

Abstract
Over the past thirty years, the importance of chemokines and their seven-transmembrane G protein-coupled receptors (GPCRs) has been increasingly recognized. Chemokine interactions with receptors trigger signaling pathway activity to form a network fundamental to diverse immune processes, including host homeostasis and responses to disease. Genetic and nongenetic regulation of both the expression and structure of chemokines and receptors conveys chemokine functional heterogeneity. Imbalances and defects in the system contribute to the pathogenesis of a variety of diseases, including cancer, immune and inflammatory diseases, and metabolic and neurological disorders, which render the system a focus of studies aiming to discover therapies and important biomarkers. The integrated view of chemokine biology underpinning divergence and plasticity has provided insights into immune dysfunction in disease states, including, among others, coronavirus disease 2019 (COVID-19). In this review, by reporting the latest advances in chemokine biology and results from analyses of a plethora of sequencing-based datasets, we outline recent advances in the understanding of the genetic variations and nongenetic heterogeneity of chemokines and receptors and provide an updated view of their contribution to the pathophysiological network, focusing on chemokine-mediated inflammation and cancer. Clarification of the molecular basis of dynamic chemokine-receptor interactions will help advance the understanding of chemokine biology to achieve precision medicine application in the clinic.
Keywords: Chemokine, Chemokine receptor, Migration, Homeostasis, Genetics, Epigenetics
Subject terms: Chemokines, Predictive markers
Leukocyte migration is a central component of physiological and pathological responses [1–9]. Chemokines are the largest family of cytokines and have chemotactic activity that is essential for host responses in homeostasis and diseases. Chemokines activate cell-surface G-protein-coupled receptors (GPCRs) to generate a regulatory network and play indispensable roles in many processes in immunobiology [10–17]. Imbalances and defects in this system alter host susceptibility to diseases, including diverse inflammatory disorders, infections and malignancies [17–20]. In this article, we highlight the most recent findings related to chemokines and receptors regarding their genetic variations and nongenetic heterogeneity. Our review provides molecular insights for chemokine biology to realize precision medicine.
Background
History
Since the discovery of the human chemokine CXCL8 or IL-8 (CXCL8/IL-8) in the last century [21–23], chemokines have been recognized to exist in a complicated mega system [10–12, 14–17]. The rather short but rich history in the field includes two waves of chemokine identification [11, 23–26]: the first discovery of inflammatory chemokines and receptors that mainly attract neutrophils and macrophages (Mφs) in the early 1990s and the second round of chemokines and receptor discovery after the mid-1990s, which identified those chemokines and receptors related to homeostasis and the trafficking of lymphocytes and dendritic cells (DCs). However, chemokine research was really initiated in 1977 after the discovery of platelet factor 4 (PF4), also called CXCL4, which was the first identified peptide containing a prototypical chemokine structure with uncharacterized chemoattractant activity [24, 26–28]. The discovery that CXCL8 and CCL2 (originally called MCP-1) [21, 22] have chemotactic activity was nevertheless a landmark finding in immunology [23, 24].
Recently, rapid advances in technologies, such as next-generation sequencing (NGS), mass spectrometry and nuclear magnetic resonance (NMR), have created abundant datasets allowing integrative multiomics analysis of chemokines even at single-cell resolution [29–40]. Additionally, increasing divergence of chemokines and their receptors has been revealed at multiple omics levels, likely underlying the functional heterogeneity and regulatory plasticity [20, 29–32, 36, 41–43]. Thus, the focus of chemokine research has been shifting from cell biology to a global perspective in life sciences, academia, and the pharmaceutical industry [37, 44–51]. Unfortunately, despite extensive pharmaceutical research, relatively few drugs are currently approved for clinical treatment [41, 44–47, 52]. An important reason is the undefined molecular basis of multiple chemokine-receptor interactions in various microenvironments [15, 20, 29, 41, 42, 45, 52–54]. Therefore, it is critical to distinguish functionally indispensable relationships from redundant ones by providing an in-depth understanding of chemokine-receptor relationships so that they can be targeted by genetic and nongenetic means. This will allow chemokine-based therapeutics to be more efficiently developed, thus likely generating a third wave of chemokine biology research.
Cell migration and leukocyte trafficking
Cell migration
Migration is not only a hallmark of many normal cells that enables them to participate in diverse physiological processes, such as development, immune responses and host defense [4, 5, 7, 55–58], but is also hijacked by malignant tumor cells for dissemination [4, 6–9, 59, 60]. Notably, four commutative principles to define directed cell migration were recently proposed (e.g., chemotaxis, haptotaxis, durotaxis and topotaxis): signal generation, sensing, transmission and signal execution [1].
Chemotaxis and leukocyte trafficking
Chemokines are best known for their chemotactic activity, which enables them to guide cell migration: gradually increasing the concentration gradient will attract cells toward the source of the chemokine, generally the site of inflection or tissue injury. Leukocyte trafficking, homing and recirculation are pivotal to proper immune responses and immunosurveillance. Leukocyte trafficking is also an indispensable process for immune cell maturation and tissue development and homeostasis and is regulated by chemokines in concert with other cytokines and adhesion molecules [2, 4, 6–8, 14]. As a consequence, infectious or other pathological agents disrupt normal leukocyte trafficking, resulting in uncontrolled flux of immune cells through the endothelial lymph nodes and bone marrow [7–9, 17, 19, 59–61]. In addition, neutrophils also move from the sites of injury back to the vasculature by following chemokine gradients in mice. This so-called neutrophil reverse migration may play a dual role in both local damage protection and systemic inflammation spread [62–65].
Understanding the spatiotemporal migration of immune cells is vital for comprehensively understanding the significance of chemokine-receptor activities and will enable more specific utilization of chemokines [1–3, 7]. However, the biological heterogeneity of chemokines may be underestimated by current state-of-the-art tools, such as superresolution tissue-clearing techniques and real-time analyses of migratory behavior [2, 3, 7, 14, 20, 29, 30]. Therefore, determining how chemokines efficiently bind to GPCRs to initiate signaling cascades and direct migration and desensitize chemokine receptors to impede cell motility for self-limitation within the injured tissue microenvironment, which has been reshaped by chemokines and innate cell recruitment, is a challenge.
Chemokine‒receptor system
Chemokines
Chemokine subfamilies
During the past 30 years, chemokines have been found to be one of the largest subfamilies of cytokines based on systematic nomenclature analyses (Table 1) [10–12, 14–18, 26, 66]. Chemokines are divided into four groups (CC, CXC, XC, and CX3C). The CXC chemokines are subdivided into two categories based on the presence of a glutamyl acid-lysine-arginine (ELR) motif, which determines the unique functions of the members. For example, ELR-containing CXCLs (e.g., CXCL8) are chemotactic for neutrophils, whereas ELR-negative CXC chemokines (e.g., CXCL13) tend to chemoattract lymphocytes but not neutrophils.
Table 1.
Overview of human and mouse chemokines
| Human | Mouse | |||||
|---|---|---|---|---|---|---|
| Symbol | Location | Aliases | Receptor(s) | Symbol | Aliases | Receptor(s) |
| CC | ||||||
| CCL1 | 17q12 | I-309, TCA3,P500,SISe | CCR8, ACKR1 | Ccl1 | TCA-3 | Ccr8 |
| CCL2 | 17q12 | MCP1, MCP-1, SCYA2, MCAF, SMC-CF, GDCF-2, HC11, MGC9434 | CCR2, CCR4, CCR5, ACKR1, ACKR2, ACKR4 | Ccl2 | JE, MCP-1 | Ccr2, Ccr4, Ackr1, Ackr2 |
| Ccl12 | MCP-5, Scya12 | Ccr2 | ||||
| CCL3 | 17q12 | MIP-1-alpha, MIP1A, SCY3, G0S19-1, LD78ALPHA | CCR1, CCR2, CCR4, CCR5, ACKR2 | Ccl3 | MIP-1 alpha | Ccr1, Ccr4, Ccr5, Ackr2 |
| CCL3L1 | 17q12 | MIP1AP, LD78BETA, G0S19-2 | CCR1, CCR3, CCR5, ACKR2 | |||
| CCL3L3 | 17q12 | LD78BETA, MGC12815 | CCR1, CCR3, CCR5, ACKR2 | |||
| CCL4 | 17q12 | MIP-1-beta, ACT-2, AT744.1 | CCR1, CCR3, CXCR4, CCR5, CCR8, ACKR2 | Ccl4 | MIP-1 beta, AT744.1, Act-2 | CCR1, CCR5 |
| CCL4L1 | 17q12 | LAG-1, MIP-1-beta, AT744.2 | CCR1, CCR5 | |||
| CCL4L2 | 17q12 | AT744.2, CCL4 L, SCYA4 L | CCR1, CCR5 | |||
| CCL5 | 17q12 | RANTES, SISd, TCP228, MGC17164 | CCR1, CCR3, CCR4, CCR5, ACKR1, ACKR2 | Ccl5 | Rantes | Ccr1, Ccr3, Ccr4, Ccr5 |
| CCL7 | 17q12 | MCP-3, NC28, FIC, MARC, MCP3 | CCR1, CCR2, CCR3, CCR5, CXCR3, ACKR1, ACKR2 | Ccl7 | MARC, FIC, MCP-3 | Ccr1, Ccr2, Ccr3 |
| CCL8 | 17q12 | MCP-2, HC14 | CCR1, CCR2, CCR3, CCR5, ACKR2, ACKR1, ACKR4 | Ccl8 | MCP-2, HC14, Scya8 | Ccr8, Ackr1, Ackr2 |
| CCL11 | 17q12 | Eotaxin | CCR3, CCR5, ACKR1, ACKR2, CXCR3 | Ccl11 | Eotaxin | Ccr3, Ackr1 |
| CCL13 | 17q12 | MCP-4, NCC-1, SCYL1, CKb10 | CCR1, CCR2, CCR3, CCR5, ACKR2, ACKR1, ACKR4 | |||
| CCL14 | 17q12 | HCC-1, HCC-3, NCC-2, SCYL2 CKb1, MCIF | CCR1, CCR3, CCR5, ACKR1, ACKR2, ACKR4 | |||
| CCL15 | 17q12 | HCC-2, NCC-3, SCYL3, MIP-5, LKN-1, MIP-1D, HMRP-2B | CCR1, CCR3 | Ccl9 | MIP-1 gamma, CCF18, MRP-2 | Ccr1, Ccr3 |
| CCL16 | 17q12 | HCC-4, SCYL4, LEC, NCC-4, LMC, LCC-1, CKb12, Mtn-1 | CCR1, CCR2, CCR3, CCR5, CCR8, ACKR1 | |||
| CCL17 | 16q21 | TARC, ABCD-2 | CCR4, CCR8, ACKR1, ACKR2 | Ccl17 | Tarc, Abcd-2 | Ccr4 |
| CCL18 | 17q12 | PARC, DC-CK1, AMAC-1, DCCK1, MIP-4, CKb7 | CCR8, PITPNM3, CCR3 | |||
| CCL19 | 9p13.3 | ELC, MIP-3b,exodus-3,CKb11 | CCR7, ACKR4, CCRL2 | Ccl19 | MIP-3 beta, ELC, Exodus-3 | Ccr7, Ackr4, CcrL2/LCCR |
| CCL20 | 2q36.3 | LARC, MIP-3a, exodus-1, ST38, CKb4 | CCR6 | Ccl20 | MIP-3 alpha, LARC, Exodus-1, | Ccr6 |
| CCL21 | 9p13.3 | SLC, exodus-2, TCA4, 6Ckine, ECL | CCR7, ACKR4 | Ccl21a | 6Ckine, Exodus-2, SLC, TCA-4, CK beta 9 | Ccr7, Ackr4 |
| Ccl21b | Ccr7 | |||||
| Ccl21d | Ccr7 | |||||
| CCL22 | 16q21 | MDC, STCP-1, ABCD-1, DC/B-CK | CCR4, ACKR2 | Ccl22 | ABCD-1, MDC, DC/beta-CK | Ccr4 |
| CCL23 | 17q12 | Ckb-8, MPIF-1, MIP-3, CKb8 | CCR1, CCR3, ACKR2 | Ccl6 | C10, MRP-1 | Ccr1 |
| CCL24 | 7q11.23 | Ckb-6, MPIF-2, Eotaxin-2, MPIF2 | CCR3, ACKR2 | Ccl24 | Eotaxin-2, MPIF-2, CK beta 6 | Ccr3 |
| CCL25 | 19p13.2 | TECK, Ckb15 | CCR9, ACKR4 | Ccl25 | TECK, CKbeta 15 | Ccr9, Ackr4 |
| CCL26 | 7q11.23 | Eotaxin-3, IMAC, MIP-4a | CCR3, CX3CR1, CCR2, CCR5 | Ccl26 | Ccl26 l, eotaxin-3 | |
| CCL27 | 9p13.3 | CTACK, ALP ILC, ESKINE, ESKY, CTAK | CCR10 | Ccl27a | Ccl27, CTACK, ALP, ILC, PESKY, Eskine | Ccr10 |
| Ccl27b | Ctack, Scya27b | Ccr3 | ||||
| CCL28 | 5p12 | SCYA28, MEC, CCK1 | CCR3, CCR10 | Ccl28 | MEC | Ccr10 |
| CXC | ||||||
| CXCL1 | 4q13.3 | SCYB1, GROa, MGSA-a, NAP-3 | CXCR2, ACKR1 | Cxcl1 | KC, Fsp, Gro1, GRO-alpha | Cxcr2, Ackr1 |
| CXCL2 | 4q13.3 | SCYB2, GROb, MIP-2a, MGSA-b, CINC-2a | CXCR2, ACKR1 | Cxcl2 | CINC-2a, Gro2, MIP-2 | Cxcr2, Ackr1 |
| CXCL3 | 4q13.3 | SCYB3, GROg, M IP-2b, CINC-2b | CXCR2, ACKR1 | Cxcl3 | Dcip1, Gm1960 | Cxcr2 |
| PF4 | 4q13.3 | CXCL4, oncostatin-A, iroplact | CXCR3, CXCR3B, ACKR1 | Pf4 | Cxcr3 | |
| PF4V1 | 4q13.3 | PCXCL1, CXCL4V1, PF4-ALT, PF4A | CXCR3, CXCR3B, ACKR1 | |||
| CXCL5 | 4q13.3 | ENA-78 | CXCR2, CXCR3B, ACKR1 | Cxcl5 | AMCF-II, Cxcl6, LIX, ENA-78, | Cxcr1, Cxcr2, Ackr1 |
| CXCL6 | 4q13.3 | GCP-2, CKA-3 | CXCR1, CXCR2, ACKR1 | |||
| CXCL7 | 4q13.3 | PPBP, THBGB1, NAP-2, CTAPIII, beta-TG | CXCR1, CXCR2, ACKR1 | Cxcl7 | Ppbp, NAP-2, CTAPIII, beta-TG | Cxcr1, Cxcr2 |
| CXCL8 | 4q13.3 | IL-8, SCYB8, LUCT, LECT, MDNCF, TSG-1, NAP-1,3-10 C, MONAP, AMCF-I, LYNAP, NAF, b-NAP, GCP-1, K60, GCP1, NAP1 | CXCR1, CXCR2, ACKR1 | |||
| CXCL9 | 4q21.1 | Mig, SCYB9, Humig, crg-10 | CXCR1, CXCR2, CXCR3, ACKR1, CCR3 | Cxcl9 | MIG, CRG-10 | Cxcr3 |
| CXCL10 | 4q21.1 | IFI10, IP-10, Crg-2, mob-1,C7,gIP-10 | CXCR3, CCR3 | Cxcl10 | CRG-2, IP-10 | Cxcr3 |
| CXCL11 | 4q21.1 | H174, b-R1,I-TAC,IP-9 | CXCR3, CXCR7, ACKR1, ACKR3, CCR3, CCR5 | Cxcl11 | I-TAC, beta-R1, H174, IP-9 | Cxcr3, Cxcr7 |
| CXCL12 | 10q11.21 | SCYB12, SDF-1, SDF-1b, PBSF, TLSF-a, TLSF-b, TPAR1 | CXCR4, ACKR2 | Cxcl12 | SDF-1, PBSF | Cxcr4, Cxcr7 |
| CXCL13 | 4q21.1 | BLC, BCA-1, BLR1 LANGIE, ANGIE2 | CXCR3, CXCR5, ACKR1 | Cxcl13 | BCA-1, BLC | Cxcr5 |
| CXCL14 | 5q31.1 | BRAK, NJAC, bolekine Kec, MIP-2 g, BMAC, KS1 | CXCR4 [14] | Cxcl14 | BRAK, BMAC Bolekine | Unknown |
| Cxcl15 | Lungkine, Weche | Unknown | ||||
| CXCL16 | 17p13.2 | SR-PSOX, CXCLG16, SRPSOX | CXCR6 | Cxcl16 | SR-PSOX | Cxcr6 |
| CXCL17 | 19q13.2 | Dcip1, UNQ473, DMC, VCC1 | Unknown | Cxcl17 | DMC, VCC-1 | Unknown |
| XC | ||||||
| XCL1 | 1q24.2 | Lymphotactin, LPTN, ATAC, SCM-1a, SCM-1 | XCR1 | Xcl1 | Lymphotactin | Xcr1 |
| XCL2 | 1q24.2 | SCM-1 beta | XCR1 | |||
| CX3C | ||||||
| CX3CL1 | 16q21 | Fractalkine, NTN, C3Xkine, ABCD-3, CXC3C, CXC3, DMC, VCC-1 | CX3CR1 | Cx3cl1 | Fractalkine, Neurotactin | Cx3cr1 |
Official gene names in which all letters are uppercase letters refer to human chemokines (left panel), and official gene names in which the first letter is uppercase and the rest are lowercase refer to murine chemokines (right panel). Alternate names in the ALIASES column shown in BOLD represent the most commonly recognized names. Receptors shown in BOLD are active or main receptor(s). The mouse chemokines homologous to human genes are listed in Table 2 [10–12, 14, 17–19, 46]
Chemokine gene orthologs
There are more than 48 human chemokines, with 53 murine counterparts (Table 1). While some chemokines have different names, e.g., murine Ccl6 and Ccl9 versus human CCL15 and CCL23, some chemokines are only present in either humans (such as CXCL8) or mice (e.g., Ccl6 and Ccl12). Table 2 shows that not all chemokines in humans have exact orthologs in mice. For instance, human CXCL1 is not homologous to Cxcl1, and mouse Cxcl5 (LIX) appears more orthologous to human CXCL6 (GCP-2) than CXCL5. Moreover, the numbers of chemokines may not be accurate due to the presence of nonallelic splice variants (SVs) and isoforms. They create considerable genetic and nongenetic heterogeneity, impacting immunosurveillance and susceptibility to a number of diseases. For example, CXCL4L1, a nonallelic variant of CXCL4, is more angiostatic than CXCL4 [67] and is found in humans but not in mice. Additionally, three SVs of Ccl27 (Ccl27a, b, c) are found in mice but not in humans (Table 1). Clarification of orthologous chemokine genes will make it easier to reliably interpret or predict their functionality in mice versus humans [68].
Table 2.
Orthologous chemokine genes between humans and mice
| Orthologous gene pair | Functional information | |
|---|---|---|
| Human gene | Murine gene | Shared recptor(s) |
| CCL1 | Ccl1 | CCR8 |
| CCL2 | Ccl12 | CCR2 |
| CCL3 | \ | \ |
| CCL3L1 | \ | \ |
| CCL3L3 | Ccl3 | CCR1; CCR5 |
| CCL4 | Ccl4 | CCR1; CCR5 |
| CCL4L1 | \ | \ |
| CCL4L2 | \ | \ |
| CCL5 | Ccl5 | CCR1; CCR3; CCR4; CCR5 |
| CCL7 | \ | \ |
| CCL8 | \ | \ |
| CCL11 | Ccl11 | CCR3 |
| CCL13 | Ccl2 | CCR2; D6 |
| CCL14 | \ | \ |
| CCL15 | \ | \ |
| CCL16 | \ | \ |
| CCL17 | Ccl17 | CCR4 |
| CCL18 | \ | \ |
| CCL19 | Ccl19 | CCR7; CCR11 |
| CCL20 | Ccl20 | CCR6 |
| CCL21 |
Ccl21a Ccl21b Ccl21c |
CCR7; CCR11 |
| CCL22 | Ccl22 | CCR4 |
| CCL23 | \ | \ |
| CCL24 | Ccl24 | CCR3 |
| CCL25 | Ccl25 | CCR9; CCR11 |
| CCL26 | \ | \ |
| CCL27 | Ccl27b | CCR10 |
| CCL28 | Ccl28 | CCR10 |
| CXCL1 | \ | \ |
| CXCL2 | Cxcl1 | CXCR2 |
| CXCL3 | Cxcl2 | CXCR2 |
| PF4 | Pf4 | CXCR3 |
| PF4V1 | \ | \ |
| CXCL5 | \ | \ |
| CXCL6 | Cxcl5 | CXCR1; CXCR2 |
| CXCL7 | \ | \ |
| CXCL8 | \ | \ |
| CXCL9 | \ | \ |
| CXCL10 | Cxcl10 | CXCR3 |
| CXCL11 | \ | \ |
| CXCL12 | Cxcl12 | CXCR4; CXCR7 |
| CXCL13 | Cxcl13 | CXCR5 |
| CXCL14 | Cxcl14 | Unknown |
| CXCL16 | Cxcl16 | CXCR6 |
| CXCL17 | Cxcl17 | Unknown |
| XCL1 | Xcl1 | XCR1 |
| XCL2 | \ | \ |
| CX3CL1 | Cx3cl1 | CX3CR1 |
Orthologous chemokine genes between humans and mice were extracted from the NCBI HomoloGene database (https://www.ncbi.nlm.nih.gov/homologene/) via the R package “homologene”, which were mainly based on genetic information. Orthologous chemokine genes pairs with inconsistent names are BOLD. “Shared Receptor(s)” means that both human and murine ligands in the orthologous pair can bind to the same receptor(s), which reflects the functional similarity of homologous genes
Characteristic structure of chemokines
Chemokines are mostly low molecular weight proteins (~8–14 kDa) produced as pro-peptides with a signal peptide that is cleaved to produce active or mature secreted proteins. Most human CXC and CC chemokine-encoding genes are located within clusters on chromosomes 4 and 17, respectively (Table 1 and Fig. 1). Although sequence identity between chemokines varies from approximately 20% to 90%, they are highly conserved overall. The conserved amino acids among chemokines are important for creating their characteristic 3-dimensional and tertiary structures [11, 19, 66, 69]. Some chemokines, such as CCL6, CCL9, CCL23, and CXCR7, contain an extended N-terminus that is proteolytically removed to enhance receptor interaction. Some other chemokines, such as CCL21, contain an extended C-terminus that can also be proteolytically removed to enhance receptor interaction. A few chemokines, such as CX3CL1 (fractalkine) and CXCL16 (SR-PSOX), exist both as cell surface-bound proteins and in soluble forms and elicit immune cell migration and adhesion based on their specific structure (which contains a mucin-like stalk that tethers the chemokine domain to a single transmembrane spanning region). This general structure suggests that chemokine-like factor 1 (CKLF-1) is a novel cytokine, and its chemoattractant capacity is crucial for neutrophils, monocytes and lymphocytes in immune and inflammatory responses [70].
Fig. 1.

Chromosome location of chemokines and receptors. The locations of chemokines and receptors on human chromosomes. The diagrams of chromosomes were adapted from the NCBI website. The different subclasses of chemokines and receptors are highlighted with different colors
Chemokine receptors (CKRs)
CKRs share the seven-transmembrane GPCR architecture that mediates chemotactic signaling. Given that over one-third of clinical drugs function through GPCRs, dissecting the structure–function relationship of GPCRs that contributes to the differences in chemotactic regulatory pathways and mechanisms is crucial for better understanding human physiology and disease etiology and for rational chemokine drug design [37, 38, 44, 45, 47–52].
Chemokines exert their biological activities by interacting with two types of receptors (Table 3). The first so-called classical or conventional chemokine receptors (cCKRs) are a family of Gαi-protein-coupled GPCRs including 10 CCRs for CC chemokines, 6 CXCRs for CXC chemokines, XCR1 for XCL1 and XCL2, and CX3CR1 for CX3CL1 [11, 16, 18, 19, 46, 69]. Chemokines binding GPCRs typically trigger the pertussis toxin-sensitive Gαi G-protein signaling pathway. The second receptor group consists of atypical chemokine receptors (ACKRs), which include six members: ACKR1-4, CCRL2 (ACKR5) and PITPNM3 (ACKR6/NIR1) [12, 19, 71]. ACKRs are also seven-transmembrane receptors that mostly couple with β-arrestins to exert diverse roles. ACKRs apparently act as chemokine scavengers or decoy receptors to negatively regulate immune responses.
Table 3.
The definitive nomenclature of chemokine receptors
| Symbol | Locus | Previous symbols | Alias symbols |
|---|---|---|---|
| CC | |||
| CCR1 | 3p21.31 | SCYAR1, CMKBR1 | CKR-1, MIP1aR, CD191 |
| CCR2 | 3p21.31 | CMKBR2 | CC-CKR-2, CKR2, MCP-1-R, CD192, FLJ78302 |
| CCR3 | 3p21.31 | CMKBR3 | CC-CKR-3, CKR3, CD193 |
| CCR4 | 3p22.3 | CC-CKR-4, CMKBR4, CKR4, k5-5, ChemR13, CD194 | |
| CCR5 | 3p21.31 | CMKBR5 | CKR-5, CC-CKR-5, CKR5, CD195, IDDM22 |
| CCR6 | 6q27 | STRL22 | CKR-L3, GPR-CY4, CMKBR6, GPR29, DRY-6, DCR2, BN-1, CD196 |
| CCR7 | 17q21.2 | CMKBR7, EBI1 | BLR2, CDw197, CD197 |
| CCR8 | 3p22.1 | CMKBRL2, CMKBR8 | CY6, TER1, CKR-L1, GPR-CY6, CDw198 |
| CCR9 | 3p21.31 | GPR28 | GPR-9-6, CDw199 |
| CCR10 | 17q21.2 | GPR2 | |
| CXC | |||
| CXCR1 | 2q35 | CMKAR1, IL8RA | CKR-1, CDw128a, CD181 |
| CXCR2 | 2q35 | IL8RB | CMKAR2, CD182 |
| CXCR3 | Xq13.1 | GPR9 | CKR-L2, CMKAR3, IP10-R, MigR, CD183 |
| CXCR4 | 2q22.1 | LESTR, NPY3R, HM89, NPYY3R, D2S201E, fusin, HSY3RR, NPYR, CD184 | |
| CXCR5 | 11q23.3 | BLR1 | MDR15, CD185 |
| CXCR6 | 3p21.31 | TYMSTR, STRL33, BONZO, CD186 | |
| XC | |||
| XCR1 | 3p21.31 | GPR5, CCXCR1 | |
| CX3C | |||
| CX3CR1 | 3p22.2 | GPR13, CMKBRL1 | CMKDR1, V28, CCRL1 |
| ACK | |||
| ACKR1 | 1q23.2 | FY, DARC | CCBP1, GPD, Dfy, CD234 |
| ACKR2 | 3p22.1 | CMKBR9, CCBP2 | CCR10, D6, CCR9 |
| ACKR3 | 2q37.3 | CMKOR1, CXCR7 | RDC1, GPR159 |
| ACKR4 | 3q22.1 | CCRL1 | CCR11, CCBP2, VSHK1, CCX-CKR, PPR1 |
| CCRL2 | 3p21.31 | HCR, CRAM-B, CKRX, CRAM-A, ACKR5 | |
| PITPNM3 | 17p13.2-p13.1 | CORD5 | NIR1, RDGBA3, ACKR6 |
G protein-mediated signaling and β-arrestin-mediated signaling have generally been considered separate. However, recent findings show direct formation of Gαi:β-arrestin signaling complexes that are distinct from other canonical GPCR signaling complexes, suggesting that G proteins and β-arrestins are cooperative instead of competitive [72, 73].
