

Abstract
Mitochondrial dysfunction is a hallmark of heart failure. Mitochondrial transplantation has been demonstrated to be able to restore heart function, but its mechanism of action remains unresolved. Using an in-house optimized mitochondrial isolation method, we tested efficacy of mitochondria transplantation in two different heart failure models. First, using a doxorubicin-induced heart failure model, we demonstrate that mitochondrial transplantation before doxorubicin challenge protects cardiac function in vivo and prevents myocardial apoptosis, but contraction improvement relies on the metabolic compatibility between transplanted mitochondria and treated cardiomyocytes. Second, using a mutation-driven dilated cardiomyopathic human induced pluripotent stem cell-derived cardiomyocyte model, we demonstrate that mitochondrial transplantation preferentially boosts contraction in the ventricular myocytes. Last, using single-cell RNA-seq, we show that mitochondria transplantation boosts contractility in dystrophic cardiomyocytes with few transcriptomic alterations. Together, we provide evidence that mitochondria transplantation confers myocardial protection and may serve as a potential therapeutic option for heart failure.
Keywords: mitochondria delivery, doxorubicin, dilated cardiomyopathy, iPSC
Graphical abstract
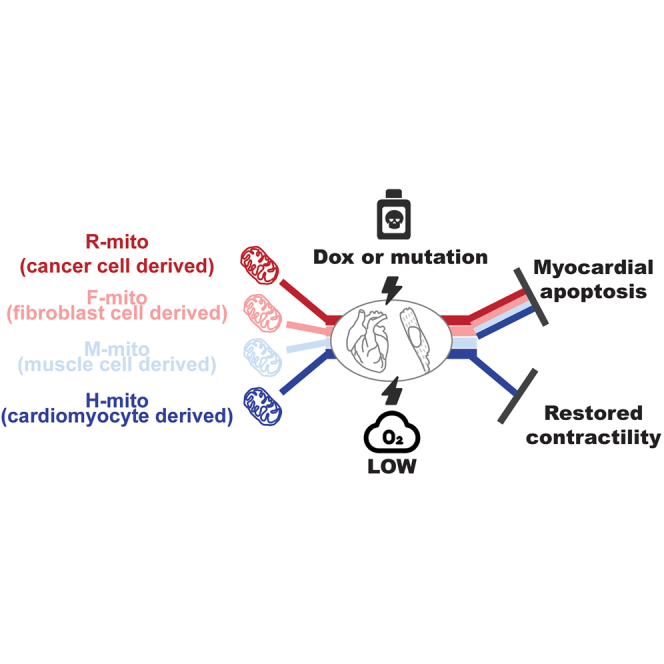
Zhang et al. demonstrate that mitochondria transplantation confer efficacy by two means: mitochondria numbers and metabolic compliance. Mitochondria transplantation was able to block doxorubicin-induced cardiotoxicity, restore ischemia reperfusion-induced mitochondrial membrane potential loss, and boost contractility in dystrophic induced pluripotent stem cell-derived cardiomyocytes.
Introduction
Heart failure (HF) is defined as the structural or functional impairment of ventricular filling or the ability to pump blood associated with various heart diseases.1 The prevalence of HF seems to be 1%–2% of adults.2,3,4 The prevalence of HF increases with age: approximately 1% for those aged less than 55 years to more than 10% in those aged 70 years or over.5,6 With the aging population and increasing prevalence of cardiovascular risk factors, it is estimated that hospital admissions due to HF is expected to increase by as much as 50% in the next 25 years.7
Currently, HF patients are treated with classical neuroendocrine antagonists targeting hemodynamics. β-Blockers, angiotensin-converting enzyme inhibitors/angiotensin II receptor blockers, angiotensin receptor-neprilysin inhibitors, aldosterone receptor antagonists, and sodium-dependent glucose transporters-2 inhibitors are recommended for HF patients.8,9 Although these drugs delay the progression of HF, quality of life remains poor. Importantly, mortality rates remain higher in observational studies than in clinical trials.10 In the Olmsted County cohort, the 1-year and 5-year mortality rates for all types of HF patients were 20% and 53%, respectively.11 A study pooling the Framingham Heart Study and Cardiovascular Health Study cohorts reported a 67% mortality rate within 5 years after diagnosis.12 Therefore, new strategies that can improve patients’ outcomes are urgently needed.
Mitochondria occupy up to approximately 20%–35% of myocardial volume and are key in providing energy for cardiomyocyte contractions.13,14 Disruption to mitochondrial homeostasis can directly result in cardiomyopathies15,16,17,18 and mitochondrial dysfunction has been shown to play a key role in HF, including anthracycline cardiomyopathy, ischemia-reperfusion injuries, and genetic cardiomyopathies.19,20,21 Although mitochondria transplantation has been shown to confer myocardial function,22,23 the exact mechanisms remain to be elucidated. Currently unresolved is whether calcium overloaded mitochondria are still capable of boosting myocardial function.24
Using anthracycline-induced and genetic dilated cardiomyopathy models, we investigated how mitochondria transplantation restores cardiac function. Using an optimized mitochondria isolation method, we first characterized mitochondria quality isolated from different sources with varying metabolic status. We tested whether mitochondria transplantation before doxorubicin challenge confers cardioprotection both in vivo and in vitro. We assayed if transplantation of mitochondria from different cell sources with varying metabolic states protected doxorubicin-induced apoptosis, myocardial contraction, and myocardial mitochondrial respiration in murine cardiomyocytes. Next, using human induced pluripotent stem cell derived cardiomyocytes (hiPSC-CMs) from dilated cardiomyopathy (DCM) patients and its CRISPR-corrected isogenic cells,25 we evaluated whether isogenic mitochondria transplantation can boost mitochondrial respiration and myocardial contraction at the single-cell level. We probed whether mitochondria transplantation preferentially increased contractile function in ventricular versus atrial cardiomyocytes, and single cell RNA-seq was used to examine transcriptomic changes.
