

Abstract
The capture of tumour‐derived extracellular vesicles (TEVs) by cells in the tumour microenvironment (TME) contributes to metastasis and notably to the formation of the pre‐metastatic niche (PMN). However, due to the challenges associated with modelling release of small EVs in vivo, the kinetics of PMN formation in response to endogenously released TEVs have not been examined. Here, we have studied the endogenous release of TEVs in mice orthotopically implanted with metastatic human melanoma (MEL) and neuroblastoma (NB) cells releasing GFP‐tagged EVs (GFTEVs) and their capture by host cells to demonstrate the active contribution of TEVs to metastasis. Human GFTEVs captured by mouse macrophages in vitro resulted in transfer of GFP vesicles and the human exosomal miR‐1246. Mice orthotopically implanted with MEL or NB cells showed the presence of TEVs in the blood between 5 and 28 days after implantation. Moreover, kinetic analysis of TEV capture by resident cells relative to the arrival and outgrowth of TEV‐producing tumour cells in metastatic organs demonstrated that the capture of TEVs by lung and liver cells precedes the homing of metastatic tumour cells, consistent with the critical roles of TEVs in PMN formation. Importantly, TEV capture at future sites of metastasis was associated with the transfer of miR‐1246 to lung macrophages, liver macrophages, and stellate cells. This is the first demonstration that the capture of endogenously released TEVs is organotropic as demonstrated by the presence of TEV‐capturing cells only in metastatic organs and their absence in non‐metastatic organs. The capture of TEVs in the PMN induced dynamic changes in inflammatory gene expression which evolved to a pro‐tumorigenic reaction as the niche progressed to the metastatic state. Thus, our work describes a novel approach to TEV tracking in vivo that provides additional insights into their role in the earliest stages of metastatic progression.
Keywords: exosomes, extracellular vesicles, inflammation, metastasis, microRNA, pre‐metastatic niche, tumour microenvironment
1. INTRODUCTION
Metastasis is a non‐random multistep process and the major cause of cancer‐related deaths (Ganesh & Massagué, 2021). It has been difficult to predict and when discovered it is–unless isolated–always associated with an unfavourable prognosis and a lack of therapeutic response (Anderson et al., 2019; Steeg & Theodorescu, 2008). It involves close and complex interactions between tumour cells (the seeds) and host cells in the metastatic organs (the soil) (Fidler, 2003; Peinado et al., 2017). Our understanding of metastasis has significantly improved over the years in particular with the evidence that factors secreted by the primary tumour can induce the formation of a supportive microenvironment in metastatic organs prior to their homing, a process known as pre‐metastatic niche (PMN) formation (Kaplan et al., 2005; Liu & Cao, 2016).
Extracellular vesicles (EVs) released by all living cells have multiple functions in intercellular communication in many physiological and pathological processes (Malloci et al., 2019; Tkach et al., 2018; Yanez‐Mo et al., 2015), including innate and acquired immunity (Buzas, 2022; Cioffi et al., 2021), and cancer progression (Chang et al., 2021). EVs represent a heterogeneous group of vesicles that includes large apoptotic bodies (1 μm), large EVs (>200 nm), small EVs (200–50 nm), vesicle‐like particles exomeres (20 nm) and supermeres (Zhang et al., 2021) that contain lipids, proteins and nucleic acids, including non‐coding regulatory microRNA (miR) (Tkach & Thery, 2016). Cancer cells release EVs (tumour‐derived EVs or TEVs) which when captured by stromal cells in the tumour microenvironment (TME) promote angiogenesis, inflammation, tumour cell survival, drug resistance, and immune escape (Azmi et al., 2013; Becker et al., 2016; Ludwig & Whiteside, 2018; Mashouri et al., 2019). TEVs have a specific integrin‐based organotropism that reflects the predominant metastatic sites of the tumour they derive from (Costa‐Silva et al., 2015; Hoshino et al., 2015) and is the basis for their postulated contribution to the PMN (Peinado et al., 2012). The mechanisms by which TEVs contribute to the PMN are however incompletely understood and hampered by technological limitations. Studies have pointed to a role in increasing immune surveillance by patrolling monocytes (Plebanek et al., 2017), inducing vascular permeability (Zeng et al., 2018), and activating endothelial cells toward inflammation (Keklikoglou et al., 2019), all contributing to metastasis. The capture of TEVs by myeloid cells in the bone marrow induces metabolic changes (Henrich et al., 2020), promotes osteoclast activation and osteolytic metastasis (Yuan et al., 2021), activates cancer‐associated fibroblasts (CAF) (Du et al., 2020; Wu et al., 2020), and polarizes macrophages (Shao et al., 2018; Zhao et al., 2020).
Thus far, most studies have used the exogenous in vitro or in vivo (intravenous, intracardiac or intratumoral) administration of EVs purified from cells in culture by a variety of techniques and tagged with reagents such as lipophilic dyes, membrane dyes, and luminescent markers that are the subject of significant artifacts (Choi & Lee, 2016; Costa‐Silva et al., 2015; Hoshino et al., 2015; Witwer et al., 2013; Stenovec et al., 2018; Zeng et al., 2018). Few studies have described models of endogenous EV release using innovative labelling strategies and reporter systems (Verweij et al., 2019) to identify tumour cells taking up EVs in vivo (Androuin et al., 2021; Verweij et al., 2021; Zomer et al., 2015), but no studies have reported the role of endogenous TEVs in the PMN. Therefore, we set out to track, for the first time, endogenous TEV release and capture throughout metastatic progression, from the earliest, pre‐metastatic phase to macro metastasis.
Here, combining in vivo murine models to track the endogenous release of TEVs tagged with a green fluorescent protein (GFTEVs) with a gene expression analysis of TEV‐capturing cells at distant metastatic sites, we provide a comprehensive temporal and molecular analysis of TEV contributions to the pre‐metastatic and metastatic niches. To our knowledge, this is the first report demonstrating the effect of endogenously released TEVs on the evolution of the PMN to the metastatic niche (MN).
2. MATERIAL AND METHODS
2.1. Reagents
All antibodies used in this study are listed in Table S1.
2.2. Cells
Luciferase‐expressing human NB cell lines CHLA‐136‐Luc, and SK‐N‐BE(2)‐Luc were obtained from Dr. C.P. Reynolds (Texas Tech University, Lubbock, TX) and Dr. R. Seeger (Children's Hospital Los Angeles, Los Angeles, CA). We have generated a liver‐tropic variant of the SK‐N‐BE(2) parental cell line by five successive rounds of tail vein injection, followed by liver tumour harvest, in vitro expansion, and re‐injection. When indicated, these cells were used in the study for their preferred tropism to the liver and referred to as SK‐N‐BE(2)Liv. M24met human melanoma cells were obtained from Dr. R. Reisfeld (The Scripps Research Institute, La Jolla, CA). The murine macrophage cell line RAW 264.7 was purchased from ATCC. Cell line authentication was done by genotype analysis using AmpFISTR® Identifier® PCR Amplification Kit and Gene Mapper ID v3.2 (Applied Biosystems). Cells were cultured at 37°C and 5% CO2 in Iscove's Modified Dulbecco's Medium (IMDM) supplemented with 10% (v/v) foetal bovine serum (FBS), L‐glutamine (2 mM), penicillin (100 units/mL) and streptomycin (0.1 mg/mL) (all from Omega Scientific). Cultures were periodically assessed for the absence Mycoplasma using MycoAlert™ PLUS Mycoplasma Detection Kit (Lonza). Cancer cells were cultured for in vivo orthotopic implantations and for in vitro co‐culture with RAW 264.7 cells. Contact co‐cultures were performed at 1:1 ratio for 3 days in 8‐well slide chambers (Tissue‐Tek) for imaging or in 10 cm dishes for FACS sorting. No‐contact co‐cultures were performed in 12‐well or 6‐well plates at 1:1 ratio (2 × 105 or 5 × 105 cells respectively) for 3–7 days with RAW 264.7 cells in the bottom well and cancer cells in the Transwell insert (0.4 μm pores).
2.3. Transduction
MEL and NB cells were plated in a 24‐well plate at 75,000 cells per well and cultured for 24 h or up to 70%–80% confluency. Cell growth medium was replaced with culture medium containing TransDux transduction reagent (System Biosciences), and cells were transduced with XPack CMV‐XP‐GFP‐EF1‐Puro lentivirus at 1:100 dilution from the commercial stock of 2.378 infection units/mL (System Biosciences) according to manufacturer instructions. Transduction medium was replaced with complete growth medium after 72 h and cells were grown for an additional 48 h before puromycin selection (2 μg/mL). Transduction efficiency was estimated at 70%–80% by fluorescence microscopy and GFP+ transfectants were sorted by flow cytometry.
2.4. Preparation and characterization of TEVs
TEVs were prepared and characterized following the recommendations of the International Society of Extracellular Vesicles, Minimal Information for the Studies of Extracellular Vesicles (Théry et al., 2018). EVs were obtained by Differential UltraCentrifugation (DUC), by OptiPrep (Sigma‐Aldrich) Density Gradient Centrifugation (ODGC), or by Size Exclusion Chromatography (SEC). Cells were maintained in complete growth medium containing 10% FBS until they reached 70%–80% confluency then rinsed three times in PBS to eliminate bovine EVs and proteins and incubated in serum‐free medium for one to two days until TEV isolation. For DUC, 24–48 h serum‐free conditioned medium (CM) was centrifuged at 2000 × g (2 K) for 5 min to remove floating cells and apoptotic bodies. The supernatant was collected and centrifuged at 10,000 × g (10 K) (Eppendorf 5810 R centrifuge with F‐34‐6‐38 rotor) for 30 min at 4°C to eliminate microvesicles and further ultra‐centrifuged at 100,000 × g (100 K) (SW32Ti rotor, Optima XE‐90 ultracentrifuge, Beckman) for 3 h at 4°C to collect smaller vesicles. The pellet was resuspended in 10.5 mL of PBS, ultra‐centrifuged at 100,000 × g for 1 h, resuspended in 25−50 μL in PBS and stored at −20°C prior to testing. For ODGC, after centrifugation at 100,000 × g, the CM was concentrated on a 100 K membrane cutoff concentrator, loaded on the top of a OptiPrep gradient (2.5 mL of 5% (w/v), 3.0 mL of 20% and 4.8 mL of 30% in PBS), and separated by ultra‐centrifugation at 100,000 × g for 16 h at 4°C in a SW40Ti rotor. Individual fractions of 1 mL were collected from top to bottom, diluted in 10 mL of PBS and re‐centrifuged at 100,000 × g for 2 h at 4°C. The pellets were resuspended in 25–50 μL of PBS and stored at −20°C before being analysed. SEC was performed with Sephacryl S‐300 High Resolution (GE Healthcare) packed on a glass chromatography column (BioRad) (10 cm height, 0.5 cm diameter). The column was equilibrated with PBS/0.32% sodium citrate and the sample was loaded onto the column and allowed to enter the resin by gravity flow. The eluate was collected in 500 μL fractions on a Model 2110 fraction collector (BioRad). The presence of EVs in the void volume was evaluated by Nanoparticle Tracking Analysis (NTA) and EV‐containing fractions were pooled and concentrated through an Amicon Ultra‐2 centrifugal filter (membrane cutoff 100 KD; Millipore) to 50 μL. Aliquots of TEVs isolated from CM of M24met XPack‐GFP and SK‐N‐BE(2) XPack‐GFP were also treated with RNase A (4 μg/mL ThermoFisher) for 1 h at 37°C before qRT‐PCR to differentiate miR‐1246 present inside versus outside TEVs.
