatures of specific macrophage clusters responsive to miR-126-enriched tumor-derived EVs were revealed. Topical tissue nanotransfection (TNT) delivery of an oligonucleotide comprising an anti-miR against miR-126 resulted in significant knockdown of miR-126 in the tumor tissue. miR-126 knockdown resulted in complete involution of the tumor and improved survival rate of tumor-affected mice. This work identifies a novel tumorigenic mechanism that relies on tumorigenic state change of TAM caused by tumor-originating EV-borne angiomiR. This disease process can be effectively targeted by topical TNT of superficial tumors.
Keywords: extracellular vesicles, macrophage, single-cell RNA sequencing, angiomiRs, miR-126, tumor, hemangioma, EOMA
Graphical abstract
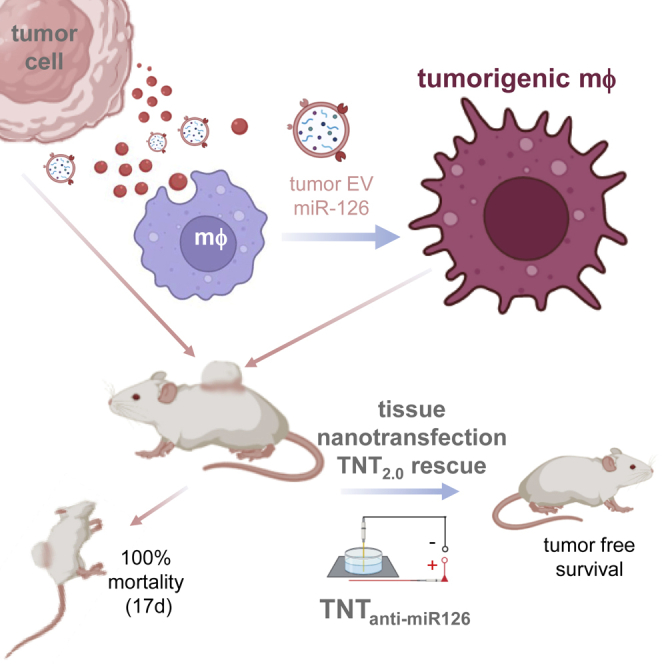
Tumor cells release extracellular vesicles (EVs) containing miR-126 angiomiR cargo, which, when delivered to tumor-associated macrophages (TAMs), generate a tumorigenic TAM subset as identified by single-cell RNA sequencing (RNA-seq). Nanoscopic imaging reveals miR-126 in single EVs. Tissue nanotransfection intervention to inhibit miR-126 (TNTanti-miR-126) prevents tumor-related death and ensures tumor-free survival.
Introduction
A compound multi-cellular microenvironment contributes to tumor development.1 Tumor-associated macrophages (TAMs) play a major role in this process.2,3,4 TAMs promote angiogenesis, tumor growth, and metastasis.5,6,7 Recent works have shown that macrophages (Mɸ) are diverse and phenotypically plastic cells. Specifically, TAMs contribute to blood vessel sprouting and remodeling. TEK receptor tyrosine kinase (TIE2), mostly identified as endothelial marker, is expressed by classically activated pro-inflammatory M1 macrophages with angiogenic function.8 TAMs are capable of forming non-endothelial blood vessels in response to the tumor microenvironment by a process known as vascular mimicry.9 Additionally, that monocytes/macrophages may convert to endothelial-like cells was proposed many years ago based on studies performing co-culture with endothelial cells.10,11 Human monocyte-derived multipotential cells differentiate along the endothelial lineage, thus providing an autologous transplantable cell source for therapeutic neovasculogenesis.12 Today, human peripheral blood monocytes serve as a robust alternative to generating endothelial cells required for vascular graft production.13 Active crosstalk between macrophages and endothelial cells precedes and enables vascularization.14,15 When human tissue-engineered blood vessel networks were implanted into athymic mice, macrophages formed vessel-like structures themselves.16 These observations point toward a heightened significance of TAMs in the development of endothelial cell tumors.
Endothelial cell tumors represent a pure form of unregulated angiogenesis17 and are the most common soft tissue tumor of infancy, affecting 3%–10% of live births.18 Most tumors are harmless; however, some are associated with significant morbidity and may be life threatening. Judah Folkman established the validated model of experimental murine hemangioendotheliomas (HEs) to study endothelial cell tumors.18 Subcutaneous inoculation of tumorigenic endothelial cells results in cell proliferation and development of blood-cell-filled vascular tissue characteristic of HEs, including development of the Kasabach-Merritt phenomenon (KMP).19,20 All mice die within 2 weeks.21 In a prospective longitudinal study on children with hemangiomas, we reported elevated urinary levels of the angiomiR microRNA-126 (miR-126).22 Although our previous works have consistently established a central role of TAMs in the development of murine HEs and related mortality,19 mechanisms underlying the potential angiogenic role of TAMs remain unclear. This work demonstrates for the first time how extracellular vesicular genetic cargoes originating from tumor cells are capable of directing the angiogenic state of TAMs in a way that is critical for tumor development. While it is known that tumor-derived EVs alter macrophages into TAMs,2,3,4 this work presents the first evidence demonstrating that such TAMs themselves can acquire tumorigenicity. That finding is leveraged to successfully achieve regression of otherwise fatal tumor using tissue nanotransfection (TNT) technology.
Results
EOMA miR-126: Therapeutic target
Specifically expressed in the vasculature, miR-126 is enriched in endothelial cells and responsible for vascular formation by regulating vascular integrity and angiogenic signaling.23 In contrast to this physiological function of miR-126, recent reports recognize a key role of this miR in a wide range of vascular diseases. The impact of miR-126 on disease states is context dependent. Attenuated abundance of miR-126 has been evident under disease conditions such as type 2 diabetes,24,25 chronic kidney disease,26 coronary artery disease,27 and sepsis,28 and elevated levels of this miR have proved to be beneficial.27,28,29,30 This pattern of finding has also been evident in several studies related to cancer. Low miR-126 has been associated with cholangiocarcinoma.31 Elevated miR-126 inhibited epithelial-mesenchymal transition and reduced cell invasion of gastric cancer.32 Upregulation of miR-126 inhibited the proliferation of skin squamous cell carcinoma.32 Exosomal miR-126 improved non-small cell lung cancer outcomes.33 In oral squamous cell carcinoma, miR-126 inhibited cell migration and invasion.34 In children with hemangioma in the proliferative phase, urinary levels of miR-126 are elevated. When the proliferative phase passes, urinary miR-126 levels are lower and comparable with those of healthy age-matched controls.22 In the murine model of HE, the miR-126 levels in tumor-forming mouse HE endothelial (EOMA) cells were three orders of magnitude higher compared with those in normal murine aortic endothelial (MAE) cells (Figure S1A). This tumor has 100% mortality by day 17 (d17) of EOMA inoculation.35 This is caused by development of the KMP characterized by red blood cell and platelet sequestration within HE tumors, anemia, and high-output heart failure.36 This is also evident in humans with HE.36 Findings of this work causatively link vascular miR-126 to the development of HE, identifying it as a direct therapeutic target for regression of the tumor.
To determine the significance of miR-126 in HE tumor regression, oligonucleotide comprising an anti-miR against miRNA-126 (α-miR-126) was delivered to the tumors directly using TNT as reported by us (Figures 1A and S1B).6,37,38 TNT successfully delivered α-miR-126 (TNTα-miR-126) and significantly lowered miR-126 abundance in HE tumor tissue (Figures 1B–1D and S1C). Laser capture microdissection (LCM)-based spatial study of tumor tissue established that TNT-based delivery of α-miR-126 decreased miR-126 abundance in both tumor perimeter and core compared with tumor-associated skin (both epidermis and dermis) (Figures S1D–S1F). In adults, α-miR-126 is known not to exhibit off-target effects,39 especially as it relates to functional blood flow (Figures S1G and S1H). miR-126 knockdown resulted in complete involution of HE (Figures 1E–1G). Furthermore, decreased tumor progression and improved survival rate were noted in the TNTα-miR-126 group (Figures 1H and S1I–S1K). These favorable outcomes were supported by consistent improvements in blood flow velocity in the TNTα-miR-126 group (Figures 1I and 1J). Histological studies demonstrated tumor regression. Lymphatic vessel endothelial hyaluronan receptor 1 or LYVE1+ is an HE tumor marker.40 LYVE1+ staining was attenuated in mice treated with TNTα-miR-126 (Figures 1K–1M). Thus, data establish lowering of miR-126 as an effective therapeutic strategy for the management of HE.
Figure 1.

TNTα-miR-126 inhibits murine HE tumor growth in vivo
(A) Schematic diagram of in vivo tissue nanotransfection technology-mediated delivery of oligonucleotides based on the voltage gradient. (B) RNA in situ detection of miR-126 in the HE tumor sections (d10) treated with scrambled oligonucleotides (TNTsham) or antagomir-126 (TNTα-miR-126). (C) Intensity calculation of RNA in situ images determined the significant reduction of miR-126 level in the TNTα-miR-126 group compared with TNTsham. ∗p < 0.05 (n = 4), Mann Whitney test. (D) Level of miR-126 from the biopsy samples isolated from non-tumor (NT), HE tumor (T), and skin from HE tumor region treated with antagomir-126 (T + TNTα-126). ∗p < 0.05 (n = 4), Mann Whitney test. (E) Representative images of 129P3/J mice treated with TNTsham and TNTα-miR-126 at d12 and d29 showing decrease in tumor growth in TNTα-miR-126-treated mice. (F) Tumor volume of day-matched animals for the treatment of TNTsham and TNTα-miR-126. ∗p < 0.05 (n = 5), Mann Whitney test. (G) TNT-based delivery (twice a week for 4 weeks) of antagomir-126 (red) inhibits HE progression. Tumor volume was quantified using calipers (length ×·width ×·height). In TNTsham mice, tumor volume sharply rose, causing death on d12–d17. (H) Kaplan-Meier survival curve of TNTsham and TNTα-miR-126 groups monitored for 90 days shows tumor-free survival for TNTα-miR-126 group. (I) Ultrasound imaging of animals from each group (n = 5) was performed on a Vevo-2100 system using high-frequency linear array transducers operating between 8 and 17 MHz. (J) Tumor blood flow analyzed using Vevo-2100 system shows significant difference at d10 after TNTα-miR-126 treatment compared with TNTsham groups. (K) H&E images show complete regression of tumor with TNTα-miR-126 treatment on d29 compared with d12 TNTα-miR-126 group. (L) Day-matched immunohistochemical analysis showing decrease in ratio of CD31+ (red) and LYVE1+ (green) cells in TNTα-miR-126-treated HE tumor at d12 compared with TNTsham group-treated HE tumor, and (M) its quantification. ∗p < 0.05, n = 5, Mann Whitney test. Data presented as mean ± SEM.
