

Summary
Different cellular compartments within a tissue present distinct cancer-initiating capacities. Current approaches to dissect such heterogeneity require cell-type-specific genetic tools based on a well-understood lineage hierarchy, which are lacking for many tissues. Here, we circumvented this hurdle and revealed the dichotomous capacity of fallopian tube Pax8+ cells in initiating ovarian cancer, utilizing a mouse genetic system that stochastically generates rare GFP-labeled mutant cells. Through clonal analysis and spatial profiling, we determined that only clones founded by rare, stem/progenitor-like Pax8+ cells can expand on acquiring oncogenic mutations whereas vast majority of clones stall immediately. Furthermore, expanded mutant clones undergo further attrition: many turn quiescent shortly after the initial expansion, whereas others sustain proliferation and manifest a bias toward Pax8+ fate, underlying early pathogenesis. Our study showcases the power of genetic mosaic system-based clonal analysis for revealing cellular heterogeneity of cancer-initiating capacity in tissues with limited prior knowledge of lineage hierarchy.
Subject areas: Biological sciences, Cell biology, Cancer, Transcriptomics
Graphical abstract
Highlights
-
•
A mouse genetic mosaic model is used for clonal analysis during premalignancy
-
•
A rare subset of stem/progenitor-like Pax8+ cells seeds ovarian cancer
-
•
Oncogenic mutations promote tumor progression via prolonged clonal expansion
-
•
Expanded mutant clones manifest biased differentiation toward Pax8+ fate
Biological sciences; Cell biology; Cancer; Transcriptomics
Introduction
Cancer initiation is an intricate process. Although oncogenic mutations are key drivers for cancer initiation, a permissive context within the cells where these mutations occur is also indispensable. Therefore, different cellular compartments within a tissue could manifest vastly different cancer-initiating capacities on acquiring the same mutations.1,2,3 For example, APC deletion in mouse intestinal stem cells causes adenomas in 3–5 weeks. However, when the same mutation occurs in the transit-amplifying cells, adenomas fail to form.4 Therefore, revealing the heterogeneous cancer-initiating capacity of cells within a tissue is crucial for pinpointing the cancer cell of origin, whose inherent signaling context contributes substantially to tumor development and pathological transformation.2,3
Although mouse models can be used to rigorously interrogate the cellular heterogeneity in cancer-initiating capacity if genetic targeting tools are available to introduce mutations into each cell type, this approach is often hampered by our limited knowledge of tissue lineage hierarchy and consequently the lack of precise genetic tools. To circumvent this problem, we deploy a mouse genetic system called Mosaic Analysis with Double Markers (MADM) (Figure 1A1).5,6 MADM contains a pair of knock-in cassettes of chimeric GFP and RFP coding sequences separated by a loxP-containing intron, each of which is linked with the wildtype or mutant alleles of a tumor suppressor gene residing on the same chromosome, respectively, resulting in a heterozygous, unlabeled mouse. Through Cre/loxP-mediated inter-chromosomal mitotic recombination that occurs at a low frequency (0.1%–1% or even lower), sporadic homozygous mutant cells labeled with GFP are generated in the MADM mouse (Figure 1A1).5,6 The rarity of mutant cells and the unequivocal GFP labeling provide clonal resolution to trace the expansion of individual mutant cells (Figure 1A2).5,7,8 When the MADM recombination is catalyzed by a pan-tissue Cre transgene, it can generate scattered GFP+ mutant cells in all cell types within that tissue. Via clonal tracing of individual mutant cells, we can pinpoint cells with a high cancer-initiating potential based on the extent of clonal expansion, allowing us to subsequently interrogate their identity, behavior, and molecular features using multi-spectral immunostaining and spatial profiling technologies.
Figure 1.

Induction of sparse mutant cells in the fallopian tube through MADM
(A) A1: MADM mechanism: MADM contains a pair of knock-in cassettes of chimeric GFP and RFP (tdTomato, tdT) coding sequences, separated by a loxP-containing intron. Cre-mediated inter-chromosomal recombination in mitotic cells followed by X segregation generates a single GFP+ cell (Green) that is a homozygous mutant, and a sibling tdTomato+ cell (Red) that is wildtype, or by Z segregation which generates a GFP+ and tdTomato+ double-positive heterozygous cell (Yellow) and a sibling colorless heterozygous cell. A2: MADM could be used to induce sporadic single GFP+ mutant cells in the tissue, which allows the study of cancer initiation and early progression at the clonal level.
(B) Induction of sporadic GFP+ mutant cells in the mouse fallopian tube with MADM. Pax8-rtTA; TetO-Cre system was introduced to drive Cre expression in the fallopian tube Pax8+ population (secretory cells) under the control of doxycycline. Cre-mediated recombination of MADM cassettes generates GFP+ cells with homozygous loss of Trp53, Brca1, and Nf1. See also Figure S1.
(C) Sparse and scattered GFP+ cells in the mouse fallopian tube after Cre induction. Four MADM-mutant mice were given doxycycline between post-natal days 0–21 (P0-21). Fallopian tubes were harvested right after the doxycycline administration at P21 for cryo-sectioning and confocal imaging. Arrows indicate GFP+ cells. Scale bar = 100 μm.
(D) The abundance of GFP+ cells in the fallopian tube is low after P0-21 doxycycline administration. The fallopian tubes from four mice at P21 were cut through continuously, and every fifth slide was used to quantify GFP+% versus total fallopian tube epithelial cells (counted by DAPI along the epithelium). Each dot indicates data from one mouse (n = 4). Data are represented as mean ± SEM.
(E) The MADM-labeled GFP+ cells were stained positive for Pax8. Fallopian tubes from three mice harvested at P21 were cryo-sectioned for immunofluorescence staining of Pax8. The arrow indicates the same cell as the GFP+ cell shown, which presents Pax8+.Scale bar = 50 μm.
(F) Percentage of MADM-labeled GFP+ cells that were also Pax8+. Fallopian tubes from three mice harvested at P21 were cryo-sectioned for immunofluorescence staining of Pax8. Pax8+% overall GFP+ cells were quantified (n =3). Data are represented as mean ± SEM.
(G) Representative images of GFP+ mutant clones post-induction at the indicated ages. The fallopian tubes from MADM-mutant mice (n =3 for each age) were sectioned for imaging. Scale bar = 100 μm. See also Figures S1–S3.
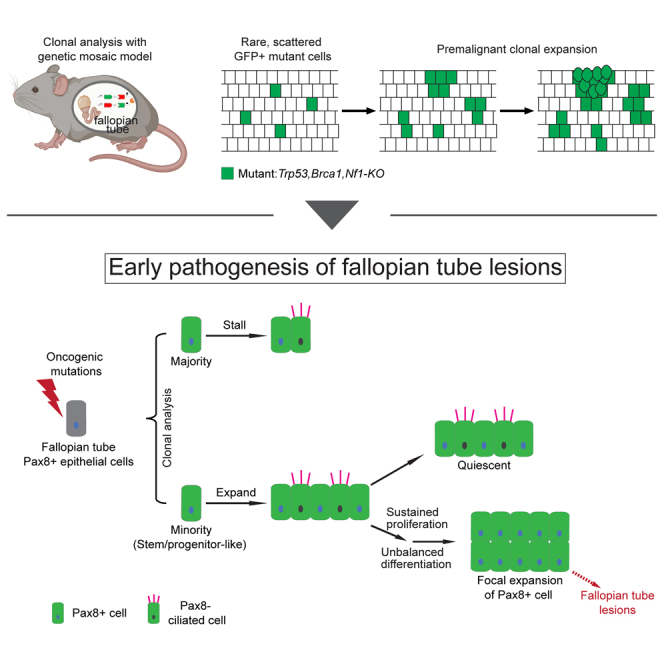
Utilizing this paradigm, we sought to finely dissect the cancer-initiating capacity of the cellular compartment within the fallopian tube (FT), which has emerged as an origin site for high-grade serous ovarian cancer (HGSOC). HGSOC is the most prevalent and aggressive ovarian cancer subtype, which causes about 14,000 deaths annually in the US.9,10 Although HGSOC was initially thought to arise solely from the ovarian surface epithelium, an increasing body of epidemiologic, clinical, molecular, and mouse modeling studies suggest the FT as an alternative origin site for a considerable portion of HGSOCs. In human studies, FT precursor lesions such as serous tubal intraepithelial lesions (STILs) and serous tubal intraepithelial carcinomas (STICs) were frequently detected in HGSOC patients.11,12,13,14,15 These precursor lesions shared identical TP53 mutations16,17,18,19 and similar transcriptomic profiles with the concurrent tumors in the same patients,20,21,22,23 implying their close lineage relationship. The FT is known to consist of ciliated cells (Acetyl-Tubulin+) that facilitate gamete transportation and secretory cells (Pax8+/Ovgp1+) that produce nutrient-rich fluid.24 Studies with genetically engineered mouse models showed that transforming oviduct (analogous to the fallopian tubes in humans, referred to as fallopian tube hereafter) secretory cells with clinically relevant mutations (TP53, BRCA1, RB1, PTEN, NF1, etc.) leads to FT precursor lesions and ovarian tumors that resemble human disease.25,26,27,28,29,30 Therefore, the secretory cells are pinpointed as the cancer cell of origin. Recent single-cell profiling revealed vast heterogeneity among FT Pax8+ cells,31,32,33 which corroborated well with their greatly varied organoid-forming potential in vitro.34,35 These findings raise the question of whether all or only some FT Pax8+ cells possess the cancer-initiating capacity. Addressing this question is critical for understanding early pathogenesis in the FT. However, such efforts have been hindered by the lack of definitive markers that can separate FT Pax8+ cells into functional subsets.
Here, taking advantage of MADM’s strengths in generating scattered single mutant cells to enable clonal tracing, we interrogated the cellular heterogeneity of FT Pax8+ cells in their capacity to initiate precursor lesions. We found that only a rare subpopulation enriched in the distal FT provides the permissive context for cancer initiation, which likely represents stem/progenitor-like cells in FT. We also delineated the process of early pathogenesis from a successfully initiated cell to FT lesions, shedding light on the early cellular alterations. In summary, this study showcases the power of genetic mosaic system-based clonal analysis for revealing stark cellular heterogeneity of cancer-initiating capacity in tissues lacking a well-understood lineage hierarchy.
