

Abstract
Maintenance of genomic methylation patterns at DNA replication forks by DNMT1 is the key to faithful mitotic inheritance. DNMT1 is often overexpressed in cancer cells and the DNA hypomethylating agents azacytidine and decitabine are currently used in the treatment of hematologic malignancies. However, the toxicity of these cytidine analogs and their ineffectiveness in treating solid tumors have limited wider clinical use. GSK-3484862 is a newly-developed, dicyanopyridine containing, non-nucleoside DNMT1-selective inhibitor with low cellular toxicity. Here, we show that GSK-3484862 targets DNMT1 for protein degradation in both cancer cell lines and murine embryonic stem cells (mESCs). DNMT1 depletion was rapid, taking effect within hours following GSK-3484862 treatment, leading to global hypomethylation. Inhibitor-induced DNMT1 degradation was proteasome-dependent, with no discernible loss of DNMT1 mRNA. In mESCs, GSK-3484862-induced Dnmt1 degradation requires the Dnmt1 accessory factor Uhrf1 and its E3 ubiquitin ligase activity. We also show that Dnmt1 depletion and DNA hypomethylation induced by the compound are reversible after its removal. Together, these results indicate that this DNMT1-selective degrader/inhibitor will be a valuable tool for dissecting coordinated events linking DNA methylation to gene expression and identifying downstream effectors that ultimately regulate cellular response to altered DNA methylation patterns in a tissue/cell-specific manner.
Graphical Abstract
Graphical Abstract.
INTRODUCTION
DNA methylation (i.e. 5-methylcytosine or 5mC) is an important epigenetic modification that influences chromatin structure and gene expression. In mammals, there are three active DNA methyltransferases, DNMT1, DNMT3A and DNMT3B, which belong to two structurally and functionally distinct DNMT families and act primarily at CpG dinucleotides [reviewed in (1,2)]. DNMT3A and DNMT3B establish the initial cytosine methylation pattern de novo, whereas DNMT1 maintains the pattern on newly replicated DNA (3,4). Cancer cells generally exhibit abnormal DNA methylation patterns, including both global hypomethylation and regional hypermethylation, with hypermethylation being linked to the silencing of tumor suppressor genes (5). Although the precise mechanisms leading to aberrant DNA methylation patterns are complex and not fully understood, researchers and clinicians have nonetheless been investigating DNA methylation inhibition as a therapeutic strategy for cancer treatment.
The nucleoside cytidine analogs 5-azacytidine (Vidaza®) and 5-aza-2′-deoxycytidine (decitabine, Dacogen®) are FDA-approved DNA demethylating agents for treating myelodysplastic syndrome (MDS), acute myeloid leukemia (AML) and chronic myelomonocytic leukemia (CMML) (6–12). These nucleoside analogs incorporate into DNA, where they trap DNMTs through the formation of an irreversible suicide complex (13–15), leading to substantial DNA damage and cellular toxicity. The anti-cancer effects of these drugs are thought to be due, in part, to the demethylation and reactivation of endogenous retroviruses, thus provoking an interferon response (16).
The dose-limiting toxicity of and limited patient tolerance for cytidine analogs, as well as their ineffectiveness in treating solid tumors (17,18), have led to a persistent search for non-nucleoside DNMT inhibitors. This exploration has led to the discoveries of RG-108 (19), the quinoline-based SGI-1027 (20), and its analogs MC3343 and MC3353 (21–24), quinazoline derivatives (25) and quinazoline–quinoline linked derivatives (26), as well as other small-molecule compounds (27). However, none of these inhibitors are specific for DNMT1 or DNMT3A/3B with a clear translation from in vitro to in vivo activity.
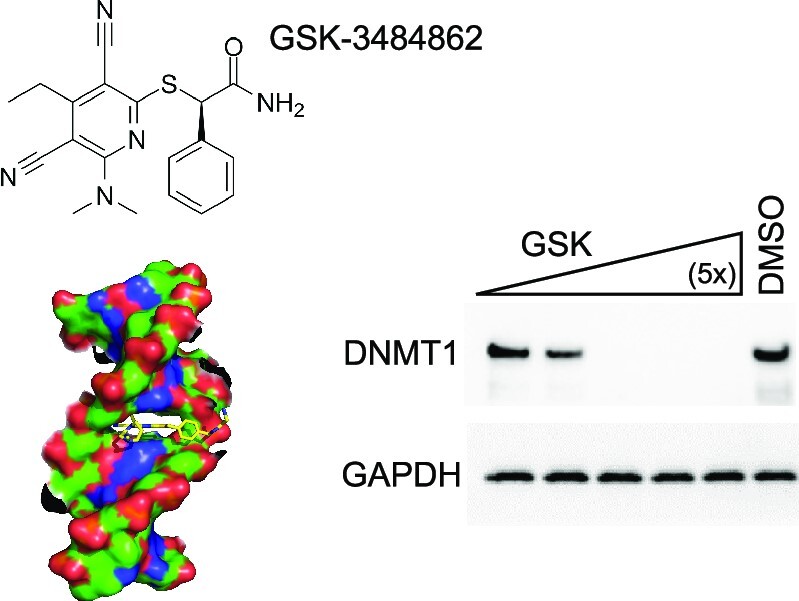
Recently, GlaxoSmithKline (GSK) reported a new class of reversible DNMT1-selective inhibitors containing a dicyanopyridine moiety, including GSK-3484862 (left panel in Figure 1A) (28). This new class of DNMT1-selective inhibitors is less cytotoxic than current cytidine analogs and when tested against a panel of >300 protein kinases and 30 other methyltransferases including DNMT3A/3B (28), showed remarkable DNMT1 specificity, making it a strong therapeutic contender.
Figure 1.

GSK(1) targets DNMT1 for degradation in A549 cells. (A) Chemical structures of GSK(1) and GSK(2) and DNMT1-bound GSK(2) with the inhibitor shown in yellow and the DNMT1 active-site loop shown in magenta (initially reported in (28,38)). (B) Western blots showing endogenous levels of DNMT1, DNMT3A and DNMT3B following treatment with 2 and 4 μM GSK(1) for 24 h. While DNMT3A appeared to be slightly reduced, subsequent experiments revealed no significant changes in DNMT3A level (Supplementary Figure S1C and Figure 2E). (C) Relative gene expression of DNMT1 in the presence and absence of GSK(1) were analyzed by RT-qPCR and normalized relative to IPO8 (N = 3). The corresponding western blot is shown in lower panel for three repeats of DMSO (in blue dots) and GSK(1)-treated samples (in red dots). (D) Cell growth as determined by cell viability assay over 2 days. (E) Time-dependent depletion of DNMT1 in cells treated with GSK(1). (F) Comparison of the potency of GSK(1) and GSK(2) in inducing DNMT1 depletion. (G) GSK(2) showed an enhanced cell toxicity compared to GSK(1) at 4 μM in A549 cells. (H) Concentration-dependent effect of GSK(1). (I) Time-dependent effect of GSK(1) at 80 nM. (J) Little effect of GSK(1) at low concentrations (3.2 and 16 nM). In panels H–J, the two DMSO lanes as indicated by a bracket are the 2× dilution (1/2) of cell lysates.
In a transgenic mouse model of sickle cell disease, where azacytidine and decitabine have been shown to induce fetal hemoglobin expression (29–34), oral GSK3482364 was well tolerated and boosted both fetal hemoglobin levels and the percentage of erythrocytes expressing fetal hemoglobin (35). GSK-3484862 (the purified R-enantiomer of GSK-3482364) inhibits Dnmt1 in murine pre-implantation Dnmt3a/3b knockout (KO) embryos (36), and results in DNA hypomethylation to nearly the extent of Dnmt1 KO murine embryonic stem cells (mESCs) (37). Due to its improved in vivo tolerability and pharmacokinetic properties compared with decitabine, GSK-3685032 (a closely related chemical to GSK-3484862; middle panel in Figure 1A) was superior to decitabine in tumor regression in a mouse model of AML (28). Further, MV4-11 leukemia cells treated with GSK-3685032 exhibited a relatively slow onset (≥3 days) of growth inhibition but with increasing potency observed over a 6-day time course. By day 6, GSK-3685032 showed an enhanced anti-proliferative effect compared with GSK-3484862 (28).
Mechanistically, these new GSK compounds contain a planar dicyanopyridine moiety that competes with the DNMT1-active site loop for intercalation specifically into the DNMT1-bound DNA positioned between the two base pairs of CpG dinucleotide (28,38) (right panel in Figure 1A). The bound dicyanopyridine-containing compound displaces the DNMT1 active-site loop (magenta in Figure 1A) from the substrate DNA and prevents it from intercalating into the same CpG site, reflecting a mechanism of DNMT inhibition distinct from those of traditional active-site inhibitors.