Tumor EV modifies macrophage state
All eukaryotic cells secrete a range of membrane-bound vesicles of different sizes and content.41,42 Extracellular vesicles (EVs) include biological nanovesicles that actively contribute to intercellular communications by several mechanisms, including facilitating the transfer of macromolecules, including miRs.43 In the tumor microenvironment, EVs play a key role in enabling the cell-cell communication network.21 In the current study, it was found that EVs from tumor cells (EVEOMA) contained elevated miR-126 levels compared with EVs from normal endothelial cells (EVMAE) (Figures S2A–S2D). Elevated levels of miR-126 were also detected in EVs isolated from the urine of children with hemangiomas compared with healthy age-matched children (Figures 2A and S2E). A similar pattern was observed in HE mice urine, where miR-126 abundance increased with the tumor progression (Figures 2B, 2C, and S2F–S2H). TNT α-miR-126 treatment lowered the abundance of miR-126 in EVs of HE tumors (Figures 2D and 2E). Direct stochastic optical reconstruction microscopy (dSTORM), super-resolution microscopy, enabled the nanoscale visualization of immunolabeled EVs carrying the miR-126 cargo (Figures 2F and 2G).44,45,46
Figure 2.

Characterization of EOMA-derived EVs and their preferential uptake by TAMs
(A) miR-126 expression in the extracellular vesicles (EVs) isolated from urine samples from age-matched healthy and HE patients (6 months old, n = 6), ∗p < 0.05, Student’s t test. (B) Abundance of EVs from mouse urine isolated from different days after tumor progression, n = 6, ∗p < 0.05, one-way ANOVA. (C) miR-126 abundance in EVs isolated from murine urine samples in different days of tumor development. ∗p < 0.05 n = 6, one-way ANOVA. (D) Quantification of EVs, estimated from tumor tissue (d10) after treatment with scrambled oligonucleotides (TNTsham) or antagomir-126 (TNTα-miR-126). ∗p < 0.05 (n = 4), Mann Whitney test. (E) Abundance of miR-126 from the isolated EV samples collected from tissue after TNTα-miR-126 or TNTsham treatment. ∗p < 0.05, n = 4, Mann Whitney test. (F) Western blot analysis of exosome markers Alix, HSP90, CD9, CD63, CD81, and exosome-negative marker GRP94. (G) Super-resolution microscopic image shows Cy3-labeled miR-126 mimic cargo present inside Atto 488-labeled exosome isolated from EOMA cell culture media. Red represents Alexa 647-tagged CD81. Eighty-two percent fraction of EVEOMA containing miR-126 cargo. Cy3 scrambled control oligonucleotide for miRNA and Alexa Fluor 647-labeled IgG antibody-stained EOMA EVs were used as negative controls. (H) Injection of CDH5-CD81-GFP-transduced EOMA cells into mice formed HE tumors in which EVs secreted by EOMA cells expressed GFP. Immunohistochemical analysis showing co-expression of F4/80 (Mφ marker, red), LYVE1 (EOMA cell marker, blue) with GFP expressed by EVEOMA (green). Nucleus is counter-stained with ToPro3. Representative confocal image is taken at 10× magnification (left) and its expansion (right) representing different regions of tumor, (i) MφEOMA-deficient tumor core region (tumor core), (ii) intermediary LYVE1+ TAM-populated region (TAMLYVE1), and (iii) MφEOMA-deficient tumor perimeter region (tumor perimeter). (I) Panel represents magnification (63×) of these three regions and extent of colocalization between F4/80 and LYVE1. Single-channel images of F4/80/LYVE1 colocalization is provided in Figure S4C. (J) Laser capture microdissection (LCM) images from different regions of tumor tissue sections before and after cut. (K) Abundance of miR-126 in different regions of HE tumor captured using LCM. ∗p < 0.05, n = 6, one-way ANOVA. (L) Heatmap representing the expression of M1 and M2 macrophage markers in different HE tumor regions captured through LCM. Data presented as mean ± SEM.
To study trafficking of EVs of EOMA origin in vivo, tumor cells were transfected with plasmid wherein the expression of EV-specific CD81-GFP reporter was driven by endothelial CDH5 promoter (Figures S3A–S3D). Immunocytochemistry studies of the HE tumor revealed colocalization of F4/80 (Mɸ marker) and EVEOMA. A subset of these Mɸ were observed to be LYVE1+, a characteristic marker of EOMA (Figures 2H, 2I, and S4A–S4E). For study purposes, the HE tumor was divided into three regions: (1) MφEOMA-deficient tumor core region (tumor core), (2) intermediary LYVE1+ TAM-populated region (TAMLYVE1), and (3) MφEOMA-deficient tumor perimeter region (tumor perimeter) (Figures 2H, 2I, and S4A–S4E). The tumor core and TAMLYVE1 regions were also enriched in GFP, indicative of the presence of CD81+ EOMA cell-derived EVs. LCM-based studies demonstrated increased abundance of miR-126 in these two regions (tumor core and TAMLYVE1; Figures 2J and 2K). Further characterization of these TAMLYVE1 tissue elements using hallmark markers of classically or alternative activation state of Mɸ revealed a mixed activation state with increase in classical activation markers inducible nitric oxide synthase (iNOS) and interleukin (IL) 6 as well as alternative activation marker IL10 (Figure 2L). Interactions between HE-derived EVs and TAMs were further analyzed using GW4869, which inhibits EV release by blocking the inward budding of the multivesicular bodies.47 GW4869 pre-treated EOMA cells were deficient in tumor-forming ability. This was manifested as attenuated tumor growth, smaller tumor volume, and increase in the survival rate of the mice (Figures 3A–3C). HE formation requires the subcutaneous inoculation of a critical number (5 × 106) of EOMA cells.35,48 Such GW4869 treatment led to the appearance of alternative active (M2) macrophage markers (Figures 2L and S5A). As expected, inoculation of half that number of cells failed to produce a tumor. However, if this sub-critical number of EOMA cells were mixed with equal number of Mɸ (MɸEOMA EV) pre-treated with EOMA-derived EVs, tumor formation was evident. In contrast, control experiments with an equal number of Mɸ pre-treated with EVMAE (MɸMAE EV) was ineffective in tumor formation (Figures 3D–3F). These findings establish the critical significance of TAMs in HE formation. In the tumor microenvironment, such TAMs are exposed to EOMA-derived EVs, making them ready to support tumor formation. Tumor cells release heterogeneous populations of EVs containing proteins, lipids, and nucleic acids. EVs are known to transfer functional RNA in the tumor microenvironment in a way that supports tumor development.49,50 Intravital imaging has shown that uptake of tumor-derived EVs by cells can enhance their tumorigenic properties.51 In several forms of cancer, miR-126 shuttled by exosomes is biologically active in the target cells, contributing to disease progression.21,52,53 In this work, conversion of Mɸ to LYVE+ tumorigenic TAMs by EOMA-originating EVs may be viewed as an angiogenic state change that supports tumorigenicity. Previously, such an angiogenic state has been assigned to TIE2-expressing TAMs.54,55 Identification of the LYVE+ tumorigenic TAMs represents an important addition to that repertoire. Importantly, this work reveals for the first time that such angiogenic switch of TAMs can be achieved via tumor-originating EVs. That EVs are capable of changing cell state was evident in our previous work reporting on keratinocyte-originating EV-dependent conversion of wound-site Mɸ to fibroblast-like cells.56 In humans, the Kaposiform HE (KHE) shows the presence of CD68+ Mɸ expressing LYVE1+ and CDH5 supporting the translational relevance of the murine LYVE+ TAMs reported in that work (Figures S5B and S5C). A similar activation pattern was noted in Mɸ associated with HE tumors isolated from mice (Figures S5D and S5E). Ex vivo treatment of Mɸ with EVEOMA led to gain of EOMA cell-like characteristics in isolated Mɸ (Figures 3G–3I). Such gain was significantly attenuated in Mɸ exposed to EVEOMA derived from miR-126-inhibited EOMA cells. These data establish that EVEOMA carrying miR-126 cargo is crucial for the change of Mɸ activation state in a way that shares LYVE+ EOMA characteristics (Figures 3J–3M). miR-126 knockdown in EOMA using α-miR-126 attenuated angiogenic properties and depleted the miR in EVs (Figures S6A–S6M).
Figure 3.