Results
Isolating mitochondria panel with varying metabolic states
To ensure mitochondria purity and ultrastructure, we tested various isolation methods and evaluated the intactness of mitochondrial cristae by transmission electron microscopy (TEM) (Figure S1). As evident by high presence of cell debris and compressed cristae structures using previously established methods,26,27,28,29 we optimized the isolation protocol (see the materials and methods section) and was able to purify intact mitochondria with little cell debris (Figure S1). Next, we tested our mitochondrial isolation method in various cell lines to answer two questions: (1) how robust is our mitochondrial isolation protocol and (2) if mitochondrial activity (mainly membrane potential and superoxide production) is maintained after mitochondria isolation. Here, we used four different cell sources to evaluate mitochondrial amount as well as metabolic preference ranging from glycolytic to fatty acid oxidation: human colon carcinoma cell line (RKO, R-mito), mouse fibroblast (NIH 3T3, F-mito), mouse skeletal muscle (C2C12, M-mito), and mouse neonatal cardiomyocytes (H-mito). Using our optimized isolation protocol, we tested mitochondrial isolation efficiency using 1 × 107 cells of the four various cell types (Table 1). As measured by flow cytometry (Table 1), mitochondrial counts measured by Mitotracker green (MTG, for mitochondrial number) and mitochondrial membrane potential (TMRM) dyes are in good agreement. Next, we wanted to address the unresolved debate as to whether isolated mitochondria are dysfunctional due to isolation procedure and buffers. We measured the mitochondrial amount (MTG), membrane potential (TMRM), and oxidative status (MitoSox) in intact cells, as well as in isolated mitochondrial (Figure 1A). As shown by flow cytometry, intact cardiomyocytes possessed the most mitochondrial amount per cell with the highest mitochondrial membrane potential compared with other cell types yet with mean mitochondrial superoxide levels (Figure 1B). However, when assayed in isolated mitochondria, both mitochondrial membrane potential and oxidative status differences were lost, suggesting that the suspension buffer is insufficient to maintain mitochondrial membrane potential (Figure 1B). This observation, which is in keeping with a previous study,24 raises the question as to whether isolated mitochondria can re-establish function after uptake by receiving cells.
Table 1.
Mitochondria isolation and quantification
| Cell type (1 × 107 cells) | Mito count per MTG | Mito count per TMRM |
|---|---|---|
| RKO (R) | 5.98 × 106 | 6.74 × 106 |
| NIH 3T3 (F) | 3.56 × 106 | 3.51 × 106 |
| C2C12 (M) | 1.3 × 106 | 0.8 × 106 |
| NMVM (H) | 2.15 × 107 | 2.19 × 107 |
Figure 1.

Isolation and quantification of mitochondria using flow cytometry
(A) Immunoelectron microscopy and immunofluorescence images of isolated mitochondria. (B) Mitochondrial amount (Mitotracker green), mitochondrial membrane potential (TMRM) and mitochondrial superoxide levels (MitoSox) in cells and isolated mitochondria were evaluated using flow cytometry (n = 3 independent cultures; 30,000–100,000 events per run). Data are shown as mean ± standard error of the mean. Significance was determined by one-way ANOVA for the remaining panels. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Transplanted mitochondria are capable of preventing doxorubicin-induced cardiotoxicity
To test if transplanted mitochondria can be absorbed by murine cardiomyocytes, we first injected human R-mito into wild-type murine hearts and isolated genomic DNA 2 and 24 h after injection. Using human and mouse mtDNA-specific primers and mouse nuclear copy number primers, we evaluated presence of human and murine mtDNA levels using reverse transcriptase quantitative PCR (RT-qPCR). The presence of human mtDNA confirms the presence of transplanted R-mito in murine hearts; moreover, we observed a significant increase in endogenous murine mtDNA, suggesting that mitochondrial transplantation stimulated mtDNA replication (Figure 2A). Next, we tested if mitochondria transplantation before doxorubicin (DOX) challenge is capable of preventing cardiotoxicity; we performed single dose intramyocardial mitochondria transplantation (M-mito + DOX) or phosphate-buffered saline (PBS) (PBS + DOX) before DOX challenge in 8-week-old C57/Bl6J male mice, as previously described.30 Untreated animals were used as wild-type controls. Upon DOX challenge, we observed a significant decrease in cardiac function (Table 2, Figures S2A and S2B) and weight loss (Figure S2C). However, these functional declines were reversed when animals were pre-treated with mitochondria injections (Table 2, Figures S2A–S2C). Next, we purified cardiomyocytes and stained the cells with either CellROX (total cellular reactive oxygen species [ROS]), MitoSox (mitochondrial oxidation), and TMRM (mitochondrial membrane potential) (Figures 2B–2D). Compared with sham (Con) cardiomyocytes, PBS + DOX cardiomyocytes exhibited increased cellular ROS (Figure 2B), decreased mitochondrial oxidation (Figure 2C), and decreased mitochondrial membrane potential (Figure 2D). Mitochondria pre-treatment prevented these changes (Figures 2B–2D). Together, these results demonstrate that mitochondria pre-treatment confers cardioprotection in DOX-induced HF.
Figure 2.

Mitochondrial transplantation protects cardiomyocytes from DOX-induced mitochondrial dysfunction
(A) Human R-mito was injected into wild-type murine hearts and amount of transplanted R-mito mitochondria were measured using human specific mtDNA primers normalized to mouse copy gene in isolated cardiomyocytes (N = 3 animals each). Data are shown as mean ± standard error of the mean. After 4 weeks of weekly DOX injection and 2 weeks of recovery, animals were subjected to Langendorff isolation. (B–D) Purified cardiomyocytes were stained for intracellular ROS (CellROX, N = 6 animals, total N = 50 cells), mitochondrial oxidative status (MitoSox, N = 6 animals, total n = 50 cells), and mitochondrial membrane potential (TMRM, N = 6 animals, total n = 100 cells). Data are shown as violin plots. Scale bar, 20 μm. Significance was determined by one-way ANOVA for the remaining panels. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Table 2.
