

Keywords: apoptosis, caspase-3, kinases, MAPKAPK2, nuclear translocation
Abstract
We have previously identified mitogen-activated protein kinase-activated protein kinase 2 (MK2) is required for caspase-3 nuclear translocation in the execution of apoptosis; however, little is known of the underlying mechanisms. Therefore, we sought to determine the role of kinase and nonkinase functions of MK2 in promoting nuclear translocation of caspase-3. We identified two non-small cell lung cancer cell lines for use in these experiments based on low MK2 expression. Wild-type, enzymatic and cellular localization mutant MK2 constructs were expressed using adenoviral infection. Cell death was evaluated by flow cytometry. In addition, cell lysates were harvested for protein analyses. Phosphorylation of caspase-3 was determined using two-dimensional gel electrophoresis followed by immunoblotting and in vitro kinase assay. Association between MK2 and caspase-3 was evaluated using proximity-based biotin ligation assays and co-immunoprecipitation. Overexpression of MK2 resulted in nuclear translocation of caspase-3 and caspase-3-mediated apoptosis. MK2 directly phosphorylates caspase-3; however, phosphorylation status of caspase-3 or MK2-dependent phosphorylation of caspase-3 did not alter caspase-3 activity. The enzymatic function of MK2 was dispensable in nuclear translocation of caspase-3. MK2 and caspase-3 associated together and a nonenzymatic function of MK2, chaperoned nuclear trafficking, is required for caspase-3-mediated apoptosis. Taken together, our results demonstrate a nonenzymatic role for MK2 in the nuclear translocation of caspase-3. Furthermore, MK2 may function as a molecular switch in regulating the transition between the cytosolic and nuclear functions of caspase-3.
INTRODUCTION
Apoptosis is a highly regulated form of programmed cell death, a process mediated by caspases, a family of cysteine proteases. Upon activation (i.e., cleavage), caspases act to amplify death stimuli via initiator caspases, and cell dismantling proceeds via the executioner caspases (1). Traditionally, activation of caspase-3, the major executioner caspase, is perceived as the terminal step of the apoptotic cascade and has heralded cell death (1–3). However, our group recently identified a disconnect between the activation of caspase-3, and the execution of apoptosis (4, 5).
We observed that endotoxin induces caspase-3 activation, endothelial apoptosis and endothelial permeability. Notably, loss of the signaling molecule mitogen-activated protein kinase-activated protein kinase 2 (MK2) resulted in the prevention of both apoptosis and endothelial barrier dysfunction; yet, to our surprise, caspase-3 activation was preserved. Despite being activated, when MK2 was absent, caspase-3 remained in the cytoplasm, preventing endotoxin-induced apoptosis (4). Furthermore, using thrombin stimulation of endothelial cells [an insult known to impact cytoskeletal changes (6) and not induce apoptosis], we noted caspase-3 is activated but remains in the cytoplasm where it exerts barrier protective effects to the endothelium (5). These observations suggest nuclear translocation of caspase-3 is required for the execution of apoptosis.
A specific role for nuclear translocation of caspase-3 in apoptosis has been postulated based on the transfection of active/cleaved subunits of caspase-3 fused to a nuclear localization signal (NLS) moiety, resulting in massive apoptosis of transfected cells (7). Furthermore, apoptosis induced by microinjection of active/cleaved caspase-3 was prevented by co-injection with wheat germ agglutinin, a compound known to inhibit all nuclear transport, suggesting that interfering with caspase-3 nuclear translocation prevents apoptosis, despite activation of the apoptotic pathway (8). The classic paradigm for active nuclear transport involves the binding of an NLS region within a target protein to a specific family of soluble karyopherin α (9) carrier proteins, which in turn facilitate nuclear transport of the target protein via the nuclear pore complexes (10). Since caspase-3 does not have an identifiable NLS, an alternative mechanism for nuclear translocation of caspase-3 must exist. However, the fundamental mechanism(s) by which caspase-3 translocates to the nucleus are poorly understood.
Based on our previous findings of MK2 deficiency and cytosolic sequestration of caspase-3 and the fact that MK2 is a kinase, which phosphorylates substrates to propagate downstream signaling, we hypothesized that MK2-dependent phosphorylation of caspase-3 is required for nuclear translocation of caspase-3 and subsequent apoptosis.
MATERIALS AND METHODS
The Johns Hopkins University Institutional Animal Care and Use Committee approved all animal protocols.
Male C57BL/6J (wild type, WT) mice aged 10–12 wk (Jackson Laboratory, Bar Harbor, ME) and MK2−/− mice, C57BL/6J background (11) were exposed to intravenous (IV) PBS or lipopolysaccharide (LPS, 0127:B8, product # L3129, Sigma) via retro-orbital injection (12) for up to 6 h. After exposure to the experimental conditions, lungs were flushed free of blood, removed, and homogenized in cell lysis buffer (CST 9803 s, Cell Signaling, Boston, MA) supplemented with protease inhibitors cocktail (Sigma P8340), PMSF (1 mM, Thermo Fisher 36978), NaF (1 mM, Sigma, 201154), and NaOV (1 mM, Sigma, S6508). Lung lysates were frozen by immersion into liquid nitrogen and subsequently stored for later analyses.
Cell Lines
Non-small cell lung carcinoma (NSCLC) cell lines—H23 and A549 cell lines and small cell lung carcinoma (SCLC) cell line—H446 were purchased from ATCC (Manassas, VA). H23 and H446 cells were cultured in RPMI 1640 media (Thermo Fisher A1049101) supplemented with 10% (vol/vol) FBS (Hyclone). A549 cells were cultured in F-12K medium (Thermo Fisher 21127022) supplemented with 10% (vol/vol) FBS (Hyclone). Cells were maintained in full growth media in 75-cm2 flasks. Cells were grown at 37°C with 5% CO2.
Our previous experiments used a loss of function strategy to determine the importance of MK2 in nuclear translocation of caspase-3 (4). In our current experiments, we seek to understand the molecular mechanisms by which MK2 promotes nuclear translocation of caspase-3, necessitating gain of function experiments. Using previously published work (13) and the Broad Institute’s The Cancer Cell Line Encyclopedia (14), we identified H23 and A549 cell lines as suitable for our experiments, based on low MK2 expression.
Adenoviral Vectors
Adenoviral vectors encoding wild-type MK2 (Ad-WT-MK2), constitutively active MK2 (Ad-Active-MK2; T222E, T334E) (15), dominant negative MK2 (Ad-Dom Neg-MK2; K93R)(16), a mutated nuclear export sequence MK2 (Ad-Mut-NES; L360A) (17) and a mutated nuclear localization sequence MK2 (Ad-Mut-NLS-MK2; K372A, K373A, K388A, K389A) (18, 19) (Supplemental Fig. S1); wild-type MK2 fused with a mutant of the Escherichia coli biotin protein ligase BirA (20, 21) on the c-terminus (Ad-WT-MK2-BioID-C) or the n-terminus (Ad-WT-MK2-BioID-N); phospho-mimicking HSP27 (Ad-PM-HSP27; S15D, S78D, S82D) (22, 23) were directly purchased from Vector Builder (Chicago, IL). The sequences of the plasmids encoding these vectors were verified by Sanger sequencing (The Genetics Resources Core Facility, Johns Hopkins University). Table 1 provides a name and brief description of each viral vector.
Table 1.
Adenoviral constructs used
| Construct | Function |
|---|---|
| Ad-eGFP | Control for infection |
| Ad-WT-MK2 | Wild-type MK2 |
| Ad-Dom Neg-MK2 | Dominant negative MK2 |
| Ad-Active-MK2 | Constitutively active MK2 |
| Ad-WT-MK2-BioID-C | Wild-type MK2, biotin ligase fused to C-terminal |
| Ad-WT-MK2-BioID-N | Wild-type MK2, biotin ligase fused to N-terminal |
| Ad-Mut-NLS-MK2 | MK2 w/ mutated NLS (cytosolic predominant) |
| Ad-Mut-NES-MK2 | MK2 w/ mutated NES (nuclear predominant) |
Infections
Cells were seeded in 6-well plates at a cell density of 5 × 105 cells per well. Cells were grown at 37°C with 5% CO2 for ∼6 h to allow adherence. After adherence, media was replaced with media plus viral vector. Cells were incubated with viral vector to have a final plaque-forming unit number of up to 100. Cells were left to incubate with the viral media at 37°C with 5% CO2 for ∼24 h. Viral media was then replaced with the appropriate subsequent media, depending on experimental conditions.