EOMA-derived EVs containing miR-126 are responsible for macrophage conversion to LYVE+ tumorigenic TAMs cells
(A) Representative images of mice shows 6- to 8-week-old female 129P3/J mice received subcutaneous injection of EOMA cells pre-treated with GW4869 (2.5 μg/mL × 48 h) or vehicle control. (B) Tumor volume was decreased on d10 compared with control group and tumor progression was slow, as control group mice died on day17. ∗p < 0.05, n = 11, Student’s t test. (C) Kaplan-Meier survival curves show GW4869 pre-treatment of cells significantly increased life span of HE-affected mice. Volume quantified using calipers (length ×·width ×·height). (D) Mice (129P3/J) injected with a combination of EOMA cells and MφEOMA EV. Sham cells were used from pair-matched mφ from the same isolation as for EVEOMA-treated cells except that Mφ were treated with EVMAE. Here, (i) threshold dose of EOMA cells caused tumor, (ii) sub-threshold of EOMA cells failed to cause tumor, (iii) sub-threshold dose caused tumor in presence of MφEOMA EV, (iv) sub-threshold dose failed to cause tumor in presence of sham cells. (E) Flipped skin shows subcutaneous tumor. (F) Tumor volumes were measured until d10 after injecting the above-mentioned cell quantities. ∗p < 0.05, n = 3 and 4), one-way ANOVA. (G) Schematic diagram demonstrating the study design of macrophage isolation and treatment with EVEOMA after delivering the antagomir-126 or scrambled oligonucleotides in EOMA cells by nano-electroporation (NEP). (H) Representative images of isolated wound macrophages on d7 using CD11b magnetic sorting was stained with LYVE1, F4/80, and CDH5, eNOS after incubation with EVEOMA (107/mL) for 7 days. (I) Percentage of colocalization of fluorescence intensity of different endothelial markers. Images were taken on three individual repeats. Results are expressed as mean ± SD, ∗p < 0.05, n = 6, two-way ANOVA. (J and K) d7 wound macrophages treated with EVEOMA+ NEP α-miR126 showed less intensity of endothelial markers compared with macrophages treated with EVEOMA+ NEP ci. Quantification of Pearson colocalization coefficient (K) shows significant decrease in endothelial marker intensity in treated group compared with control group. Results are expressed as mean ± SD, ∗p < 0.05, n = 6, two-way ANOVA. (L and M) Macrophages isolated from HE tumor after delivering the antagomir-126 by TNT showed a similar pattern in expressing endothelial and tumor markers to NEP, and colocalization quantification (M) confirms it. Results are expressed as mean ± SD, ∗p < 0.05, n = 6, two-way ANOVA. Data presented as mean ± SEM (B,F) or mean ± SD (I,K,M).
Characterization of macrophage subsets responsive to tumor EVs
Single-cell RNA-seq (scRNA-seq) was conducted to study the angiogenic Mɸ subsets in response to EV-borne miR-126 (Figure 4A). To address the effects of miR-126-independent factors of the tested EVs, additional controls were used wherein macrophages were treated with miR-126 null EVs derived from endothelial cells (Figures S6N). A total of 23,250 cells that passed the quality control parameters were subjected to downstream analyses (see section “materials and methods”; Figures S7A and S7B). Clustering of single cells from the three samples combined resulted in identification of 14 distinct clusters (Figure 4B) with unique markers for each, as shown in Figure S7C. The analysis uncovered two transcriptionally distinct Mɸ populations out of a total of 14 clusters identified. Unique gene expression signatures of these clusters formed in response to exposure to miR-126-enriched EVEOMA were revealed. We identified subpopulation CD11b+ Mɸ (cluster 4) enriched in Serpine1, collagen, type1, alpha 1 (Col1a1), and Col1a257,58 as well as for transgelin (Tagln), actin alpha 2 (Acta2), and S100 calcium-binding protein A4 (S100a4) in wound-associated macrophages. This subset belonged to highly plastic and functionally diverse steady-state condition as reported by us and others.56,58,59 Cluster 7 was identified to be specifically populated in response to EVEOMA. Of the 1,177 cells in this cluster, 1,013 were derived from the EVEOMA-treated sample. In response to EVEOMA, cluster 4 was also responsive but in a contrasting manner compared with cluster 7. In cluster 4, the number of cells decreased (1,823→390) in response to EVEOMA. The number of cells in each cluster is listed in Table S1. Cluster 7 was high in transcripts, as shown in Figure S7D. All positive cluster markers are provided in Table S2. Our findings reveal a heterogeneity of angiogenic transcriptional responses in resident Mɸ not previously appreciated by traditional bulk mRNA-seq approaches (Figure 4A).60 Differential expression analysis using Wilcoxon rank-sum test resulted in the identification of a total of 2,074 genes significantly different in MɸEOMA EV (adjusted p value <0.05; Table S3). Among them, 393 genes were found to be upregulated (logFC cutoff >0.2) and 481 genes were found to be downregulated (logFC < −0.2). The top 10 upregulated and 10 downregulated genes are depicted as a heatmap in Figure S8A. Gene set enrichment analysis (GSEA) resulted in the identification of six upregulated gene sets and seven downregulated gene sets (Figure S8B). A comparison of cluster 7 versus 4 identified 246 upregulated and 187 downregulated genes using cutoff logFC ±0.2 and adjusted p value <0.05 (Figure 4C; Table S4). The top 10 upregulated and 10 downregulated genes are depicted as a heatmap in Figure S8C. GSEA results for cluster 7 versus 4 led to the identification of a SNF5 gene set that includes genes upregulated in mouse embryonic fibroblast (MEF) cells with knockout of the tumor suppressor SNF5.61 The downregulated gene sets are similar to the gene sets downregulated when comparing all MɸEOMA EV versus Mɸ cells, as shown in Figure S8D. GSEA results are listed in Table S5.
Figure 4.

Single-cell RNA sequencing data analysis of LYVE+ tumorigenic TAMs cells identified the cellular trajectories responsible for tumor growth
(A) Schematic diagram of sample collection and single-cell analysis workflow. (B) Uniform Manifold Approximation and Projection (UMAP) of the filtered data (23,250 cells) clustered into 14 clusters. Each cell is represented as a dot. (C) Schematic representation of the differential expression analysis performed between all EVEOMA treated versus all untreated Mφ and between cluster 7 versus cluster 4. (D) Trajectory inference of cells from clusters 4 and 7 colored by clusters (left), pseudotime (middle), or the originating sample (right). (E) Violin plots representing expression level of Ccl2 in Mφ, MφmiR126 null EV, and in MφEOMA EV cells (upper). Bar plots of PCR results for Ccl2 (below). (F) Network of upregulated genes found in both comparisons Mφ EOMA EV versus Mφ or in cluster 7 versus cluster 4 cells. Nodes with red colors represent genes found to be upregulated in both comparisons MφEOMA EV versus mφ and cluster 7 cells versus cluster 4 cells. Nodes with blue color represent genes found to be upregulated in MφEOMA EV versus Mφ cells, but not in cluster 7 cells versus cluster 4 cells. Nodes with golden color represent genes found to be upregulated in cluster 7 cells versus cluster 4 cells, but not in MφEOMA EV versus Mφ cells. Data presented as mean ± SEM.
Monocle3 trajectory inference algorithm-based analyses of the scRNA-seq data identified a path in which cells from cluster 4 pass along a defined trajectory to reach cluster 7 gene expression profile states, as shown in Figure 4D. The middle part of the trajectory includes cells from cluster 7 of the Mɸ sample and cluster 4 of the MɸEOMA EV sample. Cluster 4 cells present in the MɸEOMA EV sample were distributed differently along the trajectory than cluster 4 cells present in the Mɸ sample. These cells were scattered along the path of the trajectory in between cluster 4 and cluster 7. This pseudotime trajectory-powered analysis indicated that the highly plastic cluster 4 upon exposure to EVEOMA changed its state and gave rise to a new cluster (cluster 7). This new cluster was enriched in genes such as allograft inflammatory factor 1 (Aif1) and chemokine (C-C motif) ligand 5 (Ccl5), known to cause progression of hemangioma.62,63,64,65 Interestingly, in a previous report, Aif1 expression was detected in 89% of hemangioma specimens, with a specific location in endothelial cells.62 Characterization of Aif1hiCcl5hi cells using Gene Ontology (GO) analyses indicated the increased expression of Ccl2 along with other genes involved in the vasculature development process (Figures 4E, 4F, and S9; Table S6). Our previous work has directly implicated Ccl2 in HE development by recruitment of Mɸ to the tumor site.35 Inhibition of inducible Ccl2 expression improved HE tumor outcomes.66 Using Ccl2 knockout mice, we demonstrated that Ccl2 is required for HE proliferation and may promote the growth of these lesions by bolstering angiogenic behavior of endothelial cells. Anti-CCL2 therapy in animals with intact immune systems was effective in HE management.19 Suppression of inducible Ccl2 in the HE by dietary factors also inhibited tumor growth, assigning high value to this tumor Mɸ-recruiting protein with respect to its role in HE development.48,67 Other chemokines, such as Ccl3 and Ccl4, were also increased in the cluster 7, suggestive of the presence of inflammatory stimulus conferred by EVEOMA.68 Flow sorting of AIF1hi MɸEOMA EV (MɸAIF1+) cells also demonstrated the temporal gain of Ccl2 and Ccl4 compared with macrophages treated with miR-126 null EVs (MɸmiR-126 null EV) (Figure S10A). A previous report has demonstrated that endothelial cell-originating EVs enhance monocyte activation by shifting the monocyte/macrophage balance from anti-inflammatory activation state to a pro-inflammatory activation state.54 Presence of such pro-inflammatory cellular clusters contributes to tumor progression.6,69 This was validated by significant increase in tumorigenic colony formation of these flow-sorted MɸAIF1+ compared with MɸmiR-126 null EV (Figures S10B–S10E). To characterize the Mɸ state change, we studied the residual macrophage properties of the LYVE+ macrophage. No significant departure from Mɸ signature was observed in MɸEOMA EV cells (65 genes out of total 88 detected genes remained changed, logFC ±0.2) (Figures S11A and S11B). Mixed macrophage phenotype with increased levels of tumor-promoting IL10 was evident (Figure S11C). Taken together, these scRNA-seq studies identify specific Mɸ subsets responsive to EVEOMA. The retention of macrophage characteristics in TAMLYVE1 argue in favor of a state change as opposed to a fate change.
Discussion
This work establishes a direct tumorigenic role of tumor-derived EVs. This role is enacted by a specific miR cargo of the EV. EVs, originating from the tumor, are loaded with miR-126, which is known to be specifically enriched in vascular tissue. EV-borne miR-126 is taken up by TAMs, resulting in an angiogenic state change of such macrophages. Macrophages, affected by such change of state, are capable of enabling tumor formation under conditions of sub-critical tumor cell inoculation that are otherwise known to be unable to form tumor. Topical tissue nanotransfection of the tumor in vivo was effective in successfully delivering α-miR-126, thus significantly lowering miR-126 abundance in tumor tissue and causing complete involution of the tumor. This was followed by improved survival of tumor-affected mice. TNT-dependent knockdown on tumor miR-126 depleted this miR in the EV and intercepted the angiogenic state change of macrophages necessary for tumor formation.
Materials and methods
Cell culture
EOMA cells were maintained under the same conditions as previously described.70 In brief, cells were grown in Dulbecco’s high-glucose (4.5 g/L) modified Eagle’s medium (DMEM) (Gibco, Life Technologies) enriched with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin, in a humidified incubator at 37°C and 5% CO2.