Cardiac function of control, PBS + DOX, and M-Mito + DOX animals
| Control | PBS + DOX | M-Mito + DOX | |
|---|---|---|---|
| Diameter; s | 1.88 ± 0.25 | 2.29 ± 0.43 | 1.69 ± 0.30## |
| Diameter; d | 3.45 ± 0.28 | 3.47 ± 0.42 | 3.21 ± 0.21 |
| Volume; s | 11.64 ± 3.55 | 19.01 ± 8.92∗ | 8.91 ± 3.86# |
| Volume; d | 50.02 ± 9.87 | 50.92 ± 14.95 | 41.78 ± 6.47 |
| Stroke volume | 38.37 ± 7.82 | 31.91 ± 6.27 | 32.87 ± 4.31 |
| Ejection fraction | 78.29 ± 4.37 | 64.2 ± 6.53∗∗ | 78.19 ± 8.09## |
| Fractional shortening | 46.72 ± 4.26 | 34.39 ± 4.44∗∗ | 46.38 ± 7.99## |
| Cardiac output | 15.83 ± 3.33 | 13.29 ± 2.6 | 14.01 ± 1.78 |
| LV mass | 96.98 ± 10.5 | 93.05 ± 30.31 | 98.22 ± 18.21 |
| LV mass cor | 77.58 ± 8.4 | 74.44 ± 24.25 | 78.57 ± 14.57 |
| LVAW; s | 1.37 ± 0.12 | 1.19 ± 0.22 | 1.54 ± 0.23## |
| LVAW; d | 0.88 ± 0.14 | 0.79 ± 0.22 | 1 ± 0.1 |
| LVPW; s | 1.4 ± 0.08 | 1.16 ± 0.14∗∗ | 1.3 ± 0.11# |
| LVPW; d | 0.760.08 | 0.76 ± 0.08 | 0.79 ± 0.08 |
Compared with the control group: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Compared with the PBS + DOX group: #p < 0.05; ##p < 0.01; ###p < 0.001.
Next, we asked whether mitochondrial transplantation confers cardioprotection against DOX challenge through the inhibition of apoptosis or metabolic restoration. To accomplish this, we used neonatal mouse ventricular myocytes (NMVMs) cultures challenged with DOX. First, we tested the level of mitochondria absorption of the four types of mitochondria with varying concentrations (Figure S3). R-mito, F-mito, M-mito, and H-mito showed absorption saturations around 2 h at 0.5 × 104, 1 × 104, and 2 × 104 mito/100 μL concentrations (Figure S3). Next, we transplanted 1 × 104 mito/100μL mitochondria into NMVMs in culture and challenged with 1 μM DOX (Figure 3A).31 Using propidium iodide/annexin V double staining, DOX challenge increased NMVM apoptosis (Figure 3B). To our surprise, regardless of mitochondrial source, mitochondria pre-treatment before DOX challenge was capable of preventing apoptosis in cardiomyocytes (Figure 3B). This observation suggests that the DOX-induced apoptotic signal may be diluted by an increasing mitochondria amount. Next, we examined contraction and mitochondrial respiratory functions of these NMVMs. Unlike the anti-apoptotic observation, we found that metabolically matched mitochondria were necessary for improving contractile function (Figures 3C and 3D). Further, mitochondria pre-treatment restored basal mitochondrial respiration (Figures 3E and 3F) upon DOX challenge, but only H-mito treatment restored maximal mitochondrial respiration (Figure 3G). Next, we evaluated with mitochondria transplantation affected mitofusion/mitofission. Compared with controls, H-mito treatment slightly induced mitofusion Opa1 levels, but not Mfn2 or mitofission Drp1 levels (Figure S4). DOX challenge greatly induced Drp1 and Mfn2 while decreasing Opa1 levels; H-mito pretreatment alleviated DOX-induced changes (Figure S4). Together, these data demonstrate that, in the DOX-induced HF model, mitochondrial transplantation confers cardioprotection through both mitochondrial abundance and metabolic restoration.
Figure 3.

Mitochondrial transplantation ameliorates DOX-induced myocardial dysfunction
(A) Experimental setup testing if mitochondrial pre-transplantation can prevent DOX-induced cardiotoxicity. (B) Quantification of myocardial apoptosis and necrosis upon DOX challenge (N = 3–5 neonatal hearts were pooled per sample prep; technical n = 3; 30,000–100,000 events per run). (C, D) Analysis of cardiomyocyte contraction (n = 20 cells per group). (E–G) Analysis of mitochondrial respiration (N = 3–5 neonatal hearts were pooled per sample prep; technical n = 3–8). Data are shown as mean ± standard error of the mean. Significance was determined by one-way ANOVA for the remaining panels. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001.
Metabolically compatible mitochondrial transplantation restores contractility in dilated cardiomyopathic cardiomyocytes
Given that the membrane potential of isolated mitochondria is greatly diminished compared with its source and DOX treatment affects both endogenous and transplanted mitochondria, we have not answered the question of whether transplanted mitochondria is capable of re-establishing membrane potential and function. To answer this, we transplanted isogenic mitochondria to ischemic reperfused hiPSC-CMs (Table 3) and evaluated the mitochondrial membrane potential. Using microprinted hiPSC-CMs,25 the ischemic reperfusion challenge greatly diminishes mitochondrial membrane potential as measured by TMRM (Figure S5A). Using live cell microscopy, we stained the microprinted hiPSC-CM with TMRM, induced ischemia, and transplanted Mitotracker green-labeled mitochondrial upon reperfusion (Figure S5B). Here, we observe that the absorbed green mitochondria restored the mitochondrial membrane potential in receiving hiPSC-CMs (Figure S5C). This observation suggests that, despite diminished membrane potential in isolation buffer, transplanted mitochondria is capable of re-establishing membrane potential inside cardiomyocytes.
Table 3.
hiPSC used
| Mutation | Age | Tissue | Gender | Source | |
|---|---|---|---|---|---|
| Healthy | isogenic control of DMD | 16 | urine mesenchymal | male | University of Washington |
| DCM | DMD (c.19delG) | 16 | urine mesenchymal | male | University of Washington |
Next, we tested if mitochondria transplantation can rescue mitochondrial dysfunction and myocardial contractility in a DCM hiPSC-CM model.21,25 To test this, we used hiPSC-CMs generated from a Duchenne muscular dystrophy (DMD) patient as a genetic DCM model25 (Figure 4A). Mitochondria were isolated from isogenic hiPSC-CMs to avoid genetic background or species differences. When healthy hiPSC-CM mitochondria were transplanted to DMD hiPSC-CM monolayer cultures, we observed an improvement of contractile velocity and a decrease in beating frequency (Figure 4B). Given that our differentiation protocol results in the production of both atrial and ventricular cardiomyocytes, we collected contractile velocity changes before and after mitochondria transplantation of single microprinted DMD hiPSC-CMs and performed immunofluorescence staining for ventricular marker MLC2v32 (Figure 4C). To our surprise, mitochondrial transplantation conferred contractile improvement only in ventricular hiPSC-CMs, not atrial hiPSC-CMs (Figure 4D), despite equal mitochondrial uptake rates in isolated murine ventricular and atrial cardiomyocytes (Figure S6). In keeping with our NMVM DOX findings, H-mito transplantation into DCM hiPSC-CM monolayers increased maximal mitochondrial respiration (Figure 4E). Together, these results provide support that mitochondrial transplantation preferentially restores contractile function in ventricular cardiomyocytes, while keeping atrial cardiomyocytes intact.