Immunoblot Analyses
Cell cultures were lysed using cell lysis buffer (CST 9803 s, Cell Signaling, Boston, MA) supplemented with protease inhibitors cocktail (Sigma P8340), PMSF 1 mM, Thermo Fisher 36978), NaF (1 mM, Sigma, 201154), and NaOV (1 mM, Sigma, S6508). Protein lysates were denatured using Laemmli Sample buffer (Bio-Rad 1610747), 2-mercaptoethanol (Millipore Sigma M6250), and 100°C heat (5-min exposure). Proteins were separated by SDS-PAGE (Thermo Fisher XP00122BOX), and transferred to PVDF membranes (Bio-Rad 1620177). Membranes were blocked in 5% nonfat dry milk (Bio-Rad 1706404) in TBS (Quality Biological 50983267) with 0.5% Tween-20 (Thermo Fisher BP337-500). Membranes were incubated with primary antibodies at 1:1,000 dilution overnight in 2.5% nonfat dry milk. PVDF membranes were then incubated with horseradish peroxidase-linked secondary antibodies, anti-mouse (CST 7076) or anti-rabbit (CST 7074), at 1:5,000 dilution for 1 h in 1% nonfat dry milk. Immunoblots for Streptavidin conjugated to horseradish peroxidase used bovine serum albumin instead of nonfat dry milk. Approximately 20 µg of total protein was loaded for immunoblots in experiments investigating total protein lysates. Approximately 1 µg of total protein was loaded in immunoblot experiments when investigating nuclear and cytosolic fractions, given the reduced protein yield within nuclear fractions. All immunoblot experiments used the same amount of protein for each sample and all membranes were probed for a loading control to ensure a similar amount of protein, when appropriate. Protein bands were visualized using chemiluminescent detection methods. Band intensities were quantified using ImageJ software.
Phospho-specific anti-total antibodies directed at HSP27(p-HSP27- CST-2401; t-HSP27- CST-2402) and anti-total antibodies directed at MK2 (CST-3042), caspase-3 (CST-9662), β-tubulin (CST-5346), GAPDH (CST-3683), PARP1 (CST-9542), HDAC2 (CST-2540) (Cell Signaling, Boston, MA) were used. Streptavidin conjugated to horseradish peroxidase was also used (CST-3999).
Nuclear and Cytosolic Fractionation
Cells were trypsinized and re-suspended in PBS. Nuclear and cytosolic fractions of the resulting cell suspensions were generated using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher 78833, Rockford, IL). The purity of cytosolic fractions was assessed by lack of PARP1 or HDAC2 immunoreactivity and the purity of the nuclear fraction was assessed by lack of GAPDH immunoreactivity (Cell Signaling, Boston, MA) using standard immunoblotting techniques.
Bright-Field Microscopy
Six hours after sub-confluent plating, H23 cells were infected with Ad-eGFP or Ad-WT MK2 and imaged over time. A mark was placed under side each dish to allow for sequential imaging of the same region. All images were anonymized and number of cells per image were counted by blinded investigators (A.S., G.S., M.K.). Cell counts obtained by each investigator were subsequently averaged together.
In Vitro Kinase Assay
Reactions were performed in 50 mM sodium β-glycerophosphate containing 10 mM Mg-acetate, 0.1 mM ATP, 2 uCi γ-32P-ATP (New England Nuclear, PerkinElmer, Waltham, MA), and 170 ng (6 U) of recombinant active MK2 (Millipore, Dundee, UK). Substrates were recombinant active caspase-3 (250 ng, Enzo, Farmingdale, NY) or recombinant HSP27 (14 ng, Enzo, Farmingdale, NY). Reactions were incubated at 30°C for 30 or 60 min, and resolved on 4%–12% NUPAGE gels (Life Technologies, Carlsbad, CA). After staining with Coomassie Blue, gels were dried and phosphorylated proteins were detected on a Molecular Imager FX phosphorimager (Bio-Rad, Hercules, CA) using Quantity One software.
Bio-ID Assay
H23 cells were infected with Ad-WT MK2, Ad-WT-MK2-BioID-C, or Ad-WT-MK2-BioID-N for 24 h after which the media was changed to include biotin (50 μM) (Thermo Fisher, 29129) for an additional 18 h, after which cell lysates were prepared. Cell lysates were incubated with 50 μL of Streptavidin Sepharose High-Performance Beads (GE Healthcare, 17511301) overnight at 4°C with gentle shaking. Beads were washed four times with buffer to elute off non-specific proteins bound to the sepharose beads. The buffer was aspirated and beads were re-suspended in Laemmli buffer for Western blotting.
Co-Immunoprecipitation
H23 cells were infected with Ad-eGFP, Ad-WT MK2, Ad-Mut-NLS-MK2 or Ad-Mut-NES-MK2 and 48 h afterward cell lysates were prepared. Appropriate cell lysates (50 µg) were treated with anti-MK2 (CST-3042, 1:100 dilution) or isotype control antibody (SC-2027X, 1:100) for overnight incubation at 4°C with gentle shaking. Protein A sepharose beads (GE Healthcare 17-5280-01; 10 µL) were added on the next day and incubated with shaking for 3 h at 4°C. Beads were washed two times with Krebs+ buffer to elute off non-specific proteins bound to the protein A sepharose beads. The buffer was aspirated and beads were re-suspended in 20 µl 2× Laemmli sample buffer. The samples were separated by 4%–12% SDS-PAGE and immunoblotted.
Flow Cytometry
Following experimental exposures, cell cultures were trypsinized and single-cell suspension was generated. 4',6-Diamidino-2-phenylindole, dihydrochloride (DAPI, Thermo Fisher D21490) was used to stain DNA of cells as a way to quantify condensed and fragmented nuclei, a hallmark of apoptosis (24). Data acquisition was performed on a custom FACSAria II instrument running FACSDiva acquisition software (BD Biosciences, San Jose, CA). A singly stained aliquot of H23 cells for the DAPI fluorochrome and unstained cells were used to compensate for background auto-fluorescence. 5 × 104 events were obtained per sample and analyzed using FCS Express 6 (De Novo Software, Pasadena, CA).
Statistical Analysis
Data are shown as means ± SD. Since data are obtained using cell lines, biological replicates are not feasible. Data from separate individual cultures (each individual culture represents N of 1) are plotted for each condition. Sample size is identified in Figure Legends. A combination of parametric and nonparametric tests was used. The specific statistical test and post hoc testing performed are identified within each Figure Legend. A P value of less than 0.05 was considered significant. Data were analyzed using GraphPad Prism 8 (La Jolla, CA).
RESULTS
MK2 Expression in H23 NSCLC Cells Results in Caspase-3-Mediated Apoptosis
Joseph et. al. (13) showed that NSCLCs are able to activate the apoptotic machinery in response to chemotherapy, even to the point of activating caspase-3. However, they observed that active caspase-3 was sequestered in the cytoplasm of NSCLC cell lines, unlike SCLC cell lines, where active caspase-3 translocated to the nucleus. This cytoplasmic sequestering of caspase-3 in chemo-resistant NSCLCs mirror our findings in human lung microvascular endothelial cells of cytoplasmic caspase-3 localization under conditions of MK2 deficiency (4). Therefore, we obtained previously characterized NSCLC and SCLC cell lines (13) and measured MK2 protein expression. There is markedly reduced MK2 expression in H23 cells compared with H446 cells (Fig. 1A). Using H23 cells (given endogenously low MK2), we verified adenoviral-mediated expression of MK2 (Fig. 1B). As shown in Fig. 1C (Supplemental Fig. S2), H23 cells infected with Ad-eGFP continue to grow over time, whereas those infected with Ad-WT-MK2 failed to grow and have significantly reduced cell numbers at 144 h, suggesting increased cell death. Using flow cytometry (24) (Supplemental Fig. S3), we observed increasing cell death at 48 h based on Ad-WT-MK2 multiplicity of infection (Fig. 1D).
Figure 1.