Functional characteristics of chemokines and CKRs
Subtypes of chemokines and CKRs
Chemokines are classified into homeostatic (or constitutive), inflammatory, and dual function (homeostatic/inflammatory) subtypes based on their expression patterns and functions [11–20, 26, 46, 47]. CKRs are also classified into inflammatory (which control both inflammation and homeostasis) and homeostatic subfamilies [14]. However, accumulated evidence suggests that nonchemokine functions that are also controlled by chemokine ligands and receptors needs to be considered [14, 19, 20, 74]. Homeostatic chemokines and receptors participate in tissue development and basal leukocyte localization, while inflammatory chemokines and receptors regulate immune cell trafficking to sites of inflammation, infection, tissue injury and cancer. The dual subtype chemokines can have either inflammatory and homeostatic activities depending on pathophysiological conditions (Fig. 2) [11, 12, 14, 17, 18, 46].
Fig. 2.

The functional roles mediated by interactions of chemokines with receptors expressed on immune cells. The RNA-seq data were derived from HPA. The relative mRNA expression of chemokines (left hand columns) and receptors (upper right-hand columns) in selected immune cells is shown in the heatmap, with the color based on their transcript per million (TPM) values. The inflammatory and homeostatic chemokines and receptors are shown in red and green, respectively. Chemokines with dual functions are indicated in blue [11, 14, 18, 46]. Chemokine receptors with dual functions are classified into inflammatory families [14]; for example, CCR10/CCL27-CCL28 have been shown to have homeostatic functions [11, 46, 352–354], and several mechanisms have been reported to be involved in inflammation [354]. The atypical chemokine receptors are shown in black. For instance, the platelet chemokine PF4/CXCL4 is quickly released as the first-line inflammatory mediator upon vascular injury and platelet activation. PF4 is also secreted by a variety of immune cells and has also been implicated in the pathology of a variety of inflammatory and autoimmune diseases and cancer [11, 355]. The association of chemokines with receptors was analyzed using STRING (https://string-db.org/), and their interaction networks identified based on the STRING analysis and published reviews [11, 14, 18, 46] are shown in the lower-right hand table, highlighted in purple
Nomenclature
In general, chemokines with the same name from different species are functional orthologs [11, 66, 75]. Cross-interactions between multiple chemokines and their receptors help to increase the plasticity and specificity of chemotactic functions (Fig. 2). A restricted ligand‒receptor relationship, such as a single receptor interacting with only one or two ligands, is common for chemokines primarily involved in homeostatic cell migration. Thus, the chemokine nomenclature can be helpful for understanding the functional relevance (Table 4) [10–12, 14, 26]. For instance, inflammatory chemokines (e.g., CXCL6, CXCL8, CCL2, CCL3, CCL4, and CCL5) are induced in cells or tissues upon exposure to various stimuli, and their genes are located in clusters (e.g., CCL on chromosome 17q12 and CXCL on 4q13) (Table 1 and Fig. 1). This is in contrast with the constitutive expression of homeostatic chemokines (e.g., CCL18 and CXCL13) involved in maintaining the migration and positioning of leukocytes in a steady state. Dual chemokines (e.g., CXCL12) are inducible in many tissues in response to inflammatory stimulants and are also constitutively expressed in primary lymphoid tissues. Moreover, knockout of one of the inflammatory chemokines in a cluster often induces less dramatic phenotypes than knockout of individual homeostatic chemokines. Inflammatory chemokine genes, as a product of evolution, are less stable, which may facilitate host survival and evolution [11, 12, 14, 26, 66, 75]. Since chemokines interacting with each other (chemokine interactome) and coupling with different receptors in a complicated crosstalk network can divergently modulate signal transduction [76, 77], understanding the evolution of the chemokine system may make it easier to analyze potential interactions between chemokine receptor pairs underpinning unique biological functions and to discover novel therapeutic targets.
Table 4.
Logical nomenclature: global insights into the chemokine ligand‒receptor system
| Subfamily | Inflammatory | Homeostatic |
|---|---|---|
| Location of genes * | Clustered | Isolated |
| Expression of genes | Conditional upon inflammation | Constitutive |
| Ligand‒receptor relationship | Multiple ligands for one receptor (e.g., CCL19/CCL21 bind CCR7) | Restrict (one to one) |
| Chemotactic | Neutrophils (CXC), macrophages, activated lymphocytes | Lymphocytes, dendritic cells, non-activated (homing) lymphocytes |
| Phenotype (KO) | Alternative | More dramatic |
| Genomic arrangement (evolution) | Offspring, evolutionary (mutable), dynamic | Oldest, conservative or static |
| Benefits (Host survival) | Immune responses | Homeostasis and development |
| Examples | Lack of CCR5 surface expression due to mutation: susceptible to West Nile virus but not HIV | CXCL12: fetal development across various organs |
The expression of chemokines and receptors
Bulk expression
Chemokines quantitatively dominate the chemical gradients that recruit cells expressing paired receptors. Therefore, precise assessment of chemokine expression in a spatial-temporal manner is critical for defining their functional properties. As large-scale characterization of sequence-function relations has been achieved, high-throughput, informative data are available for deciphering the normal transcriptomic landscapes of chemokine ligands and receptors. Bioinformatic analysis of these data will provide comprehensive insights into the functional diversity and complexity of the regulatory network of chemokines and receptors (Fig. 2), generating a map of chemokines and receptors that are aligned for “easy indexing” of their expression-function relationship. For example, a CCR6-expressing cell will migrate to a site where the ligand CCL20 is produced, while cells with CCR7 expression may migrate toward a site with increased expression of the ligands CCL19 and CCL21.
Single cell-based transcriptomic landscapes
The integrative analysis of data from large-scale transcriptome and single-cell RNA sequencing (scRNA-seq) analyses helps to discriminate the transcriptomic heterogeneity and phenotypic divergence of chemokines and receptors underlying their protective and destructive effects [29–36, 78–82]. As shown in Fig. 2, CKR is present on a cell and interacts with one or multiple chemokines to illustrate the complexity of the chemokine network in microenvironment sites, such as, the tumor immune microenvironment (TIME) [19, 41, 45, 83] and inflammatory sites [84], in severe acute respiratory syndrome coronavirus (SARS-CoV-2) infection [35, 36, 85–88]. The landscape heatmap shown in Fig. 3 shows the patterns of chemokine and receptor genes in multiple single cells, including immune cells.
Fig. 3.

Single-cell expression of chemokines and receptors. A summary of single-cell sequencing analyses of the expression of chemokines and receptors in human tissue cells, including immune cells and total peripheral blood mononuclear cells (PBMCs). Color coding is based on cell type, and each cell type group consists of cell types with common functions. The data were extracted from HPA (https://www.proteinatlas.org/)
The broad expression of chemokines and CKRs has been thought to be redundant, which may be a reason that targeted drugs have not been successfully developed. For instance, multiple myeloma is a clonal plasma cell proliferative malignancy characterized by an abnormal increase in monoclonal paraprotein in the bone marrow. The application of transcriptome sequencing to reveal single-cell patterns in multiple myeloma patients at different disease stages showed distinct tumor cell populations and microenvironments during disease progression [89]. To recapitulate three populations of natural killer (NK) cells (CXCR4 + , CX3CR1+ and CD56 + ), the CXCR4+ cell-dominated primary NK population is replaced by the CD56+ population during the pretransplant stage. After autologous hematopoietic cell transplantation, the CX3CR1 + NK cell population becomes dominant, and the immune profile remains stable until the first relapse. However, in the second relapse stage, a decrease in the CX3CR1+ population was found to be accompanied by the re-emergence of the CD56 + NK cell population. Such observations highlight the highly dynamic microenvironment during disease initiation and progression, which could not have been unraveled by bulk analysis. However, mounting evidence shows that there is specificity for cell migration and nonredundancy in homeostasis [14, 90].
Chemokine network
The interactions of multiple chemokines with multiple receptors, and vice versa, are considered a functional axis mediating different signaling events (Fig. 2). The data have illustrated a complex and dynamic chemokine network underlying the regulation of feedback loops, which confers chemotaxis-based cell behaviors in a spatial-temporal manner [1–3, 7, 14–16, 20, 76, 77, 91–94]. Various posttranslation modifications also affect the network to increase its heterogeneity under diverse extracellular and intracellular conditions. For instance, N-terminal or C-terminal truncation of chemokines catalyzed by proteases alters chemokine-receptor interactions, thus influencing the feedback of chemokine networks [16, 91, 95].
The proper migration of immune cells during infection relies on a balance of positive (rapid initiation of protective immunity) and negative (self-shutdown or limitation) feedback from chemokine networks. Compared to bacterial chemotaxis networks, which form negative feedback loops, eukaryotic chemokine networks appear to be “incoherent feedforward loops” [96], representing more complex regulatory networks. The balance between positive migratory cues and negative arrest signals is critical for the directed migration of leukocytes to sites of damage or infection; e.g., T-cell migration in inflamed tissue is shaped by the competition between T-cell receptor (TCR)-induced migratory arrest (‘stop’) and chemokine (‘go’) signals [7].
In addition to well-established positive feedback signaling, recent studies have revealed the mechanisms underlying the formation of chemokine-related “circuit breakers”, e.g., neutrophil swarming and circadian rhythms.
Swarming
Neutrophils are the most abundant leukocytes in peripheral blood, and their migratory dynamics in tissues are important for host homeostasis and defense [97–101]. Neutrophils also communicate with each other to be recruited to the site of infection or tissue damage through “swarming” in injured tissues to defend against various invading pathogens. However, an overresponse by neutrophils or other immune cells causes healthy tissue damage and the development of various inflammatory and degenerative diseases [97, 102]. Compared to the well-studied positive feedback loop in immune cell swarming [103], how the autoamplifying responses are eventually turned off to restore the delicate balance between protection and destruction is less clear.
GPCR-mediated negative feedback controls excessive swarm formation based on initial neutrophil activation followed by dynamic arrest in a mouse model. Neutrophils release the mediators Ltb4 and Cxcl2 as well as CAMP/CRAMP to amplify cell swarming and clustering [104, 105]. Neutrophils respond to these high concentrations of swarm mediators by desensitizing the corresponding receptors Ltbr1 and Cxcr2. Desensitization is controlled by the GPCR kinase Grk2 and involves Cxcr2 internalization, whereas desensitized Ltbr1 remains on the plasma membrane of the cells. Grk2 desensitizes Ltb4/Cxcl2-driven signaling pathways in activated neutrophils. Thus, neutrophil aggregation is limited while neutrophil bacteria killing is enhanced, a shutdown mechanism that allows them to deactivate their own receptors that respond to swarm signals [105, 106]. In addition to an interesting finding revealing that B-cell subtypes functionally enriched in the lung microvasculature by CXCL13 and CXCR5 can diminish neutrophil responses [107], another strategy to reduce excessive neutrophil recruitment in inflammatory diseases is targeting downstream regulatory element antagonist modulator (DREAM), a multifunctional transcriptional repressor promoting neutrophil recruitment in vascular inflammation by activating IKKβ and NF-κB and enhancing β2 integrin adhesiveness [108, 109].
Circadian rhythms
Leukocyte trafficking around the body and the interstitial migration of immune cells in tissues can be regulated by chemokines and other chemoattractants, and circadian rhythms are essential for all aspects of the relevant biological processes [5, 55–58, 110–112]. The diurnal programming of neutrophils is coordinated by the circadian-related protein Bmal1 (basic helix-loop-helix ARNT like 1, encoded by Arntl)-driven production of CXCL2, which controls neutrophil aging through CXCR2 autocrine signaling [58]; in contrast, Bmal1 coordination with CXCR4, a negative regulator of CXCR2 signaling, results in unrestrained aging. In light of the pervasive effects of circadian time on immune function [57], it is not surprising that targeting the Cxcl12-Cxcr4 axis with G-CSF to mobilize hematopoietic stem and progenitor cells was demonstrated to have more potent effects in mice in the afternoon [113], though the response in humans remains unknown.
Decoding the molecular basis of the chemokine-receptor interactions underlying the regulation of the network architecture will lead to a more comprehensive and precise interpretation of the functional redundancy and specificity of chemokines under various micromovements [20] and resolve other paradoxical aspects of chemokine biology. This may be beneficial for precise therapeutic intervention, e.g., to suppress unwanted inflammation while still enabling appropriate immune responses.
Genetic and nongenetic alterations of chemokines and receptors
Recently developed analytical techniques and statistical capabilities have enabled integration of multiomics biological information with high-resolution quantitative data of chemokines.
Genetic variation
Disease-associated variants of chemokines and receptors
The current understanding of the genomic landscape regarding heterogeneity proposes that multiple genomic alterations rather than a single genomic driver should be used in the molecular classification of diseases or as health risk factors [114–116]. The increased availability of transgenic mouse models (Table S1) and human disease-associated genetic data (Table S2) may make it easier to define genetic aberrations related to chemokines as potential standalone targets [54] or combined biomarkers [117–119]. For instance, omics-based approaches such as genome-wide association studies (GWASs) have been applied to detect numerous genetic variants of chemokines and receptors, among which single nucleotide polymorphisms (SNPs) are the main type of aberration associated with susceptibility to diseases, and some have been identified as host genetic risk factors for clinical testing (Table 5, references shown in Table S2). Figure 4A shows the genetic variants of chemokines and receptors, such as single nucleotide variants (SNVs) and deletions (Dels), found in different health conditions and diseases based on the most recent literature (since 2019). These data suggest that many variants of chemokines and receptors are present in metabolic disorders, and the relationship of these variants with immunity has recently been identified [20, 30, 33, 35, 37–39, 42, 43, 120, 121]. Most variants of chemokines and receptors are associated with multiple diseases or disorders, suggesting their contribution to genetic heterogeneity. Figure 4B shows health disorder-associated chemokine or receptor SNVs, some of which have been used in the clinic for standalone or combined tests (Table 5).
Table 5.
Clinical interpretation of the genomic variants of chemokines and receptors
| Gene | Variant(s) | Type | Disease name | CLNSIG | Status | ID |
|---|---|---|---|---|---|---|
| CXCR4 | NM_003467.3:c.893_1034dup (p.Glu345_Ser346insProHisProLeuCysPheProTrpSerGlnIleTer) | Dup | Warts, hypogammaglobulinemia, infections, and myelokathexis | Likely pathogenic | 1 Star | 1513755 |
| CXCR4 | NM_003467.3:c.1025_1028del (p.Thr342fs) | Del | Warts, hypogammaglobulinemia, infections, and myelokathexis | Likely pathogenic | 1 Star | 1319371 |
| CXCR4 | NM_003467.3:c.1027 G>T (p.Glu343Ter) | SNV | WHIM syndrome 1 | Pathogenic | none | 14022 |
| CXCR4 | NM_003467.3:c.1016_1017del (p.Ser339fs) | Del | Warts, hypogammaglobulinemia, infections, and myelokathexis|WHIM syndrome 1 | Likely pathogenic | 1 Star | 14021 |
| CXCR4 | NM_003467.3:c.1012_1015dup (p.Ser339fs) | Dup | not provided | Pathogenic | 1 Star | 1338437 |
| CXCR4 | NM_003467.3:c.1013 C>G (p.Ser338Ter) | SNV | Warts, hypogammaglobulinemia, infections, and myelokathexis|WHIM syndrome 1|not provided | Pathogenic | 2 Stars | 14023 |
| CXCR4 | NM_003467.3:c.1013 C>A (p.Ser338Ter) | SNV | WHIM syndrome 1 | Likely pathogenic | 1 Star | 1685294 |
| CXCR4 | NM_003467.3:c.1006 G>T (p.Gly336Ter) | SNV | Warts, hypogammaglobulinemia, infections, and myelokathexis | Pathogenic | 1 Star | 1453229 |
| CXCR4 | NM_003467.3:c.1003 G>A (p.Gly335Ser) | SNV | Warts, hypogammaglobulinemia, infections, and myelokathexis|not provided | Conflicting interpretations of pathogenicity | 1 Star | 372600 |
| CXCR4 | NM_003467.3:c.1000 C>T (p.Arg334Ter) | SNV | Warts, hypogammaglobulinemia, infections, and myelokathexis|WHIM syndrome 1|not provided | Pathogenic | 2 Stars | 14020 |
| CXCR4 | NM_003467.3:c.994 G>T (p.Gly332Ter) | SNV | Warts, hypogammaglobulinemia, infections, and myelokathexis | Likely pathogenic | 1 Star | 574352 |
| CXCR4 | NM_003467.3:c.988_989del (p.Ser330fs) | MS | not provided | Pathogenic | 1 Star | 1163801 |
| CXCR4 | NM_003467.3:c.959_960del (p.Val320fs) | MS | Warts, hypogammaglobulinemia, infections, and myelokathexis | Likely pathogenic | 1 Star | 1067193 |
| CXCR4 | NM_003467.3:c.950_953del (p.Leu317fs) | Del | Inherited Immunodeficiency Diseases | Pathogenic | 1 Star | 827702 |
| CXCR4 | NM_003467.3:c.952dup (p.Thr318fs) | Dup | not provided | Pathogenic | 1 Star | 988527 |
| CXCR4 | NM_003467.3:c.582 G>C (p.Leu194Phe) | SNV | Warts, hypogammaglobulinemia, infections, and myelokathexis|not provided | Conflicting interpretations of pathogenicity | 1 Star | 624148 |
| CXCR4 | NM_003467.3:c.219 G>A (p.Thr73 = ) | SNV | Warts, hypogammaglobulinemia, infections, and myelokathexis|not specified | Conflicting interpretations of pathogenicity | 1 Star | 1096326 |
| CXCR4 | NM_003467.3:c.157 A>C (p.Ile53Leu) | SNV | Warts, hypogammaglobulinemia, infections, and myelokathexis|WHIM syndrome 1|not specified|not provided | Conflicting interpretations of pathogenicity | 1 Star | 709395 |
| CXCR4 | NM_003467.3:c.153 T>A (p.Thr51 = ) | SNV | Warts, hypogammaglobulinemia, infections, and myelokathexis|not provided | Conflicting interpretations of pathogenicity | 1 Star | 374560 |
| CXCR2 | NM_001557.4:c.458 G>A (p.Arg153His) | SNV | not provided | Conflicting interpretations of pathogenicity | 1 Star | 809151 |
| CXCR2 | NM_001557.4:c.472 A>T (p.Lys158Ter) | SNV | not provided | Likely pathogenic | 1 Star | 809152 |
| CXCR2 | NM_001557.4:c.623 G>A (p.Arg208Gln) | SNV | WHIM syndrome 2 | Likely pathogenic | 1 Star | 1339556 |
| CXCR2 | NM_001557.4:c.968del (p.His323fs) | Del | WHIM syndrome 2 | Pathogenic | none | 1177051 |
| CCR2 | NM_001123396.4:c.190 G>A (p.Val64Ile) | SNV | Susceptibility to HIV infection | Protective | none | 8267 |
| CCR5 | NM_000579.4:c.-301 + 246 A > G | SNV | CCR5 PROMOTER POLYMORPHISM|Susceptibility to HIV infection|Acquired immunodeficiency syndrome, delayed progression to | Conflicting interpretations of pathogenicity; Protective | none | 8189 |
| CCR5 | NM_001394783.1:c.180 G>T (p.Arg60Ser) | SNV | Susceptibility to HIV infection | Protective | none | 8191 |
| CCR5 | NM_001394783.1:c.303 T>A (p.Cys101Ter) | SNV | Susceptibility to HIV infection | Protective | none | 8188 |
| CXCL12 | NM_199168.4:c.*531 G > A | SNV | Susceptibility to HIV infection | Protective | none | 8762 |
| CCL2 | NG_012123.1:g.2493 A > G | SNV | Spina bifida, susceptibility to|Mycobacterium tuberculosis, susceptibility to|Coronary artery disease, modifier of|Coronary artery disease, development of, in hiv | Pathogenic; risk factor | none | 14207 |
| CCL2 | NG_012123.1:g.2936= | SNV | Susceptibility to HIV infection | Protective | none | 14205 |
| CCL2 | NM_002982.4:c.77-109= | SNV | Susceptibility to HIV infection | Protective | none | 14206 |
| CCL11 | NG_012212.1:g.3760= | SNV | Susceptibility to HIV infection | Protective | none | 8367 |
| CCL5 | NM_001278736.2:c.76+231 T > C | SNV | Human immunodeficiency virus type 1, rapid disease progression with infection by | Pathogenic | none | 12740 |
| CCL5 | NG_015990.1:g.4973 C > G | SNV | Human immunodeficiency virus type 1, delayed disease progression with infection by | Pathogenic | none | 12739 |
The clinically significant genetic variants in chemokine genes were searched from the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar), which is a freely accessible, public archive of reports of the relationships among human variations and phenotypes, with supporting evidence. In the Type column, Dup: duplication; Del: deletion; SNV: single nucleotide variant; and MS: microsatellite. In the Disease Name column, different diseases are separated by “|”. CLNSIG: clinical significance. The column ID ID: clinvar access ID
Fig. 4.

Genetic alterations of chemokine ligands and receptors associated with diseases. A, B. Clinically relevant single nucleotide variations (SNVs) affecting phenotype, as provided in recently published literature. A Chemokine- and receptor-associated SNVs affecting phenotype involved in health and disease. B Health- or disease-related SNVs of chemokine genes (left panel) or chemokine receptor genes (right panel) are highlighted with different colors. The predicted three-dimensional (3-D) structure models of the receptors were downloaded from AlphaFold DB (https://alphafold.ebi.ac.uk/). The inflammatory, homeostatic, and dual chemokine receptors are shown in red, blue, and green, respectively. C Genetic variations in the CCR5/CCR2 gene cluster at 3p21.31. D The structure of the CCL3L gene cluster in 17q12, showing common genetic variations. The 17q location contains the genes encoding most of the CCL subfamily members, including CCL1-5, 7, and 8, indicating their functional relevance. CCL3L, CCL3L3 and the pseudogene C-C motif chemokine ligand 3 pseudogene 1 (CCL3P1, gene ID: 390788, previous name: CCL3L2 (upper panels)) are also found in this location. The amino acid alignments and protein domains (lower panels) of CCL3 (gene ID: 6348), CCL3L1 (gene ID: 6349), and CCL3L3 (gene ID: 414062) are shown
Genetic variants of chemokines and receptors in viral infection
Understanding the genetic basis of the host immune response to viral infection and host resistance will help delineate the plausible genetic determinants of immune diseases and cancer. For example, CCR5 plays an essential role in lymphocyte migration to sites of inflammation and immunosurveillance by binding its natural agonist ligands, including CCL3, CCL3L1, CCL4/MIP-1β and CCL5 (Fig. 2). CCR5, CCR2, CCR3, and CXCR4 are the genes encoding viral coreceptors, and the allelic variants and natural ligands (e.g., CCL3 transcripts and CXCL12/SDF-1) of these genes have been well studied in correlation with natural susceptibility or resistance to human immunodeficiency virus (HIV) infection [122]. Genetic loss-of-function of CCR5/RANTES (CCR5-Δ32, a 32-bp natural deletion resulting in a nonfunctional receptor) confers HIV-1 resistance [123, 124], although CCR5-Δ32 was not shown to be a factor protecting against HIV infection in an analysis of ClinVar data (Table 5). CXCR4, a specific receptor for CXCL12/SDF-1, plays an essential role in hematopoiesis and carcinogenesis (Fig. 4A). Mutations in its gene have been associated with WHIM syndrome. CCR5 and CXCR4 are major coreceptors (CD4 being the primary receptor) for HIV to enter host cells, and these genetic variants have been targeted for antiretroviral therapy interruption, attracting R&D interest [125–131].
CCR5/CCR2 gene cluster and HIV
The CCR5/CCR2 gene cluster, which spans 20 kb on chromosome 3p21.31, has been found to be a highly diverse region with many phenotypic SNVs (Fig. 1 and Fig. 4C); thus, CCR5/CCR2 haplotypes are used for analysis of the association of candidate genes with HIV-1 infection [132, 133]. For instance, CCR2-V64I (rs1799864) has an association with certain SNPs (e.g., rs1799987) in the CCR5 cis-regulatory region (Fig. 4C) and plays a beneficial role during HIV-1 infection [133, 134]. Genotyping of multiple variants (9 in CCR5/CCR2, 2 in CCL3 and 2 in CCL5) was performed in HIV-seropositive individuals, and the results showed that specific combinations of variants in genes from the same signaling pathway could define an HIV-1 resistant phenotype [135]. As shown in a longitudinal case-controlled study of 502 adult HIV-positive participants, the circulating concentrations and gene expression patterns of CXCL12 (rs1801157) and CCL2 (rs1799864) were associated with immune recovery status; furthermore, strong linkage disequilibrium (LD) between CCR2 rs1799864 and CCR5 rs1800024 and between CCR2 rs1799864 and CCR5 rs333 determined the baseline plasma CCR2 and CCR5 concentrations in participants with poor immune response. This suggests that dual blockade (CXCL12 and CCL2, CCR2 and CCR5) may be a useful therapeutic strategy for future clinical trials [117]. Further integrated genome and transcriptome analyses of antibody response and viral antigen positivity elucidated novel genetic determinants related to viral infection and the immune response, and CXCR5 was identified as one of 7 novel genes associated with viral antibody response. This indicates that chemokine genes beyond the human leukocyte antigen (HLA)-class II region not only contribute to host‒virus interactions but dominate the landscape of the viral antibody response [119].
