

Keywords: adaptation, pulmonary hypertension, right ventricle
Abstract
Right ventricular (RV) failure is the major determinant of outcome in pulmonary hypertension (PH). Calves exposed to 2-wk hypoxia develop severe PH and unlike rodents, hypoxia-induced PH in this species can lead to right heart failure. We, therefore, sought to examine the molecular and structural changes in the RV in calves with hypoxia-induced PH, hypothesizing that we could identify mechanisms underlying compensated physiological function in the face of developing severe PH. Calves were exposed to 14 days of environmental hypoxia (equivalent to 4,570 m/15,000 ft elevation, n = 29) or ambient normoxia (1,525 m/5,000 ft, n = 25). Cardiopulmonary function was evaluated by right heart catheterization and pressure volume loops. Molecular and cellular determinants of RV remodeling were analyzed by cDNA microarrays, RealTime PCR, proteomics, and immunochemistry. Hypoxic exposure induced robust PH, with increased RV contractile performance and preserved cardiac output, yet evidence of dysregulated RV-pulmonary artery mechanical coupling as seen in advanced disease. Analysis of gene expression revealed cellular processes associated with structural remodeling, cell signaling, and survival. We further identified specific clusters of gene expression associated with 1) hypertrophic gene expression and prosurvival mechanotransduction through YAP-TAZ signaling, 2) extracellular matrix (ECM) remodeling, 3) inflammatory cell activation, and 4) angiogenesis. A potential transcriptomic signature of cardiac fibroblasts in RV remodeling was detected, enriched in functions related to cell movement, tissue differentiation, and angiogenesis. Proteomic and immunohistochemical analysis confirmed RV myocyte hypertrophy, together with localization of ECM remodeling, inflammatory cell activation, and endothelial cell proliferation within the RV interstitium. In conclusion, hypoxia and hemodynamic load initiate coordinated processes of protective and compensatory RV remodeling to withstand the progression of PH.
NEW & NOTEWORTHY Using a large animal model and employing a comprehensive approach integrating hemodynamic, transcriptomic, proteomic, and immunohistochemical analyses, we examined the early (2 wk) effects of severe PH on the RV. We observed that RV remodeling during PH progression represents a continuum of transcriptionally driven processes whereby cardiac myocytes, fibroblasts, endothelial cells, and proremodeling macrophages act to coordinately maintain physiological homeostasis and protect myocyte survival during chronic, severe, and progressive pressure overload.
INTRODUCTION
Originating from pathological remodeling and/or obstruction of the pulmonary vasculature, pulmonary hypertension (PH) imposes a hemodynamic load on the cardiac right ventricle (RV) that leads to remodeling and hypertrophy, and ultimately, progression to RV failure and death (1). RV function is a critical determinant of PH outcome and an independent, negative predictor of outcome in left heart failure. Despite increasing research efforts and sophistication of hemodynamic and imaging metrics, understanding of the RV in response to PH remains incomplete (2, 3). The RV differs substantially from the left ventricle (LV) in its embryonic origin and development, contractile function, and biochemical responses (4). Approved therapies for PH target pulmonary vasoconstriction but do not directly act on the pathological RV remodeling that is a critical determinant of clinical outcome. Approved cardioprotective therapies that directly target RV are not available.
The healthy thin-walled RV coupled with the low-pressure, high-capacity pulmonary circulation, displays an impressive ability to increase its performance and match cardiac output to metabolic demand in response to a wide range of physiological requirements such as exercise or pregnancy (5–7). The initial responses of the RV to PH are likely compensatory and include rapid and transient increases in volume, increased contractility, and right ventricular hypertrophy. With PH progression, declining RV function no longer meets physiological demand resulting in symptomatic heart failure, characterized by RV dilation and increased filling pressures (right atrial pressure and right ventricular end-diastolic pressure), decreased RV-pulmonary artery (PA) mechanical coupling efficiency, and reduced cardiac output. However, for many patients with advanced PH, removal of the pulmonary hemodynamic overload by lung transplant or pulmonary endarterectomy leads to rapid recovery of RV function, suggesting a high degree of protective mechanisms in the pressure-overloaded and failing RV (8).
Our group has extensively characterized the responses of bovines to hypobaric hypoxia as a large animal model of PH and RV pressure overload (9, 10). Use of a large animal model to study RV responses to hypoxic PH provides advantages over rodent models that include a myocardium that shares many similarities to humans including myocyte composition [e.g., β-myosin heavy chain (MHC), titin isoforms], structure, and function (contraction-relaxation kinetics) (11, 12). Similar to humans and in contrast to rodents and other species, calves exposed to 14 days of environmental hypoxia develop severe PH, resulting in structural remodeling and profound molecular reprogramming within the pulmonary vasculature, accompanied by evidence of organ-level RV dysfunction and hypertrophic remodeling (13–15). Importantly, isolated single-cell mechanics show that calf cardiomyocytes have preserved (or even enhanced) contractility, and mitochondrial function and integrity are preserved despite evidence of whole organ dysfunction (16–18). Furthermore, recovery of calves at normoxic atmosphere leads to reversal of PH and restored RV function (19).
Based on these considerations, we hypothesized that molecular characterization of RV phenotype in calves with hypoxia-induced PH could identify mechanisms underlying compensated physiological function. We combined hemodynamic analysis of cardiopulmonary function, transcriptomic analysis of gene expression, proteomic analysis, and immunohistochemical visualization of structural extracellular matrix (ECM) remodeling. Our results focused on four major determinants of cardiac function: 1) hypertrophic and prosurvival mechanotransduction through YAP-TAZ signaling, 2) initiation of ECM remodeling, 3) inflammatory cell activation, and 4) angiogenesis. We suggest that these basic elements of RV remodeling remain active to preserve cardiac function during PH progression.
MATERIALS AND METHODS
Experimental Animals and Study Protocol
All animal procedures were performed under supervision of the Colorado State University Institutional Animal Care and Use Committee. The calf model of hypoxia-induced pulmonary hypertension (14-day exposure to hypobaric hypoxia, PB = 430 mmHg, atmosphere equivalent to 15,000 ft/4,570 m, PH, compared with ambient normoxia, PB = 630 mmHg, 5,000 ft/1,525 m, control) has been described previously and demonstrated to produce robust pulmonary hypertension and right ventricular hypertrophic remodeling (10, 14). These conditions simulate high elevations encountered naturally in the western United States and preserve cardiopulmonary chemoreceptor reflex mechanisms intact. Data in this report were obtained from a total of 54 consecutive animals studied by the laboratory (PH = 29, control = 25). Male Holstein calves were procured from a local dairy (postnatal days 1–3, 45–50 kg body wt), selected based on veterinary health examination, and randomized to two experimental groups: 1) pulmonary hypertension (PH), housed in a hypobaric hypoxic environmental chamber at Colorado State University Hypo/hyperbaric Facility, equivalent to 4,570 m/15,000 ft elevation (PB = 430 mmHg) for 14 days; and 2) control, age-matched control animals were maintained in ambient atmosphere (Ft. Collins, CO, elevation 1,525 m). Animals were typically studied in pairs (e.g., 2 PH or 2 control) to provide social interactions during the exposure period. The chamber was vented daily to ambient atmosphere as needed to perform animal husbandry and veterinary care.
Hemodynamics
General procedures for right heart catheterization and hemodynamic measurements with Swan-Ganz pressure catheters were performed as described previously (10, 14). Briefly, hemodynamic measurements were performed on nonsedated and manually restrained animals. Calves were placed in lateral recumbency and given lidocaine subcutaneously to place a sheath introducer (8 F) into the jugular vein for repetitive catheter placement. Catheters were advanced into the cardiopulmonary vasculature as indicated. Catheter location was verified by appearance of the pressure waveforms (20). Standard pressure traces from right atrium, right ventricle, pulmonary artery, and pulmonary capillary wedge were recorded with a Swan-Ganz balloon-tipped thermodilution catheter (6 F) hooked to a pressure transducer and interfaced to a ix/228 Data Acquisition System (DAQ) module (iWorx Systems). Digital RV pressure records for single-beat hemodynamic analysis were captured with a high-frequency solid-state catheter (Millar Instruments). Pressure-volume loop analysis for RV function was performed with a solid-state admittance catheter (Scisense-Transonic; 5-Fr) and data acquisition module (SciSense Advantage, VSL Catheter Interface Module). Note that this procedure was performed in only a subset of calves (n = 10 control and n = 14 PH). Systemic mean arterial pressures were recorded with a separate 22 ga. catheter inserted into the auricular artery. Hemodynamic measurements were obtained under the same environmental conditions that were used for 14-day experimental treatment, that is hypoxia (HX) or normoxia (NX), respectively, PH-HX versus Con-NX. For selected animals, hemodynamic testing was also performed upon transient (≤1 h) exposure of control animals to hypoxia (Con-HX) to test for acute elevation of PA pressure due to hypoxic vasoconstriction or upon transient return of hypoxic animals to normoxia (PH-NX) to test for nonreversible structural remodeling. Digital recordings of pressure and volume waveforms were stored on a personal computer for offline analysis. Hemodynamic parameters were extracted with the vendors’ proprietary software or custom code developed with Matlab.
Hypoxia exposure (4 wk, 5,800 m/18,000 ft) and hemodynamic analysis of hypoxia-induced PH in rats were performed as described according to protocols approved by University of Colorado Institutional Animal Care and Use Committee (21).
Tissue Sampling
Following the hemodynamic measurements, animals were deeply anesthetized with pentobarbitol (30 mg/kg iv) and euthanized by exsanguination for tissue harvest. The chest was opened and the RV-LV ventricular block was separated from the atria and outflow tract and removed. The RV and LV free walls were dissected from the interventricular septum. RV and LV free walls were held on ice for collection of tissue samples. Replicate transmural samples were collected from the meridional area of the free wall at the level of the papillary muscle attachment or toward the apex for RNA and protein analysis, immunohistochemistry, and histopathology, as follows. Samples were collected with a surgical prep blade or biopsy punch (8 mm diameter, Research Products) as indicated.