MK2 expression induces apoptosis in H23 cells. A: cell lysates from NSCLC cell line, H23 and the SCLC cell line, H446, were immunoblotted with antibodies recognizing total MK2. There is markedly less MK2 expression in H23 cells. B: H23 NSCLC cells were infected with adenovirus encoding MK2 or eGFP (Ad-WT-MK2 and Ad-eGFP, respectively) for 48 hrs and then harvested for protein analyses. H23 cells infected with Ad-MK2 leads to marked increase in MK2 protein expression. C: H23 NSCLC cells were infected with Ad-WT-MK2 or Ad-eGFP for up to 144 h for analyses. Time lapse bright-field microscopy was used to show the effect of MK2 expression on H23 cells. Quantification of cell counts demonstrates Ad-eGFP infected cells continue to grow over time compared Ad-WT-MK2 infected cells. H23 cells infected with Ad-WT-MK2 resulted in reduced cell counts compared to baseline. n = 22 individual cultures per group were analyzed over time. *P < 0.05 vs. Ad-WT-MK2 by unpaired Mann–Whitney test. #P < 0.05 vs. Ad-WT-MK2 6 h by unpaired Mann–Whitney test. D: after 48 h of infection, there is a dose-dependent increase in cell death based on Ad-WT-MK2 multiplicity of infection. *P < 0.05 vs. Ad-eGFP and 50 MOI using Dunnett’s multiple comparison test. E: H23 cells were infected with Ad-eGFP for 24 h and then treated with vehicle or etoposide 96 μg/mL for 24 h or Ad-WT-MK2. Cell lysates were analyzed for caspase-3 activity. There is significant increase in caspase-3/7 activity with MK2 expression. Ad-eGFP infected H23 cells treated with etoposide exhibited the greatest increase in caspase-3/7 activity. #P < 0.05 vs. Ad-eGFP. *P < 0.05 vs. all others. All post hoc analyses using Tukey’s multiple comparisons test. n = 6–21 per group. F: H23 cells were infected with Ad-eGFP for 24 h and then treated with vehicle or etoposide 96 μg/mL for 24 h or Ad-WT-MK2. Cells were then analyzed for cell death using flow cytometry. There is significant increase in cell death with Ad-WT-MK2. There is no difference in cell death between Ad-WT-MK2 infected cells and Ad-eGFP infected cells treated with etoposide. *P < 0.05 vs. Ad-eGFP. All post hoc analyses using Tukey’s multiple comparisons test. n = 6–12 per group. G: H23 cells were infected with Ad-WT-MK2 for 24 h and then pretreated with vehicle or DEVD (caspase-3 inhibitor, 50 μg/mL, 1 h) and then exposed to etoposide 96 μg/mL for 24 h and then analyzed for cell death using flow cytometry. Pretreatment with DEVD abrogated the MK2 expression-induced cell death. *P < 0.05 vs. all others. All post hoc analyses using Tukey’s multiple comparisons test. n = 3–9 per group. MK2, mitogen-activated protein kinase-activated protein kinase 2; MOI, multiplicity of infection; NSCLC, non-small cell lung carcinoma; SCLC, small cell lung carcinoma; WT, wild type.
Since tumor cell death following chemotherapy administration, i.e., etoposide, is thought to proceed, in large part, via apoptosis—a caspase-3-mediated mechanism of programmed cell death, we sought to demonstrate that the cell death observed from overexpression of MK2 is via apoptosis, and if overexpression of MK2 could potentiate etoposide-induced cell death. We initially explored whether MK2 expression induces caspase-3-mediated cell death, i.e., apoptosis. Although Ad-WT-MK2 infected H23 cells increased caspase-3 activity, Ad-eGFP infected cells exposed to etoposide (dosing that induces cell death, Supplemental Fig. S4A) demonstrated higher caspase-3 activity than with Ad-WT-MK2 infection alone (Fig. 1E). Despite having a significantly lower amount of caspase-3 activity, Ad-WT-MK2 infected cells exposed to vehicle had a similar amount of cell death when compared with Ad-eGFP infected H23 cells exposed to etoposide (Fig. 1F). In Ad-eGFP and Ad-WT-MK2 infected cells exposed to etoposide, there was similar caspase-3 activation, but greater cell death in those infected with Ad-WT-MK2 (Supplemental Fig. S4, B and C). Pretreatment with DEVD, a caspase-3 specific inhibitor, completely abrogated MK2 expression-induced cell death (Fig. 1G). These data demonstrate induction of MK2 expression in H23 cells resulted in caspase-3-mediated cell death, thereby validating H23 cells as an appropriate cell culture model to mechanistically test the role of MK2 on caspase-3-mediated cell death.
MK2 Phosphorylates Caspase-3 but Does Not Affect Its Activity
We next explored potential mechanistic interactions between MK2 and caspase-3 that might explain the synergy between MK2 and caspase-3 with regards to apoptosis. Because MK2 is a kinase, we first sought to determine if MK2 could phosphorylate caspase-3. In wild-type mice, intravenous LPS exposure activates MK2 and induces caspase-3-mediated cell death (4). In lung homogenates obtained from LPS-exposed mice, two-dimensional immunoblotting for caspase-3 demonstrated a shift toward the positive electrode on an isoelectric gradient (Supplemental Fig. S5). Treatment of lung homogenates of WT mice exposed to IV LPS with active recombinant serine/threonine phosphatase, PP2A, reversed these charge-based shifts, suggestive of an LPS-induced phosphorylation event on caspase-3. In addition, two-dimensional immunoblotting for caspase-3 of lung homogenates from MK2−/− mice exposed to LPS is similar to that of lung homogenates from WT animals exposed to LPS and treated with active recombinant PP2A, suggestive of a MK2-dependent phosphorylation event on caspase-3, as MK2 is a serine/threonine kinase (Supplemental Fig. S5). Given this suggestive data, we sought to test the hypothesis that synergistic interaction between MK2 and caspase-3 is mediated by direct phosphorylation of caspase-3. Although these data are suggestive, they do not provide conclusive proof that MK2 directly phosphorylates caspase-3, as other potential serine/threonine kinases may be directly or indirectly involved. To that end using a reductionist approach, we initially tested whether MK2 could directly phosphorylate caspase-3 in a cell-free system. Using recombinant proteins in cell-free assays, we observed that activated MK2 phosphorylates caspase-3 (Fig. 2A). We next tested whether MK2 could phosphorylate caspase-3 in vitro using H23 cells. Two-dimensional gel immunoblotting of caspase-3 in Ad-WT MK2 infected H23 cells demonstrated a leftward shift in the isoelectric focus toward the positive electrode (Fig. 2B) compared with Ad-eGFP infected cells. In addition, treatment of Ad-WT MK2 cell lysates with PP2A reversed the leftward shift of caspase-3 (Fig. 2B). To isolate the effects of MK2’s kinase activity on caspase-3, we generated functional mutations of MK2 that alter its enzymatic activity: a constitutively active mutant of MK2 (Ad-Active-MK2) and a dominant negative mutant of MK2 (Ad-Dom-Neg-MK2), as previously described (15, 16). As shown in Fig. 2C, lysates of H23 cells infected with Ad-Active-MK2 demonstrated a significant phosphorylation of HSP27, whereas lysates of H23 cells infected with Ad-Dom-Neg-MK2 have essentially no phosphorylated HSP27 (Fig. 2C). As seen in Fig. 2D, there is a leftward shift toward the positive electrode on an isoelectric gradient in Ad-Active-MK2 infected H23 cells that is not present in Ad-Dom Neg-MK2 infected cells, suggestive of an MK2-dependent phosphorylation event on caspase-3. However, the consequences of MK2-dependent phosphorylation of caspase-3 are unknown. To that end, we sought to determine whether the phosphorylation status of caspase-3 altered its enzymatic activity by using the serine/threonine phosphatase PP2A to de-phosphorylate caspase-3. As shown in Fig. 2E, treatment of lysates of H23 cells infected with Ad-eGFP or Ad-WT MK2 with PP2A did not change caspase-3 activity. Furthermore, infection of H23 cells with either Ad-Active-MK2 or Ad-Dom-Neg-Mk2 does not differentially affect caspase-3 activity (Fig. 2F), despite having a differential effect on caspase-3 phosphorylation (Fig. 2D). In summary, these data suggest that while MK2 phosphorylates caspase-3, this function does not affect caspase-3 activity.
Figure 2.