Here, we show that GSK-3484862 and GSK-3685032 target DNMT1 for degradation in a wide-range of cancer cell lines, as well as mESCs, with no detectable reduction in DNMT1 mRNA levels. Compound-induced DNMT1 degradation is proteasome-dependent and, in mESCs, requires the presence of Uhrf1, an accessory factor of Dnmt1 (39,40). For simplicity, we refer to compounds GSK-3484862 and GSK-3685032 as GSK(1) and GSK(2), respectively. Our experiments were designed to examine the effects of these compounds at early time points (<3 days), before the cells showed any signs of toxicity, with special focus on GSK(1), which at a concentration as high as 50 μM, had minimal effects on cell viability over the 3-day treatment.
MATERIALS AND METHODS
Chemicals
Chemicals used in this study include: GSK-3484862 (MedChemExpress, HY-135146), GSK-3685032 (MedChemExpress, HY-139664), Decitabine (DAC, 5-Aza-2′-deoxycytidine, Millipore Sigma, A3656), MG132 (Millipore Sigma, 474787), and Aphidicolin (Cayman Chemicals, 14007).
Antibodies
The primary antibodies used in this study were: DNMT1 [(Cell Signaling Technology (CST), #5032), DNMT3A (CST, #3598), DNMT3B (CST, #67259), H3 (CST, #14269), GAPDH (CST, #2118), Oct4 (CST, #4286), PCNA (CST, #13110), Uhrf1 (CST, #12387), Actin (Sigma-Aldrich, A2228), Myc tag (Sigma-Aldrich, M4439), Flag tag (Sigma-Aldrich, F3165) and 5- methylcytosine (5mC) (Active Motif, #39649). The secondary antibodies used were: HRP-conjugated anti-rabbit-IgG (CST, #7074) and HRP-conjugated anti-mouse-IgG (Abcam, ab6820).
Cell lines and culture
A549-Luc2, MCF7, U2OS, PC3, MOLM13, THP1, MV4-11 and GDM-1 cell lines were purchased from the American Type Culture Collection (ATCC) and validated at The University of Texas MD Anderson Cancer Center (Houston, TX). All cells were incubated at 37°C with 5% CO2.
A549-Luc2 cells were cultured in ATCC-formulated F-12K Medium (Catalog No. 30-2004) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich) and 1% penicillin/streptomycin.
MCF7 and U2OS cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) with l-glutamine and 4.5 g glucose/l but without sodium pyruvate (Mediatech) supplemented with 10% FBS and 1% penicillin/streptomycin.
PC3, MOLM13, THP1, MV4-11 and GDM-1 cells were cultured in RPMI1640 Medium supplemented with 10% FBS, 1% penicillin/streptomycin, 2 mM l-glutamine (Mediatech) and 10 mM HEPES (Mediatech).
Wild-type (WT), Dnmt1−/− (41) and Uhrf1−/− J1 mESCs, as well as Uhrf1−/− mESCs reconstituted with Flag-tagged mouse Uhrf1 or the ‘E3 ligase-dead’ H730A mutant (42), were cultured in gelatin-coated petri dishes in DMEM supplemented with 15% FBS, 0.1 mM nonessential amino acids, 0.1 mM β-mercaptoethanol, 1% penicillin/streptomycin, and 103 U/ml leukemia inhibitory factor (LIF).
Generation of stable clones in mESCs was described previously (43). In brief, bicistronic plasmid vectors (44) expressing the Blasticidin S-resistant gene and Myc-tagged mouse Dnmt1 or the catalytically inactive PC:SF (P1228S; C1229F) mutant (generated by introducing an HindIII site into the cDNA, see primers in Supplementary Table S1) were transfected into Dnmt1−/− mESCs, and individual clones were picked after seven days of selection with Blasticidin S HCl (Thermo Fisher Scientific, A1113903). Clones expressing Myc-Dnmt1 levels comparable to that of endogenous Dnmt1 in WT mESCs were used for experiments.
Chemical compound treatment
Cells were seeded onto 6-well plates at a cell density of ∼5 × 105/well. The next day, cells were treated with 0.1% DMSO (control) or compound GSK(1) or GSK(2) or DAC at the indicated concentrations. MG132 (4 μM) was used in Figure 2D. Aphidicolin (20 μg/ml) was used to treat A549 cells for 24 h prior to treatment with GSK(1) or DAC (Figure 2F and G), according to the established procedure for using the DNA synthesis inhibitor (45).
Figure 2.

GSK(1) induces DNA hypomethylation and proteasome-dependent and DNA synthesis-dependent loss of DNMT1 protein. (A) Dot blot assay using an anti-5mC antibody to detect 5mC in genomic DNA. The lower panel shows the same membrane used in top panel stained with methylene blue to verify DNA loading. (B) GSK(1)-induced hypomethylation at three genomic loci by pyrosequencing. Three technical replicates were measured for each sample. The three DMSO controls at 12, 24 and 48 h were averaged. (C) Mass spectrometry analysis of total 5mC content expressed as a percentage (i.e. number of 5mC-modified residues divided by the total number of cytosine residues × 100). (D) A549 cells were treated with GSK(1) (left), MG132 (middle) or both (right) up to 24 h and subjected to immunoblotting. (E, F) For treatment of 12 h, the effects of GSK(1) or DAC on their own (panel E) as compared with cells that had been treated with the DNA synthesis inhibitor aphidicolin for 24 h prior to treatment with GSK(1) or DAC (panel F). (G) As in panel E and F, the effect of GSK(1) or DAC treatment of 24 h, in the absence (left) and presence of aphidicolin (right). In panels F and G, the two DMSO lanes as indicated by a bracket are the 2× dilution (1/2) of cell lysates.
Cells were collected at the indicated time points for western blot, RT-qPCR and DNA methylation analysis as described below. Because GSK(1) is stable in MV4-11 cells for six days [Extended Data Figure 1A of reference (28)], cells were treated only once at the indicated concentrations.
For recovery experiments, WT J1 mESCs were treated with 2 μM of GSK(1) for 24 h, and the cells were washed three times with PBS and cultured in the absence of the compound for an additional 4 days.
Western blot
Cells were lysed with sodium dodecyl sulfate (4xSDS) sample buffer. The lysates were separated by Bis-Tris sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) using 4–20% precast polyacrylamide gel (BioRad, #4561096). The proteins were transferred to low fluorescence polyvinylidene difluoride (PVDF) membranes (BioRad, #1620261), which were blocked with 5% non-fat dry milk in Tris-buffered saline with Tween 20 (TBST) at room temperature for 1 h and then probed with primary followed by secondary antibodies. The signals were detected with Clarity Western ECL substrate (Bio-Rad Laboratories, #1705061) and imaged using a ChemiDoc imaging system (Bio-Rad Laboratories).
RNA isolation and RT-qPCR assay
Following treatment with DMSO or GSK compounds, total RNA was isolated from A549, MOLM13, THP1, MV4-11 cells and mESCs using TRIzol reagent (Invitrogen; 15596–018). For RT-qPCR assays, RNA samples were pretreated with TURBO DNase (Ambion) to remove genomic DNA contamination and then reverse-transcribed into cDNAs using a high-capacity reverse transcription kit (Applied Biosystems, 4368814). An aliquot (20–50 ng) of cDNA was used in each PCR reaction. Real-time PCR was performed in triplicate or quadruplicate using 2× SYBR Green qPCR Master Mix (Bimake.com, B21203) with specific primers and normalized to reference genes (Supplementary Table S1).
Cell viability assay
Human cancer cells (A549, MOLM13, THP1 and MV4-11) were seeded onto 96-well plates at a cell density of ∼1 × 104/well, and mESCs (WT and Dnmt1−/−) were seeded onto 6-well plates at a cell density of ∼5 × 105/well. The following day, 0.1% DMSO or GSK compounds at the indicated concentrations were added and incubated with the cells for the indicated periods of time. The viability was measured either by counting the cell numbers or by using the CellTiter-Blue Cell Viability Assay (Promega, G8080 or G9242).
Dot blot and southern blot DNA methylation assays
Genomic DNA was extracted using a Quick-DNA Miniprep kit (Zymo Research, #D3025). Dot blot assay was performed as described previously (43,46). In brief, ∼600 ng of each DNA sample was serially diluted 2-fold in TE buffer, and heat-denatured in 1M NaOH/25mM EDTA at 95°C for 10 min. After neutralizing with 2 M ammonium acetate (pH 7.0, on ice), DNA samples were loaded onto an Amersham Hybond-N+ membrane (GE Healthcare, RPN119B) using a Bio-Dot apparatus (Bio-Rad, #170-6545). After being cross-linked with UV for 1 min, the membrane was processed for Western blot (see above) with 5mC antibody (overnight at 4°C) and HRP-conjugated anti-rabbit IgG secondary antibody (1 h at room temperature).
Analysis of DNA methylation at the minor satellite repeats in mESCs was carried out as described previously (47). In brief, genomic DNA was digested with the methylation-sensitive restriction enzyme HpaII and analyzed by Southern hybridization with a specific biotin-labeled probe (Supplementary Table S1). Detection was performed using the North2South Chemiluminescent Hybridization and Detection Kit (Thermo Fisher Scientific).