Frozen tissue for RNA and protein analysis.
Transmural samples were excised from the free wall and snap-frozen in liquid nitrogen.
Immunohistochemistry.
Samples were collected with a surgical blade or biopsy punch, placed in standard cryomolds filled with OCT compound, and frozen on dry ice. The samples were oriented either transverse or longitudinal to the sectioning plane to allow orthogonal viewing of tissue structure.
Histopathology.
Samples were collected similar to the OCT-immunohistochemistry procedure, but placed into histology cassettes and immersed in formalin for fixation and paraffin embedding by the UCD Research Histology Core Laboratories.
Immunohistochemistry, Morphometry, and Histopathology
Immunohistochemistry.
Immunohistochemistry was performed on 5-μm frozen sections using indirect immunofluorescence. Images were acquired on a Zeiss fluorescence microscope with AxioVision imaging system. Additional imaging was performed on a Leica-Aperio Versa 8 scanning system. Comparison control and PH images for each experiment were obtained in the same session under identical conditions (e.g., exposure time, magnification, etc.). Microscope images were adjusted for brightness and contrast if needed, and the adjustments were made equally to both PH and control images. Antibody reagents used for immunohistochemistry are listed in Supplemental Table S9 (all Supplemental material is available at https://doi.org/10.6084/m9.figshare.22185838). Indirect immunofluorescence staining was performed with a biotin-streptavidin detection system using Alexa-488 or -594 fluorochromes according to manufacturer’s instructions (Molecular Probes/Invitrogen, Frederick, MD). Stained sections were mounted with VectaShield embedding medium with DAPI (Vector Laboratories, Burlingame, CA). Immunohistochemistry for Ki67 with detection by horse radish peroxidase-diaminobenzidine (Ki67-HRP, Fig. 7A) was performed on formalin-fixed/paraffin-embedded sections.
Myocyte morphometry.
Histochemical staining with fluorophore-conjugated wheat germ agglutinin and DAPI was performed on formalin-fixed sections. Stained sections were mounted with VectaShield and images were acquired with the Zeiss fluorescent microscope and AxioVision imaging system. Myocyte cross-sectional area was quantitated from the digitized images with Metamorph software as previously described (22). For each RV specimen, we analyzed three tissue sections, and five high-power fields per section.
Histopathology.
Sections were stained with standard hematoxylin-eosin (H&E) or Masson’s trichrome. Histopathological evaluations were performed by a board-certified veterinary pathologist (G.M.K.).
Real-Time PCR
RNA isolation and real-time PCR were performed as previously described (23). Total RNA was isolated by homogenizing tissue in TRIzol Reagent (Ambion) according to the manufacturer’s instructions. RNA integrity was determined by BioAnalyzer (Agilent). RIN ≥ 7 was used as a criterion for quality. First-strand cDNA synthesis was performed using an iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). Quantitative real-time PCR was performed using SybrGreen primers according to the manufacturer’s instructions. Gene expression abundance was calculated after normalization to Hprt using the ΔΔCt method. Primers are shown in Supplemental Table S8.
cDNA Oligonucleotide Arrays and Bioinformatic Analysis
Animals were selected for oligonucleotide array and proteomic analyses (see Proteomics) according to the criteria 1) animals with the highest quartile of mean PA pressure (mPAP) (>105 mmHg) and 2) animals showing evidence of substantial pulmonary vascular remodeling consistent with developed PH. Supplemental Table S10 shows mPAP values for animals used in cDNA microarrays and proteomic analysis. Three of identical control and PH animals were used for both procedures. There were no significant differences between mPAP of the corresponding experimental groups between the two experiments.
RNA was isolated with TRIzol and further purified with Qiagen RNEasy spin columns according to the manufacturer’s instructions. Oligonucleotide microarray analysis (n = 5 each PH, control, Affymetrix GeneChip Bovine Genome Array) was performed by the UCD Genomics Core Laboratory. The raw .CEL files were imported into Partek analysis software and log2 normalized with the Partek defaults (RMA, adjusted GC content, summarize probes). Statistical filtering of the dataset was performed with open-source software to normalize the data for background correction and remove genes called absent. Multiple sample comparisons were performed with Student’s t test combined with the Benajmini-Hochberg false discovery rate (FDR) correction at 0.1 to produce fold change and adjusted P value. The final dataset contained a total of 652 unique annotated genes. Bioinformatic analysis was performed with the Ingenuity Pathway Analysis (IPA) platform. Microarray data are deposited in the Gene Expression Omnibus (GEO), accession number GSE217438.
Proteomics
Tissue proteomics was performed on RV tissues from five control and four PH calves using a compartment-resolved method that improves extracellular matrix characterization. The method generates a cellular and chaotrope soluble extracellular matrix fraction followed by chemical digestion of the insoluble extracellular matrix (iECM) (24). These fractions are digested with trypsin before injection onto a nano LC-MS/MS system for data-dependent tandem MS analysis. Raw data were converted to .mgf format and queried against the Uniprot sequence database. Results were filtered at 1% FDR at the peptide and protein level. Protein abundances were normalized using the online freeware NormalizerDE (25), by the LoessG normalization method (limma package) (26). Features with greater than 50% missing values were deleted. Remaining features with missing values had missing values imputed using k-nearest neighbors in Perseus (27). Proteomic data analysis and results visualization were carried out in Metaboanalyst (28). Pearson correlation analysis was performed with GraphPad Prism version 9 (GraphPad Software).
Statistics
Values are expressed as means ± SD. For simple comparisons of two Gaussian-distributed sample sets, we performed Student’s unpaired, two-tail t tests. Multiple group comparisons were performed by one-way ANOVA with Sidak’s multiple comparisons test. Statistical analyses were performed with GraphPad Prism. Differences with P values less than 0.05 were considered statistically significant.
RESULTS
Hemodynamic Assessment of RV Response to Severe PH
RV and pulmonary vascular pressures.
Function of the RV-PA unit was evaluated by right heart catheterization with standard Swan-Ganz pressure catheters as shown in Supplemental Fig. S1 and Supplemental Table S1. The principal hemodynamic findings are summarized as follows. Exposure of calves to 14-day environmental hypoxia induces robust pulmonary hypertension. (Supplemental Fig. S1A). The elevated PA pressures following 14-day hypoxia are greater than those observed upon acute hypoxic challenge and do not recover to baseline following transient return to normoxia, consistent with a component of structural pulmonary vascular remodeling induced by prolonged hypoxic exposure (see Supplemental Fig. S2.3). Pulmonary vascular resistance and pulmonary vascular capacitance are also increased by 14-day hypoxia, further supporting pulmonary vascular structural remodeling (Supplemental Fig. S1B). Moreover, hypoxic exposure selectively alters pulmonary vascular and right heart function without global effects on venous return or the left heart and systemic circulation (Supplemental Fig. S1C). Right atrial pressure was not significantly affected by 14-day hypoxic exposure, although a trend toward increased pressure was noted. Pulmonary artery wedge pressure (PAWP), a surrogate of left atrial pressure, was elevated by 14-day hypoxia but promptly returned to the normal range upon restoration of ambient atmosphere. Mean arterial pressure was not altered by 14-day hypoxia.
RV pressure-volume relationships.
We analyzed RV pressure-volume relationships to provide a load-independent assessment of RV contractile function. Figure 1A shows representative P-V loops for a normoxic control calf (mPAP = 21.0, green lines) compared with a calf with hypoxia-induced PH (hypoxic PH, mPAP = 120.4, dark blue lines) and upon transient return to ambient normoxia (PH normoxic, mPAP = 56.6, light blue lines). Table 1 summarizes parameters from P-V analysis of control (n = 10) and PH calves (n = 14). The following results are noted.
Figure 1.

Right ventricular (RV) pressure-volume (P-V) analysis. Right heart catheterization and measurement of RV P-V relationships were performed as described in materials and methods. Quantitative values are summarized in Table 1. *P < 0.05 , pulmonary hypertension (PH)-hypoxia vs. control-normoxia. A: representative pressure-volume tracings from a control calf in normoxic conditions (green lines); and from a calf exposed with hypoxia-induced PH, measured in hypoxic conditions (dark blue lines) and following transient return to normoxia (light blue lines). B: RV contractility. C: RV output. bpm, beats/min. D: parameters of RV filling and diastolic function. E: component analysis of RV ventricular-vascular coupling (VVCR). Left: single-beat hemodynamic analysis of ventricular vascular coupling ratio, Ees/Ea. Digital recordings of RV pressures from indicated animals were analyzed to determine Ees/Ea as described in materials and methods. Middle: pulmonary artery (PA) end-arterial elastance. PA end-arterial elastance (Ea) was determined directly from P-V loop relationships. The relationship to mean PA pressure was determined by linear regression. Right: RV end-systolic elastance (Ees) was calculated from animals for which both P-V analysis and determination of ventricular-vascular coupling ratio were performed.
Table 1.
Pressure-volume loop analysis summary
| Parameter | Control | PH | P Value |
|---|---|---|---|
| n | 10 | 14 | |
| Heart rate | 125.5 (22.7) | 162.4 (24.9) | 1.2E-03 |
| End-systolic pressure | 45.3 (8.7) | 118.3 (25.2) | 1.3E-08 |
| End-diastolic pressure | 11.1 (9.5) | 18.5 (8.3) | 0.11 NS |
| End-systolic volume | 24.8 (9.8) | 36.1 (19.8) | 0.11 NS |
| End-diastolic volume | 61.8 (13.6) | 73.7 (23.6) | 0.16 NS |
| Stroke volume | 37.0 (8.9) | 37.6 (7.6) | 0.85 NS |
| Cardiac output | 4.6 (1.7) | 6.0 (1.4) | 0.034 |
| dP/dtmax | 958 (421) | 2,002 (600) | 1.0E-04 |
| dP/dtmin | −697 (135) | −1,878 (426) | 2.4E-08 |
| Tau (Weiss) | 26.5 (18.0) | 26.5 (5.6) | 0.99 NS |
| Ejection fraction | 60.5 (11.5) | 53.8 (14.7) | 0.24 NS |
| Stroke work | 1,533 (447) | 3,454 (977) | 8.3E-06 |
| End-arterial elastance | 1.33 (0.35) | 3.42 (0.92) | 7.3E-07 |
Values are means (SD); n, number of animals. Parameters were obtained from analysis of pressure-volume relationships in control vs. calves with pulmonary hypertension (PH) as described in materials and methods. dP/dtmax and dP/dtmin, first derivative of left ventricular pressure increase and decline, respectively; NS, not significant.
