

ABSTRACT
Human immunodeficiency virus (HIV) treatment with antiretroviral regimens containing integrase strand transfer inhibitors such as dolutegravir (DTG) and bictegravir (BIC) offers high levels of protection against the development of drug resistance mutations. Despite this, resistance to DTG and BIC can occur through the development of the R263K integrase substitution. Failure with DTG has also been associated with the emergence of the G118R substitution. G118R and R263K are usually found separately but have been reported together in highly treatment-experienced persons who experienced treatment failure with DTG. We used cell-free strand transfer and DNA binding assays and cell-based infectivity, replicative capacity, and resistance assays to characterize the G118R plus R263K combination of integrase mutations. R263K reduced DTG and BIC susceptibility ~2-fold, in agreement with our previous work. Single-cycle infectivity assays showed that G118R and G118R plus R263K conferred ~10-fold resistance to DTG. G118R alone conferred low levels of resistance to BIC (3.9-fold). However, the G118R plus R263K combination conferred high levels of resistance to BIC (33.7-fold), likely precluding the use of BIC after DTG failure with the G118R plus R263K combination. DNA binding, viral infectivity, and replicative capacity of the double mutant were further impaired, compared to single mutants. We propose that impaired fitness helps to explain the scarcity of the G118R plus R263K combination of integrase substitutions in clinical settings and that immunodeficiency likely contributes to its development.
KEYWORDS: HIV drug resistance, dolutegravir, bictegravir, integrase inhibitors, integrase, R263K, G118R
INTRODUCTION
Current treatment for human immunodeficiency virus (HIV) infection consists of combinations of two or three antiretroviral drugs that can be taken orally daily or intramuscularly every 2 months (1). In addition, every-6-month subcutaneous injections of lenacapavir were approved recently in combination with other antiretroviral drugs for the treatment of highly treatment-experienced individuals with limited therapeutic options (2). Regardless of their form of administration, for all regimens, treatment failure occurs in some cases when viral suppression cannot be achieved or maintained, and drug resistance mutations can emerge (3). The ability of an antiretroviral treatment regimen, or of a particular drug, to protect against the development of HIV resistance mutations upon treatment failure is called its barrier to resistance. Although antiretroviral drugs with a low barrier to resistance can be combined to form regimens sufficient to maintain virological suppression in the context of optimal treatment adherence, drugs with high barriers to resistance may improve clinical outcomes on population and individual levels. Advantages associated with a high barrier to resistance include cost-effectiveness associated with reduced genotyping and the preservation of future treatment options. The accumulation of multidrug resistance mutations, in contrast, can lead to the exhaustion of treatment options and death.
Integrase plays a critical role in the HIV-1 infectious cycle, mainly by mediating the integration of the reverse-transcribed retroviral DNA into human DNA. After reverse transcription is complete, the two DNA ends correspond to long terminal repeats (LTRs), which integrase binds minimally in the form of dimers. This binding to the LTR is specific and sufficient for integrase to catalyze the 3′ processing of the two LTRs, removing a GT dinucleotide and creating a reactive hydroxyl group at the 3′ terminus. Next, at least two integrase dimers bound to the two 3′-processed LTRs assemble minimally as a tetramer, which catalyzes the coordinated nucleophilic attack of the two 3′-hydroxyl groups into the host DNA. However, it is suspected that HIV-1 integrase forms higher-order multimer complexes with DNA (4–8). Cryo-electron microscopy has shown that integrase from the prototype foamy virus (PFV) can form cleaved synaptic complexes called intasomes with 3′-processed DNA in the form of two dodecamers (6). Similarly, the cleaved synaptic complex intasome of the maedi-visna virus contains a tetramer of tetramers of integrase proteins (7, 8). Similar structures have been observed for integrase isolated from the simian immunodeficiency virus (SIV) of the red-capped mangabeys (5). The nucleophilic attack step described above is called strand transfer, and it depends on the ability of integrase to bind the host DNA that serves as a target for integration of the viral DNA, i.e., the target DNA. Thus, integrase binding to both the LTR and the target DNA is important for integration.
Two integrase strand transfer inhibitors, namely, dolutegravir (DTG) and bictegravir (BIC), have high barriers to resistance, particularly when used as part of first-line antiretroviral drug regimens, with only a few cases of resistance having been reported (9–11). In these rare cases, the R263K integrase substitution is the most common (10, 12). However, other integrase resistance substitutions deserve clinical vigilance, including S153Y/F, S230R, and G118R (10, 13). We previously identified G118R as a resistance substitution emerging in cell-based selection experiments with HIV and in viruses isolated from two individuals using DTG monotherapy (14, 15). We found this substitution in combination with R263K in SIV-infected rhesus macaques also treated with DTG monotherapy (16). The same G118R plus R263K combination was found in one treatment-naive person and three highly treatment-experienced individuals who suffered treatment failure with DTG (17, 18). The latter study confirmed that G118R and R263K could coexist on single genomes together with E138K (18). The G118R plus R263K and E138K combination of substitutions was found to confer 13-fold resistance against DTG in a subtype C genetic background (18). The G118R plus R263K combination was not otherwise characterized. As DTG use rises worldwide, it becomes pressing to better understand its resistance profile (19). Therefore, we characterized the effects of the G118R plus R263K combination of integrase substitutions on integrase strand transfer activity, viral infectivity, and susceptibility to DTG and BIC.
RESULTS
Effects of G118R and R263K substitutions on integrase strand transfer activity.
To characterize the integrase proteins carrying the G118R, R263K, and G118R plus R263K substitutions, we performed two types of assays, which are schematized in Fig. 1. We used preprocessed LTR DNA for the strand transfer assay and blunt LTR DNA for the LTR DNA binding assay. Strand transfer assays with 300 nM preprocessed LTR DNA, 60 nM target DNA, and various concentrations of integrase proteins (0, 25, 50, 100, 200, 400, 800, and 1,600 nM) showed that wild-type (WT), G118R, R263K, and G118R plus R263K proteins displayed maximal integration activities between 100 and 400 nM (Fig. 2A). Additional strand transfer assays with 300 nM preprocessed LTR DNA, 400 nM integrase proteins, and various concentrations of target DNA substrate (0, 1.88, 3.75, 7.5, 15, 30, 60, and 120 nM) showed that R263K and G118R individually decreased target DNA binding affinity (pseudo-Km) and maximal strand transfer activity (pseudo-Vmax) (Fig. 2B and Table 1), in agreement with previous reports (14, 20). Combining G118R with R263K further decreased target DNA binding, compared to G118R (Table 1). Specifically, the increase in pseudo-Km values was 2.5-fold for the G118R protein and 4.4-fold for the G118R plus R263K protein, compared to the WT protein. Maximal integration (pseudo-Vmax) decreased by 60% for the R263K protein and by 85% for the G118R protein. However, maximal integration was significantly higher for the G118R plus R263K protein than for the G118R protein. Overall, enzymatic efficiencies were similar for G118R and G118R plus R263K integrase proteins.