MK2-dependent phosphorylation of caspase-3 does not alter caspase-3 activity. A: kinase activity assays of recombinant activated MK2 (1 μM) and recombinant caspase-3 (100 μM) with 32P-ATP. Coomassie stained gel shows relative protein amounts. Phosphorimaging shows phosphorylation of caspase-3. Lanes—1: 30-min reaction; 2: 60-min reaction; 3: 4–100 nM of HSP27, 30-min reaction as positive control. B: H23 cells were infected with Ad-eGFP or Ad-WT-MK2 and cell lysates underwent two-dimensional immunoblotting for caspase-3. As shown, there is a shift toward the positive electrode on an isoelectric gradient in Ad-MK2 infected H23 cells (red dash arrows). In addition, a band is present toward the negative electrode on an isoelectric gradient in Ad-eGFP infected cells that is not present in Ad-MK2 infected cells (red dash circles). Treatment of cell lysates from Ad-MK2 infected H23 cells with active recombinant serine/threonine phosphatase, PP2A, reverses these charge-based shifts. C: H23 cells were infected with Ad-eGFP, Ad-WT-MK2, Ad-Active-MK2 (constitutively active mutant of MK2) or Ad-Dom Neg-MK2 (a dominant negative mutant of MK2) and 48 h afterwards, cell lysates were analyzed for protein expression. There is significant phosphorylation of HSP27 (the canonical substrate of MK2) in Ad-WT-MK2 and Ad-Active-MK2 but not Ad-eGFP and Ad-Dom Neg-MK2 infected cells. D: H23 cells were infected with Ad-Dom Neg-MK2 or Ad-Active-MK2 and cell lysates underwent two-dimensional immunoblotting for caspase-3. A shift toward the positive electrode on an isoelectric gradient in Ad-Active-MK2 infected H23 cells is not present in Ad-Dom Neg-MK2 infected cells (red dash arrows). E: H23 cells were infected with Ad-eGFP or Ad-WT-MK2 for 48 hours and cell lysates were treated with active recombinant PP2A or vehicle and analyzed for caspase-3/7 activity. There is no interaction between MK2 expression and PP2A treatment on caspase-3/7 activity. There is no within group effect of PP2A treatment on caspase-3/7 activity using post-hoc Tukey’s multiple comparisons test. n = 9–18 per group. F: H23 cells were infected with Ad-eGFP, Ad-Active-MK2 or Ad-Dom Neg-MK2 for 48 h and cell lysates were analyzed for caspase-3 activity. Both MK2 constructs resulted in significant increase caspase-3 activity compared with Ad-eGFP infected cells. There was no difference in caspase-3 activity when comparing Ad-Active-MK2 or Ad-Dom Neg-MK2. @, P < 0.05 vs. all others. All post hoc analyses using Dunn’s multiple comparisons test. n = 9 per group. MK2, mitogen-activated protein kinase-activated protein kinase 2.
Enzymatic Function of MK2 Is Dispensable in Nuclear Translocation of Caspase-3 and Resultant Apoptosis
We have previously shown that MK2 is required for nuclear translocation of caspase-3 (4). We sought to determine if the enzymatic activity of MK2 was required for nuclear translocation of caspase-3. Infection of H23 cells with Ad-WT-MK2 resulted in increased nuclear compared with cytosolic caspase-3 activity (Fig. 3A) demonstrating nuclear translocation of caspase-3. Increased nuclear caspase-3 activity resulted in cleaved PARP1, a nuclear substrate of caspase-3 critical in the execution of apoptosis (2, 25–27) (Fig. 3B). As shown in Fig. 3C, there was a significant increase in nuclear compared with cytosolic caspase-3 activity with both Ad-Active-MK2 and Ad-Dom-Neg-MK2 conditions, which resulted in increased PARP1 cleavage (Fig. 3D, Supplemental Fig. S6A). H23 cells infected with Ad-WT-MK2, Ad-Active-MK2, or Ad-Dom-Neg-MK2 all resulted in similarly high cell death when compared with cells infected with Ad-eGFP (Fig. 3E). Collectively, these data show phosphorylation of caspase-3 by MK2 and MK2’s enzymatic activity is dispensable in MK2-mediated caspase-3 nuclear translocation and resultant apoptosis.
Figure 3.

Enzymatic activity of MK2 is dispensable in nuclear translocation of caspase-3. A: H23 cells were infected with Ad-eGFP or Ad-WT-MK2 and 48 h afterwards nuclear and cytosolic fractions were separated for analyses. H23 cells infected with Ad-eGFP show a reduced nuclear caspase-3/7 activity compared to the cytosolic compartment. Whereas, H23 cells infected with Ad-WT-MK2 exhibited significantly increased nuclear caspase-3/7 activity compared to the cytosolic compartment. Purity of nuclear and cytosolic fractions was assessed by exclusion of GAPDH and cleaved PARP1, respectively. N, nuclear. C, cytosolic. *P < 0.05 vs. cytosolic compartment by unpaired t test. n = 12 per group. B: representative immunoblot of lysates from H23 cells infected with either Ad-eGFP2 or Ad-WT-MK2 for 48 h indicates an increase in cleavage of PARP1 in Ad-WT-MK2 infected H23 cells compared with Ad-eGFP infected H23 cells, confirmed by densitometric analysis. *P < 0.05 by unpaired t test. n = 11–12 per group. C: H23 NSCLC cells infected with either Ad-Active-MK2 or Ad-Dom Neg-MK2 for 48 h exhibited significantly increased nuclear compared to cytosolic caspase-3/7 activity. N, nuclear. C, cytosolic. *P < 0.05 vs. cytosolic compartment by unpaired t test. n = 9 per group. D: H23 cells infected with either Ad-Active-MK2 or Ad-Dom Neg-MK2 exhibited significantly increased cleavage of PARP1 compared with cells infected with Ad-eGFP. *P < 0.05 vs. all others in post hoc testing using Tukey’s multiple comparisons test. n = 3 per group. E: H23 cells infected with Ad-WT-MK2, Ad-Active-MK2 or Ad-Dom Neg-MK2 for 48 hrs resulted in significantly higher amount of cell death compared to H23 cells infected with Ad-eGFP. *P < 0.05 vs. all others in post hoc testing using Tukey’s multiple comparisons test. n = 9 per group. MK2, mitogen-activated protein kinase-activated protein kinase 2; NSCLC, non-small cell lung carcinoma; SCLC, small cell lung carcinoma; WT, wild type.
MK2 Associates with Caspase-3 and MK2’s Nuclear Translocation is Required for Increased Caspase-3 Activation and Resultant Apoptosis
MK2 has both a nuclear localization sequence (NLS) and a nuclear export sequence (NES) (17). Since caspase-3 lacks an NLS, we reasoned that caspase-3 may associate with MK2 to gain access to the nucleus. Using proximity-based biotinylation assays, we show that caspase-3 is preferentially detected following pulldown with streptavidin when the biotin ligase is fused to the C-terminus of MK2 (Fig. 4A, Supplemental Fig. S7), demonstrating association between caspase-3 and MK2’s C-terminal tail. We next generated MK2 constructs with a mutated NLS, Ad-Mut-NLS-MK2, or a mutated NES, Ad-Mut-NES-MK2 (17–19) (Fig. 4B). As shown in Fig. 4C, caspase-3 is detected following pulldown with MK2. There was no difference in amount of caspase-3 detected between cells infected with Ad-WT-MK2, Ad-Mut-NLS-MK2, or Ad-Mut-NES-MK2; these mutations do not influence MK2’s association with caspase-3 (Fig. 4C). Infection of H23 cells with Ad-WT-MK2 led to a significant increase in caspase-3 activity, which is prevented with Ad-Mut-NLS-MK2 (Fig. 4D). In addition, preventing nuclear translocation of MK2, as with Ad-Mut-NLS-MK2, completely protected against MK2 expression induced apoptosis in H23 cells (Fig. 4E).
Figure 4.

MK2 associates with caspase-3 and MK2’s nuclear translocation is required for increased caspase-3 activation and resultant apoptosis. A: H23 cells were infected with Ad-WT-MK2, Ad-WT-MK2-BioID-C or Ad-WT-MK2-BioID-N (MK2 fused to biotin ligase on the c-terminus or n-terminus) and following incubation with biotin, cell lysates were precipitated with beads conjugated to streptavidin and then immunoblotted using antibodies directed against caspase-3. There is increased biotinylated caspase-3 with biotin ligase fused to MK2 on the c-terminus. B: H23 cells were infected with Ad-eGFP, Ad-WT-MK2, Ad-Mut-NLS-MK2 (mutated nuclear localization sequence, rendering cytosolic sequestration), Ad-Mut-NES-MK2 (mutated nuclear export sequence, limiting nuclear export) and 48 h afterwards nuclear and cytosolic fractions were separated for analyses. Representative immunoblot shows wild-type and mutant NES-MK2 constructs translocate to the nuclear compartment, whereas the mutant NLS-MK2 construct is sequestered in the cytosolic fraction and unable to translocate to the nucleus. C: H23 cells were infected with Ad-WT-MK2, Ad-Mut-NLS-MK2, Ad-Mut-NES-MK2 and 48 h afterwards cell lysates were immunoprecipitated with antibodies against MK2 and then immunoblotted using antibodies directed against caspase-3. As shown, caspase-3 is associated with MK2. There is no difference in association based on localization mutations. D: H23 cells were infected with Ad-eGFP, Ad-WT-MK2, Ad-Mut-NLS-MK2 and Ad-Mut-NES-MK2 and 48 h afterwards cell lysates were analyzed for caspase-3 activity. Cells infected with wild-type and mutant NES-MK2 constructs exhibited a significant increase in caspase-3/7 activity compared with H23 cells infected with Ad-eGFP or Ad-Mut-NLS-MK2. *P < 0.05 vs. Ad-WT-MK2 and Ad-Mut-NES-MK2 in post hoc testing using Tukey’s multiple comparisons test. n = 3–6 per group. E: H23 cells were infected with Ad-eGFP, Ad-WT-MK2, Ad-Mut-NLS-MK2 and Ad-Mut-NES-MK2 and 48 h afterwards cells were analyzed for cell death using flow cytometry. Cells infected with wild-type and mutant NES-MK2 constructs exhibited a significant increase in cell death compared with H23 cells infected with Ad-eGFP or Ad-Mut-NLS-MK2. *P < 0.05 vs. Ad-WT-MK2 and Ad-Mut-NES-MK2 in post hoc testing using Tukey’s multiple comparisons test. n = 9 per group. MK2, mitogen-activated protein kinase-activated protein kinase 2; NLS, nuclear localization sequence; NES, nuclear export sequence.