Bisulfite pyrosequencing methylation analysis
One microgram of genomic DNA was treated with sodium bisulfite using the EZ DNA Methylation-Gold Kit (Zymo Research) according to the manufacturer's protocol. Samples were eluted in 40 μl of M-Elution Buffer, and 5 μl were used for each PCR reaction. Bisulfite conversion and subsequent pyrosequencing analysis were performed by the Epigenomics Profiling Core at The University of Texas MD Anderson Cancer Center.
PCR primers for pyrosequencing methylation analysis were designed using the Pyromark Assay Design SW 1.0 software (Qiagen) (Supplementary Table S1). Optimal annealing temperatures for each of these primers were tested using gradient PCR. Controls for high methylation (SssI-treated DNA), low methylation (WGA-amplified DNA) and no-DNA template were included in each reaction. PCR reactions were performed in a total volume of 20 μl, and the entire volume was used for each pyrosequencing reaction as described (48). Briefly, PCR product purification was done with streptavidin-sepharose high-performance beads (GE Healthcare), and co-denaturation of the biotinylated PCR products and sequencing primer (3.6 pmol/reaction). Sequencing was then performed on a PyroMark Q96 ID instrument with the PyroMark Gold Q96 Reagents (Qiagen). The degree of methylation for each individual CpG site was calculated using the PyroMark Q96 software. The average methylation of all sites within the sequence to analyze and replicate PCR reactions were reported for each sample.
Mass spectrometry (MS) analysis
DNA samples were digested to nucleosides using Nucleoside Digestion Mix (New England Biolabs, M0649S). LC-MS/MS analysis was performed by injecting digested DNAs on an Agilent 1290 Infinity II UHPLC equipped with a G7117A diode array detector and a 6495C triple quadrupole mass detector operating in the positive electrospray ionization mode (+ESI). UHPLC was carried out on a Waters XSelect HSS T3 XP column (2.1 × 100 mm, 2.5 μm) with a gradient mobile phase consisting of methanol and 10 mM aqueous ammonium acetate (pH 4.5). MS data acquisition was performed in the dynamic multiple reaction monitoring (DMRM) mode. Each nucleoside was identified in the extracted chromatogram associated with its specific MS/MS transition: dC [M+H]+ at m/z 228.1→112.1, 5mdC [M+H]+ at m/z 242.1→126.1, and dT [M+H]+ at m/z 243.1→127.1. External calibration curves with known amounts of the nucleosides were used to calculate their ratios within the analyzed samples.
RESULTS
GSK(1) targets DNMT1 for degradation in A549 cells
We first examined the effects of the DNMT1 inhibitor GSK-3484862 [GSK(1)] in A549 human lung adenocarcinoma cells. Treatment of A549 cells with either 2 μM or 4 μM GSK(1) for 24 h resulted in drastically reduced DNMT1 protein levels with little to no change in DNMT3A and DNMT3B protein levels (Figure 1B). Importantly, we found that DNMT1 mRNA levels were unchanged despite the loss of DNMT1 protein (Figure 1C), suggesting that the loss of DNMT1 was not due to a reduction in DNMT1 transcription. We noted that while DNMT3A appeared to be slightly reduced, subsequent experiments revealed no significant changes in DNMT3A level (See Supplementary Figure S1C and Figure 2E).
To determine whether GSK(1) affected the viability of A549 cells, we performed cell viability assays and found that the growth of A549 cells treated with GSK(1) was indistinguishable from control cells at 24 h, although growth was slightly impeded in the treated cells by 48 h (Figure 1D). However, DNMT1 protein levels had already decreased after only 3 h of treatment and were barely detectable after 12 h (Figure 1E). It seems interesting that GSK(1) not only inhibits DNMT1 activity in vitro with a half-maximal inhibitory concentration (IC50) of 0.23 μM (28), but also targets DNMT1 for degradation in cells—a dual mechanism for ridding cells of DNMT1 activity.
We extended the treatments in A549 cells to include GSK(2). In a 24 h treatment, we observed that GSK(1) was more potent than GSK(2) in inducing DNMT1 depletion at the same concentrations (Figure 1F). However, GSK(2) was more toxic than GSK(1) in luminescence-based cell viability assays (Figure 1G).
Next, we investigated the concentration-dependent effect of GSK(1) in a range of inhibitor concentrations (Figure 1H). Starting from 2 μM with a 5-fold dilution, after a 2-day treatment, DNMT1 protein level in A549 cells was significantly reduced at 80 nM and higher concentrations, but it remained unchanged at 16 nM and 3.2 nM (Figure 1H). At 80 nM of GSK(1), DNMT1 was detectably reduced at 12 h and severely diminished at 24 and 48 h (Figure 1I). In contrast, at 16 nM and 3.2 nM, the compounds failed to induce DNMT1 depletion even after prolonged—up to 6 days—treatment (Figure 1J). We thus conclude that 80 nM is the effective concentration for GSK(1) to induce DNMT1 protein degradation in A549 cells.
The observation of drastic DNMT1 degradation in A549 cells treated with GSK compounds was unexpected, because previous work showed that treatment with GSK-3685032 [GSK(2)] led to only a modest reduction of DNMT1 protein levels in GDM-1 myelomonoblastic leukemia cells following 2 days of treatment at the highest concentration examined (10 μM) (28). Thus, we repeated the same experiments in GDM-1 cells. While, after two days of treatment with either GSK(1) or GSK(2), DNMT1 levels in GDM-1 cells showed dose-dependent decreases, the depletion was obviously less severe compared to A549 cells treated with similar concentrations of the compounds (Supplementary Figure S1A and B).
GSK(1) induces DNA hypomethylation
Next, we tested whether the loss of DNMT1 affected global DNA methylation levels using dot blot assays probed with an anti-5mC antibody. We found that cells treated with GSK(1) displayed global hypomethylation at 24 h after treatment with GSK(1), but not detectable at 12 h (lane 7 of Figure 2A). To examine the effects of GSK(1) treatment on DNA methylation at specific genomic loci, we performed bisulfite pyrosequencing using the same genomic DNA samples as in our dot blot analysis. We examined DNA methylation of LINE-1 repetitive elements (49), the differentially methylated region (DMR) proximal to the imprinted H19 gene (50), and the Treg-specific demethylated region (TSDR) within intron 1 of FOXP3 gene (51–54). These sites were selected because of their known DNA methylation (partially or fully methylated) status in most cancer cell lines, and because LINE-1 elements have been used to quantitate demethylation in leukemia patients following decitabine treatment (55). For the DMSO-treated controls, DNA methylation for LINE-1 elements, FOXP3-TSDR and H19-DMR remained steady over time at ∼61%, ∼97% and ∼52%, respectively (Figure 2B). We note that the DMR region of H19, a paternally imprinted gene, is fully methylated on the paternal allele and unmethylated on the maternal allele. Indeed, the observed methylation level (52%) is consistent with the expected level (50%).
In contrast, GSK(1)-treated cells showed a progressive loss of DNA methylation compared to controls (∼50% by 48 h) (Figure 2B). We observed demethylation as early as 12 h in the pyrosequenced samples. For FOXP3, the most noticeable loss of methylation had already occurred by 12 h, decreasing only slightly more between 24–48 h.
Next, we determined the percentage of cytosines bearing the 5mC mark by MS using fully digested genomic DNA from the same DMSO and GSK(1)-treated cells. In DMSO-treated control cells, on average ∼3.5% of cytosines were 5mC (Figure 2C), whereas over time, GSK(1)-treated cells showed a reduction in total 5mC from 3.5% to 2.6%, 1.8% and 1.5% at 12, 24 and 48 h, respectively (Figure 2C). By 48 h, global DNA methylation had decreased to about 43% relative to the pretreatment levels (1.5% versus 3.5% 5mC). Overall, DNA methylation results from all the assays were concordant; however, dot blot analysis failed to detect 5mC loss at 12 h, likely due to its lower sensitivity.
GSK(1) induced DNMT1 degradation is proteasome-dependent and DNA synthesis dependent
DNMT1, which has a half-life of ∼12–14 h, is highly expressed in proliferating cells, with expression peaking during the S and G2 phases of the cell cycle (56). To explore possible mechanisms leading to GSK(1)-induced DNMT1 depletion, we treated A549 cells with GSK(1) in the absence and presence of the proteasome inhibitor MG132 for up to 24 h, and assessed DNMT1 protein levels. As expected, DNMT1 protein levels decreased and eventually disappeared in cells treated with GSK(1) (lanes 2–5 in Figure 2D); however, treatment with MG132 prevented GSK(1)-induced DNMT1 depletion (lanes 10–13 in Figure 2D), suggesting that loss of DNMT1 is proteasome dependent.