2.5. Nanoparticle tracking analysis
NTA was done using a NanoSight NS300 instrument version 2.3 (Malvern Instruments). For data acquisition, samples were diluted in PBS to a particle concentration between 2 × 108 and 1 × 109 particles/mL and examined at a temperature of 20–25°C with a camera level of 13 and a gain of 250–300 over 30 or 60 s in triplicate. A detection threshold of three was used and Blur/track length/expected size were on AUTO.
2.6. Transmission electron microscopy (TEM)
The EV pellets obtained by DUC followed by ODGC were fixed with 2% (w/v) glutaraldehyde, post‐fixed with 1% (w/v) osmium tetroxide, dehydrated, and embedded in Eponate 12 Resin. Ultra‐thin sections of the samples were cut, placed on carbon coated grids and stained in 5% (w/v) uranyl acetate solution and 3% (w/v) lead solution (all reagents from Ted Pella Inc.). Samples were examined using a Morgagni 268 transmission electron microscope (Field Emission Inc.) and digital images of the samples were recorded.
2.7. Western blot analysis
Total EV protein determination was performed using a Bradford assay. Five micrograms of EV proteins were separated by sodium dodecyl sulphate (SDS) polyacrylamide gel electrophoresis (PAGE) under reducing conditions, at the exception of samples used to detect CD63, using 4%–15% (w/v) gradient Tris‐Glycine gels (TGX™, BioRad) and transferred onto cellulose membranes (BioRad). Membranes were blocked for 1 h in LI‐COR blocking buffer then incubated with primary antibodies against exosomal and non‐exosomal protein markers (Table S1). Proteins were detected with near‐infrared conjugated secondary antibodies and an Odyssey Imager equipped with Image Studio V3.1 software (LI‐COR Biosciences).
2.8. NanoString miR screening
EVs were isolated from cancer cell lines and mouse bone marrow by SEC. RNA was extracted from EVs and analysed through nCounter® miR expression panels screening for 800 miRs (NanoString). Rosalind and nSolver platforms were used to analyse nCounter data sets to identify miRs specific to TEVs. Volcano plots were generated by comparing the miR expression in human TEVs versus mouse myeloid cell EVs.
2.9. Animal experiments
Eight to twelve‐week‐old male and female immunodeficient NOD SCID Gamma (NSG) mice were orthotopically implanted with M24met XPack‐GFP or with XPack‐GFP NB cell lines following a protocol approved by the Institutional Animal Care Utilization Committee at the Saban Research Institute of Children's Hospital Los Angeles (Protocol #41‐20 approved on October 25, 2020).
Mice were anesthetized using 3%–5% inhaled isoflurane. For the MEL model, mice were injected subcutaneously with 1.0 × 106 M24met XPack‐GFP in 100 μL of PBS with a ½ mL U‐100 insulin syringe and a 28G1/2 needle. For the NB model, an incision was made through the skin and the musculature, and the left kidney was exposed. The adrenal fat pad or the subcapsular space was injected with 1.5 × 106 SK‐N‐BE(2)‐Luc XPack‐GFP or CHLA‐136‐Luc XPack‐GFP cells resuspended in 25–30 μL of Matrigel (BD Bioscience). The kidney was then repositioned in the retroperitoneal space. The flank musculature incision was closed with a single 5‐0 absorbable suture and the skin incision was closed with two staples. Following tumour implantation, mice were monitored once a week using bioluminescence imaging (Xenogen, Caliper Life Science) to measure luciferase intensity and tumour growth. Mice were sacrificed at specific time points and organs harvested as indicated below. The number of mice sacrificed at each time point varied with the type of experiments and is indicated in the legends to the figures. For each time point, one non‐tumour‐bearing mouse (littermate) was used as negative control.
Bone marrow and organs including liver, lung, kidney and brain were harvested and processed for flow cytometry and/or image analysis. Bone marrow was flushed out of the femur and tibia with PBS using a 1 mL U‐100 insulin syringe with 28G1/2 needle. Organs were embedded in Surgipath FCS 22 compound (Leica Biosystems) for cryosection and/or subjected to enzymatic digestion to obtain cell suspensions. Briefly, tissues were minced with a scalpel and placed in gentleMACS C tubes (Miltenyi Biotech) containing appropriate buffers and enzymes provided in the tissue dissociation kits (Miltenyi Biotech). The C tubes were placed onto the gentleMACS Octo Dissociator to shear the tissue at 37°C using the tissue appropriate programs recommended by the manufacturer. Samples were then passed through 70 μm cell strainers, centrifuged at 300 x g for 10 min and resuspended in PBS for immunofluorescence or flow cytometry.
2.10. Murine plasma EV isolation and qPCR
Upon sacrifice, mouse blood was collected in heparinized tubes, and EVs were isolated from the plasma and analysed for presence of miR. Plasma (50–400 μL) was clarified by a 10,000 × g centrifugation step at 4°C to pellet larger and denser microvesicles. EVs were then isolated by SEC for characterization and RNA extraction. EV‐RNA was extracted using a plasma/serum RNA purification Mini Kit (Norgen Biotek Corp) following the manufacturer's protocol. For qRT‐PCR, equal amounts (20 ng) of EV‐RNA were reverse transcribed after addition of the cel‐miR‐39 spike‐in miRNA using TaqMan miRNA Reverse Transcription kit (ThermoFisher). Quantitative real‐time PCR (qPCR) of the cDNA was performed in a CFX96 Touch Real‐Time PCR Detection System (Bio‐Rad) with TaqMan Fast Advanced Master Mix and addition of commercially available (TaqMan microRNA assay) primer and probe mixes to detect miR‐1246 according to the company's instructions. The qPCR data was analysed by the 2−ΔCt method and the expression of the spike‐in RNA cel‐miR‐39 was used to normalize the qPCR results across samples. Plasma obtained from non‐tumour‐bearing mice at day 0 before implantation were used as controls and reference values and the qPCR results obtained for tumour‐bearing mice at ulterior time points were expressed as fold increase.
2.11. ExoView imaging
Single vesicle analysis was performed using ExoView R100 and the ExoView Human Tetraspanin Kit (NanoView Biosciences). Plasma EVs isolated by SEC were incubated for 16 h at room temperature on ExoView Tetraspanin microarray chips coated with antibodies for the tetraspanins CD9, CD81 and CD63, as well as IgG negative control. After capture, the chips were washed three times with the provided solution and incubated for 1 h in the dark with an antibody mixture containing fluorescently labelled anti‐CD9, anti‐CD81, and anti‐CD63 provided in the kit. The chips were then washed with the provide wash buffer, dried, and scanned with the ExoView R100 reader using the ExoView Scanner 3.0 acquisition software. The data were analysed using ExoView Analyser 3.0.
2.12. Immunofluorescence
Suspensions of liver, and lung cells were plated in 8‐well slide chambers (Tissue‐Tek) and incubated overnight for adhesion. Cells were then fixed with 4% paraformaldehyde (w/v) in PBS for 5 min, rinsed in PBS, and blocked with 15% (v/v) FBS in PBS for 10 min. Cells were incubated overnight at 4°C with primary antibodies against mouse F4/80, desmin, or albumin (Table S1). Slides were washed three times for 5 min with 0.1% (v/v) tween in PBS and incubated with fluorochrome‐conjugated secondary antibodies for 45 min at room temperature. Slides were then washed in PBS‐tween as above and mounted with Vectashield mounting medium containing 4,6‐diamino‐2‐phenylindole (DAPI) (Vector Laboratories). Immunofluorescence analysis on frozen tissues was done on 10 μm sections fixed 5 min in 4% (w/v) paraformaldehyde and rinsed in PBS. Negative controls included tissue sections incubated without primary antibodies as well as tissue sections from non‐tumour‐bearing littermate control mice. Multiple sections were obtained through the depth of the organs for 3–4 mice for each cancer model. Quantitative analyses were done with ImageJ on 40× images, with 1–2 images per section for a total of 21 and 13 images analysed for MEL and NB models respectively. Fluorescence images were acquired on a DMI6000B microscope equipped with 20×/0.7 HC PLAN APO Ph2 and 40×/1.25 oil HCX PL APO CS lenses (Leica Microsystems, Buffalo Grove, IL) and an ORCA‐Flash4.0 LT camera (Hamamatsu Corporation, Bridgewater, NJ). The system was controlled by Leica LAS X 3.6.0 software.
2.13. Flow cytometry
Cells from contact and Transwell co‐cultures were harvested with dissociation buffer (Gibco), centrifuged, resuspended in 100 μL of PBS containing 1% FBS (v/v) (staining buffer) and incubated with fluorochrome‐conjugated primary antibodies against mouse F4/80 and human CD298 or resuspended in 100 μL of Annexin binding buffer and incubated with fluorochrome‐conjugated Annexin V (BioLegend). Cell suspensions obtained from organ dissociation were subjected to red cell haemolysis by the addition of Lysis Buffer (BD Biosciences), washed in staining buffer, centrifuged at 300 × g for 5 min, resuspended in 100 μL of staining buffer, and incubated with fluorochrome‐conjugated antibodies against mouse F4/80, mouse albumin, mouse desmin, human GD2, and human CD298 (Table S1) for 1 h in the dark at 4°C. Following the incubation, samples were washed in staining buffer, strained through a 40 μm nylon filter, and stained with DAPI. Additional flow analyses were performed on different liver cell subpopulations. Briefly, after enzymatic digestion as above, liver cell suspensions were filtered through a 70 μm nylon strainer and centrifuged 2 min at 30 × g. The pellet was collected, and the supernatant was further centrifuged for an additional 2 min at 30 × g. The pellet was collected, mixed with the first pellet, and considered enriched in liver parenchymal cells (hepatocytes, detected by flow with an anti‐albumin antibody). The supernatant was collected and centrifuged for an additional 10 min at 775 × g. The pellet was resuspended in PBS, collected, and considered enriched in non‐parenchymal cells (Kupffer cells detected with an antibody against F4/80, and stellate cells detected with an antibody against desmin). Data acquisition was done on a LSRII flow cytometer and a FACSymphony S6 Cell Sorter (BD Biosciences). A reading <10−4 events was considered below the limit of the sensitivity of the instrument. Negative controls included cells stained with fluorochrome‐conjugated isotype antibodies for each sample and cells isolated from non‐tumour‐bearing control mice.
2.14. TaqMan microarrays and qPCR on sorted cells
Sorted cells were collected in TRIzol LS (Sigma) and RNA was isolated using a standard protocol of chloroform extraction followed by a column purification (ThermoFisher). An aliquot of RNA was kept for qPCR miR detection and cDNA synthesis was performed using SuperScript IV VILO Master Mix (ThermoFisher) for the gene expression analysis.
Gene expression analysis was done using TaqMan Low Density Array cards for a mouse immune panel (ThermoFisher) following the provided protocol. For miR analysis, reverse transcription of the RNA was performed with the TaqMan miRNA Reverse Transcription kit and the cDNA was subsequently pre‐amplified with TaqMan PreAmp Master Mix and addition of commercially available (TaqMan microRNA assay) primer and probe mixes to detect miR‐1246 following manufacturer's instructions (ThermoFisher). Quantitative real‐time PCR was performed in a CFX96 Touch Real‐Time PCR Detection System (Bio‐Rad) with TaqMan Fast Advanced Master Mix (ThermoFisher) and analysed by the 2−ΔCt method. Amplification of the human and mouse bi‐specific small nuclear RNA U6 was used to normalize qPCR results across samples.