For collection of wound macrophages, 8-mm round sterile polyvinyl alcohol (PVA) sponges were implanted subcutaneously on the backs of 8-week-old 129P/3 mice. On day7, Mɸ (CD11b+) were isolated from sponges by using magnetic CD11b beads (Miltenyi Biotec, CA) as described previously.71 The isolated cells were cultured in RPMI-1640 medium (Gibco, Life Technologies) supplemented with 10% Hi-FBS and 1% antibiotic-antimycotic for single passage. The cells were cultured in a standard cell culture incubator at 5% CO2. This is a routine technique in the laboratory and has been set up under conditions that do not change cell phenotype.71,72,73
Animals
All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the Indiana University, Indianapolis, Indiana. Mice were maintained under standard conditions at 22°C ± 2°C with access to food and water ad libitum. 129P/3 mice (6–8 weeks old, female) were purchased from Jackson Laboratories. HE tumors were produced as previously described.35 Briefly, 5 million live EOMA cells (per mouse) were injected subcutaneously into dorsum of the 129P/3 mice. Trypan blue stain is used to quantify the live cells to ensure that each mouse gets the same number of live cells. Tumor volume was determined by using calipers to measure length × width × height of each tumor, as previously described.35 Tumors were harvested on day 10 after EOMA cell injection, and tissues were collected frozen in OCT (optimal cutting temperature) compound for histological analyses and liquid nitrogen for RNA isolation.
Human subjects
Human samples were collected from the Institutional Review Board (IRB)-approved study (ClinicalTrials.gov identifier NCT01598116). Declaration of Helsinki protocols was followed, and patients gave their written informed consent. Urine and tumor samples were collected from children with hemangioma at the Hemangioma and Vascular Malformation Clinic and the Primary Care Clinic at Nationwide Children’s Hospital, Columbus, Ohio using inclusion and exclusion criteria as reported previously.22 Urine samples from age-matched and healthy volunteers were collected and analyzed as controls.
Isolation of EVs from cell culture medium
EOMA cell conditioned medium from the culture plates was centrifuged at 500 × g for 5 min to remove cell debris. The supernatant was again centrifuged at 3,400 × g for 15 min and 10,000 × g for 45 min to remove cell organelles. The supernatant from the differential centrifugation was used for ultracentrifugation (Beckman Coulter Optima Max-XP Ultracentrifuge, Rotor TLA 120.2) at 75,000 rpm for 2 h. The pellet was resuspended in phosphate-buffered saline (PBS) and pelleted again by ultracentrifugation. These EVs were characterized for size and concentration by Nanosight S300, Malvern Panalytical.
Isolation of EVs from mouse tumor tissue
After being collected from the mouse, HE tumor tissue was ground in liquid nitrogen and dissolved in PBS. After a brief vortex and mini-centrifugation, supernatant was collected and centrifuged at 5,000 × g for 15 min followed by 20,000 × g for 45 min. The supernatant was used for ultracentrifugation at 75,000 rpm for 2 h. The pellet was resuspended in PBS and centrifuged again at 75,000 × g for 2 h. These EVs were characterized for size and concentration by Nanosight S300, Malvern Panalytical, and used for transmission electron microscopy (TEM) imaging.
Nanoparticle tracking analysis
Extracellular vesicles were characterized for size and concentration by using Nanosight S300 (Malvern Panalytical). After dilution in PBS 100 times, each EV sample was loaded into a microfluidics chamber by using a syringe pump and captured at 20× magnification. Five representative videos of 30 s were documented for each sample and particle size, and concentrations were averaged.
Oxford nanoimaging
To visualize EVs under super-resolution microscopy, we followed manufacturer’s protocol. EVs were isolated from Cy3-labeled miR126 (catalog [cat.] no. # CS-MmiR-SN0099-SN-01-20, GeneCopoeia) transfected EOMA cells conditioned media, incubated with NHS ester 488 for 30 min at room temperature (RT), and dialyzed overnight in carbonate buffer. Next day, EVs were incubated with Alexa Fluor 647-conjugated CD81 antibody at 4°C. Next day, the surface of each lane of the capture chip was prepared by using surface solutions. A mixture of EV samples and capture supplement was added to the lanes and incubated for 50 min at RT by shielding from light. After washing three times with the wash solution, samples were fixed with the fixation buffer for 10 min at RT. Before imaging under super-resolution microscopy, BCubed imaging buffer was added to the lanes. Images were obtained at 100× magnification. Cy3 scrambled control oligonucleotide for miRNA (cat. no. # CS-CmiR-SN0001-SN-01-20, GeneCopoeia) and Alexa Fluor 647-labeled immunoglobulin (Ig) G (antibody conjugation kit, cat. no. # A10475, Invitrogen) were used as negative controls.
Transfection
EOMA cells were seeded (1 × 105 cells/well) in a 12-well plate. After achieving the ∼70% confluency, transfection was performed by delivering miRCURY LNA miRNA hsa-miR126-3p inhibitor (QIAGEN, cat. No. YI04109148) or miRIDIAN miRNA hairpin inhibitor negative control (cat. no. IH-001005-01-20) using DharmaFECT transfection reagent (GE Dharmacon) and OptiMEM serum-free medium (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA). Fresh medium was added after 4 h of treatment. EOMA cells were collected after 72 h of transfection to look for miR-126 abundance. EVs were isolated from the conditioned medium as described above and used to treat the wound Mɸ, and 107 EVs were used to treat 0.5 million Mɸ on d1, d3, and d7 after isolating from the PVA sponges. These Mɸ were used for immunocytochemistry (ICC) staining. For generation of miR-126 null EVs, MAE cells were transfected with miRCURY LNA miRNA hsa-miR126-3p inhibitor using DharmaFECT transfection reagent. EVs were isolated from conditioned medium by using differential ultracentrifugation and analyzed for the miR-126 levels (Figure S6N), and 107 EVs were used to treat 0.5 million Mɸ until d7.
Transduction and stable cell-line creation
EOMA cells (1 × 105) were seeded in a 10-cm-diameter dish 24 h before transduction in DMEM media with 10% FBS and 1% penicillin/streptomycin. Cells were treated with 10 μL of virus (MOI-10) having Cdh5-CD81-copGFP plasmid vector (Applied Biological Materials, Richmond, BC, Canada). After 3 h, fresh medium was added. Cells were observed under confocal microscope (63× magnification) to check for successful transduction. After 72 h, cells were subjected to different concentrations (1 μg/mL, 2.5 μg/mL, 5 μg/mL, and 10 μg/mL) of puromycin to optimize it for stable cell-line creation. EOMA cells were used as control for standardization of puromycin concentration. After standardization, 2.5 μg/mL of puromycin was used for stable cell-line creation. These transduced EOMA cells (5 million per mouse) were injected subcutaneously into 129P3/J mice for HE tumor formation. Tumors were harvested on d10 in OCT compound for immunohistochemistry (IHC) staining.
Nanochannel electroporation
In vitro nanochannel electroporation (NEP) was performed as previously described.38 Briefly, 1× 105 EOMA cells were seeded in Corning 12-mm Transwell membrane cell culture inserts in high-glucose (4.5 g/L glucose) DMEM medium supplemented with 10% FBS and 1% penicillin/streptomycin. After 24 h, medium was replaced with sterile PBS and inserts were placed on a gold-coated electrode in direct contact with the plasmid solution. A counter-electrode was then immersed in the PBS of the insert, and a square wave pulse (275 V, 35-ms pulse duration, 10 pulses) was applied across the electrodes using Bio-Rad Gene Pulser Xcell power supply. Fresh medium was added after the delivery. Cells were delivered with either LNA miRNA hsa-miR126-3p inhibitor (50 μM) or control inhibitor (50 μM). After 72 h, EVs were isolated and characterized as described above, and 107 EVs were used to treat 0.5 million Mɸ on d1, d3, and d7 after isolating from the PVA sponges. These Mɸ were used for ICC staining and RT-PCR for miR-126 abundance.
TNT
In vivo TNT was performed as previously described.74,75,76,77 Briefly, after the first sign of tumor (d3 after injecting the EOMA cells), either 50 μM LNA miRNA hsa-miR126-3p inhibitor (TNTα-miR-126) or control inhibitor (TNTsham) was delivered into the tumor by placing the TNT chip directly over the tumor. A gold-coated electrode (the cathode) was immersed in the plasmid solution and a 24G needle (the anode) was inserted intradermally, juxtaposed to the TNT platform surface (Figure S1A). To nanoporate the cell membranes, electrical stimulation (250 V, duration 10 ms, 10 pulses) was applied across the electrodes. Three doses were given on d3, d7, and d10. Laser speckle imaging (PeriCam PSI High Resolution, PeriMed) was performed to analyze cutaneous perfusion. Tumors were collected on d10 in either OCT for IHC or in liquid nitrogen for RT-PCR or used to isolate TAMs.
In vitro angiogenesis assay
Four-well plates were coated with 100 μL of Matrigel (Cultrex Basement Membrane Extract reduced growth factor; R&D Systems, Minneapolis, MN) and allowed to solidify for 30 min at 37°C. EOMA cells transfected with antagomir-126 and control inhibitor were seeded (50,000 cells/well) on top of the Matrigel and maintained in a cell culture incubator. After 12 h, cells were stained with 3 μM calcein-AM (Invitrogen) for 15 min at 37°C in a cell culture incubator. Tube formation was observed, and images were captured at 10 × magnification under laser-scanning confocal system (CARL ZEISS confocal microscope LSM 888, Zeiss, Germany). Tube length was quantified by using ZEN software (Zen blue) as reported.78
TAM isolation
After being disrupted mechanically, tumors were digested with 40 μL of DNase 1 (15 mg/mL, Roche, Germany) along with 40 μL of Liberase DL (Roche, Germany) and 80 μL of Liberase DL (Roche, Germany) for 1 h at 37°C. Red blood cells were lysed with RBC lysis buffer (Invitrogen, CA). TAMs (CD11b+) were isolated by using magnetic CD11b beads (Miltenyi Biotec, CA). The isolated cells were cultured in RPMI-1640 medium (Gibco, Life Technologies) supplemented with 10% Hi-FBS and 1% antibiotic-antimycotic. The cells were cultured in a standard cell culture incubator at 5% CO2 and used for ICC and RT-PCR for miR-126 abundance.