Because molecular chaperoning is not a classic function for MK2, we sought to test a potential role for HSP27 in chaperoning caspase-3 to the nucleus. HSP27 is a well-described downstream substrate for MK2 (16, 28–30) and a molecular chaperone (31) and is known to bind to caspase-3 (32, 33). After its activation, MK2 forms a complex with HSP27 (30, 34) and phosphorylates HSP27 at amino acids 15, 78, and 82 (16, 29) in response to numerous stressors. HSP27 generally exists as a large oligomeric unit of up to 800 kDa, and its size is dependent on a number of parameters including the degree of phosphorylation of the individual monomers (23). In addition, phosphorylation of HSP27 has been shown to alter its cellular localization, with phospho-HSP27 translocating to the nucleus (22, 35) and non-phosphorylatable mutants of HSP27 remaining in the cytosol even after stimulation (22). Overexpressing phospho-mimicking HSP27, failed to increase nuclear caspase-3 activity (Supplemental Fig. S6, B and C), demonstrating an MK2 independent effect is responsible for nuclear translocation of caspase-3. In total, these data suggest that MK2 functions as a molecular chaperone for caspase-3 during nuclear translocation of both proteins and resultant caspase-3-mediated apoptosis.
To ensure the results of our mechanistic studies performed in H23 cells, a non-small cell lung cancer cell line, were not a consequence of the numerous mutations likely present in cancer cells, we replicated key findings using A549 cells. As shown in Fig. 5, A–D, increased MK2 expression results in increased caspase-3 activity, increased nuclear caspase-3 activity, as evidenced by increased PARP1 cleavage and subsequent cell death.
Figure 5.

MK2 expression in A549 cells leads to increased nuclear caspase-3 activity and cell death. A: A549 cells infected with Ad-WT-MK2 leads to marked increase in MK2 protein expression. B: A549 cells were infected with Ad-eGFP or Ad-WT-MK2 and 48 h afterwards lysates were assessed for caspase-3/7 activity. Ad-WT MK2 infected A549 cells exhibited significantly increased caspase-3/7 activity compared to cells infected with Ad-eGFP. *P < 0.05 using unpaired t test. n = 6 per group C: A549 cells were infected Ad-eGFP or Ad-WT-MK2 and 48 h afterwards cells were assessed for protein expression. Ad-WT-MK2 infected A549 cells exhibited significantly increased cleavage of PARP1, a nuclear target of caspase-3. n = 11 per group. D: A549 cells were infected Ad-eGFP or Ad-WT-MK2 and 48 h afterwards cells were assessed for cell death using flow cytometry. Ad-WT-MK2 infected A549 cells exhibited significantly increased cell death compared with cells infected with Ad-eGFP. *P < 0.05 using unpaired t test. n = 15 per group. MK2, mitogen-activated protein kinase-activated protein kinase 2; WT, wild type.
DISCUSSION
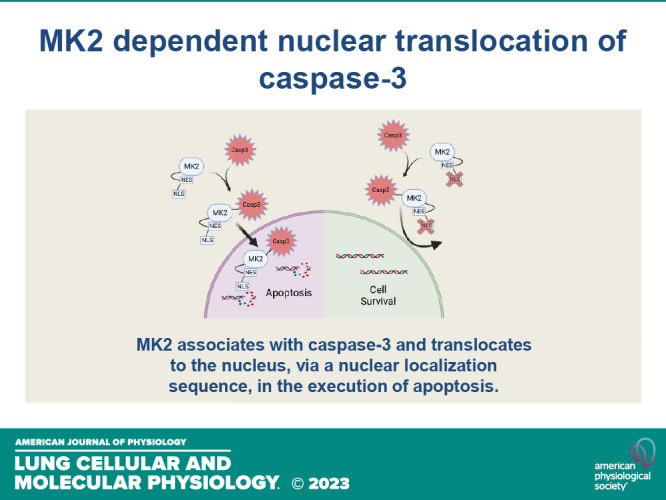
Despite some of the experiments within this manuscript demonstrating “negative” results, we feel the overall findings are novel and fundamental to understanding the mechanisms by which MK2 potentiates apoptosis. Because MK2 is a kinase, the most logical and expected function of its downstream signaling would be expected to relate to its enzymatic properties, which in fact was our hypothesis. Indeed, in Fig. 2, A and B, we show that MK2 directly phosphorylates caspase-3, which to our knowledge has not been previously shown. However, to our surprise, although MK2 phosphorylates caspase-3, this MK2-dependent phosphorylation did not alter caspase-3 activity (Fig. 2, E and F). Instead, our data revealed an unexpected, noncanonical function of MK2 that is critical for nuclear translocation of caspase-3. Our data strongly illustrate that rather than an enzymatic function, it is a molecular chaperone function of MK2 that is required for nuclear translocation of caspase-3 and subsequent apoptosis (Fig. 4). Indeed, the enzymatic function of MK2 (on caspase-3 or otherwise) is completely dispensable in nuclear translocation of caspase-3 (Fig. 3, A–D). In contrast, MK2’s nuclear localization signal is essential for promoting nuclear translocation of MK2 and preventing nuclear translocation of MK2, completely protects against MK2-induced caspase-3-mediated apoptosis (Fig. 6).
Figure 6.

Schematic of mechanism of MK2 dependent nuclear translocation of caspase-3. MK2 associates with caspase-3 and translocates to the nucleus in the execution of apoptosis. Interfering with MK2’s NLS signaling prevents nuclear translocation and apoptosis. MK2, mitogen-activated protein kinase-activated protein kinase 2; NLS, nuclear localization sequence; NES, nuclear export sequence. [Image created with BioRender.com and published with permission.]
MK2 is part of the canonical p38 mitogen-activated protein kinase pathway, where p38 phosphorylates MK2, which in turn phosphorylates HSP27 to control many complex biological processes (4, 36, 37). Our data show that dominant negative and constitutively active MK2 constructs (with differential HSP27 phosphorylation) function similarly, leading to increased nuclear caspase-3 activity and subsequent apoptosis (Fig. 3, C and D). Overexpressing phospho-mimicking HSP27, in the background of MK2 deficiency, fails to increase nuclear caspase-3 activity (Supplemental Fig. S6B). In addition, a mutant nuclear localization sequence construct of MK2, still resulted in HSP27 phosphorylation but not caspase-3 activation or cell death (Supplemental Fig. S7C; Fig. 4, D and E). In total, our work suggests that nuclear translocation of caspase-3 is an independent function of MK2 apart from downstream activation of the canonical p38 pathway.
Being a serine/threonine kinase, MK2 is primarily thought to phosphorylate substrates (28, 38–40); however, enzymes, and kinases in particular, can also function as chaperones (41–43). Our data clearly show that a nonkinase function of MK2 is responsible for the observed sensitization to cell death, and that the enzymatic function of MK2 is dispensable in the execution of apoptosis. To date, there have been mixed results regarding the chaperoning function of MK2. The protein structure of MK2 reveals a docking region within the C-terminal tail, amino acid residues 370’-400’ (44–46). Extensive work has identified a conserved amino acid sequence motif for binding to MK2: three negatively charged amino acids, i.e., aspartic acid (D), in close proximity which binds to MK2’s docking region anchored by the positively charged R386 of MK2 (45, 46). The complementary positive and negative charges form the electrostatic bond for protein-protein binding. P38α, a known binding partner of MK2, has the conserved amino acid binding sequence motif (44–46). In addition, MK2 complexes with p38 and co-localizes intracellularly with p38 during nuclear export following stimulation with arsenite (18), suggesting a potential chaperoning effect of MK2. In contrast, in other experiments, leptomycin B prevented the nuclear export of MK2 but not p38 (47), suggesting separate pathways or stimulus-specific pathways. Interestingly, our data show an association between MK2 and caspase-3, with caspase-3 preferentially associating to the C-terminal tail of MK2, where its docking region is located (45, 46), suggesting a direct binding.