Figure 4.

Mitochondrial transplantation ameliorates contractile function in DCM hiPSC-CMs
(A) Evaluating the efficacy of mitochondria transplantation for DCM using hiPSC-CM model. (B) Isogenic healthy hiPSC-CMs were used for mitochondria isolation. Mitochondrial transplantation into DCM hiPSC-CMs improves contractile speed and lowers beating frequency (n = 5). (C) Identification of ventricular hiPSC-CMs by immunofluorescence staining. (D) Mitochondria transplantation confers cardiac improvement in ventricular, not atrial, DCM cardiomyocytes (n = 5). (E) Analysis of DCM hiPSC-CM mitochondrial respiration (n = 5–6). Data are shown as mean ± standard error of the mean. Significance was determined by two-tailed, unpaired Student’s t-test for (B) or one-way ANOVA for (D). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To examine how mitochondria transplantation affects ventricular cardiomyocytes, we performed single cell RNA sequencing (RNA-seq) on H-mito-treated and -untreated DMD hiPSC-CMs. Based on uniform manifold approximation and projection (UMAP) analysis, we identified 1 hepatocyte population, 1 atrial hiPSC-CM population, and 10 ventricular hiPSC-CM populations in control and mitochondrial transplanted DMD hiPSC-CMs, respectively (Figure 5A). Hepatocytes, which also originate from the mesoderm during embryonic development, were ignored in the following Gene Ontology (GO) term analyses (Figure 5A). Differentially expressed genes from ventricular hiPSC-CMs (pooled) and atrial hiPSC-CMS were used as input and top three GO terms are shown (Figure 5B and Table S1). Clearly, mitochondrial transplantation induced gene networks associated with cell-cycle status (mitosis, cell cycle, and cell division) (Table S1) and structural reinforcement (cytoskeleton, actin-binding, and muscle protein) (Table S1) in ventricular DMD hiPSC-CMs (Figure 5B). Mitochondrial transplantation into atrial DMD hiPSC-CMs induced RNA-associated gene networks (Figure 5B), but cytoskeletal remodeling and mitochondrial respiration pathways remain unchanged; however, bulk RT-qPCR validation showed few differentially expressed genes (Figure 5C). These results suggest that, rather than working by inducing transcriptomic changes, mitochondrial transplantation strengthens ventricular myocardial function, likely through the restoration of mitochondrial respiration.
Figure 5.

Mitochondrial transplantation induces transcriptomic changes in DCM hiPSC-CMs
(A) UMAP analysis of single cell RNA-seq transplanted with or without H-mitochondria. Number of cells per subpopulation are shown. (B) Identification of cardiomyocytes using myocardial markers. (C) Top three significantly over-represented GO terms for ventricular and atrial DCM hiPSC-CMs differentially expressed genes (DEGs) are shown. (D) Potential mitochondria transplantation regulated targets were evaluated by RT-qPCR in mitochondria transplanted DCM hiPSC-CMs. Data are shown as mean ± standard error of the mean. Significance was determined by two-tailed, unpaired Student’s t-test. ∗p < 0.05.
Discussion
The protective role of mitochondria transplantation for the heart has been demonstrated in several studies22,23; it remains to be demonstrated as to how isolated mitochondria when transplanted can provide functional improvement to diseased cardiomyocytes. Using various HF models, we demonstrate that mitochondria transplantation confers protection against DOX challenge, allows the re-establishment of mitochondrial membrane potential after ischemia-reperfusion challenge and can boost myocardial function in dilated cardiomyopathic cardiomyocytes.
It has been shown that mitochondrial calcium overload may occur when injected into tissues, and the cardioprotective effect may not be due to the absorption of transplanted mitochondria.24 It has also been demonstrated that mitochondrial calcium overload may trigger aberrant calcium transients.33,34 To address this, we compared mitochondrial membrane potential and superoxide generation both in cells and isolated mitochondria. We confirm that mitochondrial membrane potential is lost during the purification process. However, we demonstrate that, when transplanted, mitochondria are capable of preventing DOX-induced apoptosis (likely through a dilution effect) and restore myocardial contraction (intact mitochondria machinery). Importantly, in our ischemic reperfusion challenge, hiPSC-CMs that received mitochondria re-established mitochondrial membrane potential faster compared with untreated. Although we acknowledge that intramuscular injections are not common for treating HF, our results provide proof of concept of preventing DOX-induced cardiotoxicity via mitochondria protection. Our results also caution the preparation process for generating intact mitochondria for transplantation, but provide support for using mitochondria transplantation in treating HF.
It is important to point out that our study attempted to characterize the absorption and degradation kinetics of transplanted mitochondria without tracing capabilities; therefore, these results have their own limitations. By leverage human versus mouse differences, we show that human mitochondrial DNA accumulation in murine hearts drives endogenous murine mtDNA increase. Using live cell imaging, we show that transplanted mitochondria can be taken up by both murine and human hiPSC-CMs in a time-dependent manner. However, given that the live mitochondrial dyes do not distinguish between transplanted and endogenous mitochondria and that mitofusion is induced after mitochondrial transplantation, we are unable to determine the level of mitochondrial protein and DNA degradation rates. In our DMD hiPSC-CM experiments, mitochondrial supplementation alleviated mitochondrial dysfunction and boosted myocardial contraction. Yet mouse studies have demonstrated that different cells have varying tolerance for mitochondrial heteroplasmy,35 so further characterization of exogenous mitochondria stability is needed. Further, one needs to caution that mtDNA mutations have been shown to drive severe cardiomyopathies36; thus, spontaneous mitochondrial mutations will need to monitored in the design of an off-the-shelf mitochondria therapy. Although our scRNA-seq suggest mitochondrial transplantation is likely to transcriptomic changes, our RT-qPCR validation results suggest mitochondrial transplantation confers efficacy through restoration of mitochondrial respiration.