Nucleoside analogs induce DNMT depletion in a DNA synthesis-dependent manner (45). To determine whether the DNMT1 degradation induced by GSK(1) is also DNA synthesis dependent, we used aphidicolin, a small-molecule inhibitor of DNA polymerase alpha (57), which has been shown to markedly inhibit DNA synthesis and dramatically reduce incorporation of decitabine in HCT116 and SW620 cells (45). While A549 cells treated with either GSK(1) or decitabine (DAC) showed similar DNMT1 depletion (Figure 2E and G), prior treatment with aphidicolin (20 μg/ml) for 24 h prevented the effect of both compounds on DNMT1 degradation (Figure 2F and G). This finding is consistent with DNMT1 being a maintenance methyltransferase that acts on the newly synthesized daughter strand during DNA synthesis. Notably, DAC, in addition to affecting DNMT1, induced overt DNMT3A and DNMT3B depletion (28), whereas GSK(1) was specific for DNMT1 (Figure 2E).
GSK(1) and GSK(2) have similar effects in a wide range of cancer cells
Due to the unexpected reduction of DNMT1 protein levels in the GSK-treated A549 cells, we set out to compare the effects of GSK(1) and GSK(2) on DNMT1 levels in additional cancer cell lines derived from solid tumors: sarcoma-derived U2OS cells; breast cancer-derived, estrogen receptor positive MCF7 cells; and prostate cancer-derived, androgen-insensitive PC3 cells. Using the same condition that we treated A549 cells (4 μM for 24 h), we observed severe DNMT1 depletion in all three cell lines, comparable to that seen in A549 cells (Figure 3A).
Figure 3.

Effect of the GSK compounds in a range of cancer cells. (A) Western blot confirms decreased DNMT1 levels in four cell lines derived from solid tumors. (B) Cell viability (relative to DMSO treatment) after the myeloid leukemia cell lines MOLM13 (top panel) and THP1 (bottom panel) were treated with GSK(1) with concentrations starting from 50 μM (with 2x dilution until 48 nM) for three days (N = 3 independent experiments with quadruplicate; mean ± SD). (C) As in panel B but with GSK(2) with concentrations starting from 25 μM with 2x dilution until 24 nM. (D) Western blot showing decreased levels of DNMT1 in MOLM13 and THP1, treated with GSK(1) or GSK(2) at two different concentrations with duplications. (E–G) Treatment with GSK(1) at 4 μM for 1 day in the indicated myeloid leukemia cell lines showing cell viability (E), relative DNMT1 mRNA (F) and DNMT1 protein levels (G). DMSO samples are in blue dots and GSK(1)-treated samples are in red dots.
Next, we compared the effects of GSK(1) and GSK(2) on cell viability, DNMT1 protein, and DNMT1 mRNA levels in myeloid leukemia cell lines MOLM13, THP1, and MV4-11 cells. Treatment of MOLM13 and THP1 cells with GSK(1) (titrated from 50 μM to 48 nM) revealed no obvious effects on viability throughout a three-day time course (Figure 3B). In contrast, GSK(2) treatment led to ∼50% reduction in cell viability at the highest concentrations tested (titrated from 25 μM to 24 nM) (Figure 3C). These findings were consistent with previous data obtained from MV4-11 cells showing that GSK(2) has an enhanced anti-proliferative effect compared with GSK(1) at six days of treatment (28). At the protein level, we observed DNMT1 depletion regardless of compound in both MOLM13 and THP1 cells (Figure 3D). Finally, we observed that the reduction in DNMT1 protein levels (Figure 3G) after treatment with 4 μM GSK(1) was not at the transcriptional level, as the relative mRNA levels of DNMT1 showed no change in MV4-11 and MOLM13 cells, and may have been slightly increased in THP1-treated cells compared with controls (Figure 3F), while the cell viability was not affected (Figure 3E).
Effect of GSK(1) in mESCs
Unlike other mammalian cell types, which are sensitive to DNA methylation loss, mESCs do not require DNA methylation for survival and proliferation (4,44,58) and, thus, are often used as a cellular system for studying DNA methylation regulators. Similar to the cancer cell lines we tested, mESCs treated with GSK(1) at concentrations as low as 0.1 μM for as little as 24 h showed severe depletion of Dnmt1 (lowercase for mouse protein; Figure 4A). Over a 3-day treatment period, mESCs showed no changes in morphology, suggesting that Dnmt1 depletion was not the consequence of differentiation. Indeed, the pluripotency marker Oct4 was not altered in treated cells (Figure 4A).
Figure 4.

Effect of GSK(1) in mESCs. (A) Western blot showing GSK(1)-induced Dnmt1 depletion. (B) Southern blot after digestion of genomic DNA from cells in panel A with the methylation-sensitive restriction enzyme HpaII, which shows GSK(1)-induced hypomethylation of the minor satellite repeats. (C) RT-qPCR data showing no change in Dnmt1 mRNA after treatment with GSK(1). DMSO samples are in blue dots and GSK(1)-treated samples are in red dots. (D) Cell viability (relative to DMSO treatment) after WT and Dnmt1−/− mESCs were treated with GSK(1) (top panel) or GSK(2) (bottom panel) with concentrations ranging from 20 nM to 12.5 μM (1:5 serial dilutions starting at 12.5 μM) for three days (N = 3 independent experiments with triplicate; mean ± SD). (E) mESCs were treated with 2 μM of GSK(1) for 24 h and then cultured in the absence of the compound for 4 additional days. Western blot shows complete recovery of DNMT1 protein by 1 d after removal of the compound. (F) Southern blotting analysis of the minor satellite repeats in cells from panel E showing gradual recovery of DNA methylation after removal of the compound. The two blots were from the same membrane with different exposure times. (G) Mouse Dnmt1, with the conserved catalytic motifs (I-X) and the location of the PC:SF mutation being shown. (H) Western blotting analysis of Dnmt1−/−mESCs reconstituted with Myc-tagged Dnmt1 or the PC:SF mutant showing degradation of both WT and mutant Dnmt1 after GSK(1) treatment (2 μM for 24 h). Two stable clones for each construct were tested. (I) Southern blotting analysis of the minor satellite repeats in cells from panel H showing restoration of DNA methylation by WT Dnmt1 (lanes 4 and 5), but not by the mutant (lanes 8 and 9), and the effect of GSK(1) on DNA methylation.
Repetitive sequences such as the minor satellite repeats in centromeric regions are heavily methylated in mESCs (59), and their methylation status reflects global methylation. To determine whether depletion of Dnmt1 protein would lead to a loss of DNA methylation in mESCs, we performed Southern blot analysis of genomic DNA after digestion with the methylation-sensitive restriction enzyme HpaII. Our analysis revealed substantial hypomethylation of the minor satellite repeats in GSK(1)-treated cells (Figure 4B). RT-qPCR analysis confirmed that GSK(1)-induced Dnmt1 depletion in mESCs was not due to changes in Dnmt1 mRNA levels (Figure 4C).
We noticed that, compared to GSK(1), GSK(2) was less effective in inducing DNMT1 degradation but more toxic when tested in human cancer cell lines (Figure 1F, G, 3B and C). To assess the extent to which the observed toxicity was related to hypomethylation, we treated mESCs, which are insensitive to methylation loss, with GSK(1) or GSK(2) for three days at concentrations ranging from 20 nM to 12.5 μM. As shown in Figure 4D, both compounds exhibited dose-dependent effects on WT mESC viability, with GSK(2) being consistently more severe than GSK(1). Interestingly, Dnmt1−/− mESCs were resistant to both GSK(1) and GSK(2), except a slight effect of GSK(2) at 12.5 μM. These results suggest that both compounds have certain degrees of toxicity that is unrelated to hypomethylation but seems to be dependent on the presence of Dnmt1, their target.
Consistent with the finding that GSK(1) induces Dnmt1 protein degradation without affecting its mRNA level, removal of the compound resulted in complete recovery of Dnmt1 protein levels within 1 day (Figure 4E). Southern blot analysis of the minor satellite repeats also showed obvious recovery of DNA methylation after GSK(1) removal, although the recovery was not complete until after 4 days (Figure 4F). Gradual recovery of DNA methylation is expected, considering that Dnmt1 acts during DNA replication.
To determine whether Dnmt1 catalytic activity would be required for its degradation by GSK(1) treatment, we performed reconstitution experiments in Dnmt1−/− mESCs by stably expressing Myc-tagged mouse Dnmt1 or a catalytically inactive mutant (PC:SF) with the conserved proline-cysteine residues in the active center being substituted with serine and phenylalanine (Figure 4G). GSK(1) treatment resulted in depletion of both WT and mutant Dnmt1 (Figure 4H). As shown in Figure 4I, WT Dnmt1 restored DNA methylation, to a great extent, in Dnmt1−/−mESCs [compare lanes 4 and 5 with lane 1 (Dnmt1−/−) and lane 2 (WT)], whereas the PC:SF mutant did not (lanes 8 and 9), and that the recovered DNA methylation in the Myc-Dnmt1 stable clones was lost again after GSK(1) treatment (lanes 6 and 7). Thus, GSK(1) induces degradation of Dnmt1 protein, regardless of its catalytic activity. This observation is in agreement of in vitro structural work that the GSK inhibitor intercalates into DNMT1-bound DNA between two CpG base pairs, causing conformational movement of the DNMT1 active-site loop (28,38).