2.15. Integrin screening by mass spectrometry
EV samples (5 μg) were dried by vacuum centrifugation and re‐dissolved in 30–50 μL 8 M Urea/50 mM ammonium bicarbonate/10 mM DTT. Following lysis and reduction, proteins were alkylated using 20 or 30 mM iodoacetamide (Sigma). Proteins were digested with Endopeptidase Lys C (Wako) in <4 M urea followed by trypsination (Promega) in <2 M Urea. Peptides were desalted and concentrated using Empore C18‐based solid phase extraction prior to analysis by high resolution/high mass accuracy reversed phase (C18) nano‐LC‐MS/MS. Peptides were separated on a C18 column (12 cm/75 μm, 3 μm beads, Nikkyo Technologies) at 200 or 300 nL/min with a gradient increasing from 1% Buffer B/95% buffer A to 40% buffer B/60% Buffer A in typically 90 or 120 min (buffer A: 0.1% formic acid, buffer B: 0.1% formic acid in 80% acetonitrile). Mass spectrometers (Q‐Exactive, Q‐Exactive Plus, Q‐Exactive‐HF or Fusion Lumos, Thermo Scientific) were operated in data dependent (DDA) positive ion mode.
2.16. Statistics
All graphic representations of the data are expressed as mean of biological replicates as specified in figure legends. Statistical analyses were conducted with Prism (GraphPad Software) using the 1‐tail Student t‐test.
3. RESULTS
3.1. Engineered human melanoma and neuroblastoma cells release GFP‐tagged TEVs that are captured in vitro by mouse macrophages
Human melanoma (MEL) cells (Montgomery et al., 1994) and human neuroblastoma (NB) cells were engineered to express a TurboGFP linked to the myristoylation domain of Lyn tyrosine kinase which anchored GFP to the internal membrane of phospholipid bilayer entities (Ingley, 2012). Stable transduced cells (designated XPack‐GFP) were selected for their green fluorescence and examined by fluorescence microscopy which revealed the presence of green fluorescence at the level the plasma membrane and in intracytoplasmic vesicles (Figure 1a). TEVs released in the culture medium were then isolated, characterized, and examined for the presence of GFP. An initial isolation by Differential Ultra‐Centrifugation (DUC) and analysis by Western blot indicated the presence of GFP in EVs with a diameter ranging from >200 nm to <50 nm (Figure 1b and Figure S1a,b). The presence of GFP in exosomes was documented by Western blot analysis of TEVs isolated from the 100,000 × g DUC pellet byODGCor by SEC (Figure 1c). This analysis revealed the presence of GFP in fractions containing syntenin, ALIX, and the tetraspanins CD63, CD81, and CD9 in the absence of non‐exosomal proteins, such as GM‐130 (a mitochondrial marker) or calnexin (an endoplasmic reticulum marker), consistent with these EVs being small EVs (Kowal et al., 2016; Mathieu et al., 2019). Visualization of these vesicles by transmission electron microscopy (TEM) (Figure S1c) indicated the presence of circular structures with a double lipid membrane and a mean size of 94.7 nm confirming their exosomal nature. Thus, the GFP was present in all TEVs, including exosomes. The presence of microRNA (miR) in total RNA isolated from these TEVs (purified by SEC) was examined by Nanostring analysis and compared with the miR content of mouse bone marrow myeloid cell‐derived EVs (Figure 1d). This analysis revealed the presence of several miRs significantly more abundant in human TEVs than mouse EVs including miR‐1246, an exosomal onco‐miR present in many human cancers, including MEL and NB (Cooks et al., 2018; Ghafouri‐Fard et al., 2021; Xu et al., 2014). An analysis of TEVs treated with RNAse indicated the presence of miR‐1246 inside but also at the surface of TEVs (Figure S1d). We therefore selected miR‐1246 as a marker of TEVs and as proof of EV cargo delivery in the capturing cells. Contact co‐culture experiments of XPack‐GFP MEL cells and XPack‐GFP NB cells with murine RAW 264.7 macrophages (Figure 1e left) demonstrated the transfer of vesicular GFP from tumour cells to RAW 264.7 cells which were then isolated from GFP+ tumour cells based on the expression of F4/80 (mouse macrophage marker) and absence of expression of human marker CD298 (Lawson et al., 2015) (Figure 1e middle and S1e) and from GFP‐ RAW 264.7 cells based on the presence of green fluorescence (4.2 ± 2.6% and 15.4 ± 5.8% of total RAW 264.7 cells in MEL and NB model respectively). qRT‐PCR on total RNA isolated from sorted cells revealed that GFTEVs transferred miR‐1246 to RAW 264.7 cells (Figure 1e right). The flow analysis also revealed the presence of cells positive for both F4/80 and CD298 markers, likely representing tumour cell‐macrophage trogocytosis or phagocytosis (Erickson & Hu, 1979; Velmurugan et al., 2016). Because in contact co‐cultures GFP could have been transferred by other mechanisms than EV uptake, such a phagocytosis, trogocytosis of membrane fragments from apoptotic cells, or even nanotubes, co‐culture experiments were also performed in contact‐independent culture conditions (Transwell) (Figure 1f left). These experiments revealed the presence of miR‐1246 in RAW 264.7 cells and a flow cytometry analysis revealed 0.022% and 0.016% of GFP+ RAW 264.7 cells co‐cultured with M24met XPack‐GFP or SK‐N‐BE(2) XPack‐GFP cells respectively. The lower GFP transfer observed in contact‐independent conditions indicates that other mechanisms than TEV‐mediated mechanisms contribute to GFP and miR‐1246 transfer in contact co‐cultures. To evaluate the transfer of GFP and miR‐1246 due to apoptotic bodies from the cancer cells in these co‐culture experiments, we performed Annexin V staining after a three‐day co‐culture and measured by flow cytometry an average of 2.9% and 4% of apoptotic cells for M24met XPack‐GFP and SK‐N‐BE(2) XPack‐GFP respectively (Figure 1f right).
FIGURE 1.
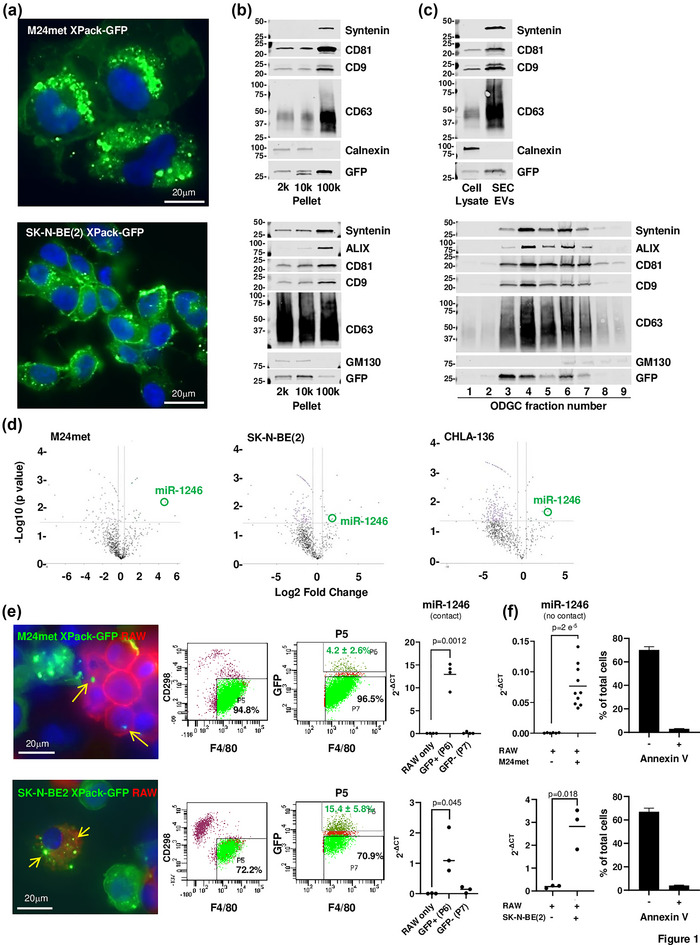
Engineered human melanoma and neuroblastoma cells release GFP‐tagged TEVs that are captured in vitro by mouse macrophages. (a) Immunofluorescence microscopy of M24met XPack‐GFP and SK‐N‐BE(2) XPack‐GFP cells showing GFP‐labelled vesicles (green). Nuclei are stained with DAPI (blue). (b) Cultures media were collected and TEVs were purified as indicated in materials and methods and illustrated in Figure S1a. Western blot was performed on pellets obtained by DUC for the indicated exosomal and non‐exosomal proteins. (c) Western blot analysis of TEVs isolated by SEC or ODGC fractions obtained from the 100K pellet. (d) TEVs from the culture medium of indicated cell lines and EVs from mouse myeloid cells were purified by SEC and ODGC, and total RNA was analysed by NanoString nCounter to determine the miR profile. Volcano plots of a Rosalind analysis show fold changes in miR expression in TEVs versus mouse EVs (x axis) and the p values (y axis). (e) Fluorescent microscopic images of M24met XPack‐GFP and SK‐N‐BE(2) XPack‐GFP cells co‐cultured with mouse RAW 264.7 macrophages stained with an anti‐F4/80 antibody (left). Flow cytometry acquisition plots during sorting of F4/80+ RAW 264.7 cells from CD298+ cancer cells (middle). RT‐PCR analysis for miR‐1246 presence in RNA isolated from sorted F4/80+ GFP+ (P6) and F4/80+ GFP‐ (P7) RAW 264.7 cells compared to RAW 264.7 cells cultured alone (right). Each point represents the average of technical triplicates of an experiment done with three or four biological replicates and the graph is representative of an experiment repeated four times. (f) RT‐PCR analysis on RNA isolated from RAW 264.7 cells co‐cultured three to seven days in Transwell with M24met XPack‐GFP or SK‐N‐BE(2) XPack‐GFP showing the presence of TEV‐derived miR‐1246. Each point represents the average of technical triplicates of an experiment done with three or four biological replicates. The top graph represents the combined results of three separate experiments and the bottom graph is representative of an experiment repeated three times (left). Percentage of apoptotic and non‐apoptotic cancer cells after a three‐day co‐culture with RAW 264.7 cells (right).