It is interesting that expressing WT MK2 in H23 cells increased caspase-3 activity, especially given that MK2 is not required for the activation of caspase-3, as we have shown previously (4). It is unlikely that MK2 expression alone is cytotoxic to H23 cells as infection with Ad-Mut-NLS-MK2 does not increase caspase-3 activity. The most likely explanation is that potential inflammatory insult following adenoviral infection, or basal endogenous stressors commonly present in cancers (48, 49), coupled with a chaperoning function of MK2 via its NLS, leads to nuclear translocation of caspase-3 and resultant DNA damage, further propagating the apoptotic signaling cascade (50), i.e., increasing caspase-3 activation. This hypothesis is supported by our data showing infection with Ad-WT-MK2 results in phosphorylation of HSP27 (Fig. 2C; Supplemental Fig. S7C), indicating activation of stress-induced pathways (36, 37). Similar stress is noted following infection with Ad-Mut-NLS-MK2 (Supplemental Fig. S7C) but does not result in increased caspase-3 activity (Fig. 4D).
Our laboratory’s identification of a disconnect between the activation of caspase-3 and the execution of apoptosis in the context of MK2 deficiency has several implications. First, this implies the possibility of regulating apoptosis downstream of activation of the apoptotic cascade. Next, we previously found loss of MK2 resulted in the prevention of both apoptosis and endothelial barrier dysfunction despite activation of caspase-3 (4). Interestingly, in these experiments, active caspase-3 was sequestered in the cytoplasm. This brings out the possibility for a nonapoptotic, noncanonical role for cytosolic caspase-3; which we have confirmed. We identified a nonapoptotic, barrier-protective function for cytosolic caspase-3 in endothelial cells via effects on cytoskeleton (5, 51). Recently, others have identified that caspase-3 activity promotes the proliferation of cells in sebaceous glands (52); however, intracellular localization of caspase-3 was not investigated. These data suggest dual and divergent, location-specific functions of active caspase-3, with cytosolic caspase-3 promoting endothelial barrier integrity [in endothelial cells (5, 51)] or proliferation (52) and nuclear caspase-3 promoting apoptosis (4). Finally, our data suggest that MK2 functions as a molecular switch to convert the cytoprotective effects of cytosolic caspase-3 to apoptosis inducing by potentiating nuclear translocation of caspase-3.
There are several limitations to our studies. First, it is not clear if MK2 directly binds to caspase-3 or requires an intermediary scaffolding protein. Because molecular chaperoning is not a classic function for MK2, we sought to identify a potential role for HSP27, a downstream substrate for MK2 (16, 28–30), a well-described molecular chaperone (31) and known to bind directly to caspase-3 (33). Furthermore, the phosphorylation of HSP27 alters its cellular localization, with phospho-HSP27 translocating to the nucleus (22, 35) and nonphosphorylatable mutants of HSP27 remaining in the cytosol even after stimulation (22). However, our data show that overexpressing constructs containing either a dominant negative MK2 (HSP27 is not phosphorylated) or a constitutively active MK2 (HSP27 is phosphorylated) (Fig. 2C), results in a similar amount of higher caspase-3 activity (Fig. 2F), nuclear translocation of caspase-3, and cell death (Fig. 3, C–E). However, overexpression of a construct containing a mutated nuclear localization sequence on MK2 is able to phosphorylate HSP27 (Supplemental Fig. S7C) but does not result in increased caspase-3 activity or cell death (Fig. 4, D and E), indicating that phosphorylated HSP27 is not the key controller of caspase-3 activity/localization. In addition, our data show overexpressing phospho-mimicking HSP27, in the absence of MK2, is insufficient to translocate caspase-3 to the nucleus (Supplemental Fig. S6B). Taken together, these findings appear to rule out HSP27 as the chaperone for caspase-3. In contrast, using two different modalities, proximity-based biotinylation and co-immunoprecipitation (Fig. 4), our data shows MK2 associates with caspase-3. It is possible that other proteins may be associated with MK2 in a chaperone complex to assist with nuclear translocation of caspase-3. Our data support this possibility, as other proteins come in close proximity to MK2 (Supplemental Fig. S7B). We are in the process of interrogating potential binding sites between MK2 and caspase-3 to assess if there is a direct binding between MK2 and caspase-3 as well as performing an unbiased mass spectrometry screen for potential alternative interacting proteins. Second, although our data show direct phosphorylation of caspase-3 by MK2, the consequences of that phosphorylation event are unclear. Our data show that the phosphorylation status of caspase-3 does not impact its activity (Fig. 2, E and F), nor does it affect its ability to translocate to the nucleus and subsequent apoptosis (Fig. 3). Although it is possible that the phosphorylation of caspase-3 is irrelevant, we are intrigued that MK2-dependent phosphosites on caspase-3 identified by mass spectrometry (data not shown) are consistent with predicted 14-3-3 phospho-binding motifs (53, 54). 14-3-3 proteins are a group of adaptor proteins that bind specific phosphorylated amino acid sequence motifs (54). 14-3-3 proteins regulate myriad signaling events through the binding of phosphopeptide motifs of effector molecules (53). This brings about the possibility that phosphorylation of caspase-3, by MK2, may alter its association to different isoforms of 14-3-3 proteins to regulate downstream signaling. This is an active focus of investigation for our laboratory. Third, the majority of our experiments were performed in non-small cell lung cancer cell lines. We have previously identified that MK2 is necessary for nuclear translocation and subsequent apoptosis in murine models and primary lung human microvascular endothelial cells (4). However, the studies from our previous work used a loss of function strategy. In our current studies, we used a more nuanced strategy to identify the specific chaperone mechanism of MK2. Because all of our mechanistic studies were performed in H23 cells, a non-small cell lung cancer cell line, we were concerned these results could be cell-type specific as a consequence of the numerous mutations likely present in cancer cells. As such, we replicated key elements of our experiments in a different cell line, A549 cells, that also had reduced MK2 expression (Fig. 5). The replication of our key findings in two separate cell lines help validate the mechanistic role for MK2 in nuclear translocation of caspase-3. The relevance of these molecular mechanisms in specific disease states will likely require additional validation in more physiologically relevant model systems. At present, there is a suggestion that MK2 may be an important mediator of non-small cell lung cancer cell death following chemotherapy and may predict outcomes in patients with non-small cell lung cancers; an ongoing effort of our laboratory. Demonstrating a similar mechanistic role for MK2 in nuclear translocation of caspase-3 in endothelial cells will require the development of novel animal and cell culture models lacking in MK2. Our laboratory is actively generating these novel reagents.
In summary, this study provides mechanistic insight into how MK2 promotes nuclear translocation of caspase-3. Specifically, our data show a noncanonical, nonkinase, molecular chaperoning function of MK2 is required for nuclear translocation of caspase-3 and subsequent cell death. Because of caspase-3’s canonical (pro-apoptotic) and noncanonical (pro-proliferative) functions, it may be an ideal candidate to mediate the transition between injury (pro-apoptotic) and recovery (pro-proliferative) phases of inflammatory-dependent conditions, i.e., acute respiratory distress syndrome. Increased understanding of MK2’s role in regulating this transition between these divergent functions of caspase-3 could allow for therapeutic targeting.
DATA AVAILABILITY
Data will be made available upon reasonable request.
SUPPLEMENTAL DATA
Supplemental Figs. S1–S7: https://doi.org/10.6084/m9.figshare.21308235.
GRANTS
This study was supported by the National Institutes of Health (R01HL133413 to M.D., R01HL159906 to L.A.S. and M.D., R01HL151530 to K.S., and K08HL132055 to K.S.).