Uhrf1 deficiency prevents GSK(1)-induced Dnmt1 degradation in mESCs
Uhrf1, a multi-domain protein, is essential for the maintenance of DNA methylation patterns by directing Dnmt1 to newly replicated DNA (39,40). In addition to several epigenetic ‘reader’ domains (SRA, TTD and PHD) that recognize hemimethylated DNA and specific histone marks, respectively (60–63), Uhrf1 contains a RING domain with E3 ubiquitin ligase activity that mediates ubiquitination of several lysine residues on the N-terminal tail of histone H3, creating binding sites for Dnmt1 (64–68). Notably, Uhrf1 has also been implicated in Dnmt1 ubiquitination and degradation (42,69–71). Therefore, we asked whether Uhrf1 participated in GSK(1)-induced Dnmt1 degradation. Treatment of Uhrf1−/− mESCs with 2 μM of GSK(1) for 48 h resulted in no changes in Dnmt1 level, in contrast to the overt effect in WT mESCs (Figure 5A), indicating Uhrf1-dependent Dnmt1 degradation. The genome in Uhrf1−/− mESCs is already severely hypomethylated, and no further loss of methylation was detected following GSK(1) treatment (Figure 5B).
Figure 5.

Uhrf1 is required for GSK(1)-induced Dnmt1 degradation in mESCs. (A) Western blot showing that Dnmt1 is degraded in WT, but not Uhrf1−/−, mESCs after GSK(1) treatment (2 μM for 48 h). (B) Southern blotting analysis of the minor satellite repeats in cells from panel A showing that GSK(1) induced hypomethylation in WT mESCs but did not induce further loss of methylation in Uhrf1−/− mESCs. (C) Mouse Uhrf1, with the functional domains – ubiquitin-like (UBL) domain, tandem Tudor domain (TTD), plant homeodomain (PHD), SET- and RING-associated (SRA) domain, and Really Interesting New Gene (RING) domain – and the location of the H730A mutation being shown. (D) Western blotting analysis of Uhrf1−/-mESCs reconstituted with Flag-tagged Uhrf1 or the H730A mutant showing that GSK(1) treatment (2 μM for 24 h) resulted in partial Dnmt1 degradation in the Flag-Uhrf1 stable clone, but not in the Flag-H730A stable clone. (E) Western blot showing that Uhrf1 deficiency also prevented Dnmt1 depletion induced by decitabine treatment (1–5 μM for 48 h).
To assess the importance of Uhrf1′s E3 ubiquitin ligase activity in GSK(1)-induced Dnmt1 degradation, we treated Uhrf1−/− mESCs that had been reconstituted with Flag-tagged mouse Uhrf1 or a ‘E3 ligase-dead’ mutant (42), which harbors a point mutation, H730A, in the RING domain (Figure 5C). While re-expression of WT Uhrf1 in Uhrf1-deficient mESCs partially, but markedly, restored the sensitivity of Dnmt1 to GSK(1) treatment, re-expression of the H730A mutant did not (Figure 5D), indicating the involvement of the E3 ubiquitin ligase activity of Uhrf1 in Dnmt1 depletion. Although the level of Flag-Uhrf1 was similar to that of endogenous Uhrf1 in WT mESCs (Figure 5D), exogenously introduced Uhrf1 might not be able to fully recapitulate the regulation and functions of endogenous Uhrf1, which could explain the partial effect on Dnmt1 degradation.
We showed that, in A549 cells, GSK(1) and decitabine had similar effects on DNMT1 depletion, which depends on DNA synthesis (Figure 2E–G). To assess whether Uhrf1 deficiency would also affect decitabine-induced Dnmt1 depletion, we compared the sensitivity of Dnmt1 to decitabine treatment in WT and Uhrf1−/− mESCs. Unlike the results in GDM-1 cells, where GSK compounds seem to be less effective than decitabine in DNMT1 depletion (28), and in A549 cells, where GSK(1) and decitabine have similar effect (Figure 2E and G), GSK(1) appeared to be more effective than decitabine in inducing Dnmt1 depletion in mESCs, which express high levels of Dnmt1. An obvious decrease in Dnmt1 level in WT mESCs was not observed until 48 h of decitabine treatment at 5 μM (Figure 5E), in comparison with a more striking effect of GSK(1) on Dnmt1 level at lower concentrations (as low as 0.1 μM) for shorter time periods (as short as 24 h) of treatment (Figure 4A). In contrast, treatment of Uhrf1−/− mESCs with 5 μM of decitabine for 48 h showed little, if any, effect on Dnmt1 level (Figure 5E), suggesting the participation of Uhrf1 in Dnmt1 degradation induced by nucleoside analogs as well.
DISCUSSION
Here, we examined GSK(1), and to a lesser extent GSK(2), two examples of a distinct chemotype containing a planar dicyanopyridine core, and found that they are both potent and selective in inducing DNMT1 protein degradation, which in turn led to a loss of maintenance methylation at hemimethylated CpG sites. Though not initially expected, the effect of GSK(1) on DNMT1 protein stability was rapid, taking effect within hours after compound treatment. In the case of A549 cells, DNMT1 loss was followed by progressive loss of global methylation, consistent with DNA replication-dependent, passive demethylation. We noted that the depletion of DNMT1 protein, and the resultant reduced DNA methylation, was not initially associated with cell toxicity, suggesting that there are additional factors that ultimately determine the cellular response. This is in stark contrast to the currently used DNA demethylating agents (azacytidine and decitabine) which are incorporated into the genome and trap DNMTs through an irreversible nucleoprotein complex, leading to substantial genomic damage and cellular toxicity. Indeed, to minimize cytotoxicity, low doses of decitabine (or azacytidine) are more effective than higher doses in treating hematopoietic malignancies (72–76). Our comparative experiments show that GSK(1) and decitabine have both similarities and differences. They are similar in that their effects on DNMT1 degradation are proteasome- and DNA synthesis-dependent, involving the participation of UHRF1. They are different in that decitabine affects all DNMTs, whereas GSK(1) is specific for DNMT1.
Although we do not yet understand how GSK(1) initially directs DNMT1 for degradation, we showed that DNMT1 depletion is proteasome dependent and, at least in mESCs, requires Uhrf1 and its E3 ubiquitin ligase activity. We note that the dicyanopyridine-based series of DNMT1-selective compounds bind preferentially to the DNMT1-DNA complex but bind neither DNMT1 alone nor DNA nonspecifically (28). This observation indicates that these compounds have a mechanism of action that is distinct from those of traditional active-site inhibitors. One possibility that the long residency of genomic occupancy by the GSK compound-induced DNMT1 nucleoprotein complexes trigger the ubiquitin–proteasome pathway. Considering the previous finding that UHRF1 can ubiquitinate DNMT1 for degradation (69,70), it is tempting to speculate that the GSK compounds facilitate UHRF1-mediated DNMT1 ubiquitination. However, given that chromatin association of DNMT1 depends on UHRF1 and that the interaction of the GSK compounds with DNMT1 requires DNMT1 to be bound to DNA (28,38), it is also possible that Dnmt1 is unaffected by GSK(1) in Uhrf1−/− mESCs because it cannot be recruited to DNA.
DNMT1, a ∼200-kDa protein with multiple functional domains, is frequently overexpressed in cancer cells (77–80). Whereas the enzymatic activity of DNMT1 is important in maintaining the epigenetic DNA 5mC modification, the intact full-length DNMT1 protein may have additional, non-catalytic functions in gene repression (e.g. a scaffolding function) that could potentially be compromised upon protein depletion but not by enzymatic inhibitors. For example, at replication foci, DNMT1 interacts with multiple partners, including PCNA (81), UHRF1 (39,40), DNA ligase 1 (82) and protein (histone) lysine methyltransferases G9a/GLP (83), among others. These interactions may override DNMT1′s intrinsic enzymatic activity by maintaining DNMT1-mediated complex formation. It is conceivable that agents that lead to depletion of the entire DNMT1 molecule might be more effective than agents that inhibit DNMT1 enzymatic activity. Further studies are necessary to better understand the potential advantages and disadvantages of DNMT1 degraders and enzymatic inhibitors as cancer therapeutics.
We have shown that in cells, the dicyanopyridine-containing DNMT1-specific inhibitors/degraders GSK(1) and GSK(2) are stable (28), have relatively low toxicity, and their effects can be reversed. This makes them ideal for dissecting the coordinated events linking alterations of DNA methylation to changes in gene expression patterns in a tissue and cell-specific manner. However, the lack of cell-killing effect of these compounds, particularly GSK(1) in cancer-derived cell lines, suggests that GSK(1) might not be effective as a stand-alone cancer therapy, although GSK(2) displayed efficacy in a preclinical model of acute myeloid leukemia (28). In mESCs, we show that both GSK(1) and GSK(2) have certain degrees of toxicity that is unrelated to DNA hypomethylation, with GSK(2) being consistently more toxic than GSK(1), consistent with results from cancer cells. Interestingly, Dnmt1-deficient mESCs are almost completely resistant to the GSK compounds, raising the possible involvement of the compound-DNMT1-DNA ternary complex in mediating toxicity. A previous study showed that Dnmt1-deficient mESCs are also more resistant to decitabine treatment than WT mESCs (84). These results, together with the other similarities between GSK(1) and decitabine, as described above, suggest shared mechanisms in Dnmt1 degradation induced by dicyanopyridine-containing compounds and nucleoside analogs. Further studies of the GSK compounds will be required to understand their effects at the molecular and cellular level in vivo, which may reveal specific pathways that can be further exploited for combination therapies that will ultimately benefit patients.