Results
MADM generates GFP-labeled mutant cells at the clonal density within FT Pax8+ population
To generate and track individual mutant cells within the FT Pax8+ population using MADM, two critical prerequisites need to be considered: 1) a Cre transgene that specifically targets the FT Pax8+ population, 2) HGSOC-relevant tumor suppressor genes reside on the telomeric side of the MADM cassettes. We incorporated the Pax8-rtTA; TetO-Cre system to target the FT Pax8+ population under the control of doxycycline (Dox).25,36 To select clinical-relevant tumor suppressor genes, we analyzed TCGA datasets of human HGSOC patients. We found that the loss of TP53 (63%), BRCA1/2 (78%), and the mutations that activate the Ras-MAPK pathway (37%, some through the loss of NF1) are among the most prevalent genetic alterations and often co-occur (Figure S1A).37,38,39,40,41 Because Trp53, Brca1, and Nf1 in mice all reside on chromosome 11, where the MADM cassettes had been knocked in (Figure S1B),42,43 we selected these mutations to establish the MADM model. After incorporating the Cre transgene and these mutations into two MADM stock lines through multi-generational breeding (Figure S1C, see STAR Methods for details), we obtained MADM-mutant mice in which rare GFP+ mutant cells could be generated within the FT Pax8+ population (Figure 1B). This breeding scheme also generated MADM-wildtype littermates as control (Figure S1C).
To assess whether the frequency of GFP-labeled cells is low enough for clonal tracing, we induced Cre activity between post-natal days 0 and 21 (P0-21) with Dox in the drinking water. We chose this period for clone induction because this is the most proliferative period of FT development that satisfies the reliance of MADM on mitotic recombination to generate mutant cells. When we assessed the labeling efficiency, we found that around 0.15% of FT epithelial cells were GFP+, sparse enough for clonal tracing (Figures 1C and 1D, clonality is further discussed in Figure S4). RFP+ wildtype cells were generated at a similar frequency. Notably, double-positive (yellow) heterozygous cells were also generated but rarely included in this study because they do not share a lineage relationship with GFP+ and RFP+ cells (Figures 1A1,S1D, and S1E). Finally, nearly all MADM-labeled cells were Pax8+ (Figures 1E and 1F), demonstrating the faithfulness of Pax8-rtTA; TetO-Cre transgenes.
As mice aged, some of these isolated GFP+ mutant cells expanded into sizable clones, occupying continuous regions of FT epithelium (Figure 1G). As clones increased in size, they gradually lost well-organized epithelial structure and eventually formed serous tubal intraepithelial lesions (STILs), characterized by nuclear enlargement, a partial loss of cilia, elevated mitotic figures (Figure S2A), a high level of Pax8 expression, and a slightly increased proliferative index based on Ki67 staining (Figure S2B). In 10 MADM mutant mice at 12 months of age, 7 out of 14 highly expanded mutant clones analyzed were diagnosed as STILs, and another 4 displayed abnormal over-crowded cyto-architecture but slightly fell short of STIL diagnosis (Figure S2C). It should be noted that aging these mice beyond one year for the onset of full-blown ovarian tumors was not possible for this model because the animals succumbed to invasive tumors in the uterus, which also contains Pax8+ cells with transforming potential44 (Figures S3A and S3B). Therefore, our model is uniquely suitable for probing into unprecedented early phases of ovarian cancer initiation with clonal resolution before STIL formation but not for studying malignant ovarian tumors.
Dichotomous expansion of individual mutant cells revealed by clonal analysis
Next, to determine whether all Pax8+ cells share similar cancer-initiating capacities, we evaluated the extent of clonal expansion of individual mutant cells. We induced GFP+ mutant clones (which all start from one cell according to the MADM design) with four MADM-mutant mice through P0-21 Dox administration. We then analyzed the clonal expansion of mutant cells at five months of age when mutant cells have expanded but not yet formed pathologically manifested lesions. To gain a comprehensive view of all mutant clones, we optically cleared the FTs with the CUBIC method,45 imaged the entire FT in 3D with light-sheet microscopy, and then assessed the size of each mutant clone by reconstructing the image stacks (Figure 2A). Of interest, the sizes of mutant clones were highly divergent: ∼70% of the clones contained <10 cells (mostly one cell), yet a small fraction of clones expanded to hundreds of cells or more (Figures 2B and 2C). To exclude the possibility that the dichotomy resulted from stochastic fluctuations, we implemented a mathematical analysis that evaluates whether the observed clonal size distribution can be explained by one single stochastic group. We estimated the clonal size distribution from the majority clones and asked whether the frequency of observed outlier large clones is within that predicted by the majority distribution. The observed clonal sizes were best fit by a negative binomial model for clones below 50 cells (KS test, pvalue<0.1, see STAR Methods for details.46 Under the null hypothesis, this one-group model predicts one clone larger than 50 cells. In contrast, we observed more than 30 clones larger than 50 cells, suggesting the existence of an outlier group (Figure 2D) (p< 10−8, Fisher Exact Test) that has a higher expansion potential than most clones. Recognizing that an alternative interpretation for the large clones is the merge of multiple smaller clones, we estimated the minimal clonal size required for two clones to merge. Based on the average distance between two mutant cells right after clonal induction and the cellular diameter (Figures S4A–S4C) for two spherically expanding clones to merge, each of the two clones needs to grow to more than 10,000 cells (Figure S4D), which is never observed in our clonal size quantification. Therefore, the chance for clonal merging is minimal.
Figure 2.

Dichotomous expansion of fallopian tube mutant clones
(A) Approach for whole-fallopian-tube 3D imaging. The harvested reproductive tract, including the fallopian tube, was first fixed with 4% paraformaldehyde (PFA) for 24 h (Day0–1), followed by shaking in CUBIC clearing reagent 1 (CUBIC-1) at 37°C,110rpm for three days (Day1–4). The tissues were then washed in PBS for 6 h to remove the CUBIC-1 reagent and shaken in CUBIC clearing reagent 2 (CUBIC-2) until day 6. The whole fallopian tubes were imaged with light-sheet microscopy.
(B) Distinct clonal size among GFP+ mutant clones showed by 3D imaging of cleared whole FT from 5 months old MADM mutant mice (n = 4). The yellow box shows a region with a non-expanded GFP+ clone (arrow), and the magenta box shows a highly expanded GFP+ clone. Scale bar = 50 μm.
(C) Violin plot of the clonal size distribution of mutant clones in four independent FTs from mice at 5 months of age, hinting at the existence of two density peaks. The magenta line indicates the 75 percentiles.
(D) The clonal size does not fit into one group distribution, suggesting the existence of distinct groups. The observed clonal size of mutant clones (green bar) from four mice at 5 months was first fitted into one negative binomial distribution (yellow dots), which predicts the observation of one clone >50 cells. Because the observed number of clones >50 cells is 27, significantly higher than predicted, the one distribution hypothesis is rejected. Fisher Exact Test, p < 10−8.
(E) At 1 and 2 months old, the clonal size dichotomy also exists. FTs from 1-month-old or 2-month-old MADM mutant mice (n = 4 for each) were cleared for 3D imaging. The yellow box shows a region with a non-expanded GFP+ clone (arrow), and the magenta box shows a highly expanded GFP+ clone. Scale bar = 50 μm.
(F) Violin plot of the clonal size distribution of mutant clones at 1 and 2 months. Data were pooled from 4 mice for each indicated age. A total of 129 GFP+ mutant clones from 1-month-old mice and 233 clones from 2-month-old mice were analyzed for plotting.
(G) The portion of outlier large clones at the indicated age. Each dot indicates data from one mouse (n = 4 for each age). Data are represented as mean ± SD. Mann-Whitney U test was used.See also Figures S4–S6.
To determine when these outlier large clones first emerged, we repeated the clonal size measurement with MADM-mutant mice at one and two months of age. To our surprise, greatly expanded clones already existed among minute ones shortly after Cre induction (Figures 2E and 2F). We ruled out the trivial explanation that such clonal size dichotomy simply reflects their differential birth date (Figure S5A) over the 21 days of Dox administration by showing a large variance of clonal sizes among age-synchronized clones (Figures S5B and S5C). We then compared the frequency of expanded large clones from 1, 2, and 5 months of age. Because the variance of clonal sizes at younger ages was smaller overall, a Poisson model was used to calculate the portion of outlier large clones for all ages (see STAR Methods for details). We found the proportion of the outlier large clones are comparable across the ages (Figure 2G), suggesting that the expansion potential of initiated Pax8+ cells was determined early.
In addition to the temporal pattern, we also noted a spatial pattern of clonal expansion. The FT comprises three distinct segments based on their relative distance to the ovary and uterus (Figure S6 Aupper panel). The distal end close to the ovary is found to be the hotspot for FT lesions in humans.14,47 To ask whether the highly expanded mutant clones demonstrate similar spatial preference toward the distal end, we stretched the FTs from 2-month-old MADM-mutant mice, performed whole-mount imaging, and found that large clones almost exclusively reside in the distal FTs, whereas small ones are distributed in all three FT segments (Figures S6A and S6B). Such spatial preference was not caused by the biased generation of more clones in the distal FT because the initial labeling frequency was similar throughout FT segments (Figure S6C). Such spatial preference of highly clonogenic cells was maintained in adult FT, as when we induce single-cell clones between 5 and 6 weeks of age48 (Figure S6D upper panel), expanded large clones emerged weeks after in the distal FT (Figure S6Dlower panel).
Collectively, these data indicate that on acquiring oncogenic mutations, initiated FT Pax8+ cells present dichotomous expansion potential: the majority immediately stall while a small, distally enriched fraction profoundly expand.
The large clones likely originate from a stem/progenitor-like population
The nearly immediate divergence of clonal size prompted us to ask if the initial oncogenic mutations are required or if it is rooted in the intrinsic heterogeneity of the clonogenic potential of Pax8+ cells. To address this question, we induced clones in the MADM-wildtype mice, in which clones of all colors are free of initial oncogenic mutations. When we examined the size of these wildtype clones at 1, 2, and 5 months of age, we observed evident clonal size divergence as early as one month of age (Figures 3A and 3B), indicating that the divergence of clonal expansion does not depend on the oncogenic mutations and is likely caused by intrinsic heterogeneity of the clonogenic potential of Pax8+ cells. Because the high clonogenic potential is reminiscent of stem/progenitor cells,49,50 we further asked whether the large clones originate from a stem/progenitor-like founder cell.
Figure 3.

The large clones originate from a stem/progenitor-like FT population
(A) Co-existence of small and large clones in wildtype fallopian tubes. Fallopian tubes from MADM-wildtype mice at the indicated ages (n =3 for each age) were cleared for whole tissue 3D imaging. The yellow box shows non-expanded small clones, and the magenta box shows expanded large clones. Scale bar = 25 μm.