In summary, by optimizing and transplanting mitochondria with varying metabolic states, we show that mitochondria transplantation therapy confers cardioprotection through two mechanisms: abundance and metabolic fitness. Using both murine and human cardiomyocyte systems, we show that metabolically matched mitochondria restore mitochondrial membrane potential and contraction function. In ventricular cardiomyocytes, mitochondrial transplantation can also boost cell cycle status and cytoskeletal pathways, which strongly warrant further development toward clinical application.
Materials and methods
Mitochondria isolation and TEM
All mitochondria isolation from cells or tissue were performed as previously described22 with minor adjustments. Briefly, fresh tissue or cells were collected and stored and washed once with 1× PBS on ice. Tissues were cut up using scissors. Samples were transferred to Glass/Teflon Potter Elvehjem homogenizers with 3 mL ice-cold Homogenizing Buffer (300 mM sucrose, 10 mM K-HEPES, and 1 mM K-EGTA, pH adjusted to 7.2, stored at 4°C), homogenized, and transferred to centrifuge tube. We added 250 μL Subtilisin A Solution (1 mg Subtilisin A in 250 μL homogenizing buffer) to the homogenate, mix by inversion, and incubate on ice for 10 min. Homogenate was filtered into a 50-mL conical centrifuge tube using a 40-μm mesh on ice and filter and 250 μL freshly prepared bovine serum albumin (BSA) Stock Solution (5 mg BSA in 250 μL homogenizing buffer) was added and mix by inversion. Repeat homogenate filtration into a 50-mL conical centrifuge tube using a 10-μm filter on ice. Transfer the filtrate to two pre-chilled 1.5-mL microfuge tubes and centrifuge at 9,000×g for 10 min at 4°C. Remove supernatant and re-suspend in 1 mL homogenizing buffer, and centrifuge at 7,000×g for 10 min at 4°C. Remove the supernatant, resuspend in PBS, and store at 4°C and use within 2 h.
For TEM morphological analyses, fresh mitochondrial pellets were fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) at 4°C for 2 h. After being washed with 0.1 M sodium cacodylate buffer, samples were post-fixed with 1% osmium tetroxide for 2 h and dehydrated through a graded ethanol series. Finally, the samples were embedded in epoxy resin at room temperature for 4 h. Ultrathin sections (70 nm) were assembled to a copper grid and observed using electron microscopy (Hitachi).
Doxorubicin-induced HF mouse model
All mouse protocols were approved by the Institutional Animal Care and Use Committee (SH9H-2020-A55-1). Male C57BL/6 mice (6–8 weeks old; weighing 18–22 g) were purchased from Shanghai Jiesjie Laboratory Animal Co., LTD. After a 1-week acclimation period, mice were randomly divided into three groups (n = 6 per group): Con group, DOX-treated group (DOX) and DOX-treated plus mitochondrial injection group (M-mito + DOX). An intraperitoneal injection of DOX (5 mg/kg; MedChem Express)30 were given for the DOX group and M-mito + DOX group or an equivalent volume of PBS for the Con group once a week for 4 weeks. Mice assigned to M-mito + DOX group received mitochondrial transplantation 1 day before DOX (total of 50,000 isolated mitochondria suspended in 80 μL; 20 μL per injection site), as previously described.37 Transthoracic echocardiography was implemented on Vevo3100 high-resolution Imaging System (FUJIFILM Visual Sonics) by an experienced investigator. 2% isoflurane was used to induce anesthesia and 0.5%–1.0% isoflurane to maintain a heart rate in the range of 410–450 beats per minutes. M-mode from the left ventricular long axis view was obtained at the mid-papillary muscle level. Echocardiographic analysis was performed using VevoLAB Version 3.0 software package (FUJIFILM Visual Sonics) and the left ventricular ejection fraction and fractional shortening were measured. At sixth week (4 weekly DOX injection + 2 weeks rest); echocardiography was performed and ventricular tissues were obtained from euthanized mice. Hearts were subjected to Langendorff perfusion as previously descrived38 and, before digestion, atria and ventricles were surgically excised and subjected to the digestion protocol separately. Ventricular cardiomyocytes were seeded onto laminin-coated (1:100 in dH2O, L2020; Sigma) 96-well confocal glass bottom plates and used for CellROX, MitoSox, and TMRM live cell imaging using an Operetta CLS High Content Imaging System (PerkinElmer). For the mitochondrial absorption assay, atrial and ventricular cardiomyocytes were first labeled with Mitotracker Green (green, endogenous mitochondria) and 1.0 × 104/100 μL TMRM-labeled H-mito (red) were added and cells were imaged over time using an Operetta CLS High Content Imaging System (PerkinElmer).
Neonatal mouse ventricular myocytes
Neonatal murine ventricles were surgically excised and NMVMs were isolated as previously described.39 After NMVMs beat, cells were treated with DOX (1 μM), as previously described,31 for 24 h in the DOX group and the mito + DOX group. For mitochondrial transplantation absorption assay, 10,000 NMVMs were seeded into 96-well confocal glass bottom plates. Mitochondria donor cells were stained with TMRM and subjected to mitochondria isolation. NMVM endogenous mitochondria were stained with Mitotracker Green before mitochondria transplantation. Purified mitochondria were added at 0.5 × 104/100 μL (low), 1.0 × 104/100 μL (medium), or 2.0 × 104/100μL (high) concentrations and endogenous and transplanted mitochondrial fluorescence were tracked over time using an Operetta CLS High Content Imaging System (PerkinElmer). For assaying the effect of mitochondrial transplantation into DOX treated NMVMs, mito + DOX group cells were incubated with freshly isolated mitochondrial (1.0 × 104/100 μL) from NMVMs for 2 h before DOX treatment. Contraction videos (12 s for each video, 50 frames per second) of NMVMs and hiPSC-CMs in different conditions were recorded under bright field mode using a 40× objective lens on an Olympus IX83 inverted microscope (Olympus). Each cell location was recorded and repeated every 1 h. Contraction speed and frequency were extrapolated using an established algorithm in MATLAB.40 Data statistics were compared with 0 h (NG condition) for normalization. Mitochondrial respiration (oxygen consumption rates [OCRs]) of NMCMs were measured using on a Seahorse XFe96 Extracellular Flux Analyzer in conjunction with Seahorse Cell Mito Stress Test Kit (Agilent). We seeded 10,000 NMVMs onto Seahorse cell culture plates 1–2 days before measurement. The completed culture media were changed to Seahorse XF DMEM supplemented with 5 μM glucose, 1 μM pyruvate, and 10 μM glutamine 1 h before measurement. After 1 h incubation at 37°C in a CO2-free incubator, OCRs were measured at basal, oligomycin (1.5 μM), FCCP (1 μM), and rotenone/antimycin A (0.5 μM) time points in triplicates. OCRs were normalized to total cell number determined by Hoechst 33342 staining, which is quantified at the end of the Seahorse experiment.