DATA AVAILABILITY
The data underlying this article are available in the article and in its online supplementary material.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Yu Cao and Swanand Hardikar for technical assistance, and Kimie Kondo of MD Anderson Cancer Center's Epigenomics Profiling Core for performing pyrosequencing. We thank Dr. Briana Dennehey for editing the manuscript and insightful comments. The work was supported by the U.S. National Institutes of Health (R35GM134744 to X.C. and R01AI1214030A1 to T.C.), the Cancer Prevention and Research Institute of Texas (RR160029 to X.C.), and The University of Texas SPORE in Lung Cancer Enhancement Research (FP00018564 to X.C and T.C.). This research was also partly supported by funds from the Texas Tobacco Settlement – Molecular Mechanisms of Tobacco Carcinogenesis. M.A.S., T.C. and X.C. are CPRIT Scholars in Cancer Research.
Authors contributions: Q.C. performed experiments in A549, U2OS, MCF7 and PC3 cells. B.L. and Y.Z. performed experiments in mESCs. J.W.H. performed experiments in hematologic cells. N.D. and I.R.C. performed mass spectrometry. M.R.E. supervised bisulfite pyrosequencing. X.Z. supervised and contributed to discussions throughout the course of the work. M.A.S., T.C. and X.C conceptualized the experiments, organized and designed the scope of the study, drafted and edited the manuscript, and acquired funding. All authors were involved and contributed to preparing the manuscript.
Contributor Information
Qin Chen, Department of Epigenetics and Molecular Carcinogenesis, University of Texas MD Anderson Cancer Center, Houston, TX, 77030, USA.
Bigang Liu, Department of Epigenetics and Molecular Carcinogenesis, University of Texas MD Anderson Cancer Center, Houston, TX, 77030, USA.
Yang Zeng, Department of Epigenetics and Molecular Carcinogenesis, University of Texas MD Anderson Cancer Center, Houston, TX, 77030, USA; Program in Genetics and Epigenetics, The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences, Houston, TX, 77030, USA.
Jee Won Hwang, Department of Epigenetics and Molecular Carcinogenesis, University of Texas MD Anderson Cancer Center, Houston, TX, 77030, USA.
Nan Dai, New England Biolabs, Inc, Ipswich, MA 01938, USA.
Ivan R Corrêa, Jr., New England Biolabs, Inc, Ipswich, MA 01938, USA
Marcos R Estecio, Department of Epigenetics and Molecular Carcinogenesis, University of Texas MD Anderson Cancer Center, Houston, TX, 77030, USA.
Xing Zhang, Department of Epigenetics and Molecular Carcinogenesis, University of Texas MD Anderson Cancer Center, Houston, TX, 77030, USA.
Margarida A Santos, Department of Epigenetics and Molecular Carcinogenesis, University of Texas MD Anderson Cancer Center, Houston, TX, 77030, USA; Program in Genetics and Epigenetics, The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences, Houston, TX, 77030, USA.
Taiping Chen, Department of Epigenetics and Molecular Carcinogenesis, University of Texas MD Anderson Cancer Center, Houston, TX, 77030, USA; Program in Genetics and Epigenetics, The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences, Houston, TX, 77030, USA.
Xiaodong Cheng, Department of Epigenetics and Molecular Carcinogenesis, University of Texas MD Anderson Cancer Center, Houston, TX, 77030, USA; Program in Genetics and Epigenetics, The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences, Houston, TX, 77030, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Cancer Online.
FUNDING
National Institutes of Health [R35GM134744 and R01AI1214030A1]; Cancer Prevention and Research Institute of Texas [RR160029].
Conflict of interest statement. N.D. and I.R.C. are employees of New England Biolabs, Inc, a manufacturer and vendor of molecular biology reagents, including several enzymes and buffers used in this study. This affiliation does not affect the authors’ impartiality, adherence to journal standards and policies, or availability of data. The other authors declare no competing interests.
REFERENCES
- 1. Bestor T.H. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000; 9:2395–2402. [DOI] [PubMed] [Google Scholar]
- 2. Cheng X., Blumenthal R.M. Mammalian DNA methyltransferases: a structural perspective. Structure. 2008; 16:341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Okano M., Bell D.W., Haber D.A., Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999; 99:247–257. [DOI] [PubMed] [Google Scholar]
- 4. Li E., Bestor T.H., Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992; 69:915–926. [DOI] [PubMed] [Google Scholar]
- 5. Wang L.H., Wu C.F., Rajasekaran N., Shin Y.K. Loss of tumor suppressor gene function in Human cancer: an overview. Cell. Physiol. Biochem. 2018; 51:2647–2693. [DOI] [PubMed] [Google Scholar]
- 6. Jones P.A., Taylor S.M. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980; 20:85–93. [DOI] [PubMed] [Google Scholar]
- 7. Yoo C.B., Jones P.A. Epigenetic therapy of cancer: past, present and future. Nat. Rev. Drug Discov. 2006; 5:37–50. [DOI] [PubMed] [Google Scholar]
- 8. Flotho C., Claus R., Batz C., Schneider M., Sandrock I., Ihde S., Plass C., Niemeyer C.M., Lubbert M. The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia. 2009; 23:1019–1028. [DOI] [PubMed] [Google Scholar]
- 9. Silverman L.R., Demakos E.P., Peterson B.L., Kornblith A.B., Holland J.C., Odchimar-Reissig R., Stone R.M., Nelson D., Powell B.L., DeCastro C.M. et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J. Clin. Oncol. 2002; 20:2429–2440. [DOI] [PubMed] [Google Scholar]
- 10. Oki Y., Jelinek J., Shen L., Kantarjian H.M., Issa J.P. Induction of hypomethylation and molecular response after decitabine therapy in patients with chronic myelomonocytic leukemia. Blood. 2008; 111:2382–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fenaux P., Mufti G.J., Hellstrom-Lindberg E., Santini V., Finelli C., Giagounidis A., Schoch R., Gattermann N., Sanz G., List A. et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009; 10:223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lubbert M., Suciu S., Baila L., Ruter B.H., Platzbecker U., Giagounidis A., Selleslag D., Labar B., Germing U., Salih H.R. et al. Low-dose decitabine versus best supportive care in elderly patients with intermediate- or high-risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J. Clin. Oncol. 2011; 29:1987–1996. [DOI] [PubMed] [Google Scholar]
- 13. Santi D.V., Norment A., Garrett C.E. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc. Natl. Acad. Sci. U.S.A. 1984; 81:6993–6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sheikhnejad G., Brank A., Christman J.K., Goddard A., Alvarez E., Ford H. Jr, Marquez V.E., Marasco C.J., Sufrin J.R., O’Gara M. et al. Mechanism of inhibition of DNA (cytosine C5)-methyltransferases by oligodeoxyribonucleotides containing 5,6-dihydro-5-azacytosine. J. Mol. Biol. 1999; 285:2021–2034. [DOI] [PubMed] [Google Scholar]
- 15. Ganesan A., Arimondo P.B., Rots M.G., Jeronimo C., Berdasco M. The timeline of epigenetic drug discovery: from reality to dreams. Clin. Epigenetics. 2019; 11:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chiappinelli K.B., Strissel P.L., Desrichard A., Li H., Henke C., Akman B., Hein A., Rote N.S., Cope L.M., Snyder A. et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015; 162:974–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sato T., Issa J.J., Kropf P. DNA hypomethylating drugs in cancer therapy. Cold Spring Harb. Perspect. Med. 2017; 7:a026948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stomper J., Rotondo J.C., Greve G., Lubbert M. Hypomethylating agents (HMA) for the treatment of acute myeloid leukemia and myelodysplastic syndromes: mechanisms of resistance and novel HMA-based therapies. Leukemia. 2021; 35:1873–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brueckner B., Garcia Boy R., Siedlecki P., Musch T., Kliem H.C., Zielenkiewicz P., Suhai S., Wiessler M., Lyko F. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res. 2005; 65:6305–6311. [DOI] [PubMed] [Google Scholar]