Western blot
EVs were lysed with RIPA buffer containing a proteinase and phosphatase inhibitors (Cell Signaling) and the protein concentration determined using a standard BCA protein assay. Samples (25 μg of protein/lane) were separated using 4%–12% sodium dodecyl sulfate/polyacrylamide gel electrophoresis. The membranes were blocked with 5% milk in Tris-HCL Tween buffer for 1 h at RT and incubated overnight at 4°C with Alix (Novus Biologicals, JM 85-31, 1:1,000), HSP-90 (Abcam, ab59459, 1:1,000), CD9 (Novus Biologicals, SA35-08, 1:1,000), CD63 (Novus Biologicals, SY21-02, 1:1,000), and CD81 (Invitrogen, MA5-32333, 1:1,000). Next day, membranes were incubated with secondary antibodies, horseradish peroxidase-conjugated donkey anti-rabbit-IgG (1:2,000, cat. no. NA934V, lot no. 9583369; Amersham Biosciences, Piscataway, NJ) and anti-mouse-IgG (1:2,000, cat. no. NA931V, lot no. 6652622; Amersham Biosciences) for 1 h at RT.79 Blots were developed by using enhanced chemiluminescence plus western blotting detection reagents (Amersham Biosciences) according to the manufacturer’s instructions.
RNA extraction and quantitative RT-PCR
Total RNA from cells or exosomes was extracted using an miRVana miRNA isolation kit according to the manufacturer’s protocol (Ambion, Life Technologies).80 To measure the miRNA abundance, Taqman MicroRNA Assays hsa-miR-126 (assay ID 0022280), snoRNA202 (assay ID 001232), TaqMan miRNA reverse transcription kit (Applied Biosystems), and Taqman universal PCR Master Mix (Applied Biosystems) were used. The quantification of gene expression was performed by using the comparative Ct (cycle threshold) method.
IHC and microscopy
IHC was performed on cryosections from mouse tumor samples using specific antibody as described previously.81 In brief, tissue specimens were frozen in optimum cutting temperature compound (Miles). Frozen tissues were sectioned at 10-μm thickness and fixed for 5 min in acetone at 4°C. Then sections were blocked with 10% normal goat serum and incubated with specific antibodies Lyve1 (Abcam, ab14917; 1:200), F4/80 (Bio-Rad, MCA497R; 1:200), and GFP (Abcam, ab13970; 1:500) overnight at 4°C. Next day, sections were washed and incubated with the Alexa Fluor 488 goat anti-rabbit antibody (1:200 dilution; Invitrogen), Alexa Fluor 568 goat anti-rat antibody (1:200 dilution; Invitrogen), and Alexa Fluor 405 goat anti-rabbit antibody (1:200 dilution; Invitrogen) for 1 h at RT. After incubation, the sections were incubated with ToPro3 for 5 min and mounted in without DAPI (Vector Laboratories). Images were obtained at 10× and 63× magnification by using a laser-scanning confocal system (CARL ZEISS confocal microscope LSM 888, Zeiss, Germany). Fluorescent intensity quantitation and colocalization was performed by using ZEN software (Zen blue). Pearson correlation coefficient was determined for each cell as a measure of colocalization and was expressed as mean ± standard deviation (SD).
ICC and imaging
Macrophages (1 × 105 cells/well) were seeded on a coverslip placed in 12-well plates for 24 h. Cells were washed with PBS, then fixed with 4% paraformaldehyde solution for 20 min at RT. After fixation, cells were incubated in a permeabilized buffer (0.1% Triton X-) for 10 min at RT. After washing with PBS, cells were blocked with 10% normal goat serum for 1 h at RT and stained with the Lyve1 (Abcam, ab14917; 1:200), F4/80 (Bio-Rad, MCA497R; 1:200), VE-Cadherin (Abcam, ab33168; 1:200), and eNOS (Invitrogen, PA3-031A, 1:200) and incubated overnight at 4°C. Next day, cells were washed and incubated with the same secondary antibody used for tissue section for 1 h at RT. After incubation, the cells were counterstained with DAPI (aqueous mount; Vector Laboratories). The images were captured by confocal microscopy (CARL ZEISS confocal microscope LSM 888, Zeiss, Germany) at 63× magnification. Quantification of the fluorescence intensity of an image and colocalization was analyzed using ZEN software (Zen blue). Pearson correlation coefficient was determined for each cell as a measure of colocalization.
LCM and quantitative RT-PCR
LCM was performed using the laser microdissection system from PALM Technologies (Zeiss, Germany).82 The mouse tissues sections (10 μm) on membrane slides were subjected to a quick histological staining. Briefly, sections were fixed in cold acetone for 2 min. After washing twice with DEPC (Diethyl pyrocarbonate)-H2O, sections were incubated with primary (F4/80 and GFP) and secondary antibodies in the dark for 5 min. Subsequently, sections were washed twice with PBS and dehydrated with increasing ethanol concentrations (75%, 95%, and 100% for 2 min each). Tissue sections were cut under 20× ocular lens and samples were collected in 25 μL of direct cell lysis extraction buffer (Invitrogen) and stored at −80°C for further processing.
Ultrasound
A high-frequency, high-resolution ultrasound imaging system (Vevo 2100; Visual-Sonics, Toronto, Canada) with an MS 550D (22–55 MHz) linear array transducer was used as described previously.21 Mice were anesthetized using a mixture of 1.5% isoflurane inhalant anesthesia with 95% oxygen, and animals were placed in the supine position on a moveable heated stage maintained at 37°C. Hair was removed from tumors using a depilatory cream. Ultrasound scan transmission gel was applied to the tumor surface and the probe was positioned on a fixed stand perpendicular to the stage. The image resolution was maintained at 100 μm laterally and 50 μm axially. B-mode mapping was performed until clearly visualizing the tumor anatomical location. Each targeted tumor was visualized in two-dimensional (2D) imaging for the visualization of the tumor. Fine position adjustment was done for the highest signal intensity with continuous adjustment of contrast dynamic range and gain until the minimal noise appeared in the background. For tumor volume measurement, different locations on the tumor were set up with the transducer and B-mode recordings were made with the ultrasound beam perpendicular to the targeted tumor. Several recordings (cine loops), each up to 5 s, of B mode were saved. Color Doppler flowmetry of tumor vasculature and tumor feeder vessel flow were also recorded using color Doppler flow imaging mode in 2D mode.
The tumor volume was measured during postprocessing. VevoLab software (Visual-Sonics) was used to compile each 2D image slice with other acquired slices and trace the area and depth of the wound, resulting in volumetric data. From the real-time B-mode observation, the framewise tracing of tumor edge borders was performed.
Single-cell sequencing and data analysis
Macrophage treatment
Mɸ (1 × 105 cells/well) were either treated or not treated with EVEOMA (107/well). Treatment was given in three doses on d1, d3, and d7. EOMA cells were used as controls.
Raw data processing and quality control for cell exclusion
Seurat package (v.3.1.1) in R (v.3.5.1)83,84 was used to preprocess and visualize the data. The initial dataset, containing 26,617 cells from three samples (untreated cells, Mɸ, 8,723 cells; cells treated with EVEOMA containing miR-126, MɸEOMA EV, 7,700 cells; and EOMA cells, 10,194 cells), was integrated using canonical correlational analysis (CCA) as described previously.85 Gene expression values were log normalized and scaled to 10,000 transcripts per cell. The top 2,000 variable genes were identified using the FindVariableFeatures function in Seurat using the vst method. The algorithm first fits a line to the relationship of the variance log and the mean log using polynomial regression. Then, it standardizes the feature values using the observed mean and expected variance, which was given by the fitted line. Feature variance is then calculated on the standardized values. Then principal-component analysis (PCA) was performed. For quality control, cells with more than 10% mitochondrial RNA, less than 100 or more than 7,500 detected transcripts, and cells with total number of counts more than 30,000 were excluded. Finally, genes that were detected in fewer than three cells were excluded. Integration of samples was performed again after excluding low-quality cells, and the top 20 principal components were used for clustering the cells, resulting in 14 clusters. The Uniform Manifold Approximation and Projection (UMAP) algorithm was used for dimensionality reduction and visualization.
Identification of clusters markers and differential expression analysis
After excluding low-quality cells, cluster markers were identified by comparing each cluster with the rest of the cells. Additionally, all cells from the treated sample were compared with all untreated cells. Furthermore, differential expression analysis was performed to compare cluster 7 cells with cluster 4 cells. All the differential expression analyses were performed using Wilcoxon rank-sum test by Seurat while excluding genes that were not expressed (gene level <0) in 20% of cells in either group during each comparison. GSEA was performed for the upregulated and downregulated genes resulting from comparing all treated cells versus untreated cells and for the genes resulting from comparing cluster 7 cells versus cluster 4 cells using adjusted p value <0.05.86 GSEA was performed against the C6 collection (oncogenic signature gene sets), which includes 189 gene sets that represent signatures of cellular pathways, which are often dis-regulated in cancer. Those gene sets were a collection of mainly microarray data from NCBI GEO or from internal unpublished profiling experiments that involved perturbation of known cancer genes. In addition, a small number of oncogenic signatures were curated from scientific publications.87
Vasculature development analysis
The vasculature development gene set (GO: 0001944) was retrieved from the MGI database including 787 genes.88 The differentially expressed genes with adjusted p value <0.05 and logFC >0.2, which were found to be upregulated while comparing treated cells versus untreated cells or cluster 7 versus cluster 4, were annotated using the vasculature development gene set. To identify whether treated cells have significant alteration in macrophage activation function, a macrophage activation gene set was retrieved from MGI database (GO: 0042116), including 101 genes.88 We next identified the percentage of genes in the macrophage activation gene set to be differentially expressed among all the identified genes in the single-cell sequencing.
Pseudotime analysis of clusters 4 and 7
Subsetting cluster 4 and 7 cells from the main Seurat object was performed and the monocle3 package in R was used to infer the trajectory.89,90 PCA was chosen for preprocessing. Then, UMAP was performed while passing cosine as the distance matrix, maximum components to be 2, a minimum distance of 0.6, and the number of neighbors to use during KNN (k-nearest neighbors) graph of 15L to the UMAP function.