FIG 1.

Schematic representation of the strand transfer and DNA binding assays. (A) The strand transfer assay was performed with a preprocessed LTR DNA (red) that lacked a GT dinucleotide and carried a reactive hydroxyl group (-OH) at the 3′ terminus. The complete method is described in Materials and Methods. Briefly, immobilized preprocessed LTR DNA (red) was incubated with integrase (multicolor) and different concentrations of biotin-labeled target DNA (black) for 1 h at 37°C. After washes, the amount of integrated target DNA was measured via biotin (green) quantification with europium-labeled streptavidin (dark blue). Time-resolved fluorescence was plotted against the target DNA concentrations to produce pseudo-Vmax (maximum integration) and pseudo-Km, which accounted for target DNA binding affinity. (B) DNA binding assays were performed with a blunt LTR DNA (red) labeled with rhodamine (green). The complete method is described in Materials and Methods. Immobilized integrase proteins (multicolor) were incubated with various concentrations of LTR DNA for 1 h at 37°C. After washes, the amount of bound DNA was measured by quantifying fluorescence. Results were plotted against LTR DNA concentrations to produce Kd values.
FIG 2.

Strand transfer activities of WT, R263K, G118R, and G118R plus R263K recombinant HIV-1 integrase proteins. (A) Relative strand transfer activity in the presence of various protein concentrations. Results from two separate experiments performed with two different protein batches (four experiments) were compiled after normalization against the mean fluorescence value (from triplicates) obtained with the WT protein at 400 nM, arbitrarily set at 100%. Means ± standard deviations are presented. (B) Relative strand transfer activity in the presence of increasing target DNA concentrations (0, 1.8, 3.75, 7, 15, 30, 60, and 120 nM). Results from three separate experiments performed with two different protein batches (six experiments) were compiled after normalization against the mean fluorescence value (from triplicates) obtained with the WT protein and 120 nM target DNA, arbitrarily set at 100%. Means ± standard deviations are presented.
TABLE 1.
Enzymatic parameters, LTR DNA binding affinity, infectivity, replicative capacity, and resistance to DTG and BIC for WT, R263K, G118R, and G118R plus R263K integrase proteins and viruses
| Genotype | Data from cell-free assaysa |
Data from cell-based assaysa |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Strand transfer assaysb |
LTR DNA binding assay Kd(nM) (IC95) | Infectivity |
Relative replicative capacity (IC95) (%) | FC in resistance (IC95) |
|||||
| Pseudo-Vmax (%) (IC95) | Pseudo-Km (nM) (IC95) | Enzymatic efficiency (pseudo-Vmax/pseudo-Km) | Relative half-infectious titer (IC95) | Relative infectivity (%)c | DTG | BIC | |||
| WT | 116 (108–125) | 24.8 (21.8–28.11) | 4.8 | 15.2 (13.1–17.2) | 1 (9.3–1.1) | 100 (83–117) | 1 (0.7–1.5) | 1 (0.8–1.2) | |
| R263K | 42 (39.8–44)d | 23.4 (22.2–24.8) | 1.8 | 30.3 (23–37.6)d | 1.5 (1.3–1.7)d | 67 | 91 (79–104) | 1.9 (1–3.4) | 2.4 (2.2–2.7)d |
| G118R | 17.6 (16.2–19.2)d,e | 61.5 (49–77)d,e | 0.3 | 27.7 (19.9–35.5)d | 2.8 (2.6–2.9)d,e | 36 | 34 (29–40)d,e | 10.9 (6.3–18.5)d,e | 3.9 (2–7.7)d |
| G118R plus R263K | 34 (29.8–38.8)d,e,f | 110 (63–206)d,e | 0.3 | 77 (63–91)d,e,f | 3.3 (3.1–3.4)d,e,f | 30 | 10 (3–18)d,e,f | 13.9 (7.8–25.7)d,e | 33.7 (9.7–106)d,e,f |
IC95, 95% confidence interval.
Enzymatic parameters were derived from binding saturation curve fitting.
Calculated from relative half-infectious titers.
Significantly different from WT (P < 0.05).
Significantly different from R263K (P < 0.05).
Significantly different from G118R (P < 0.05).
Combining G118R and R263K reduces LTR DNA binding more than single substitutions.
We previously demonstrated that R263K and G118R individually decreased integrase-viral DNA binding (14, 20). To further explore the effects of these two substitutions combined, we measured the direct interaction between WT, R263K, G118R, and G118R plus R263K integrase proteins and the viral DNA, using a binding assay that we developed previously with an LTR-mimetic DNA as ligand (21). In agreement with previous work, our results showed that R263K and G118R individually decreased integrase-viral DNA binding affinity 2- and 1.8-fold, respectively (Table 1). Combining R263K with G118R reduced integrase-viral DNA binding 5-fold.
Combining G118R and R263K decreases HIV-1 infectivity and replicative capacity and increases resistance to DTG and BIC.