3.2. The capture of TEVs by resident stromal cells in metastatic organs precedes the homing of tumour cells
A contribution of TEVs to the PMN assumes that their capture by stromal cells in a metastatic organ precedes the homing of tumour cells, a point that has not been so far demonstrated with experiments using an exogenous administration of TEVs but could be tested in our two distinct orthotopic models of endogenous release that metastasize to two sites that are the most common sites of metastasis in human cancers. To monitor the release of TEVs in tumour‐bearing mice, TEVs were isolated from the plasma by SEC and analysed by NanoSight and ExoView. They had a mean size of 92.9 nm consistent with small EVs and their human origin was confirmed by the presence of human tetraspanins CD9, CD63 and CD81 (Figure S2). We initially tested our hypothesis in a lung metastatic model with M24met XPack‐GFP MEL cells (Figure S3a). The release of TEVs over time in mice orthotopically implanted with MEL cells was further documented by the presence of miR‐1246 in plasma TEVs between day 5 and day 25 post‐implantation (Figure 2a). A time course analysis by immunofluorescence microscopy of the lungs in mice implanted with MEL cells (Figure 2b) revealed the presence of isolated F4/80+ GFP+ macrophages in the absence of GFP+ tumour cells at day 10 followed by the progressive accumulation of GFP+ tumour cells between day 15 and day 25, an observation that was confirmed by flow cytometry (Figure 2c). At day 5, no GFP signal was detected. At day 10, F4/80+ GFP+ represented 0.099% ± 0.016 of total lung cells and CD298+ GFP+ cells were not detected. Between day 15 and 25, whereas the percentage of GFP+ F4/80+ cells varied from 0.029% ± 0.015 to 0.095% ± 0.08, the percentage of CD298+ GFP+ cells progressively increased from 0.0027% ± 0.0018 at day 15 to 0.18% ± 0.15 at day 25. The hypothesis was then tested in a liver metastatic model with SK‐N‐BE(2)Liv XPack‐GFP NB cells (Figure S3b). The presence of miR‐1246 in plasma EVs was documented at day 28 post‐implantation (Figure 3a). A time course analysis by immunofluorescence microscopy showed GFP+ TEV‐capturing F4/80+ macrophages in the liver at day 14 in the absence of GFP+ tumour cells which were only detected starting at day 28 (Figure 3b). Similarly, flow cytometry analysis (using the NB tumour marker GD2) revealed the presence of GFP+ GD2‐ cells (0.07% ± 0.09) in the near absence of GFP+ GD2+ tumour cells at day 14 and a progressive increase in GFP+ GD2+ tumour cells at day 21 (0.24% ± 0.4) and day 28 (1.85% ± 2.67) (Figure 3c). Additional sets of mice were analysed by flow cytometry using CD298 as tumour cell marker to detect the progressive homing of NB and F4/80 as Kupffer cells marker to detect TEV capture. As in the melanoma model, the analysis revealed the presence of F4/80+ GFP+ cells in the liver prior to the detection of CD298+ GFP+ tumour cells (Figure 3d left). Similar data were obtained in another liver metastatic model with CHLA‐136 XPack‐GFP NB cells (Figure S3c). In all models, there was no obvious correlation between the number of TEV‐capturing cells in the PMN or MN and the number of tumour cells detected in the MN. In addition, flow cytometry plots for both mouse macrophage F4/80 and human cell marker CD298 markers revealed the presence of rare dual positive cells, which therefore indicated a low rate of trogocytosis or phagocytosis of GFP+ tumour cells by the Kupffer cells in the liver (Figure 3d right). Thus, we show that the endogenous TEV uptake by resident macrophages is reproducible regardless of tumour type and organ site and that the capture of TEVs occurs prior to the homing of metastatic tumour cells, an observation that supports a contributory role of TEVs in the PMN.
FIGURE 2.
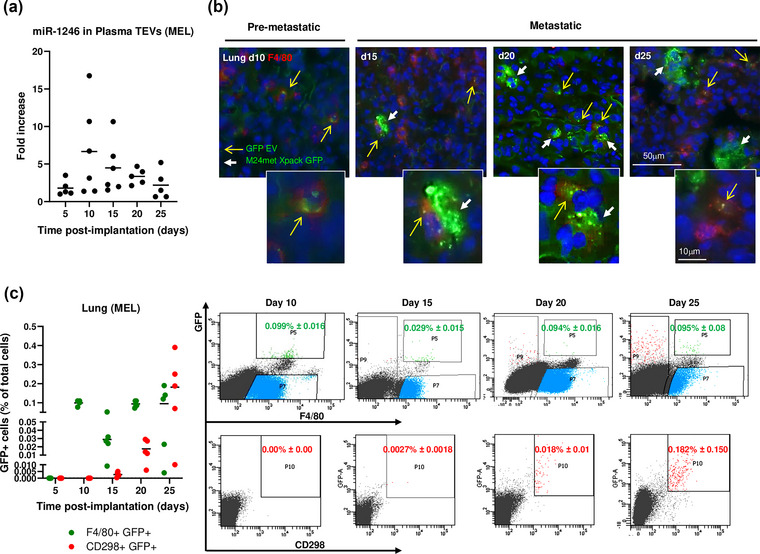
Release of GFP TEVs from primary melanoma tumour and detection in lung precedes the onset of metastasis. (a) Detection of miR‐1246 in the plasma EVs of M24met XPack‐GFP tumour‐bearing mice over time (left). (b) Immunofluorescence on frozen lung sections obtained at the indicated time points and stained with an anti‐F4/80 antibody (red). Yellow arrows indicate F4/80+ cells containing GFTEVs, and white arrows indicate M24met XPack‐GFP metastatic tumour cells. (c) Cells harvested from the lungs were analysed by flow cytometry for the presence of F4/80+/GFP+ GFTEV‐containing macrophages and the presence of CD298+/GFP+ tumour cells over time (Day 5 n = 4; Day 10 n = 4; Day 15 n = 5; Day 20 n = 5; Day 25 n = 5) (left). Representative flow analysis for the detection of F4/80+/GFP+ and CD298+/GFP+ cells in the lung of M24met XPack‐GFP tumour‐bearing mice obtained at the indicated time points (right).
FIGURE 3.
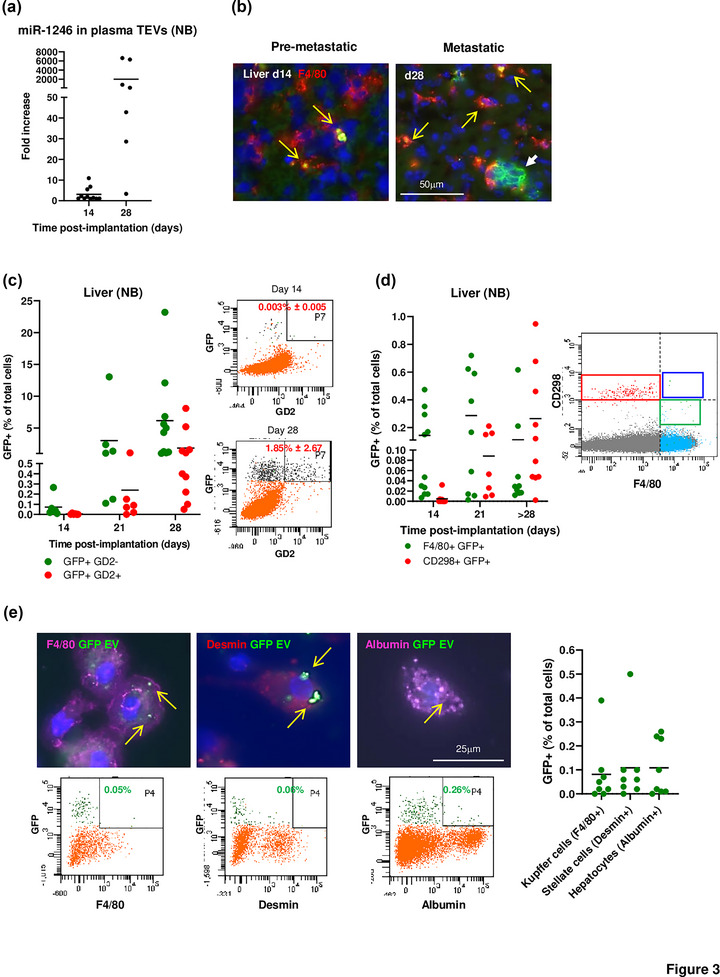
The capture of TEVs from primary neuroblastoma tumours in the liver precedes the homing of tumour cells. (a) Detection of miR‐1246 in the plasma EVs of SK‐N‐BE(2)Liv XPack‐GFP tumour‐bearing mice over time. (b) Immunofluorescence on frozen liver sections obtained at the indicated time points and stained with an anti‐F4/80 antibody (red). Yellow arrows indicate F4/80+ cells containing GFTEVs, and white arrows indicate SK‐N‐BE(2) XPack‐GFP metastatic tumour cells. (c) Cells harvested from the liver were analysed by flow cytometry for the presence of GD2‐/GFP+ TEV‐containing cells and the presence of GD2+/GFP+ tumour cells over time (left) (Day 14 n = 5; Day 21 n = 6; Day 28 n = 10). Representative flow analysis for the detection of GD2+/GFP+ cells in the liver of SK‐N‐BE(2) XPack‐GFP tumour‐bearing mice obtained at the indicated time points (right). (d) Additional flow cytometry quantitative analysis using CD298 as tumour cell maker and F4/80 as Kupffer cell marker (left) (Day 14 n = 11; Day 21 n = 11; Day 28 n = 10). Flow cytometry on liver cells from a tumour‐bearing mouse showing the separation between human CD298+ tumour cells (red box) and mouse F4/80+ macrophage (green box), and the absence of dual positive cells (blue box) (right). (e) Immunofluorescence images of cells isolated from the liver and stained with antibodies against F4/80, desmin, and albumin, to identify Kupffer cells, stellate cells, and hepatocytes respectively (top). Representative flow analysis for the detection of F4/80+/GFP+, desmin+/GFP+, and albumin+/GFP+ cells in the liver of SK‐N‐BE(2) XPack‐GFP tumour‐bearing mice (bottom). Quantification by flow cytometry of GFP+ cells in three selected liver cell populations (right) (n = 8).
Next, we characterized GFP+ cells in the liver by immunofluorescence and flow cytometry to determine whether cells other than Kupffer cells were able to capture TEVs (Figure 3e). Mice were examined between day 21 and 28 after implantation and liver tissues were processed to obtain populations enriched in Kupffer cells, hepatic stellate cells (HSC) and hepatocytes. GFTEV capture was observed in F4/80+ GFP+ Kupffer cells (0.08% ± 0.13), in desmin+ GFP+ HSC (0.11% ± 0.16), and albumin+ GFP+ hepatocytes (0.11% ± 0.12).
3.3. The capture of TEVs in the PMN is organ‐specific
We then asked whether the capture of TEVs by host cells was restricted to metastatic organs, consistent with their contribution to PMN generation. This was first tested in the lung metastasis MEL model. F4/80+ GFP+ mouse macrophages and GFP+ tumour cells were detected by fluorescence microscopy 28 days post‐implantation in the lungs, but not in the liver, kidney, or brain of the same mice (Figure 4a top). Quantitative analysis on a series of frozen sections from those organs revealed the presence of GFTEV‐capturing macrophages almost exclusively in the lung (0.68% ± 0.42 of the total cells counted per microscopic field or 9.76% ± 5.33 of all F4/80+ cells in the lung). Accordingly, M24met tumour cells in the lung represented 1.15% ± 1.28 of all cells counted (Figure 4a bottom left). Neither TEV‐capturing macrophages nor tumour cells were detected by fluorescence microscopy in the liver, kidney or brain. Quantitative flow cytometry analysis performed on similar samples confirmed the presence of F4/80+ GFP+ cells (0.017% ± 0.003) and of CD298+ tumour cells (0.069% ± 0.035) in the lung but not in the liver (Figure 4a bottom, right). In contrast, in the NB liver metastasis model, F4/80+ GFP+ mouse macrophages and GFP+ tumour cells were detected in the liver at 28 days post‐implantation (1.54% ± 0.91 and 0.79% ± 0.44 respectively) but not in the lung, the kidney, and the brain. GFTEV‐capturing macrophages represented 10.28% ± 5.56 of all macrophages counted in liver sections. A quantitative flow cytometry analysis confirmed the presence of GFP+ F4/80+ and GFP+ CD298+ cells in the liver (0.017% ± 0.009 and 0.217% ± 0.28, respectively) and their absence in the lung (Figure 4b). However, in the NB models, low levels of GFP+ GD2‐ cells (between 0.018 and 0.028% in the SK‐N‐BE(2) model and between 0.04 and 0.11% in the CHLA‐136 model) were detected in the absence of detectable GFP+ GD2+ tumour cells in the bone marrow (Figure S3d), a frequent site of metastasis in NB. Thus, with the exception of bone marrow, the capture of endogenously released TEVs occurred selectively in organs that became colonized by tumour cells.
FIGURE 4.