Soft agar colony-forming assay
Wound-site macrophages (Mɸ) were treated with EVEOMA for different time points (d4, d7, d10). Next, based on the cluster markers identified for specific clusters in a single-cell RNA seq experiment (Figure S7C), Aif1 was used to sort cluster 7 out from the total cell population. The colony-forming potential of such AIF1+ sorted cells (representing cluster 7) were then quantified using CytoSelect 96-well cell transformation assay kit (Cell Biolabs, San Diego, CA, USA).91,92 Mɸs treated with miR-126 null EVs were used as controls (Figure S6N). Briefly, a base agar layer of 0.6% agar, RPMI medium, and 10% FBS was added into each well and covered with a cell agar layer of the same composition containing 5,000 cells/well. After 7 days, the agar was solubilized, and the mixture was incubated with CyQuant working solution. Fluorescence was measured at 485/520 nm using a Biotek Synergy 96-well microplate reader.
Statistical analysis
GraphPad Prism (GraphPad Software) 9 was used for statistical analyses. The ΔΔCt value was used for statistical analysis of all RT-qPCR data. Statistical analysis between multiple groups was performed using one-way analysis of variance with the Bonferroni multiple comparison test. Statistical analysis between two groups was performed using unpaired Student’s two-sided t tests or Mann Whitney test, and p < 0.05 was considered statistically significant. Data for independent experiments were presented as mean ± SEM unless otherwise stated. Pearson correlation coefficient was determined for each cell as a measure of colocalization and was expressed as mean ± SD.
Acknowledgments
This study was primarily supported in part by NIH grants GM095657 to G.G.; DK114718 to S.R.; DK129592 to S.G.; DK128845 and DK125835 to C.K.S.; and Department of Defense grants W81XWH-21-1-0097, W81XWH-21-1-0033, and W81XWH-20-1-251 to C.K.S. and W81XWH-22-1-0146 to K.S. We thank Xiaoping Gu for technical assistance and Dr. Sheng Liu for the alignment of scRNA-seq data.
Author contributions
Conceptualization, C.K.S., G.G., and S.R.; methodology, C.K.S., G.G., K.S., and S.R.; investigation and validation, P.R.G., K.S., A.B., A.S., Y.R., A.D., and M.K.; formal analysis, K.S., A.S.A., S.K., and P.R.G.; writing – original draft, C.K.S., G.G., S.R., K.S., S.G., and P.R.G.; writing – review & editing, C.K.S., G.G., S.R., K.S., and S.G.; supervision, C.K.S, G.G., and S.R.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.11.003.
Contributor Information
Gayle M. Gordillo, Email: gmgordil@iu.edu.
Sashwati Roy, Email: roysa@iu.edu.
Supplemental information
References
- 1.Wang H., Yung M.M.H., Ngan H.Y.S., Chan K.K.L., Chan D.W. The impact of the tumor microenvironment on macrophage polarization in cancer metastatic progression. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms22126560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Soncin I., Sheng J., Chen Q., Foo S., Duan K., Lum J., Poidinger M., Zolezzi F., Karjalainen K., Ruedl C. The tumour microenvironment creates a niche for the self-renewal of tumour-promoting macrophages in colon adenoma. Nat. Commun. 2018;9:582. doi: 10.1038/s41467-018-02834-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Higuchi H., Yamakawa N., Imadome K.I., Yahata T., Kotaki R., Ogata J., Kakizaki M., Fujita K., Lu J., Yokoyama K., et al. Role of exosomes as a proinflammatory mediator in the development of EBV-associated lymphoma. Blood. 2018;131:2552–2567. doi: 10.1182/blood-2017-07-794529. [DOI] [PubMed] [Google Scholar]
- 4.Kudo K., Miki Y., Carreras J., Nakayama S., Nakamoto Y., Ito M., Nagashima E., Yamamoto K., Higuchi H., Morita S.Y., et al. Secreted phospholipase A(2) modifies extracellular vesicles and accelerates B cell lymphoma. Cell Metab. 2022;34:615–633.e8. doi: 10.1016/j.cmet.2022.02.011. [DOI] [PubMed] [Google Scholar]
- 5.Qian B.Z., Pollard J.W. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu L.Q., Du W.L., Cai M.H., Yao J.Y., Zhao Y.Y., Mou X.Z. The roles of tumor-associated macrophages in tumor angiogenesis and metastasis. Cell Immunol. 2020;353:104119. doi: 10.1016/j.cellimm.2020.104119. [DOI] [PubMed] [Google Scholar]
- 7.Yamaguchi T., Fushida S., Yamamoto Y., Tsukada T., Kinoshita J., Oyama K., Miyashita T., Tajima H., Ninomiya I., Munesue S., et al. Tumor-associated macrophages of the M2 phenotype contribute to progression in gastric cancer with peritoneal dissemination. Gastric Cancer. 2016;19:1052–1065. doi: 10.1007/s10120-015-0579-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.García S., Krausz S., Ambarus C.A., Fernández B.M., Hartkamp L.M., van Es I.E., Hamann J., Baeten D.L., Tak P.P., Reedquist K.A. Tie2 signaling cooperates with TNF to promote the pro-inflammatory activation of human macrophages independently of macrophage functional phenotype. PLoS One. 2014;9:e82088. doi: 10.1371/journal.pone.0082088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei X., Chen Y., Jiang X., Peng M., Liu Y., Mo Y., Ren D., Hua Y., Yu B., Zhou Y., et al. Mechanisms of vasculogenic mimicry in hypoxic tumor microenvironments. Mol. Cancer. 2021;20:7. doi: 10.1186/s12943-020-01288-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bohgaki M., Kitaguchi H. Conversion of cultured monocytes/macrophages into endothelial-like cells through direct contact with endothelial cells. Int. J. Hematol. 2007;86:42–48. doi: 10.1532/ijh97.06217. [DOI] [PubMed] [Google Scholar]
- 11.Tigges U., Hyer E.G., Scharf J., Stallcup W.B. FGF2-dependent neovascularization of subcutaneous Matrigel plugs is initiated by bone marrow-derived pericytes and macrophages. Development. 2008;135:523–532. doi: 10.1242/dev.002071. [DOI] [PubMed] [Google Scholar]
- 12.Kuwana M., Okazaki Y., Kodama H., Satoh T., Kawakami Y., Ikeda Y. Endothelial differentiation potential of human monocyte-derived multipotential cells. Stem Cells. 2006;24:2733–2743. doi: 10.1634/stemcells.2006-0026. [DOI] [PubMed] [Google Scholar]
- 13.Smith R.J., Jr., Andreadis S.T. Generating monocyte-derived endothelial-like cells for vascular regeneration. Methods Mol. Biol. 2022;2375:13–19. doi: 10.1007/978-1-0716-1708-3_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Y., Guo Z., Chen W., Wang X., Cao M., Han X., Zhang K., Teng B., Cao J., Wu W., et al. M2 macrophage-derived exosomes promote angiogenesis and growth of pancreatic ductal adenocarcinoma by targeting E2F2. Mol. Ther. 2021;29:1226–1238. doi: 10.1016/j.ymthe.2020.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gil Z., Billan S. Crosstalk between macrophages and endothelial cells in the tumor microenvironment. Mol. Ther. 2021;29:895–896. doi: 10.1016/j.ymthe.2021.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Graney P.L., Ben-Shaul S., Landau S., Bajpai A., Singh B., Eager J., Cohen A., Levenberg S., Spiller K.L. Macrophages of diverse phenotypes drive vascularization of engineered tissues. Sci. Adv. 2020;6:eaay6391. doi: 10.1126/sciadv.aay6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 18.O'Reilly M.S., Brem H., Folkman J. Treatment of murine hemangioendotheliomas with the angiogenesis inhibitor AGM-1470. J. Pediatr. Surg. 1995;30:325–329. doi: 10.1016/0022-3468. discussion 329–330. [DOI] [PubMed] [Google Scholar]
- 19.Gordillo G.M., Onat D., Stockinger M., Roy S., Atalay M., Beck F.M., Sen C.K. A key angiogenic role of monocyte chemoattractant protein-1 in hemangioendothelioma proliferation. Am. J. Physiol. Cell Physiol. 2004;287:C866–C873. doi: 10.1152/ajpcell.00238.2003. [DOI] [PubMed] [Google Scholar]
- 20.Dubois-Stringfellow N., Kolpack-Martindale L., Bautch V.L., Azizkhan R.G. Mice with hemangiomas induced by transgenic endothelial cells. A model for the Kasabach-Merritt syndrome. Am. J. Pathol. 1994;144:796–806. [PMC free article] [PubMed] [Google Scholar]
- 21.Gordillo G.M., Biswas A., Singh K., Sen A., Guda P.R., Miller C., Pan X., Khanna S., Cadenas E., Sen C.K. Mitochondria as target for tumor management of hemangioendothelioma. Antioxid. Redox Signal. 2021;34:137–153. doi: 10.1089/ars.2020.8059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biswas A., Pan X., Meyer M., Khanna S., Roy S., Pearson G., Kirschner R., Witman P., Faith E.F., Sen C.K., Gordillo G.M. Urinary excretion of MicroRNA-126 is a biomarker for hemangioma proliferation. Plast. Reconstr. Surg. 2017;139:1277e–1284e. doi: 10.1097/prs.0000000000003349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fish J.E., Santoro M.M., Morton S.U., Yu S., Yeh R.F., Wythe J.D., Ivey K.N., Bruneau B.G., Stainier D.Y., Srivastava D. miR-126 regulates angiogenic signaling and vascular integrity. Dev. Cell. 2008;15:272–284. doi: 10.1016/j.devcel.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pramanik S., Saha C., Chowdhury S., Bose C., Bhattacharyya N.P., Mondal L.K. Decreased levels of miR-126 and miR-132 in plasma and vitreous humor of non-proliferative diabetic retinopathy among subjects with type-2 diabetes mellitus. Diabetes Metab. Syndr. Obes. 2022;15:345–358. doi: 10.2147/dmso.S346097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zampetaki A., Kiechl S., Drozdov I., Willeit P., Mayr U., Prokopi M., Mayr A., Weger S., Oberhollenzer F., Bonora E., et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ. Res. 2010;107:810–817. doi: 10.1161/circresaha.110.226357. [DOI] [PubMed] [Google Scholar]