DISCLOSURES
Larissa Shimoda is an editor of American Journal of Physiology-Lung Cellular and Molecular Physiology and was not involved and did not have access to information regarding the peer-review process or final disposition of this article. An alternate editor oversaw the peer-review and decision-making process for this article. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
K.S., N.P., M.B.M., F.D., M.S., A.B., L.A.S., M.M., M.J.R., C.E.M., J.M., R.H., A.L.K., N.M.P., T.M.K., and M.D. conceived and designed research; O.D.R, K.S., M.K., G.S., A.S., L.Z., X.Y., N.M.P., M.B.M., H.J., M.J.R., C.E.M., and M.D. performed experiments; K.S., M.J.R., J.M., R.H., N.M.P., and M.D. analyzed data; O.D.R., K.S., M.J.R., and M.D. interpreted results of experiments; N.M.P., M.J.R., C.E.M., and M.D. prepared figures; O.D.R., K.S., M.J.R., C.E.M., and M.D. drafted manuscript; O.D.R., K.S., M.K., G.S., A.S., L.Z., X.Y., N.M.P., N.P., M.B.M., H.J., F.D., M.S., A.B., L.A.S., M.M., M.J.R., C.E.M., J.M., R.H., A.L.K., N.M.P., T.M.K., and M.D. edited and revised manuscript; O.D.R., K.S., M.K., G.S., A.S., L.Z., X.Y., N.M.P., N.P., M.B.M., H.J., F.D., M.S., A.B., L.A.S., M.M., M.J.R., C.E.M., J.M., R.H., A.L.K., N.M.P., T.M.K., and M.D. approved final version of manuscript.
ACKNOWLEDGMENTS
Preprint is available at https://www.biorxiv.org/content/10.1101/2021.11.30.470656v1.
REFERENCES
- 1. Boatright KM, Salvesen GS. Mechanisms of caspase activation. Curr Opin Cell Biol 15: 725–731, 2003. doi: 10.1016/j.ceb.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 2. Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, , et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 19: 107–120, 2012. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med 361: 1570–1583, 2009. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Damarla M, Parniani AR, Johnston L, Maredia H, Serebreni L, Hamdan O, Sidhaye VK, Shimoda LA, Myers AC, Crow MT, Schmidt EP, Machamer CE, Gaestel M, Rane MJ, Kolb TM, Kim BS, Damico RL, Hassoun PM. Mitogen-activated protein kinase-activated protein kinase 2 mediates apoptosis during lung vascular permeability by regulating movement of cleaved caspase 3. Am J Respir Cell Mol Biol 50: 932–941, 2014. doi: 10.1165/rcmb.2013-0361OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Suresh K, Carino K, Johnston L, Servinsky L, Machamer CE, Kolb TM, Lam H, Dudek SM, An SS, Rane MJ, Shimoda LA, Damarla M. A nonapoptotic endothelial barrier-protective role for caspase-3. Am J Physiol Lung Cell Mol Physiol 316: L1118–L1126, 2019. doi: 10.1152/ajplung.00487.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol (1985) 91: 1487–1500, 2001. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- 7. White MK, Amini S, Khalili K, Kogan M, Donaldson K, Darbinian N. Development of a bidirectional caspase-3 expression system for the induction of apoptosis. Cancer Biol Ther 7: 945–954, 2008. doi: 10.4161/cbt.7.6.5969. [DOI] [PubMed] [Google Scholar]
- 8. Yasuhara N, Eguchi Y, Tachibana T, Imamoto N, Yoneda Y, Tsujimoto Y. Essential role of active nuclear transport in apoptosis. Genes Cells 2: 55–64, 1997. doi: 10.1046/j.1365-2443.1997.1010302.x. [DOI] [PubMed] [Google Scholar]
- 9. Fujihara SM, Nadler SG. Modulation of nuclear protein import: a novel means of regulating gene expression. Biochem Pharmacol 56: 157–161, 1998. doi: 10.1016/s0006-2952(98)00049-5. [DOI] [PubMed] [Google Scholar]
- 10. Lange A, Mills RE, Lange CJ, Stewart M, Devine SE, Corbett AH. Classical nuclear localization signals: definition, function, and interaction with importin alpha. J Biol Chem 282: 5101–5105, 2007. doi: 10.1074/jbc.R600026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kotlyarov A, Neininger A, Schubert C, Eckert R, Birchmeier C, Volk HD, Gaestel M. MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat Cell Biol 1: 94–97, 1999. doi: 10.1038/10061. [DOI] [PubMed] [Google Scholar]
- 12. Yardeni T, Eckhaus M, Morris HD, Huizing M, Hoogstraten-Miller S. Retro-orbital injections in mice. Lab Anim (NY) 40: 155–160, 2011. doi: 10.1038/laban0511-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Joseph B, Ekedahl J, Lewensohn R, Marchetti P, Formstecher P, Zhivotovsky B. Defective caspase-3 relocalization in non-small cell lung carcinoma. Oncogene 20: 2877–2888, 2001. doi: 10.1038/sj.onc.1204402. [DOI] [PubMed] [Google Scholar]
- 14. Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER, , et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569: 503–508, 2019. doi: 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Engel K, Schultz H, Martin F, Kotlyarov A, Plath K, Hahn M, Heinemann U, Gaestel M. Constitutive activation of mitogen-activated protein kinase-activated protein kinase 2 by mutation of phosphorylation sites and an A-helix motif. J Biol Chem 270: 27213–27221, 1995. doi: 10.1074/jbc.270.45.27213. [DOI] [PubMed] [Google Scholar]
- 16. Kayyali US, Pennella CM, Trujillo C, Villa O, Gaestel M, Hassoun PM. Cytoskeletal changes in hypoxic pulmonary endothelial cells are dependent on MAPK-activated protein kinase MK2. J Biol Chem 277: 42596–42602, 2002. doi: 10.1074/jbc.M205863200. [DOI] [PubMed] [Google Scholar]
- 17. Engel K, Kotlyarov A, Gaestel M. Leptomycin B-sensitive nuclear export of MAPKAP kinase 2 is regulated by phosphorylation. EMBO J 17: 3363–3371, 1998. doi: 10.1093/emboj/17.12.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ben-Levy R, Hooper S, Wilson R, Paterson HF, Marshall CJ. Nuclear export of the stress-activated protein kinase p38 mediated by its substrate MAPKAP kinase-2. Curr Biol 8: 1049–1057, 1998. doi: 10.1016/s0960-9822(98)70442-7. [DOI] [PubMed] [Google Scholar]
- 19. Kelly KF, Spring CM, Otchere AA, Daniel JM. NLS-dependent nuclear localization of p120ctn is necessary to relieve Kaiso-mediated transcriptional repression. J Cell Sci 117: 2675–2686, 2004. [Erratum in J Cell Sci 117: 3405, 2004]. doi: 10.1242/jcs.01101. [DOI] [PubMed] [Google Scholar]
- 20. Choi-Rhee E, Schulman H, Cronan JE. Promiscuous protein biotinylation by Escherichia coli biotin protein ligase. Protein Sci 13: 3043–3050, 2004. doi: 10.1110/ps.04911804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim DI, Jensen SC, Noble KA, Kc B, Roux KH, Motamedchaboki K, Roux KJ. An improved smaller biotin ligase for BioID proximity labeling. Mol Biol Cell 27: 1188–1196, 2016. doi: 10.1091/mbc.E15-12-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Geum D, Son GH, Kim K. Phosphorylation-dependent cellular localization and thermoprotective role of heat shock protein 25 in hippocampal progenitor cells. J Biol Chem 277: 19913–19921, 2002. doi: 10.1074/jbc.M104396200. [DOI] [PubMed] [Google Scholar]
- 23. Rogalla T, Ehrnsperger M, Preville X, Kotlyarov A, Lutsch G, Ducasse C, Paul C, Wieske M, Arrigo A-P, Buchner J, Gaestel M. Regulation of Hsp27 oligomerization, chaperone function, and protective activity against oxidative stress/tumor necrosis factor alpha by phosphorylation. J Biol Chem 274: 18947–18956, 1999. doi: 10.1074/jbc.274.27.18947. [DOI] [PubMed] [Google Scholar]
- 24. Wallberg F, Tenev T, Meier P. Analysis of apoptosis and necroptosis by fluorescence-activated cell sorting. Cold Spring Harb Protoc 2016: pdb.prot087387, 2016., doi: 10.1101/pdb.prot087387. [DOI] [PubMed] [Google Scholar]