Based on our cell-free assays, we suspected that combining G118R with R263K might further impair the infectivity of HIV-1, compared to single mutants. To test this hypothesis, we performed single-cycle infectivity and multicycle replication assays. Short-term infectivity assays in TZM-bl reporter cells showed that G118R and R263K reduced infectivity by 64% and 33%, respectively (Fig. 3 and Table 1). The G118R plus R263K combination of substitutions was associated with a 70% reduction in infectivity. We measured the replicative capacity of the WT, G118R, R263K, and G118R plus R263K HIV-1 viruses in multicycle infections of PM1 cells, as published previously (22, 23). We monitored HIV-1 replication by measuring the release of active reverse transcriptase (RT) enzymes in the cell culture fluids over 20 days. The results were plotted against time, and areas under the curves were calculated to estimate the overall relative replicative capacity of the viruses (Table 1). This analysis showed that R263K reduced HIV-1 replicative capacity minimally (91%, compared to the WT virus). G118R significantly decreased the replicative capacity (34%; P < 0.05). G118R plus R263K further impaired viral replication (<10% of the value for the WT virus), compared to G118R. RT values peaked at day 11 for WT and R263K viruses and at day 20 for G118R and G118R plus R263K viruses (Fig. 4). Since G118R plus R263K virus has been reported in patients failing DTG-based antiretroviral therapy, we performed resistance assays with DTG. However, we also included BIC since this drug may be considered after failure with DTG, given that resistance levels can be lower for BIC than for DTG (24, 25) (Table 1). We confirmed that R263K confers low-level resistance against DTG (1.9-fold) and BIC (2.4-fold), in agreement with previous reports (25, 26). G118R conferred higher resistance levels against DTG (10.9-fold) and BIC (3.9-fold) (27, 28). G118R plus R263K further increased resistance levels against DTG (13.9-fold) and BIC (33.7-fold), compared to G118R, but only the latter increase was statistically significant (Table 1).
FIG 3.

Relative infectivity of WT, R263K, G118R, and G118R plus R263K HIV-1. TZM-bl reporter cells were infected for 24 h with various HIV-1 forms at different concentrations. Results from two separate experiments with two different viral stocks were compiled after normalization against the mean maximal and minimal RLU values (from triplicates), arbitrarily set at 100% and 0%, respectively. Infectivity curves were plotted against the log viral concentrations in RT units using GraphPad Prism v9.4.1. Means ± standard deviations are presented.
FIG 4.
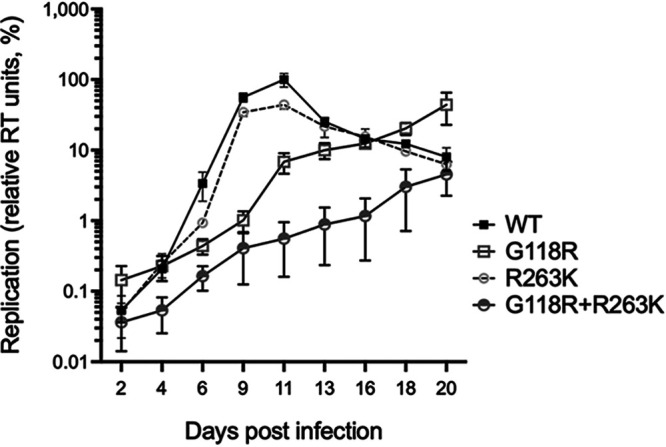
Multicycle replication assay results. PM1 cells were infected with WT, R263K, G118R, and G118R plus R263K HIV-1, and replication was monitored by quantifying the RT activity in the cell culture fluids 2, 4, 6, 9, 11, 13, 16, 18, and 20 days postinfection. Results from two separate experiments with two different viral stocks were compiled after normalization against the maximal RT values obtained with the WT virus after 11 days of infection, arbitrarily set at 100%. A logarithmic scale was used to allow visualization of the G118R plus R263K virus replication curve. Areas under the curves were calculated using GraphPad Prism v9.4.1, and results are presented in Table 1. Means ± standard deviations are presented.
DISCUSSION
Our observation that combining G118R and R263K was detrimental to integration and viral infectivity helps to explain why this combination has been found only rarely in clinical settings. We already showed that G118R and R263K reduced HIV-1 subtype B integrase strand transfer activity when considered individually (14, 27) and that cell-free integration and infectivity assays typically showed similar trends (29). When we carefully examined our data, we found that adding R263K to G118R (from G118R to G118R plus R263K) reduced HIV-1 replicative capacity by 24% (Table 1). Reciprocally, adding G118R to R263K (from R263K to G118R plus R263K) reduced the replicative capacity by 81%. The larger negative impact of G118R on integration and fitness is explained by in silico structural modeling showing that G118R occludes the integrase catalytic site. The G118R substitution also clashed conformationally with BIC when this inhibitor was bound to integrase (Fig. 5). These observations are consistent with previous observations (14, 18, 20). Congruent with our cell-based and cell-free experiments, molecular modeling did not reveal conformational changes that could suggest that G118R and R263K compensate for each other. Reciprocally to integration and fitness, the increase in resistance against DTG associated with adding R263K to G118R (from G118R to G118R plus R263K) (+3-fold) was smaller than that caused by adding G118R to R263K (from R263K to G118R+R263K) (+12-fold). Given the latter significant benefit in terms of resistance, it seems difficult to explain why G118R does not arise from R263K-positive viruses more often in clinical settings without considering the critical connection between viral fitness and immunological pressure.
FIG 5.

Effect of G118R on BIC binding to the HIV-1 integrase catalytic site. The WT protein is shown in red and the in silico-modeled G118R mutant in green. BIC is colored by standard elements (nitrogen in blue, carbon in gray, oxygen in red, and fluorine in green). The overlap between G118R and BIC suggests a conformational clash between the substituted amino acid and the small molecule.
The rarity of cases with G118R plus R263K is not merely a consequence of the individual scarcity of G118R and R263K. For example, although they were individually reported for 6 participants, R263K and G118R were not found in combination in the IMPAACT P1093 clinical trial (30). Similarly, none of the 9 participants in the NADIA clinical trials who developed DTG resistance mutations, including 5 with G118R and 4 with R263K, had a G118R plus R263K combination (31). G118R plus R263K was previously reported without evidence of the two substitutions coexisting in single genomes (17). Instead, G118R and R263K representation in the retroviral population was compatible with their segregation on distinct genomes. G118R occurrence rates varied from 0% to 45% after emergence, while R263K rates antagonistically varied from 99% to 30%. The total representation of the two mutations was consistently <100% (17).
In contrast, three participants in the DAWNING clinical trial experienced virological failure with G118R plus R263K, and clonal analysis found the two mutations together in single HIV-1 genomes in two cases (18). In those cases, high viral loads (1,248,517 and 852,142 HIV RNA copies/mL of plasma in the DAWNING trial) and non-B subtypes (subtypes C and A1) might have favored the emergence of this combination of mutations (18).