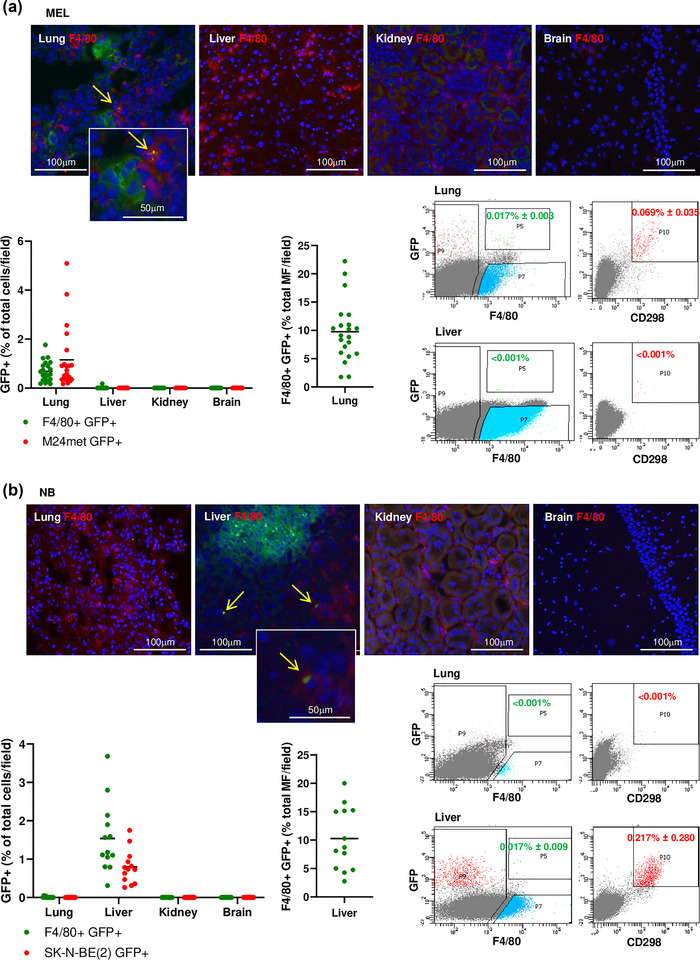
The capture of TEVs in the PMN is organ specific. (a) Immunofluorescence microscopy on frozen sections of lung, liver, kidney, and brain tissues obtained from a MEL tumour‐bearing mouse at day 28 post‐implantation of M24met XPack‐GFP cells (metastatic stage) and stained with an anti‐F4/80 antibody (red). Only the lung sections showed metastatic MEL cells (green) and the presence of F4/80+ cells containing GFTEVs (yellow arrows) (top). Quantification of F4/80+ GFP+ and GFP+ MEL cells in frozen sections of indicated organs for four tumour‐bearing mice as specified in the methods. Each data point represents the percentage of GFP+ cells of the total cells counted per microscopic field (between 290–750 cells) (bottom left). The number of GFTEV‐capturing cells expressed as a percentage of the total number of macrophages (MF) counted per field (bottom middle). Representative flow cytometry analysis on lung (metastatic organ) and liver (non‐metastatic organ) of a mouse, 28 days post‐implantation (bottom right). (b) Immunofluorescence microscopy on frozen sections of indicated tissues as in (a) obtained from a NB tumour‐bearing mouse at day 28 post‐implantation of SK‐N‐BE(2)Liv XPack‐GFP cells and stained with an anti‐F4/80 antibody (red). Only the liver sections showed metastatic NB cells (green) and the presence of F4/80+ cells containing GFTEVs (yellow arrows) (top). Quantification of F4/80+ GFP+ and GFP+ NB in frozen sections of indicated organs of three mice as in (a) (between 270–423 cells counted per field) (bottom left). Representative flow cytometry analysis done on lung (non‐metastatic organ) and liver (metastatic organ) of a mouse, 28 days post‐implantation (bottom right).
To assess the possible involvement of integrins in the organotropism of the metastasis, we investigated integrin packaging in TEVs from lung‐tropic M24met MEL and liver‐tropic SK‐N‐BE(2) NB (Figure S4). A proteomic analysis revealed the presence of distinct sets of integrins, with M24met TEV expressing high levels of integrin β1 (ITGB1), and several of its partners, integrin α2 (ITGA2), α3 (ITGA3) and α6 (ITGA6), among others. SK‐N‐BE(2)Liv TEVs also predominantly contained β1 (ITGB1) but with partners α1 and α4. These observations suggest that those distinct integrin heterodimers may mediate lung versus liver tropism of TEVs released by M24met and SK‐N‐BE(2)Liv primary tumours respectively, given their preferred affinity for distinct extracellular matrix (ECM) components.
3.4. TEV‐capturing cells undergo dynamic changes in inflammatory gene expression during pre‐metastatic to metastatic niche progression
The rare TEV‐capturing cells (F4/80+ macrophages from the lung in the MEL model and F4/80+ Kupffer cells and Desmin+ HSC from the liver in the NB model) were isolated by flow activated cell sorting and pooled from several tumour‐bearing mice to allow bulk RNA analysis. Separated collections were obtained from organs before homing of tumour cells at the PMN stage and after their homing at the MN stage (Figure 5a). With a few exceptions, the presence of GFP in these cells matched the detection of miR‐1246, which was not detected in cells isolated from control non‐tumour‐bearing mice. miR‐1246 was however detected in two samples of F4/80+ GFP‐ cells, in which the GFP may have been proteolytically degraded or alternatively in which miR‐1246 was transferred independently of TEV capture (Figure 5b). A gene expression analysis of 96 mouse genes (immune cell markers, chemokines and receptors, cytokines/growth factors, regulatory signalling genes, ECM, metabolism, apoptosis) was performed by Taqman low density array on RNA extracted from these cells comparing RNA expression in GFTEV‐capturing cells and GFP‐ non‐TEV‐capturing cells with RNA expression in corresponding cells obtained from non tumour‐bearing mice. This analysis revealed cancer‐specific signatures among the two cancer models and dynamic changes between the PMN and the MN (Figure 5c). In the PMN of the MEL model, cytokine/growth factor genes like Csf1, Il1α, Il18, Tnfα and Ski (a negative regulator of TGFβ) and Bcl2l1 were upregulated in TEV‐capturing macrophages whereas Cd38 (a M1 marker in inflammatory macrophages), Ccl3, Ccl5 and Cxcl10 were upregulated in non‐TEV‐capturing macrophages. With the exception of Il1α, Il18 and Ski, genes overexpressed in the PMN were no longer upregulated in the MN. In the PMN of the NB model, genes for macrophage markers (Cd4, Cd68, and Cd86, Icos), cytokines such as Il10 and Il18, Hmox1, Tfrc and adhesion molecules such as Vcam1 were upregulated in TEV‐capturing macrophages while Cd4, Cd68, Icos, Il18, Tnfα, Fn1, Hmox1 and Vcam1 genes were upregulated in TEV‐capturing HSC and at the exception of Cd4, no changes in gene expression were observed in non‐TEV‐capturing HSC. As in the MEL model, most of these genes were no longer upregulated in the MN, while other genes (Il12, Bcl2, and Edn1 in macrophages and Cxcr3, Il6 and Vegfa in HSC) became upregulated instead. Thus, these data suggest that capture of TEVs by PMN macrophages and, in the case of liver, HSC, generates an inflammatory reaction to which both TEV‐capturing and non‐TEV‐capturing cells contribute characterized by the upregulation of several cytokines, chemokines, and regulatory genes that recede as the PMN evolves toward a MN with the colonization of tumour cells.
FIGURE 5.

TEV‐capturing cells undergo dynamic changes in inflammatory gene expression during PMN to MN progression. (a) Number of GFTEV‐containing cells isolated by flow sorting before and after the homing of tumour cells in the metastatic organs for both MEL and NB cancer models, and number of mice from which those cells were pooled. The experiment was repeated twice for MEL and once on two cell types for NB. (b) RT‐PCR analysis for miR‐1246 presence in RNA isolated from sorted F4/80+ GFP‐ and F4/80+ GFP+ lung macrophages (MEL) or F4/80+ GFP‐ and F4/80+ GFP+ liver Kupffer cells and desmin+ GFP‐ and desmin+ GFP+ HSC (NB) of tumour‐bearing mice compared to RNA from corresponding cells of non‐tumour‐bearing control mice. (c) Heat map representing the results of an mRNA analysis using TaqMan Mouse Immune Microarray cards to compare GFTEV‐capturing cells and GFP‐ non‐capturing cells to control cells from a non‐tumour‐bearing mouse in PMN vs MN for both cancer models for a total of 36 mice. The results were normalized to 18S, Actinb, and Gapdh housekeeping genes and were expressed as relative quantity (RQ) compared to gene expression in control mice.
4. DISCUSSION
Our work provides new evidence that TEVs are critical for orchestrating the inflammatory microenvironment characteristic of the PMN in two different models of endogenous release of TEVs by primary tumours. We demonstrate that (1) TEVs are captured by host cells, mostly resident macrophages, prior to the homing of tumour cells in the PMN, (2) that TEVs are not captured by host cells in non‐metastatic organs, and (3) that the capture of TEVs induces in host cells dynamic changes in the expression of inflammatory genes.
Using cells engineered to produce a GFP reporter anchored to the TEV internal membrane has eliminated the requirement to purify EVs and thus altering their structure and function. This tagging method also obviated the use of reagents residing within the TEV membrane but having the ability to diffuse outside and be captured by other cells (Takov et al., 2017). The method we have used however presents some limitations that were overcome in this study. Using a transmembrane protein like tetraspanin to tag the EVs could disrupt ligand‐receptor interaction and organotropism, a problem we avoided by using the myristoylation domain of an internal kinase. The method however does not only specifically tag small EVs but also large EVs and apoptotic bodies having a lipid bilayer, an important aspect in this study. Our data thus support a role for any type of large and small TEVs. Another limitation is that the reporter protein can be the subject of proteolytic degradation, and this was likely the case in some of our studies where we detected miR‐1246 in GFP‐ Kupffer cells in the liver of tumour‐bearing mice. Thus, the low number of TEV‐capturing cells detected in metastatic organs in most of our experiments (varying between 0.1% and 5%) likely underestimates capture levels in the metastatic organs. The rare case where miR‐1246 was present in GFP‐ Kupffer cells however suggests that proteolytic degradation did not systematically occur. The transfer of the GFP alone attached to a membrane fragment from tumour cells was another possibility that we tried to eliminate by demonstrating the transfer of miR‐1246 TEV cargo to GFP+ mouse cells. However, we cannot eliminate the possibility that miR‐1246 may also be present in the membrane of tumour cells (as we show it is present in the external membrane of TEVs). This possibility is less likely in the PMN where no tumour cells were detected. We also cannot entirely exclude the possibility that free miR or miR‐associated with lipid vesicles may have been taken by GFP‐macrophages. The use of miR‐1246 as a marker of TEV capture of tumour‐bearing mice has therefore its limitations. We demonstrated the presence of miR‐1246 inside TEVs purified by SEC but also at the surface of TEVs, allowing the identification of circulating TEVs in tumour‐bearing mice. Finally, the possibility that in some samples the presence of GFP in macrophages in vivo could be the result of phagocytosis, trogocytosis or fusion with tumour cells that we had observed in vitro, cannot be entirely eliminated but was shown to be a very rare event.
The use of endogenously labelled TEVs made necessary the separation of GFTEV‐producer tumour cells from GFTEV‐capturing host cells. This difficulty was circumvented using a human to mouse model and the selection of human and mouse specific surface markers that allowed the separation of GFP+ tumour cells identified by their positivity for a human specific CD298 marker (MEL and NB models) or for the disialoganglioside D2 (GD2) (NB model) from GFP+ murine cells positive for the mouse macrophage marker F4/80 and for mouse desmin (HSC marker). None of the markers selected and antibodies used had cross‐species specificity. In the case of SK‐N‐BE (Steeg & Theodorescu, 2008), where a population of cells with low GD2 expression was detected, some GFP+/GD2‐ cells could have been identified as GFPTEV‐capturing cells rather than being tumour cells with low GD2 expression in our early experiments. This was not the case in our subsequent experiments in which cells were isolated based on their CD298 expression.