RV contractile performance is increased. RV end-systolic pressure is increased in PH, with a concomitant increase in stroke work and rate of contraction (dP/dtmax; −dP/dtmin increased similarly, not shown) indicating increased RV contractility to match the pressure overload (Fig. 1B).
RV output is physiologically compensated. RV stroke volume measured from pressure-volume analysis was unchanged by hypoxia-induced PH. The preserved stroke volume together with the reflexive increase in heart rate resulted in modest but significantly increased RV cardiac output (Fig. 1C).
RV filling pressures and volumes are preserved in PH. Parameters indicative of normal RV filling pressures and volumes through the cardiac cycle were not significantly altered in PH calves, including RV end-systolic volume, end-diastolic volume, end-diastolic pressure, and ejection fraction. In addition, the RV diastolic time constant Tau (Weiss), a measure of diastolic function, also remained constant in PH calves (Fig. 1D).
Analysis of RV ventricular-vascular coupling. The efficiency of RV-PA coupling, measured as the ratio of RV end-systolic elastance to PA end-arterial elastance (Ees/Ea), is a key parameter to evaluate the efficiency of biomechanical coupling between the right ventricle and the pulmonary arterial circulation, where values > 1 indicate normal RV-PA coupling and values < 1 suggest dysfunction (6). Conventionally this metric can be determined from simultaneous right ventricular pressure and volume measurements but for technical reasons, this was not feasible in the bovine model. Therefore, we performed single-beat hemodynamic analysis from high-frequency digitized RV pressure recordings to determine this parameter. This method has been validated previously to predict PH outcomes in human pediatric patients (29). As shown in Fig. 1E, left, Ees/Ea was significantly reduced in hypertensive calves compared with normoxic controls (0.53 ± 0.26, n = 11, vs. 1.11 ± 0.51, n = 9, P = 0.0042), indicating a significant defect in mechanical coupling efficiency despite the maintained cardiac output. Figure 1E, middle, shows that end-arterial elastance, Ea, determined from the pressure-volume analysis, was increased in PH calves, consistent with anatomic pulmonary vascular remodeling described in next section, Histopathological features of early RV remodeling. The increase in Ea followed a linear relationship with developed PH measured separately as mean PA pressure in our cohort. In a limited set of animals where we determined Ea from pressure-volume analysis, together with the ratio Ees/Ea from single-beat pressure analysis, it was possible to calculate RV end-systolic elastance, Ees, as a load-independent parameter of RV contractility (Fig. 1E, right). This calculation shows that Ees is significantly increased in PH calves compared with normoxic controls. Thus, the observed decrease in ventricular-vascular coupling ratio results from the increase in Ea driven by vascular remodeling, whereas RV contractility measured as Ees is actually increased to compensate. These results underscore the conclusion that increased RV contractile performance compensates for the hypoxia-induced pressure overload to maintain cardiac output in severe PH.
Structural and Transcriptomic Reorganization in Early RV Adaptation to Severe PH
Histopathological features of early RV remodeling.
Exposure of calves to 14-day environmental hypoxia elicits histopathological features of RV remodeling, as shown in Supplemental Fig. S2. Principal findings were evidence of myocyte hypertrophy (Supplemental Fig. S2.1), combined with diffuse and modest interstitial and perivascular deposition of fibrillar collagen observed with Masson’s Trichrome stain (Supplemental Fig. S2.2). However, frank regions or tracts of replacement fibrosis were not appreciated. There was mild increase in perivascular cellularity, particularly around medium-sized coronary vessels, suggestive of inflammatory infiltrates. These histopathological alterations do not appear to result in global alterations of RV structure seen in more severe inflammatory-fibrotic diseases. Consistent with previous reports from our group, exposure of calves to 14-day environmental hypoxia elicits robust remodeling of the entire pulmonary arterial circulation including adventitial expansion and medial hypertrophy of small distal arterioles, medium-sized pulmonary arteries, and larger intralobular vessels (Supplemental Fig. S2.3) (10).
Transcriptomic landscape of early RV remodeling.
Hemodynamic characterization demonstrates augmented and physiologically adapted RV contractile performance in calves with PH. Accordingly, we pursued a global approach to identify determinants of RV remodeling using cDNA microarray analysis of RV gene expression combined with informatic analysis by Ingenuity Pathway Analysis (IPA) on control and hypertensive animals (n = 5 each) from our study cohort. The gene expression between the two groups was filtered at a false discovery rate of 0.1, yielding 652 unique, annotated, and differentially expressed genes. Figure 2A shows principal component analysis of the dataset, and Fig. 2B shows hierarchical cluster analysis of the individual samples. PH causes substantial and reproducible reprogramming of gene expression in the calf RV.
Figure 2.

cDNA microarray analysis of global gene expression in right ventricle of hypertensive neonatal calves. RNA was isolated from right ventricular tissues of calves with hypoxia-induced pulmonary hypertension (PH) or normoxic controls (n = 5 each) and analyzed by cDNA microarrays as described in materials and methods. A: principal component analysis comparing PH vs. control animals. B: hierarchical cluster map of differentially expressed genes for PH vs. control animals.
Gene-ontology and canonical signaling pathways in early RV remodeling.
Initial assessment of gene expression from cDNA microarrays was performed with standardized algorithms in the Ingenuity knowledge base. Clustering of differential gene expression according to gene-ontological (GO) categories of disease and cell function is shown in Fig. 3A. Significant enrichment (overlap P < 0.05) and net activation (|Z-score| ≥ 2) of gene expression is concentrated in a fingerprint of specific functions suggesting cell migration, intercellular interactions, cytoskeletal rearrangement, cell proliferation, and inhibition of cell death. Assignment of gene expression data according to canonical signaling pathways is shown in Fig. 3B. We infer that these patterns of activated signaling pathways underlie downstream cellular functions related to cytoskeletal rearrangement, inflammation, cardiac hypertrophy, and vascular remodeling.
Figure 3.

Transcriptomic landscape of right ventricular (RV) remodeling. Data from cDNA microarrays from pulmonary hypertension (PH) vs. control calves was performed with Ingenuity Pathway Analysis as described in materials and methods. A: gene-ontology (GO) analysis of RV remodeling. Gene networks linked to the indicated cellular functions are shown, filtered for significance by log (enrichment P value) > 1.3 (P < 0.05). B: canonical signaling pathways in RV remodeling. Signaling pathways were filtered by log (enrichment P value > 1.3). Size of the symbol is proportionate to number of genes in the cluster. C: functional categories of gene expression: aggregated analysis. Major gene-ontological categories of gene expression were identified (Supplemental Fig. S3). The average z-scores and P values of all functional gene clusters within the category having enrichment P value < 0.05 are shown. Symbol size is proportionate to the number of functional clusters within each category.
Important context is provided by considering these individual networks of gene expression representing the most significantly activated processes in RV remodeling within the larger context of alterations in the RV transcriptomic landscape. As shown in overview in Supplemental Fig. S3 and quantitated in Fig. 3C, clusters of activated gene networks tend to be most strongly associated with the major functional categories of 1) cellular movement, 2) intercellular signaling, and 3) inflammation, including the subcategories of immune cell trafficking, hematological system development, and inflammatory response, with average z-scores = 1.0–1.5 for these categories. A second tier of functional categories with activated z-scores 0.5–1.0, encompasses the functional categories of 4) cell structure and function, including the subcategories of cellular development, cellular function and maintenance, and cellular assembly and organization and 5) cell proliferation, including subcategories cell growth and proliferation, cell death and survival, and cell cycle. We note a clear locus of inhibition of gene clusters related to cell death within this group, in contrast to general activation of most other processes. Finally, many of these gene networks are also associated with broader categories of 6) cardiovascular system development and function; 7) organismal injury and abnormality, including subcategory cardiovascular disease; and 8) cancer, as might be expected for processes of tissue reorganization and remodeling in response to physiological stress (average z-scores ∼0.5). Together these results demonstrate clear qualitative trends for the major cellular processes involved in compensatory RV remodeling, including strong signatures of intercellular signaling, cell movement, cell structural reorganization, and immune-inflammatory cell activation.
Transcriptomic Signatures and Cellular Correlates of Early RV Adaptation to Severe PH
It was of interest to further investigate aspects of the RV transcriptome with specific relevance to remodeling processes important to PH pathophysiology. For this purpose, we used IPA to interrogate our dataset and design custom networks of gene expression focused on the areas of hypertrophy-mechanotransduction, ECM remodeling, inflammatory cell activation, and angiogenesis. These studies were complemented by targeted measurements of key gene products with real-time PCR. Immunohistochemical localization of key cellular components was used to determine the relationships among these molecular processes within the structure of the remodeling RV.
Cardiac hypertrophy and Yap-TAZ signaling.