When the combination of G118R plus R263K occurs, the current report and previous publications suggest that none of the other integrase inhibitors may be used. Indeed, we show that this combination of substitutions confers high levels of resistance against BIC. In addition, neither raltegravir nor elvitegravir should be considered, since G118R alone confers resistance to both drugs (27, 32). This situation is different from that when R263K is found alone, since that substitution is predicted to retain susceptibility to raltegravir (33). We cannot speculate on the effectiveness of twice-daily 50 mg DTG, or higher doses, after G118R plus R263K has emerged. Further characterization of HIV resistance pathways evolving from G118R and R263K is required to improve the treatment management of the few individuals who will progress through these pathways.
Based on the DAWNING observations and given that viral fitness is critical to the control of HIV-1 (34), we hypothesize that low CD4+ cell counts and high viral loads provide a weak immunological environment favorable to the development of highly drug-resistant, poorly replicative combinations of mutations such as G118R plus R263K. Reciprocally, however, we also think that developing such combinations of substitutions may not preclude future virological suppression with DTG- or BIC-based antiretroviral regimens once immune competency has been restored. Clinical data are needed to test this hypothesis, which is supported by our previous publication, in which progression to high levels of resistance to DTG through the S153F plus R263K combination (fold change [FC] in resistance to DTG of >100) did not lead to treatment failure (23).
MATERIALS AND METHODS
Cells and antiviral compounds.
TZM-bl and PM1 cells were obtained from the NIH AIDS Research and Reference Reagent Program, and 293T cells were obtained from the American Type Culture Collection (ATCC) (CRL-11268). TZM-bl and 293T cells were maintained in Dulbecco’s minimal essential medium (DMEM) with 10% fetal bovine serum (FBS) (Gibco), 50 units/mL penicillin, and 50 μg/mL streptomycin. PM1 cells were grown in Roswell Park Memorial Institute (RPMI) 1640 medium with 10% FBS and penicillin-streptomycin. DTG and BIC were purchased from Toronto Research Chemicals (Toronto, Canada) (D528800 and B382095, respectively).
Recombinant integrase protein expression and purification.
R263K and G118R coding mutations were combined via site-directed mutagenesis using the previously described G118R primers and pET15b-INBR263K plasmid (27). Plasmids were amplified in XL-10 Gold bacteria (Stratagene), and proper mutagenesis was verified by Sanger sequencing. The resulting pET15b-INBG118R+R263K plasmid was transformed in parallel with pET15b-INBR263K, pET15b-INBG118R, and pET15b-INBWT into BL21(DE3) bacterial cells to produce recombinant integrase proteins, using methods described previously (13, 14, 23, 27, 28, 35–37). Transformed BL21 cells were grown overnight at 37°C in Luria-Bertani (LB) broth with 100 μg/mL ampicillin, in 5-mL shaken precultures. Sixteen to 20 h later, the precultures were used to seed cultures in 500 mL LB with ampicillin. Bacterial growth was monitored until the optical density at 600 nm (OD600) reached 0.4 to 0.6, when 1 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) was added to the culture to induce integrase protein expression for 2 h at 37°C. Bacterial cells were pelleted by centrifugation, dried, and frozen at −80°C for 16 to 20 h. All subsequent procedures were conducted on ice or at 4°C. Recombinant integrase proteins were expressed in fusion with a polyhistidine tag that permitted their purification via its interaction with nickel-nitriloacetic acid (Ni-NTA) agarose beads. Pellets were thawed and lysed by sonication in prechilled 1 M NaCl, 20 mM imidazole, 50 mM Tris (pH 7.5), 500 μM MgCl2, 500 μM MnCl2, 2 mM dithiothreitol (DTT), 50 μM ZnCl2, supplemented with complete EDTA-free protease inhibitors (Roche). Cell lysates were bound to Ni-NTA agarose beads for 1 h with agitation at 4°C. Elution was performed using polypropylene columns and increasing concentrations of imidazole (0 to 2 M) at 4°C. Fractions with purified full-length integrase proteins were identified by 10% SDS-PAGE and Coomassie staining, digested with thrombin (Sigma) as described previously (38), and dialyzed against a storage buffer (20 mM HEPES [pH 7.5], 1 M NaCl, 1 mM EDTA, 5 mM DTT, 10% glycerol) for 16 to 20 h, using a molecular weight cutoff value of 30 kDa to remove the His tag. Dialyzed proteins were checked by SDS-PAGE followed by Coomassie staining, which showed that full-length recombinant integrase proteins were consistently >90% pure (see Fig. S1 in the supplemental material). Purified proteins were stored at −80°C for several months without loss of activity. The WT, G118R, R263K, and G118R plus R263K recombinant proteins were purified simultaneously in parallel to ensure that the same buffers and conditions applied to different enzymes. We purified two separate batches of WT, G118R, R263K, and G118R plus R263K integrase proteins to ensure the reproducibility of our cell-free experiments. Strand transfer and DNA binding assays were performed with the two protein batches, and results were compiled after normalization, which was performed as detailed below.
Integrase strand transfer assays.