(B) Violin plot of the clonal size distribution of wildtype clones at 1, 2, and 5 months indicates similar clonal size dichotomy as the mutant clones. Data were pooled from 3 mice for each age. A total of 211 clones from 1-month-old mice, 310 clones from 2-month-old mice, and 284 clones from 5-month-old mice were assessed for plotting.
(C) The experimental scheme of fluorescence-guided laser-capture microdissection of small and large clones from 1-month-old MADM-wildtype mice (n = 6) and cDNA library preparation for sequencing. The arrow indicates small clones and the dashed circle outlines a large clone.
(D) Small and large clones show different expression patterns. log2 +1 transformed TMPs of 132 differentially expressed genes were subjected to Zscore normalization, and clustering was performed using Euclidian distance and the Ward.D method. See also Table S1 for the list of differentially expressed genes.
(E) Gene set enrichment assay showing upregulation of cilia-related gene sets in small clones.
(F) Gene set enrichment assay showing upregulation of proliferation and stem cell-related gene sets in large clones. See also Figures S7 and S8 and Table S1.
Speculating that a portion of the cells within the large clones might retain the stem/progenitor-like property of their founders that are absent in small clones, we isolated large clones and small clones by fluorescence-guided laser capture microdissection and performed RNA-sequencing. We purposefully chose FTs from 1-month-old MADM-wildtype mice for this experiment, with the following considerations: 1) large clones are readily distinguishable from small clones by size but have not yet overly expanded to the extent that signatures of the stem/progenitor-like cells would be completely overshadowed by differentiated cells within clones51; 2) a young age to minimize compound effects caused by ovulation; 3) wildtype clones to avoid the additional impact of oncogenic mutations. From each of the six mice, we micro-dissected small clones and large clones to collect ∼200 cells each (Figure 3C). These six pairs of samples underwent RNA extraction, cDNA preparation, and linear amplification followed by RNA sequencing at a depth of ∼4 million aligned reads per sample52,53(see STAR Methods for details). We found small and large clones are readily distinguishable based on differential expression patterns (Figure 3D, see Table S1 for the list of differentially expressed genes). Through gene set enrichment analysis, we found the upregulation of multiple ciliated-cell-related signatures in the small clones, indicating their highly differentiated status, which is further supported by an upregulation of the Ezh2 signature that is known for transcriptional repression54,55 and a down-regulation of the stem-cell signature (Figure 3E). In contrast, the large clones showed slightly upregulated proliferative and stem cell signature56(Figure 3F). However, we noted that the p values are borderline, likely because of both the intra-clonal heterogeneity (most cells within the large clones have differentiated while only a small fraction retains stem/progenitor-like properties) and inter-clonal heterogeneity (not all large clones continue to progress). Nevertheless, it appears that large clones are more proliferative and less differentiated compared to small clones. Reasoning that multi-potency is another hallmark of stem/progenitor cells, we assessed the cell composition in large clones: the presence of both AcTUB+ ciliated cells and Pax8+ “secretory” cells (Figure S7)furthersupports the stem/progenitor nature of the clone founders. Finally, as stem/progenitor cells often serve as the cancer cell of origin,3 we performed principal component analysis57 by projecting our data onto normal mouse fallopian tube and ovarian tumor datasets from the literature,30 and indeed found that the large clones lean significantly closer toward the tumors (Figure S8). In summary, our data indicate that the large clones likely originate from a stem/progenitor-like Pax8+ subset.
Oncogenic mutations prolonged the expansion and persistence of large clones
To understand how oncogenic mutations alter the clonal behavior of those FT Pax8+ cells with high clonogenic potential, we compared the proliferative activity of large clones with or without oncogenic mutations in a time course. To label proliferating cells, we treated the MADM-mutant and MADM-wildtype mice with EdU in the drinking water for seven days before harvest at 1, 2, or 5 months of age. The wildtype large clones showed peak proliferative rate at one month (EdU+:27.6 ± 2.8%), which then dropped precipitously at two months (1.9 ± 0.6%) and stayed low at five months (2.0 ± 0.5%). (Figures 4A, 4C, and4D), suggesting that the initial expansion capacity diminished quickly at the end of FT development.51 For mutant clones, although the overall diminishing trend is similar to wildtype ones, we noted two important differences. First, the overall proliferation of mutant clones was maintained at a higher level than the wildtype ones at 2 and 5 months (Figures 4B and 4C). Second, the proliferative rate of large mutant clones at 2 and 5 months of age showed a notably great variance: Although some clones were nearly as proliferative as one month, others almost completely stopped (Figures 4B–4D). A closer examination of 27 large clones from seven 5-month-old mice revealed that 10 mutant clones maintaining active proliferation manifested an abnormal nodular organization, in contrast to a single-layer organization of 17 quiescent clones (Figure S9). These data indicate that oncogenic mutations could prolong proliferation in the large clones after the developmental window, albeit not at full penetrance. Consequently, although mutant and wildtype large clones are comparable in size at one month, a portion of the mutant clones became much larger than wildtype ones at older ages (Figure 4E).
Figure 4.

Large mutant clones show prolonged expansion and advantageous long-term survival
(A) Wildtype large clones lost their proliferation potential post fallopian tube development. Three mice at each indicated age were treated with 0.5 mg/ml EdU in their drinking water for 7 days before harvest. Fallopian tubes were continuously sectioned for staining of EdU and confocal imaging. The dashed circle outlines a wildtype large clone. Scale bar = 20 μm.
(B) A fraction of the mutant large clones maintained their proliferation potential post fallopian tube development. Three MADM-mutant mice at each indicated age were treated with 0.5 mg/ml EdU in their drinking water for 7 days before harvest. Fallopian tubes were continuously sectioned for staining of EdU and confocal imaging. The dashed circle outlines a GFP+ large mutant clone. Scale bar = 20 μm.
(C) EdU+% in large wildtype and mutant clones at the indicated ages. Each dot represents one large clone. Clones from three mice of each age were pooled for plotting.
(D) Number of EdU+ cells in each individual large wildtype and mutant clone at the indicated ages. Each dot represents one large clone. Clones from three mice of each age were pooled for plotting.
(E) The size of large mutant clones was similar to that of wildtype clones at one month, whereas mutant large clones become significantly larger at 2 and 5 months. Three fallopian tubes of each indicated age were continuously cut and imaged to quantify the clonal size. Each dot represents one large clone. Clones from three fallopian tubes were pooled for plotting. Data are represented as mean ±SD. Mann-Whitney U test was used, ∗∗<0.01∗∗∗∗<0.0001.
(F) F1: GFP+ large clones co-exist with sibling RFP+ large clones in 1-, 2-, and 5-month-old MADM-wildtype mice. F2: Although the GFP+ large mutant clones co-exist with sibling RFP+ large clones at one month, only the GFP+ mutant clones survived at 2 and 5 months, whereas the RFP+ wildtype large clones were lost. Four MADM-wildtype/MADM-mutant mice at each indicated age were harvested, and the fallopian tubes were cut for imaging. The arrowheads indicate GFP+ large clones. The arrows indicate RFP+ large wildtype clones. Scale bar = 50 μm.
(G) A comparable number of GFP+ and RFP+ large wildtype clones co-exist in fallopian tubes from MADM-wildtype mice at 1, 2, and 5 months. Fallopian tubes from four mice at each indicated age were harvested for continuous cutting to quantify the number of GFP/RFP+ large clones. Data are represented as mean ±SEM.
(H) GFP+ large mutant clones persisted, whereas sibling RFP+ large wildtype clones were lost in fallopian tubes from MADM-mutant mice at 2 and 5 months. Fallopian tubes from four mice at each indicated age were harvested for continuous cutting to quantify the number of GFP/RFP+ large clones. Data are represented as mean ±SEM.See also Figure S9.
One of the unique strengths of genetic mosaic with MADM is that GFP+ mutant cells and RFP+ wildtype sibling cells are simultaneously generated in the same MADM-mutant animal (known as twin spots, Figure 1A1), providing the opportunity to directly compare their clonal behaviors. Meanwhile, the MADM-mutant animals also have MADM-wildtype littermates (Figure S1C). Both types of animals contain wildtype clones, but one is surrounded by heterozygous/homozygous-mutant cells, whereas the other is surrounded by wildtype cells, allowing us to dissect how different surroundings affect the wildtype clones. In MADM-wildtype animals, we found the large wildtype clones persisted (Figure 4F1) as the number of GFP+ and RFP+ large wildtype clones are comparable at 1, 2, and 5 months. The total number of GFP+/RFP+ large clones also didn’t significantly change over time (Figure 4G). In stark contrast, in MADM-mutant animals, although RFP+ large wildtype clones were present initially at one month of age, they were seldom found in 2- and 5-month-old mice when we examined more than 20 animals (Figures 4F2 and 4H), suggesting that wildtype large clones are not able to persist for the long-term when accompanied by mutant clones. Collectively, these results suggest that the oncogenic mutations prolong the proliferation of large clones and provide advantages over their sibling wildtype clones for long-term persistence.
Large mutant clones show unbalanced differentiation during their progression
The epithelium of the normal FT at the distal end, where large clones locate, consists of interspersed secretory cells (Pax8+, ∼30%) and ciliated cells (Ac-Tubulin+, ∼70%) (Figures S10A and S10B).58 Abnormal expansion of secretory cells has been found in both benign FT epithelial hyperplasia (e.g., “secretory-cell-outgrowths”, SCOUTs) and FT precursor lesions (e.g., STILs and STICs).51,59,60 Therefore, we asked whether the expanded mutant clones, as a probable intermediate between normal and disease, could manifest this drift of cell composition, which we assessed along a time course at 2, 5, and 12 months of age with immunostaining of Pax8 and ciliated cell marker (Ac-TUB). We found that although the small clones comprise very few Pax8+ cells at all ages (Figure 5A upper panel, Figure S10C), the Pax8+% within large mutant clones significantly increased from 2 to 5 months of age, which persisted at 12 months of age (Figure 5A lower panel, B). The Pax8+ cell in large mutant clones often formed continuous stretches, contrasting with the interspersed Pax8+ and Pax8-cells in the adjacent non-mutant regions (Figure 5C). Naturally, the percentage of Pax8+ cells in the large mutant clones is significantly higher than their adjacent non-mutant regions (Figure 5D). In summary, large clones appear to be at a premalignant stage preceding clinically identifiable lesions.
Figure 5.