- 20. Datta J., Ghoshal K., Denny W.A., Gamage S.A., Brooke D.G., Phiasivongsa P., Redkar S., Jacob S.T. A new class of quinoline-based DNA hypomethylating agents reactivates tumor suppressor genes by blocking DNA methyltransferase 1 activity and inducing its degradation. Cancer Res. 2009; 69:4277–4285. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21. Valente S., Liu Y., Schnekenburger M., Zwergel C., Cosconati S., Gros C., Tardugno M., Labella D., Florean C., Minden S. et al. Selective non-nucleoside inhibitors of human DNA methyltransferases active in cancer including in cancer stem cells. J. Med. Chem. 2014; 57:701–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gros C., Fleury L., Nahoum V., Faux C., Valente S., Labella D., Cantagrel F., Rilova E., Bouhlel M.A., David-Cordonnier M.H. et al. New insights on the mechanism of quinoline-based DNA methyltransferase inhibitors. J. Biol. Chem. 2015; 290:6293–6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Manara M.C., Valente S., Cristalli C., Nicoletti G., Landuzzi L., Zwergel C., Mazzone R., Stazi G., Arimondo P.B., Pasello M. et al. A quinoline-based DNA methyltransferase inhibitor as a possible adjuvant in osteosarcoma therapy. Mol. Cancer Ther. 2018; 17:1881–1892. [DOI] [PubMed] [Google Scholar]
- 24. Zwergel C., Schnekenburger M., Sarno F., Battistelli C., Manara M.C., Stazi G., Mazzone R., Fioravanti R., Gros C., Ausseil F. et al. Identification of a novel quinoline-based DNA demethylating compound highly potent in cancer cells. Clin. Epigenetics. 2019; 11:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rotili D., Tarantino D., Marrocco B., Gros C., Masson V., Poughon V., Ausseil F., Chang Y., Labella D., Cosconati S. et al. Properly substituted analogues of BIX-01294 lose inhibition of G9a histone methyltransferase and gain selective anti-DNA methyltransferase 3A activity. PLoS One. 2014; 9:e96941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Halby L., Menon Y., Rilova E., Pechalrieu D., Masson V., Faux C., Bouhlel M.A., David-Cordonnier M.H., Novosad N., Aussagues Y. et al. Rational design of bisubstrate-type analogues as inhibitors of DNA methyltransferases in cancer cells. J. Med. Chem. 2017; 60:4665–4679. [DOI] [PubMed] [Google Scholar]
- 27. Huang S., Stillson N.J., Sandoval J.E., Yung C., Reich N.O. A novel class of selective non-nucleoside inhibitors of human DNA methyltransferase 3A. Bioorg. Med. Chem. Lett. 2021; 40:127908. [DOI] [PubMed] [Google Scholar]
- 28. Pappalardi M.B., Keenan K., Cockerill M., Kellner W.A., Stowell A., Sherk C., Wong K., Pathuri S., Briand J., Steidel M. et al. Discovery of a first-in-class reversible DNMT1-selective inhibitor with improved tolerability and efficacy in acute myeloid leukemia. Nat. Cancer. 2021; 2:1002–1017. [PMC free article] [PubMed] [Google Scholar]
- 29. Dover G.J., Charache S.H., Boyer S.H., Talbot C.C. Jr, Smith K.D 5-Azacytidine increases fetal hemoglobin production in a patient with sickle cell disease. Prog. Clin. Biol. Res. 1983; 134:475–488. [PubMed] [Google Scholar]
- 30. Ley T.J., Anagnou N.P., Noguchi C.T., Schechter A.N., DeSimone J., Heller P., Nienhuis A.W. DNA methylation and globin gene expression in patients treated with 5-azacytidine. Prog. Clin. Biol. Res. 1983; 134:457–474. [PubMed] [Google Scholar]
- 31. Lowrey C.H., Nienhuis A.W. Brief report: treatment with azacitidine of patients with end-stage beta-thalassemia. N. Engl. J. Med. 1993; 329:845–848. [DOI] [PubMed] [Google Scholar]
- 32. Saunthararajah Y., Hillery C.A., Lavelle D., Molokie R., Dorn L., Bressler L., Gavazova S., Chen Y.H., Hoffman R., DeSimone J. Effects of 5-aza-2′-deoxycytidine on fetal hemoglobin levels, red cell adhesion, and hematopoietic differentiation in patients with sickle cell disease. Blood. 2003; 102:3865–3870. [DOI] [PubMed] [Google Scholar]
- 33. Akpan I., Banzon V., Ibanez V., Vaitkus K., DeSimone J., Lavelle D. Decitabine increases fetal hemoglobin in Papio anubis by increasing gamma-globin gene transcription. Exp. Hematol. 2010; 38:989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stomper J., Ihorst G., Suciu S., Sander P.N., Becker H., Wijermans P.W., Plass C., Weichenhan D., Bisse E., Claus R. et al. Fetal hemoglobin induction during decitabine treatment of elderly patients with high-risk myelodysplastic syndrome or acute myeloid leukemia: a potential dynamic biomarker of outcome. Haematologica. 2019; 104:59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gilmartin A.G., Groy A., Gore E.R., Atkins C., Long E.R., Montoute M.N., Wu Z., Halsey W., McNulty D.E., Ennulat D. et al. In vitro and in vivo induction of fetal hemoglobin with a reversible and selective DNMT1 inhibitor. Haematologica. 2021; 106:1979–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Haggerty C., Kretzmer H., Riemenschneider C., Kumar A.S., Mattei A.L., Bailly N., Gottfreund J., Giesselmann P., Weigert R., Brandl B. et al. Dnmt1 has de novo activity targeted to transposable elements. Nat. Struct. Mol. Biol. 2021; 28:594–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Azevedo Portilho N., Saini D., Hossain I., Sirois J., Moraes C., Pastor W.A. The DNMT1 inhibitor GSK-3484862 mediates global demethylation in murine embryonic stem cells. Epigenetics Chromatin. 2021; 14:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Horton J.R., Pathuri S., Wong K., Ren R., Rueda L., Fosbenner D.T., Heerding D.A., McCabe M.T., Pappalardi M.B., Zhang X. et al. Structural characterization of dicyanopyridine containing DNMT1-selective, non-nucleoside inhibitors. Structure. 2022; 30:793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bostick M., Kim J.K., Esteve P.O., Clark A., Pradhan S., Jacobsen S.E. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007; 317:1760–1764. [DOI] [PubMed] [Google Scholar]
- 40. Sharif J., Muto M., Takebayashi S., Suetake I., Iwamatsu A., Endo T.A., Shinga J., Mizutani-Koseki Y., Toyoda T., Okamura K. et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007; 450:908–912. [DOI] [PubMed] [Google Scholar]
- 41. Lei H., Oh S.P., Okano M., Juttermann R., Goss K.A., Jaenisch R., Li E. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development. 1996; 122:3195–3205. [DOI] [PubMed] [Google Scholar]
- 42. Dan J., Rousseau P., Hardikar S., Veland N., Wong J., Autexier C., Chen T. Zscan4 Inhibits maintenance DNA methylation to facilitate telomere elongation in mouse embryonic stem cells. Cell Rep. 2017; 20:1936–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zeng Y., Ren R., Kaur G., Hardikar S., Ying Z., Babcock L., Gupta E., Zhang X., Chen T., Cheng X. The inactive Dnmt3b3 isoform preferentially enhances Dnmt3b-mediated DNA methylation. Genes Dev. 2020; 34:1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen T., Ueda Y., Dodge J.E., Wang Z., Li E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell. Biol. 2003; 23:5594–5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Patel K., Dickson J., Din S., Macleod K., Jodrell D., Ramsahoye B. Targeting of 5-aza-2′-deoxycytidine residues by chromatin-associated DNMT1 induces proteasomal degradation of the free enzyme. Nucleic Acids Res. 2010; 38:4313–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yu D., Zhou J., Chen Q., Wu T., Blumenthal R.M., Zhang X., Cheng X. Enzymatic characterization of In vitro activity of RNA methyltransferase PCIF1 on DNA. Biochemistry. 2022; 61:1005–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Veland N., Lu Y., Hardikar S., Gaddis S., Zeng Y., Liu B., Estecio M.R., Takata Y., Lin K., Tomida M.W. et al. DNMT3L facilitates DNA methylation partly by maintaining DNMT3A stability in mouse embryonic stem cells. Nucleic Acids Res. 2019; 47:152–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Estecio M.R., Gharibyan V., Shen L., Ibrahim A.E., Doshi K., He R., Jelinek J., Yang A.S., Yan P.S., Huang T.H. et al. LINE-1 hypomethylation in cancer is highly variable and inversely correlated with microsatellite instability. PLoS One. 2007; 2:e399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chalitchagorn K., Shuangshoti S., Hourpai N., Kongruttanachok N., Tangkijvanich P., Thong-ngam D., Voravud N., Sriuranpong V., Mutirangura A. Distinctive pattern of LINE-1 methylation level in normal tissues and the association with carcinogenesis. Oncogene. 2004; 23:8841–8846. [DOI] [PubMed] [Google Scholar]
- 50. Jinno Y., Sengoku K., Nakao M., Tamate K., Miyamoto T., Matsuzaka T., Sutcliffe J.S., Anan T., Takuma N., Nishiwaki K. et al. Mouse/human sequence divergence in a region with a paternal-specific methylation imprint at the human H19 locus. Hum. Mol. Genet. 1996; 5:1155–1161. [DOI] [PubMed] [Google Scholar]