The initial response of the heart to pressure or volume overload is hypertrophy, increasing free wall thickness, and contractility (30). This reflects net deposition of myofibrils within cardiac myocytes and consequently increased myocyte size. These changes in cellular and ventricular morphometry are accompanied by reprogramming of myocyte contractile, Ca handling, and natriuretic peptide gene expression (31). These observations led us to evaluate cell and molecular correlates of RV hypertrophy. As shown in Fig. 4A, histochemical staining with wheat germ agglutinin to visualize cardiac myocyte perimeters shows increased myocyte cross-sectional area in the RV of a PH calf compared with a control calf. Morphometric analysis demonstrates quantitative increases in RV myocyte size in linear proportion to developed PA pressure. We further quantitated mRNAs typical of pathological cardiac hypertrophy in relation to PA pressure as shown in Fig. 4B. β-Myosin heavy chain (MYH7) is the principal myosin heavy chain isoform expressed in the hearts of large animals including humans and bovines, and its expression increases relative to α-myosin heavy chain in human heart failure. Skeletal muscle α-actin and brain natriuretic peptide are not expressed postnatally in normal ventricles but are reexpressed upon induction of pathological hypertrophy; moreover, elevated circulating brain natriuretic peptide (BNP) is an established biomarker of human heart disease. Expression of each of these mRNAs is selectively increased in the RV in linear proportion to developed PH, whereas expression in the companion LV is minimally or nonresponsive. These results are consistent with hypertrophic remodeling of the cardiac myocyte that underlies the adaptation of the RV to hemodynamic load in PH.
Figure 4.

Molecular and cellular correlates of right ventricular (RV) hypertrophic remodeling in hypoxia-induced pulmonary hypertension (PH). A: myocyte cross-sectional area determined by quantitative morphometry. Histochemical staining with wheat germ agglutinin and morphometric analysis of myocyte size were performed as described in materials and methods. Right: representative images of RV sections from control (n = 9) and PH (n = 11) calves. Left: relationship between myocyte size and mean pulmonary artery (PA) pressure determined by linear regression. B: quantitative PCR evaluation of hypertrophic gene expression. Abundances of the indicated mRNAs were determined by real-time PCR from RV or left ventricular (LV) tissues of control and PH calves. The relationship of mRNA abundance to mean PA pressure was determined by linear regression. ■, RV; □, LV. C: functional responses mediated by YAP-TAZ signaling. C: predicted functional responses mediated by YAP-TAZ signaling. Differentially expressed genes predicted to interact with YAP-TAZ were identified in the RV microarray dataset (Supplemental Table S2) and gene-ontology analysis of functional responses was performed with Ingenuity Pathway Analysis. The responses shown are filtered at z-score > 2, P < 0.05. Symbols are color coded according to major functional category as indicated (red, cell-cell signaling and interaction; orange, cell proliferation; green, cell movement).
The analyses of cardiopulmonary function and quantitative gene expression suggest that hypoxia-induced PH drives RV function and gene expression in large part by hemodynamic pressure overload. Previous reports have established YAP-TAZ as an important mechanotransduction signaling pathway in PH (32). We therefore interrogated the microarray dataset for differentially expressed genes linked to YAP-TAZ activation. We identified a YAP-TAZ interaction network of 44 differentially expressed genes, including YAP1 itself, as tabulated in Supplemental Table S2. Functional analysis of this network with IPA showed concerted activation of gene-ontological categories related to cell movement, cell-cell signaling and interaction, cellular growth and proliferation, cell development, and cardiovascular system development (Fig. 4C and Supplemental Fig. S4). Interestingly, the principal cluster of downregulated functions connected to this network was focused on inhibition of cell death pathways.
ECM remodeling.
It will be recalled that histopathological evaluation (Supplemental Fig. S2) showed minimal frank fibrosis in the RV. Nonetheless, the initiation of ECM remodeling is confirmed by increased immunohistochemical staining for tenascin C and ED-A fibronectin, proteins involved in provisional matrix formation, as shown in Fig. 5A (ED-A, extracellular domain-A, is a cell-associated isoform distinct from circulating plasma fibronectins). Both ECM markers show patchy, nonuniform, and widely distributed immunostaining. Importantly, increased tenascin C immunostaining colocalizes with cardiac vascular endothelium visualized with Tomato lectin, demonstrating ECM remodeling within the perivascular-interstitial compartment (Supplemental Fig. S5). Figure 5B quantitates expression with qPCR for the ECM remodeling genes TnC, ED-A fibronectin, the matrikine osteopontin, and Col1a1, the principal fibrillar collagen of the heart. In each case, RV gene expression increases in proportion with increasing PA pressure, whereas the corresponding gene expression in LV was not affected. Finally, a network of 46 differentially expressed genes in our dataset was identified with IPA to associate with the ontological function terms “extracellular matrix” or “fibrosis” as shown in Supplemental Table S3. Reanalysis of this gene expression subset with IPA predicts downstream responses related to cell movement and cell-cell interactions, suggesting the interaction of ECM remodeling gene expression with functional mobilization of inflammatory cells (Fig. 5C). Additional predicted functions are related to organ morphology and growth, as expected for a structural remodeling process.
Figure 5.

Molecular and cellular correlates of right ventricular (RV) extracellular matrix remodeling in hypoxia-induced pulmonary hypertension (PH). A: extracellular matrix (ECM) remodeling in hypertensive calf RV. Cryosections of RV free wall were collected from PH and control calves and stained for extracellular domain-A (ED-A) fibronectin, or tenascin C as described in materials and methods. Representative images from PH and control animals (n = 4 each) are shown with wide field and higher resolution views, scale as indicated. B: quantitative PCR evaluation of ECM gene expression. Abundances of the indicated mRNAs were determined by real-time PCR from RV or left ventricular (LV) tissues of control and PH calves. The relationship of mRNA abundance to mean pulmonary artery pressure was determined by linear regression. Black square, RV; white square, LV. C: functional responses associated with ECM and profibrotic genes. Genes associated with the terms cardiovascular “ECM” or “fibrosis” were identified and their functional associations with gene-ontological categories were determined with Ingenuity Pathway Analysis. Functional subcategories are listed according to –log (enrichment P value), all z-scores > 2.0. Major functional categories are indicated by color: green square, cell movement; red square, cell-cell interaction; blue square, organ injury and abnormality; brown square, embryonic and organismal development; tan square, cell death and survival.
Proteomic analysis of ECM remodeling.
Many ECM proteins have low rates of synthesis, long half-lives, and are extensively regulated posttranscriptionally and posttranslationally. To complement our transcriptomic ECM analysis, we therefore used proteomic analysis with compartment-specific mass spectrometry to quantitate the identities and abundances of ECM proteins. A set of 174 proteins was identified in calf RV ECM, comprising core ECM (collagens, glycoproteins, and proteoglycans) and ECM-associated fractions (ECM regulators, ECM-affiliated proteins, and secreted factors, Fig. 6A). ECM remodeling in PH is confirmed by significantly altered expression (P ≤ 0.1) of numerous core ECM (n = 24) and ECM-associated proteins (n = 30), including elastin microfibril components (FBN1, EMILIN2, LTBP1/2, and ADAMTSL4), specific basement membrane components (COL4A4, COL18A1, and FLBLN2), and diverse ECM signaling molecules (THBS4, IGFBP7, VCAN, TGFBI, and S100A2/11) (Fig. 6B, Supplemental Table S4). Additional proteins are associated with collagen biosynthesis and maturation (PLOD1/3, BGN, SERPINH1, TGM2, and COL12A1); we further note upregulation of proteolytic enzymes (MMP2 and CTSZ) and downregulation of an ensemble of SERPINs (A1, B2, B6, F2, D1, C1, and A5). Together, these alterations suggest remodeling and reorganization of ECM rather than fibrotic collagen deposition. Moreover, there are positive and negative correlations between a number of these components with mPAP and likewise with a subset of lectin-binding proteins (galectins-1,3 and lectin mannose binding 1), basement membrane collagen COL4A4, cathepsins D, Z, and ADAMTSL4 that binds fibrillin 1 to promote microfibril biogenesis (Fig. 6C). Consistent with immunohistochemical staining in Fig. 5A, tenascin C and ED-A fibronectin, proteins involved in provisional matrix formation begin to increase at the protein level at this early time also but not significant due to variability of measurements (FC 16 and 2.4). However, related matrix protein tenascin XB is significantly increased and correlates with increased mPAP. We identified a total of 17 proteins common to our transcriptomic and proteomic datasets; 11 of these were concordantly regulated in PH RV, demonstrating reasonable qualitative agreement (Fig. 6D). In an effort to evaluate the functional correlates of ECM remodeling in PH RV, we combined the transcriptomic (n = 47) and proteomic (n = 54) signatures of ECM gene expression and performed pathway analysis with Ingenuity as shown in Fig. 6E. The results are strongly directed toward functions involved in cell movement, cell-cell signaling and interactions, development, and survival, suggesting a dynamic environment facilitating cell recruitment and migration.
Figure 6.