Large mutant clones show unbalanced differentiation along their progression
(A) The Pax8 status of small and large mutant clones in an age cohort. Fallopian tubes from four MADM-mutant mice at each indicated age were continuously cut for Pax8 staining. Representative images of 23 small mutant clones, 8 large mutant clones at 2 months, 24 small mutant clones, 15 large mutant clones at 5 months, 15 small mutant clones, 11 large mutant clones at 12 months. The dashed circle indicates a small/large GFP+ mutant clone. Scale bar = 50 μm.
(B) Increased Pax8+% in large mutant clones from older mice. Each dot represents one large mutant clone. 8 large mutant clones were plotted at 2 months, 15 at 5 months, and 11 at 12 months from four mice at each age. See also Figure S9A.Data are represented as mean ±SEM. Mann-Whitney U test, ∗∗∗<0.001.
(C) The mutant clones showed a continuous stretch of Pax8+ cells (lower panel), whereas Pax8+ and Pax8-cells were interspersed in non-mutant adjacent regions (upper panel). Fallopian tubes from three MADM-mutant mice at 5 months old were harvested and sectioned for Pax8 staining. The dashed lines outline regions of interspersed Pax8+ and Pax8-cells (upper panel) or continuous stretches of Pax8+ cells (lower panel). Scale bar = 20 μm.
(D) The mutant clones contained a higher portion of Pax8+ cells than adjacent non-mutant regions. Three mutant/adjacent non-mutant pairs were quantified from each MADM-mutant mouse at 5 months old, and three mice were assessed in total. Mann-Whitney U test, ∗∗<0.01.
(E) The scheme to perform clonal tracing of proliferating cells within large mutant clones through EdU pulse-chase assay and the potential outcomes.
(F) EdU+ subclones within large mutant clones mainly consist of non-ciliated cells. The upper panel shows a four-cell subclone with non-ciliated cells only. The lower panel shows a subclone with one ciliated cell (+) and three non-ciliated cells (−). Fallopian tubes were harvested from six 5-month-old MADM-mutant mice that were treated with the EdU pulse-chase scheme, shown in Figure 5E. These fallopian tubes were cut continuously and stained for EdU and cilia and imaged to quantify the ciliated and non-ciliated cell number in each EdU+ subclone. Representative images from a total of 18 EdU+ subclones from six mice were shown. The arrow indicates positive cilia staining. The dashed circle indicates an EdU+ subclone. Scale bar = 25 μm.
(G) The EdU+ subclones (>2 cells) within the large mutant clones mainly consist of non-ciliated cells. In contrast, in wildtype distal FT, ciliated cells account for ∼70% (See also Figures S9B and S9C). A total of 18 EdU+ subclones from six MADM-mutant mice at 5 months old. C1 in the yaxis is short for Clone 1. See also Figures S10, and S11.
We further asked whether the elevated Pax8+% in large mutant clones reflects a biased propensity of proliferating mutant cells to differentiate toward a non-ciliated fate. To test this hypothesis, we performed an EdU pulse-chase experiment to determine the fate of the newly born cells within large mutant clones (Figure 5E). First, we mapped the length of EdU pulse and found that 3-day administration led to clonal labeling of proliferating cells (Figures S11A–S11E). After a 4-day chase, the initial EdU-labeled cells within mutant clones were able to divide further and form EdU+ subclones as large as 7 cells (Figures S11F–S11H). When we examined the cell composition of the EdU+ subclones, we found that, whereas 70% of cells in wildtype EdU+ subclones had cilia, substantially fewer cells in mutant EdU+ subclones were ciliated (Figures 5F and 5G), suggesting a higher propensity of mutant cells to adopt a non-ciliated cell fate. This biased cell differentiation may underscore the progression of some mutant clones toward FT lesions.
Discussion
Precisely identifying the cancer cell of origin in a tissue type with limited knowledge of normal lineage hierarchy is challenging. Although FT Pax8+ cells have been identified as the cell of origin of HGSOC; accumulating evidence point out the cellular heterogeneity among the Pax8+ cells. It is unknown whether all or only a specific subset of FT Pax8+ cells holds cancer-initiating potential. In this study, to investigate the possible heterogeneity of cancer-initiating potential among FT Pax8+ cells, we used a mouse genetic mosaic system called MADM to sparsely induce Pax8+ mutant cells and then followed their clonal progression. We found that individual mutant cells went through dichotomous clonal expansion—only a rare subset of clones progressed, whereas others stalled immediately on acquiring oncogenic mutations. Further spatial profiling and marker staining revealed that the expanded clones are less differentiated, more proliferative, and bipotential, indicative of stem/progenitor-like cells as their founders. Finally, we found that oncogenic mutations conferred cells in the expanded clones sustained proliferation and biased differentiation toward the Pax8 cell fate that led to long-term clonal persistence, representing premalignant progression toward FT precursor lesions such as STILs.
The primary approach to identify cancer cell-of-origin is using genetically engineered conditional knockout mouse models to examine the transforming potential of each cell lineage in a tissue of interest. Unfortunately, this approach is feasible only when a well-established tissue lineage hierarchy and lineage-specific Cre transgenes are available. Considering the absence of such knowledge and tools in many tissues, here we demonstrated an alternative approach to tackle this problem, deploying a mouse genetic system called Mosaic Analysis with Double Markers (MADM). With MADM, one could induce sparsely scattered single GFP+ mutant cells in all cell types within a tissue, then track clonal progression to pinpoint highly expanded clones, and finally use multi-spectral immunostaining and spatial profiling to thoroughly interrogate the identity, cellular behavior, and molecular features of the founder cells in these clones.61
It should be noted that, although clonal level labeling can also be achieved with inducible Cre systems (e.g., CreER or tetracycline-inducible Cre),62 the MADM system has unique advantages. First, using inducible Cre systems, the mutant cells are often visualized by a separate Cre reporter transgene.63,64 The stochastic nature of two independent recombination events (the excision of the floxed gene and the excision of the floxed “stop signal” in the Cre reporter) leads to unreliable coupling, especially when the induction of Cre activity is tuned down to ensure clonality.6,65 In contrast, MADM achieves simultaneous gene knockout and reporter expression in a single mitotic recombination event, guaranteeing a faithful coupling of mutant genotype and fluorescence labeling. Furthermore, the analytical power of MADM is enhanced by the internal control, i.e., RFP-labeled wildtype sibling cells, which provide the opportunity to directly compare the clonal behavior of mutant cells with their wildtype siblings side by side, allowing one to detect even the subtlest abnormality.66,67 Taking advantage of this unique feature, we were able to find that mutations confer an advantage for long-term clonal persistence, as large RFP+ wildtype sibling clones that co-existed with mutant ones at the beginning disappeared at older ages (Figures 4F2 and 4H). Finally, as a modular system, MADM-based clonal analysis of premalignant progression can be applied to all cancer types, as a genome-wide library of MADM mice has recently been made available68 that allows one to study almost all genes.
Although FT Pax8+ cells have been identified as a cancer cell-of-origin for HGSOC, several questions remain to be answered. First, the Pax8+ cells account for more than 50% of the FT epithelium, could all or only a subset of them initiate cancer? Through the MADM-based clonal tracing of individual mutant Pax8+ cells, we found a dichotomous progression of mutant cells: Only a small subset of Pax8+ cells was capable of clonal expansion whereas others barely expanded on acquiring oncogenic mutations, providing in vivo evidence of the heterogeneous cancer-initiating potential of the FT Pax8+ cells. Second, our study added to the understanding of the distal-biased presentation of FT lesions. Although repeated exposure to ovulation-induced inflammatory cytokines has been the main proposed explanation for this distal-bias,69,70 our data show that the highly expanded clones are already enriched in the distal FT before the puberty age (Figure S6). Therefore, as a complementary explanation to the ovulation hypothesis, the distal FT may be inherently enriched for cells uniquely susceptible to cancer initiation, such as FT stem/progenitor-like cells.34,71,72 The presence of dichotomous clonal size among wildtype clones, the histological data showing the bipotency, and the LCM/RNAseq data showing the less-differentiated status of large clones further support the existence of a subset of stem/progenitor-like cells within the Pax8+ population. Admittedly, although we strived to pinpoint specific signatures of these elusive stem cells, the tremendous intra- and inter-clonal heterogeneity made it extremely difficult. This is actually expected based on recent single-cell profiling studies of the human fallopian tube: although these studies inferred the existence of a stem/progenitor-like subset,31,33,73 putative stem/progenitor-like signatures from these studies had very limited concordance likely because of the rarity of these cells. In the future, our model may provide an excellent opportunity to solve this problem because these stem/progenitor-like cells are most likely enriched in large clones, especially mutant ones. Focused single-cell spatial profiling of these clones may enable us to pinpoint their definitive signatures.
It is known that oncogenic mutations co-opt the inherent properties of the cell of origin for successful cancer initiation.3,74,75 We reported that oncogenic mutations prolonged the proliferation of the highly clonogenic cells: Although the wildtype ones halted proliferation after the normal developmental window, mutant ones were able to maintain proliferation, thus, grew significantly larger than wildtype ones at older ages. Besides cell proliferation, the oncogenic mutations also confer an advantage for long-term persistence: Although sibling pairs of large GFP+ mutant clones and large RFP+ wildtype clones initially co-existed in young MADM-mutant mice, only the mutant ones were observed in older mice. This advantage of long-term persistence might have enabled the accumulation of more genetic/epigenetic changes for further progression. Finally, the large mutant clones also manifested biased differentiation toward the Pax8+ fate, resulting in stretches of uninterrupted Pax8+ cells and a significantly increased proportion of Pax8+ cells, a tendency also found in human FT precursor lesions (i.e., STICs).51,59,76 Because the previous lineage tracing studies showed that the Pax8+ population contains FT stem/progenitor-like cells,51 this biased cell fate might reflect a tendency to preserve stemness, a feature often found during the progression of many cancer types, such as esophageal cancer and skin cancer.75,77,78 These observations warrant further investigations into the molecular mechanisms of how oncogenic mutations extend proliferation, augment survival, and bias differentiation, for further delineating the process of premalignant progression of ovarian cancer in the FT.
In summary, our study demonstrates an application of a mouse genetic mosaic system to dissect cellular heterogeneity of cancer-initiating capacity in tissues with limited prior knowledge of their lineage hierarchy. For fallopian tube-derived ovarian cancer, our work shed light on the cancer cell of origin and revealed critical cellular events underlying early pathogenesis. Follow-up studies on stem/progenitor-like cells in FT should deepen our understanding of ovarian cancer initiation and progression, paving the way for the development of early detection and prevention strategies.