The target DNA for integration was prepared by annealing the oligonucleotides 5′-TGACCAAGGGCTAATTCACT-3′-biotin and 5′-AGTGAATTAGCCCTTGGTCA-3′-biotin (target DNA) using low-chelating Tris-EDTA (TE) buffer. The same method was used to produce preprocessed HIV-1 LTR DNA with the oligonucleotides 5′-Am-MC12-ACCCTTTTAGTCAGTGTGGAAAATCTCTAGCA-3′ and 5′-ACTGCTAGAGATTTTCCACACTGACTAAAAG-3′, where Am-MC12 indicates a reactive amino group linked to the DNA by 12 carbon atoms. This LTR DNA had a 2-nucleotide 5′ staggered end and mimicked the 3′-processed HIV-1 DNA LTR that is the target for integrase. By using this substrate, we ensured that our assay measured only strand transfer. Another enzymatic activity of integrase, called 3′-processing, was not assessed. First, annealed preprocessed LTR DNA (100 nM) was linked covalently to DNA-BIND 96-well plates (2498; Corning) in PBS (pH 7.4) for 4 h at room temperature. Unbound DNA was removed, and the plates’ reactive surface was blocked by incubation with 0.5% FBS in PBS overnight at 4°C. The following day, DNA-coated plates were washed twice with 200 μL of PBS (pH 7.4) and assay buffer (50 mM morpholinopropanesulfonic acid [MOPS] [pH 6.8], 50 μg/mL bovine serum albumin [BSA], 50 mM NaCl, 30 mM MgCl2, 0.15% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate [CHAPS]). Purified integrase proteins were thawed on ice, diluted to their working concentrations in assay buffer with 5 mM DTT, and added to the DNA-coated plates within 15 min (100 μL/well), with three wells per condition. Annealed target double-stranded DNA (dsDNA) was diluted in assay buffer and added to the plates (50 μL). Strand transfer reactions were allowed to proceed for 1 h at 37°C. Negative controls included wells without LTR DNA, integrase proteins, or target DNA. After strand transfer, the plates were washed three times with 200 μL 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.05% Tween 20, 2 mg/mL BSA, to remove the free target DNA. Covalently bound biotinylated target DNA was detected by adding 0.025 μg/mL Eu-labeled streptavidin (1244-360; Perkin Elmer) and 50 μM diethylenetriaminepentaacetic dianhydride (DTPA) (284-025; Sigma-Aldrich) diluted in wash buffer. After two washes with the wash buffer, 150 μL DELFIA enhancement solution (4001-0010; Perkin Elmer) was added to the plate, and the time-resolved fluorescence was measured on a FLUOStar Optima plate reader. Experiments were performed with two separate batches of purified recombinant proteins, twice for experiments with different integrase protein concentrations (25, 50, 100, 200, 400, 800, and 1,600 nM) plus 60 nM target DNA and three times for experiments with a fixed integrase protein concentration (400 nM) and different target DNA concentrations (1.875, 3.75, 7.5, 15, 30, 60, and 120 nM). Fluorescence values were normalized against results obtained with 400 nM WT protein (arbitrarily set at 100%) for each experiment in which we varied protein concentrations. For the experiments in which we varied the target DNA concentrations, fluorescence values were normalized against those obtained with the WT protein plus 120 nM target DNA (arbitrarily set at 100%) for each experiment. This normalization allowed us to compile the results from repeat experiments with two different batches of recombinant proteins. The combined normalized results obtained with various target DNA concentrations were fitted to binding saturation curves and used to determine the enzymatic parameters reported in Table 1, namely, the maximal integration (pseudo-Vmax), half-maximal substrate concentration (pseudo-Km), and enzymatic efficiency (pseudo-Vmax/pseudo-Km).
DNA binding assay.
We measured the direct interactions between purified recombinant integrase proteins and HIV LTR DNA using a previously published protocol (20, 21, 23). The HIV LTR-mimetic dsDNA was prepared by annealing the oligonucleotides 5′-CTTTTAGTCAGTGTGGAAAATCTCTAGCAGT-3′ and 5′-RhoR-ACTGCTAGAGATTTTCCACACTGACTAAAAG-3′, where RhoR stands for rhodamine red. Annealing was performed in low-chelating TE buffer, as described previously (27). Purified recombinant integrase proteins were thawed on ice and immobilized on 96-well high-bind microplates (3925; Corning) overnight at 4°C. Unbound proteins were washed away twice with PBS (pH 7.4), and plates were blocked with 5% BSA (Sigma-Aldrich) in PBS for 2 h. Excess BSA was removed with two PBS washes, and plates were equilibrated with 150 μL binding buffer (20 mM MOPS [pH 7.2], 20 mM NaCl, 7.5 mM MgCl2, 5 mM DTT). Various concentrations of dsDNA substrate (0 to 535 nM in 1:2 serial dilutions) were added, followed by 1 h of incubation at room temperature. After three washes with PBS, 100 μL PBS was added to each well, and RhoR-mediated fluorescence was measured at 544-nm excitation and 590-nm emission wavelengths using a FLUOStar Optima plate reader. Background fluorescence from integrase-negative wells was subtracted to yield specific relative fluorescent units. Each data point was produced in triplicate (three separate wells). Two batches of enzymes were used, and results were compiled via normalization against DNA binding values obtained with the WT protein and 120 nM viral DNA, arbitrarily set at 1. Results were plotted against DNA concentrations, binding saturation curves were produced using GraphPad Prism v9 software, and binding affinity (Kd) values were derived from these curves.
Virus production.
The pNL4.3-INBG118R+R263K plasmid was derived from the pNL4.3-INBR263K plasmid by site-directed mutagenesis, as described above. Proper site-directed mutagenesis was validated by Sanger sequencing. The pNL4.3-INBG118R and pNL4.3-INBR263K plasmids have been described elsewhere (14). WT, G118R, R263K, and G118R plus R263K HIV-1 viruses were produced by transfection in 293T cells (13). Briefly, 7.5 million 293T cells were seeded on day 0 in a 75-cm2 flask in Opti-MEM supplemented with 10% FBS without antibiotics. Eighteen to 24 h later, cells were checked to have reached approximately 75% confluence. Twenty-five micrograms of proviral plasmid DNA was transfected using 150 μL of Lipofectamine 2000, according to the manufacturer’s recommendations. Four hours after transfection, cell culture medium was renewed with Opti-MEM with 10% FBS. Virus production for 48 h was followed by the harvesting of the cell culture fluids (12 mL). Supernatants were centrifuged for 10 min at 3,500 rpm, decanted to a new 15-mL tube, and filtered gently through a 0.2-μm filter with a 15-mL syringe. Filtered viruses were treated with benzonase (Millipore), aliquoted into 500 μL aliquots, and frozen at −80°C for at least 24 h before they were used for infectivity assays and RT quantification.
Cell-based single-cycle infectivity assays.