- 26.Fourdinier O., Schepers E., Metzinger-Le Meuth V., Glorieux G., Liabeuf S., Verbeke F., Vanholder R., Brigant B., Pletinck A., Diouf M., et al. Serum levels of miR-126 and miR-223 and outcomes in chronic kidney disease patients. Sci. Rep. 2019;9:4477. doi: 10.1038/s41598-019-41101-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sheikh M.S.A., Almaeen A., Alduraywish A., Alomair B.M., Salma U., Fei L., Yang T.L. Overexpression of miR-126 protects hypoxic-reoxygenation-exposed HUVEC cellular injury through regulating LRP6 expression. Oxid Med. Cell Longev. 2022;2022:3647744. doi: 10.1155/2022/3647744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zou Q., Liu C., Hu N., Wang W., Wang H. miR-126 ameliorates multiple organ dysfunction in septic rats by regulating the differentiation of Th17/Treg. Mol. Biol. Rep. 2022;49:2985–2998. doi: 10.1007/s11033-022-07121-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu B., Jiang Y., Wang X., Wang S. An integrated hypothesis for miR-126 in vascular disease. Med. Res. Arch. 2020;8 doi: 10.18103/mra.v8i5.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suresh Babu S., Thandavarayan R.A., Joladarashi D., Jeyabal P., Krishnamurthy S., Bhimaraj A., Youker K.A., Krishnamurthy P. MicroRNA-126 overexpression rescues diabetes-induced impairment in efferocytosis of apoptotic cardiomyocytes. Sci. Rep. 2016;6:36207. doi: 10.1038/srep36207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poleto Spinola L., Gabriel F.V., Fernandes Ferreira R., Calastri M.C.J., Greciele D.T., Aguiar F.L., Santana Ferreira Boin I.F., Larissa B.E.D.C., Chaim Correia M.F., Zanovelo E.M., et al. Underexpression of miR-126-3p in patients with cholangiocarcinoma. Asian Pac. J. Cancer Prev. 2021;22:573–579. doi: 10.31557/apjcp.2021.22.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang B., Lari Najafi M. Resveratrol inhibits skin squamous cell carcinoma proliferation, migration and invasion through up-regulating miR-126. Cell Mol. Biol. 2020;66:142–147. [PubMed] [Google Scholar]
- 33.Li M., Wang Q., Zhang X., Yan N., Li X. Exosomal miR-126 blocks the development of non-small cell lung cancer through the inhibition of ITGA6. Cancer Cell Int. 2020;20:574. doi: 10.1186/s12935-020-01653-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qin W.J., Lv L.H., Zhang M., Zhou X., Liu G.Q., Lu H.J. MiR-126 inhibits cell migration and invasion by targeting ADAM9 in oral squamous cell carcinoma. Eur. Rev. Med. Pharmacol. Sci. 2019;23:10324–10331. doi: 10.26355/eurrev_201912_19670. [DOI] [PubMed] [Google Scholar]
- 35.Gordillo G.M., Atalay M., Roy S., Sen C.K. Hemangioma model for in vivo angiogenesis: inducible oxidative stress and MCP-1 expression in EOMA cells. Methods Enzymol. 2002;352:422–432. doi: 10.1016/s0076-6879(02)52038-3. [DOI] [PubMed] [Google Scholar]
- 36.Hoak J.C., Warner E.D., Cheng H.F., Fry G.L., Hankenson R.R. Hemangioma with thrombocytopenia and microangiopathic anemia (Kasabach-Merritt syndrome): an animal model. J. Lab Clin. Med. 1971;77:941–950. [PubMed] [Google Scholar]
- 37.Gallego-Perez D., Pal D., Ghatak S., Malkoc V., Higuita-Castro N., Gnyawali S., Chang L., Liao W.C., Shi J., Sinha M., et al. Topical tissue nano-transfection mediates non-viral stroma reprogramming and rescue. Nat. Nanotechnol. 2017;12:974–979. doi: 10.1038/nnano.2017.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roy S., Sen C.K., Ghatak S., Higuita-Castro N., Palakurti R., Nalluri N., Clark A., Stewart R., Gallego-Perez D., Prater D.N., Khanna S. Neurogenic tissue nanotransfection in the management of cutaneous diabetic polyneuropathy. Nanomedicine. 2020;28:102220. doi: 10.1016/j.nano.2020.102220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarvestani S.T., Stunden H.J., Behlke M.A., Forster S.C., McCoy C.E., Tate M.D., Ferrand J., Lennox K.A., Latz E., Williams B.R., Gantier M.P. Sequence-dependent off-target inhibition of TLR7/8 sensing by synthetic microRNA inhibitors. Nucleic Acids Res. 2015;43:1177–1188. doi: 10.1093/nar/gku1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dadras S.S., Skrzypek A., Nguyen L., Shin J.W., Schulz M.M., Arbiser J., Mihm M.C., Detmar M. Prox-1 promotes invasion of kaposiform hemangioendotheliomas. J. Invest. Dermatol. 2008;128:2798–2806. doi: 10.1038/jid.2008.176. [DOI] [PubMed] [Google Scholar]
- 41.Raposo G., Stahl P.D. Extracellular vesicles: a new communication paradigm? Nat. Rev. Mol. Cell Biol. 2019;20:509–510. doi: 10.1038/s41580-019-0158-7. [DOI] [PubMed] [Google Scholar]
- 42.Lipinski S., Tiemann K. Extracellular vesicles and their role in the spatial and temporal expansion of tumor-immune interactions. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms22073374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zietzer A., Hosen M.R., Wang H., Goody P.R., Sylvester M., Latz E., Nickenig G., Werner N., Jansen F. The RNA-binding protein hnRNPU regulates the sorting of microRNA-30c-5p into large extracellular vesicles. J. Extracell Vesicles. 2020;9:1786967. doi: 10.1080/20013078.2020.1786967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Santos M.F., Rappa G., Karbanová J., Fontana S., Bella M.A.D., Pope M.R., Parrino B., Cascioferro S.M., Vistoli G., Diana P., et al. Itraconazole inhibits nuclear delivery of extracellular vesicle cargo by disrupting the entry of late endosomes into the nucleoplasmic reticulum. J. Extracell Vesicles. 2021;10:e12132. doi: 10.1002/jev2.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van den Broek B., Wuyts C., Sisto A., Pintelon I., Timmermans J.P., Somers V., Timmerman V., Hellings N., Irobi J. Oligodendroglia-derived extracellular vesicles activate autophagy via LC3B/BAG3 to protect against oxidative stress with an enhanced effect for HSPB8 enriched vesicles. Cell Commun. Signal. 2022;20:58. doi: 10.1186/s12964-022-00863-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang C., Zhang C., Liu L., Chen B., Li Y., Du J. Macrophage-derived mir-155-containing exosomes suppress fibroblast proliferation and promote fibroblast inflammation during cardiac injury. Mol. Ther. 2017;25:192–204. doi: 10.1016/j.ymthe.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X., Huang W., Liu G., Cai W., Millard R.W., Wang Y., Chang J., Peng T., Fan G.C. Cardiomyocytes mediate anti-angiogenesis in type 2 diabetic rats through the exosomal transfer of miR-320 into endothelial cells. J. Mol. Cell Cardiol. 2014;74:139–150. doi: 10.1016/j.yjmcc.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gordillo G., Fang H., Park H., Roy S. Nox-4-dependent nuclear H2O2 drives DNA oxidation resulting in 8-OHdG as urinary biomarker and hemangioendothelioma formation. Antioxid. Redox Signal. 2010;12:933–943. doi: 10.1089/ars.2009.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ridder K., Sevko A., Heide J., Dams M., Rupp A.K., Macas J., Starmann J., Tjwa M., Plate K.H., Sültmann H., et al. Extracellular vesicle-mediated transfer of functional RNA in the tumor microenvironment. Oncoimmunology. 2015;4:e1008371. doi: 10.1080/2162402x.2015.1008371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kalita-de Croft P., Sharma S., Sobrevia L., Salomon C. Extracellular vesicle interactions with the external and internal exposome in mediating carcinogenesis. Mol. Aspects Med. 2021;87:101039. doi: 10.1016/j.mam.2021.101039. [DOI] [PubMed] [Google Scholar]
- 51.Zomer A., Maynard C., Verweij F.J., Kamermans A., Schäfer R., Beerling E., Schiffelers R.M., de Wit E., Berenguer J., Ellenbroek S.I.J., et al. In Vivo imaging reveals extracellular vesicle-mediated phenocopying of metastatic behavior. Cell. 2015;161:1046–1057. doi: 10.1016/j.cell.2015.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taverna S., Amodeo V., Saieva L., Russo A., Giallombardo M., De Leo G., Alessandro R. Exosomal shuttling of miR-126 in endothelial cells modulates adhesive and migratory abilities of chronic myelogenous leukemia cells. Mol. Cancer. 2014;13:169. doi: 10.1186/1476-4598-13-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pontis F., Roz L., Mensah M., Segale M., Moro M., Bertolini G., Petraroia I., Centonze G., Ferretti A.M., Suatoni P., et al. Circulating extracellular vesicles from individuals at high-risk of lung cancer induce pro-tumorigenic conversion of stromal cells through transfer of miR-126 and miR-320. J. Exp. Clin. Cancer Res. 2021;40:237. doi: 10.1186/s13046-021-02040-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Atanasov G., Pötner C., Aust G., Schierle K., Dietel C., Benzing C., Krenzien F., Bartels M., Eichfeld U., Schmelzle M., et al. TIE2-expressing monocytes and M2-polarized macrophages impact survival and correlate with angiogenesis in adenocarcinoma of the pancreas. Oncotarget. 2018;9:29715–29726. doi: 10.18632/oncotarget.25690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Turrini R., Pabois A., Xenarios I., Coukos G., Delaloye J.F., Doucey M.A. TIE-2 expressing monocytes in human cancers. Oncoimmunology. 2017;6:e1303585. doi: 10.1080/2162402x.2017.1303585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sinha M., Sen C.K., Singh K., Das A., Ghatak S., Rhea B., Blackstone B., Powell H.M., Khanna S., Roy S. Direct conversion of injury-site myeloid cells to fibroblast-like cells of granulation tissue. Nat. Commun. 2018;9:936. doi: 10.1038/s41467-018-03208-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rodero M.P., Legrand J.M., Bou-Gharios G., Khosrotehrani K. Wound-associated macrophages control collagen 1α2 transcription during the early stages of skin wound healing. Exp. Dermatol. 2013;22:143–145. doi: 10.1111/exd.12068. [DOI] [PubMed] [Google Scholar]