Limitations of the study
The MADM model used here is uniquely applicable for studying the initiation and early pathogenesis of cancer in the fallopian tube but not for malignant ovarian tumors. Because the Pax8 promoter also expresses in the uterine epithelium, mice always succumb to invasive uterine tumors before the onset of full-blown ovarian tumors. In the future, switching to a more specific promoter should make our mouse model applicable for studying the entire tumorigenic process, from initiation to the full-blown tumor stage. Our spatial profiling of the highly expanded clones indicates stem/progenitor-like cells as their origin, but failed to pinpoint definitive markers or signatures. Further spatial profiling at single-cell resolution would be required to identify candidate markers of these elusive clonal founder cells.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Pax8 | Proteintech | Cat#10336-1-AP; RRID: AB_2236705 |
| Anti-Acetylated Tubulin | Sigma-Aldrich | Cat# T7451; RRID: AB_609894 |
| Anti-Ki67 | Abcam | Cat# ab15580; RRID: AB_443209 |
| Alexa-555 Donkey Anti-Rabbit IgG (H+L) | Thermo Fisher Scientific | Cat#A31572; RRID: AB_162543 |
| Alexa-647 Donkey Anti-Mouse IgG (H+L) | Thermo Fisher Scientific | Cat#A31571; RRID: AB-162542 |
| Alexa 488 Donkey Anti-Mouse IgG (H+L) | Thermo Fisher Scientific | Cat#A21202; RRID: Ab_141607 |
| Alexa-647 Donkey Anti- Rabbit IgG (H+L) | Thermo Fisher Scientific | Cat#711-605-152; RRID: AB_2492288 |
| Chemicals, peptides, and recombinant proteins | ||
| Doxycycline | Sigma-Aldrich | D9891 |
| Sucrose | Fisher Scientific | BP220-10 |
| Paraformaldehyde | Sigma-Aldrich | 158127 |
| Hematoxylin | Vector Laborotories | ZH1027 |
| Cytoseal 60 | Fisher Scientific | 23-244257 |
| Urea | Sigma-Aldrich | U5128 |
| N,N,N',N'-Tetrakis(2-hydroxypropyl)ethylenediamine | Alfa Aesar | L16280 |
| Triethanolamine | Sigma-Aldrich | T58300 |
| α-Thioglycerol | TCI | T0905 |
| Agarose | Lonza | 50011 |
| DAPI (4′,6-Diamidino-2-phenylindole dihydrochloride) | Sigma-Aldrich | 32670 |
| LCM caps | Applied Biosystems | LCM0214 |
| Proteinase K | Sigma | P2308 |
| SuperScript III | Invitrogen | 18080-044 |
| Streptavidin Magnetic Beads | Thermo Fisher | 88816 |
| SuperMagnet Plate | Alpaqua | A001322 |
| Terminal transferase | Roche | 3333575001 |
| 5x terminal transferase buffer | Invitrogen | 16314-015 |
| dNTP | Roche | 11277049001 |
| High Fidelity PCR system | Roche | 11732650001 |
| AMPure XP beads | Beckman Coulter | A63881 |
| EdU (5-ethynyl-2′-deoxyuridine) | Thermo Fisher | A10044 |
| Alexa Fluor™ 647 Azide | Thermo Fisher | A10277 |
| Critical commercial assays | ||
| Qubit™ dsDNA HS and BR Assay Kits | Thermo Fisher | Q32850 |
| Nextera XT DNA Library Preparation Kit | illumina | FC-131-1024 |
| NextSeq 500/550 v2.5 Kits | illumina | 20024907 |
| Deposited data | ||
| All code necessary to define the dichotomy of clonal size distribution and the cutoff size of small and large clones | This study | https://github.com/JanesLab/ZengJ_MADMovarian/ |
| LCM small and large fallopian tube clones_RNAseq | This study | PRJNA865696; GEO: GSE210409 |
| Mouse normal fallopian tube and ovarian tumor_RNAseq | McCool et al.,202030 | GEO: GSE135590 |
| Experimental models: Organisms/strains | ||
| GT11ML | The Jackson Laboratory | Jax#002976 |
| TG11ML | The Jackson Laboratory | Jax#002977 |
| p53-KO | The Jackson Laboratory | Jax#002101 |
| Brca1flox | NCI Mouse Repository | Strain#01XC8 |
| Nf1flox | NCI Mouse Repository | Strain#01XM4 |
| Pax8-rtTA | The Jackson Laboratory | Jax#007176 |
| TetO-Cre | The Jackson Laboratory | Jax#006234 |
| Oligonucleotides | ||
| See Table S2 | This study | N/A |
| Software and algorithms | ||
| GraphPad Prism 9 | GraphPad | https://www.graphpad.com/ |
| FIJI | Open resource | https://imagej.net/software/fiji/ |
| R software | R CRAN | https://cran.r-project.org/ |
| RStudio | Open resource | https://www.rstudio.com/ |
| GSEA | Broad Institute | https://www.broadinstitute.org/gsea/index.jsp |
| Biorender | Biorender | https://biorender.com |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Hui Zong (hz9s@virginia.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Mice
The following mouse lines were crossed to establish the MADM-mutant and MADM-wildtype mice: TG11ML (stock NO. 022976; JAX), GT11ML (stock NO. 022977; JAX), Brca1flox (strain NO. 01XC8; NCI), Nf1flox (strain no. 01XM4; NCI), Trp53KO (stock no. 002101; JAX), Pax8-rtTA (stock NO. 007176; JAX),36 TetO-Cre (stock NO. 006234; JAX).79 The breeding scheme is shown in Figure S1. Female mice were used for all experiments. Age-matched mice were chosen randomly for control and experimental groups.
Mice were maintained under specific pathogen-free conditions at the animal facility of the University of Virginia. All animal procedures are approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Virginia in accordance with national guidelines to ensure the humanity of all animal experiments.
Method details
Doxycycline administration
To induce fallopian tube clones, doxycycline was dissolved at 2 mg/ml in sterile water with 30 mg/ml sucrose added. The doxycycline-containing water was used as the drinking water for the nursing mother, which transmits to pups through the milk and induces Cre expression in the pulps. Doxycycline was given between post-natal days 0–21 of the pulps.
Immunostaining
Mouse reproductive tracts were fixed overnight in 4% paraformaldehyde (PFA) at 4°C. For immunofluorescence staining, tissues were then washed with PBS to remove recessive PFA, soaked with 30% sucrose, embedded in optimal cutting temperature (OCT), and sectioned at 18 μm thickness. Tissue slides were incubated in Permeabilization/blocking buffer (0.3% Triton-X 100 in PBS [PBST] plus 5% normal donkey serum) for 20 min, then primary antibodies (Pax8,1:50,10336-1-AP, Proteintech; AcTUB,1:500, T7451, Sigma) diluted in Permeabilization/block buffer were added and incubated at 4°C overnight. Secondary antibody incubation was performed for 1 hour at RT in PBT. To stain nuclei, slides were incubated in DAPI solution (1ug/mL in PBST, 28718-90-3, Sigma-Aldrich) for 5 mins before mounting with 70% glycerol and covered.
For immunohistochemistry (IHC) staining of Ki67, PFA fixed tissues were embedded in paraffin and sectioned at 4 μm thickness. The slides were deparaffinized with xylene and gradually rehydrated with ethanol and water. Antigen retrieval was performed in citrate buffer (0.01M sodium citrate, 0.05% Tween-20, pH 6.0). Slides were boiled for 20 mins in buffer with a pressure cooker, then cooled down at room temperature. Endogenous peroxidase was blocked by 3% hydrogen peroxide through incubating for 15 mins. Slides were then washed with PBST, blocked in 5% serum for 20 mins, and incubated with anti-Ki67 (1:250, Abcam, ab15580) diluted in PBST overnight at 4°C. The Ki67 signal was developed using Anti-Rabbit HRP-DAB Cell & Tissue Staining Kit (R&D systems, CST005) following the manufacturer’s protocol. Slides were counterstained with hematoxylin (Vector Laboratories, ZH1027), air dried, and mounted with Cytoseal 60 (Fisher Scientific, 23-244257).
Tissue slides were imaged with Zeiss LSM 700/710 confocal microscope and Nikon Eclipse Ni-U microscope. Images were processed with Zeiss Zen (Blue Edition) and Fiji.
Tissue clearing with the CUBIC method and 3D imaging
The PFA fixed ovary and fallopian tube were cleared for large-scale 3D imaging with the standard CUBIC method.45 Tissues were first immersed in 50% reagent-1 and shaken at 110 rpm, 37°C for 6h, and then transferred to 100% reagent-1 with DAPI (1ug/ml) to shake for 2–3 days until becoming transparent. After reagent-1, tissues were washed three times with PBS, 30 mins each, with shaking to remove the reagent-1. Tissues were immersed in 50% reagent-2 for 6h with shaking at 37°C; after that, the buffer was exchanged for 100% reagent-2 with shaking for 24h. The Zeiss Z.1 light-sheet microscopy system was used to acquire images. Tissues were embedded in 2% (w/w) agarose gel; the agarose was dissolved in a modified reagent-2 (10% urea, 50% sucrose, 30% H2O, hot stir at 80°C until fully dissolved, then add 10% Tri-ethanolamine). The warm agarose gel solution, together with tissue, was aspirated into a 1 ml syringe with the neck cut off, and then the syringe was placed on ice for quick solidification. The syringe was placed on the holder of the light-sheet microscope, and the tissue was pushed out for imaging.
Counting clone size
The whole FT was cleared and imaged at 5-um intervals with light-sheet microscopy at single-cell resolution through the entire tissue. Three-dimensional reconstruction of clones was performed; labeled cells found at the exact location through multiple image stacks were counted as one clone. The size of clones was measured manually by counting the DAPI-stained nuclei.
Mathematical detection of outlier clones
The clonal size distribution along the fallopian tube was evaluated in terms of cell division events. With n as the number of cells per clone, there will be n-1 cell divisions. For clonal size distribution at five months, we fitted with a negative binomial distribution, which captures the behavior of the over-dispersed data observed in our mouse models by allowing the mean and variance to be different. Clonal size distribution at 1 and 2 months, which showed a smaller range, were fitted with a Poisson distribution. To estimate the statistical significance of our fitting, we generated null distributions for the Kolmogorov-Smirnov (KS) statistic between an ideal probability mass function and a random negative binomial/Poisson distribution over 1.000 iterations. The parameters used for generating the null distributions were obtained by merging all datasets and taking a maximum of 44/8-9-10, which showed significance in our first general screening. Then, the empirical nominal p-value of the observed KS for the merged data was calculated relative to the null distribution until a cutoff with significance was observed. Our final cutoff was set as the maximum number of divisions for which the probability of observing our KS was p >= 0.1. The validity of this cutoff was confirmed by evaluating the individual datasets over the null distributions. Then, the maximum expected clonal sizes (n) in the fallopian tube were calculated, and any clone larger than n was taken as the outlier. Subsequently, we computed the expected clone sizes for all the models, and we tested their significance against the observed values in our datasets using Fisher exact/Chi-square tests. Finally, we defined over-proliferative clones (Expected - observed outliers) for the size distributions observed under the Poisson model and tested their significance between 1, 2, and 5 months by an Anova with Tukey’s Honest Significant Difference test.