hiPSC-CM and cell cultures
The use of hiPSC was approved by Institutional Stem Cell Committee (SH9H-2018-T83-3) and hiPSC and hiPSC-CM differentiations were carried out as previously described (Table 3).25 RKO cells (human colon carcinoma cell line) were a kind gift from Prof. Shiyan Yu. Fibroblast cell line and C2C12 (rat myoblast cell line) were acquired from the Chinese Academy of Science Cell Bank and cultured in Dulbecco’s modified Eagle medium containing 10% fetal bovine serum (Gibco). Mito Tracker Green, TMRM (tetramethylrhodamine, methyl ester), MitoSox (Thermo Fisher Scientific), and FITC Annexin V Apoptosis Detection Kit (BD Biosciences) were and used per manufacturer’s instructions. Stained cells and mitochondria were assayed on CytoFLEX S (Beckman) and analyzed with Flow Jow10. To mimic myocardial ischemia-reperfusion injury, hiPSC-CMs were placed in a 0.5% hypoxic incubator for 90 min followed by re-oxygenation; membrane potentials were evaluated at 30, 60, and 120 min.
hiPSC-CM mitochondrial respiration (OCRs) of DCM hiPSC-CMs were measured using on a Seahorse XFe96 Extracellular Flux Analyzer in conjunction with Seahorse Cell Mito Stress Test Kit (Agilent), as previously described.25 Day 28 DCM hiPSC-CMs were seeded at 15,000 cells/well onto Seahorse cell culture plates 2 days before measurement. For mitochondrial transplantation, hiPSC-CMs were incubated with 1.0 × 104 H-mito or PBS for 1 h. The completed culture media were changed to Seahorse XF DMEM supplemented with 5 μM glucose, 1 μM pyruvate, and 10 μM glutamine 1 h before measurement. After 1 h incubation at 37°C in a CO2-free incubator, OCRs were measured at basal, oligomycin (2.5 μM), FCCP (1 μM), and rotenone/antimycin A (2 μM) time points in triplicate. OCRs were normalized to total cell number determined by Hoechst 33342 staining, which is quantified at the end of the Seahorse experiment.
For gene expression analyses, 1 μg total RNA was extracted using Trizol (Invitrogen) reagent per manufacture’s protocol. HiScript II Reverse Transcriptase kit (Vazyme) was used to reverse transcribe and generate single stranded cDNA. For mtDNA copy number assay, 100 ng total DNA was isolated using the cardiac tissue or cells Genome DNA Extract kit (Solarbio) according to the manufacturer’s instructions. Primers used can be found in Table S2. Real-time qPCRs were carried out on an AbiQuantStudioTM 6 Flex machine (Applied Biosystems). Samples were assayed in technical duplicates and averaged for final fold enrichment calculations.
Single-cell RNA-seq
DCM hiPSC-CM treated with H-mito or PBS were dissociated using 290 U/mg of pre-warmed collagenase type II (Invitrogen) in Hank’s balanced salt solution. Single-cell suspensions (2 × 105 cells/mL) with PBS were loaded onto microwell chips using the Singleron Matrix Single Cell Processing System (Singleron). Individual libraries were diluted to 4 nM, and sequenced on Illumina novaseq 6000 with 150 bp paired end reads. Raw reads from scRNA-seq were processed to get gene expression matrixes using CeleScope (https://github.com/singleron-RD/CeleScope) v1.1.7 pipeline. STAR v2.6.1a42 was used according to the reference genome GRCm38 (ensembl version 92 annotation) to map reads. UMI counts and gene counts of each cell were acquired with featureCounts43 v2.0.1 software, then used to generate expression matrix files for subsequent analysis. The batch effect between samples was removed by Harmony44 and a UMAP algorithm was applied to visualize cells in a two-dimensional space. Seurat FindMarkers function based on the Wilcox likelihood ratio test with default parameters was used to identify differentially expressed genes (DEGs). Genes expressed in more than 10% of the cells in a cluster and with an average log (fold change) value of more than 0.25 were selected as DEGs. For cell type annotation of each cluster, we combined expression of the canonical markers with knowledge from literatures. To investigate the potential functions of DEGs, GO, and Kyoto Encyclopedia of Genes and Genomes analysis were used with “clusterProfiler” R package. Pathways with p_adj values of less than 0.05 were considered as significantly enriched.
Statistical analysis
All experimental data are presented as mean ± standard error of the mean. Statistical significance between two groups was determined using two-tailed Student’s t-test. For multiple group comparison, One-way ANOVA with Tukey’s multiple comparison was used. A p value of less than 0.05 was considered statistically significant. Data were analyzed and presented with GraphPad Prism.
Acknowledgments
We thank the Electron Microscopy center of Shanghai Institute of Precision Medicine, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, for their technical support and assistance in the electron microscopy. We thank staff at Electron Microscopy Center, Bioimaging Facility, and Protein Facility of Shanghai Institute of Precision Medicine for their expert assistance. The authors also thank all members of the Chang laboratory for valuable suggestions related to experimental design and interpretation. This research was supported by the National Natural Science Foundation of China (82070248 to A.C.Y.C., 82000327 to Y.L.); Shanghai Pujiang Program (19PJ1407000 to A.C.Y.C., 2020PJD028 to Y.L.); The Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning (SHSMU-ZLCX20211700 to A.C.Y.C.); Innovative Research Team of High-Level Local Universities in Shanghai (A.C.Y.C.); The SHIPM-mu fund from Shanghai Institute of Precision Medicine and the Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine (jc201905 to A.Z.).
Author contributions
A.Z., Y.L., and A.C.Y.C. designed the research; A.Z., Y.L., J.P., F.P., A.C.C., H.W., and S.G. performed the experiments and analyzed the results. A.Z., Y.L., C.W., and A.C.Y.C. drafted the manuscript. C.W. and A.C.Y.C. supervised the project.
Declaration of interests
None.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.02.016.
Contributor Information
Changqian Wang, Email: wcqian@hotmail.com.
Alex CY. Chang, Email: alexchang@shsmu.edu.cn.