Infectivity and resistance assays were performed as described via single-round infections of reporter TZM-bl cells (23). For infectivity assays, 30,000 TZM-bl cells were plated in 50 μL DMEM supplemented with 10% FBS plus penicillin-streptomycin in 96-well white opaque cell culture plates. Sixteen to 24 h later, viral stocks were rapidly thawed at 37°C and serially diluted 1:3 in DMEM with 10% FBS and penicillin-streptomycin 10 times, and 50 μL/well was added to TZM-bl cells in triplicate. Outside wells were filled with 200 μL/well of sterile PBS to limit evaporation. Twenty-four hours later, cell culture medium was removed by aspiration, and 60 μL reporter lysis buffer (Promega) was added to each well. Within 15 min, 30 μL lysate was transferred to a 96-well white opaque plate, and 30 μL of luciferase reagent (Promega) was added. Plates were immediately analyzed for luminescence on a MicroBetaTriLux microplate luminescence counter (Perkin-Elmer). Each data point was produced in triplicate. Relative luminescence units (RLUs) were plotted against viral titers and normalized using GraphPad Prism v 9.4.1 (GraphPad Software, LLC), with maximal and minimal luminescence arbitrarily set at 100% and 0%, respectively. This allowed us to compile the results from three separate experiments, two with the same virus batch and another one with a separate batch of viruses. In all cases, experiments were performed with all viruses simultaneously.
Antiretroviral drug susceptibility.
For antiretroviral drug susceptibility assays, a similar approach was used with some modifications. Thirty thousand TZM-bl cells per well were plated on a 96-well cell culture plate. Eighteen to 24 h later, 1:10 serial dilutions of DTG (0.16 pM to 1.6 μM) or BIC (0.14 pM to 1.4 μM) were added to the cells in triplicate. After 1 h of incubation, the cells were infected with the equivalent of 30,000 RT units of WT, G118R, R263K, or G118R plus R263K virus per well, as reported previously (23). After 24 h, luciferase production was measured as described above, and RLU values were plotted against drug concentrations. Normalization was performed as described above for infectivity assays using GraphPad Prism. The results from three separate experiments (two with one batch of viruses and one with another batch of viruses) were compiled thanks to normalization. This compilation was used to calculate FCs in 50% inhibitory concentrations (IC50s) relative to the WT virus using GraphPad Prism.
Multicycle replication assays.
The ability of the WT, G118R, R263K, and G118R plus R263K viruses to replicate over time was tested by infecting PM1 cells and measuring the RT activity released in the cell culture fluids 2, 4, 6, 9, 11, 13, 16, 18, and 20 days postinfection. Briefly, 30,000 PM1 cells per well were plated in triplicate in a 96-well plate. Eighteen to 20 h later, the cells were infected with the equivalent of 30,000 RT units per well for 1 h. Unbound viruses were removed, and 200 μL fresh complete RPMI 1640 medium was added to the cells. At the indicated time, 100 μL of cell-free medium was collected for RT quantification, and 10,000 uninfected PM1 cells in 100 μL fresh medium were added. This experiment was performed twice, with two different viral preparations. To compile the results of the two experiments, RT values were normalized against results for the WT virus at day 11, arbitrarily set at 100%. Compiled normalized RT results were plotted against time, and areas under the curves were calculated using GraphPad Prism and expressed relative to the value for the WT virus as relative replicative capacity.
Molecular modeling.
The tetrameric integrase complex with DNA and BIC (PDB code 6PUW) was used as a template to examine the effects of G118R on the integrase structure (6). In silico modeling effects of the individual R263K and G118R substitutions have been published previously (14, 18, 27). We used a published approach to examine the structure of the G118R mutant by modeling the mutated protein against chain A from PDB code 6PUW (18, 27). Proper conformation was validated by pairwise structure alignment on the RCSB PDB website (https://www.rcsb.org/alignment). We used AceDock to model BIC binding to the G118R-mutated integrase (39). Visualization and illustration creation were performed using Maestro v2022-2 software (Schrodinger LLC).
ACKNOWLEDGMENTS
This work was supported by the Canadian Institutes for Health Research (grant HB1 164063 to T.M.).
We are grateful to Maureen Oliveira for virus quantification.
We declare that we have no competing interests.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Saag MS, Gandhi RT, Hoy JF, Landovitz RJ, Thompson MA, Sax PE, Smith DM, Benson CA, Buchbinder SP, Del Rio C, Eron JJ, Fätkenheuer G, Günthard HF, Molina J-M, Jacobsen DM, Volberding PA. 2020. Antiretroviral drugs for treatment and prevention of HIV infection in adults: 2020 recommendations of the International Antiviral Society-USA Panel. JAMA 324:1651–1669. doi: 10.1001/jama.2020.17025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Segal-Maurer S, DeJesus E, Stellbrink H-J, Castagna A, Richmond GJ, Sinclair GI, Siripassorn K, Ruane PJ, Berhe M, Wang H, Margot NA, Dvory-Sobol H, Hyland RH, Brainard DM, Rhee MS, Baeten JM, Molina J-M. CAPELLA Study Investigators. 2022. Capsid inhibition with lenacapavir in multidrug-resistant HIV-1 infection. N Engl J Med 386:1793–1803. doi: 10.1056/NEJMoa2115542. [DOI] [PubMed] [Google Scholar]
- 3.Wainberg MA, Zaharatos GJ, Brenner BG. 2011. Development of antiretroviral drug resistance. N Engl J Med 365:637–646. doi: 10.1056/NEJMra1004180. [DOI] [PubMed] [Google Scholar]