Fluorescence-guided laser-capture micro-dissection of fallopian tube epithelial cells
Cryo-embedded fresh FTs were sectioned at 8 μm thickness and were then dehydrated with 70% ethanol (30 sec), 95% ethanol (30 sec), and 100% ethanol for 1 min, followed by clearing with xylene (2 min). After air drying, slides were micro-dissected with Arcturus XT LCM instrument (Applied Biosystems) and Capsule HS LCM caps. The typical instrument settings of ∼50 mW power and ∼2 ms duration with the smallest spot size were used.
RNA extraction, reverse transcription, and amplification
The procedures were described before.52 In brief, RNA from the micro-dissected cells was extracted through enzymatic digestion with proteinase K, then reverse transcribed with 5′-biotin modified oligo(dT)24 (IDT) and SuperScript III (50°C, 30 min; then heat inactivation at 70°C for 15 min). The first-strand products were purified with Streptavidin magnetic beads and SuperMagnet Plate to remove genomic DNA. Then the cDNA was poly(A)-tailed and amplified with 25 cycles of PCR with AL1 primer.
RNA sequencing and analysis
For sequencing, the cDNA samples were re-amplified following protocols described before.52 100 μl PCR reactions were set up for each sample: 1) 5 μg AL1 primer, 2) 1x High-Fidelity buffer (Roche), 3.5 mM MgCl2, 200 μM dNTPs, 100 μg/ml BSA, 3) 1 μl cDNA template and 4) 1 μl Expand High Fidelity polymerase (Sigma). The following PCR program was used: 1minat 94°C, 2minat 42°C, and 3minat 72°C. 10 cycles were run to keep an exponential phase of re-amplification.53 The products were purified with AMPure XP beads (Beckman Coulter) according to the manufacturer’s protocol to remove excessive primers. The concentration of purified cDNA libraries was measured with the Qubit dsDNA BR Assay Kit (Thermo Fisher). Libraries were diluted to 0.2 ng/μl for tagmentation with the NXTR XT DNA SMP Prep Kit (Illumina). Libraries were multiplexed at an equimolar ratio, and 1.3 pM of the multiplexed pool was sequenced on a NextSeq 500 instrument with a NextSeq 500/550 Mid/High Output v2.5 kit (Illumina) to obtain 75-bp paired-end reads and an average of 3.6 million alignments per sample. From the sequencing reads, adapters were trimmed using fastq-mcf in the ea-utils package (version ea-utils.1.04.636), and with the following options:-q 10-t 0.01-k 0 (quality threshold 10, 0.01% occurrence frequency, no nucleotide skew causing cycle removal). Datasets were aligned to the mouse transcriptome (GRCm38.95), using RSEM80 (version 1.3.0) with the following options:--bowtie2--single-cell-prior--paired-end (Bowtie2 transcriptome aligner, single-cell prior to account for dropouts, paired-end reads). RSEM read counts were converted to transcripts per million (TPM) by dividing each value by the total read count for each sample and multiplying by 106.
DESeq281 (version 1.37.0) was used to identify differentially expressed transcripts between the small and large clone groups. Genes with an Padj <0.1 were considered significantly up- or downregulated between the two groups. To generate a heatmap for the differentially expressed genes between small and large clones, their TPMs were transformed with log2 +1 and then were subjected to Z-score normalization. Clustering was performed using Euclidian distance and the Ward.D method.
Gene-Set Enrichment Analysis (GSEA)82 was implemented to determine molecular signatures for the differential expressed transcripts ("h.all.v7.4.symbols.gmt"). Sequencing data were deposited to the GEO database: GSE210409.
Principal component analysis57 was performed in R (version 4.1.1) using the prcomp package. We used 16 transcriptomic datasets (TPMs data) representing 4 normal fallopian tubes and 12 ovarian tumors from mice downloaded from the GEO database (GSE135590). PCA was performed on the 48526 genes for which reads were identified in all the GEO samples and separately on the 129 genes found to be differentially expressed in our samples Finally, we projected our sample data into the PC1 and PC2 obtained from the GEO subset to identify clustering patterns associated with the large- and small-clones relative to the normal fallopian tubes and ovarian tumors. Centroids were calculated, and the Euclidean distances to these centroids were computed for individual small- and large-clone samples using R (dist function).
EdU assay of cell proliferation
5-ethynyl-2′–deoxyuridine (EdU, Invitrogen, Cat# A10044) was administered through drinking water (0.5 mg/ml). EdU staining was performed following standard procedures.83
Quantification and statistical analysis
Statistical analysis was performed with GraphPad Prism software. Data were presented as the mean ± SD or mean ± standard error of the mean (SEM) as indicated in the figure legends. Statistical significance is noted by ∗p<0.05; ∗∗p<0.01; ∗∗∗p<0.001; ∗∗∗∗p<0.0001; ns, not significant. The tests used are indicated in the figure legends.
Acknowledgments
We thank Dr. Zhe Li, Dr. Bing Xu, and Xiaoyu Zhao for providing critical feedback on the manuscript. We also thank Dr. Stacey Criswell at the Advanced Microscopy Facility, Dr. Pat Pramoonjago at the Biorepository and Tissue Research Facility, and Sheri Vanhoose at the Research Histology Core, and Shelly Verling at the vivarium for their assistance on the project. These core facilities are supported by UVA Cancer Center grant #P30-CA044579. We are grateful to Dr. Ammasi Periasamy and Dr. Ruofan Cao at the Keck Center for the usage of the Light-sheet Z.1 microscopy system. This work was partly supported by the Department of Defense Ovarian Cancer Research Program #W81XWH-17-1-0174 (J.K. S-D. & H.Z.), the Rivkin Center (H.Z.), the National Cancer Institute #R01-CA256199 (K.A.J. & H.Z.) and #R50-CA265089 (L.W.), the UVACancer Center Seed Grant (J.K.S-D. & H.Z.), the UVA Systems & Biomolecular Data Sciences Training Grant (T32-GM145443 to K.A.J.; supporting A.C.A-Y.), and the UVACancer Center Training Grant (J.Z.).
Author contributions
Conceptualization, J.Z., J.K.S-D., and H.Z.; Methodology, J.Z., E.C., Y.J., L.W., and B.E.K.; Investigation, J.Z., A.C.A-Y., and E.C.; Formal Analysis, J.Z., A.C.A-Y., E.K., T.J., and K.A.A., Writing–Original Draft, J.Z. and H.Z.; Writing–Review and Editing, J.Z., A.C.A-Y., K.A.J., J.K.S-D., and H.Z.; Funding Acquisition: J.K.S-D. and H.Z.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: April 25, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106742.
Contributor Information
Jill K. Slack-Davis, Email: jks6a@virginia.edu.
Hui Zong, Email: hz9s@virginia.edu.
Supplemental information
Data and code availability
-
•
RNA sequencing data of small and large clones have been deposited to the GEO database: (GSE210409, PRJNA865696) and are publicly available as of the date of publication.