To the SNP rs7082209 affects an area upstream of CXCL12 and is associated with decreased susceptibility to tuberculosis (TB) in HIV-positive individuals [136]. CCR5 promoter polymorphisms, including rs2734648 and rs1799987, in the Chinese Han population were shown to confer an extraordinarily increased risk of susceptibility to pulmonary TB and TB progression, possibly because they affect transcription factor-binding sites to regulate CCR5 expression [137]. Deficiency of the GATA-1 binding site in the ACKR1/DARC promoter, which abolishes erythroid gene expression in Duffy-negative individuals, thus conferring resistance to Plasmodium vivax, was demonstrated to be the underlying mechanism [138, 139]. Another novel mechanism of an SNP in the regulation of HIV-1 infection was recently uncovered by Kulkarni et al. [140]. The SNP rs1015164A/G maps downstream of CCR5 (approximately 34 kilobases) and leads to variation in an activating transcription factor 1 (ATF1)-binding site that controls the expression of CCR5AS (Fig. 4C). CCR5AS blocks interactions between the RNA-binding protein Raly and the CCR5 3ʹ untranslated region, protecting CCR5 mRNA from Raly-mediated degradation. Reduced CCR5 expression induced by inhibition of CCR5AS diminished infection of CD4 + T cells with CCR5-tropic HIV, thus influencing HIV disease outcome [140]. Since the genetic factors affecting these chemokines and receptors are located in noncoding regions, such as promoters, enhancers and intergenic regions, their alterations may increase the transcriptional regulatory plasticity of chemokine molecules. This is evidenced by the common super-enhancer (SE) located in the genomic region for XCR1 and CCR1; the SE is near the CCR1 gene locus and is linked to high transcriptional activity of CCR1 [141]. Differential polymorphisms occurring at splicing sites may lead to aberrant alternative splicing variants (SVs) with functional divergence and even opposing activities. However, this possibility remains to be further explored.
The CCL3/CCL3L1-CCR5 axis in HIV
CCL3 is a natural ligand for the HIV-1 coreceptor CCR5, colocalizing with CCL3L3 and the pseudogene C-C motif chemokine ligand 3 pseudogene 1 (CCL3P1) in a region of human 17q12 containing most of the CCL chemokines (Fig. 1, Fig. 4D), indicating their functional relevance. CCL3 has three SVs, but only CCL3-V1 encodes the 92-aa chemokine CCL3. CCL3L1 (SCYA3 L/MIP1A) in the 17q12 alternate locus shares ~96% nucleotide sequence identity with CCL3 and encodes a 93-amino acid preprotein with differences in several key amino acid residues. CCL3L3 is a centromeric copy of CCL3L1 with identical amino acids (Fig. 4D).
The affinity of CCL3L1 binding to CCR5 was much higher than that to CCL3 and CCL5, and CCL3L1 is the most potent agonist of CCR5 and suppresses HIV-1 infection [142, 143], whereas CCL7/MCP-3 is the main antagonistic ligand of CCR5. The inhibitory effect of CCL3L1 on the entry of HIV-1 into CCR5-expressing cells is due to the proline (P) that is visible in position 2 of mature CCL3L1 (after removal of the signaling peptide). Moreover, individuals tend to have distinct copy number variations (CNVs) of CCL3L1, whereas there is typically only a single copy of CCL3 per haploid genome. Thus, CCL3L1 may be a dominant HIV-suppressive chemokine. Generic variants such as CNVs of CCL3L1 have been implicated in HIV-1 susceptibility [144]. Interestingly, CCL3 antisense RNA 1 (CCL3-AS1) has several SVs and was found to map near CCL3 in 17q12, with yet to be clarified patterns of expression and function.
Chemokine variants in COVID-19
An understanding of the genetic and immunological determinants of resistance to infection (e.g., autosomal recessive deficiencies of CCR5 in HIV-1 infection and deficiency of ACKR1 in Plasmodium vivax infection) may provide a road map for identifying monogenic or common determinants of resistance or susceptibility to infection with SARS-CoV-2 [54, 118]. In addition to a suggestive association between CCL2-A2518G gene variants and the severity of COVID-19 [145], a genome-wide study showed associations between the risk of severe COVID-19 and a multigene locus at 3p21.31 and the ABO blood group locus at 9q34.2. Regarding the locus at 3p21.31, the frequency of the rs11385942 insertion–deletion GA or G variant is related to predisposition to the most severe forms of COVID-19; and the gene cluster including CCR9, CXCR6 and XCR1 (Fig. 5D) is involved in T-cell and dendritic cell function. The identified 3p21.31 (CCR5/CCR2) gene cluster may thereby act as a genetic biomarker for susceptibility to COVID-19 infection [146]. Exploring the effect of chemokine gene variants on SARS-CoV-2 infection and disease severity will provide important insights into the immune mechanisms preventing infection.
Fig. 5.

Regulatory chromatin markers and health- and disease-associated CpG methylation. A Chromatin in nondividing cells can be divided into euchromatin and heterochromatin, and the two chromatin states refer to areas that are transcriptionally active and inactive, respectively. Epigenetic factors include DNA/RNA methylation and histone modifications, RNA transcript variations (e.g., different splice forms of RNA as epigenetic regulators), and noncoding RNAs (ncRNAs, such as miRNAs, sRNAs, and ncRNAs as well as RNAi and AS), as well as chromatin architecture remodeling [150, 157–166]. Covalent epigenetic modifications of histones and DNA are the most common epigenetic marks, and they alter neighboring nucleosomes to impact the accessibility of loci for transcription factors and coregulators. The gene or regulatory element associated with these epigenetic modification marks indicates the status (active, repressive or poised). These epigenetic marks can be determined using epigenetic analyses. Examples include chromatin immunoprecipitation (ChIP), micrococcal nuclease (MNase) and DNase I hypernasality site (DHS) assays with PCR or sequencing techniques [152, 161, 162, 169–174]. B Heatmap showing the differentially methylated chemokine genes associated with health and disease. C Chemokine genes with differential CpG methylation associated with normal processes such as aging, body weight control, immune responses, metabolism and diseases such as neurological and mental disorders. D Health- and disease-associated CpG methylation is found in the CCR5/CCR2 gene cluster
However, a growing number of studies have revealed that pervasive somatic mutations may occur in nonmalignant tissues, and not all genetic abnormalities lead to functional changes or increased susceptibility to diseases [147, 148]. Unlike the monogenetic determinants affecting CCR5, some genetic variants may act as “noise” and may not be good markers of disease conditions or biomarkers, resulting in poorly targeted immunotherapies [15, 20, 29, 41, 42, 45, 52–54].
Epigenetic alterations in the regulation of chemokine genes
Nongenetic heterogeneity propagated by epigenomic and transcriptomic alterations facilitates cellular functional plasticity, tissue specificity and phenotypic diversity [6, 20, 30–33, 35, 37–39, 42, 43, 120, 121, 149, 150]. Many novel sequencing-based approaches have been developed to unravel the heterogeneous and diverse epigenetic mechanisms, which has increased the understanding of the evolutionary and ecological roles of ‘nongenetic’ inheritance (NGI) [151–155]. The identification of epigenetic markers and distinct epigenotypes related to health and disease conditions can help identify promising strategies for disease management. Here, we summarize recent findings and discuss current concepts related to the role of chemokine epigenetics in the regulation of immune surveillance, host protection and tissue development.
Epigenetic regulation of gene expression
Common epigenetic mechanisms
Among the numerous intracellular mechanisms and mediators, epigenetic alterations, that is, nongenetic heritable alterations, play an indispensable role in regulating chemokine molecules; some epigenetic factors are key determinants of immune cell migration and memory, development and homeostasis [6, 31–33, 149, 150, 156], thus being defined as the “epiregulome” [149, 150]. Epigenetic events affect diverse gene regulation mechanisms leading to epigenetic modifications, as well as remodeling and modification of the conformation of chromatin architecture [150, 157–166]. Chemokine epigenetic marks can be combined with reference epigenomes to define cell function and identity with high resolution and spatiotemporal dynamics and in a cell type/tissue-specific manner [31–33, 167, 168]. Cell type/tissue-specific epigenomic patterns and transcriptional patterns define immune cell lineages and can be used in future studies of the role of chemokines in immune dysregulation in diseases and aging (Fig. 5A).
Epigenetic technologies
Many novel computational strategies can be used for analysis of data derived from chromatin immunoprecipitation (ChIP), micrococcal nuclease (MNase) and DNase I hypernasality site assays with next-generation sequencing (Fig. 5B) [152, 161, 162, 169–174]. ChIP assays and related technologies, such as chromosome conformation capture (3 C) coupled to sequencing (Hi-C), Hi-ChIP technologies, and chromatin interaction analysis by paired-end tag sequencing (ChIA-PET), are more accurate assays for detecting chromatin architecture at the genome scale [164, 175–179]. For instance, Hi-ChIP technologies have been employed to identify topologically associating domains (TADs), genomic regions organized by preferential interactions between chromatin and DNA sequences that play important roles in the proper control of chemokine gene expression by inducing the formation of chromatin loops. e.g., via promoter–enhancer interactions and super-enhancer (SEs).
DNA methylation
The levels of CpG methylation and demethylation
DNA methylation (DNAm), also called CpG methylation (CpGm) or 5-methylcytosine (5mC) modification, is a dynamic process catalyzed by members of the DNA methyltransferase (DNMT) enzyme family, which add methyl groups to the 5ʹ carbon of cytosine bases to create 5mC. Notably, demethylation of 5mC can occur throughout different physiological processes and is involved in many pathological conditions: 5mC is oxidized by ten-eleven translocation methylcytosine dioxygenases (TETs) to produce 5-hydroxymethylcytosine (5hmC), which has been shown to regulate the pluripotency of embryonic stem cells, neuron development, and tumorigenesis [180, 181].
Regulator of DNA methylation
In mammals, DNMT3A and DNMT3B respond to de novo methylation patterns early in development, while DNA methylation is maintained during cellular replication by DNMT1 interacting with ubiquitin-like with PHD and RING finger domain 1 (UHRF1), a key epigenetic regulator [182]. Recently, UHRF1 has been identified as a modulator suppressing multiple exacerbating factors in rheumatoid arthritis (RA) and found to contribute to negative feedback mechanisms that suppress multiple pathogenic events in arthritis, including epigenetic silencing of CCL20, a common UHRF1 target gene among cytokine-, RA-, and antiapoptosis-related genes. This suggests that the epigenetic mechanisms associated with the induction of RA-specific aberrations should be elucidated so that they can be controlled by epigenetic drugs for RA therapy [183].
The cooccurrence of DNMT-associated methylation and TET-associated demethylation confers methylation heterogeneity and is related to tumorigenesis; for example, tumor suppressor genes can be repressed by methylation rather than hypermethylation [184]. Therefore, cooccurrence of several factors, such as DNA methylation, may represent a unique layer of epigenetic regulation of gene expression that may facilitate breaking of symmetry during differentiation [181, 184–186]. Although an increasing number of studies have reported a role of reagent-induced or TET-mediated demethylation of chemokines in various disorders, such as CXCL8 [187] in osteoarthritis, Cxcl1 [188] in lung inflammation, and CCL2 [189] and CXCL12 [190] in carcinogenesis, further studies should consider the cooccurrence of several factors related to methylation, such as the ratio between the levels of methylation and demethylation (including 5-mC and 5hmC levels), to precisely interpret the regulatory effect of DNA methylation on chemokine expression in immune cells [181, 191–193].
Although many DNA methylation-associated chemokines have been found to be related to epigenetically driven pathways in the context of the specific immune microenvironment, few studies have focused on 5hmC modification of chemokines. A study using immunohistochemistry to detect 5hmC and T-cell-attracting chemokines in different-grade cervical lesions demonstrated that 5hmC was positively associated with the expression of T-cell-attracting chemokines (including CXCL9, CXCL10, and CXCL11) but negatively associated with the severity of cervical lesions, indicating that immunosuppression was present in precancerous cervical lesions [194]. Furthermore, 5hmC levels were increased in CXCR4 gene bodies in colorectal cancer (CRC) compared to adjacent mucosa, although differential CXCR4 methylation was not found [195]. Considering the therapeutic potential of the CXCL12-CXCR4/ACKR3 axis in cancer, 5hmC is a promising biomarker for precision medicine [196–198]. However, the challenge that remains is to develop innovative tools to reveal the differences between 5mC and 5hmC modification, which will enable more accurate data interpretation, as these modifications have different effects (5mC is a repressive mark, while 5hmC is an intermediate form of demethylation), and especially aid the development of techniques to interrogate circulating cell-free DNA (cfDNA) [191, 199].
Localization of DNA methylation underpins immune cell and tissue type specificity
That disruption of DNA methylation, not only CpG methylation density but also CpG methylation position, occurs early in tumors makes DNA methylation the best epigenetic marker, as it conveys information about health conditions and diseases, and targeting DNA methylation is a promising approach for disease management [158]. In addition to the well-known epigenetic silencing of tumor suppressive chemokines that results from promoter CpG island (CpGI) hypermethylation, CpG methylation can occur in CpGI shores, CpGI shelves, and open seas. Different methylation statuses exist in differentially methylated regions (DMRs), which contain multiple consecutive methylated CpGs and have implications for disease development and progression. These differentially methylated positions (DMPs) and/or DMRs are vital for tissue development and cell differentiation in a tissue-/cell-specific manner [200].
The DMPs and DMRs scattered throughout the genome also have functional implications that remain to be explored. For instance, CpG or CpGI methylation (iCpGIm) in the gene-body has opposite effects to pCpGIm, which affects mRNA splicing, contributing to transcriptome diversity [191, 201]. More tissue-specific DMRs are found in CpGI shores (~2 kb away from islands), the methylation of which shows a higher correlation with gene expression than the methylation of CpG islands [202]. In general, DNA methylation and demethylation regulate spatial and temporal gene expression (e.g., CpGI methylation silencing of tumor suppressor genes), impact chromatin remodeling (hypermethylated heterochromatin repeats), and are critical for embryonic development, lineage identity and cellular differentiation processes. Since epigenetic regulation of myeloid and lymphoid cell differentiation and function is important for appropriate host defense and organ homeostasis, which shape innate and adaptive immune responses, DNA methylation was proposed as “a transcriptional regulator of the immune system” [203]. The immune system has thus become a prototypical model for studying epigenetic effects on immune cell type- and stimulus-specific transcriptional programs, and relevant studies have generated a wealth of data [31, 161, 169, 170, 203]; furthermore, integrated analysis focusing on chemokine epigenetics may provide in-depth opinions about immune surveillance and homeostasis development. For instance, Roy et al. observed that differentially methylated sites were hypomethylated in innate immune cells but hypomethylated in adaptive immune cells [31]. These cell-specific differential methylation patterns may be used to define epigenetic states and gene expression profiles of innate and adaptive immune cell types that may underpin the functional differences of developmentally distinct cell types. Interestingly, that CXCR5 has B-cell-specific DMRs reveals that cell-specific differentially methylated sites are associated with enhancer-related epigenetic marks (e.g., DNase I hypernasality sites, H3K4me1, and H3K27ac) but not with H3K4me3.
Differential CpG site methylation in health conditions and diseases
The distribution of DNA methylation is a main consideration when selecting methodology, designing experiments and performing bioinformatic analysis [200, 204]. Epigenome-wide association studies (EWAS) have increased ability to measure global CpG methylation and are thus useful for uncovering context-dependent regulatory roles of chemokines [205–207]. Using system-level approaches, relevant studies of epigenetic epidemiology have revealed extensive DMPs in chemokine genes that are phenotypically associated with different health conditions and diseases (Table S3, Table 6). Furthermore, these DMPs could be combined to develop aging- or perinatal-related risk factors for chemical hazard (such as air pollution) assessment. These methylation-driven chemokine gene signatures may be prognostic biomarkers in immune and genetic, metabolic, neurological and mental disorders and cancer (Fig. 5B, C) (Table 7).
Table 6.
Differentially methylated CpG sites occurring in chemokines and receptors are associated withhealth conditions and diseases
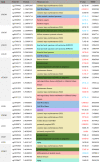 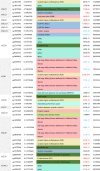 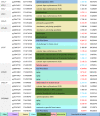
|
The categorization of health condition or disease-associated phenotypes/traits is highlighted with different colors as above
The data were selected from the literature since 2019, and detailed information is shown in Table S3. In the phenotypes/trait, RE: the correlation with positive, BLAC: negative, or BLUE: no indication (NA).
CAD coronary artery disease, CD Crohns disease, HDP hypertensive disorders in pregnancy, HNSCC head and neck squamous cell carcinoma, SLE systemic lupus erythematosus
Table 7.
Overview of selected clinical studies of agents targeting chemokines and receptors
| Target | Drug | Mechanism of Action | NCT | Status | Condition or Disease | Phase |
|---|---|---|---|---|---|---|
| CCR2/CCR5 | Cenicriviroc (CVC) (TAK-652; TBR-652) | dual antagonist of CCR2/CCR5 | NCT04593940 | Completed | Covid19 | Phase 3 |
| CCR4 | Mogamulizumab (KW-0761) | humanized monoclonal antibody that binds to CCR4 | NCT01728805 | Completed | Cutaneous T-Cell Lymphoma | Phase 3 |
| CCR5 | Vicriviroc | antagonist of CCR5 | NCT00523211 | Completed | HIV Infections|Acquired Immunodeficiency Syndrome | Phase 3 |
| NCT00474370 | Completed | HIV Infections|Acquired Immunodeficiency Syndrome | Phase 3 | |||
| CCR5 | Maraviroc | CCR5 antagonist | NCT02881762 | Completed | Hepatitis C|Human Immunodeficiency Virus | Phase 4 |
| NCT02159027 | Completed | AIDS Dementia Complex | Phase 2|Phase 3 | |||
| NCT01389063 | Unknown | Endothelial Dysfunction | Phase 4 | |||
| NCT01866267 | Completed | Human Immunodeficiency Virus|AIDS | Phase 4 | |||
| NCT01190293 | Completed | HIV Infection | Phase 4 | |||
| NCT01449006 | Completed | Human Immunodeficiency Virus (HIV) | HIV Associated Neurocognitive Disorders (HAND) | Phase 4 | |||
| NCT03402815 | Completed | HIV Infection With Other Conditions|Cardiovascular Risk Factor|Atherosclerosis|Inflammation | Phase 4 | |||
| NCT01235013 | Unknown | HIV-1 Infection | Phase 4 | |||
| NCT01348308 | Completed | HIV-1 Infection|AIDS | Phase 3 | |||
| NCT00884858 | Completed | HIV Infections | Phase 4 | |||
| NCT00666705 | Completed | Healthy | Phase 4 | |||
| NCT00735072 | Completed | HIV Infection | Phase 4 | |||
| NCT00853840 | Completed | AIDS | Phase 4 | |||
| NCT01896921 | Completed | HIV | Phase 3 | |||
| NCT00875368 | Completed | HIV Infections | Phase 4 | |||
| NCT03178084 | Completed | HIV/AIDS | Phase 3 | |||
| NCT01327547 | Completed | HIV Coinfection | Phase 4 | |||
| NCT01384682 | Completed | HIV | Phase 4 | |||
| NCT00966329 | Completed | HIV | HIV Infections | Phase 4 | |||
| NCT01275625 | Completed | HIV | Phase 4 | |||
| NCT00870363 | Completed | HIV Infections | Phase 4 | |||
| NCT00426660 | Completed | HIV Infections | Phase 3 | |||
| NCT01680536 | Completed | HIV | Phase 4 | |||
| NCT03129113 | Completed | Hepatic Steatosis|HIV-1-infection | Phase 2|Phase 3 | |||
| NCT01013987 | Unknown | HIV-1 Adults Patients|AIDS|Triple Class Failure | Phase 4 | |||
| NCT00478231 | Completed | Acquired Immunodeficiency Syndrome|HIV Infection | Phase 3 | |||
| NCT00925756 | Completed | HIV Infections | Phase 4 | |||
| NCT00808002 | Completed | HIV Infections | Phase 3 | |||
| NCT00844519 | Completed | HIV Infection|Cardiovascular Disease|Inflammation|HIV Infections | Phase 3 | |||
| NCT01533272 | Completed | HIV Infection | Phase 4 | |||
| NCT00717067 | Completed | Human Immunodeficiency Virus (HIV) Infection | Phase 4 | |||
| NCT02519777 | Completed | HIV Infections | Phase 4 | |||
| NCT01060618 | Completed | HIV Infections | Phase 2|Phase 3 | |||
| NCT00098293 | Completed | HIV-1 | Phase 3 | |||
| NCT00098722 | Completed | HIV Infections | Phase 2|Phase 3 | |||
| NCT00098306 | Completed | HIV Infections | Phase 2|Phase 3 | |||
| NCT00098748 | Completed | HIV Infections | Phase 2|Phase 3 | |||
| NCT03218592 | Completed | HIV/AIDS | Phase 4 | |||
| NCT01154673 | Completed | Acute HIV Infection | Phase 2|Phase 3 | |||
| NCT01637259 | Completed | Proteinuria|HIV | Phase 4 | |||
| NCT01367236 | Completed | HIV|Impaired Cognition | Phase 4 | |||
| NCT04965662 | Completed | HIV-1-infection | Phase 4 | |||
| NCT01033760 | Completed | HIV-1 Infections | Phase 3 | |||
| NCT01378910 | Completed | HIV | Phase 4 | |||
| NCT00935480 | Completed | HIV Infections | Phase 3 | |||
| NCT00624195 | Completed | HIV Infections | Phase 2|Phase 3 | |||
| NCT02302547 | Completed | HIV | Phase 3 | |||
| NCT02588820 | Unknown | HIV Infections | Phase 3 | |||
| NCT00537394 | Completed | HIV Infections | Phase 3 | |||
| NCT02016924 | Recruiting | Acquired Immune Deficiency Syndrome (AIDS) | HIV Infections | Phase 2|Phase 3 | |||
| NCT03631732 | Completed | HIV-1 Infection | Phase 3 | |||
| NCT02121795 | Completed | HIV-1 Infection | Phase 3 | |||
| NCT02469246 | Completed | HIV-1 Infection | Phase 3 | |||
| NCT00708162 | Completed | HIV Infection | Phase 3 | |||
| NCT02859961 | Active, | HIV | Phase 2|Phase 3 | |||
| CCR5 | Leronlimab (PRO140) | a humanized monoclonal antibody to CCR5 | NCT04901676 | Suspended | COVID-19 Pneumonia | Phase 3 |
| NCT04901689 | Suspended | COVID-19 Pneumonia | Phase 3 | |||
| NCT03902522 | Active, | HIV-1-infection | Phase 2|Phase 3 | |||
| NCT02859961 | Active, | HIV | Phase 2|Phase 3 | |||
| NCT02990858 | Active, | Hiv|Human Immunodeficiency Virus | Phase 2|Phase 3 | |||
| NCT02483078 | Completed | HIV | Phase 2|Phase 3 | |||
| NCT05271370 | Active, | HIV-1-infection | Phase 2|Phase 3 | |||
| CCR9 | Vercirnon (CCX282-B; GSK1605786) | antagonist of CCR9 | NCT01277666 | Completed | Crohn’s Disease | Phase 3 |
| CXCR1/CXCR2 | Ladarixin | dual CXCR1 and CXCR2 antagonist | NCT04628481 | Recruiting | Drug: Ladarixin|Drug: Placebo | Phase 3 |
| CXCR1/CXCR2 | Reparixin | CXCR1/2 antagonist | NCT05254990 | Recruiting | COVID-19 Pneumonia|Sars-CoV-2 Infection | Phase 3 |
| NCT04878055 | Completed | Pneumonia, Viral | Phase 3 | |||
| NCT01967888 | Completed | Pancreatectomy for Chronic Pancreatitis | Phase 2|Phase 3 | |||
| NCT01817959 | Completed | Islet Transplantation in Diabetes Mellitus Type 1 | Phase 3 | |||
| CXCR4 | Plerixafor (SDZ-SID-791; JLK-169; SID-791; AMD3100, AMD-3100, JM-3100, JM 3100; trade name Mozobil) | antagonist of CXCR4 | NCT02056210 | Completed | Diabetes | Phase 4 |
| NCT05087212 | Recruiting | Autologous Haematopoietic Stem Cell Transplant | Phase 4 | |||
| NCT00838357 | Completed | Lymphoma (Non-Hodgkin’s Lymphoma)|Hodgkin’s Disease or Multiple Myeloma|Front Line Mobilization|Transplantation | Phase 3 | |||
| NCT01164475 | Completed | Non-Hodgkin’s Lymphoma | Phase 4 | |||
| NCT02006225 | Unknown | Autologous Stem Cell Transplantation | Phase 4 | |||
| NCT01767714 | Completed | Non-Hodgkin’s Lymphoma | Phase 3 | |||
| NCT02231879 | Completed | Myelokathexis|Infections|Neutropenia|Warts|Hypogammaglobulinemia | Phase 2|Phase 3 | |||
| NCT00103662 | Completed | Multiple Myeloma | Phase 3 | |||
| NCT00103610 | Completed | Lymphoma, Non-Hodgkin | Phase 3 | |||
| NCT01146834 | Completed | Multiple Myeloma | Phase 3 | |||
| NCT04000698 | Recruiting | Refractory Acute Myeloid Leukemia|Refractory Acute Lymphoblastic Leukemia | Phase 3 | |||
| NCT04293185 | Recruiting | Sickle Cell Disease | Phase 3 | |||
| CXCR4 | AMD-070 (AMD11070; AMD070; X4P-001; Mavorixafor) | antagonist of CXCR4 | NCT03995108 | Active, | WHIM Syndrome | Phase 3 |
| BL-8040 (Motixafortide; TF-14016; BKT-140; T-140) | antagonist/inhibitor of CXCR4 | NCT03246529 | Active, | Multiple Myeloma | Phase 3 | |
| CXCR4 | POL6326 (Balixafortide TFA) | CXCR4 Antagonist | NCT03786094 | Active, | Metastatic Breast Cancer|Locally Recurrent Breast Cancer | Phase 3 |
| CCL5 | OTR4120 (CACICOL20) | Glycomimetic | NCT02119039 | Completed | Keratoconus | Phase 4 |
Data source: clinicaltrials.gov (https://www.clinicaltrials.gov/). The selected drug was ongoing over phase III or completed. In the column of NCT, green: completed; black: ongoing
In the column of mechanism, bold black: antagonist, bold blue: antibody
Studying the DMPs in chemokine clusters will help to elucidate relevant epigenetic mechanisms underlying their effects on immune gene regulation, and the results will highlight the importance of accounting for cellular heterogeneity and phenotypic diversity in chemokine biology. As shown in Fig. 5D, most of the differentially methylated CpGs in the CCR5/CCR2 gene cluster are located in intergenic regions of CCR genes, which may contain interspersed repetitive sequences (IRSs) or functional elements (e.g., tissue-specific enhancers or SEs). Their epigenetic disruption may affect the expression of chemokines that are linked to diseases. IRSs (e.g., LINE-1, SINE-1, and Alu elements) are identical or nearly identical tandem DNA repeats that are disseminated throughout the genome; they are often packaged in heterochromatin or exist in regulatory and intragenic regions as a result of transposition or retrotransposition events. These elements were originally called “junk” repeats, but they are now recognized to represent a large source of individual variation among humans, and long stretches of these elements are usually called CNVs. Aberrant methylation of IRSs has been shown to alter chromosomal stability and cause genetic variations and abnormal RNA splicing and expression, thus playing a role in chemokine-mediated immune disorders and carcinogenesis [208, 209]. For instance, LINE-1 and other repeats were found to be hypomethylated in lymphocytes and neutrophils from patients with systemic lupus erythematosus (SLE) [210], possibly affecting SLE-related genes, and this finding may have implications for diagnosis or immune system modification in immunity and inflammation.