- 4.Hare S, Maertens GN, Cherepanov P. 2012. 3′-processing and strand transfer catalysed by retroviral integrase in crystallo. EMBO J 31:3020–3028. doi: 10.1038/emboj.2012.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cook NJ, Li W, Berta D, Badaoui M, Ballandras-Colas A, Nans A, Kotecha A, Rosta E, Engelman AN, Cherepanov P. 2020. Structural basis of second-generation HIV integrase inhibitor action and viral resistance. Science 367:806–810. doi: 10.1126/science.aay4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Passos DO, Li M, Jóźwik IK, Zhao XZ, Santos-Martins D, Yang R, Smith SJ, Jeon Y, Forli S, Hughes SH, Burke TR, Craigie R, Lyumkis D. 2020. Structural basis for strand-transfer inhibitor binding to HIV intasomes. Science 367:810–814. doi: 10.1126/science.aay8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ballandras-Colas A, Chivukula V, Gruszka DT, Shan Z, Singh PK, Pye VE, McLean RK, Bedwell GJ, Li W, Nans A, Cook NJ, Fadel HJ, Poeschla EM, Griffiths DJ, Vargas J, Taylor IA, Lyumkis D, Yardimci H, Engelman AN, Cherepanov P. 2022. Multivalent interactions essential for lentiviral integrase function. Nat Commun 13:2416. doi: 10.1038/s41467-022-29928-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ballandras-Colas A, Maskell DP, Serrao E, Locke J, Swuec P, Jónsson SR, Kotecha A, Cook NJ, Pye VE, Taylor IA, Andrésdóttir V, Engelman AN, Costa A, Cherepanov P. 2017. A supramolecular assembly mediates lentiviral DNA integration. Science 355:93–95. doi: 10.1126/science.aah7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anstett K, Brenner B, Mesplede T, Wainberg MA. 2017. HIV drug resistance against strand transfer integrase inhibitors. Retrovirology 14:36. doi: 10.1186/s12977-017-0360-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cevik M, Orkin C, Sax PE. 2020. Emergent resistance to dolutegravir among INSTI-naive patients on first-line or second-line antiretroviral therapy: a review of published cases. Open Forum Infect Dis 7:ofaa202. doi: 10.1093/ofid/ofaa202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pham HT, Mesplede T. 2019. Bictegravir in a fixed-dose tablet with emtricitabine and tenofovir alafenamide for the treatment of HIV infection: pharmacology and clinical implications. Expert Opin Pharmacother 20:385–397. doi: 10.1080/14656566.2018.1560423. [DOI] [PubMed] [Google Scholar]
- 12.Rhee S-Y, Grant PM, Tzou PL, Barrow G, Harrigan PR, Ioannidis JPA, Shafer RW. 2019. A systematic review of the genetic mechanisms of dolutegravir resistance. J Antimicrob Chemother 74:3135–3149. doi: 10.1093/jac/dkz256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pham HT, Labrie L, Wijting IEA, Hassounah S, Lok KY, Portna I, Goring ME, Han Y, Lungu C, van der Ende ME, Brenner BG, Boucher CA, Rijnders BJA, van Kampen JJA, Mesplède T, Wainberg MA. 2018. The S230R integrase substitution associated with virus load rebound during dolutegravir monotherapy confers low-level resistance to integrase strand-transfer inhibitors. J Infect Dis 218:698–706. doi: 10.1093/infdis/jiy175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quashie PK, Mesplède T, Han Y-S, Oliveira M, Singhroy DN, Fujiwara T, Underwood MR, Wainberg MA. 2012. Characterization of the R263K mutation in HIV-1 integrase that confers low-level resistance to the second-generation integrase strand transfer inhibitor dolutegravir. J Virol 86:2696–2705. doi: 10.1128/JVI.06591-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brenner BG, Thomas R, Blanco JL, Ibanescu R-I, Oliveira M, Mesplède T, Golubkov O, Roger M, Garcia F, Martinez E, Wainberg MA. 2016. Development of a G118R mutation in HIV-1 integrase following a switch to dolutegravir monotherapy leading to cross-resistance to integrase inhibitors. J Antimicrob Chemother 71:1948–1953. doi: 10.1093/jac/dkw071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Rompay KKA, Hassounah S, Keele BF, Lifson JD, Ardeshir A, Watanabe J, Pham HT, Chertova E, Sowder R, Balzarini J, Mesplède T, Wainberg MA. 2019. Dolutegravir monotherapy of simian immunodeficiency virus-infected macaques selects for several patterns of resistance mutations with variable virological outcomes. J Virol 93:e01189-18. doi: 10.1128/JVI.01189-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lübke N, Jensen B, Hüttig F, Feldt T, Walker A, Thielen A, Däumer M, Obermeier M, Kaiser R, Knops E, Heger E, Sierra S, Oette M, Lengauer T, Timm J, Häussinger D. 2019. Failure of dolutegravir first-line ART with selection of virus carrying R263K and G118R. N Engl J Med 381:887–889. doi: 10.1056/NEJMc1806554. [DOI] [PubMed] [Google Scholar]
- 18.Underwood M, Horton J, Nangle K, Hopking J, Smith K, Aboud M, Wynne B, Sievers J, Stewart EL, Wang R. 2022. Integrase inhibitor resistance mechanisms and structural characteristics in antiretroviral therapy-experienced, integrase inhibitor-naive adults with HIV-1 infection treated with dolutegravir plus two nucleoside reverse transcriptase inhibitors in the DAWNING Study. Antimicrob Agents Chemother 66:e01643-21. doi: 10.1128/AAC.01643-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lancet HIV. 2020. End resistance to dolutegravir roll-out. Lancet HIV 7:e593. doi: 10.1016/S2352-3018(20)30231-9. [DOI] [PubMed] [Google Scholar]
- 20.Quashie PK, Mesplède T, Han Y-S, Veres T, Osman N, Hassounah S, Sloan RD, Xu H-T, Wainberg MA. 2013. Biochemical analysis of the role of G118R-linked dolutegravir drug resistance substitutions in HIV-1 integrase. Antimicrob Agents Chemother 57:6223–6235. doi: 10.1128/AAC.01835-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han Y-S, Xiao W-L, Quashie PK, Mesplède T, Xu H, Deprez E, Delelis O, Pu J-X, Sun H-D, Wainberg MA. 2013. Development of a fluorescence-based HIV-1 integrase DNA binding assay for identification of novel HIV-1 integrase inhibitors. Antiviral Res 98:441–448. doi: 10.1016/j.antiviral.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 22.van Kampen JJA, Pham HT, Yoo S, Overmars RJ, Lungu C, Mahmud R, Schurink CAM, van Boheemen S, Gruters RA, Fraaij PLA, Burger DM, Voermans JJC, Rokx C, van de Vijver DAMC, Mesplède T. 2022. HIV-1 resistance against dolutegravir fluctuates rapidly alongside erratic treatment adherence: a case report. J Glob Antimicrob Resist 31:323–327. doi: 10.1016/j.jgar.2022.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pham HT, Alves BM, Yoo S, Xiao MA, Leng J, Quashie PK, Soares EA, Routy J-P, Soares MA, Mesplède T. 2021. Progressive emergence of an S153F plus R263K combination of integrase mutations in the proviral DNA of one individual successfully treated with dolutegravir. J Antimicrob Chemother 76:639–647. doi: 10.1093/jac/dkaa471. [DOI] [PubMed] [Google Scholar]