-
•
All code necessary to define the dichotomy of clonal size distribution and the cutoff size of small and large clones can be found at https://github.com/JanesLab/ZengJ_MADMovarian/
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Perez-Losada J., Balmain A. Stem-cell hierarchy in skin cancer. Nat. Rev. Cancer. 2003;3:434–443. doi: 10.1038/nrc1095. [DOI] [PubMed] [Google Scholar]
- 2.Garraway L.A., Sellers W.R. Lineage dependency and lineage-survival oncogenes in human cancer. Nat. Rev. Cancer. 2006;6:593–602. doi: 10.1038/nrc1947. [DOI] [PubMed] [Google Scholar]
- 3.Visvader J.E. Cells of origin in cancer. Nature. 2011;469:314–322. doi: 10.1038/nature09781. [DOI] [PubMed] [Google Scholar]
- 4.Barker N., Ridgway R.A., van Es J.H., van de Wetering M., Begthel H., van den Born M., Danenberg E., Clarke A.R., Sansom O.J., Clevers H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- 5.Zong H., Espinosa J.S., Su H.H., Muzumdar M.D., Luo L. Mosaic analysis with double markers in mice. Cell. 2005;121:479–492. doi: 10.1016/j.cell.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 6.Muzumdar M.D., Luo L., Zong H. Modeling sporadic loss of heterozygosity in mice by using mosaic analysis with double markers (MADM) Proc. Natl. Acad. Sci. USA. 2007;104:4495–4500. doi: 10.1073/pnas.0606491104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu C., Sage J.C., Miller M.R., Verhaak R.G.W., Hippenmeyer S., Vogel H., Foreman O., Bronson R.T., Nishiyama A., Luo L., Zong H. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell. 2011;146:209–221. doi: 10.1016/j.cell.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hippenmeyer S. Dissection of gene function at clonal level using mosaic analysis with double markers. Front. Biol. 2013;8:557–568. doi: 10.1007/s11515-013-1279-6. [DOI] [Google Scholar]
- 9.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2020. CA. Cancer J. Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 10.Vaughan S., Coward J.I., Bast R.C., Jr., Berchuck A., Berek J.S., Brenton J.D., Coukos G., Crum C.C., Drapkin R., Etemadmoghadam D., et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat. Rev. Cancer. 2011;11:719–725. doi: 10.1038/nrc3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kindelberger D.W., Lee Y., Miron A., Hirsch M.S., Feltmate C., Medeiros F., Callahan M.J., Garner E.O., Gordon R.W., Birch C., et al. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: evidence for a causal relationship. Am. J. Surg. Pathol. 2007;31:161–169. doi: 10.1097/01.pas.0000213335.40358.47. [DOI] [PubMed] [Google Scholar]
- 12.Medeiros F., Muto M.G., Lee Y., Elvin J.A., Callahan M.J., Feltmate C., Garber J.E., Cramer D.W., Crum C.P. The tubal fimbria is a preferred site for early adenocarcinoma in women with familial ovarian cancer syndrome. Am. J. Surg. Pathol. 2006;30:230–236. doi: 10.1097/01.pas.0000180854.28831.77. [DOI] [PubMed] [Google Scholar]
- 13.Visvanathan K., Shaw P., May B.J., Bahadirli-Talbott A., Kaushiva A., Risch H., Narod S., Wang T.L., Parkash V., Vang R., et al. Fallopian tube lesions in women at high risk for ovarian cancer: a multicenter study. Cancer Prev. Res. 2018;11:697–706. doi: 10.1158/1940-6207.CAPR-18-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee Y., Miron A., Drapkin R., Nucci M.R., Medeiros F., Saleemuddin A., Garber J., Birch C., Mou H., Gordon R.W., et al. A candidate precursor to serous carcinoma that originates in the distal fallopian tube. J. Pathol. 2007;211:26–35. doi: 10.1002/path.2091. [DOI] [PubMed] [Google Scholar]
- 15.Przybycin C.G., Kurman R.J., Ronnett B.M., Shih I.-M., Vang R. Are all pelvic (nonuterine) serous carcinomas of tubal origin? Am. J. Surg. Pathol. 2010;34:1407–1416. doi: 10.1097/PAS.0b013e3181ef7b16. [DOI] [PubMed] [Google Scholar]
- 16.Kuhn E., Kurman R.J., Vang R., Sehdev A.S., Han G., Soslow R., Wang T.L., Shih I.M. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma--evidence supporting the clonal relationship of the two lesions. J. Pathol. 2012;226:421–426. doi: 10.1002/path.3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soong T.R., Howitt B.E., Miron A., Horowitz N.S., Campbell F., Feltmate C.M., Muto M.G., Berkowitz R.S., Nucci M.R., Xian W., Crum C.P. Evidence for lineage continuity between early serous proliferations (ESPs) in the Fallopian tube and disseminated high-grade serous carcinomas. J. Pathol. 2018;246:344–351. doi: 10.1002/path.5145. [DOI] [PubMed] [Google Scholar]
- 18.Labidi-Galy S.I., Papp E., Hallberg D., Niknafs N., Adleff V., Noe M., Bhattacharya R., Novak M., Jones S., Phallen J., et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 2017;8:1093. doi: 10.1038/s41467-017-00962-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ardighieri L., Mori L., Conzadori S., Bugatti M., Falchetti M., Donzelli C.M., Ravaggi A., Odicino F.E., Facchetti F. Identical TP53 mutations in pelvic carcinosarcomas and associated serous tubal intraepithelial carcinomas provide evidence of their clonal relationship. Virchows Arch. 2016;469:61–69. doi: 10.1007/s00428-016-1933-x. [DOI] [PubMed] [Google Scholar]
- 20.Marquez R.T., Baggerly K.A., Patterson A.P., Liu J., Broaddus R., Frumovitz M., Atkinson E.N., Smith D.I., Hartmann L., Fishman D., et al. Patterns of gene expression in different histotypes of epithelial ovarian cancer correlate with those in normal fallopian tube, endometrium, and colon. Clin. Cancer Res. 2005;11:6116–6126. doi: 10.1158/1078-0432.CCR-04-2509. [DOI] [PubMed] [Google Scholar]
- 21.Tone A.A., Begley H., Sharma M., Murphy J., Rosen B., Brown T.J., Shaw P.A. Gene expression profiles of luteal phase fallopian tube epithelium from BRCA mutation carriers resemble high-grade serous carcinoma. Clin. Cancer Res. 2008;14:4067–4078. doi: 10.1158/1078-0432.CCR-07-4959. [DOI] [PubMed] [Google Scholar]
- 22.Beirne J.P., McArt D.G., Roddy A., McDermott C., Ferris J., Buckley N.E., Coulter P., McCabe N., Eddie S.L., Dunne P.D., et al. Defining the molecular evolution of extrauterine high grade serous carcinoma. Gynecol. Oncol. 2019;155:305–317. doi: 10.1016/j.ygyno.2019.08.029. [DOI] [PubMed] [Google Scholar]
- 23.Ducie J., Dao F., Considine M., Olvera N., Shaw P.A., Kurman R.J., Shih I.M., Soslow R.A., Cope L., Levine D.A. Molecular analysis of high-grade serous ovarian carcinoma with and without associated serous tubal intra-epithelial carcinoma. Nat. Commun. 2017;8:990. doi: 10.1038/s41467-017-01217-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stewart C.A., Behringer R.R. In: Mouse Development: From Oocyte to Stem Cells. Kubiak J.Z., editor. Springer Berlin Heidelberg; 2012. Mouse oviduct development; pp. 247–262. [DOI] [Google Scholar]
- 25.Perets R., Wyant G.A., Muto K.W., Bijron J.G., Poole B.B., Chin K.T., Chen J.Y.H., Ohman A.W., Stepule C.D., Kwak S., et al. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer Cell. 2013;24:751–765. doi: 10.1016/j.ccr.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang S., Dolgalev I., Zhang T., Ran H., Levine D.A., Neel B.G. Both fallopian tube and ovarian surface epithelium are cells-of-origin for high-grade serous ovarian carcinoma. Nat. Commun. 2019;10:5367. doi: 10.1038/s41467-019-13116-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu R., Zhai Y., Kuick R., Karnezis A.N., Garcia P., Naseem A., Hu T.C., Fearon E.R., Cho K.R. Impact of oviductal versus ovarian epithelial cell of origin on ovarian endometrioid carcinoma phenotype in the mouse. J. Pathol. 2016;240:341–351. doi: 10.1002/path.4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sherman-Baust C.A., Kuhn E., Valle B.L., Shih I.-M., Kurman R.J., Wang T.-L., Amano T., Ko M.S.H., Miyoshi I., Araki Y., et al. A genetically engineered ovarian cancer mouse model based on fallopian tube transformation mimics human high-grade serous carcinoma development. J. Pathol. 2014;233:228–237. doi: 10.1002/path.4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhai Y., Wu R., Kuick R., Sessine M.S., Schulman S., Green M., Fearon E.R., Cho K.R. High-grade serous carcinomas arise in the mouse oviduct via defects linked to the human disease. J. Pathol. 2017;243:16–25. doi: 10.1002/path.4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCool K.W., Freeman Z.T., Zhai Y., Wu R., Hu K., Liu C.J., Tomlins S.A., Fearon E.R., Magnuson B., Kuick R., Cho K.R. Murine oviductal high-grade serous carcinomas mirror the genomic alterations, gene expression profiles, and immune microenvironment of their human counterparts. Cancer Res. 2020;80:877–889. doi: 10.1158/0008-5472.can-19-2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ulrich N.D., Shen Y.-c., Ma Q., Yang K., Hannum D.F., Jones A., Machlin J., Randolph J.F., Smith Y.R., Schon S.B., et al. Cellular heterogeneity of human fallopian tubes in normal and hydrosalpinx disease states identified using scRNA-seq. Dev. Cell. 2022;57:914–929.e7. doi: 10.1016/j.devcel.2022.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu Z., Artibani M., Alsaadi A., Wietek N., Morotti M., Shi T., Zhong Z., Santana Gonzalez L., El-Sahhar S., Carrami E.M., et al. The repertoire of serous ovarian cancer non- genetic heterogeneity revealed by single-cell sequencing of normal fallopian tube epithelial cells. Cancer Cell. 2020;37:226–242.e7. doi: 10.1016/j.ccell.2020.01.003. [DOI] [PubMed] [Google Scholar]
- 33.Dinh H.Q., Lin X., Abbasi F., Nameki R., Haro M., Olingy C.E., Chang H., Hernandez L., Gayther S.A., Wright K.N., et al. Single-cell transcriptomics identifies gene expression networks driving differentiation and tumorigenesis in the human fallopian tube. Cell Rep. 2021;35 doi: 10.1016/j.celrep.2021.108978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paik D.Y., Janzen D.M., Schafenacker A.M., Velasco V.S., Shung M.S., Cheng D., Huang J., Witte O.N., Memarzadeh S. Stem-like epithelial cells are concentrated in the distal end of the fallopian tube: a site for injury and serous cancer initiation. Stem Cell. 2012;30:2487–2497. doi: 10.1002/stem.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rose I.M., Bidarimath M., Webster A., Godwin A.K., Flesken-Nikitin A., Nikitin A.Y. WNT and inflammatory signaling distinguish human Fallopian tube epithelial cell populations. Sci. Rep. 2020;10:9837. doi: 10.1038/s41598-020-66556-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Traykova-Brauch M., Schönig K., Greiner O., Miloud T., Jauch A., Bode M., Felsher D.W., Glick A.B., Kwiatkowski D.J., Bujard H., et al. An efficient and versatile system for acute and chronic modulation of renal tubular function in transgenic mice. Nat. Med. 2008;14:979–984. doi: 10.1038/nm.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walsh T., Casadei S., Lee M.K., Pennil C.C., Nord A.S., Thornton A.M., Roeb W., Agnew K.J., Stray S.M., Wickramanayake A., et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Nati Acad Sci USA. 2011;108:18032–18037. doi: 10.1073/pnas.1115052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahmed A.A., Etemadmoghadam D., Temple J., Lynch A.G., Riad M., Sharma R., Stewart C., Fereday S., Caldas C., Defazio A., et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010;221:49–56. doi: 10.1002/path.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patch A.M., Christie E.L., Etemadmoghadam D., Garsed D.W., George J., Fereday S., Nones K., Cowin P., Alsop K., Bailey P.J., et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015;521:489–494. doi: 10.1038/nature14410. [DOI] [PubMed] [Google Scholar]