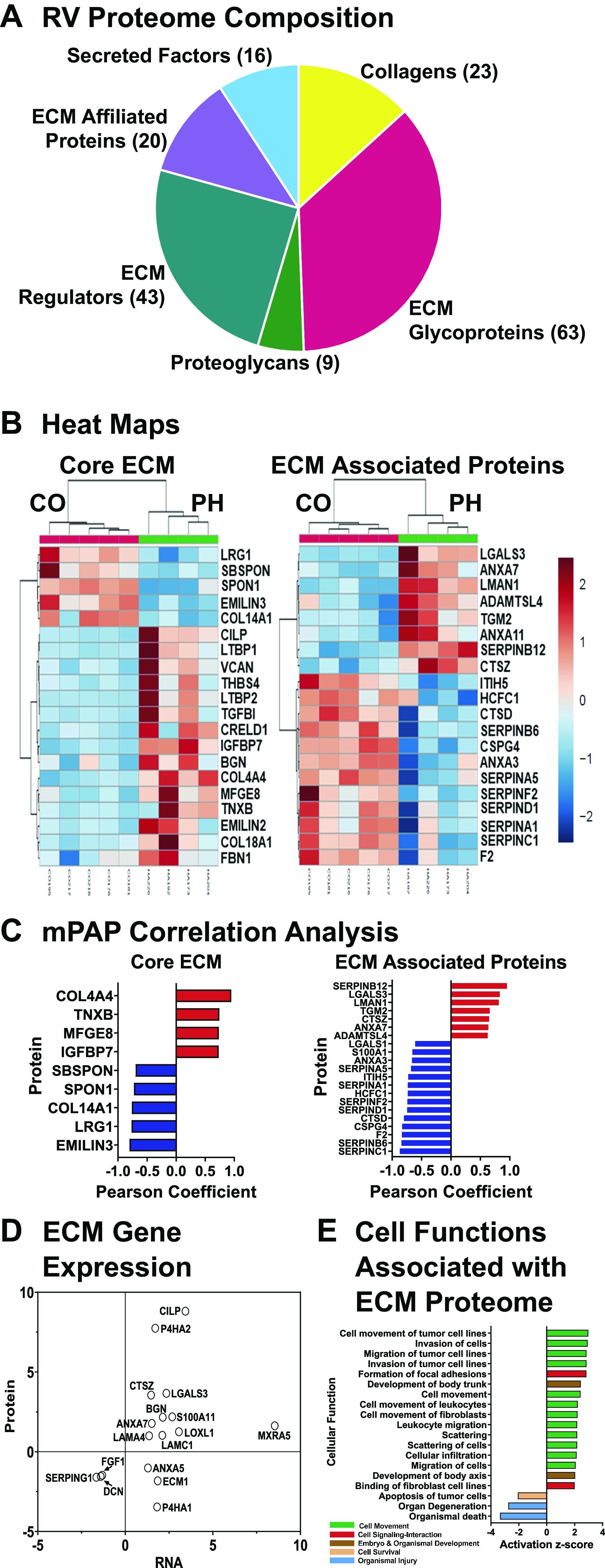
Proteomic analysis of the right ventricular (RV) extracellular matrix (ECM) matrisome. Proteomic analysis was performed as described in materials and methods. A: matrisome composition (174 proteins). B: differentially expressed proteins (P < 0.1) in core ECM (n = 20) and ECM-associated matrisomal protein (n = 34) compartments from pulmonary hypertension (PH) Calf RV. Top 20 proteins are shown. C: Pearson correlation analysis of mean PA pressure (mPAP) dependence of differentially expressed proteins. D: correlations between transcriptomic and proteomic determinations in PH RV. E: integrated transcriptomic-proteomic gene ontological analysis of differentially expressed ECM genes.
Immune-inflammatory cell activation.
We next evaluated RV inflammatory gene expression at the onset of PH. Figure 7A shows immunohistochemical markers of activated monocyte-macrophage lineage cells in the RV of hypertensive calves visualized by increased immunostaining with CD45 (pan-leukocyte), CD14 (monocyte), and CD163 (alternately activated macrophage). Supplemental Fig. S7 shows full-section scanning digital images from RV specimens stained with the same markers. The results showing immunoreactivity uniformly distributed throughout the myocardium are consistent with the higher magnification images provided in Fig. 7A. We extended these observations by quantitation of proinflammatory mRNAs using qPCR as shown in Fig. 7B. We observed increased expression of the proinflammatory genes in linear relation to PA pressure. Figure 7B shows the proinflammatory cytokine IL-1β together with the IL-1 receptor antagonist (IL1RA), which is coexpressed at higher abundance. In addition, we show MCP-1, a major chemokine for macrophage recruitment, and TLR2, involved in innate immunity. In each case, RV gene expression increases in proportion with increasing PA pressure, whereas the corresponding gene expression in LV was not affected. These results were extended to investigate inflammatory gene expression within the RV transcriptome. As shown in Supplemental Fig. S6 and Supplemental Table S5, a network of 60 differentially expressed RV genes associated with gene-ontology categories related to inflammation was identified with IPA. This gene network overlaps significantly with functional responses related to leukocyte-monocyte movement, migration, or binding, suggesting inflammatory cell activation in the context of compensatory RV remodeling.
Figure 7.

Molecular and cellular correlates of right ventricular (RV) inflammation in hypoxia-induced PH. A: inflammatory cell activation in hypertensive calf RV. Cryosections of RV free wall were collected from pulmonary hypertension (PH) (n = 8) and control (n = 4) calves and stained for the indicated immune-inflammatory cell markers as described in materials and methods. Data show representative images. B: quantitative PCR evaluation of inflammatory gene expression. Abundances of the indicated mRNAs were determined by real-time PCR from RV or left ventricular (LV) tissues of control and PH calves. The relationship of mRNA abundance to mean pulmonary artery pressure was determined by linear regression. Black square, RV; white square, LV.
VEGF signaling and angiogenesis.
We hypothesized that RV hypertrophy in PH includes compensatory angiogenesis to support enhanced RV contractility. Immunohistochemical colocalization of Tomato lectin with Ki67, a marker for cell proliferation, was performed. As shown in Fig. 8A, the RV from calves with PH shows a substantial increase in cell proliferation colocalizing to vascular endothelium. This observation was confirmed with Ki67-HRP immunohistochemistry showing robust cell proliferation (Fig. 8A, bottom). Supporting this interpretation, Ingenuity analysis identifies a network of 76 differentially expressed genes connected with the functional term “Angiogenesis” (Fig. 8B, Supplemental Table S6, z = 2.5, −logP = 8.5). We further explored potential upstream signaling pathways controlled by major regulators of angiogenesis, including hypoxia (HIF1-HIF2), VEGF, and mechanical force (YAP-TAZ, see above). Significant gene networks are present in the dataset for each of these regulators, and they each share genes in common with the angiogenesis signaling network (Fig. 8B, Supplemental Table S6). The integrated signaling pathway constructed from these gene clusters clearly supports a proangiogenic response to hypoxia-induced PH in the calf RV. In sum, our data confirm that activation of RV hypertrophy, inflammation, ECM remodeling, and angiogenesis occur concomitantly in PH.
Figure 8.

Molecular and cellular correlates of right ventricular (RV) angiogenesis in hypoxia-induced pulmonary hypertension (PH). A: vascular endothelial cell proliferation in hypertensive RV. Top: cryosections of RV free wall were collected from PH (n = 5) and control (n = 4) calves and costained with the cell proliferation marker Ki67, and with fluorescent tomato lectin to visualize the vascular endothelium, as described in materials and methods. Representative images are shown. Arrows indicate colocalization of cell proliferation with RV vasculature. Bottom: formalin-fixed sections were stained with Ki67 antibody and visualized with HRP-DAB histochemistry. B: angiogenesis regulatory gene networks in hypertensive RV. The networks of differentially expressed genes connected with the gene-ontology category, “angiogenesis,” and shared genes with the regulatory gene networks for VEGF, HIF1-HIF2, and TAP-TAZ signaling were identified with Ingenuity Pathway Analysis. Interactions and shared genes among all these networks are indicated, where black text shows the total number of genes in the regulatory network and blue text shows the number of genes shared with the angiogenesis signaling network.
Role of cardiac fibroblasts in RV remodeling.
Taken together, the foregoing results suggest that interactions among proremodeling macrophages, fibroblasts, and endothelial cells direct interstitial remodeling in hypertensive RV. Recent results from our laboratories showed that cardiac fibroblasts from the PH calf RV express a transcriptomic signature enriched in gene expression related to ECM remodeling, hypoxia signaling, and production and secretion of cytokines and growth factors. Mediators released from the PH calf cardiac fibroblasts were shown to promote survival of cardiac myocytes in a dedifferentiated state in vitro (33). Mechanisms of myocyte dedifferentiation or hibernation initially may be protective and a prerequisite for myocyte recovery upon release of pressure overload as is observed following lung transplant for PH. As an initial approach, we evaluated the transcriptomic datasets identified in cultured PH-activated cardiac fibroblasts (PH c-Fib) or naive ventricular cardiac myocytes treated in vitro with PH cardiac fibroblast conditioned medium, compared with our microarray data from PH-RV. Figure 9A shows gene overlaps among these datasets (RV vs. fibroblast, n = 136; RV vs. myocyte, n = 358). We focused specifically on genes that were concordantly up- or downregulated in RV and in the respective cultured cell datasets (RV vs. fibroblast, n = 42 up, n = 14 down, of 136 total; RV vs. myocyte, n = 125 up, n = 80 down, of 358 total). Pathway analysis results with IPA are shown in Fig. 9, B and C, respectively. The cardiac fibroblast gene expression signature within the RV dataset is enriched in functions related to cell movement, tissue differentiation, and angiogenesis. Fibroblast-directed gene expression in cardiac myocytes that is shared with the RV dataset is enriched in functions of cell movement, inflammatory cell activation, and cell survival. These results point to critical roles for cardiac fibroblasts as mediators of intercellular communication in RV remodeling (34).
Figure 9.

Transcriptomic signatures associated with pulmonary hypertension (PH)-activated right ventricular (RV) fibroblasts. A: Venn analysis of shared genes differentially expressed in PH neonatal calf RV, in vitro cultured PH neonatal calf RV fibroblasts, and in vitro cultured rat ventricular myocytes treated with conditioned medium from PH neonatal calf RV fibroblasts (myocytes). RV data from this study; in vitro cell culture data from Bruns et al. (33). B: gene-ontological analysis of concordantly expressed genes in RV and in cultured PH neonatal calf RV fibroblasts. C: gene-ontological analysis of concordantly expressed genes in RV and in cultured rat ventricular myocytes treated with conditioned medium from PH neonatal calf RV fibroblasts. Major functional categories of annotated gene functions are indicated by color: green square, cell movement; yellow square, cell structure, function, development; purple square, cardiovascular system development, function; tan square, cell survival and proliferation; red square, cell signaling and interaction.