- 58.Simões F.C., Cahill T.J., Kenyon A., Gavriouchkina D., Vieira J.M., Sun X., Pezzolla D., Ravaud C., Masmanian E., Weinberger M., et al. Macrophages directly contribute collagen to scar formation during zebrafish heart regeneration and mouse heart repair. Nat. Commun. 2020;11:600. doi: 10.1038/s41467-019-14263-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Poczobutt J.M., De S., Yadav V.K., Nguyen T.T., Li H., Sippel T.R., Weiser-Evans M.C., Nemenoff R.A. Expression profiling of macrophages reveals multiple populations with distinct biological roles in an immunocompetent orthotopic model of lung cancer. J. Immunol. 2016;196:2847–2859. doi: 10.4049/jimmunol.1502364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kumar M.P., Du J., Lagoudas G., Jiao Y., Sawyer A., Drummond D.C., Lauffenburger D.A., Raue A. Analysis of single-cell RNA-seq identifies cell-cell communication associated with tumor characteristics. Cell Rep. 2018;25:1458–1468.e4. doi: 10.1016/j.celrep.2018.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Isakoff M.S., Sansam C.G., Tamayo P., Subramanian A., Evans J.A., Fillmore C.M., Wang X., Biegel J.A., Pomeroy S.L., Mesirov J.P., Roberts C.W. Inactivation of the Snf5 tumor suppressor stimulates cell cycle progression and cooperates with p53 loss in oncogenic transformation. Proc. Natl. Acad. Sci. USA. 2005;102:17745–17750. doi: 10.1073/pnas.0509014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jia J., Bai Y., Fu K., Sun Z.J., Chen X.M., Zhao Y.F. Expression of allograft inflammatory factor-1 and CD68 in haemangioma: implication in the progression of haemangioma. Br. J. Dermatol. 2008;159:811–819. doi: 10.1111/j.1365-2133.2008.08744.x. [DOI] [PubMed] [Google Scholar]
- 63.Aldinucci D., Colombatti A. The inflammatory chemokine CCL5 and cancer progression. Mediators Inflamm. 2014;2014:292376. doi: 10.1155/2014/292376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sun J., Huang J., Lan J., Zhou K., Gao Y., Song Z., Deng Y., Liu L., Dong Y., Liu X. Overexpression of CENPF correlates with poor prognosis and tumor bone metastasis in breast cancer. Cancer Cell Int. 2019;19:264. doi: 10.1186/s12935-019-0986-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rao S., Mondragón L., Pranjic B., Hanada T., Stoll G., Köcher T., Zhang P., Jais A., Lercher A., Bergthaler A., et al. AIF-regulated oxidative phosphorylation supports lung cancer development. Cell Res. 2019;29:579–591. doi: 10.1038/s41422-019-0181-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Atalay M., Gordillo G., Roy S., Rovin B., Bagchi D., Bagchi M., Sen C.K. Anti-angiogenic property of edible berry in a model of hemangioma. FEBS Lett. 2003;544:252–257. doi: 10.1016/s0014-5793(03)00509-x. [DOI] [PubMed] [Google Scholar]
- 67.Gordillo G., Fang H., Khanna S., Harper J., Phillips G., Sen C.K. Oral administration of blueberry inhibits angiogenic tumor growth and enhances survival of mice with endothelial cell neoplasm. Antioxid. Redox Signal. 2009;11:47–58. doi: 10.1089/ars.2008.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ridiandries A., Tan J.T., Bursill C.A. The role of CC-chemokines in the regulation of angiogenesis. Int. J. Mol. Sci. 2016;17 doi: 10.3390/ijms17111856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wan S., Zhao E., Kryczek I., Vatan L., Sadovskaya A., Ludema G., Simeone D.M., Zou W., Welling T.H. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. 2014;147:1393–1404. doi: 10.1053/j.gastro.2014.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Biswas A., Khanna S., Roy S., Pan X., Sen C.K., Gordillo G.M. Endothelial cell tumor growth is Ape/ref-1 dependent. Am. J. Physiol. Cell Physiol. 2015;309:C296–C307. doi: 10.1152/ajpcell.00022.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Das A., Abas M., Biswas N., Banerjee P., Ghosh N., Rawat A., Khanna S., Roy S., Sen C.K. A modified collagen dressing induces transition of inflammatory to reparative phenotype of wound macrophages. Sci. Rep. 2019;9:14293. doi: 10.1038/s41598-019-49435-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Das A., Datta S., Roche E., Chaffee S., Jose E., Shi L., Grover K., Khanna S., Sen C.K., Roy S. Novel mechanisms of Collagenase Santyl Ointment (CSO) in wound macrophage polarization and resolution of wound inflammation. Sci. Rep. 2018;8:1696. doi: 10.1038/s41598-018-19879-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Das A., Ghatak S., Sinha M., Chaffee S., Ahmed N.S., Parinandi N.L., Wohleb E.S., Sheridan J.F., Sen C.K., Roy S. Correction of MFG-E8 resolves inflammation and promotes cutaneous wound healing in diabetes. J. Immunol. 2016;196:5089–5100. doi: 10.4049/jimmunol.1502270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhou X., Brown B.A., Siegel A.P., El Masry M.S., Zeng X., Song W., Das A., Khandelwal P., Clark A., Singh K., et al. Exosome-mediated crosstalk between keratinocytes and macrophages in cutaneous wound healing. ACS Nano. 2020;14:12732–12748. doi: 10.1021/acsnano.0c03064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rustagi Y., Abouhashem A.S., Verma P., Verma S.S., Hernandez E., Liu S., Kumar M., Guda P.R., Srivastava R., Mohanty S.K., et al. Endothelial phospholipase Cγ2 improves outcomes of diabetic ischemic limb rescue following VEGF therapy. Diabetes. 2022;71:1149–1165. doi: 10.2337/db21-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Singh K., Rustagi Y., Abouhashem A.S., Tabasum S., Verma P., Hernandez E., Pal D., Khona D.K., Mohanty S.K., Kumar M., et al. Genome-wide DNA hypermethylation opposes healing in chronic wound patients by impairing epithelial-to-mesenchymal transition. J. Clin. Invest. 2022 doi: 10.1172/jci157279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xuan Y., Ghatak S., Clark A., Li Z., Khanna S., Pak D., Agarwal M., Roy S., Duda P., Sen C.K. Fabrication and use of silicon hollow-needle arrays to achieve tissue nanotransfection in mouse tissue in vivo. Nat. Protoc. 2021;16:5707–5738. doi: 10.1038/s41596-021-00631-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Singh K., Sinha M., Pal D., Tabasum S., Gnyawali S.C., Khona D., Sarkar S., Mohanty S.K., Soto-Gonzalez F., Khanna S., et al. Cutaneous epithelial to mesenchymal transition activator ZEB1 regulates wound angiogenesis and closure in a glycemic status-dependent manner. Diabetes. 2019;68:2175–2190. doi: 10.2337/db19-0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gordillo G.M., Biswas A., Khanna S., Pan X., Sinha M., Roy S., Sen C.K. Dicer knockdown inhibits endothelial cell tumor growth via microRNA 21a-3p targeting of Nox-4. J. Biol. Chem. 2014;289:9027–9038. doi: 10.1074/jbc.M113.519264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Singh K., Pal D., Sinha M., Ghatak S., Gnyawali S.C., Khanna S., Roy S., Sen C.K. Epigenetic modification of MicroRNA-200b contributes to diabetic vasculopathy. Mol. Ther. 2017;25:2689–2704. doi: 10.1016/j.ymthe.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gordillo G.M., Biswas A., Khanna S., Spieldenner J.M., Pan X., Sen C.K. Multidrug resistance-associated protein-1 (MRP-1)-dependent glutathione disulfide (GSSG) efflux as a critical survival factor for oxidant-enriched tumorigenic endothelial cells. J. Biol. Chem. 2016;291:10089–10103. doi: 10.1074/jbc.M115.688879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bhamidipati T., Sinha M., Sen C.K., Singh K. Laser capture microdissection in the spatial analysis of epigenetic modifications in skin: a comprehensive review. Oxid. Med. Cell Longev. 2022;2022:4127238. doi: 10.1155/2022/4127238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Satija R., Farrell J.A., Gennert D., Schier A.F., Regev A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015;33:495–502. doi: 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A.R., Kamitaki N., Martersteck E.M., et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liberzon A., Subramanian A., Pinchback R., Thorvaldsdóttir H., Tamayo P., Mesirov J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Blake J.A., Richardson J.E., Bult C.J., Kadin J.A., Eppig J.T. MGD: the mouse genome database. Nucleic Acids Res. 2003;31:193–195. doi: 10.1093/nar/gkg047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N.J., Livak K.J., Mikkelsen T.S., Rinn J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cao J., Packer J.S., Ramani V., Cusanovich D.A., Huynh C., Daza R., Qiu X., Lee C., Furlan S.N., Steemers F.J., et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science. 2017;357:661–667. doi: 10.1126/science.aam8940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee A.R., Lee S., Shin J.Y., Kim J.Y., Moon K.S., Jung J. Biomarker LEPRE1 induces pelitinib-specific drug responsiveness by regulating ABCG2 expression and tumor transition states in human leukemia and lung cancer. Sci. Rep. 2022;12:2928. doi: 10.1038/s41598-022-06621-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang Y., Li L., Zhang X., Zhao X. Long non-coding RNA OIP5-AS1 suppresses microRNA-92a to augment proliferation and metastasis of ovarian cancer cells through upregulating ITGA6. J. Ovarian Res. 2022;15:25. doi: 10.1186/s13048-021-00937-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.