- 41.Toss A., Tomasello C., Razzaboni E., Contu G., Grandi G., Cagnacci A., Schilder R.J., Cortesi L. Hereditary ovarian cancer: not only BRCA 1 and 2 genes. BioMed Res. Int. 2015;2015:341723. doi: 10.1155/2015/341723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tasic B., Miyamichi K., Hippenmeyer S., Dani V.S., Zeng H., Joo W., Zong H., Chen-Tsai Y., Luo L. Extensions of MADM (mosaic analysis with double markers) in mice. PLoS One. 2012;7 doi: 10.1371/journal.pone.0033332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hippenmeyer S., Youn Y.H., Moon H.M., Miyamichi K., Zong H., Wynshaw-Boris A., Luo L. Genetic mosaic dissection of Lis1 and Ndel1 in neuronal migration. Neuron. 2010;68:695–709. doi: 10.1016/j.neuron.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tacha D., Zhou D., Cheng L. Expression of PAX8 in normal and neoplastic tissues: a comprehensive immunohistochemical study. Appl. Immunohistochem. Mol. Morphol. 2011;19:293–299. doi: 10.1097/PAI.0b013e3182025f66. [DOI] [PubMed] [Google Scholar]
- 45.Susaki E.A., Tainaka K., Perrin D., Yukinaga H., Kuno A., Ueda H.R. Advanced CUBIC protocols for whole-brain and whole-body clearing and imaging. Nat. Protoc. 2015;10:1709–1727. doi: 10.1038/nprot.2015.085. [DOI] [PubMed] [Google Scholar]
- 46.Bliss C.I., Fisher R.A. Fitting the negative binomial distribution to biological data. Biometric. 1953;9:176–200. doi: 10.2307/3001850. [DOI] [Google Scholar]
- 47.Crum C.P., Drapkin R., Miron A., Ince T.A., Muto M., Kindelberger D.W., Lee Y. The distal fallopian tube: a new model for pelvic serous carcinogenesis. Curr. Opin. Obstet. Gynecol. 2007;19:P3–P9. doi: 10.1097/GCO.0b013e328011a21f. [DOI] [PubMed] [Google Scholar]
- 48.Yamanouchi H., Umezu T., Tomooka Y. Reconstruction of oviduct and demonstration of epithelial fate determination in mice. Biol. Reprod. 2010;82:528–533. doi: 10.1095/biolreprod.109.078329. [DOI] [PubMed] [Google Scholar]
- 49.Fuchs E., Segre J.A. Stem cells: a new lease on life. Cell. 2000;100:143–155. doi: 10.1016/s0092-8674(00)81691-8. [DOI] [PubMed] [Google Scholar]
- 50.Post Y., Clevers H. Defining adult stem cell function at its simplest: the ability to replace lost cells through mitosis. Cell Stem Cell. 2019;25:174–183. doi: 10.1016/j.stem.2019.07.002. [DOI] [PubMed] [Google Scholar]
- 51.Ghosh A., Syed S.M., Tanwar P.S. In vivo genetic cell lineage tracing reveals that oviductal secretory cells self-renew and give rise to ciliated cells. Development. 2017;144:3031–3041. doi: 10.1242/dev.149989. [DOI] [PubMed] [Google Scholar]
- 52.Singh S., Wang L., Schaff D.L., Sutcliffe M.D., Koeppel A.F., Kim J., Onengut-Gumuscu S., Park K.S., Zong H., Janes K.A. In situ 10-cell RNA sequencing in tissue and tumor biopsy samples. Sci. Rep. 2019;9:4836. doi: 10.1038/s41598-019-41235-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang L., Janes K.A. Stochastic profiling of transcriptional regulatory heterogeneities in tissues, tumors and cultured cells. Nat. Protoc. 2013;8:282–301. doi: 10.1038/nprot.2012.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Viré E., Brenner C., Deplus R., Blanchon L., Fraga M., Didelot C., Morey L., Van Eynde A., Bernard D., Vanderwinden J.M., et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871–874. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- 55.Cao R., Wang L., Wang H., Xia L., Erdjument-Bromage H., Tempst P., Jones R.S., Zhang Y. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 56.Musa J., Aynaud M.M., Mirabeau O., Delattre O., Grünewald T.G. MYBL2 (B-Myb): a central regulator of cell proliferation, cell survival and differentiation involved in tumorigenesis. Cell Death Dis. 2017;8 doi: 10.1038/cddis.2017.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pizato N., Luzete B.C., Kiffer L., Corrêa L.H., de Oliveira Santos I., Assumpção J.A.F., Ito M.K., Magalhães K.G. Omega-3 docosahexaenoic acid induces pyroptosis cell death in triple-negative breast cancer cells. Sci. Rep. 2018;8:1952. doi: 10.1038/s41598-018-20422-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harwalkar K., Ford M.J., Teng K., Yamanaka N., Yang B., Burtscher I., Lickert H., Yamanaka Y. Anatomical and cellular heterogeneity in the mouse oviduct-its potential roles in reproduction and preimplantation development. Biol. Reprod. 2021;104:1249–1261. doi: 10.1093/biolre/ioab043. [DOI] [PubMed] [Google Scholar]
- 59.Chen E.Y., Mehra K., Mehrad M., Ning G., Miron A., Mutter G.L., Monte N., Quade B.J., McKeon F.D., Yassin Y., et al. Secretory cell outgrowth, PAX2 and serous carcinogenesis in the Fallopian tube. J. Pathol. 2010;222:110–116. doi: 10.1002/path.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Meserve E.E.K., Brouwer J., Crum C.P. Serous tubal intraepithelial neoplasia: the concept and its application. Mod. Pathol. 2017;30:710–721. doi: 10.1038/modpathol.2016.238. [DOI] [PubMed] [Google Scholar]
- 61.Yao M., Ventura P.B., Jiang Y., Rodriguez F.J., Wang L., Perry J.S.A., Yang Y., Wahl K., Crittenden R.B., Bennett M.L., et al. Astrocytic trans-differentiation completes a multicellular paracrine feedback loop required for medulloblastoma tumor growth. Cell. 2020;180:502–520.e19. doi: 10.1016/j.cell.2019.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cheon D.J., Orsulic S. Mouse models of cancer. Annu. Rev. Pathol. 2011;6:95–119. doi: 10.1146/annurev.pathol.3.121806.154244. [DOI] [PubMed] [Google Scholar]
- 63.Novak A., Guo C., Yang W., Nagy A., Lobe C.G. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis. 2000;28:147–155. [PubMed] [Google Scholar]
- 64.Mao X., Fujiwara Y., Chapdelaine A., Yang H., Orkin S.H. Activation of EGFP expression by Cre-mediated excision in a new ROSA26 reporter mouse strain. Blood. 2001;97:324–326. doi: 10.1182/blood.v97.1.324. [DOI] [PubMed] [Google Scholar]
- 65.Holzenberger M., Lenzner C., Leneuve P., Zaoui R., Hamard G., Vaulont S., Bouc Y.L. Cre-mediated germline mosaicism: a method allowing rapid generation of several alleles of a target gene. Nucleic Acids Res. 2000;28:E92. doi: 10.1093/nar/28.21.e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Beattie R., Postiglione M.P., Burnett L.E., Laukoter S., Streicher C., Pauler F.M., Xiao G., Klezovitch O., Vasioukhin V., Ghashghaei T.H., Hippenmeyer S. Mosaic analysis with double markers reveals distinct sequential functions of Lgl1 in neural stem cells. Neuron. 2017;94:517–533.e3. doi: 10.1016/j.neuron.2017.04.012. [DOI] [PubMed] [Google Scholar]
- 67.Terry T.T., Cheng T., Mahjoub M., Zong H. Mosaic Analysis with Double Markers reveals IGF1R function in granule cell progenitors during cerebellar development. Dev. Biol. 2020;465:130–143. doi: 10.1016/j.ydbio.2020.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Contreras X., Amberg N., Davaatseren A., Hansen A.H., Sonntag J., Andersen L., Bernthaler T., Streicher C., Heger A., Johnson R.L., et al. A genome-wide library of MADM mice for single-cell genetic mosaic analysis. Cell Rep. 2021;35 doi: 10.1016/j.celrep.2021.109274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu N.Y., Huang H.S., Chao T.H., Chou H.M., Fang C., Qin C.Z., Lin C.Y., Chu T.Y., Zhou H.H. Progesterone prevents high-grade serous ovarian cancer by inducing necroptosis of p53-defective fallopian tube epithelial cells. Cell Rep. 2017;18:2557–2565. doi: 10.1016/j.celrep.2017.02.049. [DOI] [PubMed] [Google Scholar]
- 70.King S.M., Hilliard T.S., Wu L.Y., Jaffe R.C., Fazleabas A.T., Burdette J.E. The impact of ovulation on fallopian tube epithelial cells: evaluating three hypotheses connecting ovulation and serous ovarian cancer. Endocr. Relat. Cancer. 2011;18:627–642. doi: 10.1530/ERC-11-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kessler M., Hoffmann K., Brinkmann V., Thieck O., Jackisch S., Toelle B., Berger H., Mollenkopf H.J., Mangler M., Sehouli J., et al. The Notch and Wnt pathways regulate stemness and differentiation in human fallopian tube organoids. Nat. Commun. 2015;6:8989. doi: 10.1038/ncomms9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xie Y., Park E.S., Xiang D., Li Z. Long-term organoid culture reveals enrichment of organoid-forming epithelial cells in the fimbrial portion of mouse fallopian tube. Stem Cell Res. 2018;32:51–60. doi: 10.1016/j.scr.2018.08.021. [DOI] [PubMed] [Google Scholar]
- 73.Qin G., Park E.-S., Chen X., Han S., Xiang D., Ren F., Liu G., Chen H., Yuan G.-C., Li Z. Distinct niche structures and intrinsic programs of fallopian tube and ovarian surface epithelial cells. iScience. 2022;26 doi: 10.1016/j.isci.2022.105861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Greaves M., Maley C.C. Clonal evolution in cancer. Nature. 2012;481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lytle N.K., Barber A.G., Reya T. Stem cell fate in cancer growth, progression and therapy resistance. Nat. Rev. Cancer. 2018;18:669–680. doi: 10.1038/s41568-018-0056-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tao T., Lin W., Wang Y., Zhang J., Chambers S.K., Li B., Lea J., Wang Y., Wang Y., Zheng W. Loss of tubal ciliated cells as a risk for "ovarian" or pelvic serous carcinoma. Am. J. Cancer Res. 2020;10:3815–3827. [PMC free article] [PubMed] [Google Scholar]
- 77.Alcolea M.P., Greulich P., Wabik A., Frede J., Simons B.D., Jones P.H. Differentiation imbalance in single oesophageal progenitor cells causes clonal immortalization and field change. Nat. Cell Biol. 2014;16:615–622. doi: 10.1038/ncb2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Driessens G., Beck B., Caauwe A., Simons B.D., Blanpain C. Defining the mode of tumour growth by clonal analysis. Nature. 2012;488:527–530. doi: 10.1038/nature11344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Perl A.K.T., Wert S.E., Nagy A., Lobe C.G., Whitsett J.A. Early restriction of peripheral and proximal cell lineages during formation of the lung. Proc. Natl. Acad. Sci. USA. 2002;99:10482–10487. doi: 10.1073/pnas.152238499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Salic A., Mitchison T.J. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl. Acad. Sci. USA. 2008;105:2415–2420. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
RNA sequencing data of small and large clones have been deposited to the GEO database: (GSE210409, PRJNA865696) and are publicly available as of the date of publication.
-
•
All code necessary to define the dichotomy of clonal size distribution and the cutoff size of small and large clones can be found at https://github.com/JanesLab/ZengJ_MADMovarian/
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.