- 24.Cheung PK, Shahid A, Dong W, Lepik KJ, Montaner JSG, Brockman MA, Brumme ZL, Brumme CJ. 2022. Impact of combinations of clinically observed HIV integrase mutations on phenotypic resistance to integrase strand transfer inhibitors (INSTIs): a molecular study. J Antimicrob Chemother 77:979–988. doi: 10.1093/jac/dkab498. [DOI] [PubMed] [Google Scholar]
- 25.Tsiang M, Jones GS, Goldsmith J, Mulato A, Hansen D, Kan E, Tsai L, Bam RA, Stepan G, Stray KM, Niedziela-Majka A, Yant SR, Yu H, Kukolj G, Cihlar T, Lazerwith SE, White KL, Jin H. 2016. Antiviral activity of bictegravir (GS-9883), a novel potent HIV-1 integrase strand transfer inhibitor with an improved resistance profile. Antimicrob Agents Chemother 60:7086–7097. doi: 10.1128/AAC.01474-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oliveira M, Ibanescu R-I, Anstett K, Mésplède T, Routy J-P, Robbins MA, Brenner BG. Montreal Primary HIV (PHI) Cohort Study Group. 2018. Selective resistance profiles emerging in patient-derived clinical isolates with cabotegravir, bictegravir, dolutegravir, and elvitegravir. Retrovirology 15:56. doi: 10.1186/s12977-018-0440-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quashie PK, Oliviera M, Veres T, Osman N, Han Y-S, Hassounah S, Lie Y, Huang W, Mesplède T, Wainberg MA. 2015. Differential effects of the G118R, H51Y, and E138K resistance substitutions in different subtypes of HIV integrase. J Virol 89:3163–3175. doi: 10.1128/JVI.03353-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mesplède T, Quashie PK, Osman N, Han Y, Singhroy DN, Lie Y, Petropoulos CJ, Huang W, Wainberg MA. 2013. Viral fitness cost prevents HIV-1 from evading dolutegravir drug pressure. Retrovirology 10:22. doi: 10.1186/1742-4690-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mesplède T, Leng J, Pham HT, Liang J, Quan Y, Han Y, Wainberg MA. 2017. The R263K dolutegravir resistance-associated substitution progressively decreases HIV-1 integration. mBio 8:157–163. doi: 10.1128/mBio.00157-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vavro C, Ruel T, Wiznia A, Montañez N, Nangle K, Horton J, Buchanan AM, Stewart EL, Palumbo P. 2022. Emergence of resistance in HIV-1 integrase with dolutegravir treatment in a pediatric population from the IMPAACT P1093 study. Antimicrob Agents Chemother 66:e01645-21. doi: 10.1128/AAC.01645-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paton NI, Musaazi J, Kityo C, Walimbwa S, Hoppe A, Balyegisawa A, Asienzo J, Kaimal A, Mirembe G, Lugemwa A, Ategeka G, Borok M, Mugerwa H, Siika A, Odongpiny ELA, Castelnuovo B, Kiragga A, Kambugu A. NADIA Trial Team. 2022. Efficacy and safety of dolutegravir or darunavir in combination with lamivudine plus either zidovudine or tenofovir for second-line treatment of HIV infection (NADIA): week 96 results from a prospective, multicentre, open-label, factorial, randomised, non-inferiority trial. Lancet HIV 9:e381. doi: 10.1016/S2352-3018(22)00092-3. [DOI] [PubMed] [Google Scholar]
- 32.Munir S, Thierry E, Malet I, Subra F, Calvez V, Marcelin A-G, Deprez E, Delelis O. 2015. G118R and F121Y mutations identified in patients failing raltegravir treatment confer dolutegravir resistance. J Antimicrob Chemother 70:739–749. doi: 10.1093/jac/dku474. [DOI] [PubMed] [Google Scholar]
- 33.Oliveira M, Mesplède T, Moïsi D, Ibanescu RI, Brenner B, Wainberg MA. 2015. The dolutegravir R263K resistance mutation in HIV-1 integrase is incompatible with the emergence of resistance against raltegravir. AIDS 29:2255–2260. doi: 10.1097/QAD.0000000000000866. [DOI] [PubMed] [Google Scholar]
- 34.Du Y, Zhang T-H, Dai L, Zheng X, Gorin AM, Oishi J, Wu T-T, Yoshizawa JM, Li X, Yang OO, Martinez-Maza O, Detels R, Sun R. 2017. Effects of mutations on replicative fitness and major histocompatibility complex class I binding affinity are among the determinants underlying cytotoxic-T-lymphocyte escape of HIV-1 Gag epitopes. mBio 8:e01050-17. doi: 10.1128/mBio.01050-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mesplede T, Osman N, Wares M, Quashie PK, Hassounah S, Anstett K, Han Y, Singhroy DN, Wainberg MA. 2014. Addition of E138K to R263K in HIV integrase increases resistance to dolutegravir, but fails to restore activity of the HIV integrase enzyme and viral replication capacity. J Antimicrob Chemother 69:2733–2740. doi: 10.1093/jac/dku199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mesplède T, Quashie PK, Hassounah S, Osman N, Han Y, Liang J, Singhroy DN, Wainberg MA. 2015. The R263K substitution in HIV-1 subtype C is more deleterious for integrase enzymatic function and viral replication than in subtype B. AIDS 29:1459–1466. doi: 10.1097/QAD.0000000000000752. [DOI] [PubMed] [Google Scholar]
- 37.Anstett K, Cutillas V, Fusco R, Mesplède T, Wainberg MA. 2016. Polymorphic substitution E157Q in HIV-1 integrase increases R263K-mediated dolutegravir resistance and decreases DNA binding activity. J Antimicrob Chemother 71:2083–2088. doi: 10.1093/jac/dkw109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bar-Magen T, Sloan RD, Faltenbacher VH, Donahue DA, Kuhl BD, Oliveira M, Xu H, Wainberg MA. 2009. Comparative biochemical analysis of HIV-1 subtype B and C integrase enzymes. Retrovirology 6:103. doi: 10.1186/1742-4690-6-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruiz-Carmona S, Alvarez-Garcia D, Foloppe N, Garmendia-Doval AB, Juhos S, Schmidtke P, Barril X, Hubbard RE, Morley SD. 2014. rDock: a fast, versatile and open source program for docking ligands to proteins and nucleic acids. PLoS Comput Biol 10:e1003571. doi: 10.1371/journal.pcbi.1003571. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download aac.01386-22-s0001.tif, TIF file, 12.9 MB (12.9MB, tif)