Supplemental information
Data availability
Single-cell RNA-seq data have been deposited at GEO database (GEO: PRJNA901916). Raw data generated are available from the corresponding author (A.C.Y.C.) on reasonable request.
References
- 1.Conrad N., Judge A., Tran J., Mohseni H., Hedgecott D., Crespillo A.P., Allison M., Hemingway H., Cleland J.G., McMurray J.J.V., et al. Temporal trends and patterns in heart failure incidence: a population-based study of 4 million individuals. Lancet. 2018;391:572–580. doi: 10.1016/s0140-6736(17)32520-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. James S.L., Abate D., Abate K.H., Abay S.M., Abbafati C., Abbasi N., Abbastabar H., Abd-Allah F., Abdela J., Abdelalim A., et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392:1789–1858. doi: 10.1016/s0140-6736(18)32279-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Virani S.S., Alonso A., Benjamin E.J., Bittencourt M.S., Callaway C.W., Carson A.P., Chamberlain A.M., Chang A.R., Cheng S., Delling F.N., et al. Heart disease and stroke statistics—2020 update: a report from the American heart association. Circulation. 2020;141:e139–e596. doi: 10.1161/cir.0000000000000757. [DOI] [PubMed] [Google Scholar]
- 4.van Riet E.E., Hoes A.W., Wagenaar K.P., Limburg A., Landman M.A., Rutten F.H. Epidemiology of heart failure: the prevalence of heart failure and ventricular dysfunction in older adults over time. A systematic review. Eur. J. Heart Fail. 2016;18:242–252. doi: 10.1002/ejhf.483. [DOI] [PubMed] [Google Scholar]
- 5.Ceia F., Fonseca C., Mota T., Morais H., Matias F., de Sousa A., Oliveira A., EPICA Investigators Prevalence of chronic heart failure in Southwestern Europe: the EPICA study. Eur. J. Heart Fail. 2002;4:531–539. doi: 10.1016/s1388-9842(02)00034-x. [DOI] [PubMed] [Google Scholar]
- 6.Bibbins-Domingo K., Pletcher M.J., Lin F., Vittinghoff E., Gardin J.M., Arynchyn A., Lewis C.E., Williams O.D., Hulley S.B. Racial differences in incident heart failure among young adults. N. Engl. J. Med. 2009;360:1179–1190. doi: 10.1056/nejmoa0807265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Savarese G., Lund L.H. Global public Health burden of heart failure. Card. Fail. Rev. 2017;3:7–11. doi: 10.15420/cfr.2016:25:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Authors/Task Force Members. McDonagh T.A., Metra M., Adamo M., Gardner R.S., Baumbach A., Böhm M., Burri H., Butler J., Čelutkienė J., et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). With the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 2022;24:4–131. doi: 10.1002/ejhf.2333. [DOI] [PubMed] [Google Scholar]
- 9.Writing Committee Members. ACC/AHA Joint Committee Members. Aguilar D., Allen L.A., Byun J.J., Colvin M.M., Deswal A., Drazner M.H., Dunlay S.M., Evers L.R., et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure. J. Card. Fail. 2022;28:e1–e167. doi: 10.1016/j.cardfail.2022.02.010. [DOI] [PubMed] [Google Scholar]
- 10.Tahhan A.S., Vaduganathan M., Greene S.J., Fonarow G.C., Fiuzat M., Jessup M., Lindenfeld J., O’Connor C.M., Butler J. Enrollment of older patients, women, and racial and ethnic minorities in contemporary heart failure clinical trials: a systematic review. JAMA Cardiol. 2018;3:1011–1019. doi: 10.1001/jamacardio.2018.2559. [DOI] [PubMed] [Google Scholar]
- 11.Gerber Y., Weston S.A., Redfield M.M., Chamberlain A.M., Manemann S.M., Jiang R., Killian J.M., Roger V.L. A contemporary appraisal of the heart failure epidemic in olmsted county, Minnesota, 2000 to 2010. JAMA Intern. Med. 2015;175:996–1004. doi: 10.1001/jamainternmed.2015.0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsao C.W., Lyass A., Enserro D., Larson M.G., Ho J.E., Kizer J.R., Gottdiener J.S., Psaty B.M., Vasan R.S. Temporal trends in the incidence of and mortality associated with heart failure with preserved and reduced ejection fraction. JACC. Heart Fail. 2018;6:678–685. doi: 10.1016/j.jchf.2018.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neubauer S. The failing heart — an engine out of fuel. N. Engl. J. Med. 2007;356:1140–1151. doi: 10.1056/nejmra063052. [DOI] [PubMed] [Google Scholar]
- 14.Page E., McCallister L.P. Quantitative electron microscopic description of heart muscle cells Application to normal, hypertrophied and thyroxin-stimulated hearts. Am. J. Cardiol. 1973;31:172–181. doi: 10.1016/0002-9149(73)91030-8. [DOI] [PubMed] [Google Scholar]
- 15.Song M., Franco A., Fleischer J.A., Zhang L., Dorn G.W. Abrogating mitochondrial dynamics in mouse hearts accelerates mitochondrial senescence. Cell Metab. 2017;26:872–883.e5. doi: 10.1016/j.cmet.2017.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gong G., Song M., Csordas G., Kelly D.P., Matkovich S.J., Dorn G.W., 2nd Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science. 2015;350:aad2459. doi: 10.1126/science.aad2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song M., Gong G., Burelle Y., Gustafsson Å.B., Kitsis R.N., Matkovich S.J., Dorn G.W. Interdependence of parkin-mediated mitophagy and mitochondrial fission in adult mouse hearts. Circ. Res. 2015;117:346–351. doi: 10.1161/circresaha.117.306859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song M., Mihara K., Chen Y., Scorrano L., Dorn G.W. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 2015;21:273–286. doi: 10.1016/j.cmet.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dorn G.W., Vega R.B., Kelly D.P. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015;29:1981–1991. doi: 10.1101/gad.269894.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang A.C.Y., Chang A.C.H., Kirillova A., Sasagawa K., Su W., Weber G., Lin J., Termglinchan V., Karakikes I., Seeger T., et al. Telomere shortening is a hallmark of genetic cardiomyopathies. Proc. Natl. Acad. Sci. USA. 2018;115:9276–9281. doi: 10.1073/pnas.1714538115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang A.C.Y., Ong S.-G., LaGory E.L., Kraft P.E., Giaccia A.J., Wu J.C., Blau H.M. Telomere shortening and metabolic compromise underlie dystrophic cardiomyopathy. Proc. Natl. Acad. Sci. USA. 2016;113:13120–13125. doi: 10.1073/pnas.1615340113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Emani S.M., Piekarski B.L., Harrild D., Del Nido P.J., McCully J.D. Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 2017;154:286–289. doi: 10.1016/j.jtcvs.2017.02.018. [DOI] [PubMed] [Google Scholar]
- 23.O’Brien C.G., Ozen M.O., Ikeda G., Vaskova E., Jung J.H., Bayardo N., Santoso M.R., Shi L., Wahlquist C., Jiang Z., et al. Mitochondria-rich extracellular vesicles rescue patient-specific cardiomyocytes from doxorubicin injury insights into the SENECA trial. JACC. Cardiooncol. 2021;3:428–440. doi: 10.1016/j.jaccao.2021.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bertero E., O’Rourke B., Maack C. Mitochondria do not survive calcium overload during transplantation. Circ. Res. 2020;126:784–786. doi: 10.1161/circresaha.119.316291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang A.C.Y., Pardon G., Chang A.C.H., Wu H., Ong S.-G., Eguchi A., Ancel S., Holbrook C., Ramunas J., Ribeiro A.J.S., et al. Increased tissue stiffness triggers contractile dysfunction and telomere shortening in dystrophic cardiomyocytes. Stem Cell Rep. 2021;16:2169–2181. doi: 10.1016/j.stemcr.2021.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Preble J.M., Pacak C.A., Kondo H., MacKay A.A., Cowan D.B., McCully J.D. Rapid isolation and purification of mitochondria for transplantation by tissue dissociation and differential filtration. J. Vis. Exp. 2014:e51682. doi: 10.3791/51682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Azimzadeh P., Asadzadeh Aghdaei H., Tarban P., Akhondi M.M., Shirazi A., Khorram Khorshid H.R. Comparison of three methods for mitochondria isolation from the human liver cell line (HepG2) Gastroenterol. Hepatol. Bed Bench. 2016;9:105–113. [PMC free article] [PubMed] [Google Scholar]
- 28.Wieckowski M.R., Giorgi C., Lebiedzinska M., Duszynski J., Pinton P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 2009;4:1582–1590. doi: 10.1038/nprot.2009.151. [DOI] [PubMed] [Google Scholar]
- 29.Frezza C., Cipolat S., Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured filroblasts. Nat. Protoc. 2007;2:287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- 30.Li D.L., Wang Z.V., Ding G., Tan W., Luo X., Criollo A., Xie M., Jiang N., May H., Kyrychenko V., et al. Doxorubicin blocks cardiomyocyte autophagic Flux by inhibiting lysosome acidification. Circulation. 2016;133:1668–1687. doi: 10.1161/circulationaha.115.017443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pan J.-A., Tang Y., Yu J.-Y., Zhang H., Zhang J.-F., Wang C.-Q., Gu J. miR-146a attenuates apoptosis and modulates autophagy by targeting TAF9b/P53 pathway in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2019;10:668. doi: 10.1038/s41419-019-1901-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao Y., Rafatian N., Feric N.T., Cox B.J., Aschar-Sobbi R., Wang E.Y., Aggarwal P., Zhang B., Conant G., Ronaldson-Bouchard K., et al. A platform for generation of chamber-specific cardiac tissues and disease modeling. Cell. 2019;176:913–927.e18. doi: 10.1016/j.cell.2018.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mason F.E., Pronto J.R.D., Alhussini K., Maack C., Voigt N. Cellular and mitochondrial mechanisms of atrial fibrillation. Basic Res. Cardiol. 2020;115:72. doi: 10.1007/s00395-020-00827-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miragoli M., Sanchez-Alonso J.L., Bhargava A., Wright P.T., Sikkel M., Schobesberger S., Diakonov I., Novak P., Castaldi A., Cattaneo P., et al. Microtubule-dependent mitochondria alignment regulates calcium release in response to nanomechanical stimulus in heart myocytes. Cell Rep. 2016;14:140–151. doi: 10.1016/j.celrep.2015.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharpley M.S., Marciniak C., Eckel-Mahan K., McManus M., Crimi M., Waymire K., Lin C.S., Masubuchi S., Friend N., Koike M., et al. Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell. 2012;151:333–343. doi: 10.1016/j.cell.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strauss K.A., DuBiner L., Simon M., Zaragoza M., Sengupta P.P., Li P., Narula N., Dreike S., Platt J., Procaccio V., et al. Severity of cardiomyopathy associated with adenine nucleotide translocator-1 deficiency correlates with mtDNA haplogroup. Proc. Natl. Acad. Sci. USA. 2013;110:3453–3458. doi: 10.1073/pnas.1300690110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun X., Gao R., Li W., Zhao Y., Yang H., Chen H., Jiang H., Dong Z., Hu J., Liu J., et al. Alda-1 treatment promotes the therapeutic effect of mitochondrial transplantation for myocardial ischemia-reperfusion injury. Bioact. Mater. 2021;6:2058–2069. doi: 10.1016/j.bioactmat.2020.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen X., Lin H., Xiong W., Pan J., Huang S., Xu S., He S., Lei M., Chang A.C.Y., Zhang H. p53-Dependent mitochondrial compensation in heart failure with preserved ejection fraction. J. Am. Heart Assoc. 2022;11:e024582. doi: 10.1161/jaha.121.024582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wei X., Chang A.C.H., Chang H., Xu S., Xue Y., Zhang Y., Lei M., Chang A.C.Y., Zhang Q. Hypoglycemia induced mitochondrial connexin-43 accumulation aggravates diabetic cardiomyopathy. Front. Cardiovasc. Med. 2021:800185. doi: 10.21203/rs.3.rs-885699/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huebsch N., Loskill P., Mandegar M.A., Marks N.C., Sheehan A.S., Ma Z., Mathur A., Nguyen T.N., Yoo J.C., Judge L.M., et al. Automated video-based analysis of contractility and calcium Flux in human-induced pluripotent stem cell-derived cardiomyocytes cultured over different spatial scales. Tissue Eng. Part C Methods. 2015;21:467–479. doi: 10.1089/ten.tec.2014.0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell RNA-seq data have been deposited at GEO database (GEO: PRJNA901916). Raw data generated are available from the corresponding author (A.C.Y.C.) on reasonable request.