DISCUSSION
This study integrated hemodynamic, transcriptomic, proteomic, and immunohistochemical approaches to evaluate molecular and cellular correlates of RV hypertrophy, inflammation, ECM remodeling, and angiogenesis in a large animal model of severe PH. The bovine heart has greater similarity to humans than rodent models in terms of structure, function, and response to severe pressure overload. The principal findings are summarized as follows. First, we demonstrate severe hypoxia-induced PH with evidence of structural pulmonary vascular remodeling that is compensated by increased RV contractile performance accompanied by hypertrophic remodeling. Second, immune-inflammatory cell activation, ECM remodeling, and angiogenesis occur in concert and are localized to the RV interstitium at this stage of RV response. Third, processes favoring cell survival and proliferation dominate over cell death. These findings provide new insight into the protective nature of RV remodeling in severe PH. Specifically, they show the resilience of RV function in the face of severe pressure overload, the repertoire of adaptive mechanisms activated to preserve RV function and insure myocyte survival, and the coordination of RV remodeling among multiple cell types in addition to the cardiac myocyte. The novelty of our work comes from the granular detail of specific signaling and gene expression networks that present further opportunities to identify stage-specific signatures of RV gene expression which can inform development of surrogate biomarkers and identification of cardioprotective therapies. The significance lies in the appreciation that within the defined hemodynamic context of this large animal model, our findings suggest that RV remodeling during PH progression represents a continuum of transcriptionally driven processes whereby cardiac myocyte and nonmyocyte cell types interact to maintain physiological homeostasis and protect myocyte survival during chronic, severe, and progressive pressure overload.
Our hemodynamic assessment employed direct measures of cardiac contractility through pressure-volume analysis combined with single-beat hemodynamics to confirm and extend previous reports from our group using the calf model (10, 14, 35). These measurements reveal that despite increased contractility and preserved cardiac output, RV-PA mechanical coupling is impaired, likely owing to profound pulmonary vascular remodeling. These results can be contrasted to measurements of cardiac responses in a rat model of hypoxia as shown in Supplemental Table S7. Despite more severe hypoxia than this study (5,800 m, 4 wk), rats show more modest PH, maintained cardiac output, and RV-PA coupling values preserved near unity. Similar results were obtained with the rat Sugen-hypoxia PH model (36). Thus, the bovine model under these conditions allows investigation of a critical transition in human PH progression from adaptation to failure that is unavailable from rodent models.
RV hypertrophy reflects increased deposition of sarcomeres within the cardiomyocyte and results in increased myocyte contractility. Here, we quantitated that increases in myocyte size and hypertrophic gene expression occur selectively in the RV compared with LV and in proportion to the increased PA pressure. This result is consistent with our previous study showing preserved or enhanced myofilament Ca-sensitivity and pCa-force relationship in cardiac myocytes from PH calf RV (18). It is also noteworthy to us that the mPAP dependence of hypertrophic remodeling appears to be a continuous function rather than demonstrating a threshold behavior of permissive mild mPAP elevation, consistent with emerging consensus to lower the threshold mPAP value defining clinical PH (1).
Rapidly accumulating evidence supports a key role for mechanotransduction through the HIPPO-YAP-TAZ axis in regulating cardiac myocyte hypertrophy and survival during development and in response to infarction or pressure overload of the left heart (37, 38). A role for YAP-TAZ signaling in vascular remodeling in PH has also been reported (32). We find a strong transcriptomic signature of YAP-TAZ activation in the hypertensive RV that couples to prosurvival, proproliferative, and proremodeling responses, suggesting an important role in RV adaption to pressure overload. To our knowledge, this is the first reported evidence of YAP-TAZ signaling in the right ventricle response to PH. Targeting this pathway may provide important insight into RV protective mechanisms in PH. Additional studies will be needed to directly relate temporal changes in gene/protein expression with changes in myocardial stress in the bovine model to characterize the continuous interplay between the biomechanical stimulus and structural-contractile adaptation. Moreover, we previously observed RV chamber expansion and interventricular septal distortion in this model and recognized that altered RV morphology likely impacts the efficiency of RV-LV coupling (15, 39, 40). Further studies will be needed to directly measure LV torsional strain and stress.
Substantial research points to the importance of inflammation, particularly within the pulmonary vasculature, as a universal driver in the pathological remodeling that determines PH progression and outcome (41, 42). However, understanding of the specific molecular interactions of inflammatory processes in the RV and their effects on myocardial function is incomplete (42–44). Our data show a pattern of gene expression consistent with activation and migration of immune-inflammatory cells. Furthermore, we observed increased immunostaining of monocyte-macrophage lineages, with abundant CD163+ macrophages reflecting a proremodeling rather than proinflammatory phenotype. These results suggest that inflammatory activation may participate importantly in compensatory interstitial remodeling in PH. In this regard, Gorr et al. (45) reported increased numbers of macrophages and increased expression of genes related to immune function in RV compared with LV under both normoxia and 2-wk rodent hypoxia, supporting an important role for inflammatory activation specifically in compensated RV remodeling. In contrast to these reports and to other reports from our group, another recent study by Krishnan et al. (46) using a model of adaptive responses to hypoxia in rats described general immune downregulation in both lung and RV (47). These disparities may relate to differences in this model using hypoxic exposure spanning in utero and into 6-wk postnatal life.
The relationship of profibrotic ECM remodeling with RV function in PH exhibits similar variability (48–50). Adaptive RV phenotypes as seen in Eisenmenger’s syndrome feature minimal RV fibrosis and low diastolic stiffness, together with RV hypertrophy and enhanced RV contractility, similar to our results. Remodeling of interstitial ECM has been suggested to serve as structural reinforcement to support increased RV contractility. Furthermore, consistent with our data, interstitial remodeling commonly includes alterations in nonfibrillar matricellular proteins such as tenascins C and X, SPARC proteins, thrombospondins, periostin, osteopontin, CTGF, and proteoglycans (51, 52). These proteins play important roles to coordinate multiple cell types for regulation of inflammatory, fibrotic, reparative, and angiogenic pathways (51). Important to note, profibrotic and ECM remodeling can be driven by multiple cell types within the myocardium including myofibroblasts, cardiac myocytes, and polarized proremodeling M2-macrophages (see above) (48, 51).
Capillary rarefaction has been reported as an additional driver of RV ischemia and failure in PH (53, 54). RV hypertrophy due to chronic PH pressure overload reduces RV perfusion and increases the vulnerability of the RV to the effects of hypoxia and ischemia (55). However, careful stereological analysis has shown that the apparent diminished RV capillary density in hypoxic mouse PH, Sugen/HX rat PH, and human PAH in fact results mainly from RV hypertrophy that is compensated by increased endothelial cell proliferation and angiogenesis, with preserved metabolic substrate delivery and no evidence of endothelial cell apoptosis (56–58). Extensive studies have demonstrated that hypoxia and mechanical forces including stretch, shear stress, and flow, act on growth factors including VEGF, basic FGF, angiopoietin, and others, to promote angiogenesis. Preliminary studies in our group have shown that altered hemodynamic forces such as shear stress are coupled to changes in vascular dimensions and tortuosity. Our results presented here show clear evidence of endothelial cell proliferation in PH RV, which is supported by activation of genes involved in hypoxia-VEGF-angiogenesis signaling. This combination of angiogenesis and elaboration of ECM within the perivascular-interstitial space is consistent with coordinate adaptation and maturation of capillary and arteriolar coronary vasculature to pressure overload. Together these responses may contribute to compensatory remodeling in pressure overload, intermittent hypoxic exposure, or exercise training, and are prominent in the proliferative context of embryonic and postnatal development (59). These adaptations are likely to support myocardial perfusion and myocyte function and survival in the hypertrophic, hypercontractile RV.
Limitations in this study suggest directions to address in future research. A key unanswered question will be to assign transcriptomic signatures to identified cell types within the remodeling RV. It will be necessary to perform single-cell RNA-Sequencing on freshly isolated cell populations to fully address this point. Spatiotemporal profiling of gene expression in relation to structural remodeling represents an additional goal for research. A second criticism is that we have only captured a discrete point in the response of the RV to PH progression. It will be of interest to extend these studies to beef cattle raised in high-elevation environments, where individual animals may adapt or progress to frank RV failure. This naturally occurring large animal model may provide unique insights into the progression and phenotypic diversity of cardiac and pulmonary responses to chronic hypoxia-induced pulmonary hypertension (12).
In conclusion, we show that hypoxia-induced PH engages transcriptionally driven and mutually reinforcing processes of cardiac hypertrophy, inflammatory cell activation, ECM remodeling, and angiogenesis that contribute to compensatory RV function in the presence of advancing pulmonary vascular disease. Unique features of this large animal model provide insight into a critical window where therapeutic intervention may provide a path to diagnose and intercept cardiac dysfunction in PH.
DATA AVAILABILITY
Data will be made available upon reasonable request.
SUPPLEMENTAL DATA
Supplemental Figs. S1–S7 and Supplemental Tables S1–S10: https://doi.org/10.6084/m9.figshare.22185838.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants P01 HL152961 and P01 HL014985 (to K.R.S.) and Training Grant T32 HL007171 (to G.M.K. and J.W.; Principal Investigator, K.R.S.) and US Department of Defense Grants W81XWH-20-1-0249 and W81XWH-19-1-0259 (to K.R.S.).
DISCLOSURES
G.M.K. is employed by Innotiv/Bolder Biopath, Boulder, CO. M.G.E. is owner of Bioinfo Solutions, LLC, Parker, CO. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
R.D.B., K.R.S., and F.B.G. conceived and designed research; R.D.B., K.S.H., M.L., M.G.F., J.H., G.M.K., T.N.H., J.W., H.Z., S.R.R., L.A.W., and F.B.G. performed experiments; R.D.B., K.S.H., M.L., J.H., G.M.K., S.R.R., and S.K. analyzed data; R.D.B., K.S.H., M.L., M.G.F., J.H., G.M.K., J.W., S.R.R., M.G.E., C.-J.H., B.B.G., L.A.W., P.M.B., T.L., V.O.K., K.C.H., and K.R.S. interpreted results of experiments; R.D.B. prepared figures; R.D.B. drafted manuscript; R.D.B., B.B.G., L.A.W., F.B.G., P.M.B., T.L., V.O.K., K.C.H., and K.R.S. edited and revised manuscript; R.D.B., K.S.H., M.L., M.G.F., J.H., G.M.K., T.N.H., J.W., H.Z., S.R.R., M.G.E., S.K., C.-J.H., B.B.G., L.A.W., F.B.G., P.M.B., T.L., V.O.K., K.C.H., and K.R.S. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors are grateful for superb technical assistance from the following individuals: Yanmei Du, Amanda Flockton, Lauren Henry, Stephen Hofmeister, Zoe Loomis, Alexandre McKeon, Andy Poczobutt, Sandra Walchak, and to Marcia McGowan for assistance with manuscript preparation. Dr. S. Kelly Ambler and Andy Poczobutt assisted with figure preparation. R.D.B. thanks Dr. S. Kelly Ambler for numerous helpful discussions.
REFERENCES
- 1. McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, , et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation 119: 2250–2294, 2009. doi: 10.1161/CIRCULATIONAHA.109.192230. [DOI] [PubMed] [Google Scholar]
- 2. Vonk Noordegraaf A, Chin KM, Haddad F, Hassoun PM, Hemnes AR, Hopkins SR, Kawut SM, Langleben D, Lumens J, Naeije R. Pathophysiology of the right ventricle and of the pulmonary circulation in pulmonary hypertension: an update. Eur Respir J 53: 1801900, 2019. doi: 10.1183/13993003.01900-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vonk-Noordegraaf A, Haddad F, Chin KM, Forfia PR, Kawut SM, Lumens J, Naeije R, Newman J, Oudiz RJ, Provencher S, Torbicki A, Voelkel NF, Hassoun PM. Right heart adaptation to pulmonary arterial hypertension: physiology and pathobiology. J Am Coll Cardiol 62: D22–D33, 2013. doi: 10.1016/j.jacc.2013.10.027. [DOI] [PubMed] [Google Scholar]
- 4. Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, Dupuis J, Long CS, Rubin LJ, Smart FW, Suzuki YJ, Gladwin M, Denholm EM, Gail DB; National Heart L, Blood Institute Working Group on Cellular and Molecular Mechanisms of Right Heart Failure. Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation 114: 1883–1891, 2006. doi: 10.1161/CIRCULATIONAHA.106.632208. [DOI] [PubMed] [Google Scholar]
- 5. Cornwell WK, Tran T, Cerbin L, Coe G, Muralidhar A, Hunter K, Altman N, Ambardekar AV, Tompkins C, Zipse M, Schulte M, O'Gean K, Ostertag M, Hoffman J, Pal JD, Lawley JS, Levine BD, Wolfel E, Kohrt WM, Buttrick P. New insights into resting and exertional right ventricular performance in the healthy heart through real-time pressure-volume analysis. J Physiol 598: 2575–2587, 2020. doi: 10.1113/JP279759. [DOI] [PubMed] [Google Scholar]
- 6. Naeije R, Brimioulle S, Dewachter L. Biomechanics of the right ventricle in health and disease (2013 Grover Conference series). Pulm Circ 4: 395–406, 2014. doi: 10.1086/677354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pinsky MR. The right ventricle: interaction with the pulmonary circulation. Crit Care 20: 266, 2016. [Erratum in Crit Care 20: 364, 2016]. doi: 10.1186/s13054-016-1440-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reesink HJ, Marcus JT, Tulevski II, Jamieson S, Kloek JJ, Vonk Noordegraaf A, Bresser P. Reverse right ventricular remodeling after pulmonary endarterectomy in patients with chronic thromboembolic pulmonary hypertension: utility of magnetic resonance imaging to demonstrate restoration of the right ventricle. J Thorac Cardiovasc Surg 133: 58–64, 2007. doi: 10.1016/j.jtcvs.2006.09.032. [DOI] [PubMed] [Google Scholar]
- 9. Pugliese SC, Poth JM, Fini MA, Olschewski A, El Kasmi KC, Stenmark KR. The role of inflammation in hypoxic pulmonary hypertension: from cellular mechanisms to clinical phenotypes. Am J Physiol Lung Cell Mol Physiol 308: L229–L252, 2015. doi: 10.1152/ajplung.00238.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stenmark KR, Fasules J, Hyde DM, Voelkel NF, Henson J, Tucker A, Wilson H, Reeves JT. Severe pulmonary hypertension and arterial adventitial changes in newborn calves at 4,300 m. J Appl Physiol (1985) 62: 821–830, 1987. doi: 10.1152/jappl.1987.62.2.821. [DOI] [PubMed] [Google Scholar]
- 11. Milani-Nejad N, Janssen PML. Small and large animal models in cardiac contraction research: advantages and disadvantages. Pharmacol Ther 141: 235–249, 2014. doi: 10.1016/j.pharmthera.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rhodes J. Comparative physiology of hypoxic pulmonary hypertension: historical clues from brisket disease. J Appl Physiol (1985) 98: 1092–1100, 2005. doi: 10.1152/japplphysiol.01017.2004. [DOI] [PubMed] [Google Scholar]
- 13. Bartels K, Brown RD, Fox DL, Bull TM, Neary JM, Dorosz JL, Fonseca BM, Stenmark KR. Right ventricular longitudinal strain is depressed in a bovine model of pulmonary hypertension. Anesth Analg 122: 1280–1286, 2016. doi: 10.1213/ANE.0000000000001215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lemler MS, Bies RD, Frid MG, Sastravaha A, Zisman LS, Bohlmeyer T, Gerdes AM, Reeves JT, Stenmark KR. Myocyte cytoskeletal disorganization and right heart failure in hypoxia-induced neonatal pulmonary hypertension. Am J Physiol Heart Circ Physiol 279: H1365–H1376, 2000. doi: 10.1152/ajpheart.2000.279.3.H1365. [DOI] [PubMed] [Google Scholar]
- 15. Applegate TJ, Krafsur GM, Boon JA, Zhang H, Li M, Holt TN, Ambler SK, Abrams BA, Gustafson DL, Bartels K, Garry FB, Stenmark KR, Brown RD. Brief report: case comparison of therapy with the histone deacetylase inhibitor vorinostat in a neonatal calf model of pulmonary hypertension. Front Physiol 12: 712583, 2021. doi: 10.3389/fphys.2021.712583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bruns DR, Brown RD, Stenmark KR, Buttrick PM, Walker LA. Mitochondrial integrity in a neonatal bovine model of right ventricular dysfunction. Am J Physiol Lung Cell Mol Physiol 308: L158–L167, 2015. doi: 10.1152/ajplung.00270.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sharifi Kia D, Kim K, Simon MA. Current understanding of the right ventricle structure and function in pulmonary arterial hypertension. Front Physiol 12: 641310, 2021. doi: 10.3389/fphys.2021.641310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Walker LA, Walker JS, Glazier A, Brown DR, Stenmark KR, Buttrick PM. Biochemical and myofilament responses of the right ventricle to severe pulmonary hypertension. Am J Physiol Heart Circ Physiol 301: H832–H840, 2011. doi: 10.1152/ajpheart.00249.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bakerman PR, Stenmark KR, Fisher JH. Alpha-skeletal actin messenger RNA increases in acute right ventricular hypertrophy. Am J Physiol Lung Cell Mol Physiol 258: L173–L178, 1990. doi: 10.1152/ajplung.1990.258.4.L173. [DOI] [PubMed] [Google Scholar]
- 20. Holt TN, Callan RJ. Pulmonary arterial pressure testing for high mountain disease in cattle. Vet Clin North Am Food Anim Pract 23: 575–596, vii, 2007. doi: 10.1016/j.cvfa.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 21. Gu S, Mickael C, Kumar R, Lee MH, Sanders L, Kassa B, Harral J, Williams J, Hansen KC, Stenmark KR, Tuder RM, Graham BB. The role of macrophages in right ventricular remodeling in experimental pulmonary hypertension. Pulm Circ 12: e12105, 2022. doi: 10.1002/pul2.12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brown RD, Ambler SK, Li M, Sullivan TM, Henry LN, Crossno JT Jr, Long CS, Garrington TP, Stenmark KR. MAP kinase kinase kinase-2 (MEKK2) regulates hypertrophic remodeling of the right ventricle in hypoxia-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol 304: H269–H281, 2013. doi: 10.1152/ajpheart.00158.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li M, Riddle SR, Frid MG, El Kasmi KC, McKinsey TA, Sokol RJ, Strassheim D, Meyrick B, Yeager ME, Flockton AR, McKeon BA, Lemon DD, Horn TR, Anwar A, Barajas C, Stenmark KR. Emergence of fibroblasts with a proinflammatory epigenetically altered phenotype in severe hypoxic pulmonary hypertension. J Immunol 187: 2711–2722, 2011. doi: 10.4049/jimmunol.1100479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McCabe MC, Schmitt LR, Hill RC, Dzieciatkowska M, Maslanka M, Daamen WF, van Kuppevelt TH, Hof DJ, Hansen KC. Evaluation and refinement of sample preparation methods for extracellular matrix proteome coverage. Mol Cell Proteomics 20: 100079, 2021. doi: 10.1016/j.mcpro.2021.100079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Willforss J, Chawade A, Levander F. NormalyzerDE: online tool for improved normalization of omics expression data and high-sensitivity differential expression analysis. J Proteome Res 18: 732–740, 2019. doi: 10.1021/acs.jproteome.8b00523. [DOI] [PubMed] [Google Scholar]
- 26. Smyth GK. limma: linear models for microarray data. In: Bioinformatics and Computational Biology Solutions Using R and Bioconductor Statistics for Biology and Health, edited by Gentleman R, Carey V.J., Huber W., Irizarry R.A., Dudoit S.. New York, NY: Springer, 2005. [Google Scholar]
- 27. Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740, 2016. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 28. Pang Z, Zhou G, Ewald J, Chang L, Hacariz O, Basu N, Xia J. Using MetaboAnalyst 5.0 for LC-HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat Protoc 17: 1735–1761, 2022. doi: 10.1038/s41596-022-00710-w. [DOI] [PubMed] [Google Scholar]
- 29. Breeman KTN, Dufva M, Ploegstra MJ, Kheyfets V, Willems TP, Wigger J, Hunter KS, Ivy DD, Berger RMF, Truong U. Right ventricular-vascular coupling ratio in pediatric pulmonary arterial hypertension: a comparison between cardiac magnetic resonance and right heart catheterization measurements. Int J Cardiol 293: 211–217, 2019. doi: 10.1016/j.ijcard.2019.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Haddad F, Hunt SA, Rosenthal DN, Murphy DJ. Right ventricular function in cardiovascular disease, part I: anatomy, physiology, aging, and functional assessment of the right ventricle. Circulation 117: 1436–1448, 2008. doi: 10.1161/CIRCULATIONAHA.107.653576. [DOI] [PubMed] [Google Scholar]
- 31. Lowes BD, Minobe W, Abraham WT, Rizeq MN, Bohlmeyer TJ, Quaife RA, Roden RL, Dutcher DL, Robertson AD, Voelkel NF, Badesch DB, Groves BM, Gilbert EM, Bristow MR. Changes in gene expression in the intact human heart. Downregulation of alpha-myosin heavy chain in hypertrophied, failing ventricular myocardium. J Clin Invest 100: 2315–2324, 1997. doi: 10.1172/JCI119770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sun W, Chan SY. Pulmonary arterial stiffness: an early and pervasive driver of pulmonary arterial hypertension. Front Med (Lausanne) 5: 204, 2018. doi: 10.3389/fmed.2018.00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bruns DR, Tatman PD, Kalkur RS, Brown RD, Stenmark KR, Buttrick PM, Walker LA. The right ventricular fibroblast secretome drives cardiomyocyte dedifferentiation. PLoS One 14: e0220573, 2019. doi: 10.1371/journal.pone.0220573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol 45: 657–687, 2005. doi: 10.1146/annurev.pharmtox.45.120403.095802. [DOI] [PubMed] [Google Scholar]
- 35. Durmowicz AG, Orton EC, Stenmark KR. Progressive loss of vasodilator responsive component of pulmonary hypertension in neonatal calves exposed to 4,570 m. Am J Physiol Heart Circ Physiol 265: H2175–H2183, 1993. doi: 10.1152/ajpheart.1993.265.6.H2175. [DOI] [PubMed] [Google Scholar]
- 36. Jayasekera G, Wilson KS, Buist H, Woodward R, Uckan A, Hughes C, Nilsen M, Church AC, Johnson MK, Gallagher L, Mullin J, MacLean MR, Holmes WM, Peacock AJ, Welsh DJ. Understanding longitudinal biventricular structural and functional changes in a pulmonary hypertension Sugen-hypoxia rat model by cardiac magnetic resonance imaging. Pulm Circ 10: 2045894019897513, 2020. doi: 10.1177/2045894019897513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Totaro A, Panciera T, Piccolo S. YAP/TAZ upstream signals and downstream responses. Nat Cell Biol 20: 888–899, 2018. doi: 10.1038/s41556-018-0142-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ikeda S, Sadoshima J. Regulation of myocardial cell growth and death by the Hippo pathway. Circ J 80: 1511–1519, 2016. doi: 10.1253/circj.CJ-16-0476. [DOI] [PubMed] [Google Scholar]
- 39. Dufva MJ, Truong U, Tiwari P, Ivy DD, Shandas R, Kheyfets VO. Left ventricular torsion rate and the relation to right ventricular function in pediatric pulmonary arterial hypertension. Pulm Circ 8: 2045894018791352, 2018. doi: 10.1177/2045894018791352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kheyfets VO, Truong U, Ivy D, Shandas R. Structural and biomechanical adaptations of right ventricular remodeling-in pulmonary arterial hypertension-reduces left ventricular rotation during contraction: a computational study. J Biomech Eng 141: 0510021–05100210, 2019. doi: 10.1115/1.4042682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res 115: 165–175, 2014. doi: 10.1161/CIRCRESAHA.113.301141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, Jessup M, Grizzle WE, Aldred MA, Cool CD, Tuder RM. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med 186: 261–272, 2012. doi: 10.1164/rccm.201201-0164OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sun X-Q, Abbate A, Bogaard H-J. Role of cardiac inflammation in right ventricular failure. Cardiovasc Res 113: 1441–1452, 2017. doi: 10.1093/cvr/cvx159. [DOI] [PubMed] [Google Scholar]
- 44. Dewachter L, Dewachter C. Inflammation in right ventricular failure: does it matter? Front Physiol 9: 1056, 2018. doi: 10.3389/fphys.2018.01056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gorr MW, Sriram K, Chinn AM, Muthusamy A, Insel PA. Transcriptomic profiles reveal differences between the right and left ventricle in normoxia and hypoxia. Physiol Rep 8: e14344, 2020. doi: 10.14814/phy2.14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. El Kasmi KC, Pugliese SC, Riddle SR, Poth JM, Anderson AL, Frid MG, Li M, Pullamsetti SS, Savai R, Nagel MA, Fini MA, Graham BB, Tuder RM, Friedman JE, Eltzschig HK, Sokol RJ, Stenmark KR. Adventitial fibroblasts induce a distinct proinflammatory/profibrotic macrophage phenotype in pulmonary hypertension. J Immunol 193: 597–609, 2014. doi: 10.4049/jimmunol.1303048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Krishnan S, Stearman RS, Zeng L, Fisher A, Mickler EA, Rodriguez BH, Simpson ER, Cook T, Slaven JE, Ivan M, Geraci MW, Lahm T, Tepper RS. Transcriptomic modifications in developmental cardiopulmonary adaptations to chronic hypoxia using a murine model of simulated high-altitude exposure. Am J Physiol Lung Cell Mol Physiol 319: L456–L470, 2020. doi: 10.1152/ajplung.00487.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Egemnazarov B, Crnkovic S, Nagy BM, Olschewski H, Kwapiszewska G. Right ventricular fibrosis and dysfunction: actual concepts and common misconceptions. Matrix Biol 68–69: 507–521, 2018. doi: 10.1016/j.matbio.2018.01.010. [DOI] [PubMed] [Google Scholar]
- 49. Overbeek MJ, Lankhaar J-W, Westerhof N, Voskuyl AE, Boonstra A, Bronzwaer JGF, Marques KMJ, Smit EF, Dijkmans BAC, Vonk-Noordegraaf A. Right ventricular contractility in systemic sclerosis-associated and idiopathic pulmonary arterial hypertension. Eur Respir J 31: 1160–1166, 2008. doi: 10.1183/09031936.00135407. [DOI] [PubMed] [Google Scholar]
- 50. van der Bruggen CEE, Tedford RJ, Handoko ML, van der Velden J, de Man FS. RV pressure overload: from hypertrophy to failure. Cardiovasc Res 113: 1423–1432, 2017. doi: 10.1093/cvr/cvx145. [DOI] [PubMed] [Google Scholar]
- 51. Frangogiannis NG. Fibroblasts and the extracellular matrix in right ventricular disease. Cardiovasc Res 113: 1453–1464, 2017. doi: 10.1093/cvr/cvx146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Imoto K, Okada M, Yamawaki H. Expression profile of matricellular proteins in hypertrophied right ventricle of monocrotaline-induced pulmonary hypertensive rats. J Vet Med Sci 79: 1096–1102, 2017. doi: 10.1292/jvms.17-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, Ockaili R, McCord JM, Voelkel NF. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation 120: 1951–1960, 2009. doi: 10.1161/CIRCULATIONAHA.109.883843. [DOI] [PubMed] [Google Scholar]
- 54. Frump AL, Bonnet S, de Jesus Perez VA, Lahm T. Emerging role of angiogenesis in adaptive and maladaptive right ventricular remodeling in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 314: L443–L460, 2018. doi: 10.1152/ajplung.00374.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. van Wolferen SA, Marcus JT, Westerhof N, Spreeuwenberg MD, Marques KMJ, Bronzwaer JGF, Henkens IR, Gan CT-J, Boonstra A, Postmus PE, Vonk-Noordegraaf A. Right coronary artery flow impairment in patients with pulmonary hypertension. Eur Heart J 29: 120–127, 2008. doi: 10.1093/eurheartj/ehm567. [DOI] [PubMed] [Google Scholar]
- 56. Graham BB, Kumar R, Mickael C, Kassa B, Koyanagi D, Sanders L, Zhang L, Perez M, Hernandez-Saavedra D, Valencia C, Dixon K, Harral J, Loomis Z, Irwin D, Nemkov T, D'Alessandro A, Stenmark KR, Tuder RM. Vascular adaptation of the right ventricle in experimental pulmonary hypertension. Am J Respir Cell Mol Biol 59: 479–489, 2018. doi: 10.1165/rcmb.2018-0095OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kolb TM, Peabody J, Baddoura P, Fallica J, Mock JR, Singer BD, D'Alessio FR, Damarla M, Damico RL, Hassoun PM. Right ventricular angiogenesis is an early adaptive response to chronic hypoxia-induced pulmonary hypertension. Microcirculation 22: 724–736, 2015. doi: 10.1111/micc.12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Graham BB, Koyanagi D, Kandasamy B, Tuder RM. Right ventricle vasculature in human pulmonary hypertension assessed by stereology. Am J Respir Crit Care Med 196: 1075–1077, 2017. doi: 10.1164/rccm.201702-0425LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tomanek RJ, Zheng W, Yue X. Growth factor activation in myocardial vascularization: therapeutic implications. Mol Cell Biochem 264: 3–11, 2004. doi: 10.1023/b:mcbi.0000044369.88528.a3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figs. S1–S7 and Supplemental Tables S1–S10: https://doi.org/10.6084/m9.figshare.22185838.
Data Availability Statement
Data will be made available upon reasonable request.