Our data demonstrate preferential uptake of endogenous TEVs by macrophages similar to that observed with the capture of exogenous EVs, confirming their bona‐fide roles in PMN and MN biology (Costa‐Silva et al., 2015; Hyenne et al., 2019; Shao et al., 2018; Zhao et al., 2020). What is presently unknown is whether these TEV‐capturing macrophages are resident macrophages of embryonic origin like Kupffer cells and alveolar macrophages, or monocyte‐derived macrophages having captured TEVs in the bone marrow and being recruited to the PMN. Within the limit of detection, we did not observe free GFP+ TEVs in the PMN. We therefore cannot conclude that they arrested in the PMN before being captured by resident macrophages. Interestingly, in the liver, we also observed TEVs capture by HSC and hepatocytes. HSC have been shown to contribute to metastasis and are activated by colorectal TEVs to promote metastasis (Kang et al., 2011; Zhao et al., 2022). The capture of EVs by hepatocytes has not been extensively studied and was not examined here because of difficulties in isolating TEV‐capturing hepatocytes. Other cells like sinusoidal or vascular endothelial cells may also capture TEVs.
We provide important evidence that the capture of endogenously released TEVs exhibits strong organotropism, being selectively captured in the lung in the MEL model and in the liver in the NB model. Within the technical limit of immunofluorescence microscopy, we did not detect free TEVs in metastatic organs and therefore cannot conclude on the organotropism of free TEVs. The absence of capture of endogenous TEVs in the liver in the MEL model is particularly remarkable considering that the liver has been shown to preferentially capture exogenously administered EVs (Németh et al., 2021). The molecular basis for this tropism however has not been examined here in detail but could be integrin mediated. In MEL TEVs, we have shown the expression of α2β1, α3β1, and α6β1, all of which bind laminin, an ECM molecule abundant in the lung, suggesting that these integrins may mediate the lung tropism of M24met MEL cells and TEVs. Conversely, NB TEVs expressed α1β1 and α4β1. α4β1 preferentially binds fibronectin, which is increased during liver PMN formation (Costa‐Silva et al., 2015) and was overexpressed by HSC in the PMN in the NB model whereas α1β1 binds to collagen and laminin. Taken together these data suggest that TEVs may exploit multiple integrin heterodimer ‘zip codes’ for docking to lung and liver PMN and are in line with our previous observations in other organotrophic cancer models (Hoshino et al., 2015).
One exception to this organotropism however is the bone marrow where we detected the presence of a low percentage of TEV‐capturing myeloid cells but an absence of metastatic cells within the time frame of our animal experiments. These TEV‐capturing myeloid cells may represent bone marrow‐derived cells recruited to the PMN as reported (Peinado et al., 2012). Alternatively, it is conceivable that in this model, bone marrow and bone metastasis could have developed later if mice had been allowed to survive longer. The bone marrow is a common site of metastasis in NB (Ara & DeClerck, 2006) and is known to promote tumour cell dormancy (Fornetti et al., 2018; Mayhew et al., 2020). The contribution of TEVs to bone marrow and bone metastasis and dormancy will require further investigation.
The kinetic gene expression analysis performed on TEV‐capturing cells brings novel insight into the role of TEVs in metastasis evolution from the PMN to the MN. The increased expression of several surface markers of macrophages is consistent with these cells being isolated for their F4/80 positivity. The upregulation of some genes in non‐TEV‐capturing macrophages not upregulated in TEV‐capturing macrophages, suggest that these macrophages contribute to the inflammatory reaction initiated in the PMN. Whereas some markers such as CD38, CD80 and CD86 are typically observed in M1 macrophages (Jablonski et al., 2015; Larionova et al., 2020), others (ICOS) have been associated with a pro‐metastatic and immunosuppressive activity (Raineri et al., 2021). The increased expression of adhesion molecules like VCAM‐1 (a ligand of integrin α4β1), and E‐selectin (a lectin receptor) may promote TEV uptake. We also observed in the PMN of both models the upregulation of multiple factors promoting the recruitment of immune‐suppressive myeloid cells like CCL2, CCL3, CCL5, and CXCL10 (Argyle & Kitamura, 2018; Shang et al., 2022; van Deventer et al., 2013; Zhang et al., 2015), the M2 polarization of macrophages such as IL‐10 (Mirlekar, 2022) and the survival and metastatic behaviour of tumour cells like IL‐1α, IL‐6 and TNFα (Kuan & Ziegler, 2018; Johnson et al., 2018; Shang et al., 2017). The upregulation of Hmox1 (heme oxygenase 1) is of particular interest as this gene has been recently shown to induce the expression of VEGFA and IL‐10, promoting tumour cell trans‐endothelial migration (Lin et al., 2015), angiogenesis and epithelial to mesenchymal transformation (Consonni et al., 2021), and to modulate the transcriptional and epigenetic programs of macrophages toward a pro‐metastatic activity (Alaluf et al., 2020). Whereas the upregulation of some of these genes is likely a direct effect of the capture of TEVs, the upregulation of others could be a secondary reaction. For example, IL‐10, VEGFA, and VCAM1 can be upregulated by TNFα and HMOX1 (Ham et al., 2016; Lin et al., 2015).
However, some factors with anti and pro‐metastatic activities were also found upregulated in the PMN, in particular IL‐18. IL‐18 has been shown to cooperate with IL‐12 to prevent liver metastasis in a mouse model of NB treated with a dendritic vaccine (Iinuma et al., 2006). However, IL‐18 increases VCAM1 expression, and cooperates with TNFα and IL‐1 to promote liver metastasis in the B16 melanoma intrasplenic injection model (Vidal‐Vanaclocha et al., 2000). It also promotes the differentiation of myeloid cells into myeloid‐derived suppressive cells (Lim et al., 2014). Furthermore, a comparison between changes in gene expression in the PMN and the MN reveals progressive loss of anti‐metastatic inflammatory gene expression, and the persistent or new upregulation of genes with pro‐metastatic function such as Fn1, Il6, Csf1, Tgfb, Smad3, and Vegfa. The data thus suggest that a selection process in the PMN may occur where metastatic initiating cells need to overcome the anti‐metastatic activity of some TEV‐induced factors to successfully metastasize. Such a process may explain the well‐documented inefficiency of metastasis and may contribute to dormancy. A more in‐depth analysis at the single cell level will however be needed to further understand the contribution of these inflammatory changes in the PMN, and our data demonstrate the possibility to perform such analysis in models of endogenous release. Also, whether miR‐1246 has any functional role in these changes in gene expression has not been examined in this study and will require further investigation.
A final limitation to our studies is the use of immunodeficient mice. We selected xenotransplanted models because we considered it important to study the role of TEVs in human tumours and because the use of a human‐mouse model facilitated the isolation of human TEV‐producing cells from murine TEV‐capturing cells and the gene expression analysis. Our kinetic analyses were also possible because of the reproducible metastatic behaviour over time. However, our data pointing to changes in inflammation in the PMN, suggest an important contribution of the adaptative immune system raising the need to develop similar models of endogenous release of TEVs in immunocompetent mice. In this aspect our manuscript provides a proof of principle for such future studies.
In summary, using a model of endogenous release of TEVs in two xenotransplanted mouse models, we have obtained data providing a deeper insight into the evolution of the PMN to a MN. It is anticipated that these models will allow testing of novel approaches to intervene in the PMN and to possibly detect early metastasis formation.
AUTHOR CONTRIBUTIONS
Y.A.D. conceived and supervised the project. Y.A.D. and L.B. designed the study, L.B. planned and conducted the experiments and generated the figures. R.N. generated panels for Figure 1b, c and contributed to Figure S1. P.N. performed EV isolation and qRT‐PCR for micro RNAs and contributed to Figure S1d and S2. K.S. contributed to Figure 1e. H.S. contributed to Figure S1c. A.B. contributed to Figure 3e. I.M. contributed to Figure S4. I.M. and D.L. provided input and edited the manuscript. Y.A.D and L.B. analysed and interpreted the data and wrote the manuscript.
CONFLICT OF INTEREST STATEMENT
There is no conflict of interest to be declared by the authors.
Supporting information
Supplementary information
Supplementary information
Supplementary information
Supplementary information
Supplemental Table S1. Antibodies used in this study:
ACKNOWLEDGEMENTS
The authors would like to thank Mrs. J. Rosenberg for her help in the formatting of the manuscript, and the personnel of the Core Facilities of the Saban Research Institute at Children's Hospital Los Angeles (Flow Cytometry, Extracellular Vesicle, Cell Imaging, and Animal Imaging Cores) for their expertise and assistance.
This work was supported by National Institutes of Health/National Cancer Institute grant R01 CA207983 to Y.A. DeClerck.