- 25. De Vos M, Schreiber V, Dantzer F. The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochem Pharmacol 84: 137–146, 2012. doi: 10.1016/j.bcp.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 26. Decker P, Muller S. Modulating poly (ADP-ribose) polymerase activity: potential for the prevention and therapy of pathogenic situations involving DNA damage and oxidative stress. Curr Pharm Biotechnol 3: 275–283, 2002. doi: 10.2174/1389201023378265. [DOI] [PubMed] [Google Scholar]
- 27. Petrilli V, Herceg Z, Hassa PO, Patel NS, Di Paola R, Cortes U, Dugo L, Filipe HM, Thiemermann C, Hottiger MO, Cuzzocrea S, Wang ZQ. Noncleavable poly(ADP-ribose) polymerase-1 regulates the inflammation response in mice. J Clin Invest 114: 1072–1081, 2004. doi: 10.1172/JCI200421854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kotlyarov A, Yannoni Y, Fritz S, Laass K, Telliez J-B, Pitman D, Lin L-L, Gaestel M. Distinct cellular functions of MK2. Mol Cell Biol 22: 4827–4835, 2002. doi: 10.1128/MCB.22.13.4827-4835.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vertii A, Hakim C, Kotlyarov A, Gaestel M. Analysis of Properties of Small Heat Shock Protein Hsp25 in MAPK-activated Protein Kinase 2 (MK2)-deficient Cells: MK2-dependent insolubilization of Hsp25 oligomers correlates with susceptibility to stress. J Biol Chem 281: 26966–26975, 2006. doi: 10.1074/jbc.M602134200. [DOI] [PubMed] [Google Scholar]
- 30. Zheng C, Lin Z, Zhao ZJ, Yang Y, Niu H, Shen X. MAPK-activated protein kinase-2 (MK2)-mediated formation and phosphorylation-regulated dissociation of the signal complex consisting of p38, MK2, Akt, and Hsp27. J Biol Chem 281: 37215–37226, 2006. doi: 10.1074/jbc.M603622200. [DOI] [PubMed] [Google Scholar]
- 31. Concannon CG, Gorman AM, Samali A. On the role of Hsp27 in regulating apoptosis. Apoptosis 8: 61–70, 2003. doi: 10.1023/a:1021601103096. [DOI] [PubMed] [Google Scholar]
- 32. Pandey P, Farber R, Nakazawa A, Kumar S, Bharti A, Nalin C, Weichselbaum R, Kufe D, Kharbanda S. Hsp27 functions as a negative regulator of cytochrome c-dependent activation of procaspase-3. Oncogene 19: 1975–1981, 2000. doi: 10.1038/sj.onc.1203531. [DOI] [PubMed] [Google Scholar]
- 33. Voss OH, Batra S, Kolattukudy SJ, Gonzalez-Mejia ME, Smith JB, Doseff AI. Binding of caspase-3 prodomain to heat shock protein 27 regulates monocyte apoptosis by inhibiting caspase-3 proteolytic activation. J Biol Chem 282: 25088–25099, 2007. doi: 10.1074/jbc.M701740200. [DOI] [PubMed] [Google Scholar]
- 34. Wu R, Kausar H, Johnson P, Montoya-Durango DE, Merchant M, Rane MJ. Hsp27 regulates Akt activation and polymorphonuclear leukocyte apoptosis by scaffolding MK2 to Akt signal complex. J Biol Chem 282: 21598–21608, 2007. doi: 10.1074/jbc.M611316200. [DOI] [PubMed] [Google Scholar]
- 35. McClaren M, Isseroff RR. Dynamic changes in intracellular localization and isoforms of the 27-kD stress protein in human keratinocytes. J Invest Dermatol 102: 375–381, 1994. doi: 10.1111/1523-1747.ep12371798. [DOI] [PubMed] [Google Scholar]
- 36. Damarla M, Hasan E, Boueiz A, Le A, Pae HH, Montouchet C, Kolb T, Simms T, Myers A, Kayyali US, Gaestel M, Peng X, Reddy SP, Damico R, Hassoun PM. Mitogen activated protein kinase activated protein kinase 2 regulates actin polymerization and vascular leak in ventilator associated lung injury. PLoS One 4: e4600, 2009. doi: 10.1371/journal.pone.0004600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Le A, Damico R, Damarla M, Boueiz A, Pae HH, Skirball J, Hasan E, Peng X, Chesley A, Crow MT, Reddy SP, Tuder RM, Hassoun PM. Alveolar cell apoptosis is dependent on p38 MAP kinase-mediated activation of xanthine oxidoreductase in ventilator-induced lung injury. J Appl Physiol (1985) 105: 1282–1290, 2008. doi: 10.1152/japplphysiol.90689.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Menon MB, Gaestel M. MK2-TNF-signaling comes full circle. Trends Biochem Sci 43: 170–179, 2018. doi: 10.1016/j.tibs.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 39. Soni S, Anand P, Padwad YS. MAPKAPK2: the master regulator of RNA-binding proteins modulates transcript stability and tumor progression. J Exp Clin Cancer Res 38: 121, 2019. doi: 10.1186/s13046-019-1115-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stokoe D, Engel K, Campbell DG, Cohen P, Gaestel M. Identification of MAPKAP kinase 2 as a major enzyme responsible for the phosphorylation of the small mammalian heat shock proteins. FEBS Lett 313: 307–313, 1992. doi: 10.1016/0014-5793(92)81216-9. [DOI] [PubMed] [Google Scholar]
- 41. Posas F, Saito H. Osmotic activation of the HOG MAPK pathway via Ste11p MAPKKK: scaffold role of Pbs2p MAPKK. Science 276: 1702–1705, 1997. doi: 10.1126/science.276.5319.1702. [DOI] [PubMed] [Google Scholar]
- 42. Rauch J, Volinsky N, Romano D, Kolch W. The secret life of kinases: functions beyond catalysis. Cell Commun Signal 9: 23, 2011. doi: 10.1186/1478-811X-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang CC, Tsou CL. Enzymes as chaperones and chaperones as enzymes. FEBS Lett 425: 382–384, 1998. doi: 10.1016/s0014-5793(98)00272-5. [DOI] [PubMed] [Google Scholar]
- 44. Haar ET, Prabakhar P, Liu X, Lepre C. Crystal structure of the p38 alpha-MAPKAP kinase 2 heterodimer. J Biol Chem 282: 9733–9739, 2007. [Erratum in J Biol Chem 282: 14684, 2007]. doi: 10.1074/jbc.M611165200. [DOI] [PubMed] [Google Scholar]
- 45. Tanoue T, Nishida E. Molecular recognitions in the MAP kinase cascades. Cell Signal 15: 455–462, 2003. doi: 10.1016/s0898-6568(02)00112-2. [DOI] [PubMed] [Google Scholar]
- 46. White A, Pargellis CA, Studts JM, Werneburg BG, Farmer BT. 2nd. Molecular basis of MAPK-activated protein kinase 2:p38 assembly. Proc Natl Acad Sci USA 104: 6353–6358, 2007. doi: 10.1073/pnas.0701679104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gong X, Ming X, Deng P, Jiang Y. Mechanisms regulating the nuclear translocation of p38 MAP kinase. J Cell Biochem 110: 1420–1429, 2010. doi: 10.1002/jcb.22675. [DOI] [PubMed] [Google Scholar]
- 48. Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat 7: 97–110, 2004. doi: 10.1016/j.drup.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 49. Ubhi T, Brown GW. Exploiting DNA replication stress for cancer treatment. Cancer Res 79: 1730–1739, 2019. doi: 10.1158/0008-5472.CAN-18-3631. [DOI] [PubMed] [Google Scholar]
- 50. Norbury CJ, Zhivotovsky B. DNA damage-induced apoptosis. Oncogene 23: 2797–2808, 2004. doi: 10.1038/sj.onc.1207532. [DOI] [PubMed] [Google Scholar]
- 51. Suresh K, Servinsky L, Johnston L, Punjabi NM, Dudek SM, Damarla M. Comparison of polynomial fitting versus single time point analysis of ECIS data for barrier assessment. Physiol Rep 9: e14983, 2021. doi: 10.14814/phy2.14983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yosefzon Y, Soteriou D, Feldman A, Kostic L, Koren E, Brown S, Ankawa R, Sedov E, Glaser F, Fuchs Y. Caspase-3 regulates YAP-dependent cell proliferation and organ size. Mol Cell 70: 573–587.e4, 2018. doi: 10.1016/j.molcel.2018.04.019. [DOI] [PubMed] [Google Scholar]
- 53. Yang X, Lee WH, Sobott F, Papagrigoriou E, Robinson CV, Grossmann JG, Sundstrom M, Doyle DA, Elkins JM. Structural basis for protein-protein interactions in the 14-3-3 protein family. Proc Natl Acad Sci USA 103: 17237–17242, 2006. doi: 10.1073/pnas.0605779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gardino AK, Yaffe MB. 14-3-3 proteins as signaling integration points for cell cycle control and apoptosis. Semin Cell Dev Biol 22: 688–695, 2011. doi: 10.1016/j.semcdb.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figs. S1–S7: https://doi.org/10.6084/m9.figshare.21308235.
Data Availability Statement
Data will be made available upon reasonable request.