- 51. Baron U., Floess S., Wieczorek G., Baumann K., Grutzkau A., Dong J., Thiel A., Boeld T.J., Hoffmann P., Edinger M. et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur. J. Immunol. 2007; 37:2378–2389. [DOI] [PubMed] [Google Scholar]
- 52. Floess S., Freyer J., Siewert C., Baron U., Olek S., Polansky J., Schlawe K., Chang H.D., Bopp T., Schmitt E. et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007; 5:e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim H.P., Leonard W.J. CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J. Exp. Med. 2007; 204:1543–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Huehn J., Polansky J.K., Hamann A. Epigenetic control of FOXP3 expression: the key to a stable regulatory T-cell lineage?. Nat. Rev. Immunol. 2009; 9:83–89. [DOI] [PubMed] [Google Scholar]
- 55. Yang A.S., Doshi K.D., Choi S.W., Mason J.B., Mannari R.K., Gharybian V., Luna R., Rashid A., Shen L., Estecio M.R. et al. DNA methylation changes after 5-aza-2′-deoxycytidine therapy in patients with leukemia. Cancer Res. 2006; 66:5495–5503. [DOI] [PubMed] [Google Scholar]
- 56. Esteve P.O., Chin H.G., Benner J., Feehery G.R., Samaranayake M., Horwitz G.A., Jacobsen S.E., Pradhan S. Regulation of DNMT1 stability through SET7-mediated lysine methylation in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:5076–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pedrali-Noy G., Spadari S., Miller-Faures A., Miller A.O., Kruppa J., Koch G. Synchronization of HeLa cell cultures by inhibition of DNA polymerase alpha with aphidicolin. Nucleic Acids Res. 1980; 8:377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tsumura A., Hayakawa T., Kumaki Y., Takebayashi S., Sakaue M., Matsuoka C., Shimotohno K., Ishikawa F., Li E., Ueda H.R. et al. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells. 2006; 11:805–814. [DOI] [PubMed] [Google Scholar]
- 59. Chen T., Tsujimoto N., Li E. The PWWP domain of Dnmt3a and Dnmt3b is required for directing DNA methylation to the major satellite repeats at pericentric heterochromatin. Mol. Cell. Biol. 2004; 24:9048–9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hashimoto H., Horton J.R., Zhang X., Bostick M., Jacobsen S.E., Cheng X. The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature. 2008; 455:826–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rothbart S.B., Krajewski K., Nady N., Tempel W., Xue S., Badeaux A.I., Barsyte-Lovejoy D., Martinez J.Y., Bedford M.T., Fuchs S.M. et al. Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat. Struct. Mol. Biol. 2012; 19:1155–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu X., Gao Q., Li P., Zhao Q., Zhang J., Li J., Koseki H., Wong J. UHRF1 targets DNMT1 for DNA methylation through cooperative binding of hemi-methylated DNA and methylated H3K9. Nat. Commun. 2013; 4:1563. [DOI] [PubMed] [Google Scholar]
- 63. Rothbart S.B., Dickson B.M., Ong M.S., Krajewski K., Houliston S., Kireev D.B., Arrowsmith C.H., Strahl B.D. Multivalent histone engagement by the linked tandem Tudor and PHD domains of UHRF1 is required for the epigenetic inheritance of DNA methylation. Genes Dev. 2013; 27:1288–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nishiyama A., Yamaguchi L., Sharif J., Johmura Y., Kawamura T., Nakanishi K., Shimamura S., Arita K., Kodama T., Ishikawa F. et al. Uhrf1-dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature. 2013; 502:249–253. [DOI] [PubMed] [Google Scholar]
- 65. Qin W., Wolf P., Liu N., Link S., Smets M., La Mastra F., Forne I., Pichler G., Horl D., Fellinger K. et al. DNA methylation requires a DNMT1 ubiquitin interacting motif (UIM) and histone ubiquitination. Cell Res. 2015; 25:911–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ishiyama S., Nishiyama A., Saeki Y., Moritsugu K., Morimoto D., Yamaguchi L., Arai N., Matsumura R., Kawakami T., Mishima Y. et al. Structure of the Dnmt1 reader module complexed with a unique two-mono-ubiquitin mark on histone H3 reveals the basis for DNA methylation maintenance. Mol. Cell. 2017; 68:350–360. [DOI] [PubMed] [Google Scholar]
- 67. Harrison J.S., Cornett E.M., Goldfarb D., DaRosa P.A., Li Z.M., Yan F., Dickson B.M., Guo A.H., Cantu D.V., Kaustov L. et al. Hemi-methylated DNA regulates DNA methylation inheritance through allosteric activation of H3 ubiquitylation by UHRF1. Elife. 2016; 5:e17101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Vaughan R.M., Dickson B.M., Whelihan M.F., Johnstone A.L., Cornett E.M., Cheek M.A., Ausherman C.A., Cowles M.W., Sun Z.W., Rothbart S.B. Chromatin structure and its chemical modifications regulate the ubiquitin ligase substrate selectivity of UHRF1. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:8775–8780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Du Z., Song J., Wang Y., Zhao Y., Guda K., Yang S., Kao H.Y., Xu Y., Willis J., Markowitz S.D. et al. DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Sci. Signal. 2010; 3:ra80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Qin W., Leonhardt H., Spada F. Usp7 and Uhrf1 control ubiquitination and stability of the maintenance DNA methyltransferase Dnmt1. J. Cell. Biochem. 2011; 112:439–444. [DOI] [PubMed] [Google Scholar]
- 71. Mudbhary R., Hoshida Y., Chernyavskaya Y., Jacob V., Villanueva A., Fiel M.I., Chen X., Kojima K., Thung S., Bronson R.T. et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell. 2014; 25:196–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Issa J.P., Garcia-Manero G., Giles F.J., Mannari R., Thomas D., Faderl S., Bayar E., Lyons J., Rosenfeld C.S., Cortes J. et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in hematopoietic malignancies. Blood. 2004; 103:1635–1640. [DOI] [PubMed] [Google Scholar]
- 73. Kantarjian H., Oki Y., Garcia-Manero G., Huang X., O’Brien S., Cortes J., Faderl S., Bueso-Ramos C., Ravandi F., Estrov Z. et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood. 2007; 109:52–57. [DOI] [PubMed] [Google Scholar]
- 74. Saunthararajah Y., Sekeres M., Advani A., Mahfouz R., Durkin L., Radivoyevitch T., Englehaupt R., Juersivich J., Cooper K., Husseinzadeh H. et al. Evaluation of noncytotoxic DNMT1-depleting therapy in patients with myelodysplastic syndromes. J. Clin. Invest. 2015; 125:1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Duchmann M., Itzykson R. Clinical update on hypomethylating agents. Int. J. Hematol. 2019; 110:161–169. [DOI] [PubMed] [Google Scholar]
- 76. Ueda M., El-Jurdi N., Cooper B., Caimi P., Baer L., Kolk M., Brister L., Wald D.N., Otegbeye F., Lazarus H.M. et al. Low-dose azacitidine with DNMT1 level monitoring to treat post-transplantation acute myelogenous leukemia or myelodysplastic syndrome relapse. Biol. Blood Marrow Transplant. 2019; 25:1122–1127. [DOI] [PubMed] [Google Scholar]
- 77. Wong K.K. DNMT1 as a therapeutic target in pancreatic cancer: mechanisms and clinical implications. Cell Oncol. (Dordr.). 2020; 43:779–792. [DOI] [PubMed] [Google Scholar]
- 78. Wong K.K. DNMT1: a key drug target in triple-negative breast cancer. Semin. Cancer Biol. 2021; 72:198–213. [DOI] [PubMed] [Google Scholar]
- 79. Zhu A., Hopkins K.M., Friedman R.A., Bernstock J.D., Broustas C.G., Lieberman H.B. DNMT1 and DNMT3B regulate tumorigenicity of human prostate cancer cells by controlling RAD9 expression through targeted methylation. Carcinogenesis. 2021; 42:220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mohan K.N. DNMT1: catalytic and non-catalytic roles in different biological processes. Epigenomics. 2022; 14:629–643. [DOI] [PubMed] [Google Scholar]
- 81. Chuang L.S., Ian H.I., Koh T.W., Ng H.H., Xu G., Li B.F. Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science. 1997; 277:1996–2000. [DOI] [PubMed] [Google Scholar]
- 82. Ferry L., Fournier A., Tsusaka T., Adelmant G., Shimazu T., Matano S., Kirsh O., Amouroux R., Dohmae N., Suzuki T. et al. Methylation of DNA ligase 1 by G9a/GLP recruits UHRF1 to replicating DNA and regulates DNA methylation. Mol. Cell. 2017; 67:550–565. [DOI] [PubMed] [Google Scholar]
- 83. Esteve P.O., Chin H.G., Smallwood A., Feehery G.R., Gangisetty O., Karpf A.R., Carey M.F., Pradhan S. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 2006; 20:3089–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Juttermann R., Li E., Jaenisch R. Toxicity of 5-aza-2′-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:11797–11801. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material.