

Abstract
Patients and families benefit when the genetic etiology of cardiomyopathy is elucidated through a multidisciplinary approach including genetic counseling and judicious use of genetic testing. The yield of genetic testing is optimized when performed on a proband with a clear phenotype, and interrogates genes that are validated in association with that specific form of cardiomyopathy. Variants of uncertain significance are frequently uncovered and should not be overinterpreted. Identifying an impactful genetic variant as the cause of a patient’s cardiomyopathy can have important prognostic impact, and enable streamlined cascade testing to highlight at risk relatives. Certain genotypes are associated with unique potential cardiac and noncardiac risk factors and may dictate personalized approaches to treatment.
Keywords: cardiomyopathy, genetic testing, genetic counseling, genetics
Cardiomyopathies are typically classified by the primary myocardial derangement: left ventricular hypertrophy (hypertrophic cardiomyopathy [HCM]); left and/or right ventricular chamber dilatation (dilated cardiomyopathy [DCM]), including cases with prominent arrhythmia (arrhythmogenic cardiomyopathy [ACM]); or normal ventricular geometry with focal or global hypokinesia. Within these coarse phenotypic categorizations lie a range of nuanced endotypes, some classified as “genetic cardiomyopathies” warranting personalized, genotype-based management. In recent years, classification systems that distinguish between the different cardiomyopathic etiologies, including the MOGE(S) (morphofunctional phenotype [M], organ involvement [O], genetic inheritance pattern [G], etiological annotation [E], and optional information about heart failure functional status [S]) criteria,1 have been proposed, highlighting a trend toward diagnostic granularity. This review presents evidence in favor of incorporating genetic evaluations into the care of patients with cardiomyopathy, offering field guidance for best practices.
GENETIC SCREENING: OVERALL APPROACH
Genetic evaluation for cardiomyopathy ideally involves: 1) collecting and documenting a detailed family history; 2) 1-on-1 patient counseling on the genetic investigation; and 3) molecular genetic testing when appropriate (Central Illustration). Due to wide population variability in gene sequences, the probabilistic nature of genetic results, and the challenges of interpreting the impact of a particular gene variant, the yield of testing is highest when performed on individuals with a definitive phenotype. Options for genetic testing range from sequencing of a single gene, a targeted set of genes related to the phenotype in question, or in some cases, exome sequencing; genome sequencing is not routinely used in clinical practice. Testing is typically performed on a blood, saliva, or buccal swab sample using next-generation sequencing, in which DNA is extracted, purified, amplified, and fragmented, then isolated and attached to labeled beads for short-read sequencing. Sequence data are aligned against a “reference” human genome sequence, and variants present in the patient sample are further analyzed to determine likelihood of pathogenicity. This variant interpretation process differs slightly by each genetics laboratory but weighs the variant type (ie, loss-of-function variants such as frameshift mutations or variants affecting the canonical splice site are considered putatively damaging), allele frequency in the general population as well as in ethnic subgroups (using large publicly available databases of healthy control exomes or genomes such as gnomAD [Genome Aggregation Database]), the degree of evolutionary conservation of the amino acid affected by the variant, and the properties of that particular amino acid (eg, charge, hydrophobicity), informing whether a change will be tolerated by the protein or whether it resides in a known “hot spot” for damaging variants in a given gene. Because of the complexity of this process, the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology have established a set of guidelines in an attempt to standardize variant interpretation.2 Once a causative variant in a proband is identified, other relatives, including those without any known phenotype, can be screened for the presence or absence of that variant at the single site within the gene, a process known as cascade genetic testing.
CENTRAL ILLUSTRATION.
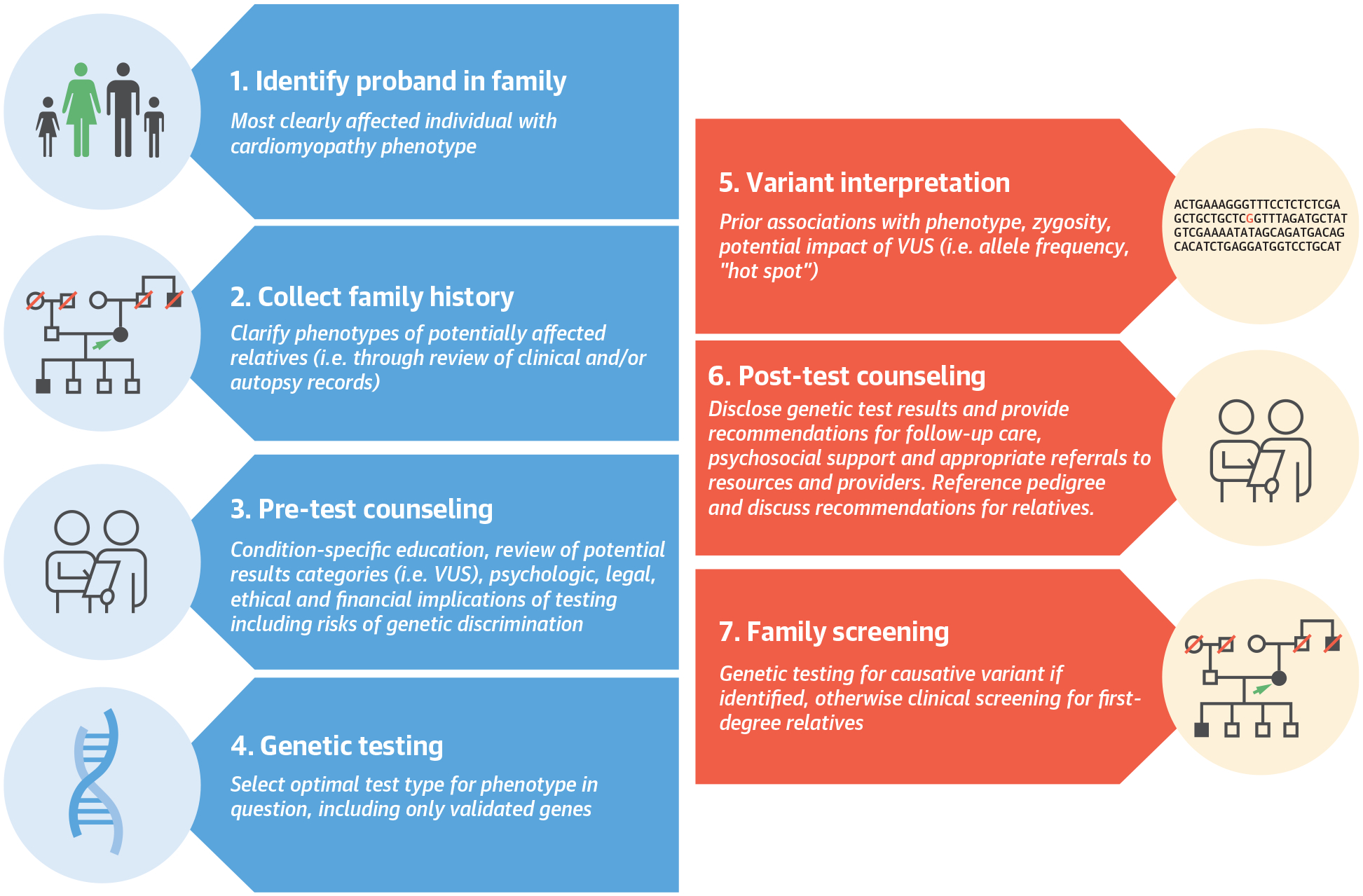
Optimal Overall Strategy for Genetic Evaluation in Patients With Cardiomyopathy
Identification of the optimal proband, collection of family history, and pretest counseling are recommended prior to initiation of genetic testing. Genetic sequencing results are analyzed for presence of important variants, interpreted in the context of the proband’s phenotype. Patients and relatives are counseled as to clinical recommendations based on each variant. VUS = variant of uncertain significance.
CHALLENGE 1: GENE-DISEASE ASSOCIATIONS
Large multigene (30–100+ genes) testing panels are most employed for genetic assessment in cardiomyopathies. Evolving understanding of gene-disease pairs has prompted formation of expert panels through the National Human Genome Research Institute–funded ClinGen Resource, which are systematically assessing the strength of evidence for associations between genes and phenotypes including ACM,3 DCM,4 and HCM;5 through such reviews, genes that are commonly included in diagnostic testing panels have been found to have limited or no evidence for disease association. These efforts will inform refined testing panels that improve the clinical utility of genetic testing for cardiomyopathies.
CHALLENGE 2: VARIANT CLASSIFICATION
Interpretation of genetic testing results is challenging in part due to the high degree of gene variation in the general population. In fact, in 1 exome series of 1,000 unrelated individuals in the United Kingdom, each individual had, on average >20,000 gene variants, including 160 unique rare (population allele frequency ≤0.1%) variants.6 Thus, the ACMG has recommended variant modifiers signifying likelihood of impact for disease (Figure 1). Variants of uncertain significance (VUSs) are those with insufficient data available to adjudicate more definitively. The ACMG variant classification matrix is designed to reduce the number of false positive “true mutation” calls and therefore inevitably skews toward VUS calls. The contemporary practice of using larger gene panels and exome sequencing raises the likelihood of VUS results. Disclosure of VUSs to patients is an actively debated topic, with some experts arguing that they should be ignored during clinical decision making on the premise that they are “innocent until proven guilty,” allocate patients to “genetic purgatory,”7 and increase the likelihood of misunderstanding ones results. VUSs are typically not useful for cascade genetic testing in relatives due to their uncertainty; overinterpretation can potentially cause harm, either by wrongly dismissing individuals from clinical surveillance or by causing unwarranted anxiety about disease risk, economic burden, or overestimation of benefit for therapies and inappropriate device implantation. Patient attitudes toward receiving uncertain genetic results may be negative (including regret, distress/anxiety, and poor comprehension/recall of their results) or optimistic (beneficence from furthering scientific understanding, enthusiasm for receiving a potential explanation for their condition).8 Importantly, VUSs may be reclassified over time; the vast majority will be downgraded to likely benign or benign status.9 HCM variants previously reported as pathogenic, especially in individuals of African ancestry, were later found to be benign when reanalyzed using population data from multiple ancestry groups.10 Still, in 1 study, approximately 8% of patients with HCM received a VUS result that upon re-examination was likely pathogenic,11 and data from SHaRe (Sarcomeric Human Cardiomyopathy Registry) revealed that even VUSs can have an impact on outcome, indicating that some VUSs are actually misclassified pathogenic variants.12 Broader population data, especially including diverse ethnic groups, increased data sharing across testing laboratories, and improved tools for functional classification of variants will lead to better diagnostic certainty, and fewer VUSs, in genetic testing.
FIGURE 1.
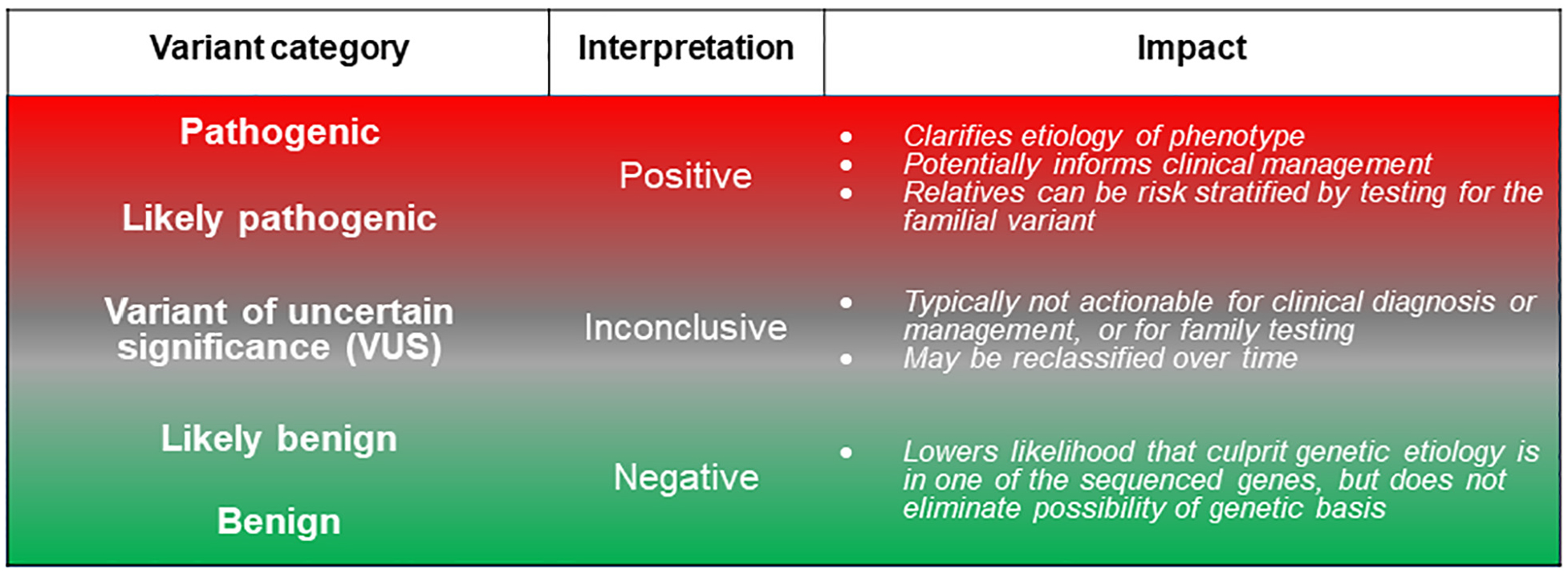
Spectrum of Genetic Testing Results
Genetic variants are graded according to likelihood of causing disease, here denoted with red shading for high-impact, disease-causing variants (actionable) and green shading for variants that are unlikely to cause disease. Gray shading highlights variants of uncertain significance (VUSs), for which insufficient data are available to definitively adjudicate as either pathogenic or benign.
CHALLENGE 3: INCLUDING GENETICS SPECIALISTS ON THE CARE TEAM
Genetic counselors are specially trained and certified to assist patients and their families in understanding the genetic causes of disease and the risks and benefits of genetic testing and should ideally be involved at every step of the genetic work-up. One key contribution is collecting a detailed family history, documented through a comprehensive 3-generation pedigree, a visual “road map” for capturing inheritance patterns and communicating testing recommendations to families. Formalizing the family history in this way affords the opportunity for patients to gather additional information from relatives, sometimes including hospital or autopsy records. When genetic testing is appropriate, genetic counselors are instrumental in selecting the optimal test (depending on the patient’s presentation, available diagnostic results, and insurance) and performing pre- and post-test counseling (Central Illustration). Genetic counselors report a higher level of confidence in counseling patients about VUS results compared with cardiologists and are more likely to recommend ongoing follow-up to ensure that patients receive information about variant reclassification.13
Several genetic testing laboratories currently offer no-charge testing for cardiomyopathy through programs with industry partners. One consequence of the apparent ease of testing through these partnership programs is that nongenetics specialists may feel emboldened to independently order the genetic test. Bypassing genetic counselors may jeopardize patients’ comprehension and decision making. The ideal care team integrates genetics specialist who can provide coordinated care for the entire family, including children and adults.
CHALLENGE 4: PREDICTIVE VALUE OF A “POSITIVE” GENETIC TEST RESULT
Genetic cardiomyopathies are associated with reduced penetrance (genotype-positive relatives do not necessarily manifest disease) and variable expressivity (differing degrees of severity in relatives at different ages). Pre- and post-test counseling for individuals undergoing familial variant genetic testing should review the potential impact of a positive result, including its limited predictive value for clinical manifestations. Relatives who are confirmed to carry a familial variant warrant baseline and ongoing clinical surveillance (including electrocardiogram, echocardiogram, long-term rhythm moni-tory, laboratory testing, and/or cardiac magnetic resonance as indicated by genotype or phenotype)14 but may or may not ever show signs of disease. This strategy enables prompt initiation of therapy in genotype-positive individuals once a phenotype emerges.
CHALLENGE 5: CLINICAL DECISION MAKING IN GENETICALLY AT-RISK INDIVIDUALS
Based on efficacy in overt heart failure, neurohormonal blockade is often initiated at the earliest signs of cardiac remodeling or reduction in systolic function. In HCM, one promising approach may involve angiotensin receptor blockers, which prevent ventricular hypertrophy and fibrosis in mice if administered early, through inhibition of transforming growth factor beta. In a randomized, double-blind, placebo-controlled trial, valsartan improved cardiac structure and function in young, genotype-positive, early-stage (mild hypertrophy) individuals with primarily Class I symptoms.15 An earlier and smaller trial tested the utility of the calcium-channel blocker diltiazem in phenotype-negative sarcomere variant carriers without hypertrophy and found benefit for stabilizing ventricular geometry, wall thickness, and biomarkers.16 Additional and larger studies are needed to determine whether these or other strategies will be clinically impactful in broader populations of genotype-positive at-risk individuals.
CHALLENGE 6: EMERGENCE OF COMPLEX GENETICS
The traditional paradigm of classic Mendelian rare variants causing monogenic cardiomyopathies may be incomplete. Increasingly, a subset of cardiomyopathy with digenic and oligogenic etiologies is recognized,17 suggesting that multiple high-impact variants can have cumulative effects on disease penetrance and associate with earlier onset. Thus, the practice of dismissing from clinical surveillance those relatives who test negative for a single familial pathogenic variant is questioned. Some experts now suggest that relatives should continue intermittent surveillance regardless of genotype status, due to the possibility that additional undetected causative variants may pose risk in families. Further research is needed to better understand the spectrum of genetic architecture in cardiomyopathies, including the role of modifier genes, polygenic loci, and interaction with environmental factors.18
GENETIC EVALUATION IN PRACTICE: DCM AND ACM
An overview of practical recommendations for genetic evaluation in cardiomyopathies is depicted in Figure 2. After ruling out ischemic, autoimmune, endocrine, medication, toxin, or tachycardia-induced factors, idiopathic DCM can be classified as familial or nonfamilial (sporadic). Genetic etiologies can be confirmed in 50% and 20% to 40% of these groups, respectively, and thus should be investigated. To date, more than 50 genes have been causally linked to DCM, with 23 genes responsible for >97% of all genetic DCM.19 These DCM genes govern structure or function of a range of cardiomyocyte elements, including the sarcomere/Z-band, desmosome, cytoskeleton, nuclear lamina, mitochondria, and ion-channel proteins. DCM is characterized by genetic and allelic heterogeneity, with many variants found to be novel or private in families. Among patients with DCM, genotype-positive status is associated with increased heart failure and arrhythmia outcomes and worse left ventricular reverse remodeling compared with genotype-negative patients.20
FIGURE 2.
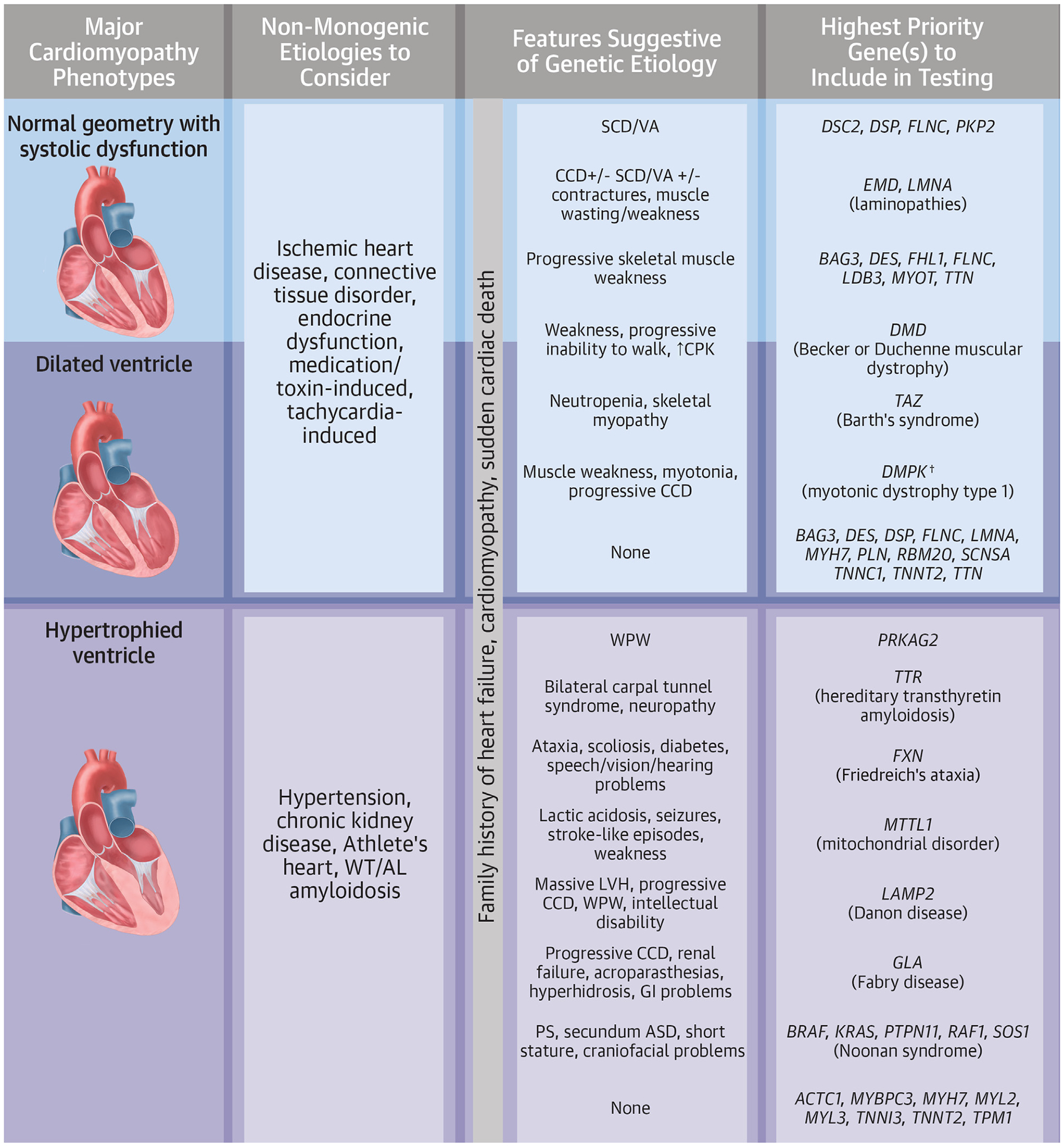
Practical Recommendations for Clinicians
Cardiomyopathy phenotypes can be roughly categorized according to ventricular morphology and function. After ruling out systemic and environmental/lifestyle causes of cardiomyopathy, clinicians should consider genetic etiologies. A family history of heart failure, cardiomyopathy, or sudden death in a first-degree relative suggests a possible monogenic familial disease. Unique additional cardiac (ie, arrhythmia, conduction defect, or congenital lesion) and extracardiac (ie, skeletal myopathy) features may guide the genetic evaluation; a genetic testing strategy may start with a focus on the gene(s) specifically linked to a single highly suspected endotype (ie, TTR gene when hereditary transthyretin amyloidosis is high on the differential, perhaps due to the presence of an abnormal nuclear scintigraphy scan), or may involve use of a broader multigene panel. Clinicians should ensure that the selected test includes the gene(s) and methodologies (ie, need for repeat expansion testing for DMPK in patients with suspected myotonic dystrophy type 1) necessary for diagnoses. The dagger indicates the requirement of repeat expansion analysis. AL = amyloid light chain; ASD = atrial septal defect; CCD = cardiac conduction disease; CPK = creatine phosphokinase; GI = gastrointestinal; LVH = left ventricular hypertrophy; PS = pulmonic stenosis; SCD = sudden cardiac death; VA = ventricular arrhythmia; WPW = Wolff-Parkinson-White syndrome; WT = wild-type.
ACM AND DCM GENOTYPES WARRANTING UNIQUE CLINICAL MANAGEMENT
For many patients with DCM, detection of a causative variant will not significantly alter clinical management; however, several notable exceptions exist. Although phenotypes and symptoms may overlap in different ACM/DCM endotypes, a number of important etiologies warrant unique clinical considerations. In the following section and in Table 1, examples are presented in which recognition of genotype influences clinical management.
TABLE 1.
Special Cases for Genotype-Specific Considerations in Clinical Management of DCM, ACM, and HCM, Including Genes Associated With Unique Cardiac and Noncardiac Features
| Dilated, Nondilated, and Arrhythmogenic Phenotypes | Hypertrophic Phenotypes | ||
|---|---|---|---|
| Gene | Genotype-Specific Features | Gene | Genotype-Specific Features |
| TTN |
|
PRKAG2 |
|
| LMNA a |
|
LAMP2 a |
|
| GLA a |
|
||
| SCN5A a |
|
GAA a |
|
| DSP, PKP2, DSC2, DSG, JUP |
|
TTR a |
|
| FLNC |
|
||
|
DMD
a
DES |
|
||
Genotype-specific drug therapy either available or under investigation.
ACM = arrhythmogenic cardiomyopathy; DCM = dilated cardiomyopathy; EF = ejection fraction; HCM = hypertrophic cardiomyopathy; ICD = implantable cardioverter-defibrillator; LQTS = long QT syndrome; SCD = sudden cardiac death.
TTN.
Truncating variants in the titin (TTN) gene are the most commonly implicated genetic cause of DCM, causing up to 25% of familial and 18% of sporadic cases.21 Progression in TTN-associated DCM is faster than non-TTN DCM, with earlier age at death, transplantation, or ventricular assist device. Due to the length of its coding sequence (about 100 kb), TTN was not included on first-generation genetic testing panels, and therefore this gene may not have been assessed in patients who underwent genetic testing prior to 2012. While an important cause of genetic DCM, truncating variants in TTN are also found in approximately 1% to 3% of the general population. Nonsense, frameshift, and canonical splice site TTN variants, as well as those occurring in the A-band domain, are enriched in DCM patients compared with healthy control subjects.22 As genetic testing for a range of applications increasingly reveals truncating TTN variants in unselected populations (ie, genomic screening programs or secondary reporting in exome sequencing performed for noncardiac indications), recognition of variant features associated with pathogenicity will improve prognostication.
LMNA.
Cardiac disease related to the nuclear envelope protein lamin A (LMNA) is associated with nearly 100% penetrance in older adults, causing a high burden of progressive malignant brady- and tachyarrhythmias frequently occurring in addition to heart failure.23 Anticipant arrangements for therapies directed at sudden death and heart failure risk are warranted, including consideration for primary prevention implantable cardioverter-defibrillator with or without resynchronization therapy in carriers of causative LMNA variants. Worse outcomes have been reported in individuals who engaged in competitive sports24 and patients with these genotypes warrant counseling on exercise modification. Furthermore, certain LMNA variants are associated with skeletal myopathy, including the limb-girdle and Emery-Dreifuss muscular dystrophies;25 therefore, carriers should be referred to a neuromuscular specialist for evaluation including measurement of creatine kinase level. Until recently, no specific drug therapy has been available for LMNA-associated disease. However, the Long-Term Efficacy and Safety of ARRY-371797 (PF-07265803) in Patients with Lamin A/C-Related Dilated Cardiomyopathy trial is studying a small molecule inhibitor of the p38α MAPK (mitogen-activated protein kinase) pathway in patients with LMNA-associated DCM,26 because this pathway has been found to be hyper-activated in rodent models.
SCN5A.
SCN5A-related DCM is another example in which genotype may dictate clinical management. SCN5A encodes the α subunit of the cardiac sodium channel, Nav1.5, and is associated with pleiotropic phenotypes including Brugada syndrome, long QT syndrome type 3, familial conduction disease, and familial atrial fibrillation,27 and with some variants such as cardiomyopathy. A specific phenotype of DCM with frequent multifocal premature ventricular contractions occurs when certain amino acid substitutions result in a new gating pore. In one of the more successful applications of precision medicine for cardiomyopathies, 87% of such cases resolve when treated with sodium-channel blockade.28 This customized treatment strategy stands in contrast to that for Brugada-associated SCN5A variants, for which sodium-channel blockers (other than quinidine) can unmask an arrhythmic phenotype and are contraindicated.29
DESMOSOME GENES.
Among individuals with genetically mediated DCM, 10% are due to damaging variants in genes encoding elements of the cardiomyocyte desmosome (desmoplakin [DSP], plakophilin-2 [PKP2], desmocollin-2 [DSC2], desmoglein-2 [DSG2], plakoglobin [JUP]).20 These genes are traditionally associated with arrhythmogenic right ventricular cardiomyopathy but are increasingly recognized to cause left-dominant or biventricular cardiomyopathy as well.30 Compared with other DCM genotypes, desmosomal genes are associated with higher incidence of malignant ventricular arrhythmia, but less left ventricular reverse remodeling.20 Individuals harboring causative variants in desmosomal genes and LMNA have the highest rates of sudden cardiac death and ventricular tachyarrhythmias, independent of left ventricular ejection fraction.31 Truncating variants in the filamin C gene (FLNC) confer a phenotype that overlaps with that caused by desmosomal and LMNA variants, with high rates of ventricular arrhythmia and frequent premature sudden death, and should be considered for implantable defibrillators, even if systolic dysfunction is only moderate.32 Pathogenic variants in phospholamban (PLN) and the cardiomyocytespecific RNA splicing factor RBM20 are also important causes of both ACM and DCM.33,34 Because of the role that endurance or frequent exercise plays in potentiating ventricular arrhythmias, heart failure, and worsening cardiomyopathy among desmosomal variant carriers, exercise restriction is advised.35
GENETIC EVALUATION IN PRACTICE: HCM
As with DCM, genetic HCM is characterized by both gene and allele heterogeneity. Damaging variants in genes related to sarcomeric structure and function (primarily myosin-binding protein C [MYBPC3], beta-myosin heavy chain [MYH7], troponin T2 [TNNT2], troponin I3 [TNNI3], alpha-actinin 2 [ACTN2], myosin light chain 3 [MYL3], tropomyosin alpha-1 chain [TPM1]) are found in 40% to 50% of patients with HCM who undergo genetic testing.36 Compared with patients with HCM with negative or uncertain genetic testing (ie, VUSs), sarcomere-positive genotypes are associated with worse outcomes, including earlier onset and greater burden of disease, with increased risk for ventricular arrhythmia, heart failure, and atrial fibrillation.12 Mortality in genotype-negative patients may not be increased above that of the general population, highlighting the prognostic value of genetic testing in HCM.
Penetrance and expressivity are also variable in HCM. Of genotype-positive/phenotype-negative relatives who carry a familial sarcomeric variant, 50% developed HCM over 15 years of follow-up.36 Furthermore, the natural history of genotype-mediated disease may be different in unselected populations. Of participants from the UK Biobank carrying likely pathogenic or pathogenic sarcomeric variants, penetrance of left ventricular hypertrophy was only 18%, although genotype-positive status was associated with increased risk of adverse cardiovascular outcomes and an attenuated cardiomyopathic phenotype, with increased wall thickness relative to genotype-negative biobank participants.37 Incidental discovery of such genotypes warrants ongoing clinical screening.
One important role for genetic testing in HCM is to differentiate sarcomeric disease from the so-called phenocopies. These are syndromic and infiltrative conditions including lysosomal and glycogen storage diseases (ie, Danon, Fabry, or Pompe disease), RASopathies (ie, Noonan, Costello, or cardiofaciocutaneous syndromes), and amyloidosis that manifest with cardiac changes mimicking classic sarcomeric HCM. In 1 center, these “mimickers” were detected in 1.45% of patients with HCM who underwent genetic testing.38 Although rare, recognition of these entities, often elucidated only through genetic testing, is essential, as the natural history and management differs significantly from that of sarcomeric HCM (Table 1). Amyloidosis is another increasingly recognized HCM phenocopy. In 1 Italian series of patients with an initial diagnosis of HCM, 9% were reclassified as having cardiac amyloidosis through either genetic testing revealing a pathogenic transthyretin gene (TTR) variant (3.2%) or clinical testing uncovering wild-type transthyretin or light chain disease.39 In the United States, this may be an even more important under-recognized cause of HCM, as 4% of African Americans harbor the cardiomyopathic TTR variant, Val142Ile (V142I).40 Transthyretin stabilizers and silencers (antisense oligonucleotide or small interfering RNA) are now approved for treatment of cardiac amyloidosis and hereditary polyneuropathy respectively.41 Early treatment appears to associate with the most favorable outcomes, supporting the need for expanded use of genetic testing, especially in African Americans with ungenotyped HCM.
OTHER CARDIOMYOPATHIES
Genetic etiologies have been described for forms of left ventricular noncompaction and restrictive cardiomyopathy, but yield of genetic testing is lower in these settings. Some forms of myopathic heart disease appear to be genetically mediated but are unmasked by specific exposures including myocarditis,42 peripartum,43 and alcoholic44 cardiomyopathies. Interestingly, the genes associated with these conditions overlap with those for other cardiomyopathies including DCM, HCM, and neuromuscular diseases with cardiac involvement. Genetic testing using a broad cardiomyopathy panel can be considered as part of the etiologic work-up for these entities, especially given the potential implications for risk in family members. A recommendation for genetic testing patients with peripartum cardiomyopathy was cited in the most recent practice guidelines.14
GENETIC TESTING FROM THE BEGINNING TO THE END OF LIFE
Because uncovering a genetic etiology in cardiomyopathies impacts prognosis, risk stratification in relatives, and potentially precision medicine approaches to clinical management, genetic testing should be performed in an affected individual (including children) as soon as a cardiomyopathy is evident. However, it is never “too late” for genetic testing. Postmortem testing on tissues obtained at autopsy from an individual with cardiomyopathy or sudden death can reveal a previously unrecognized genetic cause and inform risk in surviving relatives. Genetic testing can also guide decision making at the earliest stages of life. When a genetic cause of cardiomyopathy in a family is known, preimplantation genetic diagnosis with in vitro fertilization may be desired: embryos are screened in vitro for the known familial variant, and only those without the variant are implanted, thereby averting transmission of risk to offspring. Because homozygous variants in some genes (TTN, LMNA, DMD, DES, and FLNC) are associated with more profound and sometimes lethal phenotypes, reproductive partners of heterozygous carriers should undergo prenatal genetic testing to screen for variants in the same gene(s); couples at risk for homozygous transmission warrant further genetic counseling. In families in which a genetic cause of ACM, DCM, or HCM is identified in a proband, testing for the causative variant should be offered to children and adult relatives of all ages.
STUDY LIMITATIONS.
Importantly, the yield of contemporary genetic testing is significantly lower in certain populations due to historically lower rates of testing. In 1 series, only 4% of DCM patients of African descent compared with 27% of non-Hispanic Europeans were found to harbor an actionable variant. Additional research in diverse populations and more equitable utilization of genetic testing in clinical practice are needed to improve understanding of the genetic architecture of cardiomyopathies and address disparities in care.45
CONCLUSIONS
The genetic evaluation can yield actionable information to clarify etiology of cardiomyopathies, potentially alter clinical management, and guide risk stratification of relatives. Genetic testing results are probabilistic and require careful interpretation to avoid overattribution of risk. Accurate genotype data can inform prognostication and, in specific cases, present avenues for disease-modifying treatments. A clinical team that integrates genetics specialists in this process optimizes patient understand and coordination of care.
HIGHLIGHTS.
Cardiomyopathies can appear clinically indistinguishable but may warrant unique management considerations by genotype.
Genetic evaluations ideally involve trained geneticists or genetic counselors, with testing tailored to the phenotype under investigation.
Variants of uncertain significance should not be overinterpreted, and evidence for “causative” variants should be periodically reappraised.
FUNDING SUPPORT AND AUTHOR DISCLOSURES
The authors have reported that they have no relationships relevant to the contents of this paper to disclose.
ABBREVIATIONS AND ACRONYMS
- ACM
arrhythmogenic cardiomyopathy
- DCM
dilated cardiomyopathy
- HCM
hypertrophic cardiomyopathy
- VUS
variant of uncertain significance
Footnotes
The author attests they are in compliance with human studies committees and animal welfare regulations of the author’s institution and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the Author Center.
REFERENCES
- 1.Arbustini E, Narula N, Dec GW, et al. The MOGE(S) classification for a phenotype-genotype nomenclature of cardiomyopathy: endorsed by the World Heart Federation. J Am Coll Cardiol. 2013;62:2046–2072. [DOI] [PubMed] [Google Scholar]
- 2.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.James CA, Jongbloed JDH, Hershberger RE, et al. International evidence based reappraisal of genes associated with arrhythmogenic right ventricular cardiomyopathy using the Clinical Genome Resource Framework. Circ Genom Precis Med. 2021;14:e003273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jordan E, Peterson L, Ai T, et al. Evidence-based assessment of genes in dilated cardiomyopathy. Circulation. 2021;144:7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ingles J, Goldstein J, Thaxton C, et al. Evaluating the clinical validity of hypertrophic cardiomyopathy genes. Circ Genom Precis Med. 2019;12:e002460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruark E, Munz M, Renwick A, et al. The ICR1000 UK exome series: a resource of gene variation in an outbred population. F1000Res. 2015;4:883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ackerman MJ. Genetic purgatory and the cardiac channelopathies: exposing the variants of uncertain/unknown significance issue. Heart Rhythm. 2015;12:2325–2331. [DOI] [PubMed] [Google Scholar]
- 8.Clift K, Macklin S, Halverson C, McCormick JB, Abu Dabrh AM, Hines S. Patients’ views on variants of uncertain significance across indications. J Community Genet. 2020;11:139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quiat D, Witkowski L, Zouk H, Daly KP, Roberts AE. Retrospective analysis of clinical genetic testing in pediatric primary dilated cardiomyopathy: testing outcomes and the effects of variant reclassification. J Am Heart Assoc. 2020;9: e016195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manrai AK, Funke BH, Rehm HL, et al. Genetic misdiagnoses and the potential for health disparities. N Engl J Med. 2016;375:655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walsh R, Thomson KL, Ware JS, et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 2017;19:192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ho CY, Day SM, Ashley EA, et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation. 2018;138:1387–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muller RD, McDonald T, Pope K, Cragun D. Evaluation of clinical practices related to variants of uncertain significance results in inherited cardiac arrhythmia and inherited cardiomyopathy genes. Circ Genom Precis Med. 2020;13:e002789. [DOI] [PubMed] [Google Scholar]
- 14.Hershberger RE, Givertz MM, Ho CY, et al. Genetic evaluation of cardiomyopathy—a Heart Failure Society of America practice guideline. J Card Fail. 2018;24:281–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ho CY, Day SM, Axelsson A, et al. Valsartan in early-stage hypertrophic cardiomyopathy: a randomized phase 2 trial. Nat Med. 2021;27:1818–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ho CY, Lakdawala NK, Cirino AL, et al. Diltiazem treatment for pre-clinical hypertrophic cardiomyopathy sarcomere mutation carriers: a pilot randomized trial to modify disease expression. J Am Coll Cardiol HF. 2015;3:180–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li L, Bainbridge MN, Tan Y, Willerson JT, Marian AJ. A potential oligogenic etiology of hypertrophic cardiomyopathy: a classic single-gene disorder. Circ Res. 2017;120:1084–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cerrone M, Remme CA, Tadros R, Bezzina CR, Delmar M. Beyond the one gene-one disease paradigm: complex genetics and pleiotropy in inheritable cardiac disorders. Circulation. 2019;140:595–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haas J, Frese KS, Peil B, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. 2015;36:1123–1135a. [DOI] [PubMed] [Google Scholar]
- 20.Escobar-Lopez L, Ochoa JP, Mirelis JG, et al. Association of genetic variants with outcomes in patients with nonischemic dilated cardiomyopathy. J Am Coll Cardiol. 2021;78: 1682–1699. [DOI] [PubMed] [Google Scholar]
- 21.Herman DS, Lam L, Taylor MR, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roberts AM, Ware JS, Herman DS, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin trunca tions in health and disease. Sci Transl Med. 2015;7: 270ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar S, Baldinger SH, Gandjbakhch E, et al. Long-term arrhythmic and nonarrhythmic outcomes of lamin A/C mutation carriers. J Am Coll Cardiol. 2016;68:2299–2307. [DOI] [PubMed] [Google Scholar]
- 24.Pasotti M, Klersy C, Pilotto A, et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J Am Coll Cardiol. 2008;52: 1250–1260. [DOI] [PubMed] [Google Scholar]
- 25.Maggi L, Carboni N, Bernasconi P. Skeletal muscle laminopathies: a review of clinical and molecular features. Cells. 2016;5:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Feria A, Owens AT. Novel therapies in inherited cardiomyopathies. Prog Pediatr Cardiol. 2021;63:101444. [Google Scholar]
- 27.Wilde AAM, Amin AS. Clinical spectrum of SCN5A mutations: long QT syndrome, Brugada syndrome, and cardiomyopathy. J Am Coll Cardiol EP. 2018;4:569–579. [DOI] [PubMed] [Google Scholar]
- 28.Peters S, Thompson BA, Perrin M, et al. Arrhythmic phenotypes are a defining feature of dilated cardiomyopathy-associated SCN5A variants: a systematic review. Circ Genom Precis Med. 2022;15:e003432. [DOI] [PubMed] [Google Scholar]
- 29.Brugada J, Campuzano O, Arbelo E, Sarquella-Brugada G, Brugada R. Present status of Brugada syndrome: JACC State-of-the-Art Review. J Am Coll Cardiol. 2018;72:1046–1059. [DOI] [PubMed] [Google Scholar]
- 30.Miles C, Finocchiaro G, Papadakis M, et al. Sudden death and left ventricular involvement in arrhythmogenic cardiomyopathy. Circulation. 2019;139:1786–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gigli M, Merlo M, Graw SL, et al. Genetic risk of arrhythmic phenotypes in patients with dilated cardiomyopathy. J Am Coll Cardiol. 2019;74:1480–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ortiz-Genga MF, Cuenca S, Dal Ferro M, et al. Truncating FLNC mutations are associated with high-risk dilated and arrhythmogenic cardiomyopathies. J Am Coll Cardiol. 2016;68:2440–2451. [DOI] [PubMed] [Google Scholar]
- 33.van der Zwaag PA, van Rijsingen IA, Asimaki A, et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail. 2012;14: 1199–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parikh VN, Caleshu C, Reuter C, et al. Regional variation in RBM20 causes a highly penetrant arrhythmogenic cardiomyopathy. Circ Heart Fail. 2019;12:e005371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Al-Khatib SM, Stevenson WG, Ackerman MJ, et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2018;72:e91–e220. [DOI] [PubMed] [Google Scholar]
- 36.Lorenzini M, Norrish G, Field E, et al. Penetrance of hypertrophic cardiomyopathy in sarcomere protein mutation carriers. J Am Coll Cardiol. 2020;76:550–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Marvao A, McGurk KA, Zheng SL, et al. Phenotypic expression and outcomes in individuals with rare genetic variants of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2021;78:1097–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoss S, Habib M, Silver J, et al. Genetic testing for diagnosis of hypertrophic cardiomyopathy mimics: yield and clinical significance. Circ Genom Precis Med. 2020;13:e002748. [DOI] [PubMed] [Google Scholar]
- 39.Maurizi N, Rella V, Fumagalli C, et al. Prevalence of cardiac amyloidosis among adult patients referred to tertiary centres with an initial diagnosis of hypertrophic cardiomyopathy. Int J Cardiol. 2020;300:191–195. [DOI] [PubMed] [Google Scholar]
- 40.Quarta CC, Buxbaum JN, Shah AM, et al. The amyloidogenic V122I transthyretin variant in elderly black Americans. N Engl J Med. 2015;372:21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Emdin M, Aimo A, Rapezzi C, et al. Treatment of cardiac transthyretin amyloidosis: an update. Eur Heart J. 2019;40:3699–3706. [DOI] [PubMed] [Google Scholar]
- 42.Kontorovich AR, Patel N, Moscati A, et al. Myopathic cardiac genotypes increase risk for myocarditis. J Am Coll Cardiol Basic Trans Science. 2021;6:584–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ware JS, Li J, Mazaika E, et al. Shared genetic predisposition in peripartum and dilated cardiomyopathies. N Engl J Med. 2016;374:233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ware JS, Amor-Salamanca A, Tayal U, et al. Genetic etiology for alcohol-induced cardiac toxicity. J Am Coll Cardiol. 2018;71:2293–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morales A, Kinnamon DD, Jordan E, et al. Variant interpretation for dilated cardiomyopathy: refinement of the American College of Medical Genetics and Genomics/ClinGen Guidelines for the DCM Precision Medicine Study. Circ Genom Precis Med. 2020;13:e002480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lopez-Sainz A, Dominguez F, Lopes LR, et al. Clinical features and natural history of PRKAG2 variant cardiac glycogenosis. J Am Coll Cardiol. 2020;76:186–197. [DOI] [PubMed] [Google Scholar]
- 47.Cenacchi G, Papa V, Pegoraro V, Marozzo R, Fanin M, Angelini C. Review: Danon disease: review of natural history and recent advances. Neuropathol Appl Neurobiol. 2020;46:303–322. [DOI] [PubMed] [Google Scholar]
- 48.Lenders M, Brand E. Fabry disease: the current treatment landscape. Drugs. 2021;81:635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]