Since the CCR5/CCR2 gene cluster acts as a central regulatory region, it might be a useful model for studying disease-associated epigenetic alternations and genetic variants controlling chemokine expression and function to identify cell-specific enhancers buried in intergenic regions [207, 211]. As mentioned, dissection of global site-specific methylation patterns related to transcription factors, other epigenetic modifications, and gene expression in human immune cell types showed differential methylation sites in enhancer-related DMRs of CXCR5 that defined cell specificity [31].
RNA methylation
Chemical modifications of ncRNA and N6-methyladenosine (m6A) are novel epigenetic modifications that can be studied to decipher functional correlations between mRNAs and certain biological processes, including cell differentiation and cell fate determination, a field termed “epitranscriptomics” [160]. For instance, the hypoxia-induced m6A demethylase alkB homolog 5 (ALKBH5) removes m6A and stimulates tumor macrophage recruitment and tumor immune escape through epigenetic and epitranscriptomic upregulation of CXCL8 in glioblastoma [212]. ALKBH5 in neutrophils can be downregulated during bacterial infection. ALKBH5-mediated m6A promoted the migration capability of neutrophils by altering RNA decay, affecting the protein expression of its targets (for example, upregulating the expression of the neutrophil migration-promoting factor CXCR2 and downregulating the expression of the neutrophil migration-suppressing GPCR PTGER4). Therefore, activation or upregulation of the ALKBH-5-m6A demethylation axis is an intrinsic mechanism that drives efficient neutrophil migration [213]. Genome-phenome studies of the chemokines that dominate chemokine biological and regulatory pathways are needed to identify disease-specific epigenetic markers and targets [31, 214–217].
Epigenetic modifications
Super-enhancer regulation of chemokines and receptors
Studies of epigenetics using innovative techniques have revealed that promoter-enhancer compatibility is important in higher-order chromatin structures, e.g., three-dimensional (3D) chromatin loops known as TADs may recruit and stabilize transcription factor complexes to exert long-range gene transcriptional regulation [177, 178, 218–221], and most regulators binding distal enhancers in intronic or intergenic regions regulate tissue-specific pathways and drive condition-specific gene expression, ultimately determining cell identity [218, 220, 222]. SEs are large clusters of enhancers with aberrantly high levels of transcription factor binding and are thus critical for cell type specification and oncogenic transcription [223–226]. The epigenetic reader protein bromodomain‐containing protein 4 (BRD4) belongs to the family of bromodomain and extraterminal (BET) chromatin proteins, which are important targets for small molecule compounds [227, 228]. In addition, an in vivo study provided proof-of-concept for targeting BRD4 with a cell-permeable small molecule (JQ1) in NUT midline carcinoma (NMC), an aggressive squamous carcinoma that develops due to a fusion oncogene (e.g., NUT in frame with BRD4) [229]. I-BET, a synthetic compound that selectively binds BET, showed the capacity to interfere with the binding of BETs to acetylated histones to disrupt the formation of the chromatin complexes. For example, I-BET induced highly selective suppression of the expression of key LPS-inducible cytokines (Il6, Ifnb, Il1b, Il12a) and chemokines (Cxcl9 and Ccl12) as well as the chemokines Ccl2-5 and Cxcl1/2, but did not affect the cytokine Tnf, in bone marrow-derived macrophages (BMDMs). However, treatment of BMDMs with I-BET suppressed the expression of TNF-inducible key proinflammatory cytokine (Il1b, Il1a) and chemokine genes (Ccl5, Cxcl10, Cxcl2/3) associated with epigenetic modifications and CpG content and that contribute to sepsis pathogenesis, conferring protection against LPS-induced endotoxic shock and bacteria-induced sepsis [230, 231].
Dysregulation of the inflammatory response disrupts the tissue homeostasis resulting from coordinated epigenetic regulation of the master transcription factor NF‐κB, rapidly inducing inflammatory gene expression [232, 233]. In human umbilical vein endothelial cells (HUVECs), the key inflammatory factor TNF‐α, induces the formation of large NF‐κB‐bound enhancer clusters (NF‐κB‐SEs) associated with active histone marks (H3K27ac), and BRD4 forces the expression of proinflammatory genes, including chemokine genes [231]. A recent study showed that TNF‐α rapidly induces co‐occupancy of lysine demethylases 7 A (KDM7A) and 6 A (UTX) at NF‐κB‐associated SEs in human ECs, which is essential for activation of NF‐κB‐dependent inflammatory genes, such as demethylated KDM7A H3K9 in the target genes CXCL2 and CXCL8 and demethylated UTX H3K27 in CCL2. As exemplified by CXCL8 and other gene loci, Hi‐C in combination with ChIA‐PET revealed that TNF‐α‐responsive SE‐SE interactions were newly formed within sub‐TADs with decreased levels of H3K9me2 and H3K27me3 in SEs immediately following TNF‐α stimulation. These data suggest that coordinated demethylation of H3K9 and H3K27 by KDM7A and UTX might be functionally involved in the formation of SEs and the chromosomal conformation changes that activate their associated genes during early inflammatory responses in human ECs [234, 235]. Interestingly, the vital roles of KDM7A and UTX in the regulation of TNF-NF-κB axis-dependent inflammatory genes were found to be regulated by a TNF-responsive microRNA, miR-3679-5p. This is in line with the results of an integrative meta-analysis of the relationship between SEs and miRNA networks, which showed that SEs mark cell-type-specific miRNAs associated with cancer hallmarks, suggesting that SEs are major drivers of the tissue-specific miRNome [236].
Along the same lines, Fanucchi et al. showed that TNF-responsive genes, including chemokine genes, are arranged in TADs to form chemokine-SEs [237, 238]. These chromosome loops allow chemokines located in different chromosomes to form chemokine-SEs that are spatially available to be regulated by a subset of lncRNAs expressed within the TADs of HUVECs, termed immune gene-priming lncRNAs (IP-lncRNAs or IPLs). IPLs can direct the WD repeat-containing protein 5 (WDR5)–mixed lineage leukemia protein 1 (MLL1) complex across multiple chemokine promoters (e.g., CXCL8, CXCL1, CXCL2 and CXCL3 in human 4q21) by forming cis contacts with TNF-responsive genes associated with H3K4me3. One particular IPL, upstream master lncRNA of the inflammatory chemokine locus (UMLILO), forms the UMLILO–WDR5–MLL1 axis in the cis regulation of H3K4me3 modification at CXCL chemokine promoters within the same TAD. TNF-activated UMLILO is also related to a classic inducer of trained immunity, β-glucan, which can increase the transcription of several IPLs and chemokines to train immunity responses. Moreover, UMLILO is absent in mouse CXC-chemokine SEs, and mice lack β-glucan-trained immune responses. Insertion of UMLILO into mouse chemokine SEs resulted in training of CXCL genes with H3K4me3 epigenetic accumulation. Considering the differences in CXCL gene loci between mice and humans, this study may partly explain why mice are more resistant to inflammatory stimuli than humans. The study supports the epigenetic regulation of InscRNAs by chemokines [239] and provides strong evidence that UMLILO–WDR5–MLL1 axis-mediated chromatin looping of CXC-chemokine SEs controls immune gene priming in response to innate immune cell signaling to generate a nonspecific enhanced response to pathogen reinfection.
By using ChIP–seq and 4C-seq and analyzing published databases, a putative SE for multiple CXCLs located 20 kb upstream from the CXCL gene loci was identified in alcoholic hepatitis (AH) and found to orchestrate TNFα/NF‐κB-induced upregulation of CXCL chemokines (e.g., CXCL1, CXCL6 and CXCL8, related to neutrophil recruitment and infiltration) associated with active histone modifications in liver sinusoidal endothelial cells (LSECs), a major source of CXCL chemokines regulated by the TNFα/NF-κB signaling axis in the liver. BET inhibitors suppressed the expression of CXCLs by inhibiting transcription factor binding at CXCL SE and promoter sites. These high-throughput epigenomic studies in both humans and mice support a conserved role for CXCL SEs in regulating CXCL gene involvement in propagating inflammatory signaling by inducing chemokine expression and show the therapeutic potential of BET inhibition in AH treatment [240]. Owing to their broad activity against a large number of inflammatory genes and their specificity for their target genes, SEs are attractive candidates for pharmacological intervention [164, 218, 240].
Epigenetic modifications of chemokines in tumor-infiltrating lymphocytes (TILs)
Polycomb group (PcG) proteins are crucial epigenetic regulators that function as transcriptional repressors via two main epigenetic complexes, polycomb repressive complex 1 (PRC1) and PRC2, the aberrant activity of which is involved in carcinogenesis. The core components of PRC2 include embryonic ectoderm development (EED), suppressor of Zeste 12 homolog protein (SUZ12) and enhancer of Zeste homolog 1/2 (EZH1/2). EZH1/2 have a Su(var) 3–9, enhancer-of-zeste and trithorax (SET) domain with histone methyltransferase activity that monomethylates, dimethylates or trimethylates lysine 27 of histone H3 (H3K27me1/2/3). PRC2 exerts repressive effects by binding to the repressive marker H3K27me3 to repress expression from neighboring nucleosomes. PcG proteins can form distinct multiprotein complexes in various contexts, such as in early development, during an immune response, and cancer and play a role in proliferation-differentiation balance and metabolism. PcG proteins thus provide the basis for mechanistic divergence, and interfering with PcG functions may be a powerful strategy to counter tumor progression [241, 242].
Trafficking of T cells to tumors Tumor-infiltrating lymphocytes (TILs) are key players generating “hot” tumor microenvironment (TMEs), and chemokines direct the trafficking of T cells and other immune cells [243]. TILs are more responsive to immunotherapy combined with inhibitors of programmed cell death protein 1 (PD1) and its ligand PDL1 [244]. Impaired intratumoral accumulation of T cells in the TME leads to poor cancer immunotherapy efficacy and resistance, and chemokines, e.g., CCL5, CXCL9, CXCL10, and CX3CL1, are crucial for T-cell infiltration due to their ability to induce migration of immune cells [245–247]. An increasing number of studies have recently revealed the importance of the epigenetic modification of chemokines in the specific regulation of the trafficking of T cells to tumors.
Epigenetic modification for T-cell trafficking and PD-L1 checkpoint blockade A study showed that EZH2-mediated H3K27me3 modification and DNMT1-mediated DNA methylation block ovarian tumor production of the Th1-type chemokines CXCL9 and CXCL10 (CXCL9/10) and subsequently enable effector T-cell trafficking to the TME. Combined inhibition of EZH2 and DNMT1 augmented the expression of the inflammatory chemokines CXCL9 and CXCL10, which increased TILs and decreased tumor progression, thus improving the therapeutic efficacy of PD-L1 checkpoint blockade and adoptive T-cell transfusion in tumor-bearing mice [248]. In addition, epigenetic silencing of the Th1-type chemokine CXCL9/10 via deposition of H3K27me3 mediated by PRC2 components (EZH2, SUZ12 and EED) impaired T-cell trafficking toward colon tumors, suggesting that PRC2/H3K27me3-mediated Th1-type chemokine silencing is a novel immune evasion mechanism in human colon cancer. Therefore, epigenetic restoration of repressed Th1-type chemokine expression to enhance T-cell infiltration into tumors may improve the clinical efficacy of cancer therapy [249]. Consistent with these reports, a class of pyrimidone compounds, represented by BR-001, was recently found to exert antitumor effects by upregulating CXCL10 to trigger CD8+ T-cell trafficking toward tumor sites. This may be associated with the capacity of BR-001 to directly bind EED in the H3K27me3-binding pocket to disrupt the EED-H3K27me3 interaction. Although no synergistic effect was observed in the BR-001 and anti-PD-1 combination group, the study suggests that the regression of colon tumors may be induced by inhibiting PRC2 modulation of the tumor immune microenvironment [250].
Downregulation of interferon-γ inducible protein 16 (IFI16), a direct target of EZH2, decreases stimulator of interferon genes (STING) activation and downstream CXCL10/11 expression in response to trastuzumab treatment in HER2+ breast cancer (BC). Dual inhibition of EZH2 and histone deacetylases (HDACs) significantly activated IFI16-dependent immune responses to trastuzumab. Another combination strategy, a novel histone methylation inhibitor combined with an HDAC inhibitor, induced complete tumor eradication and long-term T-cell memory in a HER2 + BC mouse model. These findings reveal the IFI16-CXCL10/11 signaling pathway as the crucial pathway conferring anti-HER2 trastuzumab resistance, and this pathway can be epigenetically targeted by EZH2 and HDAC inhibitor combination therapy to induce complete tumor eradication through increased CD8 + T-cell infiltration and induction of long-term T-cell memory in HER2+ breast cancer [251]. An analysis of TCGA data from clinical specimens from patients with triple-negative breast cancer (TNBC) showed that the expression of immune regulatory genes, including CD8 + T-cell attracting chemokine genes (CCL5, CXCL9, CXCL10) and the gene encoding the immune checkpoint molecule PD-L1, was negatively associated with the levels of histone lysine specific demethylase 1 (LSD1). Furthermore, LSD1 inhibition resulted in H3K4me2-induced restoration of immune regulatory gene expression, which in turn increased CD8 + T-cell tumor infiltration to overcome resistance to immunotherapy [252].
Epigenetic regulation of the CCL19/21-CCR7 axis in dendritic cells (DCs)
CCR7, coupled with its natural ligands CCL19 and CCL21 (the CCL19/21-CCR7 axis), controls the trafficking of DCs and metastasis and invasion of some malignant tumor cells [6, 253–255]. Abnormal DC trafficking results in immune pathologies, including autoimmune responses, infectious diseases, allergic diseases and cancer [6, 256]. Epigenetic modifications such as the transcriptionally repressive H3K27me3 modification associated with Ccr7 were shown to determine the migratory capacity of distinct DC subsets (migratory conventional DCs vs nonmigratory bone marrow DCs) [257] and affect epigenetic alteration of CCR7 and CXCR4 in tumor cells [258], and the NAD-dependent deacetylase sirtuin 6 (SIRT6) may promote the ability of CXCR4-positive DCs to migrate to the afferent lymph nodes in the development of experimental autoimmune encephalomyelitis (EAE) [259].
A recent study was possibly inspired by the role of lncRNAs in the epigenetic regulation of chemokine signals; for example, breast cancer antigen-resistance 4 (BCAR4) mediates cooperative epigenetic regulation of the CCR7-CCL21 axis to promote tumor cell migration [239] and regulates DC differentiation by interacting with transcription factors [260]; the study identified epigenetic regulation of the timely termination of DC trafficking at the late stage to prevent unwanted inflammation [261]. CCR7 mediates rapid but transient DC migration to initiate protective immunity and maintain immune homeostasis. In addition to the well-established CCR7-triggered DC recruitment during the early stages of immune defense against invading pathogens, CCR7 stimulation also upregulates the long noncoding RNA Lnc-Dpf3 via m6A demethylation to prevent its degradation, and Lnc-Dpf3 feedback directly binds the transcription factor hypoxia-inducible factor 1-alpha (HIF-1α) and suppresses its activity to restrain CCR7-mediated DC migration and inhibiting glycolysis. This study provided important insights into the crosstalk between epigenetic mechanisms and metabolic pathways in regulating the network of DC-based immune responses. Therefore, understanding of the epigenetic regulation of CCR7-dependent DC migration is essential for developing therapeutic and vaccination strategies for inflammatory and autoimmune disease treatment.
Chromatin organization of chemokines in neutrophil extracellular traps (NETs)
NET formation and its inducing factors
Upon activation, neutrophils eliminate pathogens through phagocytosis, degranulation, and cytokine production. NETs are net-like extracellular fibers of processed chromatin (DNA-histone complexes) decorated with neutrophil-derived and adhered proteins that trap and neutralize microbes. NET formation follows a well-orchestrated cell death program called NETosis. During NETosis, neutrophils release large amounts of DNA and histones into tissues, where they can target microbes or serve as chemoattractants [99, 262–265]. The well-described role of histones as damage-associated molecular patterns (DAMPs), such as PAD4-mediated citrullinated histone H3 (citH3), contributes to the antimicrobial function and pathogenic effect of NETs. DNA, as a sticky polyanionic molecule, is capable of binding to bacterial cell walls for immobilization of pathogens on NETs to direct contact with cytotoxic molecules in the NET-DNA complex. Therefore, citH3 and cell-free DNA (cfDNA) are considered more specific NET markers under various disease conditions [264, 266–268]. Although NETs protect against infection, their inappropriate release is also implicated in the pathology associated with inflammatory and autoimmune diseases and cancer [98, 266, 267, 269–272]. As such, an understanding of NET formation and its inducing factors will enable the development of improved therapeutic targeting strategies, and NETs and their inducing factors represent a good model to study the epigenetic regulation of the inflammatory chemokines underlying dynamic changes in chromatin configuration and spatiotemporal remodeling.
NETs in SLE
The cfDNA structures released due to chromatin decondensation and spreading can also directly clog blood vessels and establish vessel-blocking thrombi or interact with anti-nuclear antibodies, forming immune complexes in SLE [273, 274]. SLE also features low-density granulocytes (LDGs) and increased levels of a pathogenic neutrophil subset. A detailed analysis of the bulk and single-cell transcriptomic, epigenetic, and functional profiles of lupus LDGs showed that lupus neutrophil subsets differed phenotypically and functionally in terms of NET formation, chemotaxis mediated by formyl peptide receptors 1 (FPR1), CXCR1 and CXCR3, and other processes, suggesting neutrophil heterogeneity and the putative role of neutrophils in the pathogenesis of SLE associated with vascular damage [274].
NETs in malignancy
NET components in cancer Experimental and clinical studies have revealed the presence of NETs and their components in a variety of cancers [275, 276]. The effect of NETs on malignancy and metastasis and the contribution of NETs to TME heterogeneity have attracted emerging interest. NET-DNA binds to CCDC25, a transmembrane DNA receptor, on tumor cells and enhances cell motility and facilitates NET-mediated distant metastases, revealing therapeutic target potential of targeting the cytoplasmic membrane DNA sensor for metastasis [277]. NETs induced by tumor-derived CXCL8 coupled with CXCR2 promoted diffuse large B-cell lymphoma (DLBCL) progression by activating Toll-like receptor 9 (TLR9), an important DNA sensor, and its downstream pathways. Aggressive interactions of tumor cells and NETs via the CXCL8–CXCR2 axis in DLBCL thus have implications for prognostication and targeting NET formation, and this crosstalk represents a new therapeutic target for this challenging disease [278] and other diseases; e.g., the HMGB1/RAGE/CXCL8 axis could be targeted to inhibit glioma progression [279]. Park et al. demonstrated that metastatic breast cancer cells can recruit neutrophils via the expression of CXCL1/2 and induce NET formation at sites of dissemination in the absence of infection. The NETs in turn support the spread of metastasis, and this could be inhibited by administration of DNase I-coated nanoparticles [280]. Inflammatory stimulants (e.g., CXCL1, CXCL2 and CXCL8) can stimulate neutrophil chemotaxis and activation to generate chromatin webs, thereby inducing NET formation in the omentum, a preferential metastasis site of ovarian cancer, while inhibition of NETs decreased the implantation of cancer cells [281].
NETs and proteases In addition to the IFI16-CXCL10/11 signaling pathway conferring anti-HER2 trastuzumab resistance [251], a study in a mouse model revealed that NET formation induced from sustained lung inflammation could convert dormant disseminated cancer cells (DCCs) into aggressive lung metastases by affecting NET-associated proteases, neutrophil elastase (NE) and matrix metalloproteinase 9 (MMP9), providing important insights into the microenvironmental control of DCC reactivation from dormancy, which could have therapeutic implications [282]. Indeed, proteases that actively degrade proinflammatory mediators have been shown to be enriched in NETs. As trypsin activation, leukocyte recruitment, and impaired microvascular perfusion participate in the pathophysiology of severe acute pancreatitis (AP) with systemic inflammation and lung damage, the relationship of NETs with trypsinogen activation-mediated inflammation and tissue injury was investigated in a mouse AP model induced by taurocholate or L-arginine [283]. Neutrophil depletion blocked taurocholate-induced deposition of NETs in the pancreas. The administration of DNase I to mice reduced neutrophil infiltration and tissue damage in the inflamed pancreas and lung, accompanied by decreased levels of blood amylase, IL-6, HMGB1 and CXCL2/MIP-2. The addition of NETs and histones to acinar cells induced the production of trypsin and STAT3. Notably, increased levels of cfDNA and DNA–histone complexes were found in the serum of AP animals and patients with severe AP. That NETs contribute to the development of AP and regulate organ inflammation and injury suggests that they might be a useful target for ameliorating local and systemic inflammation in severe AP. Therapeutic strategies directed against NET formation may provide a clinical benefit by reducing inflammatory tissue damage in patients.
The digestive activity of the trypsin enzyme may facilitate tissue inflammation and cell migration/metastasis in association with inflammatory chemokines [284, 285], and thus, it would be interesting to determine the interplay between trypsin family members and chemokines in chromatin dynamics during NETosis. A novel biohybrid platform that was recently developed by conjugating DNase I to a nonfouling microgel could be employed as a nonthrombogenic active microgel-based coating for blood-contacting surfaces to reduce NET-mediated inflammation and microthrombi formation [286], thus aiding monitoring of processes related to cell mobility, including inflammatory infiltration and cancer metastasis.
NETs for neutrophil self-limitation
In-depth studies of inflammation-related carcinogenesis have provided proof of concept for NET inhibition strategies for the prevention of thrombotic/vascular complications, cancer propagation, and severe infections, such as sepsis and COVID-19 [269, 272, 287, 288]; however, NETs are also involved in noninfectious, sterile inflammation and acute injuries associated with autoimmunity and cancer [289]. Furthermore, NETs participate in a powerful negative feedback mechanism that self-limits neutrophil activation by providing a temporary (pop-up) chemokine-degrading scaffold [268, 290, 291]. For instance, CXCL8-induced NETs have been preliminarily shown to contribute to cancer development and progression; furthermore, blockade of CXCL8 or its receptors (CXCR1 and CXCR2) is being pursued for drug development, and clinical trials of such drugs used alone or in combination with anti-PD-L1 checkpoint inhibitors are already ongoing [271]. Although NETs are highly dynamic and complicated chromatin structures, recent technological advances in strategies such as Hi-ChIP [152], spatially-resolved transcript amplicon readout mapping (STARmap), a 3D intact-tissue RNA sequencing [162, 292] may help us to dissect the epigenetic interactions between DNA and histones at high resolution and the epigenetic regulation of chemokine-mediated pathways. Owing to the central role of chemokines in the control of cell mobility, such studies will shed light on the immune response and tissue homeostasis and lead to the identification of translatable precision biomarkers and therapeutic targets.
Overall, these insights suggest that epigenetic modifications are dynamically controlled to regulate chemokine expression via specific inflammatory and homeostasis pathways and serve as reversible controls that have potential as therapeutic targets for disease prevention and management. However, researchers still need to develop convenient techniques to rapidly assess immune cell responses to treatments at single-cell resolution [293].
Abnormal expression of chemokines and receptors
Differential expression of chemokines confers phenotypic heterogeneity
Aberrant expression of chemokines and receptors has been reported in various diseases, including inflammatory diseases and cancer [14, 18, 19, 294–296]. For example, the serum levels of the IFN-α-induced chemokines CCL2, CXCL10 and CCL19 were found to correlate with lupus patient age and disease duration and thus have implications for monitoring disease activity and the determining the degree of organ damage in SLE [297]. In contrast to low expression of the favorable prognostic marker CX3CL1 induced by epigenetic silencing, expression of CCL3, CCL8, CCL15, CCL18 and CXCL9 was negatively correlated with prognosis and T-cell infiltration in nephroblastoma [298].
Analysis of TCGA data showed differential expression patterns of chemokines and receptors in cancer patients with different clinical outcomes (Fig. 6), suggesting that cancer type-related transcriptional heterogeneity may cause functional heterogeneity affecting clinical outcomes, revealing potential prognostic targets for translational studies [20]. For example, decreased expression of CXCL12 was associated with unfavorable overall survival (OS) and disease-free survival (DFS) in all types of cancer, whereas CXCR4 was highly expressed in several cancer types. For example, in stomach cancer (STAD), high CXCR4 expression was associated with favorable OS, while its high expression was associated with poor DFS in patients with kidney renal clear cell carcinoma (KIRC). An even more extreme example is that dysregulation of CCL19, a homeostatic chemokine that interacts with CCR7 to play a crucial role in the development of lymphoid organs [299] (Fig. 2), showed both tumor-suppressive and oncogenic effects in cancer. Despite there being no significant changes in CCL19 expressed in an analysis of TCGA data, higher CCL19 expression and secretion were found in metastatic nodes of patients with head and neck squamous cell carcinoma (HNSC) than in benign nodes or primary tumors, and the CCL19-CXCR5 axis was found to exert prosurvival signaling associated with tumor progression and disease relapse [300]. In contrast, CCL19 was expressed at significantly lower levels in CRC tissues. Upregulation of CCL19 expression could inhibit CRC angiogenesis by promoting inhibition of the Met/ERK/Elk-1/HIF-1α/VEGF-A pathway by miR-206, suggesting a novel therapeutic strategy for antivascular treatment in CRC [301].
Fig. 6.

Chemokines and receptor expression and its association with clinical outcomes in human cancer. The associations of chemokine expression and receptor expression (A) with clinical patient outcomes (B) in multiple cancer types was identified using the limma method and the GEPIA tool (http://gepia.cancer-pku.cn/). Red: upregulated in tumor samples (log2FC > 1 and adjusted p < 0.05), blue: downregulated in tumor samples (log2FC < -1 and adjusted p < 0.05), gray: stable. BLCA bladder urothelial carcinoma, BRCA breast invasive carcinoma, CESC cervical squamous cell carcinoma, CHOL cholangiocarcinoma, ESCA esophageal carcinoma, GBM glioblastoma multiforme, HNSC head and neck squamous cell carcinoma, KICH kidney chromophobe, KIRC kidney renal clear cell carcinoma, KIRP kidney renal papillary cell carcinoma, LIHC liver hepatocellular carcinoma, LUAD lung adenocarcinoma, LUSC lung squamous cell carcinoma, PRAD prostate adenocarcinoma, READ rectum adenocarcinoma, STAD stomach adenocarcinoma, THCA thyroid carcinoma, UCEC uterine corpus endometrial carcinoma
Notably, new findings continue to improve the understanding of chemokine biology. For instance, CCL22 is a dual chemokine constitutively expressed or induced upon inflammation, serving as an antimicrobial protein (Fig. 2). CCL22-deficient mice display partially penetrant preweaning lethality (Table S1) and increased susceptibility to inflammatory diseases [302]. T-cell-derived cytokines maintain the constitutive expression of CCL22 at high levels in lymphoid organs during homeostasis [302]. CCL22 expressed on dendritic cells (DCs) interacts with CCR4 (CCL22-CCR4 axis) to mediate DC–T-cell contacts that are crucial for immune regulation by Tregs, suggesting that the CCL22–CCR4 axis is also an immune checkpoint and that targeting the interaction of CCL22 with its receptor may be an effective but less harmful therapeutic strategy [303]. A recent study showed that CCL22 was abundantly expressed by tumor-associated macrophages (TAMs) from humans in esophageal squamous cell carcinoma (ESCC) tissues. ESCC TAM-released CCL22 promoted tumor invasion and reduced patient survival via activation of the CCR4/DGKα/FAK complex in ESCC cells, revealing opportunities for targeting the tumor-promoting microenvironment to achieve anticancer effects [304]. Thus, the differential expression and regulation patterns of chemokines contribute to the site- and cell-specific divergent pathophysiological responses. Chemokines exert dual roles and produce paradoxical effects in the TME in a context-dependent manner; these roles and effects may confer functional tumor heterogeneity and thus phenotypic plasticity.
Alternative splicing (AS) contributes to phenotypic heterogeneity
An introduction to AS
Most human protein-coding genes undergo AS, a key transcriptional and posttranscriptional process that leads to the formation of multiple transcript variants or splicing variants (SVs) that exert diverse effects via multiple mechanisms, including nonsense-mediated mRNA decay (NMD) (Fig. 7A). These splicing events are functionally important for innate and adaptive immune responses [305, 306] due to their capacity to generate tissue- and cell type-specific or stimulus-responsive SVs [307–309], which have diverse or even opposing functions [310, 311]. Abnormal SVs preferentially produced in various diseases have been proposed as biomarkers for diagnosis and treatment, and studies of such SVs have revealed precision therapy approaches to correct disease-specific defects caused by mis-splicing [312, 313].
Fig. 7.

RNA splicing of chemokines and receptors. A Schematic representation of alternative splicing (AS) and different splicing events. Human protein-coding genes undergo AS through the use of alternate acceptor (AA) sites, alternate donor (AD) sites, alternate promoters (APs), alternate terminators (ATs), exon skipping (ES), mutually exclusive exons (ME), and retained introns (RIs), and the most common form of RIs is mutually exclusive exons (MEs), which allows constitutive splicing (Fig. 6A). B Schematic of the CXCL12 and CXCR4 transcripts. C Comparison of the alternative splicing events of CXCL12 and CXCR4 between multiple types of tumor and normal tissues. The data were extracted from TCGA RNA-seq data (https://bioinformatics.mdanderson.org/TCGASpliceSeq/). For each splicing event, the percent spliced in (PSI) was compared between normal and tumor samples by the Wilcoxon rank sum test, and splicing events with significant differences (p < 0.05) are marked with red labels. For CXCL12, AT1 is an AT event affecting exon 5.2; AT2 is an AT event affecting exon 3.3; AT3 is an AT event affecting exon 4; AT4 is an AT event affecting exon 6; RI is an RI event affecting exon 3.2; and ES is an ES event affecting exons 2.2, 3.1 and 5.1. For CXCR4, AP1 is an AP event affecting exon 1, and AP2 is an AP event affecting exon 2.1
Over 77% (37 of 48) of chemokines and receptors have more than one transcript and protein isoform. Although splicing factors and the processing cascades necessary for spliceosome function are well known [307–309], most of the abnormal chemokine SVs detected at the transcriptional level can be translated into distinct protein isoforms. As confusion mounts over the role of RNA isoforms in functional diversity and phenotypic plasticity [314, 315], most chemokine transcript variants have not been studied, and their contribution to immune disorders and malignancy remains unknown [305, 306, 315]. A few notable examples of chemokine SVs with altered ligand-binding or signaling properties have been reported [316–322]. We summarize findings related to the CXCL12-CXCR4 axis as an example to illustrate the transcriptional heterogeneity that contributes to nongenetic phenotypic divergence.
The CXCL12-CXCR4 axis
CXCL12-SV CXCL12 is located on chromosome 10q11 and is broadly expressed in multiple tissues and cells (Figs. 2 and 3). CXCL12 has multiple transcript variants, five of which, CXCL12-V1 to CXCL12-V5, are currently NCBI-annotated transcripts. CXCL12-V1 to CXCL12-V4 encode CXCL12 isoforms α to δ, while CXCL12-V5 encodes CXCL12 isoform 5 or isoform ε. Another transcript, CXCL12-V6, encodes CXCL12 isoform ϕ, which is identical to isoform ε. CXCL12-V1, CXCL12-V2 to CXCL12-V6 are produced through AT, RI, ES, and their combination. Other transcripts (i.e., ENST00000395795.5) still remain to be experimentally validated (Fig. 7A, B) [323, 324]. It is interesting to note that three SVs have been identified in mice, different from the six SVs in humans.
CXCR4-SVs CXCR4, located on chromosome 2q22.1, has five transcripts. CXCR4-V3 has three exons encoding the longest CXCR4 isoform, isoform C. CXCR4-V1 (also known as CXCR4-Lo) has only one exon transcribed through alternative promoters (APs), encoding CXCR4 isoform A. CXCR4-V2 encodes CXCR4 isoform B, and CXCR4-V5 encodes CXCR4 isoform E (Fig. 7B) [325–327]. CXCR4 displays diverse expression in the BM, lymph nodes, spleen and appendix and high expression in immune cells (Figs. 2, 3).
CXCL12/CXCR4 functionality CXCL12 induces diverse effects on hematopoietic progenitor cells, endothelial cells, and leukocytes by interacting with the classical receptor CXCR4 and the atypical receptor ACKR3/CXCR7 (Fig. 2) [323, 328]. Another atypical receptor, ACKR1/DARC, has also been shown to bind the CXCL12 dimer but not the monomer, and thus its binding is dependent on the differential expression of CXCL12 isoforms [329]. Deletion of Cxcl12 or Cxcr4 in mice results in a variety of developmental abnormalities and embryonic death (Table S1), whereas genetic variants of CXCL12 or CXCR4 are associated with resistance to HIV-1 infection and the development of WHIM (warts, hypogammaglobulinemia, infections, and myelokathexis) syndrome (Table S2) [330]. Therefore, the CXCL12/CXCR4/ACKR3 interaction in the chemokine network is indispensable for the development of hematopoietic and cardiovascular organs. In addition to being an essential player in embryogenesis, hematopoiesis, and angiogenesis, CXCL12 displays inflammatory functions in immune surveillance, the inflammatory response, autoimmune diseases, and tumor growth and metastasis [328]. In fact, the CXCL12/CXCR4 axis is among the most studied chemokine axes in cancer metastasis due to its capacity to support cancer cell proliferation, migration and invasion [324, 331].
Differential expression of SVs Differences in transcriptional related to cancer type may cause functional heterogeneity and differences in clinical outcomes that make it difficult to identify potential prognostic biomarkers for translational studies. CXCL12 is the most primitive chemokine and is highly conserved through evolution, and it may have diverse cellular functions in various biological processes because it has multiple SVs capable of encoding different isoforms [323, 324]. As shown in Fig. 7C, an analysis of the AS events of CXCL12 and CXCR4 transcripts between multiple tumor and normal tissues suggested divergent expression of CCL12- or CXCR4-SVs in tumor tissues. Compared to CXCR4, which showed a smaller difference in SVs, CXCL12 displayed a significant difference in most splicing events in normal versus tumor tissues. The bulk expression of CXCL12 was decreased in HNSC tissues but was not changed in STAD tissues, and it was not associated with patient clinical outcomes (Fig. 6). This may be because of different and even opposing changes in CCL12-SVs, such as an increase in the level of alternate terminator 1 (AT1) but a decrease in the level of AT2 (both of which effected the expression of CXCL12) in HNSC and STAD tumors, making the expression ultimately no different from that in controls (Fig. 7C). If the differentially expressed SVs have functional differences and are not distinguished, it may impair the final functional output or increase uncertainty risk. Although CXCL12 is subjected to more posttranslational than transcriptional regulation [196, 332, 333], cell- or tissue-type specific RNA isoforms may be the cause of some of the controversial or paradoxical effects of chemokine‒receptors on different signaling pathways in immune and cancer cells under specific microenvironments.
Many transcripts exist per gene, most of which are thought to not be functionally relevant, and some even have opposing effects. For precise evaluation of clinical effectiveness and drug resistance, the specific expression of functionally distinct SVs, rather than their overall expression, should be considered for assay design to accurately reflect transcriptional heterogeneity. Therefore, the next round of translational studies in chemokine biology should focus on improving the understanding the differential expression and functionality of these transcript isoforms to guide the discovery and validation of biomarkers and targets.
Chemokines and receptors for precision medicine
Chemokines as noninvasive biomarkers for liquid biopsy
Chemokines have unique characteristics in cell mobility and immunity; for example, they establish concentration gradients and effect secretion under multiple layers of dynamic regulation, included genetic and epigenetic modification of chemokine genes (e.g., SNVs, chemokine-SEs and cfDNA). Due to their identifiable tissue specificity, chemokines may serve as ideal liquid biopsy-based biomarkers for early diagnosis or to guide targeted therapy for immune disorders and cancer [53, 334]. Figure 8A summarizes the differential expression of chemokines and receptors in liquid biopsy elements, including extracellular vesicles, circulating tumor cells (CTCs) and blood, of patients with several cancer types; the results suggest the potential of assessing chemokine expression by liquid biopsy. In addition, the serum levels of IFN-α-induced chemokines used to monitor SLE [297] and CX3CL1 methylation predicted T-cell infiltration in nephroblastoma [298]. As mentioned above, high serum levels of CCL3 and CCL4 and high CCR5 expression in primary specimens were found to be associated with poorer prognosis in patients with CRC [335]. The roles of CXCL13/CXCR5 and CCL22/CCR4 in multiple sclerosis (MS) and other autoimmune diseases have been reported [336, 337]. However, in an examination of a wide panel of cytokines and chemokines (CCL1, CCL2, CCL3, CCL22, CXCL11, CXCL13, and IL-16) in the cerebrospinal fluid of relapsing-remitting MS patients, only CCL3 was found to be associated with both MS diagnosis and oligoclonal IgG, a typical marker for inflammation in MS [338].
Fig. 8.

Chemokine molecules as potential noninvasive biomarkers. Heatmaps showing that differential expression of chemokines and receptors in tumor tissues from cancer patients compared to normal controls (A) or in COVID-19 specimens compared to healthy controls (B). The significant differences in between tumor tissues and normal tissues are shown in red (upregulation) or blue (downregulation) ( | log2FC | >1 & adjusted p value < 0.05). The data were downloaded from the Bbcancer database (http://bbcancer.renlab.org). The total sample number (tumor and normal samples) is shown at the bottom right. The color bars on the top indicate the sample type (yellow: CTCs; green: blood; blue: extracellular vesicles, EVs)
Induction of chemokines and receptor expression by SARS-CoV-2
The CCL2-CCR2 axis
As mentioned above, SARS-CoV-2 infection in patients with poor clinical outcomes is characterized by high levels of proinflammatory cytokines and chemokines (e.g., IL-6, CCL2/MCP1, CCL3/MIP1α, CCL4/MIP1β and CXCL10/IP-10) and cytokine storms [85, 87]. CCL2 is an inflammatory chemokine that exerts both agonistic and antagonistic effects by binding to CCR2 expressed by monocytes/macrophages, plasmacytoid dendritic cells (pDCs), T cells and natural killer T (NKT) cells (Fig. 2). Trafficking of monocytes/macrophages and T cells is impaired by Ccr2 deficiency (Table S1). The CCL2-CCR2 axis contributes to an immunosuppressive TME; thus, antagonistic drugs targeting CCR2 may be beneficial for cancer therapy or decrease undesired immune responses in COVID-19 and autoimmune diseases (i.e., nonalcoholic steatohepatitis) (Table S3). CCL2 genetic and epigenetic alterations, such as CCL2-A2518G in COVID-19 [145], CCR2 rs1799864 in HIV [117], and CCL2-SE [234, 235], are promising biomarkers for clinical translation. However, the CCR2 pathway was also found to promote viral control and restrict inflammation within the respiratory tract during SARS-CoV-2 infection [339], suggesting that CCR2 and its ligands have dual functions.
Inflammatory chemokines predicting severity of infection
Increased CXCL8 plays a key role in promoting acute SARS infection, viral bronchiolitis, severe immunopathology, and respiratory syncytial virus (HRSV) infection disease progression [340]. However, an analysis of TGCA data showed decreased expression of CCL3L1, CCL3L3, CCR4L1, CCR4L2, CXCL5, CCR4, CCR7 and CXCR5, but increased expression of CXCL10 and CXCRL2 was the most significant factor related to the host response to SARS-CoV-2 infection (Fig. 8B).
A recent study using scRNA-seq revealed differential expression of inflammatory cytokines in COVID-19 patients with different disease severities [36]. In addition to well-known cytokines (e.g., IL-1, IL-6 and IL-10), chemokines, including CCL3, CXCL10, CXCL5, and CCR2, were found to have increased expression in peripheral blood mononuclear cells (PBMCs) derived from COVID-19 patients with moderate, severe and critical disease, and the levels of CCL3 and CXCL10 were also assessed in plasma. While the expression of CCR6, CCR7 and CXCR4 in PBMCs decreased with severity, the transcript levels of XCL1, XCL2, CCL5 and CXCR3 increased from moderate to severe disease in COVID-19 patients but returned to normal with the development of critical disease. Moreover, high levels of expression of favorable chemokine genes were observed in B cells (CCL5, XCL1 and XCL2), T cells (CCL4, CXCR3 and CXCR6) and monocytes (CCL2, CXCL8 and CXCL10) in patients with moderate, severe and critical disease. Pivotal inflammatory chemokine receptor‒ligand pairs were found to mediate the intensity of interactions between CD8 effector T/NK cells and monocytes, as they were elevated in moderate and severe COVID-19 cases but diminished in critical cases. CCL3L1-DDP4 was increased in critical cases, whereas CCL3-CCR5, CCL4-CCR5, CCL4-SLC7A1 and CCL4L2-VSIR were enhanced in moderate and severe cases but decreased in critical cases. This study therefore suggests that inflammatory chemokines respond dynamically and nonredundantly to SARS-CoV-2 infection and that chemokine signatures may reflect disease severity and may be conducive to drug development [271].
Hypertension with COVID-19
Hypertensive patients are more likely to develop severe pneumonia or organ damage than patients without hypertension [341], and the cellular serine protease TMPRSS2 can prime the SARS-2-S protein for entry. An inhibitor of TMPRSS2, camostat mesylate, blocks SARS-CoV-2 infection of lung cells [342]. A recent observation showed that macrophages and neutrophils from hypertension patients with COVID-19 exhibited higher expression of the proinflammatory cytokines CCL3 and CCL4 and the chemokine receptor CCR1. Antihypertensive blockade of the renin–angiotensin–aldosterone system (RAAS), specifically with the use of an angiotensin-converting enzyme inhibitor (ACEi), might improve outcomes in patients with hypertension and COVID-19 [341, 343].
Chemokines in targeted therapy
The therapeutic targeting of chemokines and receptors has been reviewed in several recent publications [41, 44–51, 344]. Current ongoing (later phases) and completed clinical trials of drugs targeting chemokines and receptors are listed in Table S3, and as can be seen, drugs targeting chemokine receptors are the major drugs used for antiviral therapy. Several clinical trials targeting chemokines are still in the early phases [47]. Targeting chemokine-receptor axes for precision therapy will require a comprehensive understanding of their differential expression and mechanisms in different tumor microenvironments, as targeting these axes may result in effects from target pathway redundancy and context-dependent immunosuppressive actions of the antagonist [45]. Readers should also refer to excellent specific review articles for more in-depth information [41, 43–52, 95].
CXCR4 antagonists
Based on the role of the CXCL12-CXCR4 axis in cancer metastasis, many CXCR4 antagonists for cancer therapy are in clinical development (Table S3). Of these, plerixafor is a bicyclam with hematopoietic stem cell mobilizing activity that selectively and reversibly antagonizes the binding of CXCL12 to CXCR4 on bone marrow stromal cells [345]. A phase II clinical trial is in progress to evaluate its use in combination with standard temozolomide chemoradiotherapy for patients with glioblastoma (NCT03746080). Plerixafor combined with granulocyte-colony stimulating factor (GCSF) has been shown to mobilize hematopoietic stem cells more efficiently than plerixafor alone [345]. BL-8040/motixafortide, a short, high-affinity synthetic peptide antagonist for CXCR4 with longer receptor occupancy, is being tested in a phase Ib/II trial (NCT02826486). This trial is investigating the safety, pharmacokinetics and anticancer activity of a combination immunotherapy in patients with advanced or metastatic gastric/gastroesophageal junction cancer/esophageal cancer.
The CCL3-CCR5 axis
As mentioned above, CCL3, CCL4, and CCL5 are HIV-suppressive factors produced by CD8-positive T cells to modulate virus-induced inflammation. CCL3 is produced by macrophages, CD4 + T cells, CD8 + T cells, NK cells, fibroblasts and mast cells and is an important activator of both innate and adaptive responses (Fig. 2). For example, atherosclerotic plaque-resident T cells differentially express several chemokine receptors that bind with their corresponding ligands to form CCL3-CCR5 and CX3 CL1-CX3 CR1 interactions, which induce T-cell migration into human atherosclerotic plaques, where T-cell accumulation contributes to plaque destabilization and atherosclerosis [346]. Ccl3 induced by administration of the antimitotic chemotherapy drug docetaxel (DTX) promoted proinflammatory macrophage polarization to suppress tumor progression and increased DTX chemosensitivity in breast cancer via the CCR5-p38/interferon regulatory factor 5 pathway [347]. CCL3 is also considered a neutrophil chemoattractant; it activates and enhances the cytotoxicity of NK cells and plays a critical role in both immune surveillance and tolerance by regulating lymph node homing of dendritic cell subsets and inducing antigen-specific T-cell responses [348]. For example, the innate immune mediators CCL3 and CCL4 were found to be elevated in the lungs of patients with chronic beryllium disease (CBD), a granulomatous lung disorder that is triggered in susceptible individuals by inhalation of beryllium-containing particulates. These chemokine-derived peptides may serve as neoantigen epitopes that can activate specific CD4 + T cells, thus revealing a direct link between persistent innate and adaptive immune activation [349]. However, CCL3 plays roles in both antitumor and protumor activities depending on the underlying signaling cascades that are activated through binding to the receptors CCR1, CCR4 and CCR5 and/or interacting with CCL4. For instance, the β-catenin-metadherin/CEACAM1-CCL3 positive feedback cascade has been shown to lead to metastasis in ovarian cancer by increasing the level of infiltrating tumor-associated macrophages (TAMs) at the metastatic site [350]. CCL3 derived from TAMs and cancer cells in esophageal squamous cell carcinoma (ESCC) promoted tumor cell migration and invasion via the CCL3-CCR5 axis and the PI3K/Akt and MEK/ERK pathways [351]. Therefore, the CCL3-CCR5 axis represents a potential therapeutic target for cancer treatment.
Concluding remarks
Substantial progress has been achieved in chemokine biology, and multiomics data have enabled the identification of genome and metabolome profiles with complex regulatory networks and functional plasticity. A more comprehensive understanding of the chemokine interactome will not only enable more rational management of complex diseases but also promote the development of robust, convenient, sensitive, and specific assays for the noninvasive but reliable detection of chemokines for diagnosis and treatment guidance. While the rational design of cancer immunotherapies targeting disrupted epigenetic pathways related to chemokines may be a more realistic goal for pharmacological development, appropriate interpretation of the data requires an understanding of the spatial-temporal genetic variations and nongenetic heterogeneity in different microenvironments. Further proof-of-concept is warranted for translational studies of chemokine applications in precision medicine. Therefore, novel technology to be used in combination with single-cell-based 3-D imaging should be developed to allow more sensitive quantification of the complex chemokine interactome in health and diseases. In this context, processing bioinformatic analysis data with artificial intelligence (AI) systems has emerged as a major achievement in the era of chemokine biology research. Given these advances, it is time to further reveal the science behind chemokine biology to achieve precision medicine.
Supplementary information
Acknowledgements
We thank our laboratory members for technical help and insightful discussions.
Author contributions
JH, JMW and HX conceived and designed the work. HX, SL, ZZ, DL, XZ, MY, RZ, YW, JQ, XL, BL, CW, and KC collected and analyzed the data. HX, SL and JH interpreted the data, generated tables and figures and wrote the original draft. JH wrote, reviewed and edited the manuscript. HX, TY, JMW and JH reviewed and edited the formal manuscript.
Funding
This study was funded in part by the National Natural Science Foundation of China (NSFC Grant No. 81872021, 32200462); Beijing Jiaotong University undergraduate innovation and entrepreneurship training project (No. 220171097, 220171072, 220171037, 220171088, 220171104); R&D Program of Beijing Municipal Education Commission (Grant No. KM202110025004) and Beijing Hospitals Authority Youth Programme (grant No. QML20231602). JH, KC and JMW were also funded in part by Federal funds from the National Cancer Institute, National Institutes of Health (under Contract No. HHSN261200800001E) and by the Intramural Research Programs of the NCI, CCR, and NIH.
Competing interests
The authors declare no competing interests.
Consent to publish
All authors read and agreed to the submission of this work for publication.
Footnotes
These authors contributed equally: Hanli Xu, Shuye Lin.
Supplementary information
The online version contains supplementary material available at 10.1038/s41423-023-01032-x.
References
- 1.SenGupta S, Parent CA, Bear JE. The principles of directed cell migration. Nat Rev Mol Cell Biol. 2021;22:529–47. doi: 10.1038/s41580-021-00366-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shellard A, Mayor R. All roads lead to directional cell migration. Trends Cell Biol. 2020;30:852–68. doi: 10.1016/j.tcb.2020.08.002. [DOI] [PubMed] [Google Scholar]
- 3.Yamada KM, Sixt M. Mechanisms of 3D cell migration. Nat Rev Mol Cell Biol. 2019;20:738–52. doi: 10.1038/s41580-019-0172-9. [DOI] [PubMed] [Google Scholar]
- 4.Sun H, Sun C, Xiao W, Sun R. Tissue-resident lymphocytes: from adaptive to innate immunity. Cell Mol Immunol. 2019;16:205–15. doi: 10.1038/s41423-018-0192-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mueller SN. Neural control of immune cell trafficking. J Exp Med. 2022;219:e20211604. doi: 10.1084/jem.20211604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu J, Zhang X, Cheng Y, Cao X. Dendritic cell migration in inflammation and immunity. Cell Mol Immunol. 2021;18:2461–71. doi: 10.1038/s41423-021-00726-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fowell DJ, Kim M. The spatio-temporal control of effector T cell migration. Nat Rev Immunol. 2021;21:582–96. doi: 10.1038/s41577-021-00507-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ran GH, Lin YQ, Tian L, Zhang T, Yan DM, Yu JH, et al. Natural killer cell homing and trafficking in tissues and tumors: from biology to application. Signal Transduct Target Ther. 2022;7:205. doi: 10.1038/s41392-022-01058-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alon R, Sportiello M, Kozlovski S, Kumar A, Reilly EC, Zarbock A, et al. Leukocyte trafficking to the lungs and beyond: lessons from influenza for COVID-19. Nat Rev Immunol. 2021;21:49–64. doi: 10.1038/s41577-020-00470-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–7. doi: 10.1016/S1074-7613(00)80165-X. [DOI] [PubMed] [Google Scholar]
- 11.Zlotnik A, Yoshie O. The chemokine superfamily revisited. Immunity. 2012;36:705–16. doi: 10.1016/j.immuni.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bachelerie F, Ben-Baruch A, Burkhardt AM, Combadiere C, Farber JM, Graham GJ, et al. International Union of Basic and Clinical Pharmacology. [corrected]. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharm Rev. 2014;66:1–79. doi: 10.1124/pr.113.007724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schulz O, Hammerschmidt SI, Moschovakis GL, Forster R. Chemokines and chemokine receptors in lymphoid tissue dynamics. Annu Rev Immunol. 2016;34:203–42. doi: 10.1146/annurev-immunol-041015-055649. [DOI] [PubMed] [Google Scholar]
- 14.Lopez-Cotarelo P, Gomez-Moreira C, Criado-Garcia O, Sanchez L, Rodriguez-Fernandez JL. Beyond chemoattraction: multifunctionality of chemokine receptors in leukocytes. Trends Immunol. 2017;38:927–41. doi: 10.1016/j.it.2017.08.004. [DOI] [PubMed] [Google Scholar]
- 15.David BA, Kubes P. Exploring the complex role of chemokines and chemoattractants in vivo on leukocyte dynamics. Immunol Rev. 2019;289:9–30. doi: 10.1111/imr.12757. [DOI] [PubMed] [Google Scholar]
- 16.Hughes CE, Nibbs RJB. A guide to chemokines and their receptors. FEBS J. 2018;285:2944–71. doi: 10.1111/febs.14466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen K, Bao Z, Tang P, Gong W, Yoshimura T, Wang JM. Chemokines in homeostasis and diseases. Cell Mol Immunol. 2018;15:324–34. doi: 10.1038/cmi.2017.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32:659–702. doi: 10.1146/annurev-immunol-032713-120145. [DOI] [PubMed] [Google Scholar]
- 19.Ozga AJ, Chow MT, Luster AD. Chemokines and the immune response to cancer. Immunity. 2021;54:859–74. doi: 10.1016/j.immuni.2021.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saxena S, Singh RK. Chemokines orchestrate tumor cells and the microenvironment to achieve metastatic heterogeneity. Cancer Metastasis Rev. 2021;40:447–76. doi: 10.1007/s10555-021-09970-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsushima K, Yang, Oppenheim JJ. Interleukin-8: an evolving chemokine. Cytokine. 2022;153:155828. doi: 10.1016/j.cyto.2022.155828. [DOI] [PubMed] [Google Scholar]
- 22.Yoshimura T. Discovery of IL-8/CXCL8 (The Story from Frederick) Front Immunol. 2015;6:278. doi: 10.3389/fimmu.2015.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang P, Wang JM. Chemokines: the past, the present and the future. Cell Mol Immunol. 2018;15:295–8. doi: 10.1038/cmi.2018.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moser B. Editorial: history of chemoattractant research. Front Immunol. 2015;6:548. doi: 10.3389/fimmu.2015.00548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshimura T. The chemokine MCP-1 (CCL2) in the host interaction with cancer: a foe or ally? Cell Mol Immunol. 2018;15:335–45. doi: 10.1038/cmi.2017.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zlotnik A. Perspective: insights on the nomenclature of cytokines and chemokines. Front Immunol. 2020;11:908. doi: 10.3389/fimmu.2020.00908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deuel TF, Keim PS, Farmer M, Heinrikson RL. Amino acid sequence of human platelet factor 4. Proc Natl Acad Sci USA. 1977;74:2256–8. doi: 10.1073/pnas.74.6.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eisman R, Surrey S, Ramachandran B, Schwartz E, Poncz M. Structural and functional comparison of the genes for human platelet factor 4 and PF4alt. Blood. 1990;76:336–44. doi: 10.1182/blood.V76.2.336.336. [DOI] [PubMed] [Google Scholar]
- 29.Montaldo E, Lusito E, Bianchessi V, Caronni N, Scala S, Basso-Ricci L, et al. Cellular and transcriptional dynamics of human neutrophils at steady state and upon stress. Nat Immunol. 2022;23:1470–83. doi: 10.1038/s41590-022-01311-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Evers TMJ, Sheikhhassani V, Haks MC, Storm C, Ottenhoff THM, Mashaghi A. Single-cell analysis reveals chemokine-mediated differential regulation of monocyte mechanics. iScience. 2022;25:103555. doi: 10.1016/j.isci.2021.103555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roy R, Ramamoorthy S, Shapiro BD, Kaileh M, Hernandez D, Sarantopoulou D, et al. DNA methylation signatures reveal that distinct combinations of transcription factors specify human immune cell epigenetic identity. Immunity. 2021;54:2465–80.e2465. doi: 10.1016/j.immuni.2021.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gangoso E, Southgate B, Bradley L, Rus S, Galvez-Cancino F, McGivern N, et al. Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell. 2021;184:2454–70.e2426. doi: 10.1016/j.cell.2021.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kourtis N, Wang Q, Wang B, Oswald E, Adler C, Cherravuru S, et al. A single-cell map of dynamic chromatin landscapes of immune cells in renal cell carcinoma. Nat Cancer. 2022;3:885–98. doi: 10.1038/s43018-022-00391-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hornburg M, Desbois M, Lu S, Guan Y, Lo AA, Kaufman S, et al. Single-cell dissection of cellular components and interactions shaping the tumor immune phenotypes in ovarian cancer. Cancer Cell. 2021;39:928–44.e926. doi: 10.1016/j.ccell.2021.04.004. [DOI] [PubMed] [Google Scholar]
- 35.Guo C, Wu M, Huang B, Zhao R, Jin L, Fu B, et al. Single-cell transcriptomics reveal a unique memory-like NK cell subset that accumulates with ageing and correlates with disease severity in COVID-19. Genome Med. 2022;14:46. doi: 10.1186/s13073-022-01049-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie X, Cheng X, Wang G, Zhang B, Liu M, Chen L, et al. Single-cell transcriptomes of peripheral blood cells indicate and elucidate severity of COVID-19. Sci China Life Sci. 2021;64:1634–44. doi: 10.1007/s11427-020-1880-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calebiro D, Grimes JG. Protein-coupled receptor pharmacology at the single-molecule level. Annu Rev Pharm Toxicol. 2020;60:73–87. doi: 10.1146/annurev-pharmtox-010919-023348. [DOI] [PubMed] [Google Scholar]
- 38.Shimada I, Ueda T, Kofuku Y, Eddy MT, Wuthrich K. GPCR drug discovery: integrating solution NMR data with crystal and cryo-EM structures. Nat Rev Drug Disco. 2019;18:59–82. doi: 10.1038/nrd.2018.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hauser AS, Kooistra AJ, Munk C, Heydenreich FM, Veprintsev DB, Bouvier M, et al. GPCR activation mechanisms across classes and macro/microscales. Nat Struct Mol Biol. 2021;28:879–88. doi: 10.1038/s41594-021-00674-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Asher WB, Geggier P, Holsey MD, Gilmore GT, Pati AK, Meszaros J, et al. Single-molecule FRET imaging of GPCR dimers in living cells. Nat Methods. 2021;18:397–405. doi: 10.1038/s41592-021-01081-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24:541–50. doi: 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kasela S, Ortega VE, Martorella M, Garudadri S, Nguyen J, Ampleford E, et al. Genetic and non-genetic factors affecting the expression of COVID-19-relevant genes in the large airway epithelium. Genome Med. 2021;13:66. doi: 10.1186/s13073-021-00866-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lercher A, Baazim H, Bergthaler A. Systemic immunometabolism: challenges and opportunities. Immunity. 2020;53:496–509. doi: 10.1016/j.immuni.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ortiz Zacarias NV, Bemelmans MP, Handel TM, de Visser KE, Heitman LH. Anticancer opportunities at every stage of chemokine function. Trends Pharm Sci. 2021;42:912–28. doi: 10.1016/j.tips.2021.08.001. [DOI] [PubMed] [Google Scholar]
- 45.Propper DJ, Balkwill FR. Harnessing cytokines and chemokines for cancer therapy. Nat Rev Clin Oncol. 2022;19:237–53. doi: 10.1038/s41571-021-00588-9. [DOI] [PubMed] [Google Scholar]
- 46.Rosenkilde MM, Tsutsumi N, Knerr JM, Kildedal DF, Garcia KC. Viral G Protein-Coupled Receptors Encoded by beta- and gamma-Herpesviruses. Annu Rev Virol. 2022;9:329–51. doi: 10.1146/annurev-virology-100220-113942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Markl F, Huynh D, Endres S, Kobold S. Utilizing chemokines in cancer immunotherapy. Trends Cancer. 2022;8:670–82. doi: 10.1016/j.trecan.2022.04.001. [DOI] [PubMed] [Google Scholar]
- 48.Takacs GP, Flores-Toro JA, Harrison JK. Modulation of the chemokine/chemokine receptor axis as a novel approach for glioma therapy. Pharm Ther. 2021;222:107790. doi: 10.1016/j.pharmthera.2020.107790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng X, Wu Y, Bi J, Huang Y, Cheng Y, Li Y, et al. The use of supercytokines, immunocytokines, engager cytokines, and other synthetic cytokines in immunotherapy. Cell Mol Immunol. 2022;19:192–209. doi: 10.1038/s41423-021-00786-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang D, Zhou Q, Labroska V, Qin S, Darbalaei S, Wu Y, et al. G protein-coupled receptors: structure- and function-based drug discovery. Signal Transduct Target Ther. 2021;6:7. doi: 10.1038/s41392-020-00435-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gupta M, Chandan K, Sarwat M. Natural products and their derivatives as immune check point inhibitors: targeting cytokine/chemokine signalling in cancer. Semin Cancer Biol. 2022;86:214–32. doi: 10.1016/j.semcancer.2022.06.009. [DOI] [PubMed] [Google Scholar]
- 52.Saxton RA, Glassman CR, Garcia KC. Emerging principles of cytokine pharmacology and therapeutics. Nat Rev Drug Disco. 2022;22:21–37. doi: 10.1038/s41573-022-00557-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Georgakis MK, Gill D, Rannikmäe K, Traylor M, Anderson CD, Lee JM, et al. Genetically determined levels of circulating cytokines and risk of stroke. Circulation. 2019;139:256–68. doi: 10.1161/CIRCULATIONAHA.118.035905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Andreakos E, Abel L, Vinh DC, Kaja E, Drolet BA, Zhang Q, et al. A global effort to dissect the human genetic basis of resistance to SARS-CoV-2 infection. Nat Immunol. 2022;23:159–64. doi: 10.1038/s41590-021-01030-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zheng D, Ratiner K, Elinav E. Circadian influences of diet on the microbiome and immunity. Trends Immunol. 2020;41:512–30. doi: 10.1016/j.it.2020.04.005. [DOI] [PubMed] [Google Scholar]
- 56.Holtkamp SJ, Ince LM, Barnoud C, Schmitt MT, Sinturel F, Pilorz V, et al. Circadian clocks guide dendritic cells into skin lymphatics. Nat Immunol. 2021;22:1375–81. doi: 10.1038/s41590-021-01040-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.He W, Holtkamp S, Hergenhan SM, Kraus K, de Juan A, Weber J, et al. Circadian expression of migratory factors establishes lineage-specific signatures that guide the homing of leukocyte subsets to tissues. Immunity. 2018;49:1175–90.e1177. doi: 10.1016/j.immuni.2018.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Adrover JM, Del Fresno C, Crainiciuc G, Cuartero MI, Casanova-Acebes M, Weiss LA, et al. A neutrophil timer coordinates immune defense and vascular protection. Immunity. 2019;51:966–7. doi: 10.1016/j.immuni.2019.11.001. [DOI] [PubMed] [Google Scholar]
- 59.Teixido J, Hidalgo A, Fagerholm S. Editorial: leukocyte trafficking in homeostasis and disease. Front Immunol. 2019;10:2560. doi: 10.3389/fimmu.2019.02560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Buckley CD, McGettrick HM. Leukocyte trafficking between stromal compartments: lessons from rheumatoid arthritis. Nat Rev Rheumatol. 2018;14:476–87. doi: 10.1038/s41584-018-0042-4. [DOI] [PubMed] [Google Scholar]
- 61.Pick R, He W, Chen CS, Scheiermann C. Time-of-day-dependent trafficking and function of leukocyte subsets. Trends Immunol. 2019;40:524–37. doi: 10.1016/j.it.2019.03.010. [DOI] [PubMed] [Google Scholar]
- 62.Nourshargh S, Renshaw SA, Imhof BA. Reverse migration of neutrophils: where, when, how, and why? Trends Immunol. 2016;37:273–86. doi: 10.1016/j.it.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 63.de Oliveira S, Rosowski EE, Huttenlocher A. Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol. 2016;16:378–91. doi: 10.1038/nri.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ji J, Fan J. Neutrophil in reverse migration: role in sepsis. Front Immunol. 2021;12:656039. doi: 10.3389/fimmu.2021.656039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu Q, Zhao W, Yan M, Mei H. Neutrophil reverse migration. J Inflamm (Lond) 2022;19:22. doi: 10.1186/s12950-022-00320-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nomiyama H, Osada N, Yoshie O. The evolution of mammalian chemokine genes. Cytokine Growth Factor Rev. 2010;21:253–62. doi: 10.1016/j.cytogfr.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 67.Ruytinx P, Proost P, Struyf S. CXCL4 and CXCL4L1 in cancer. Cytokine. 2018;109:65–71. doi: 10.1016/j.cyto.2018.02.022. [DOI] [PubMed] [Google Scholar]
- 68.Teng MW, Galon J, Fridman WH, Smyth MJ. From mice to humans: developments in cancer immunoediting. J Clin Invest. 2015;125:3338–46. doi: 10.1172/JCI80004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.von Hundelshausen P, Wichapong K, Gabius HJ, Mayo KH. The marriage of chemokines and galectins as functional heterodimers. Cell Mol Life Sci. 2021;78:8073–95. doi: 10.1007/s00018-021-04010-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Han W, Lou Y, Tang J, Zhang Y, Chen Y, Li Y, et al. Molecular cloning and characterization of chemokine-like factor 1 (CKLF1), a novel human cytokine with unique structure and potential chemotactic activity. Biochem J. 2001;357:127–35. doi: 10.1042/bj3570127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morein D, Erlichman N, Ben-Baruch A. Beyond cell motility: the expanding roles of chemokines and their receptors in malignancy. Front Immunol. 2020;11:952. doi: 10.3389/fimmu.2020.00952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smith JS, Pack TF, Inoue A, Lee C, Zheng K, Choi I, et al. Noncanonical scaffolding of G(alphai) and beta-arrestin by G protein-coupled receptors. Science. 2021;371:eaay1833. doi: 10.1126/science.aay1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smith JS, Pack TF. Noncanonical interactions of G proteins and beta-arrestins: from competitors to companions. FEBS J. 2021;288:2550–61. doi: 10.1111/febs.15749. [DOI] [PubMed] [Google Scholar]
- 74.Rodriguez-Fernandez JL, Criado-Garcia O. A meta-analysis indicates that the regulation of cell motility is a non-intrinsic function of chemoattractant receptors that is governed independently of directional sensing. Front Immunol. 2022;13:1001086. doi: 10.3389/fimmu.2022.1001086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zlotnik A, Yoshie O, Nomiyama H. The chemokine and chemokine receptor superfamilies and their molecular evolution. Genome Biol. 2006;7:243. doi: 10.1186/gb-2006-7-12-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.von Hundelshausen P, Agten SM, Eckardt V, Blanchet X, Schmitt MM, Ippel H, et al. Chemokine interactome mapping enables tailored intervention in acute and chronic inflammation. Sci Transl Med. 2017;9:eaah6650. doi: 10.1126/scitranslmed.aah6650. [DOI] [PubMed] [Google Scholar]
- 77.Weber C, Koenen RR. Fine-tuning leukocyte responses: towards a chemokine ‘interactome’. Trends Immunol. 2006;27:268–73. doi: 10.1016/j.it.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 78.Volden R, Vollmers C. Single-cell isoform analysis in human immune cells. Genome Biol. 2022;23:47. doi: 10.1186/s13059-022-02615-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mogilenko DA, Shchukina I, Artyomov MN. Immune ageing at single-cell resolution. Nat Rev Immunol. 2021;22:484–98. doi: 10.1038/s41577-021-00646-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhao J, Zhang S, Liu Y, He X, Qu M, Xu G, et al. Single-cell RNA sequencing reveals the heterogeneity of liver-resident immune cells in human. Cell Disco. 2020;6:22. doi: 10.1038/s41421-020-0157-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dudek M, Pfister D, Donakonda S, Filpe P, Schneider A, Laschinger M, et al. Auto-aggressive CXCR6(+) CD8 T cells cause liver immune pathology in NASH. Nature. 2021;592:444–9. doi: 10.1038/s41586-021-03233-8. [DOI] [PubMed] [Google Scholar]
- 82.Levitin HM, Yuan J, Sims PA. Single-cell transcriptomic analysis of tumor heterogeneity. Trends Cancer. 2018;4:264–8. doi: 10.1016/j.trecan.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu G, He M, Yin X, Wang W, Zhou J, Ren K, et al. The pan-cancer landscape of crosstalk between TRP family and tumour microenvironment relevant to prognosis and immunotherapy response. Front Immunol. 2022;13:837665. doi: 10.3389/fimmu.2022.837665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med. 2020;383:2255–73. doi: 10.1056/NEJMra2026131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tay MZ, Poh CM, Renia L, MacAry PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;20:363–74. doi: 10.1038/s41577-020-0311-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ahmad S, Manzoor S, Siddiqui S, Mariappan N, Zafar I, Ahmad A, et al. Epigenetic underpinnings of inflammation: connecting the dots between pulmonary diseases, lung cancer and COVID-19. Semin Cancer Biol. 2022;83:384–98. doi: 10.1016/j.semcancer.2021.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ye CH, Hsu WL, Peng GR, Yu WC, Lin WC, Hu S, et al. Role of the immune microenvironment in SARS-CoV-2 infection. Cell Transpl. 2021;30:9636897211010632. doi: 10.1177/09636897211010632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zheng HY, He XY, Li W, Song TZ, Han JB, Yang X, et al. Pro-inflammatory microenvironment and systemic accumulation of CXCR3+ cell exacerbate lung pathology of old rhesus macaques infected with SARS-CoV-2. Signal Transduct Target Ther. 2021;6:328. doi: 10.1038/s41392-021-00734-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liu R, Gao Q, Foltz SM, Fowles JS, Yao L, Wang JT, et al. Co-evolution of tumor and immune cells during progression of multiple myeloma. Nat Commun. 2021;12:2559. doi: 10.1038/s41467-021-22804-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dyer DP. Understanding the mechanisms that facilitate specificity, not redundancy, of chemokine-mediated leukocyte recruitment. Immunology. 2020;160:336–44. doi: 10.1111/imm.13200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stone MJ, Hayward JA, Huang C, Huma ZE, Sanchez J. Mechanisms of regulation of the chemokine-receptor network. Int J Mol Sci. 2017;18:342. doi: 10.3390/ijms18020342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Proudfoot AE, Uguccioni M. Modulation of chemokine responses: synergy and cooperativity. Front Immunol. 2016;7:183. doi: 10.3389/fimmu.2016.00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Crijns H, Vanheule V, Proost P. Targeting chemokine-glycosaminoglycan interactions to inhibit inflammation. Front Immunol. 2020;11:483. doi: 10.3389/fimmu.2020.00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tan RZ, Chiam KH. A computational model for how cells choose temporal or spatial sensing during chemotaxis. PLoS Comput Biol. 2018;14:e1005966. doi: 10.1371/journal.pcbi.1005966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Patwardhan A, Cheng N, Trejo J. Post-translational modifications of g protein-coupled receptors control cellular signaling dynamics in space and time. Pharm Rev. 2021;73:120–51. doi: 10.1124/pharmrev.120.000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chang H, Levchenko A. Adaptive molecular networks controlling chemotactic migration: dynamic inputs and selection of the network architecture. Philos Trans R Soc Lond B Biol Sci. 2013;368:20130117. doi: 10.1098/rstb.2013.0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ley K, Hoffman HM, Kubes P, Cassatella MA, Zychlinsky A, Hedrick CC, et al. Neutrophils: New insights and open questions. Sci Immunol. 2018;3:eaat4579. doi: 10.1126/sciimmunol.aat4579. [DOI] [PubMed] [Google Scholar]
- 98.Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18:134–47. doi: 10.1038/nri.2017.105. [DOI] [PubMed] [Google Scholar]
- 99.Quail DF, Amulic B, Aziz M, Barnes BJ, Eruslanov E, Fridlender ZG, et al. Neutrophil phenotypes and functions in cancer: a consensus statement. J Exp Med. 2022;219:e20220011. doi: 10.1084/jem.20220011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Metzemaekers M, Gouwy M, Proost P. Neutrophil chemoattractant receptors in health and disease: double-edged swords. Cell Mol Immunol. 2020;17:433–50. doi: 10.1038/s41423-020-0412-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Herrero-Cervera A, Soehnlein O, Kenne E. Neutrophils in chronic inflammatory diseases. Cell Mol Immunol. 2022;19:177–91. doi: 10.1038/s41423-021-00832-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lammermann T, Kastenmuller W. Concepts of GPCR-controlled navigation in the immune system. Immunol Rev. 2019;289:205–31. doi: 10.1111/imr.12752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Glaser KM, Mihlan M, Lammermann T. Positive feedback amplification in swarming immune cell populations. Curr Opin Cell Biol. 2021;72:156–62. doi: 10.1016/j.ceb.2021.07.009. [DOI] [PubMed] [Google Scholar]
- 104.Lämmermann T, Afonso PV, Angermann BR, Wang JM, Kastenmüller W, Parent CA, et al. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature. 2013;498:371–5. doi: 10.1038/nature12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kienle K, Glaser KM, Eickhoff S, Mihlan M, Knöpper K, Reátegui E, et al. Neutrophils self-limit swarming to contain bacterial growth in vivo. Science. 2021;372:eabe7729. doi: 10.1126/science.abe7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mihlan M, Glaser KM, Epple MW, Lammermann T. Neutrophils: amoeboid migration and swarming dynamics in tissues. Front Cell Dev Biol. 2022;10:871789. doi: 10.3389/fcell.2022.871789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Podstawka J, Sinha S, Hiroki CH, Sarden N, Granton E, Labit E, et al. Marginating transitional B cells modulate neutrophils in the lung during inflammation and pneumonia. J Exp Med. 2021;218:e20210409. doi: 10.1084/jem.20210409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Li J, Kumari T, Barazia A, Jha V, Jeong SY, Olson A, et al. Neutrophil DREAM promotes neutrophil recruitment in vascular inflammation. J Exp Med. 2022;219:e20211083. doi: 10.1084/jem.20211083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gomez JC, Doerschuk CM. Neutrophil DREAM come true: The not-so-impossible quest for mechanisms of neutrophil function and heterogeneity. J Exp Med. 2022;219:jem.2021214112202021c. doi: 10.1084/jem.2021214112202021c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yoshida TM, Wang A, Hafler DA. Basic principles of neuroimmunology. Semin Immunopathol. 2022;44:685–95. doi: 10.1007/s00281-022-00951-7. [DOI] [PubMed] [Google Scholar]
- 111.Gray KJ, Gibbs JE. Adaptive immunity, chronic inflammation and the clock. Semin Immunopathol. 2022;44:209–24. doi: 10.1007/s00281-022-00919-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Olejniczak I, Oster H, Ray DW. Glucocorticoid circadian rhythms in immune function. Semin Immunopathol. 2022;44:153–63. doi: 10.1007/s00281-021-00889-2. [DOI] [PubMed] [Google Scholar]
- 113.Lucas D, Battista M, Shi PA, Isola L, Frenette PS. Mobilized hematopoietic stem cell yield depends on species-specific circadian timing. Cell Stem Cell. 2008;3:364–6. doi: 10.1016/j.stem.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bailey C, Black JRM, Reading JL, Litchfield K, Turajlic S, McGranahan N, et al. Tracking cancer evolution through the disease course. Cancer Disco. 2021;11:916–32. doi: 10.1158/2159-8290.CD-20-1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. 2014;14:535–46. doi: 10.1038/nrc3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Skoulidis F, Heymach JV. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat Rev Cancer. 2019;19:495–509. doi: 10.1038/s41568-019-0179-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yeregui E, Viladés C, Domingo P, Ceausu A, Pacheco YM, Veloso S, et al. High circulating SDF-1and MCP-1 levels and genetic variations in CXCL12, CCL2 and CCR5: Prognostic signature of immune recovery status in treated HIV-positive patients. EBioMedicine. 2020;62:103077. doi: 10.1016/j.ebiom.2020.103077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Casanova JL, Abel L. From rare disorders of immunity to common determinants of infection: following the mechanistic thread. Cell. 2022;185:3086–103. doi: 10.1016/j.cell.2022.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kaiser LM, Hunter ZR, Treon SP, Buske C. CXCR4 in Waldenstrom’s Macroglobulinema: chances and challenges. Leukemia. 2021;35:333–45. doi: 10.1038/s41375-020-01102-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Adrian-Kalchhauser I, Sultan SE, Shama LNS, Spence-Jones H, Tiso S, Keller Valsecchi CI, et al. Understanding ‘Non-genetic’ inheritance: insights from molecular-evolutionary crosstalk. Trends Ecol Evol. 2020;35:1078–89. doi: 10.1016/j.tree.2020.08.011. [DOI] [PubMed] [Google Scholar]
- 121.Malta-Santos H, Fukutani KF, Sorgi CA, Queiroz A, Nardini V, Silva J, et al. Multi-omic analyses of plasma cytokines, lipidomics, and transcriptomics distinguish treatment outcomes in cutaneous leishmaniasis. iScience. 2020;23:101840. doi: 10.1016/j.isci.2020.101840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wichukchinda N, Nakayama EE, Rojanawiwat A, Pathipvanich P, Auwanit W, Vongsheree S, et al. Effects of CCR2 and CCRS polymorphisms on HIV-1 infection in Thai females. J Acquir Immune Defic Syndr. 2008;47:293–7. doi: 10.1097/QAI.0b013e318162caab. [DOI] [PubMed] [Google Scholar]