Blavier, L. , Nakata, R. , Neviani, P. , Sharma, K. , Shimada, H. , Benedicto, A. , Matei, I. , Lyden, D. , & DeClerck, Y. A. (2023). The capture of extracellular vesicles endogenously released by xenotransplanted tumours induces an inflammatory reaction in the premetastatic niche. Journal of Extracellular Vesicles, 12, e12326. 10.1002/jev2.12326
REFERENCES
- Alaluf, E. , Vokaer, B. , Detavernier, A. , Azouz, A. , Splittgerber, M. , & Carrette, A. (2020). Heme oxygenase‐1 orchestrates the immunosuppressive program of tumor‐associated macrophages. JCI Insight, 5, e133929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, R. L. , Balasas, T. , Callaghan, J. , Coombes, R. C , Evans, J. , Hall, J. A. , Kinrade, S. , Jones, D. , Jones, P. S. , Jones, R. , Marshall, J. F. , Panico, M. B. , Shaw, J. A. , Steeg, P. S. , Sullivan, M. , Tong, W. , Westwell, A. D. , & Ritchie, J. W. A. (2019). A framework for the development of effective anti‐metastatic agents. Nature Reviews Clinical Oncology, 16, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Androuin, A. , Verweij, F. J. , & Van Niel, G. (2021). Zebrafish as a preclinical model for extracellular vesicle‐based therapeutic development. Advanced Drug Delivery Review, 176, 113815. [DOI] [PubMed] [Google Scholar]
- Ara, T. , & Declerck, Y. A. (2006). Mechanisms of invasion and metastasis in human neuroblastoma. Cancer and Metastasis Reviews, 25, 645–657. [DOI] [PubMed] [Google Scholar]
- Argyle, D. , & Kitamura, T. (2018). Targeting macrophage‐recruiting chemokines as a novel therapeutic strategy to prevent the progression of solid tumors. Frontiers in Immunology, 9, 2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azmi, A. S. , Bao, B. , & Sarkar, F. H. (2013). Exosomes in cancer development, metastasis, and drug resistance: A comprehensive review. Cancer and Metastasis Reviews, 32, 623–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, A. , Thakur, B. K. , Weiss, J. M. , Kim, H. S. , Peinado, H. , & Lyden, D. (2016). Extracellular vesicles in cancer: Cell‐to‐cell mediators of metastasis. Cancer Cell, 30, 836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzas, E. I. (2022). The roles of extracellular vesicles in the immune system. Nature Reviews Immunology, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, W. H. , Cerione, R. A. , & Antonyak, M. A. (2021). Extracellular vesicles and their roles in cancer progression. Methods in Molecular Biology, 2174, 143–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, H. , & Lee, D. S. (2016). Illuminating the physiology of extracellular vesicles. Stem Cell Research & Therapy, 7, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consonni, F. M. , Bleve, A. , Totaro, M. G. , Storto, M. , Kunderfranco, P. , Termanini, A. , Pasqualini, F. , Alì, C. , Pandolfo, C. , Sgambelluri, F. , Grazia, G. , Santinami, M. , Maurichi, A. , Milione, M. , Erreni, M. , Doni, A. , Fabbri, M. , Gribaldo, L. , Rulli, E. , … Sica, A. (2021). Heme catabolism by tumor‐associated macrophages controls metastasis formation. Nature Immunology, 22, 595–606. [DOI] [PubMed] [Google Scholar]
- Cooks, T. , Pateras, I. S. , Jenkins, L. M. , Patel, K. M. , Robles, A. I. , Morris, J. , Forshew, T. , Appella, E. , Gorgoulis, V. G. , & Harris, C. C. (2018). Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR‐1246. Nature Communications, 9, 771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa‐Silva, B. , Aiello, N. M. , Ocean, A. J. , Singh, S. , Zhang, H. , Thakur, B. K. , Becker, A. , Hoshino, A. , Mark, M. T. , Molina, H. , Xiang, J. , Zhang, T. , Theilen, T. M. , García‐Santos, G. , Williams, C. , Ararso, Y. , Huang, Y. , Rodrigues, G. , Shen, T. L. , … Lyden, D. (2015). Pancreatic cancer exosomes initiate pre‐metastatic niche formation in the liver. Nature Cell Biology, 17, 816–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, C. , Duan, X. , Yao, X. , Wan, J. , Cheng, Y. , Wang, Y. , Yan, Y. , Zhang, L. , Zhu, L. , Ni, C. , Wang, M. , & Qin, Z. (2020). Tumour‐derived exosomal miR‐3473b promotes lung tumour cell intrapulmonary colonization by activating the nuclear factor‐κB of local fibroblasts. Journal of Cellular and Molecular Medicine, 24, 7802–7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson, K. L. , & Hu, F. (1979). Cell interactions in the initial contact between cultured melanoma cells and syngeneic macrophages. American Journal of Pathology, 95, 17–28. [PMC free article] [PubMed] [Google Scholar]
- Fidler, I. J. (2003). The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nature Reviews. Cancer, 3, 453–8. [DOI] [PubMed] [Google Scholar]
- Fornetti, J. , Welm, A. L. , & Stewart, S. A. (2018). Understanding the bone in cancer metastasis. Journal of Bone and Mineral Research, 33, 2099–2113. [DOI] [PubMed] [Google Scholar]
- Ganesh, K. , & Massagué, J. (2021). Targeting metastatic cancer. Nature Medicine, 27, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghafouri‐Fard, S. , Khoshbakht, T. , Hussen, B. M. , Taheri, M. , & Samadian, M. (2021). A Review on the Role of miR‐1246 in the Pathoetiology of Different Cancers. Frontiers in Molecular Biosciences, 8, 771835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ham, B. , Fernandez, M. C. , D'Costa, Z. , & Brodt, P. (2016). The diverse roles of the TNF axis in cancer progression and metastasis. Trends in Cancer Research, 11, 1–27. [PMC free article] [PubMed] [Google Scholar]
- Henrich, S. E. , Mcmahon, K. M. , Plebanek, M. P. , Calvert, A. E. , Feliciano, T. J. , Parrish, S. , Tavora, F. , Mega, A. , De Souza, A. , Carneiro, B. A. , & Thaxton, C. S (2020). Prostate cancer extracellular vesicles mediate intercellular communication with bone marrow cells and promote metastasis in a cholesterol‐dependent manner. Journal of Extracellular Vesicles, 10, e12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino, A. , Costa‐Silva, B. , Shen, T. L. , Rodrigues, G. , Hashimoto, A. , Tesic Mark, M. , Molina, H. , Kohsaka, S. , Di Giannatale, A. , Ceder, S. , Singh, S. , Williams, C. , Soplop, N. , Uryu, K. , Pharmer, L. , King, T. , Bojmar, L. , Davies, A. E. , Ararso, Y. , , … Lyden, D. (2015). Tumour exosome integrins determine organotropic metastasis. Nature, 527, 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyenne, V. , Ghoroghi, S. , Collot, M. , Bons, J. , Follain, G. , Harlepp, S. , Mary, B. , Bauer, J. , Mercier, L. , Busnelli, I. , Lefebvre, O. , Fekonja, N. , Garcia‐Leon, M. J. , Machado, P. , Delalande, F. , López, A. A. , Silva, S. G. , Verweij, F. J. , Van Niel, G. , … Goetz, J. G. (2019). Studying the fate of tumor extracellular vesicles at high spatiotemporal resolution using the zebrafish embryo. Developmental Cell, 48, 554–572.e7.e7. [DOI] [PubMed] [Google Scholar]
- Iinuma, H. , Okinaga, K. , Fukushima, R. , Inaba, T. , Iwasaki, K. , Okinaga, A. , Takahashi, I. , & Kaneko, M. (2006). Superior protective and therapeutic effects of IL‐12 and IL‐18 gene‐transduced dendritic neuroblastoma fusion cells on liver metastasis of murine neuroblastoma. Journal of Immunology, 176, 3461–3469. [DOI] [PubMed] [Google Scholar]
- Ingley, E. (2012). Functions of the Lyn tyrosine kinase in health and disease. Cell Communication and Signaling, 10, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonski, K. A. , Amici, S. A. , Webb, L. M. , Ruiz‐Rosado, J. De. D. , Popovich, P. G. , Partida‐Sanchez, S. , & Guerau‐De‐Arellano, M. (2015). Novel markers to delineate murine M1 and M2 macrophages. PLoS One, 10, e0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, D. E. , O'Keefe, R. A. , & Grandis, J. R. (2018). Targeting the IL‐6/JAK/STAT3 signalling axis in cancer. Nature Reviews Clinical Oncology, 15, 234–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, N. , Gores, G. J. , & Shah, V. H. (2011). Hepatic stellate cells: Partners in crime for liver metastases? Hepatology, 54, 707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan, R. N. , Riba, R. D. , Zacharoulis, S. , Bramley, A. H. , Vincent, L. , Costa, C. , Macdonald, D. D. , Jin, D. K. , Shido, K. , Kerns, S. A. , Zhu, Z. , Hicklin, D. , Wu, Y. , Port, J. L. , Altorki, N. , Port, E. R. , Ruggero, D. , & Shmelkov, S. V. (2005). VEGFR1‐positive haematopoietic bone marrow progenitors initiate the pre‐metastatic niche. Nature, 438, 820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keklikoglou, I. , Cianciaruso, C. , Güç, E. , Squadrito, M. L. , Spring, L. M. , Tazzyman, S. , Lambein, L. , Poissonnier, A. , Ferraro, G. B. , Baer, C. , Cassará, A. , Guichard, A. , Iruela‐Arispe, M. L , Lewis, C. E. , Coussens, L. M. , Bardia, A. , Jain, R. K. , Pollard, J. W. , & De Palma, M. (2019). Chemotherapy elicits pro‐metastatic extracellular vesicles in breast cancer models. Nature Cell Biology, 21, 190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal, J. , Arras, G. , Colombo, M. , Jouve, M. , Morath, J. P. , Primdal‐Bengtson, B. , Dingli, F. , Loew, D. , Tkach, M. , & Théry, C. (2016). Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proceedings of the National Academy of Sciences of the United States of America, 113, E968–E977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan, E. L. , & Ziegler, S. F. (2018). A tumor‐myeloid cell axis, mediated via the cytokines IL‐1α and TSLP, promotes the progression of breast cancer. Nature Immunology, 19, 366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larionova, I. , Tuguzbaeva, G. , Ponomaryova, A. , Stakheyeva, M. , Cherdyntseva, N. , Pavlov, V. , Choinzonov, E. , & Kzhyshkowska, J. (2020). Tumor‐associated macrophages in human breast, colorectal, lung, ovarian and prostate cancers. Frontiers in Oncology, 10, 566511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson, D. A. , Bhakta, N. R. , Kessenbrock, K. , Prummel, K. D. , Yu, Y. , Takai, K. , Zhou, A. , Eyob, H. , Balakrishnan, S. , Wang, C. Y. , Yaswen, P. , Goga, A. , & Werb, Z. (2015). Single‐cell analysis reveals a stem‐cell program in human metastatic breast cancer cells. Nature, 526, 131–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, H. X. , Hong, H. J. , Cho, D. , & Kim, T. S. (2014). IL‐18 enhances immunosuppressive responses by promoting differentiation into monocytic myeloid‐derived suppressor cells. Journal of Immunology, 193, 5453–5460. [DOI] [PubMed] [Google Scholar]
- Lin, H. H. , Chiang, M. T. , Chang, Po. C. , & Chau, L. Y. (2015). Myeloid heme oxygenase‐1 promotes metastatic tumor colonization in mice. Cancer Science, 106, 299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , & Cao, X. (2016). Characteristics and significance of the pre‐metastatic niche. Cancer Cell, 30, 668. [DOI] [PubMed] [Google Scholar]
- Ludwig, N. , & Whiteside, T. L. (2018). Potential roles of tumor‐derived exosomes in angiogenesis. Expert Opinion on Therapeutic Targets, 22, 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malloci, M. , Perdomo, L. , Veerasamy, M. , Andriantsitohaina, R. , Simard, G. , , & Martínez, M. C. (2019). Extracellular vesicles: Mechanisms in human health and disease. Antioxid Redox Signaling, 30, 813. [DOI] [PubMed] [Google Scholar]
- Mashouri, L. , Yousefi, H. , Aref, A. R. , Ahadi, A. M. , Molaei, F. , & Alahari, S. K. (2019). Exosomes: Composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Molecular Cancer, 18, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu, M. , Martin‐Jaular, L. , Lavieu, G. , & Théry, C. (2019). Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell‐to‐cell communication. Nature Cell Biology, 21, 9–17. [DOI] [PubMed] [Google Scholar]
