

Keywords: LC3-interacting region, mitochondrial outer membrane proteins, mitophagy, selective autophagy receptor
Abstract
A20 binding inhibitor of nuclear factor kappa B (NF-κB)-1 (ABIN-1), a polyubiquitin-binding protein, is a signal-induced autophagy receptor that attenuates NF-κB-mediated inflammation and cell death. The present study aimed to elucidate the potential role of ABIN-1 in mitophagy, a biological process whose outcome is decisive in diverse physiological and pathological settings. Microtubule-associated proteins 1A/1B light chain 3B-II (LC3B-II) was found to be in complex with ectopically expressed hemagglutinin (HA)-tagged-full length (FL)-ABIN-1. Bacterial expression of ABIN-1 and LC3A and LC3B showed direct binding of ABIN-1 to LC3 proteins, whereas mutations in the LC3-interacting region (LIR) 1 and 2 motifs of ABIN-1 abrogated ABIN-1/LC3B-II complex formation. Importantly, induction of autophagy in HeLa cells resulted in colocalization of ABIN-1 with LC3B-II in autophagosomes and with lysosomal-associated membrane protein 1 (LAMP-1) in autophagolysosomes, leading to degradation of ABIN-1 with p62. Interestingly, ABIN-1 was found to translocate to damaged mitochondria in HeLa-mCherry-Parkin transfected cells. In line with this observation, clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9)-mediated deletion of ABIN-1 significantly inhibited the degradation of the mitochondrial outer membrane proteins voltage-dependent anion-selective channel 1 (VDAC-1), mitofusin-2 (MFN2), and translocase of outer mitochondrial membrane (TOM)20. In addition, short interfering RNA (siRNA)-mediated knockdown of ABIN-1 significantly decreased lysosomal uptake of mitochondria in HeLa cells expressing mCherry-Parkin and the fluorescence reporter mt-mKEIMA. Collectively, our results identify ABIN-1 as a novel and selective mitochondrial autophagy regulator that promotes mitophagy, thereby adding a new player to the complex cellular machinery regulating mitochondrial homeostasis.
INTRODUCTION
A20 binding inhibitor of nuclear factor kappa B (NF-κB)-1 (ABIN-1), also called tumor necrosis factor α-induced protein 3 (TNFAIP3)-interacting protein 1 (TNIP-1), genetically encoded on chromosome 5q32-33.1, is a ubiquitously expressed protein located in the cytoplasm and nuclear compartments of the cell (1). ABIN-1 shares limited sequence homology in four ABIN homology domains (AHD 1–4) with two family members, ABIN-2 and ABIN-3 (2). AHD1 binds the ubiquitin-editing protein, TNFAIP3 (A20), whereas AHD2, also called ubiquitin-binding domain (UBD) in ABIN proteins and NF-κB essential modulator (NEMO) (UBAN) domain, binds polyubiquitin and polyubiquitinated proteins (3). The functions of AHD3 and AHD4 still remain to the elucidated. Unlike ABIN-2 and ABIN-3, ABIN-1 has a C-terminal NEMO-binding domain (NBD) (4). ABIN-1 is a polyubiquitin-binding protein regulating various cellular functions, including development, immunity, and tissue homeostasis (5, 6), mediated at least in part by inhibition of NF-κB activation through the AHD1, UBAN, and NEMO domains (4, 6, 7). In addition to its anti-inflammatory role, ABIN-1 acts as a suppressor of different forms of cell death, including apoptosis (8–10) and necroptosis (11–14). The anti-inflammatory and prosurvival roles of ABIN-1 has therapeutic potential for the treatment of several inflammatory diseases, including colitis (15), systemic lupus erythematosus (SLE) (16), sepsis (17), glomerulonephritis (18), psoriasis (19), inflammatory bowel disease (20), osteoarthritis (21), asthma, chronic obstructive pulmonary disease (COPD) (22), and inflammatory liver diseases (23). ABIN-1 regulates ubiquitination and/or proteasome/lysozyme-dependent degradation of various interacting client proteins, including histone deacetylase 1 (HDAC1) (24), Tax1-binding protein 1 (TAX1BP1) (25), β-arrestin-2 (26), and nucleotide oligomerization domain (NOD)-like receptors (NLRs)P10 (27). The UBAN domain of ABIN-1 shares high sequence homology with the UBD of optineurin (OPTN), a selective autophagy receptor (SAR) (7). Accordingly, ABIN-1 was recently identified as a signal-induced autophagy receptor that attenuates NF-κB activation by recognizing linear ubiquitin chains (28).
Selective autophagy is an evolutionarily conserved catabolic process that discriminates distinct cellular components and target-specific cargo such as toxic intracellular components, damaged organelles, aggregated proteins, unwanted macromolecules, and invading pathogens through SARs for degradation by sequestration into de novo-formed autophagosomes that are subjected to lysosomal degradation to maintain cellular homeostasis (29). Till date, a number of different SARs have been identified, with those that mediate mitophagy, i.e., the selective degradation of mitochondria, being the best-studied among this group (30).
Mitochondria, also called the powerhouse of the cells, are organelles that are centrally involved in a plethora of functions, including adenosine 5′-triphosphate (ATP) production, redox homeostasis, Ca2+signaling, and apoptosis (31). Mitochondrial integrity is vital for cellular metabolism, homeostasis, and a myriad of complex signaling cascades that regulate cell survival and cell death (32). Mitochondrial homeostasis is regulated by mitophagy, a selective autophagic process of sequestering and degrading damaged mitochondria in autophagolysosomes that is mediated by either the phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-Parkin regulated ubiquitin-dependent pathway or by the ubiquitin-independent receptor-mediated pathway (29). Selective mitophagy receptors that target dysfunctional mitochondria for autophagic degradation include B-cell lymphoma 2 (BCL2) and adenovirus E1B 19-kDa-interacting protein 3 (BNIP3) (33), BNIP3-like (BNIP3L)/NIX22 (33), FUN14 domain-containing protein 1 (FUNDC1) (33), spermatogenesis associated 33 (SPATA33) (34), FK506 binding protein 8 (FKBP8) (35), and prohibitin 2 (PHB2) located on the inner mitochondrial membrane (36). Here, we demonstrate ABIN-1 as a novel regulator of mitochondrial autophagy and delineate the key functions of its light chain (LC)3-interacting region (LIR) domains in this process.
MATERIALS AND METHODS
Chemicals
The chemicals used in the present study were purchased from the following sources: Dulbecco’s modified Eagle’s medium (DMEM GlutaMax, Gibco, 61965026), Penicillin-Streptomycin (P/S, 15140122), Platinum PCR SuperMix high fidelity (Invitrogen, 12532016), pcDNA3.1(+) mammalian expression vector (Invitrogen, V79020), One Shot TOP10 chemically competent Escherichia coli (E. coli) (Invitrogen, C404003), Luria-Bertani (LB) agar plates with 100 μg/mL Ampicillin (R110204), LB agar plates with 50 μg/mL Kanamycin (J60540EQF), GeneArt site-directed mutagenesis system (A13282), AccuPrime Pfx DNA polymerase (12344024), One Shot MAX efficiency DH5α-T1R competent cells (12297016), TOPO TA cloning kit (450641), Ambion silencer select pre-designed ABIN-1 siRNA (Assay ID 13556), Lipofectamine RNAiMAX transfection reagent (13778150), Silencer select negative control No. 1 siRNA (4390843), Pierce anti-HA magnetic beads (88836), BL21(DE3) competent cells (EC0114), LB broth base (12780052), Mitochondria isolation kit for cultured cells (89874), Puromycin dihydrochloride (A1113803), and Pierce bicinchoninic acid (BCA) protein assay kit (23227) were purchased from Thermo Fisher Scientific, Darmstadt, Germany; fetal bovine serum (FBS, F7524), Oligomycin A (O, 75351), Antimycin A (A, A8674), doxycycline hyclate (DOX, D5207), pGEX-4T-1 (GE28-9545-49), isopropyl-β-d-1-thiogalactopyranoside, (IPTG, 16758), glutathion-sepharose 4B (GE17-0756-01), dimethylsulfoxide (DMSO, D2650), and methanol (M1775) were purchased from SIGMA (Merck, Darmstadt, Germany); VenorGeM classic kit (11–1025) was purchased from Minerva Biolabs GmbH, Berlin, Germany. Torin-1 (4247) was brought from Bio-Techne GmbH, Wiesbaden, Germany. Rapamycin (R-5000) was purchased from LC Laboratories, Woburn, MA; Bafilomycin A1 (BafA1, 11038) was purchased from Cayman chemical, Ann Arbor, MI; QIAquick PCR purification kit (28104), QIAprep spin miniprep kit (27106X4), Plasmid mini kit (12123), and EndoFree plasmid maxi kit (12362) were purchased from Qiagen, Hilden, Germany; BamHI (R0136L), XhoI (R0146L), SfiI (R0123L), HindIII (R0104L), BspE1 (R0540L), BbsI (R0539L), T4 DNA ligase (M0202L), Quick ligation kit (M2200L), Amylose resin (E8021L), and NEBExpress Maltose binding protein (MBP) fusion and purification system (E8201S) were purchased from New England Biolabs, Frankfurt, Germany; pSBtet-Pur (60507), pSBtet-Neo (60509), monomeric enhanced green fluorescent protein (mEGFP)-C1 (54759), mCherry-Parkin (23956), pCMV(CAT)T7-SB100 (34879), pSpCas9(BB)-2A-Puro (PX459) V2.0 (62988), pMAL-c2X (75286), and mCherry-hLC3B-pcDNA3.1 (40827) were purchased from Addgene, Teddington, UK; FuGENE HD transfection reagent (E2311) was purchased from Promega, Mannheim, Germany; GFP-Trap Magnetic Agarose (MA) (gtma-100) was brought from ChromoTek, Martinsried, Germany; Ponceau S Solution (ab270042) was purchased from Abcam, Berlin, Germany; VECTASHIELD antifade mounting medium with DAPI (H-1200-10) was purchased from Vector Laboratories, Burlingame, CA. Geneticin disulfate solution (G418, Art. No. 2039.3) was purchased from Carl Roth, Karlsruhe, Germany.
Cell Culture
HeLa cells (American Type Culture Collection, Manassas, VA) were cultured in DMEM supplemented with 10% FBS and 1% P/S and grown at 37°C, 5% CO2, and 95% relative humidity. Mycoplasma contamination was excluded using the VenorGeM classic kit according to the manufacturer’s instructions. Expression of mCherry-Parkin protein in stable HeLa-mCherry-Parkin transfected cells was induced with DOX (0.5 µg/mL).
Molecular Cloning of ABIN-1-HA-Full Length
Full length (FL) human ABIN-1 was amplified by polymerase chain reaction (PCR) from the template of an open reading frame clone (GeneScript, product ID: OHu13881D) with primers containing an N-terminal BamHI restriction site and a C-terminal human influenza hemagglutinin (HA)-tag, together with an XhoI restriction site (Table 1). PCR was performed using the platinum PCR SuperMix high fidelity and the corresponding PCR product was purified using the QIAquick PCR purification kit according to the manufacturer’s instructions. hABIN1-1-HA and the pcDNA3.1(+) vector were digested with BamHI and XhoI, and ligated at a molar ratio of 1:3 (vector:insert) using the T4 DNA ligase according to the manufacturer’s protocol. The ligation mixture was transformed into One Shot TOP10 chemically competent E. coli by heat shock, after which transformed clones were selected on LB agar plates with 100 μg/mL ampicillin, and single colonies were expanded. Plasmid DNA was isolated using the QIAprep spin miniprep kit according to the manufacturer’s instructions and was analyzed by sequencing (Eurofins Genomics, Ebersberg, Germany). DNA concentrations were determined using the Nano-Drop spectrophotometer (Peqlab Biotechnologie, Munich, Germany).
Table 1.
Primers used in this study
| Primer | Sequence 5′ to 3′ |
|---|---|
| hABIN-1 fwd | CGCGGATCCACCCTCATGGAAGGGAGAGGACCGTACCGGAAAC |
| hABIN-1-HA rev | CCGCTCGAGTCAAGCGTAATCTGGAACATCGTATGGGTACTGA |
| GGCCCCTCACGGTCATTTTTTG | |
| LIR1 [F83A/L86A] fwd | CCCTCCTTGGGCTCCGCCGACCCCGCGGCTGAGCTCACAGGA |
| LIR1 [F83A/L86A] rev | TCCTGTGAGCTCAGCCGCGGGGTCGGCGGAGCCCAAGGAGGG |
| LIR2 [F125A/V128A] fwd | GGCACCTCCTCTGAAGCTGAAGTGGCCACTCCTGAGGAGCAG |
| LIR2 [F125A/V128A] rev | CTGCTCCTCAGGAGTGGCCACTTCAGCTTCAGAGGAGGTGCC |
| UBAN [D472N] fwd | GTGAAGATCTTCGAGGAGAACTTCCAGAGGGAGCGCAGT |
| UBAN [D472N] rev | ACTGCGCTCCCTCTGGAAGTTCTCCTCGAAGATCTTCAC |
| mCherryParkin fwd | GACGGCCTCTGAGGCCGCCACCATGGTGAGCAAGGGCGAGGAGG |
| mCherryParkin rev | GACGGCCTGACAGGCCTTACACGTCGAACCAGTGGTCCCCCATG |
| ABIN-1ORF 3 fwd | GACTCCGGAATCATGGAAGGGAGAGGACCGTACC |
| ABIN-1ORF 5 rev | GACAAGCTTTCACTGAGGCCCCTCACGGTCA |
| mEGFP-C1-hABIN-1 fwd | GGCCTCTGAGGCCGCCACCATGGTGAGCAAGGGCGAGGAGC |
| mEGFP-C1-hABIN-1 rev | GGCCTGACAGGCCTCACTGAGGCCCCTCACGGTCATTTTTTGG |
| gRNA-1-sense | CACCGGCAAGGGATAAAGATGTTAG |
| gRNA-1-antisense | AAACCTAACATCTTTATCCCTTGCC |
| gRNA-2-sense | CACCGGAGCCTGGTCGCTTCCATCT |
| gRNA-2-antisense | AAACAGATGGAAGCGACCAGGCTCC |
| gRNA-3-sense | CACCGAGTCCCAGATGGAAGCGACC |
| gRNA-3-antisense | AAACGGTCGCTTCCATCTGGGACTC |
Fwd, forward; HA, hemagglutinin; hABIN-1, human A20 binding and inhibitor of nuclear factor kappa B (NF-κB)-1; LIR, LC3-interacting region; NEMO, NF-κB essential modulator; ORF, open reading frame; rev, reverse; UBAN, ubiquitin-binding domain (UBD) in ABIN proteins and NEMO.
Site-Directed Mutagenesis of ABIN-1 LIRs
The GeneArt site-directed mutagenesis system was used to generate ABIN-1 mutants with defective LIR domains as per the manufacturer’s instructions. Briefly, pcDNA3.1(+)-hABIN-1-HA plasmid DNA was mixed with overlapping primer pairs carrying the desired LIR mutations (Table 1) and AccuPrime Pfx DNA polymerase as instructed by the manufacturer. PCR cycling conditions were as follows: 37°C for 20 min, 94°C for 2 min, 18 cycles at 94°C for 20 s, 57°C for 4 min, and 68°C for 2 min, followed by 68°C for 5 min. The PCR product was transformed into One Shot MAX efficiency DH5α-T1R competent cells. The purified plasmid DNA was analyzed by Sanger sequencing (Eurofins Genomics) before being used for further applications. Accordingly, the following ABIN-1 variants with point mutations in LIR1 (Phe83 → Ala83 and Leu86 → Ala86, F83A/L86A), LIR2 (Phe125 → Ala125 and Val128 → Ala128, F125A/V128A), or both (LIR1 + 2) were used. The UBAN domain of ABIN-1 was inactivated by mutating Asp472 → Asn472 (D472N), to inhibit binding to linear and K63-linked ubiquitin chains (6). The HA-tagged hABIN-1 was overexpressed either as a full length (FL), LIR1-, LIR2-, double LIR (LIR1 + 2)-mutated, or ubiquitin-binding defective variant (UBAN) in HeLa cells.
TOPO Cloning of mCherry-Parkin and mEGFP-hABIN-1
mCherry-Parkin cDNA was PCR amplified with primers containing SfiI restriction sites (Table 1) from the mCherry-Parkin vector to generate 3′ A-overhangs necessary for subsequent TOPO cloning with the platinum PCR SuperMix high fidelity as per the manufacturer’s instructions. PCR cycling conditions were as follows: 94°C for 2 min, 35 cycles at 94°C for 30 s, 55°C for 30 s, 68°C for 2 min, followed by 68°C for 5 min.
The PCR product was cloned into a TOPO vector with the TOPO TA cloning kit and transformed into One Shot TOP10 chemically competent E. coli. Transformed clones were selected on LB agar plates with 50 μg/mL Kanamycin. Isolated plasmid DNA from single colonies was digested with SfiI. Subsequently, mCherry-Parkin cDNA was cloned into the pSBtet-Pur or pSBtet-Neo vectors. Purified plasmid DNA was verified by sequencing before performing endotoxin-free plasmid purification for subsequent experiments.
For generating mEGFP-hABIN-1 cDNA, hABIN-1 was amplified by PCR from the open reading frame (ORF) clone (GeneScript, Product ID: OHu13881D) using primers containing a HindIII and BspE1 restriction site (Table 1). The PCR protocol was as described for HA-tagged ABIN-1. The resulting PCR product and mEGFP-C1 vector were digested using HindIII and BspE1. Insert and vector were ligated at a molar ratio of 1:3 with a Quick ligation kit and transformed using One Shot TOP10 chemically competent E. coli. After plasmid purification, insert of hABIN-1 into the mEGFP-C1 vector was verified by Sanger sequencing. TOPO cloning was as described for mCherry-Parkin.
Stably transfected cell lines were generated using the sleeping beauty (SB) expression vector system (37). Briefly, pSBtet-Pur and pSBtet-Neo vectors containing either mCherry-Parkin or mEGFP-hABIN-1 were transfected into HeLa cells, in addition to the pCMV(CAT)T7-SB100. Transfection of HeLa cells was performed using FuGENE HD transfection reagent according to the manufacturer’s instructions. Transfected cells were selected based on the respective antibiotic resistance (puromycin, 3 µg/mL and G418, 800 µg/mL).
CRISPR/Cas9-Mediated Knockout of ABIN-1
To generate clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9)-mediated knockout of ABIN-1, three single gRNAs selected using the CRISPOR program (http://crispor.tefor.net) were synthesized as single-strand DNA oligonucleotides flanked by overhangs that are compatible with BbsI endonuclease-based cloning (Table 1). The gRNAs were cloned into the pSpCas9(BB)-2A-Puro (PX459) V2.0 pre-digested with BbsI, according to the manufacturer’s instructions. TOPO cloning was as described for mCherry-Parkin and transformed clones were selected based on ampicillin resistance. Bacterial clones were purified using the plasmid mini kit and EndoFree plasmid maxi kit as per the manufacturer’s instructions. pSpCas9(BB)-2A-Puro vectors carrying the gRNAs were sequenced before transfection. All three gRNAs were transfected simultaneously into HeLa cells using the FuGENE HD transfection reagent as per the manufacturer’s instructions. Transfected cells from single colonies that were expanded and screened by Western blot for successful knockdown of ABIN-1 were used for further experiments.
siRNA-Mediated Knockdown of ABIN-1
Transient knockdown of ABIN-1 was achieved using Ambion silencer select predesigned ABIN-1 siRNA (Assay ID 13556). HeLa cells were transfected with siRNAs using Lipofectamine RNAiMAX transfection reagent as per the manufacturer’s instructions. Silencer select negative control No. 1 siRNA was used as a negative control. Knockdown efficacy was confirmed after 48 h by Western blot analysis.
Coimmunoprecipitation.
For immunoprecipitation of HA-tagged proteins, hABIN-1-HA or empty plasmid control (Vector Co) was transiently overexpressed in HeLa cells for 24 h, incubated with BafA1 (200 nM) for 4 h, and immunoprecipitated with Pierce anti-HA magnetic beads. GFP-trap immunoprecipitation of GFP-tagged proteins was performed using the GFP-Trap_MA kit. About 800 µg of the protein lysate was used for immunoprecipitation and all experiments were performed as per the manufacturer’s instructions. The eluted samples were analyzed by sodium-dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting.
The Glutathione S-Transferase Pull-down Assay
LC3A, LC3B, and 4xUbiquitin (4xUb) were cloned into the pGEX-4T-1 vector, while ABIN-1 either as FL, LIR1-, LIR2-, or double LIR-mutated (LIR1 + 2) variants were cloned into the pMAL-c2X vector as per the manufacturer’s instructions. Briefly, glutathione s-transferase (GST)-tagged proteins or MBP-tagged ABIN-1 were expressed in BL21(DE3) competent cells and grown in LB broth base to an OD600 of 0.5–0.8. Protein expression was induced by 0.5 mM IPTG. Cells were incubated with washed Glutathion-Sepharose 4B beads or amylose resin for 2 h at 4°C on a rotator. Purified fusion proteins were washed and used for GST pull-down. MBP-tagged ABIN-1 was added to GST-fused protein beads and incubated for 2 h at 4°C on a rotator. Subsequently, beads were centrifuged, washed, and eluted. Purity of GST-fusion proteins were analyzed by Ponceau S staining, while binding of MBP-tagged ABIN-1 to GST-fusion proteins was detected by Western blot.
Mitochondrial Fractionation
Mitochondrial and cytosolic fractions were isolated from whole cell lysates obtained from 3.3 × 106 HeLa-mCherry-Parkin cells cultured in media containing DOX (0.5 µg/mL, 24 h) and stimulated with oligomycin/antimycin A (OA) or DMSO (10 µM, 4 h), using the mitochondria isolation kit for cultured cells according to the manufacturer’s instructions. All fractions were analyzed by Western blot.
SDS-PAGE and Western Blotting
For the analysis of ABIN-1 processing during autophagy, 0.8 × 106 HeLa-mCherry-Parkin cells were treated with rapamycin (100 nM) and/or BafA1 (4- and 8 h, 200 nM; 24 h, 10 nM) for the indicated time points. For the analysis of ABIN-1-dependent degradation of mitophagy substrates, 0.8 × 106 HeLa-mCherry-Parkin and CRISPR/Cas9-mediated ABIN-1 knockout (KO)-mCherry-Parkin cells cultured in media containing DOX (0.5 µg/mL, 24 h) were stimulated with OA (1 µM, 24 h) and the degradation of mitochondrial proteins was analyzed by Western blotting. Cell lysates were prepared as described previously (38). Briefly, cells or tissues were lysed in radioimmunoprecipitation assay (RIPA) buffer, centrifuged at 10,000 rpm for 10 min at 4°C, and the concentration of proteins in the cell lysates (supernatants) was determined using the Pierce BCA protein assay kit as per the manufacturer’s protocol. SDS-PAGE and Western blotting were as described previously (38). Anti-LC3B (1:8,000, NB100-2220) and anti-p62/SQSTM1 (1:500, NBP2-43663) were purchased from Novus Biologicals (Wiesbaden, Germany). Anti-ABIN-1 (1:1,000, 15104-1-AP) was purchased from Proteintech (Manchester, UK). Anti-GFP (1:500, sc-9996), anti-Parkin (1:500, Sc32282), anti-HA (1:500, Sc7392), and anti-translocase of outer membrane (TOM20) (1:500, sc17764) were purchased from Santa Cruz Biotechnology (Heidelberg, Germany). Anti-COXII (1:1,000, ab110258), anti-mitofusin-2 (MFN2, 1:1,000, ab56889), and anti-voltage-dependent anion-selective channel 1 (VDAC1, 1:1,000, ab14734) were purchased from Abcam. β-Actin (1:5,000, A5441) was purchased from SIGMA. The following secondary antibodies were used in the study: anti-rabbit-horseradish peroxidase (HRP) (1:5,000, ab97051; Abcam) or anti-mouse-HRP (1:5,000, sc-2005, Santa Cruz Biotechnology). Quantification with normalization to the housekeeping protein was performed using the Quantity one software (v.4.3.0; Bio-Rad Laboratories, München, Germany).
Mito-mKEIMA Assay
Mito (mt)-mKEIMA-Parkin-expressing HeLa cells transfected with siRNA control (siRNA Co) and siRNA-ABIN-1 for 48 h were treated with DMSO (10 μM), OA (10 μM) or OA (10 μM) together with BafA1 (200 nM) for 6 h. Fluorescent cells, 20,000 per sample were collected and analyzed by fluorescence-activated cell sorting (FACS) for dual-excitation at 405 (pH 8) and 561 (pH 4) nm with 610/20 nm emission filters using the BD LSRFortessa cell analyzer equipped with the BD FACSDiva software (BD Biosciences, Heidelberg, Germany). The percentage of lysosomal mt-mKEIMA cells represented by the ratio 561/405 nm was calculated.
Imaging by Laser Scanning Microscopy
HeLa cells (0.15 × 106) stably expressing mEGFP-ABIN-1 were transfected with mCherry-hLC3B-pcDNA3.1 using FuGENE HD transfection reagent as per the manufacturer’s instructions. The cells were left untransfected for colocalization studies of hABIN-1 with LAMP-1. The growth medium was supplemented with 0.5 μg/mL DOX and incubated for 24 h to induce mEGFP-hABIN-1 protein expression. The cells were then treated with BafA1 (300 nM), Torin-1 (1 μM), or DMSO (1 μM) for 4 h, rinsed, fixed with methanol for 10 min at −20°C, washed, blocked for 1 h in 5% FBS, rinsed, and stained with anti-LAMP-1 (1:400, ab24170, Abcam). mCherry-LC3B-transfected cells were incubated with goat anti-rabbit IgG Alexa-Fluor 594 (1:400, A-11012, Thermo Fisher Scientific). The stained cells were mounted using VECTASHIELD antifade mounting medium with DAPI. Images were acquired using the Leica SP8 laser scanning confocal microscope (Concord, ON, Canada) and analyzed using the Fiji- ImageJ 2.0.0 software.
Statistical Analysis
Differences between the two groups were evaluated with an unpaired, two-tailed, Student t test. For comparisons involving more than two groups, one-way analysis of variance (ANOVA) was used, followed by Bonferroni post hoc test using the software GraphPad Prism 6 (GraphPad Software, Inc., San Diego, CA). Data are expressed as means ± standard deviation (SD). Number (n) represents the number of replicates in the designed experiments. Differences were considered significant at *P < 0.05, **P < 0.01, and ***P < 0.001.
RESULTS
ABIN-1 Binds LC3B via Its LIR Motifs
Selective autophagy relies on UBAN-mediated recognition and binding of cargo by SARs and subsequent delivery to lipidated autophagosome-bound LC3 through LIRs (39). Accordingly, the role of ABIN-1 LIR domains in the autophagic process was analyzed. In line with the recently published findings (28), motif mapping of human ABIN-1 revealed two LIR motifs at amino acids 83 to 86 (FDPL) and 125 to 128 (FEVV). The canonical LIR sequence consists of the motif [W/F/Y]0-X1-X2-[L/V/I]3 conserved at positions X0 and X3, together with Phe (F) and Trp (W), frequently found at position X0 (Fig. 1A). To evaluate the binding of ABIN-1 to LC3B, HeLa cells transfected with HA-tagged hABIN-1 (hABIN-1-HA) or empty plasmid control (Vector Co), incubated with BafA1 for 4 h, was coimmunoprecipitated (Co-IP) with an anti-HA antibody. hABIN-1-HA was predominantly bound to the lipidated membrane-associated LC3B-II, rather than to the active cytosolic deacetylated LC3B-I (Fig. 1B). Binding of ABIN-1 with LC3B-II was confirmed by reciprocal Co-IP of hABIN-1-HA with EGFP-tagged LC3B (Fig. 1C). Thus, binding of ABIN-1 to LC3B-II supports its possible role as a SAR.
Figure 1.

A20 binding and inhibitor of nuclear factor kappa B (NF-κB)-1 (ABIN-1) binds 1A/1B light chain 3B (LC3B) via. LC3-interacting region (LIR) motifs. A: schematic representation of the domain architecture of ABIN-1. The alignment of LIR motifs with known autophagy receptors is illustrated underneath. LIR sequences are emphasized in bold. B: HeLa cells transiently transfected with hemagglutinin (HA)-tagged hABIN-1 (hABIN-1-HA) or empty plasmid control (Vector Co) for 24 h, incubated with Bafilomycin A1 (BafA1) (200 nM) for 4 h, immunoprecipitated (IP) with anti-HA-beads, and analyzed by immunoblotting. C: reciprocal IP of (B) using HeLa cells transfected with enhanced green fluorescent protein (EGFP)-LC3B and hABIN-1-HA and IP with anti-GFP-beads. D: HeLa cells transfected with Vector Co, plasmids encoding hABIN-1-HA full length (FL), or mutants defective in the LIR1-, LIR2-, LIR1 + 2-, or ubiquitin-binding domain in ABIN and NEMO (UBAN)-domain and IP with anti-HA antibody. β-Actin served as the loading control for the input. Immunoprecipitated ABIN-1-HA or EGFP-LC3B served as loading controls for the respective IPs. E: glutathione-S-transferase (GST)-pull-down assay of ABIN-1 or the indicated mutants. Purified GST, GST4x-Ubiquitin (Ub), GST-LC3A, or GST-LC3B purified from Escherichia coli were immobilized on GST beads and incubated with bacterial purified recombinant maltose binding protein (MBP)-tagged ABIN-1 protein. ABIN-1 fused to MBP was detected by Western blotting. Ponceau S staining was used to visualize GST-fusion proteins; n = 3 replicates. NBD, NEMO-binding domain.
The requirement of both LIR motifs for the binding of ABIN-1 to LC3B was evaluated by analyzing the binding affinities of LC3-I and LC3-II to ABIN-1 variants with point mutations in LIR1, LIR2, or both (LIR1 + 2), as well as UBAN (D472N) in transfected HeLa cells, in vitro. Mutated LIR2 significantly reduced the binding of ABIN-1 to LC3B-II compared with that of mutated LIR1, whereas mutation of both LIR domains (LIR1 + 2) completely inhibited the binding (Fig. 1D). The ubiquitin-binding defective variant of ABIN-1 (UBAN) was bound to LC3B-II with a similar affinity as that of the ABIN-1-HA FL protein (Fig. 1D). Direct interaction between ABIN-1 and LC3 proteins was confirmed by GST pull-down experiments with ABIN-1-FL and LIR-mutant proteins (Fig. 1E). GST alone or fused with either 4xUbiquitin (4xUb), LC3A, or LC3B were expressed in E. coli, immobilized on GST-agarose beads, and incubated with MBP-tagged ABIN-1. Consistent with the Co-IP results, single-point mutations in both LIR domains abrogated the interaction between ABIN-1 and LC3B (Fig. 1E). LIR2 domain of ABIN-1 was found to be more significantly involved in the binding to LC3B compared with LIR1. In contrast, both LIR domains mediated the interaction of ABIN-1 with LC3A, whereas the binding was inhibited in the double LIR1 + 2-mutant. All mutants retained their ability to bind linear ubiquitin chains fused to GST (GST-4xUb) (Fig. 1E). Thus, the LIR domains of ABIN-1 mediate its binding to LC3 proteins. Amino acids 137 to 140 of LIR2 domain had a stronger binding affinity to LC3B-II than that of LIR1 domain. Accordingly, we hypothesize that ABIN-1 acts as a SAR associated with LC3B-II on the membrane of autophagosomes.
ABIN-1 Colocalizes with LC3B and LAMP-1 and Translocates to Damaged Mitochondria
To confirm the involvement of ABIN-1 in autophagy and the autophagic flux, the colocalization of ABIN-1 with LC3B was analyzed in DOX-inducible mEGFP-hABIN-1 and mCherry-LC3B co-transfected HeLa cells by laser-scanning microscopy. Under basal conditions, mEGFP-hABIN-1 was evenly distributed in the cytosol without clustering into cytoplasmatic mCherry-LC3B containing structures (Fig. 2A). Induction of autophagy with the mammalian target of rapamycin (mTOR) inhibitor, Torin-1, and blockage of lysosomal fusion with BafA1, resulted in significant colocalization of mEGFP-hABIN-1 with mCherry-LC3B-positive vesicles (Fig. 2A). Similarly, mEGFP-hABIN-1 did not cluster with endogenous LAMP-1 containing structures under basal conditions (Fig. 2B), but Torin-1 induced its clustering with LAMP-1 containing lysosomes (Fig. 2B). Thus, autophagy induced the binding and localization of ABIN-1 to LC3B- and LAMP-1-positive cellular vesicles, supporting its potential role as a SAR.
Figure 2.

A20 binding and inhibitor of nuclear factor kappa B (NF-κB)-1 (ABIN-1) colocalizes with LC3B and lysosomal-associated membrane protein 1 (LAMP-1) and translocate to damaged mitochondria. A and B: representative confocal images of HeLa cells overexpressing monomeric enhanced green fluorescent protein (mEGFP)-hABIN-1 (green). A: cells cotransfected with mCherry-LC3B (red) for 24 h and treated with Torin-1 (1 μM) or dimethylsulfoxide (DMSO) (1 μM) and Bafilomycin A1 (BafA1, 300 nM) for 4 h. B: cells stimulated with Torin-1 and immunostained for endogenous LAMP-1 (red). Nuclei were stained with 4′, 6-diamidino-2-phenylindole (DAPI). Scale bar = 10 μm. C: mCherry-Parkin expressing HeLa cells treated with oligomycin/antimycin A (OA) (10 µM, 4 h) or DMSO (10 µM, 4 h) and Western blotted to analyze whole cell lysate, mitochondrial-, and cytosolic-fractions. Translocase of outer membrane (TOM)20 and cytochrome c oxidase subunit II (COXII) served as mitochondrial markers; n = 3 replicates. Co, empty plasmid control; DOX, doxycycline hyclate.
Next, we investigated whether ABIN-1 might have a functional role in the selective removal of damaged mitochondria by mitophagy. The localization of ABIN-1 under conditions of mitochondrial damage was analyzed in OA-treated mCherry-Parkin expressing HeLa cells. Following cellular fractionation, p62 and PARKIN were found to be localized both in the cytoplasmic and mitochondrial fractions, whereas activation of mitophagy further enhanced the overall expression of both proteins. In unstimulated cells, the expression of ABIN-1 was restricted to the cytosolic fraction and was found to be absent in the mitochondrial fraction. However, activation of mitophagy significantly enhanced the expression of ABIN-1 in the whole cell lysate and led to its enrichment in the mitochondrial fraction (Fig. 2C). Mitochondrial proteins TOM20 and COXII served as controls for effective fractionation and mitophagy activation. These findings demonstrating mitophagy-induced enhanced expression and translocation of the LC3 interactor ABIN-1 to the damaged mitochondria support the notion that ABIN-1 might act as a selective mitophagy receptor.
ABIN-1 Is Processed during Autophagy
Since SARs are commonly degraded via the autophagic flux together with their cargo, the effect of the autophagic process on the protein level of ABIN-1 was further analyzed. To this end, HeLa cells were treated with the mTOR-inhibitor rapamycin in the absence or presence of BafA1, to discriminate between increased autophagosome formation or impaired autophagosome-lysosome fusion, respectively, and analyzed by immunoblotting. The expression of both ABIN-1 and p62 were significantly enhanced 4 h post-rapamycin treatment (**P < 0.01 and **P < 0.01, respectively) and 4 h (ABIN-1, **P < 0.01; and p62, ***P < 0.001) and 24 h (ABIN-1, **P < 0.01) post-rapamycin/BafA1 treatment compared with that of the control untreated cells (Fig. 3, A–C), indicating that both proteins were stimulated during autophagy and degraded via the autophagic flux. Analysis of LC3B protein levels revealed enhanced conversion of LC3B-I to LC3B-II post-rapamycin treatment, and significant accumulation of LC3B-II post-rapamycin/BafA1 co-treatment at all time points (4 h, **P < 0.01; 8 h, ***P < 0.001; and 24 h, ***P < 0.001) compared with that of the controls (Fig. 3, A and D). Thus, similar to p62, ABIN-1 is recruited into autophagosomes for lysosomal degradation following induction of autophagy.
Figure 3.

A20 binding and inhibitor of nuclear factor kappa B (NF-κB)-1 (ABIN-1) is processed during autophagy. A: Western blot analysis of ABIN-1, p62, and 1A/1B light chain 3B (LC3B) expression in HeLa cells treated with rapamycin (100 nM) and/or Bafilomycin A1 (BafA1) (4 and 8 h, 200 nM; 24 h, 10 nM) for the indicated time points. Quantification of ABIN-1 (B), p62 (C), and LC3B-II (D). The protein levels were normalized to β-actin. Data represent means ± standard deviation (SD); n = 3 replicates; (*P < 0.05, **P < 0.01, and ***P < 0.001).
ABIN-1-Dependent Degradation of Mitophagy Substrates
The effect of ABIN-1 in mitophagy-induced degradation of mitochondrial substrates was analyzed in CRISPR/Cas9-mediated ABIN-1 KO-HeLa-mCherry-Parkin cells (ABIN-1 KO) by immunoblotting. The degradation of mitochondrial proteins MFN2 (Fig. 4, A and B, *P < 0.05), VDAC1 (Fig. 4, A and C, *P < 0.05), TOM20 (Fig. 4, A and D, *P < 0.05), and COXII (Fig. 4, A and E, *P < 0.05), were significantly inhibited following OA-induced mitophagy (24 h) in ABIN-1 KO- compared with that of HeLa-mCherry-Parkin transfected cells (wild type (WT) control cells) indicating partial inhibition of mitophagy in the absence of ABIN-1. Thus, ABIN-1 is involved in targeting mitochondrial proteins for autophagy-mediated degradation.
Figure 4.

A20 binding and inhibitor of nuclear factor kappa B (NF-κB)-1 (ABIN-1)-dependent degradation of mitophagy substrates. A: HeLa-mCherry-Parkin [wild type (WT) control] and CRISPR/Cas9-mediated ABIN-1 KO-HeLa-mCherry-Parkin cells (ABIN-1 KO) stimulated with oligomycin and antimycin A (OA, 1 µM, 24 h) and analyzed by Western blotting. Quantification of mitofusin-2 (MFN2) (B), voltage-dependent anion-selective channel 1 (VDAC1) (C), translocase of outer membrane (TOM)20 (D), and cytochrome c oxidase subunit II (COXII) (E). The protein levels were normalized to β-actin. Data represent means ± standard deviation (SD); n = 3 replicates; *P < 0.05.
Absence of ABIN-1 Partially Inhibits Mitophagy
The effect of ABIN-1 on mitophagy was further analyzed using the sensitive FACS-based mt-mKEIMA assay. Mt-mKEIMA, a fluorescence-based reporter, is specifically targeted to the mitochondria via a mitochondria-targeting signal peptide sequence. By analyzing the spectral shift from 405 (in neutral pH) to 561 nm (in acidic pH), the percentage of lysosome-engulfed mt-mKEIMA that represents ongoing mitophagy is quantified. Based on the resistance of the marker to degradation within lysosomes, it represents a robust and reliable method to quantify overall mitophagy in a cell population. Parkin-expressing mt-mKEIMA-cells were subjected to siRNA-mediated knockdown of ABIN-1 (Fig. 5C) and treated with OA and BafA1. Insignificant levels of mitophagy were observed under basal conditions, but OA treatment led to a significant increase in mitophagy, both in siRNA control (***P < 0.001) and siRNA-ABIN-1 (***P < 0.001) transfected cells. However, OA-induced mitophagy was partially inhibited in siRNA-ABIN-1-treated compared with siRNA control Parkin-expressing mt-mKEIMA-cells (8%, Fig. 5, A and B, *P < 0.05). As expected, BafA1 treatment efficiently blocked mitophagy in both cell types. Based on these findings, we propose that ABIN-1 represents a novel mitophagy receptor/adaptor recruited to damaged mitochondria mediating their autophagosomal degradation.
Figure 5.

Absence of A20 binding and inhibitor of nuclear factor kappa B (NF-κB)-1 (ABIN-1) partially inhibits mitophagy. A: representative data for mt-mKEIMA-Parkin-expressing HeLa cells transfected with siRNA control (siRNA Co) and siRNA-ABIN-1 for 48 h. Cells were treated with dimethylsulfoxide (DMSO) (10 μM), oligomycin and antimycin A (OA, 10 μM) or OA (10 μM) together with BafA1 (200 nM) for 6 h and analyzed by fluorescence-activated cell sorting for lysosomal positive mt-mKEIMA, representing cells undergoing mitophagy. B: quantification of (A). C: Western blot analysis of mt-mKEIMA-Parkin-expressing HeLa cells treated with siRNA Co or siRNA-ABIN-1 after 48 h. β-Actin served as the loading control. Data represent means ± standard deviation (SD); n = 5 replicates; *P < 0.05 and ***P < 0.001); n.s. = not significant.
DISCUSSION
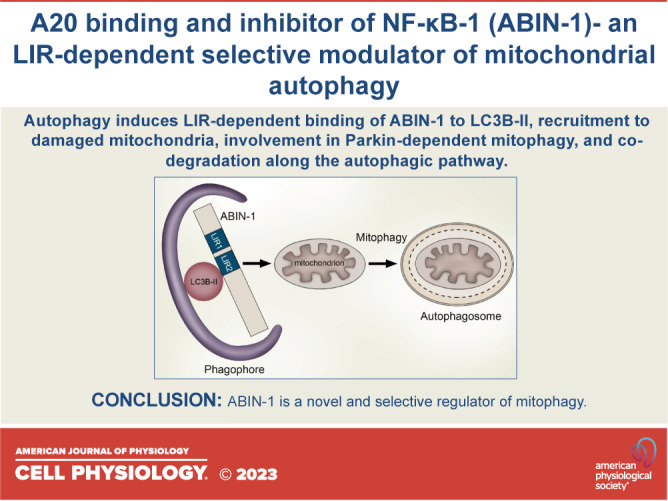
It is well established that ABIN-1 plays a crucial role in the maintenance of cellular homeostasis by regulating inflammation and cell death. In the present study, we built on the identified presence of two LIR domains in ABIN-1 and deciphered a completely novel function of this protein as a modulator and putative SAR involved in mitophagy. Stimulation of autophagy induced the direct LIR-dependent binding of ABIN-1 to LC3B-II, colocalization of ABIN-1 with autophagolysosomes, whereas mitochondrial damage triggered its translocation to mitochondria and mitophagy activation, as indicated by the degradation of the mitochondrial outer membrane proteins VDAC-1, MFN2, and TOM20 together with its own processing via the autophagic pathway (Fig. 6).
Figure 6.

A model of A20 binding and inhibitor of nuclear factor kappa B (NF-κB)-1 (ABIN-1)-mediated modulation of mitophagy. Autophagy induces the 1A/1B light chain 3 (LC3)-interacting region (LIR) 1 and 2 motifs-dependent binding of ABIN-1 to LC3B-II on the forming phagophore membrane and subsequent colocalization with LC3B-positive autophagosomes and lysosomal associated membrane protein 1 (LAMP-1)-containing autophagolysosomes. Furthermore, autophagy induces translocation of ABIN-1 to damaged mitochondria where it mediates degradation of mitochondrial outer membrane proteins mitofusin-2 (MFN2), translocase of outer membrane (TOM20), and voltage-dependent anion-selective channel 1 (VDAC1), together with its own processing via. the autophagic pathway, resulting in the selective degradation of damaged mitochondria by mitophagy.
During selective autophagy, the ATG8 proteins LC3 and GABA type A receptor-associated protein (GABARAP) recruit cargo to the growing phagophore membrane by interacting with SARs and adaptors that directly bind to the selected cargo (30). The LIR motifs of soluble and membrane-bound SARs facilitate interaction with lipidated ATG8 proteins present on the growing phagophore for internalization of the cargo (39). Several Atg8/LIR complexes have been reported, namely Atg8/Atg19 (40), LC3B/p62 (40), LC3C/NDP52 (41), and GABARAP/neighbor of BRCA1 gene 1 (NBR1) (42). TAX1BP1 and NDP52 use atypical LIR motif consisting of the tripeptide Leu-Val-Val to bind LC3C. Thus, a majority of SARs including OPTN execute autophagic degradation of their cargo in an LIR-/LC3B-dependent manner (43). The presence of LIR motifs in ABIN-1 suggests its ability to bind ATG8 family proteins. In the present study, the binding of ABIN-1 to LC3B was found to be LIR-dependent. Single-point mutations in the LIR motifs abrogated the complex formation between ABIN-1 and LC3B. By using purified bacterial proteins, we were able to show LIR-dependent direct binding of ABIN-1 to the ATG8 proteins LC3A and LC3B. As expected, the mutants were able to bind linear ubiquitin chains. Autophagy receptors need to bind lipidated ATG8 proteins present on the inner concave surface of the growing phagophore membrane for the internalization of the cargo (39). Although phosphorylation of Serine 83 is required for the binding of ABIN-1 to LC3A (28), the present study suggested the predominance of LIR2 over LIR1 in binding to LC3B. In addition, ABIN-1 showed preferential binding to the lipidated membrane-bound form of LC3B (LC3B-II) rather than the cytoplasmic LC3-I. Similar to NDP52, TAX1BP1, and NBR1 that harbor multiple LIR motifs (44), the presence of two LIR motifs in ABIN-1 might indicate the involvement of multivalent interaction with ATG8 proteins.
Selective autophagy is orchestrated by diverse SARs that mediate distinct types of selective autophagy (44). Mitophagy is coordinated by mitochondrially recruited SARs. NIX is a mitophagy receptor that binds GABARAP proteins via an N-terminal LIR (45). BNIP3 mediates LIR-dependent mitophagy and ER-phagy (46). SARs mediating mitophagy in an LC3/GABARAP-dependent manner include the outer mitochondrial membrane proteins: FUNDC1 (47), activating molecule in Beclin-1-regulated autophagy (AMBRA1) (48), FKBP8 (35), the inner mitochondrial membrane protein PHB2 (36), and the matrix proteins NIPSNAP1 and NIPSNAP2 (49). In the present study, ABIN-1 was found to be localized in the cytoplasm under basal conditions. However, upon induction of autophagy and mitochondrial stress, ABIN-1 was found to translocate to the damaged mitochondria, supporting its potential role as a selective mitophagy receptor. Furthermore, concurrent blockage of lysosomal fusion led to the clustering of ABIN-1 with LC3B positive autophagosomes and colocalization with LAMP-1-positive lysosomes, underscoring its resemblance to SARs in targeting specific cargo such as mitochondria to the autophagolysosomes.
The soluble SARs comprising the sequestosome-1-like receptors (SLRs) including p62, NBR1, NDP52, OPTN, and TAX1BP1 that act as selective mitophagy receptors are characterized by their dimerization or multimerization capabilities, presence of LIR-motifs, and UBDs, that are required for functioning as autophagic cargo receptors (50). ABIN-1 shares a similar structure and features, thereby supporting its function as a selective mitophagy receptor. The levels of SLRs are regulated by degradation through the autophagic pathway (30). Under inflammatory conditions, ABIN-1 is phosphorylated by tank-binding kinase 1 (TBK1) leading to activating of an LIR motif resulting in selective autophagy-dependent degradation (unpublished data deposited in bioRxiv) (51). In the present study, we observed alterations in ABIN-1 levels that were very consistent with the effects seen in p62: induction of autophagy increases the degradation of p62, whereas inhibition of autophagy or blockade of the autophagic flux leads to a corresponding increase in protein levels (52). Consistently, ABIN-1 protein was accumulated upon blockage of autophagy, indicating its regulation through autophagic flux in the present study. However, the levels of p62 and ABIN-1 did not decrease upon autophagy induction, probably due to parallel transcriptional upregulation. Autophagy-mediated increase in p62 levels by de novo synthesis was previously reported (53). Thus, it is highly expected that increased synthesis of ABIN-1 and p62 might have compensated the autophagic degradation of the respective proteins.
Selective removal of damaged mitochondria by mitophagy is executed by either ubiquitin-dependent or ubiquitin-independent mechanisms (32). In the ubiquitin-dependent pathway, PINK1 localized on the outer mitochondrial membrane phosphorylates Parkin and ubiquitin, thereby promoting the ubiquitination of specific outer mitochondrial membrane proteins for proteasomal degradation by mitophagy (54). Although Parkin and p62 were shown to be recruited to depolarized mitochondria in response to OA treatment in the present study, CRISPR/Cas9-mediated deletion of ABIN-1 significantly inhibited the degradation of mitochondrial outer membrane proteins, signifying a crucial role of ABIN-1 in mitophagy and supporting a previously undescribed function of ABIN-1 as a selective mitophagy receptor/adapter.
SARs rely on ubiquitin for binding to their respective cargo, while efficient binding of cargo to the forming phagophore membrane depends on multiple receptor-ATG8 interactions (39). Since all soluble SARs bind ubiquitin, they are likely to be recruited to the same cargo, thereby fulfilling either unique or redundant roles. Accordingly, p62 and NBR1 are dispensable for mitophagy in certain settings (55). In the same study, double knockout of NDP52/OPTN and triple knockout of NDP52/OPTN/TAX1BP1, but not deletion of either NDP52 or OPTN was shown to significantly inhibit mitophagy (55). ABIN-1 when ectopically targeted to mitochondria is able to induce mitophagy (unpublished data deposited in bioRxiv) (56). In the present study, the overall mitophagy level was partially but significantly reduced in the absence of ABIN-1 following induction of autophagy and mitochondrial damage by OA treatment. This could be due to the compensatory role of other SARs serving to counteract the lack of ABIN-1. Nevertheless, the identified role of ABIN-1 as a selective mitophagy receptor favors future studies to better understand the complex molecular cross talk involved in the regulation of inflammation and cell death by mitochondria (57). Based on our data shown in this study, we cannot faithfully discriminate the exact function of ABIN-1 either as a bona fide SAR or as an adaptor protein during selective autophagy. Future studies are warranted to elucidate the same.
Collectively, our data decipher LIR-dependent binding of ABIN-1 to ATG8 proteins, its recruitment to damaged mitochondria, and involvement in Parkin-dependent mitophagy, as well as its co-degradation as a mitophagy SAR/adapter along the autophagy pathway. The present study is the first to reveal a completely novel function of ABIN-1 in mitophagy that could pave a way for a better understanding of mitochondria-mediated regulation of inflammation and diverse cell death fates in different pathological settings including infectious diseases and various other types of tissue damage.
DATA AVAILABILITY
Data will be made available upon reasonable request.
GRANTS
This project is supported by grants from the German Research Council SFB 1177, 259130777, Project D08 (to C.M.), Project E01 (to I.D.), Project E02 (to L.S.), Project E09 (to D.K.); SFB 1039, Project B02 (to L.S.), the Cardio-Pulmonary Institute (CPI), EXC 2026, Project ID: 390649896 (to L.S. and M.W.), and the German Center for Lung Research 82 DZL 005A1 (to M.W.).
DISCLOSURES
Liliana Schaefer is an editor of the American Journal of Physiology-Cell Physiology and was not involved and did not have access to information regarding the peer-review process or final disposition of this article. An alternate editor oversaw the peer-review and decision-making process for this article. No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.M., H.R., I.D., C.M., and L.S. conceived and designed research; R.M., H.R., J.M., and J.L.-M. performed experiments; R.M. and H.R. analyzed data; R.M., H.R., and G.T. interpreted results of experiments; J.Z.-B. and L.S.H. prepared figures; R.M. and L.S. drafted manuscript; D.K., C.P., A.R., and M.W. edited and revised manuscript; R.M., H.R., J.Z.-B., C.P., G.T., J.M., J.L.-M., A.R., L.S.H., V.D., I.D., D.K., C.M., M.W., and L.S. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors sincerely thank Dr. Richard J. Youle for kindly providing mCherry-Parkin donor vector, Dr. Eric Kowarz for kindly providing pSBtet-Pur and pSBtet-Neo plasmids, Dr. Michael Davidson for kindly providing mEGFP-C1 vector, Dr. Karla Kirkegaard for kindly providing EGFP-LC3 vector, Dr. Zsuzsanna Izsvak for kindly providing pCMV(CAT)T7-SB100, and Dr. David Rubinsztein for kindly providing mCherry-hLC3B-pcDNA3.1.
REFERENCES
- 1. Fukushi M, Dixon J, Kimura T, Tsurutani N, Dixon MJ, Yamamoto N. Identification and cloning of a novel cellular protein Naf1, Nef-associated factor 1, that increases cell surface CD4 expression. FEBS Lett 442: 83–88, 1999. doi: 10.1016/S0014-5793(98)01631-7. [DOI] [PubMed] [Google Scholar]
- 2. G'Sell RT, Gaffney PM, Powell DW. A20-binding inhibitor of NF-κB activation 1 is a physiologic inhibitor of NF-κB: a molecular switch for inflammation and autoimmunity. Arthritis Rheumatol 67: 2292–2302, 2015. doi: 10.1002/art.39245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Heyninck K, Kreike MM, Beyaert R. Structure-function analysis of the A20-binding inhibitor of NF-kappa B activation, ABIN-1. FEBS Lett 536: 135–140, 2003. doi: 10.1016/s0014-5793(03)00041-3. [DOI] [PubMed] [Google Scholar]
- 4. Mauro C, Pacifico F, Lavorgna A, Mellone S, Iannetti A, Acquaviva R, Formisano S, Vito P, Leonardi A. ABIN-1 binds to NEMO/IKKgamma and co-operates with A20 in inhibiting NF-kappaB. J Biol Chem 281: 18482–18488, 2006. doi: 10.1074/jbc.M601502200. [DOI] [PubMed] [Google Scholar]
- 5. Hong JY, Lin SC, Kuo BJ, Lo YC. Structural and biochemical basis for higher-order assembly between A20-binding inhibitor of NF-κB 1 (ABIN1) and M1-linked ubiquitins. J Mol Biol 433: 167116, 2021. doi: 10.1016/j.jmb.2021.167116. [DOI] [PubMed] [Google Scholar]
- 6. Nanda SK, Venigalla RK, Ordureau A, Patterson-Kane JC, Powell DW, Toth R, Arthur JS, Cohen P. Polyubiquitin binding to ABIN1 is required to prevent autoimmunity. J Exp Med 208: 1215–1228, 2011. doi: 10.1084/jem.20102177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wagner S, Carpentier I, Rogov V, Kreike M, Ikeda F, Löhr F, Wu CJ, Ashwell JD, Dötsch V, Dikic I, Beyaert R. Ubiquitin binding mediates the NF-kappaB inhibitory potential of ABIN proteins. Oncogene 27: 3739–3745, 2008. doi: 10.1038/sj.onc.1211042. [DOI] [PubMed] [Google Scholar]
- 8. Oshima S, Turer EE, Callahan JA, Chai S, Advincula R, Barrera J, Shifrin N, Lee B, Benedict Yen TS, Woo T, Malynn BA, Ma A. ABIN-1 is a ubiquitin sensor that restricts cell death and sustains embryonic development. Nature 457: 906–909, 2009. [Erratum in Nature 458: 538, 2009]. doi: 10.1038/nature07575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Luyer MD, Derikx JP, Beyaert R, Hadfoune M, van Kuppevelt TH, Dejong CH, Heineman E, Buurman WA, Greve JW. High-fat nutrition reduces hepatic damage following exposure to bacterial DNA and hemorrhagic shock. J Hepatol 50: 342–350, 2009. doi: 10.1016/j.jhep.2008.08.025. [DOI] [PubMed] [Google Scholar]
- 10. Martín-Vicente M, González-Sanz R, Cuesta I, Monzón S, Resino S, Martinez I. Downregulation of A20 expression increases the immune response and apoptosis and reduces virus production in cells infected by the human respiratory syncytial virus. Vaccines (Basel) 8: 100, 2020. doi: 10.3390/vaccines8010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bao J, Ye B, Ren Y. ABIN1 inhibits inflammation through necroptosis-dependent pathway in ulcerative colitis. Genet Res (Camb) 2022: 9313559, 2022. doi: 10.1155/2022/9313559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cai J, Hu D, Sakya J, Sun T, Wang D, Wang L, Mao X, Su Z. ABIN-1 is a key regulator in RIPK1-dependent apoptosis (RDA) and necroptosis, and ABIN-1 deficiency potentiates necroptosis-based cancer therapy in colorectal cancer. Cell Death Dis 12: 140, 2021. doi: 10.1038/s41419-021-03427-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dziedzic SA, Su Z, Jean Barrett V, Najafov A, Mookhtiar AK, Amin P, Pan H, Sun L, Zhu H, Ma A, Abbott DW, Yuan J. ABIN-1 regulates RIPK1 activation by linking Met1 ubiquitylation with Lys63 deubiquitylation in TNF-RSC. Nat Cell Biol 20: 58–68, 2018. doi: 10.1038/s41556-017-0003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li M, Liu Y, Xu C, Zhao Q, Liu J, Xing M, Li X, Zhang H, Wu X, Wang L, Ou Y, Wu X, Zhao X, Liu H, Qiu L, Li F, Li J, Rong W, Luo Y, Deng J, Wang X, Wang Z, Zhao Y, Lv A, Li Q, Zhang H. Ubiquitin-binding domain in ABIN1 is critical for regulating cell death and inflammation during development. Cell Death Differ 29: 2034–2045, 2022. doi: 10.1038/s41418-022-00994-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pu T, Liu W, Wu Y, Zhao Y. ABIN1 alleviates inflammatory responses and colitis via facilitating A20 activity. Ther Adv Chronic Dis 11: 2040622320944782, 2020. doi: 10.1177/2040622320944782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang DM, Cheng LQ, Zhai ZF, Feng L, Zhong BY, You Y, Zhang N, Song ZQ, Yang XC, Chen FR, Hao F. Single-nucleotide polymorphism and haplotypes of TNIP1 associated with systemic lupus erythematosus in a Chinese Han population. J Rheumatol 40: 1535–1544, 2013. doi: 10.3899/jrheum.121391. [DOI] [PubMed] [Google Scholar]
- 17. Li H, Sun A, Meng T, Zhu Y. Expression and role of ABIN1 in sepsis: in vitro and in vivo studies. Open Med (Wars) 16: 33–40, 2021. [Retraction in Open Med (Wars) 17: 1064, 2022]. doi: 10.1515/med-2021-0008. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18. Korte EA, Caster DJ, Barati MT, Tan M, Zheng S, Berthier CC, Brosius FC 3rd, Vieyra MB, Sheehan RM, Kosiewicz M, Wysoczynski M, Gaffney PM, Salant DJ, McLeish KR, Powell DW. ABIN1 determines severity of glomerulonephritis via activation of intrinsic glomerular inflammation. Am J Pathol 187: 2799–2810, 2017. doi: 10.1016/j.ajpath.2017.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harirchian P, Lee J, Hilz S, Sedgewick AJ, Perez White BE, Kesling MJ, Mully T, Golovato J, Gray M, Mauro TM, Purdom E, Kim EA, Sbitany H, Bhutani T, Vaske CJ, Benz SC, Cho RJ, Cheng JB. A20 and ABIN1 suppression of a keratinocyte inflammatory program with a shared single-cell expression signature in diverse human rashes. J Invest Dermatol 139: 1264–1273, 2019. doi: 10.1016/j.jid.2018.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kattah MG, Shao L, Rosli YY, Shimizu H, Whang MI, Advincula R, Achacoso P, Shah S, Duong BH, Onizawa M, Tanbun P, Malynn BA, Ma A. A20 and ABIN-1 synergistically preserve intestinal epithelial cell survival. J Exp Med 215: 1839–1852, 2018. doi: 10.1084/jem.20180198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Peng K, Li Y, Lu C, Hu S. ABIN-1 protects chondrocytes from lipopolysaccharide-induced inflammatory injury through the inactivation of NF-κB signalling. Clin Exp Pharmacol Physiol 47: 1212–1220, 2020. doi: 10.1111/1440-1681.13291. [DOI] [PubMed] [Google Scholar]
- 22. El Bakkouri K, Wullaert A, Haegman M, Heyninck K, Beyaert R. Adenoviral gene transfer of the NF-kappa B inhibitory protein ABIN-1 decreases allergic airway inflammation in a murine asthma model. J Biol Chem 280: 17938–17944, 2005. doi: 10.1074/jbc.M413588200. [DOI] [PubMed] [Google Scholar]
- 23. Wullaert A, Wielockx B, Van Huffel S, Bogaert V, De Geest B, Papeleu P, Schotte P, El Bakkouri K, Heyninck K, Libert C, Beyaert R. Adenoviral gene transfer of ABIN-1 protects mice from TNF/galactosamine-induced acute liver failure and lethality. Hepatology 42: 381–389, 2005. doi: 10.1002/hep.20785. [DOI] [PubMed] [Google Scholar]
- 24. Ma Y, Yuan S, Tian X, Lin S, Wei S, Hu T, Chen S, Li X, Chen S, Wu D, Wang M, Guo D. ABIN1 inhibits HDAC1 ubiquitination and protects it from both proteasome- and lysozyme-dependent degradation. J Cell Biochem 119: 3030–3043, 2018. doi: 10.1002/jcb.26428. [DOI] [PubMed] [Google Scholar]
- 25. Gao L, Coope H, Grant S, Ma A, Ley SC, Harhaj EW. ABIN1 protein cooperates with TAX1BP1 and A20 proteins to inhibit antiviral signaling. J Biol Chem 286: 36592–36602, 2011. doi: 10.1074/jbc.M111.283762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang Y, Zhou P, Lu F, Su R, Gong Z. A20-binding inhibitor of nuclear factor-κB targets β-arrestin2 to attenuate opioid tolerance. Mol Pharmacol 100: 170–180, 2021. doi: 10.1124/molpharm.120.000211. [DOI] [PubMed] [Google Scholar]
- 27. Mirza N, Sowa AS, Lautz K, Kufer TA. NLRP10 affects the stability of Abin-1 to control inflammatory responses. J Immunol 202: 218–227, 2019. doi: 10.4049/jimmunol.1800334. [DOI] [PubMed] [Google Scholar]
- 28. Shinkawa Y, Imami K, Fuseya Y, Sasaki K, Ohmura K, Ishihama Y, Morinobu A, Iwai K. ABIN1 is a signal-induced autophagy receptor that attenuates NF-κB activation by recognizing linear ubiquitin chains. FEBS Lett 596: 1147–1164, 2022. doi: 10.1002/1873-3468.14323. [DOI] [PubMed] [Google Scholar]
- 29. Khaminets A, Behl C, Dikic I. Ubiquitin-dependent and independent signals in selective autophagy. Trends Cell Biol 26: 6–16, 2016. doi: 10.1016/j.tcb.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 30. Gubas A, Dikic I. A guide to the regulation of selective autophagy receptors. FEBS J 289: 75–89, 2022. doi: 10.1111/febs.15824. [DOI] [PubMed] [Google Scholar]
- 31. Osellame LD, Blacker TS, Duchen MR. Cellular and molecular mechanisms of mitochondrial function. Best Pract Res Clin Endocrinol Metab 26: 711–723, 2012. doi: 10.1016/j.beem.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ma K, Chen G, Li W, Kepp O, Zhu Y, Chen Q. Mitophagy, mitochondrial homeostasis, and cell fate. Front Cell Dev Biol 8: 467, 2020. doi: 10.3389/fcell.2020.00467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wei H, Liu L, Chen Q. Selective removal of mitochondria via mitophagy: distinct pathways for different mitochondrial stresses. Biochim Biophys Acta 1853: 2784–2790, 2015. doi: 10.1016/j.bbamcr.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 34. Xu X, Zhang Y, Cheng H, Zhou R. SPATA33 functions as a mitophagy receptor in mammalian germline. Autophagy 17: 1284–1286, 2021. doi: 10.1080/15548627.2021.1909836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bhujabal Z, Birgisdottir AB, Sjottem E, Brenne HB, Overvatn A, Habisov S, Kirkin V, Lamark T, Johansen T. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep 18: 947–961, 2017. doi: 10.15252/embr.201643147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wei Y, Chiang WC, Sumpter R Jr, Mishra P, Levine B. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell 168: 224–238.e10, 2017. doi: 10.1016/j.cell.2016.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kowarz E, Löscher D, Marschalek R. Optimized sleeping beauty transposons rapidly generate stable transgenic cell lines. Biotechnol J 10: 647–653, 2015. doi: 10.1002/biot.201400821. [DOI] [PubMed] [Google Scholar]
- 38. Moreth K, Brodbeck R, Babelova A, Gretz N, Spieker T, Zeng-Brouwers J, Pfeilschifter J, Young MF, Schaefer RM, Schaefer L. The proteoglycan biglycan regulates expression of the B cell chemoattractant CXCL13 and aggravates murine lupus nephritis. J Clin Invest 120: 4251–4272, 2010. doi: 10.1172/JCI42213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Johansen T, Lamark T. Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J Mol Biol 432: 80–103, 2020. doi: 10.1016/j.jmb.2019.07.016. [DOI] [PubMed] [Google Scholar]
- 40. Noda NN, Kumeta H, Nakatogawa H, Satoo K, Adachi W, Ishii J, Fujioka Y, Ohsumi Y, Inagaki F. Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells 13: 1211–1218, 2008. doi: 10.1111/j.1365-2443.2008.01238.x. [DOI] [PubMed] [Google Scholar]
- 41. von Muhlinen N, Akutsu M, Ravenhill BJ, Foeglein A, Bloor S, Rutherford TJ, Freund SM, Komander D, Randow F. LC3C, bound selectively by a noncanonical LIR motif in NDP52, is required for antibacterial autophagy. Mol Cell 48: 329–342, 2012. doi: 10.1016/j.molcel.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rozenknop A, Rogov VV, Rogova NY, Löhr F, Güntert P, Dikic I, Dötsch V. Characterization of the interaction of GABARAPL-1 with the LIR motif of NBR1. J Mol Biol 410: 477–487, 2011. doi: 10.1016/j.jmb.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 43. Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, Dotsch V, Bumann D, Dikic I. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333: 228–233, 2011. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kirkin V, Rogov VV. A diversity of selective autophagy receptors determines the specificity of the autophagy pathway. Mol Cell 76: 268–285, 2019. doi: 10.1016/j.molcel.2019.09.005. [DOI] [PubMed] [Google Scholar]
- 45. Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Löhr F, Popovic D, Occhipinti A, Reichert AS, Terzic J, Dötsch V, Ney PA, Dikic I. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 11: 45–51, 2010. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem 287: 19094–19104, 2012. doi: 10.1074/jbc.M111.322933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, Huang L, Xue P, Li B, Wang X, Jin H, Wang J, Yang F, Liu P, Zhu Y, Sui S, Chen Q. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 14: 177–185, 2012. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 48. Strappazzon F, Nazio F, Corrado M, Cianfanelli V, Romagnoli A, Fimia GM, Campello S, Nardacci R, Piacentini M, Campanella M, Cecconi F. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ 22: 419–432, 2015. [Erratum in Cell Death Differ 22: 517, 2015]. doi: 10.1038/cdd.2014.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Princely Abudu Y, Pankiv S, Mathai BJ, Håkon Lystad A, Bindesbøll C, Brenne HB, Yoke Wui Ng M, Thiede B, Yamamoto A, Mutugi Nthiga T, Lamark T, Esguerra CV, Johansen T, Simonsen A. NIPSNAP1 and NIPSNAP2 act as “eat me” signals for mitophagy. Dev Cell 49: 509–525.e12, 2019. doi: 10.1016/j.devcel.2019.03.013. [DOI] [PubMed] [Google Scholar]
- 50. Onishi M, Yamano K, Sato M, Matsuda N, Okamoto K. Molecular mechanisms and physiological functions of mitophagy. EMBO J 40: e104705, 2021. doi: 10.15252/embj.2020104705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhou J, Rasmussen NL, Olsvik HL, Akimov V, Hu Z, Evjen G, Blagoev B, Lamark T, Johansen T, Dengjel J. TBK1 phosphorylation activates LIR-dependent degradation of the inflammation repressor TNIP1 (Preprint). bioRxiv, 2022. doi: 10.1101/2022.03.02.482646. [DOI] [PMC free article] [PubMed]
- 52. Bjørkøy G, Lamark T, Pankiv S, Øvervatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 452: 181–197, 2009. doi: 10.1016/S0076-6879(08)03612-4. [DOI] [PubMed] [Google Scholar]
- 53. Sahani MH, Itakura E, Mizushima N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy 10: 431–441, 2014. doi: 10.4161/auto.27344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yoo SM, Jung YK. A molecular approach to mitophagy and mitochondrial dynamics. Mol Cells 41: 18–26, 2018. doi: 10.14348/molcells.2018.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524: 309–314, 2015. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Le Guerroué F, Werner A, Wang C, Youle R. TNIP1 inhibits mitophagy via interaction with FIP200 and TAX1BP1 (Preprint). bioRxiv, 2022. doi: 10.1101/2022.03.14.484269. [DOI]
- 57. Marchi S, Guilbaud E, Tait SWG, Yamazaki T, Galluzzi L. Mitochondrial control of inflammation. Nat Rev Immunol 25: 1–14, 2022. doi: 10.1038/s41577-022-00760-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available upon reasonable request.