- 123.Scheller SH, Rashad Y, Saleh FM, Willingham KA, Reilich A, Lin D, et al. Biallelic, selectable, knock-in targeting of CCR5 via CRISPR-Cas9 mediated homology directed repair inhibits HIV-1 replication. Front Immunol. 2022;13:821190. doi: 10.3389/fimmu.2022.821190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Xu M. CCR5-Delta32 biology, gene editing, and warnings for the future of CRISPR-Cas9 as a human and humane gene editing tool. Cell Biosci. 2020;10:48. doi: 10.1186/s13578-020-00410-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gavegnano C, Savarino A, Owanikoko T, Marconi VC. Crossroads of cancer and HIV-1: pathways to a Cure for HIV. Front Immunol. 2019;10:2267. doi: 10.3389/fimmu.2019.02267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381:661–6. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 127.Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature. 1996;381:667–73. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 128.Oberlin E, Amara A, Bachelerie F, Bessia C, Virelizier JL, Arenzana-Seisdedos F, et al. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature. 1996;382:833–5. doi: 10.1038/382833a0. [DOI] [PubMed] [Google Scholar]
- 129.Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, et al. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature. 1996;382:829–33. doi: 10.1038/382829a0. [DOI] [PubMed] [Google Scholar]
- 130.Yandrapally S, Mohareer K, Arekuti G, Vadankula GR, Banerjee S. HIV co-receptor-tropism: cellular and molecular events behind the enigmatic co-receptor switching. Crit Rev Microbiol. 2021;47:499–516. doi: 10.1080/1040841X.2021.1902941. [DOI] [PubMed] [Google Scholar]
- 131.Xu L, Wang J, Liu Y, Xie L, Su B, Mou D, et al. CRISPR-edited stem cells in a patient with HIV and acute lymphocytic leukemia. N Engl J Med. 2019;381:1240–7. doi: 10.1056/NEJMoa1817426. [DOI] [PubMed] [Google Scholar]
- 132.Ansari-Lari MA, Liu XM, Metzker ML, Rut AR, Gibbs RA. The extent of genetic variation in the CCR5 gene. Nat Genet. 1997;16:221–2. doi: 10.1038/ng0797-221. [DOI] [PubMed] [Google Scholar]
- 133.Kostrikis LG, Huang Y, Moore JP, Wolinsky SM, Zhang L, Guo Y, et al. A chemokine receptor CCR2 allele delays HIV-1 disease progression and is associated with a CCR5 promoter mutation. Nat Med. 1998;4:350–3. doi: 10.1038/nm0398-350. [DOI] [PubMed] [Google Scholar]
- 134.Fantuzzi L, Tagliamonte M, Gauzzi MC, Lopalco L. Dual CCR5/CCR2 targeting: opportunities for the cure of complex disorders. Cell Mol Life Sci. 2019;76:4869–86. doi: 10.1007/s00018-019-03255-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Vega JA, Villegas-Ospina S, Aguilar-Jimenez W, Rugeles MT, Bedoya G, Zapata W. Haplotypes in CCR5-CCR2, CCL3 and CCL5 are associated with natural resistance to HIV-1 infection in a Colombian cohort. Biomedica. 2017;37:267–73. doi: 10.7705/biomedica.v37i3.3237. [DOI] [PubMed] [Google Scholar]
- 136.Sobota RS, Stein CM, Kodaman N, Maro I, Wieland-Alter W, Igo RP, Jr, et al. A chromosome 5q31.1 locus associates with tuberculin skin test reactivity in HIV-positive individuals from tuberculosis hyper-endemic regions in east Africa. PLoS Genet. 2017;13:e1006710. doi: 10.1371/journal.pgen.1006710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Liu S, Liu N, Wang H, Zhang X, Yao Y, Zhang S, et al. CCR5 promoter polymorphisms associated with pulmonary tuberculosis in a Chinese Han population. Front Immunol. 2020;11:544548. doi: 10.3389/fimmu.2020.544548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tournamille C, Colin Y, Cartron JP, Le Van Kim C. Disruption of a GATA motif in the Duffy gene promoter abolishes erythroid gene expression in Duffy-negative individuals. Nat Genet. 1995;10:224–8. doi: 10.1038/ng0695-224. [DOI] [PubMed] [Google Scholar]
- 139.Maheshwari A, Killeen RB Duffy Blood Group System. In: StatPearls. Treasure Island (FL) 2022. [PubMed]
- 140.Kulkarni S, Lied A, Kulkarni V, Rucevic M, Martin MP, Walker-Sperling V, et al. CCR5AS lncRNA variation differentially regulates CCR5, influencing HIV disease outcome. Nat Immunol. 2019;20:824–34. doi: 10.1038/s41590-019-0406-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Schmidt SV, Krebs W, Ulas T, Xue J, Baßler K, Günther P, et al. The transcriptional regulator network of human inflammatory macrophages is defined by open chromatin. Cell Res. 2016;26:151–70. doi: 10.1038/cr.2016.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Menten P, Struyf S, Schutyser E, Wuyts A, De Clercq E, Schols D, et al. The LD78beta isoform of MIP-1alpha is the most potent CCR5 agonist and HIV-1-inhibiting chemokine. J Clin Invest. 1999;104:R1–5. doi: 10.1172/JCI7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Aquaro S, Menten P, Struyf S, Proost P, Van Damme J, De Clercq E, et al. The LD78beta isoform of MIP-1alpha is the most potent CC-chemokine in inhibiting CCR5-dependent human immunodeficiency virus type 1 replication in human macrophages. J Virol. 2001;75:4402–6. doi: 10.1128/JVI.75.9.4402-4406.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Liu S, Yao L, Ding D, Zhu H. CCL3L1 copy number variation and susceptibility to HIV-1 infection: a meta-analysis. PLoS One. 2010;5:e15778. doi: 10.1371/journal.pone.0015778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dogan S, Mart Komurcu SZ, Korkmaz MD, Kaya E, Yavas S, Dogan S, et al. Effect of chemokine gene variants on Covid-19 disease severity. Immunol Invest. 2022;51:1–10. doi: 10.1080/08820139.2022.2088383. [DOI] [PubMed] [Google Scholar]
- 146.Katz DH, Wilson JG, Gerszten RE, Jackson F. Malmo Heart Study G. Mining a GWAS of severe Covid-19. N Engl J Med. 2020;383:2589. doi: 10.1056/NEJMc2025747. [DOI] [PubMed] [Google Scholar]
- 147.Yoshida K, Gowers KHC, Lee-Six H, Chandrasekharan DP, Coorens T, Maughan EF, et al. Tobacco smoking and somatic mutations in human bronchial epithelium. Nature. 2020;578:266–72. doi: 10.1038/s41586-020-1961-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Martincorena I, Fowler JC, Wabik A, Lawson A, Abascal F, Hall M, et al. Somatic mutant clones colonize the human esophagus with age. Science. 2018;362:911–7. doi: 10.1126/science.aau3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhang Q, Cao X. Epigenetic regulation of the innate immune response to infection. Nat Rev Immunol. 2019;19:417–32. doi: 10.1038/s41577-019-0151-6. [DOI] [PubMed] [Google Scholar]
- 150.Zhang Q, Cao X. Epigenetic remodeling in innate immunity and inflammation. Annu Rev Immunol. 2021;39:279–311. doi: 10.1146/annurev-immunol-093019-123619. [DOI] [PubMed] [Google Scholar]
- 151.Casado-Pelaez M, Bueno-Costa A, Esteller M. Single cell cancer epigenetics. Trends Cancer. 2022;8:820–38. doi: 10.1016/j.trecan.2022.06.005. [DOI] [PubMed] [Google Scholar]
- 152.Ku WL, Nakamura K, Gao W, Cui K, Hu G, Tang Q, et al. Single-cell chromatin immunocleavage sequencing (scChIC-seq) to profile histone modification. Nat Methods. 2019;16:323–5. doi: 10.1038/s41592-019-0361-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gao W, Lai B, Ni B, Zhao K. Genome-wide profiling of nucleosome position and chromatin accessibility in single cells using scMNase-seq. Nat Protoc. 2020;15:68–85. doi: 10.1038/s41596-019-0243-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Preissl S, Gaulton KJ, Ren B. Characterizing cis-regulatory elements using single-cell epigenomics. Nat Rev Genet. 2022;24:21–43. doi: 10.1038/s41576-022-00509-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Rudolph KL. DNA-methylation aging at single-cell level. Nat Aging. 2021;1:1086–7. doi: 10.1038/s43587-021-00154-z. [DOI] [PubMed] [Google Scholar]
- 156.Lau CM, Wiedemann GM, Sun JC. Epigenetic regulation of natural killer cell memory. Immunol Rev. 2022;305:90–110. doi: 10.1111/imr.13031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mattei AL, Bailly N, Meissner A. DNA methylation: a historical perspective. Trends Genet. 2022;38:676–707. doi: 10.1016/j.tig.2022.03.010. [DOI] [PubMed] [Google Scholar]
- 158.Ting AH, Lee BH. 5mC: what goes on must come off. Nat Rev Mol Cell Biol. 2022;23:306. doi: 10.1038/s41580-022-00469-8. [DOI] [PubMed] [Google Scholar]
- 159.Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2019;20:608–24. doi: 10.1038/s41580-019-0168-5. [DOI] [PubMed] [Google Scholar]
- 160.Xiong X, Yi C, Peng J. Epitranscriptomics: toward a better understanding of RNA modifications. Genomics Proteom Bioinforma. 2017;15:147–53. doi: 10.1016/j.gpb.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Schones DE, Zhao K. Genome-wide approaches to studying chromatin modifications. Nat Rev Genet. 2008;9:179–91. doi: 10.1038/nrg2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Carter B, Zhao K. The epigenetic basis of cellular heterogeneity. Nat Rev Genet. 2021;22:235–50. doi: 10.1038/s41576-020-00300-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Janssen SM, Lorincz MC. Interplay between chromatin marks in development and disease. Nat Rev Genet. 2022;23:137–53. doi: 10.1038/s41576-021-00416-x. [DOI] [PubMed] [Google Scholar]
- 164.Yu M, Ren B. The three-dimensional organization of Mammalian genomes. Annu Rev Cell Dev Biol. 2017;33:265–89. doi: 10.1146/annurev-cellbio-100616-060531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Cenik BK, Shilatifard A. COMPASS and SWI/SNF complexes in development and disease. Nat Rev Genet. 2021;22:38–58. doi: 10.1038/s41576-020-0278-0. [DOI] [PubMed] [Google Scholar]
- 166.Boltsis I, Grosveld F, Giraud G, Kolovos P. Chromatin conformation in development and disease. Front Cell Dev Biol. 2021;9:723859. doi: 10.3389/fcell.2021.723859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Breeze CE, Beck S, Berndt SI, Franceschini N. The missing diversity in human epigenomic studies. Nat Genet. 2022;54:737–9. doi: 10.1038/s41588-022-01081-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hanahan D. Hallmarks of cancer: new dimensions. Cancer Disco. 2022;12:31–46. doi: 10.1158/2159-8290.CD-21-1059. [DOI] [PubMed] [Google Scholar]
- 169.Crompton JG, Narayanan M, Cuddapah S, Roychoudhuri R, Ji Y, Yang W, et al. Lineage relationship of CD8(+) T cell subsets is revealed by progressive changes in the epigenetic landscape. Cell Mol Immunol. 2016;13:502–13. doi: 10.1038/cmi.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Northrup DL, Zhao K. Application of ChIP-Seq and related techniques to the study of immune function. Immunity. 2011;34:830–42. doi: 10.1016/j.immuni.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–37. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 172.Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–31. doi: 10.1016/j.cell.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lai B, Gao W, Cui K, Xie W, Tang Q, Jin W, et al. Principles of nucleosome organization revealed by single-cell micrococcal nuclease sequencing. Nature. 2018;562:281–5. doi: 10.1038/s41586-018-0567-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Jin W, Tang Q, Wan M, Cui K, Zhang Y, Ren G, et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature. 2015;528:142–6. doi: 10.1038/nature15740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–93. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.de Laat W, Duboule D. Topology of mammalian developmental enhancers and their regulatory landscapes. Nature. 2013;502:499–506. doi: 10.1038/nature12753. [DOI] [PubMed] [Google Scholar]
- 177.Downes DJ, Beagrie RA, Gosden ME, Telenius J, Carpenter SJ, Nussbaum L, et al. High-resolution targeted 3C interrogation of cis-regulatory element organization at genome-wide scale. Nat Commun. 2021;12:531. doi: 10.1038/s41467-020-20809-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Dekker J, Belmont AS, Guttman M, Leshyk VO, Lis JT, Lomvardas S, et al. The 4D nucleome project. Nature. 2017;549:219–26. doi: 10.1038/nature23884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Han J, Zhang Z, Wang K. 3C and 3C-based techniques: the powerful tools for spatial genome organization deciphering. Mol Cytogenet. 2018;11:21. doi: 10.1186/s13039-018-0368-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Branco MR, Ficz G, Reik W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat Rev Genet. 2011;13:7–13. doi: 10.1038/nrg3080. [DOI] [PubMed] [Google Scholar]
- 181.Parry A, Rulands S, Reik W. Active turnover of DNA methylation during cell fate decisions. Nat Rev Genet. 2021;22:59–66. doi: 10.1038/s41576-020-00287-8. [DOI] [PubMed] [Google Scholar]
- 182.Li J, Li L, Wang Y, Huang G, Li X, Xie Z, et al. Insights Into the role of DNA methylation in immune cell development and autoimmune disease. Front Cell Dev Biol. 2021;9:757318. doi: 10.3389/fcell.2021.757318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Saeki N, Inoue K, Ideta-Otsuka M, Watamori K, Mizuki S, Takenaka K, et al. Epigenetic regulator UHRF1 orchestrates proinflammatory gene expression in rheumatoid arthritis in a suppressive manner. J Clin Invest. 2022;132:e150533. doi: 10.1172/JCI150533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Shi J, Xu J, Chen YE, Li JS, Cui Y, Shen L, et al. The concurrence of DNA methylation and demethylation is associated with transcription regulation. Nat Commun. 2021;12:5285. doi: 10.1038/s41467-021-25521-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Yousefi PD, Suderman M, Langdon R, Whitehurst O, Davey Smith G, Relton CL. DNA methylation-based predictors of health: applications and statistical considerations. Nat Rev Genet. 2022;23:369–83. doi: 10.1038/s41576-022-00465-w. [DOI] [PubMed] [Google Scholar]
- 186.Hurtado C, Acevedo Sáenz LY, Vásquez Trespalacios EM, Urrego R, Jenks S, Sanz I, et al. DNA methylation changes on immune cells in systemic lupus erythematosus. Autoimmunity. 2020;53:114–21. doi: 10.1080/08916934.2020.1722108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Takahashi A, de Andres MC, Hashimoto K, Itoi E, Oreffo RO. Epigenetic regulation of interleukin-8, an inflammatory chemokine, in osteoarthritis. Osteoarthr Cartil. 2015;23:1946–54. doi: 10.1016/j.joca.2015.02.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Yee KM, Shuai RW, Liu B, Huynh CA, Niu C, Lee HR, et al. TET1 controls Cxcl1 induction by DNA demethylation and promotes neutrophil recruitment during acute lung injury. bioRxiv. 2021:2021.2009.2007.459280.
- 189.Wang YF, Yu L, Hu ZL, Fang YF, Shen YY, Song MF, et al. Regulation of CCL2 by EZH2 affects tumor-associated macrophages polarization and infiltration in breast cancer. Cell Death Dis. 2022;13:748. doi: 10.1038/s41419-022-05169-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Li H, Li Q, Ma Z, Zhou Z, Fan J, Jin Y, et al. AID modulates carcinogenesis network via DNA demethylation in bladder urothelial cell carcinoma. Cell Death Dis. 2019;10:251. doi: 10.1038/s41419-019-1472-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Li L, Gao Y, Wu Q, Cheng ASL, Yip KY. New guidelines for DNA methylome studies regarding 5-hydroxymethylcytosine for understanding transcriptional regulation. Genome Res. 2019;29:543–53. doi: 10.1101/gr.240036.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ginno PA, Gaidatzis D, Feldmann A, Hoerner L, Imanci D, Burger L, et al. A genome-scale map of DNA methylation turnover identifies site-specific dependencies of DNMT and TET activity. Nat Commun. 2020;11:2680. doi: 10.1038/s41467-020-16354-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.de Mendoza A, Nguyen TV, Ford E, Poppe D, Buckberry S, Pflueger J, et al. Large-scale manipulation of promoter DNA methylation reveals context-specific transcriptional responses and stability. Genome Biol. 2022;23:163. doi: 10.1186/s13059-022-02728-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Yang X, Shen X, Li Z, Li W, Liu Y. Reduction in immune cell number and loss of 5hmC are associated with lesion grade in cervical carcinogenesis. 3 Biotech. 2021;11:486. doi: 10.1007/s13205-021-03028-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Stuckel AJ, Zhang W, Zhang X, Zeng S, Dougherty U, Mustafi R, et al. Enhanced CXCR4 expression associates with increased gene body 5-hydroxymethylcytosine modification but not decreased promoter methylation in colorectal cancer. Cancers (Basel) 2020;12:539. doi: 10.3390/cancers12030539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Khare T, Bissonnette M, Khare S. CXCL12-CXCR4/CXCR7 axis in colorectal cancer: therapeutic target in preclinical and clinical studies. Int J Mol Sci. 2021;22:7371. doi: 10.3390/ijms22147371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Xu L, Zhou Y, Chen L, Bissessur AS, Chen J, Mao M, et al. Deoxyribonucleic acid 5-hydroxymethylation in cell-free deoxyribonucleic acid, a novel cancer biomarker in the era of precision medicine. Front Cell Dev Biol. 2021;9:744990. doi: 10.3389/fcell.2021.744990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Le QV, Lee J, Lee H, Shim G, Oh YK. Cell membrane-derived vesicles for delivery of therapeutic agents. Acta Pharm Sin B. 2021;11:2096–113. doi: 10.1016/j.apsb.2021.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Zhang X, Zhao D, Yin Y, Yang T, You Z, Li D, et al. Circulating cell-free DNA-based methylation patterns for breast cancer diagnosis. NPJ Breast Cancer. 2021;7:106. doi: 10.1038/s41523-021-00316-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Campagna MP, Xavier A, Lechner-Scott J, Maltby V, Scott RJ, Butzkueven H, et al. Epigenome-wide association studies: current knowledge, strategies and recommendations. Clin Epigenetics. 2021;13:214. doi: 10.1186/s13148-021-01200-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Cain JA, Montibus B, Oakey RJ. Intragenic CpG islands and their impact on gene regulation. Front Cell Dev Biol. 2022;10:832348. doi: 10.3389/fcell.2022.832348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–86. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Morales-Nebreda L, McLafferty FS, Singer BD. DNA methylation as a transcriptional regulator of the immune system. Transl Res. 2019;204:1–18. doi: 10.1016/j.trsl.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Xiong Z, Yang F, Li M, Ma Y, Zhao W, Wang G, et al. EWAS Open Platform: integrated data, knowledge and toolkit for epigenome-wide association study. Nucleic Acids Res. 2022;50:D1004–D1009. doi: 10.1093/nar/gkab972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Zaghlool SB, Kühnel B, Elhadad MA, Kader S, Halama A, Thareja G, et al. Epigenetics meets proteomics in an epigenome-wide association study with circulating blood plasma protein traits. Nat Commun. 2020;11:15. doi: 10.1038/s41467-019-13831-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Myte R, Sundkvist A, Van Guelpen B, Harlid S. Circulating levels of inflammatory markers and DNA methylation, an analysis of repeated samples from a population based cohort. Epigenetics. 2019;14:649–59. doi: 10.1080/15592294.2019.1603962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Ahsan M, Ek WE, Rask-Andersen M, Karlsson T, Lind-Thomsen A, Enroth S, et al. The relative contribution of DNA methylation and genetic variants on protein biomarkers for human diseases. PLoS Genet. 2017;13:e1007005. doi: 10.1371/journal.pgen.1007005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Huang J, Fan T, Yan Q, Zhu H, Fox S, Issaq HJ, et al. Lsh, an epigenetic guardian of repetitive elements. Nucleic Acids Res. 2004;32:5019–28. doi: 10.1093/nar/gkh821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Ni K, Ren J, Xu X, He Y, Finney R, Braun S, et al. LSH mediates gene repression through macroH2A deposition. Nat Commun. 2020;11:5647. doi: 10.1038/s41467-020-19159-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Sukapan P, Promnarate P, Avihingsanon Y, Mutirangura A, Hirankarn N. Types of DNA methylation status of the interspersed repetitive sequences for LINE-1, Alu, HERV-E and HERV-K in the neutrophils from systemic lupus erythematosus patients and healthy controls. J Hum Genet. 2014;59:178–88. doi: 10.1038/jhg.2013.140. [DOI] [PubMed] [Google Scholar]
- 211.Do C, Shearer A, Suzuki M, Terry MB, Gelernter J, Greally JM, et al. Genetic-epigenetic interactions in cis: a major focus in the post-GWAS era. Genome Biol. 2017;18:120. doi: 10.1186/s13059-017-1250-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Dong F, Qin X, Wang B, Li Q, Hu J, Cheng X, et al. ALKBH5 facilitates hypoxia-induced paraspeckle assembly and IL8 secretion to generate an immunosuppressive tumor microenvironment. Cancer Res. 2021;81:5876–88. doi: 10.1158/0008-5472.CAN-21-1456. [DOI] [PubMed] [Google Scholar]
- 213.Liu Y, Song R, Zhao L, Lu Z, Li Y, Zhan X, et al. m(6)A demethylase ALKBH5 is required for antibacterial innate defense by intrinsic motivation of neutrophil migration. Signal Transduct Target Ther. 2022;7:194. doi: 10.1038/s41392-022-01020-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Zhao J, Cheng F, Jia P, Cox N, Denny JC, Zhao Z. An integrative functional genomics framework for effective identification of novel regulatory variants in genome-phenome studies. Genome Med. 2018;10:7. doi: 10.1186/s13073-018-0513-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Castellanos-Rubio A, Ghosh S. Functional Implications of Intergenic GWAS SNPs in Immune-Related LncRNAs. Adv Exp Med Biol. 2022;1363:147–60. doi: 10.1007/978-3-030-92034-0_8. [DOI] [PubMed] [Google Scholar]
- 216.Li H, Achour I, Bastarache L, Berghout J, Gardeux V, Li J, et al. Integrative genomics analyses unveil downstream biological effectors of disease-specific polymorphisms buried in intergenic regions. NPJ Genom Med. 2016;1:16006. doi: 10.1038/npjgenmed.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Gazal S, Weissbrod O, Hormozdiari F, Dey KK, Nasser J, Jagadeesh KA, et al. Combining SNP-to-gene linking strategies to identify disease genes and assess disease omnigenicity. Nat Genet. 2022;54:827–36. doi: 10.1038/s41588-022-01087-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–47. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Isbel L, Grand RS, Schubeler D. Generating specificity in genome regulation through transcription factor sensitivity to chromatin. Nat Rev Genet. 2022;23:728–40. doi: 10.1038/s41576-022-00512-6. [DOI] [PubMed] [Google Scholar]
- 220.Field A, Adelman K. Evaluating enhancer function and transcription. Annu Rev Biochem. 2020;89:213–34. doi: 10.1146/annurev-biochem-011420-095916. [DOI] [PubMed] [Google Scholar]
- 221.Bergman DT, Jones TR, Liu V, Ray J, Jagoda E, Siraj L, et al. Compatibility rules of human enhancer and promoter sequences. Nature. 2022;607:176–84. doi: 10.1038/s41586-022-04877-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Heinz S, Romanoski CE, Benner C, Glass CK. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol. 2015;16:144–54. doi: 10.1038/nrm3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–19. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Jia Y, Chng WJ, Zhou J. Super-enhancers: critical roles and therapeutic targets in hematologic malignancies. J Hematol Oncol. 2019;12:77. doi: 10.1186/s13045-019-0757-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Huang M, Chen Y, Yang M, Guo A, Xu Y, Xu L, et al. dbCoRC: a database of core transcriptional regulatory circuitries modeled by H3K27ac ChIP-seq signals. Nucleic Acids Res. 2018;46:D71–D77. doi: 10.1093/nar/gkx796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Zabidi MA, Arnold CD, Schernhuber K, Pagani M, Rath M, Frank O, et al. Enhancer-core-promoter specificity separates developmental and housekeeping gene regulation. Nature. 2015;518:556–9. doi: 10.1038/nature13994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Wang N, Wu R, Tang D, Kang R. The BET family in immunity and disease. Signal Transduct Target Ther. 2021;6:23. doi: 10.1038/s41392-020-00384-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Trojer P, Targeting BET. Bromodomains in cancer. Annu Rev Cancer Biol. 2022;6:313–36. doi: 10.1146/annurev-cancerbio-070120-103531. [DOI] [Google Scholar]
- 229.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–23. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–73. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Borck PC, Guo LW, Plutzky J. BET epigenetic reader proteins in cardiovascular transcriptional programs. Circ Res. 2020;126:1190–208. doi: 10.1161/CIRCRESAHA.120.315929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Zhang Q, Lenardo MJ, Baltimore D. 30 years of NF-kappaB: a blossoming of relevance to human pathobiology. Cell. 2017;168:37–57. doi: 10.1016/j.cell.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Taniguchi K, Karin M. NF-kappaB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. 2018;18:309–24. doi: 10.1038/nri.2017.142. [DOI] [PubMed] [Google Scholar]
- 234.Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–71. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 235.Higashijima Y, Matsui Y, Shimamura T, Nakaki R, Nagai N, Tsutsumi S, et al. Coordinated demethylation of H3K9 and H3K27 is required for rapid inflammatory responses of endothelial cells. EMBO J. 2020;39:e103949. doi: 10.15252/embj.2019103949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Suzuki HI, Young RA, Sharp PA. Super-enhancer-mediated RNA processing revealed by integrative MicroRNA network analysis. Cell. 2017;168:1000–14.e1015. doi: 10.1016/j.cell.2017.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Fanucchi S, Fok ET, Dalla E, Shibayama Y, Börner K, Chang EY, et al. Immune genes are primed for robust transcription by proximal long noncoding RNAs located in nuclear compartments. Nat Genet. 2019;51:138–50. doi: 10.1038/s41588-018-0298-2. [DOI] [PubMed] [Google Scholar]
- 238.Fok ET, Davignon L, Fanucchi S, Mhlanga MM. The lncRNA connection between cellular metabolism and epigenetics in trained immunity. Front Immunol. 2018;9:3184. doi: 10.3389/fimmu.2018.03184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Xing Z, Lin A, Li C, Liang K, Wang S, Liu Y, et al. lncRNA directs cooperative epigenetic regulation downstream of chemokine signals. Cell. 2014;159:1110–25. doi: 10.1016/j.cell.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Liu M, Cao S, He L, Gao J, Arab JP, Cui H, et al. Super enhancer regulation of cytokine-induced chemokine production in alcoholic hepatitis. Nat Commun. 2021;12:4560. doi: 10.1038/s41467-021-24843-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Kim JJ, Kingston RE. Context-specific polycomb mechanisms in development. Nat Rev Genet. 2022;23:680–95. doi: 10.1038/s41576-022-00499-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Parreno V, Martinez AM, Cavalli G. Mechanisms of Polycomb group protein function in cancer. Cell Res. 2022;32:231–53. doi: 10.1038/s41422-021-00606-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.van der Woude LL, Gorris MAJ, Halilovic A, Figdor CG, de Vries IJM. Migrating into the tumor: a roadmap for T cells. Trends Cancer. 2017;3:797–808. doi: 10.1016/j.trecan.2017.09.006. [DOI] [PubMed] [Google Scholar]
- 244.Chen X, Pan X, Zhang W, Guo H, Cheng S, He Q, et al. Epigenetic strategies synergize with PD-L1/PD-1 targeted cancer immunotherapies to enhance antitumor responses. Acta Pharm Sin B. 2020;10:723–33. doi: 10.1016/j.apsb.2019.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Franciszkiewicz K, Boissonnas A, Boutet M, Combadiere C, Mami-Chouaib F. Role of chemokines and chemokine receptors in shaping the effector phase of the antitumor immune response. Cancer Res. 2012;72:6325–32. doi: 10.1158/0008-5472.CAN-12-2027. [DOI] [PubMed] [Google Scholar]
- 246.Peng W, Liu C, Xu C, Lou Y, Chen J, Yang Y, et al. PD-1 blockade enhances T-cell migration to tumors by elevating IFN-gamma inducible chemokines. Cancer Res. 2012;72:5209–18. doi: 10.1158/0008-5472.CAN-12-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 248.Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527:249–53. doi: 10.1038/nature15520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Nagarsheth N, Peng D, Kryczek I, Wu K, Li W, Zhao E, et al. PRC2 epigenetically silences Th1-type chemokines to suppress effector T-cell trafficking in colon cancer. Cancer Res. 2016;76:275–82. doi: 10.1158/0008-5472.CAN-15-1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Dong H, Liu S, Zhang X, Chen S, Kang L, Chen Y, et al. An allosteric PRC2 inhibitor targeting EED suppresses tumor progression by modulating the immune response. Cancer Res. 2019;79:5587–96. doi: 10.1158/0008-5472.CAN-19-0428. [DOI] [PubMed] [Google Scholar]
- 251.Ong LT, Lee WC, Ma S, Oguz G, Niu Z, Bao Y, et al. IFI16-dependent STING signaling is a crucial regulator of anti-HER2 immune response in HER2+ breast cancer. Proc Natl Acad Sci USA. 2022;119:e2201376119. doi: 10.1073/pnas.2201376119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Qin Y, Vasilatos SN, Chen L, Wu H, Cao Z, Fu Y, et al. Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances antitumor efficacy of immune checkpoint blockade. Oncogene. 2019;38:390–405. doi: 10.1038/s41388-018-0451-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Hong W, Yang B, He Q, Wang J, Weng Q. New insights of CCR7 signaling in dendritic cell migration and inflammatory diseases. Front Pharm. 2022;13:841687. doi: 10.3389/fphar.2022.841687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Salem A, Alotaibi M, Mroueh R, Basheer HA, Afarinkia K. CCR7 as a therapeutic target in cancer. Biochim Biophys Acta Rev Cancer. 2021;1875:188499. doi: 10.1016/j.bbcan.2020.188499. [DOI] [PubMed] [Google Scholar]
- 255.Godoy-Tena G, Ballestar E. Epigenetics of dendritic cells in tumor immunology. Cancers (Basel) 2022;14:1179. doi: 10.3390/cancers14051179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Chauvistre H, Sere K. Epigenetic aspects of DC development and differentiation. Mol Immunol. 2020;128:116–24. doi: 10.1016/j.molimm.2020.10.011. [DOI] [PubMed] [Google Scholar]
- 257.Moran TP, Nakano H, Kondilis-Mangum HD, Wade PA, Cook DN. Epigenetic control of Ccr7 expression in distinct lineages of lung dendritic cells. J Immunol. 2014;193:4904–13. doi: 10.4049/jimmunol.1401104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Mori T, Kim J, Yamano T, Takeuchi H, Huang S, Umetani N, et al. Epigenetic up-regulation of C-C chemokine receptor 7 and C-X-C chemokine receptor 4 expression in melanoma cells. Cancer Res. 2005;65:1800–7. doi: 10.1158/0008-5472.CAN-04-3531. [DOI] [PubMed] [Google Scholar]
- 259.Ferrara G, Benzi A, Sturla L, Marubbi D, Frumento D, Spinelli S, et al. Sirt6 inhibition delays the onset of experimental autoimmune encephalomyelitis by reducing dendritic cell migration. J Neuroinflammation. 2020;17:228. doi: 10.1186/s12974-020-01906-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Wang P, Xue Y, Han Y, Lin L, Wu C, Xu S, et al. The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation. Science. 2014;344:310–3. doi: 10.1126/science.1251456. [DOI] [PubMed] [Google Scholar]
- 261.Liu J, Zhang X, Chen K, Cheng Y, Liu S, Xia M, et al. CCR7 chemokine receptor-inducible lnc-Dpf3 restrains dendritic cell migration by inhibiting HIF-1alpha-mediated glycolysis. Immunity. 2019;50:600–15.e615. doi: 10.1016/j.immuni.2019.01.021. [DOI] [PubMed] [Google Scholar]
- 262.Le Y, Zhou Y, Iribarren P, Wang J. Chemokines and chemokine receptors: their manifold roles in homeostasis and disease. Cell Mol Immunol. 2004;1:95–104. [PubMed] [Google Scholar]
- 263.Sollberger G, Tilley DO, Zychlinsky A. Neutrophil extracellular traps: the biology of chromatin externalization. Dev Cell. 2018;44:542–53. doi: 10.1016/j.devcel.2018.01.019. [DOI] [PubMed] [Google Scholar]
- 264.Lewis HD, Liddle J, Coote JE, Atkinson SJ, Barker MD, Bax BD, et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat Chem Biol. 2015;11:189–91. doi: 10.1038/nchembio.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, Jr, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. 2010;107:15880–5. doi: 10.1073/pnas.1005743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Zhu D, Zhang Y, Wang S. Histone citrullination: a new target for tumors. Mol Cancer. 2021;20:90. doi: 10.1186/s12943-021-01373-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Yu K, Proost P. Insights into peptidylarginine deiminase expression and citrullination pathways. Trends Cell Biol. 2022;32:746–61. doi: 10.1016/j.tcb.2022.01.014. [DOI] [PubMed] [Google Scholar]
- 268.Knopf J, Leppkes M, Schett G, Herrmann M, Munoz LE. Aggregated NETs sequester and detoxify extracellular histones. Front Immunol. 2019;10:2176. doi: 10.3389/fimmu.2019.02176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Al-Kuraishy HM, Al-Gareeb AI, Al-Hussaniy HA, Al-Harcan NAH, Alexiou A, Batiha GE. Neutrophil Extracellular Traps (NETs) and Covid-19: a new frontiers for therapeutic modality. Int Immunopharmacol. 2022;104:108516. doi: 10.1016/j.intimp.2021.108516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Lu Y, Huang Y, Li J, Huang J, Zhang L, Feng J, et al. Eosinophil extracellular traps drive asthma progression through neuro-immune signals. Nat Cell Biol. 2021;23:1060–72. doi: 10.1038/s41556-021-00762-2. [DOI] [PubMed] [Google Scholar]
- 271.Teijeira A, Garasa S, Ochoa MC, Villalba M, Olivera I, Cirella A, et al. IL8, Neutrophils, and NETs in a Collusion against Cancer Immunity and Immunotherapy. Clin Cancer Res. 2021;27:2383–93. doi: 10.1158/1078-0432.CCR-20-1319. [DOI] [PubMed] [Google Scholar]
- 272.Abrams ST, Morton B, Alhamdi Y, Alsabani M, Lane S, Welters ID, et al. A novel assay for neutrophil extracellular trap formation independently predicts disseminated intravascular coagulation and mortality in critically Ill patients. Am J Respir Crit Care Med. 2019;200:869–80. doi: 10.1164/rccm.201811-2111OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Sisirak V, Sally B, D’agati V, Martinez-Ortiz W, Özçakar ZB, David J, et al. Digestion of chromatin in apoptotic cell microparticles prevents autoimmunity. Cell. 2016;166:88–101. doi: 10.1016/j.cell.2016.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Mistry P, Nakabo S, O’neil L, Goel RR, Jiang K, Carmona-Rivera C, et al. Transcriptomic, epigenetic, and functional analyses implicate neutrophil diversity in the pathogenesis of systemic lupus erythematosus. Proc Natl Acad Sci USA. 2019;116:25222–8. doi: 10.1073/pnas.1908576116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Kwak SB, Kim SJ, Kim J, Kang YL, Ko CW, Kim I, et al. Tumor regionalization after surgery: Roles of the tumor microenvironment and neutrophil extracellular traps. Exp Mol Med. 2022;54:720–9. doi: 10.1038/s12276-022-00784-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Gonzalez-Aparicio M, Alfaro C. Influence of interleukin-8 and neutrophil extracellular trap (NET) Formation In The Tumor Microenvironment: Is There A Pathogenic Role? J Immunol Res. 2019;2019:6252138. doi: 10.1155/2019/6252138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature. 2020;583:133–8. doi: 10.1038/s41586-020-2394-6. [DOI] [PubMed] [Google Scholar]
- 278.Nie M, Yang L, Bi X, Wang Y, Sun P, Yang H, et al. Neutrophil extracellular traps induced by IL8 promote diffuse large B-cell lymphoma progression via the TLR9 signaling. Clin Cancer Res. 2019;25:1867–79. doi: 10.1158/1078-0432.CCR-18-1226. [DOI] [PubMed] [Google Scholar]
- 279.Zha C, Meng X, Li L, Mi S, Qian D, Li Z, et al. Neutrophil extracellular traps mediate the crosstalk between glioma progression and the tumor microenvironment via the HMGB1/RAGE/IL-8 axis. Cancer Biol Med. 2020;17:154–68. doi: 10.20892/j.issn.2095-3941.2019.0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med. 2016;8:361ra138. doi: 10.1126/scitranslmed.aag1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Lee W, Ko SY, Mohamed MS, Kenny HA, Lengyel E, Naora H. Neutrophils facilitate ovarian cancer premetastatic niche formation in the omentum. J Exp Med. 2019;216:176–94. doi: 10.1084/jem.20181170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. 2018;361:eaao4227. doi: 10.1126/science.aao4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Merza M, Hartman H, Rahman M, Hwaiz R, Zhang E, Renström E, et al. Neutrophil extracellular traps induce trypsin activation, inflammation, and tissue damage in mice with severe acute pancreatitis. Gastroenterology. 2015;149:1920–31.e1928. doi: 10.1053/j.gastro.2015.08.026. [DOI] [PubMed] [Google Scholar]
- 284.Lopez-Otin C, Matrisian LM. Emerging roles of proteases in tumour suppression. Nat Rev Cancer. 2007;7:800–8. doi: 10.1038/nrc2228. [DOI] [PubMed] [Google Scholar]
- 285.Dudani JS, Warren AD, Bhatia SN. Harnessing protease activity to improve cancer care. Annu Rev Cancer Biol. 2018;2:353–76. doi: 10.1146/annurev-cancerbio-030617-050549. [DOI] [Google Scholar]
- 286.Hosseinnejad A, Ludwig N, Wienkamp AK, Rimal R, Bleilevens C, Rossaint R, et al. DNase I functional microgels for neutrophil extracellular trap disruption. Biomater Sci. 2021;10:85–99. doi: 10.1039/D1BM01591E. [DOI] [PubMed] [Google Scholar]
- 287.Jiménez-Alcázar M, Rangaswamy C, Panda R, Bitterling J, Simsek YJ, Long AT, et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science. 2017;358:1202–6. doi: 10.1126/science.aam8897. [DOI] [PubMed] [Google Scholar]
- 288.Meegan JE, Bastarache JA. NET gain for sepsis research: a new approach to assess neutrophil function in patients. Am J Respir Crit Care Med. 2019;200:798–9. doi: 10.1164/rccm.201905-1074ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. 2017;23:279–87. doi: 10.1038/nm.4294. [DOI] [PubMed] [Google Scholar]
- 290.Schauer C, Janko C, Munoz LE, Zhao Y, Kienhöfer D, Frey B, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med. 2014;20:511–7. doi: 10.1038/nm.3547. [DOI] [PubMed] [Google Scholar]
- 291.Uderhardt S, Knopf J, Herrmann M. Neutrophil swarm control: what goes up must come down. Signal Transduct Target Ther. 2021;6:416. doi: 10.1038/s41392-021-00836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Wang X, Allen WE, Wright MA, Sylwestrak EL, Samusik N, Vesuna S, et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science. 2018;361:eaat5691. doi: 10.1126/science.aat5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Ren XH, He XY, Xu C, Han D, Cheng SX. Functional tumor targeting nano-systems for reprogramming circulating tumor cells with in situ evaluation on therapeutic efficiency at the single-cell level. Adv Sci (Weinh) 2022;9:e2105806. doi: 10.1002/advs.202105806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Baazim H, Antonio-Herrera L, Bergthaler A. The interplay of immunology and cachexia in infection and cancer. Nat Rev Immunol. 2022;22:309–21. doi: 10.1038/s41577-021-00624-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Raza S, Rajak S, Tewari A, Gupta P, Chattopadhyay N, Sinha RA, et al. Multifaceted role of chemokines in solid tumors: From biology to therapy. Semin Cancer Biol. 2022;86:1105–21. doi: 10.1016/j.semcancer.2021.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Wuguo, T, Lingli W, Jianjie, Z, Donglin L The pan-cancer landscape of crosstalk between chemokinesand the tumor immune microenvironment. In: Research Square; 2022.
- 297.Geneva-Popova MG, Popova-Belova SD, Gardzheva PN, Kraev KI. A study of IFN-alpha-induced chemokines CCL2, CXCL10 and CCL19 in patients with systemic lupus erythematosu. Life (Basel) 2022;12:251. doi: 10.3390/life12020251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Mi T, Jin L, Zhang Z, Wang J, Li M, Zhanghuang C, et al. DNA hypermethylation-regulated CX3CL1 reducing T cell infiltration indicates poor prognosis in wilms tumour. Front Oncol. 2022;12:882714. doi: 10.3389/fonc.2022.882714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Gowhari Shabgah A, Al-Obaidi ZMJ, Sulaiman Rahman H, Kamal Abdelbasset W, Suksatan W, Bokov DO, et al. Does CCL19 act as a double-edged sword in cancer development? Clin Exp Immunol. 2022;207:164–75. doi: 10.1093/cei/uxab039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Wang J, Seethala RR, Zhang Q, Gooding W, van Waes C, Hasegawa H, et al. Autocrine and paracrine chemokine receptor 7 activation in head and neck cancer: implications for therapy. J Natl Cancer Inst. 2008;100:502–12. doi: 10.1093/jnci/djn059. [DOI] [PubMed] [Google Scholar]
- 301.Xu Z, Zhu C, Chen C, Zong Y, Feng H, Liu D, et al. CCL19 suppresses angiogenesis through promoting miR-206 and inhibiting Met/ERK/Elk-1/HIF-1alpha/VEGF-A pathway in colorectal cancer. Cell Death Dis. 2018;9:974. doi: 10.1038/s41419-018-1010-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Piseddu I, Röhrle N, Knott MML, Moder S, Eiber S, Schnell K, et al. Constitutive expression of CCL22 is mediated by T cell-derived GM-CSF. J Immunol. 2020;205:2056–65. doi: 10.4049/jimmunol.2000004. [DOI] [PubMed] [Google Scholar]
- 303.Rapp M, Wintergerst MWM, Kunz WG, Vetter VK, Knott M, Lisowski D, et al. CCL22 controls immunity by promoting regulatory T cell communication with dendritic cells in lymph nodes. J Exp Med. 2019;216:1170–81. doi: 10.1084/jem.20170277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Chen J, Zhao D, Zhang L, Zhang J, Xiao Y, Wu Q, et al. Tumor-associated macrophage (TAM)-derived CCL22 induces FAK addiction in esophageal squamous cell carcinoma (ESCC) Cell Mol Immunol. 2022;19:1054–66. doi: 10.1038/s41423-022-00903-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Martinez NM, Lynch KW. Control of alternative splicing in immune responses: many regulators, many predictions, much still to learn. Immunol Rev. 2013;253:216–36. doi: 10.1111/imr.12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Blake D, Lynch KW. The three as: alternative splicing, alternative polyadenylation and their impact on apoptosis in immune function. Immunol Rev. 2021;304:30–50. doi: 10.1111/imr.13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Dvinge H, Guenthoer J, Porter PL, Bradley RK. RNA components of the spliceosome regulate tissue- and cancer-specific alternative splicing. Genome Res. 2019;29:1591–604. doi: 10.1101/gr.246678.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Bonnal SC, Lopez-Oreja I, Valcarcel J. Roles and mechanisms of alternative splicing in cancer - implications for care. Nat Rev Clin Oncol. 2020;17:457–74. doi: 10.1038/s41571-020-0350-x. [DOI] [PubMed] [Google Scholar]
- 309.Gordon JM, Phizicky DV, Neugebauer KM. Nuclear mechanisms of gene expression control: pre-mRNA splicing as a life or death decision. Curr Opin Genet Dev. 2021;67:67–76. doi: 10.1016/j.gde.2020.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Yu X, Munoz-Sagredo L, Streule K, Muschong P, Bayer E, Walter RJ, et al. CD44 loss of function sensitizes AML cells to the BCL-2 inhibitor venetoclax by decreasing CXCL12-driven survival cues. Blood. 2021;138:1067–80. doi: 10.1182/blood.2020006343. [DOI] [PubMed] [Google Scholar]
- 311.Wu SY, Lee CF, Lai HT, Yu CT, Lee JE, Zuo H, et al. Opposing functions of BRD4 isoforms in breast cancer. Mol Cell. 2020;78:1114–32.e1110. doi: 10.1016/j.molcel.2020.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Sonneveld S, Verhagen BMP, Tanenbaum ME. Heterogeneity in mRNA translation. Trends Cell Biol. 2020;30:606–18. doi: 10.1016/j.tcb.2020.04.008. [DOI] [PubMed] [Google Scholar]
- 313.Nakano M, Nakajima M. Significance of A-to-I RNA editing of transcripts modulating pharmacokinetics and pharmacodynamics. Pharm Ther. 2018;181:13–21. doi: 10.1016/j.pharmthera.2017.07.003. [DOI] [PubMed] [Google Scholar]
- 314.Bludau I, Aebersold R. Proteomic and interactomic insights into the molecular basis of cell functional diversity. Nat Rev Mol Cell Biol. 2020;21:327–40. doi: 10.1038/s41580-020-0231-2. [DOI] [PubMed] [Google Scholar]
- 315.Wan Y, Larson DR. Splicing heterogeneity: separating signal from noise. Genome Biol. 2018;19:86. doi: 10.1186/s13059-018-1467-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Charo IF, Myers SJ, Herman A, Franci C, Connolly AJ, Coughlin SR. Molecular cloning and functional expression of two monocyte chemoattractant protein 1 receptors reveals alternative splicing of the carboxyl-terminal tails. Proc Natl Acad Sci USA. 1994;91:2752–6. doi: 10.1073/pnas.91.7.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Wong LM, Myers SJ, Tsou CL, Gosling J, Arai H, Charo IF. Organization and differential expression of the human monocyte chemoattractant protein 1 receptor gene. Evidence for the role of the carboxyl-terminal tail in receptor trafficking. J Biol Chem. 1997;272:1038–45. doi: 10.1074/jbc.272.2.1038. [DOI] [PubMed] [Google Scholar]
- 318.Yu CR, Peden KW, Zaitseva MB, Golding H, Farber JM. CCR9A and CCR9B: two receptors for the chemokine CCL25/TECK/Ck beta-15 that differ in their sensitivities to ligand. J Immunol. 2000;164:1293–305. doi: 10.4049/jimmunol.164.3.1293. [DOI] [PubMed] [Google Scholar]
- 319.Gupta SK, Pillarisetti K. Cutting edge: CXCR4-Lo: molecular cloning and functional expression of a novel human CXCR4 splice variant. J Immunol. 1999;163:2368–72. doi: 10.4049/jimmunol.163.5.2368. [DOI] [PubMed] [Google Scholar]
- 320.Lasagni L, Francalanci M, Annunziato F, Lazzeri E, Giannini S, Cosmi L, et al. An alternatively spliced variant of CXCR3 mediates the inhibition of endothelial cell growth induced by IP-10, Mig, and I-TAC, and acts as functional receptor for platelet factor 4. J Exp Med. 2003;197:1537–49. doi: 10.1084/jem.20021897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Ehlert JE, Addison CA, Burdick MD, Kunkel SL, Strieter RM. Identification and partial characterization of a variant of human CXCR3 generated by posttranscriptional exon skipping. J Immunol. 2004;173:6234–40. doi: 10.4049/jimmunol.173.10.6234. [DOI] [PubMed] [Google Scholar]
- 322.Colobran R, Pujol-Borrell R, Armengol MP, Juan M. The chemokine network. II. On how polymorphisms and alternative splicing increase the number of molecular species and configure intricate patterns of disease susceptibility. Clin Exp Immunol. 2007;150:1–12. doi: 10.1111/j.1365-2249.2007.03489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Janssens R, Struyf S, Proost P. The unique structural and functional features of CXCL12. Cell Mol Immunol. 2018;15:299–311. doi: 10.1038/cmi.2017.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Righetti A, Giulietti M, Sabanovic B, Occhipinti G, Principato G, Piva F. CXCL12 and its isoforms: different roles in pancreatic cancer? J Oncol. 2019;2019:9681698. doi: 10.1155/2019/9681698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Sand LG, Scotlandi K, Berghuis D, Snaar-Jagalska BE, Picci P, Schmidt T, et al. CXCL14, CXCR7 expression and CXCR4 splice variant ratio associate with survival and metastases in Ewing sarcoma patients. Eur J Cancer. 2015;51:2624–33. doi: 10.1016/j.ejca.2015.08.020. [DOI] [PubMed] [Google Scholar]
- 326.Cui H, Li Z, Chen S, Li X, Chen D, Wang J, et al. CXCL12/CXCR4-Rac1-mediated migration of osteogenic precursor cells contributes to pathological new bone formation in ankylosing spondylitis. Sci Adv. 2022;8:eabl8054. doi: 10.1126/sciadv.abl8054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Steurer M, Montillo M, Scarfò L, Mauro FR, Andel J, Wildner S, et al. Olaptesed pegol (NOX-A12) with bendamustine and rituximab: a phase IIa study in patients with relapsed/refractory chronic lymphocytic leukemia. Haematologica. 2019;104:2053–60. doi: 10.3324/haematol.2018.205930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Britton C, Poznansky MC, Reeves P. Polyfunctionality of the CXCR4/CXCL12 axis in health and disease: implications for therapeutic interventions in cancer and immune-mediated diseases. FASEB J. 2021;35:e21260. doi: 10.1096/fj.202001273R. [DOI] [PubMed] [Google Scholar]
- 329.Gutjahr JC, Crawford KS, Jensen DR, Naik P, Peterson FC, Samson G, et al. The dimeric form of CXCL12 binds to atypical chemokine receptor 1. Sci Signal. 2021;14:eabc9012. doi: 10.1126/scisignal.abc9012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Kachuri L, Francis SS, Morrison ML, Wendt GA, Bossé Y, Cavazos TB, et al. The landscape of host genetic factors involved in immune response to common viral infections. Genome Med. 2020;12:93. doi: 10.1186/s13073-020-00790-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Smit MJ, Schlecht-Louf G, Neves M, van den Bor J, Penela P, Siderius M, et al. The CXCL12/CXCR4/ACKR3 axis in the tumor microenvironment: signaling, crosstalk, and therapeutic targeting. Annu Rev Pharm Toxicol. 2021;61:541–63. doi: 10.1146/annurev-pharmtox-010919-023340. [DOI] [PubMed] [Google Scholar]
- 332.Zhao R, Liu J, Li Z, Zhang W, Wang F, Zhang B. Recent advances in CXCL12/CXCR4 antagonists and nano-based drug delivery systems for cancer therapy. Pharmaceutics. 2022;14:1541. doi: 10.3390/pharmaceutics14081541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Rusetska N, Kowalski K, Zalewski K, Zięba S, Bidziński M, Goryca K, et al. CXCR4/ACKR3/CXCL12 axis in the lymphatic metastasis of vulvar squamous cell carcinoma. J Clin Pathol. 2022;75:324–32. doi: 10.1136/jclinpath-2020-206917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Olivarria G, Lane TE. Evaluating the role of chemokines and chemokine receptors involved in coronavirus infection. Expert Rev Clin Immunol. 2022;18:57–66. doi: 10.1080/1744666X.2022.2017282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Nishikawa G, Kawada K, Nakagawa J, Toda K, Ogawa R, Inamoto S, et al. Bone marrow-derived mesenchymal stem cells promote colorectal cancer progression via CCR5. Cell Death Dis. 2019;10:264. doi: 10.1038/s41419-019-1508-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Pan Z, Zhu T, Liu Y, Zhang N. Role of the CXCL13/CXCR5 axis in autoimmune diseases. Front Immunol. 2022;13:850998. doi: 10.3389/fimmu.2022.850998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Bogacka J, Pawlik K, Ciapala K, Ciechanowska A, Mika J. CC chemokine receptor 4 (CCR4) as a possible new target for therapy. Int J Mol Sci. 2022;23:15638. doi: 10.3390/ijms232415638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Puthenparampil M, Stropparo E, Zywicki S, Bovis F, Cazzola C, Federle L, et al. Wide cytokine analysis in cerebrospinal fluid at diagnosis identified CCL-3 as a possible prognostic factor for multiple sclerosis. Front Immunol. 2020;11:174. doi: 10.3389/fimmu.2020.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Vanderheiden A, Thomas J, Soung AL, Davis-Gardner ME, Floyd K, Jin F, et al. CCR2 signaling restricts SARS-CoV-2 infection. mBio. 2021;12:e0274921. doi: 10.1128/mBio.02749-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Khalil BA, Elemam NM, Maghazachi AA. Chemokines and chemokine receptors during COVID-19 infection. Comput Struct Biotechnol J. 2021;19:976–88. doi: 10.1016/j.csbj.2021.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Lim GB. ACEi reduces hypertension-induced hyperinflammation in COVID-19. Nat Rev Cardiol. 2021;18:231. doi: 10.1038/s41569-021-00512-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–80.e278. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Trump S, Lukassen S, Anker MS, Chua RL, Liebig J, Thürmann L, et al. Hypertension delays viral clearance and exacerbates airway hyperinflammation in patients with COVID-19. Nat Biotechnol. 2021;39:705–16. doi: 10.1038/s41587-020-00796-1. [DOI] [PubMed] [Google Scholar]
- 344.Sjoberg E, Meyrath M, Chevigne A, Ostman A, Augsten M, Szpakowska M. The diverse and complex roles of atypical chemokine receptors in cancer: From molecular biology to clinical relevance and therapy. Adv Cancer Res. 2020;145:99–138. doi: 10.1016/bs.acr.2019.12.001. [DOI] [PubMed] [Google Scholar]
- 345.Lidonnici MR, Aprile A, Frittoli MC, Mandelli G, Paleari Y, Spinelli A, et al. Plerixafor and G-CSF combination mobilizes hematopoietic stem and progenitors cells with a distinct transcriptional profile and a reduced in vivo homing capacity compared to plerixafor alone. Haematologica. 2017;102:e120–e124. doi: 10.3324/haematol.2016.154740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Komissarov A, Potashnikova D, Freeman ML, Gontarenko V, Maytesyan D, Lederman MM, et al. Driving T cells to human atherosclerotic plaques: CCL3/CCR5 and CX3CL1/CX3CR1 migration axes. Eur J Immunol. 2021;51:1857–9. doi: 10.1002/eji.202049004. [DOI] [PubMed] [Google Scholar]
- 347.Sheng D, Ma W, Zhang R, Zhou L, Deng Q, Tu J, et al. Ccl3 enhances docetaxel chemosensitivity in breast cancer by triggering proinflammatory macrophage polarization. J Immunother Cancer. 2022;10:e003793. doi: 10.1136/jitc-2021-003793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Ntanasis-Stathopoulos I, Fotiou D, Terpos E. CCL3 signaling in the tumor microenvironment. Adv Exp Med Biol. 2020;1231:13–21. doi: 10.1007/978-3-030-36667-4_2. [DOI] [PubMed] [Google Scholar]
- 349.Falta MT, Crawford JC, Tinega AN, Landry LG, Crawford F, Mack DG, et al. Beryllium-specific CD4+ T cells induced by chemokine neoantigens perpetuate inflammation. J Clin Invest. 2021;131:e144864. doi: 10.1172/JCI144864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.To SKY, Tang MKS, Tong Y, Zhang J, Chan K, Ip P, et al. A Selective beta-Catenin-Metadherin/CEACAM1-CCL3 Axis Mediates Metastatic Heterogeneity upon Tumor-Macrophage. Interact Adv Sci (Weinh) 2022;9:e2103230. doi: 10.1002/advs.202103230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Kodama T, Koma YI, Arai N, Kido A, Urakawa N, Nishio M, et al. CCL3-CCR5 axis contributes to progression of esophageal squamous cell carcinoma by promoting cell migration and invasion via Akt and ERK pathways. Lab Invest. 2020;100:1140–57. doi: 10.1038/s41374-020-0441-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Yang J, Hu S, Zhao L, Kaplan DH, Perdew GH, Xiong N. Selective programming of CCR10(+) innate lymphoid cells in skin-draining lymph nodes for cutaneous homeostatic regulation. Nat Immunol. 2016;17:48–56. doi: 10.1038/ni.3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Xu M, Li C, Yang J, Ye A, Yan L, Yeoh BS, et al. Activation of CD81(+) skin ILC2s by cold-sensing TRPM8(+) neuron-derived signals maintains cutaneous thermal homeostasis. Sci Immunol. 2022;7:eabe0584. doi: 10.1126/sciimmunol.abe0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Mergia Terefe E, Catalan Opulencia MJ, Rakhshani A, Ansari MJ, Sergeevna SE, Awadh SA, et al. Roles of CCR10/CCL27-CCL28 axis in tumour development: mechanisms, diagnostic and therapeutic approaches, and perspectives. Expert Rev Mol Med. 2022;24:e37. doi: 10.1017/erm.2022.28. [DOI] [PubMed] [Google Scholar]
- 355.Silva-Cardoso SC, Tao W, Angiolilli C, Lopes AP, Bekker C, Devaprasad A, et al. CXCL4 links inflammation and fibrosis by reprogramming monocyte-derived dendritic cells in vitro. Front Immunol. 2020;11:2149. doi: 10.3389/fimmu.2020.02149. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.