- Mayhew, V. , Omokehinde, T. , & Johnson, R. W. (2020). Tumor dormancy in bone. Cancer Reports (Hoboken), 3, e1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirlekar, B. (2022). Tumor promoting roles of IL‐10, TGF‐β, IL‐4, and IL‐35: Its implications in cancer immunotherapy. SAGE Open Medicine, 10, 205031212110690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery, A. M. , Mueller, B. M. , Reisfeld, R. A. , Taylor, S. M. , & DeClerck, Y. A. (1994). Effect of tissue inhibitor of the matrix metalloproteinases‐2 expression on the growth and spontaneous metastasis of a human melanoma cell line. Cancer Research, 54, 5467–5473. [PubMed] [Google Scholar]
- Németh, K. , Varga, Z. , Lenzinger, D. , Visnovitz, T. , Koncz, A. , Hegedűs, N. , Kittel, Á. , Máthé, D. , Szigeti, K. , Lőrincz, P. , O'neill, C. , Dwyer, R. , Liu, Z. , Buzás, E. I. , & Tamási, V. (2021). Extracellular vesicle release and uptake by the liver under normo‐ and hyperlipidemia. Cellular and Molecular Life Sciences, 78, 7589–7604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado, H. , Alečković, M. , Lavotshkin, S. , Matei, I. , Costa‐Silva, B. , Moreno‐Bueno, G. , Hergueta‐Redondo, M. , Williams, C. , García‐Santos, G. , Ghajar, C. M. , Nitadori‐Hoshino, A. , Hoffman, C. , Badal, K. , Garcia, B. A. , Callahan, M. K. , Yuan, J. , Martins, V. R. , Skog, J. , Kaplan, R. N. , … Lyden, D. (2021). Melanoma exosomes educate bone marrow progenitor cells toward a pro‐metastatic phenotype through MET. Nature Medicine, 18, 883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado, H. , Zhang, H. , Matei, I. R. , Costa‐Silva, B. , Hoshino, A. , Rodrigues, G. , Psaila, B. , Kaplan, R. N. , Bromberg, J. F. , Kang, Y. , Bissell, M. J. , Cox, T. R. , Giaccia, A. J. , Erler, J. T. , Hiratsuka, S. , Ghajar, C. M. , & Lyden, D. (2017). Pre‐metastatic niches: Organ‐specific homes for metastases. Nature Reviews Cancer, 17, 302. [DOI] [PubMed] [Google Scholar]
- Pelissier Vatter, F. A. , Cioffi, M. , Hanna, S. J. , Castarede, I. , Caielli, S. , & Pascual, V. (2021). Extracellular vesicle‐ and particle‐mediated communication shapes innate and adaptive immune responses. Journal of Experimental Medicine, 218, e20202579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plebanek, M. P. , Angeloni, N. L. , Vinokour, E. , Li, J. , Henkin, A. , Martinez‐Marin, D. , Filleur, S. , Bhowmick, R. , Henkin, J. , Miller, S. D. , Ifergan, I. , Lee, Y. , Osman, I. , Thaxton, C. S , & Volpert, O. V. (2017). Pre‐metastatic cancer exosomes induce immune surveillance by patrolling monocytes at the metastatic niche. Nature Communications, 8, 1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pužar Dominkuš, P. , Stenovec, M. , Sitar, S. , Lasič, E. , Zorec, R. , & Plemenitaš, A. (2018). PKH26 labeling of extracellular vesicles: Characterization and cellular internalization of contaminating PKH26 nanoparticles. Biochim Biophys Acta Biomembr, 1860, 1350–61. [DOI] [PubMed] [Google Scholar]
- Raineri, D. , Cappellano, G. , Vilardo, B. , Maione, F. , Clemente, N. , Canciani, E. , Boggio, E. , Gigliotti, C. L. , Monge, C. , Dianzani, C. , Boldorini, R. , Dianzani, U. , & Chiocchetti, A. (2021). Inducible T‐cell costimulator ligand plays a dual role in melanoma metastasis upon binding to osteopontin or inducible T‐cell costimulator. Biomedicines, 10, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang, C. , Sun, Y. , Wang, Y. , Shi, H. , Han, X. , Mo, Y. , Wang, D. , Ke, Y. , & Zeng, X. (2022). CXCL10 conditions alveolar macrophages within the premetastatic niche to promote metastasis. Cancer Letters, 537, 215667. [DOI] [PubMed] [Google Scholar]
- Shang, G. S. , Liu, L. , & Qin, Yi. W. (2017). IL‐6 and TNF‐α promote metastasis of lung cancer by inducing epithelial‐mesenchymal transition. Oncology Letters, 13, 4657–4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao, Y. , Chen, T. , Zheng, X. , Yang, S. , Xu, K. , Chen, X. , Xu, F. , Wang, L. , Shen, Y. , Wang, T. , Zhang, M. , Hu, W. , Ye, C. , Yu, X. , Shao, J. , & Zheng, S. (2018). Colorectal cancer‐derived small extracellular vesicles establish an inflammatory premetastatic niche in liver metastasis. Carcinogenesis, 39, 1368–1379. [DOI] [PubMed] [Google Scholar]
- Steeg, P. S. , & Theodorescu, D. (2008). Metastasis: A therapeutic target for cancer. Nature Clinical Practice. Oncology, 5, 206–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takov, K. , Yellon, D. M. , & Davidson, S. M. (2017). Confounding factors in vesicle uptake studies using fluorescent lipophilic membrane dyes. Journal of Extracellular Vesicles, 6, 1388731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry, C. , Witwer, K. W. , Aikawa, E. , Alcaraz, M. J. , Anderson, J. D. , Andriantsitohaina, R. , Antoniou, A. , Arab, T. , Archer, F. , Atkin‐Smith, G. K. , Ayre, D. C. , Bach, J. M. , Bachurski, D. , Baharvand, H. , Balaj, L. , Baldacchino, S. , Bauer, N. N. , Baxter, A. A. , Bebawy, M. , … Zuba‐Surma, E. K. (2018). Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of Extracellular Vesicles, 7, 1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkach, M. , Kowal, J. , , & Théry, C. (2018). Why the need and how to approach the functional diversity of extracellular vesicles. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences, 373, 20160479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkach, M. , & Théry, C. (2016). Communication by extracellular vesicles: Where we are and where we need to go. Cell, 164, 1226–1232. [DOI] [PubMed] [Google Scholar]
- Van Deventer, H. W. , Palmieri, D. A. , Wu, Q. P. , Mccook, E. C. , & Serody, J. S. (2013). Circulating fibrocytes prepare the lung for cancer metastasis by recruiting Ly‐6C+ monocytes via CCL2. Journal of Immunology, 190, 4861–4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velmurugan, R. , Challa, D. K. , Ram, S. , Ober, R. J. , & Ward, E. S (2016). Macrophage‐mediated trogocytosis leads to death of antibody‐opsonized tumor cells. Molecular Cancer Therapeutics, 15, 1879–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verweij, F. J. , Balaj, L. , Boulanger, C. M. , Carter, D. R. F. , Compeer, E. B. , D'angelo, G. , El Andaloussi, S. , Goetz, J. G. , Gross, J. C. , Hyenne, V. , Krämer‐Albers, E. M. , Lai, C. P. , Loyer, X. , Marki, A. , Momma, S. , Nolte‐‘T Hoen, E. N. M. , Pegtel, D. M. , Peinado, H. , Raposo, G. , … Van Niel, G. (2021). The power of imaging to understand extracellular vesicle biology in vivo. Nature Methods, 18, 1013–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verweij, F. J. , Revenu, C. , Arras, G. , Dingli, F. , Loew, D. , Pegtel, D. M , Follain, G. , Allio, G. , Goetz, J. G. , Zimmermann, P. , Herbomel, P. , Del Bene, F. , Raposo, G. , & Van Niel, G. (2019). Live tracking of inter‐organ communication by endogenous exosomes in vivo. Developmental Cell, 48, 573–589.e4.e4. [DOI] [PubMed] [Google Scholar]
- Vidal‐Vanaclocha, F. , Fantuzzi, G. , Mendoza, L. , Fuentes, A. M. , Anasagasti, M. J. , Martín, J. , Carrascal, T. , Walsh, P. , Reznikov, L. L. , Kim, S. H. , Novick, D. , Rubinstein, M. , & Dinarello, C. A. (2000). IL‐18 regulates IL‐1beta‐dependent hepatic melanoma metastasis via vascular cell adhesion molecule‐1. Proceedings of the National Academy of Sciences of the United States of America, 97, 734–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witwer, K. W. , Buzás, E. I. , Bemis, L. T. , Bora, A. , Lässer, C. , Lötvall, J. , Nolte‐‘T Hoen, E. N. , Piper, M. G. , Sivaraman, S. , Skog, J. , Théry, C. , Wauben, M. H. , & Hochberg, F. (2013). Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. Journal of Extracellular Vesicles, 2, 20360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X. , Zhou, Z. , Xu, S. , Liao, C. , Chen, X. , Li, B. , Peng, J. , Li, D. , & Yang, L. (2020). Extracellular vesicle packaged LMP1‐activated fibroblasts promote tumor progression via autophagy and stroma‐tumor metabolism coupling. Cancer Letters, 478, 93–106. [DOI] [PubMed] [Google Scholar]
- Xu, Li. J. , Jiang, T. , Zhao, W. , Han, J. F. , Liu, J. , Deng, Y. Q. , Zhu, S. Y. , Li, Y. X. , Nian, Q. G. , Zhang, Yu. , Wu, X. . Y. , Qin, E. De. , & Qin, C. F. (2014). Parallel mRNA and microRNA profiling of HEV71‐infected human neuroblastoma cells reveal the up‐regulation of miR‐1246 in association with DLG3 repression. PLoS One, 9, e95272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yáñez‐Mó, M. , Siljander, P. R.‐M. , Andreu, Z. , Bedina Zavec, A. , Borràs, F. E. , Buzas, E. I. , Buzas, K. , Casal, E. , Cappello, F. , Carvalho, J. , Colás, E. , Cordeiro‐Da Silva, A. , Fais, S. , Falcon‐Perez, J. M. , Ghobrial, I. M. , Giebel, B. , Gimona, M. , Graner, M. , Gursel, I. , … Wever, O. D. (2015). Biological properties of extracellular vesicles and their physiological functions. Journal of Extracellular Vesicles, 4, 27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, X. , Qian, N. , Ling, S. , Li, Y. , Sun, W. , Li, J. , Du, R. , Zhong, G. , Liu, C. , Yu, G. , Cao, D. , Liu, Z. , Wang, Y. , Qi, Z. , Yao, Y. , Wang, F. , Liu, J. , Hao, S. , Jin, X. , … Li, Y. (2021). Breast cancer exosomes contribute to pre‐metastatic niche formation and promote bone metastasis of tumor cells. Theranostics, 11, 1429–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z. , Li, Y. , Pan, Y. , Lan, X. , Song, F. , Sun, J. , Zhou, K. , Liu, X. , Ren, X. , Wang, F. , Hu, J. , Zhu, X. , Yang, W. , Liao, W. , Li, G. , Ding, Y. , & Liang, L. (2018). Cancer‐derived exosomal miR‐25‐3p promotes pre‐metastatic niche formation by inducing vascular permeability and angiogenesis. Nature Communications, 9, 5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q. , Jeppesen, D. K. , Higginbotham, J. N. , Graves‐Deal, R. , Trinh, V. Q. , Ramirez, M. A. , Sohn, Y. , Neininger, A. C. , Taneja, N. , Mckinley, E. T. , Niitsu, H. , Cao, Z. , Evans, R. , Glass, S. E. , Ray, K. C. , Fissell, W. H. , Hill, S. , Rose, K. L. , Huh, W. , … Coffey, R. J. (2021). Supermeres are functional extracellular nanoparticles replete with disease biomarkers and therapeutic targets. Nature Cell Biology, 23, 1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q. , Qin, J. , Zhong, L. , Gong, L. , Zhang, B. , Zhang, Y. , & Gao, W. Q. (2015). CCL5‐mediated Th2 immune polarization promotes metastasis in luminal breast cancer. Cancer Research, 75, 4312–4321. [DOI] [PubMed] [Google Scholar]
- Zhao, S. , Mi, Y. , Guan, B. , Zheng, B. , Wei, P. , Gu, Y. , Zhang, Z. , Cai, S. , Xu, Y. , Li, X. , He, X. , Zhong, X. , Li, G. , Chen, Z. , & Li, D. (2020). Tumor‐derived exosomal miR‐934 induces macrophage M2 polarization to promote liver metastasis of colorectal cancer. Journal of Hematology & Oncology, 13, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, S. , Mi, Y. , Zheng, B. , Wei, P. , Gu, Y. , Zhang, Z. , Xu, Y. , Cai, S. , Li, X. , & Li, D. (2022). Highly‐metastatic colorectal cancer cell released miR‐181a‐5p‐rich extracellular vesicles promote liver metastasis by activating hepatic stellate cells and remodelling the tumour microenvironment. Journal of Extracellular Vesicles, 11, e12186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zomer, A. , Maynard, C. , Verweij, F. J. , Kamermans, A. , Schäfer, R. , Beerling, E. , Schiffelers, R. M. , De‐Wit, E. , Berenguer, J. , Ellenbroek, S. I. J. , Wurdinger, T. , Pegtel, D. M. , & Van‐Rheenen, J. (2015). In vivo imaging reveals extracellular vesicle‐mediated phenocopying of metastatic behavior. Cell, 161, 1046–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information
Supplementary information
Supplementary information
Supplementary information
Supplemental Table S1. Antibodies used in this study: