

Abstract
Purpose:
Paclitaxel (PTX) is one the most potent and commonly used chemotherapies for breast and pancreatic cancer. Several ongoing clinical trials are investigating means of enhancing delivery of PTX across the blood-brain barrier for glioblastomas (GBMs). Despite the widespread use of PTX for breast cancer, and the initiative to repurpose this drug for gliomas, there are no predictive biomarkers to inform which patients will likely benefit from this therapy.
Experimental Design:
To identify predictive biomarkers for susceptibility to PTX, we performed a genome-wide CRISPR knock-out (KO) screen using human glioma cells. The genes whose KO was most enriched in the CRISPR screen underwent further selection based on their correlation with survival in the breast cancer patient cohorts treated with PTX and not in patients treated with other chemotherapies, a finding that was validated on a second independent patient cohort using progression-free survival.
Results:
Combination of CRISPR screen results with outcomes from taxane-treated breast cancer patients led to the discovery of endoplasmic reticulum (ER) protein SSR3 as a putative predictive biomarker for PTX. SSR3 protein levels showed positive correlation with susceptibility to PTX in breast cancer cells, glioma cells and in multiple intracranial glioma xenografts models. Knockout of SSR3 turned the cells resistant to PTX while its overexpression sensitized the cells to PTX. Mechanistically, SSR3 confers susceptibility to PTX through regulation of phosphorylation of ER stress sensor IRE1α.
Conclusion:
Our hypothesis generating study showed SSR3 as a putative biomarker for susceptibility to PTX, warranting its prospective clinical validation.
Keywords: Paclitaxel, biomarker, glioblastoma, breast cancer, SSR3
Introduction:
Paclitaxel (PTX) and taxanes are among the most commonly used chemotherapeutic drugs for cancer. These drugs elicit cytotoxicity to cancer cells at nanomolar concentrations. Thus, PTX is the basis of chemotherapeutic regimens for breast, pancreatic, lung and ovarian carcinomas (1–4).
Gliomas and specifically glioblastomas (GBMs) are the most common and most deadly of all primary brain tumors in adults. Unfortunately, despite extensive research and the use of multimodal therapeutic strategies, the median overall survival time is 15–20 months from diagnosis (5). PTX is one of the most potent drugs against GBM in-vitro, with an IC50 1400-fold lower than standard of care drug temozolomide (6). However, the poor penetration of PTX across the blood-brain barrier (BBB) has limited its use for brain tumors (7). In this context, there are multiple emerging strategies to deliver PTX into the brain, including peptide-drug conjugate, PTX polymer microspheres/nanoparticles, convection-enhanced delivery and focused ultrasound (6,8–12). In this context, we have launched a Phase 1–2 clinical trial for recurrent GBM to evaluate the safety and efficacy of enhancing the delivery of systemic PTX using a skull-implantable ultrasound device that transiently opens the BBB (NCT04528680).
PTX is highly potent, yet there is a spectrum of susceptibility to this drug across individual tumors within a given cancer type. This is particularly important for GBM, as these tumors are notorious for their molecular heterogeneity and unpredictable susceptibility to therapies. Thus, even if the challenge of PTX delivery across the BBB is solved, the efficacious implementation of PTX-based therapy for GBM will be significantly influenced by the identification of tumors that respond to this drug. Despite the widespread use of PTX for breast cancer, among other malignancies, and our approach to repurpose it for GBM, there are no predictive biomarkers to inform which patients will benefit from this therapy. Understanding the biological basis of individual susceptibility to this drug, and developing predictive biomarkers will allow a personalized implementation of PTX-based therapy for GBM, and refine the indication for using this drug in common scenarios such as breast cancer in which PTX is readily used. In this context, we present an unbiased approach that incorporates causality using CRISPR screen, with correlative evidence using patient datasets, to discover predictive biomarkers for susceptibility to PTX.
Materials and Methods
Cell lines
Cells were incubated at 36°C and 5% CO2. 8MGBA and AM38 cells were grown in Minimum Essential Media (Corning) with 20% Fetal Bovine Serum (GE Health Sciences) and 1% Penicillin/Streptomycin (Corning). H4 cells were grown in in Dulbecco’s Minimum Essential Media (DMEM) containing 10% FBS. TS543 cells were grown in NeuroCult NS-A proliferation kit (StemCell Technologies) with 20ng/ml recombinant human Epidermal Growth Factor (PeproTech), 20ng/ml recombinant human Platelet Derived Growth Factor-AA (PeproTech), 20ng/ml recombinant Fibroblast Growth Factor (PeproTech), and 2 ug/ml heparin sulfate (StemCell Technologies). MES83 cells were a generous gift from Ichiro Nakano (University of Alabama). MES83 cells were cultured in Dulbecco’s Minimum Essential Media (DMEM) containing 10% FBS and 1% P/S. All other GBM PDX cells were purchased from Mayo Clinic (Scottsdale, AZ, USA) and grown in Dulbecco’s Minimum Essential Media (DMEM) containing 10% FBS. Breast cancer cell lines MDA-MB-231, BT549, Hs578T and HCC1937 were purchased from ATCC. MDA-MB-468 cells were kindly gifted by Dr. Dai Horuichi (Northwestern University). The breast cancer and the glioma cell lines used in this study were profiled for STR and tested negative for mycoplasma contamination. The list of all the cell lines, primary antibodies and reagents used in this study is described in the Resources Table (supplementary Table S1).
Animal Studies
All animal studies were performed in accordance with the Northwestern’s Institutional Animal Care and Usage Committee (IACUC). Mice were housed in pathogen free conditions at a relatively constant temperature of 24°C and humidity of 30–50%. Six to eight week old male and female athymic nude mice purchased from Charles River Laboratories were used in these studies.
Intracranial Patient Derived Xenograft Mouse Model
The intracranial injection protocol was approved by the IACUC at Northwestern University. The protocol followed to generate intracranial models is as described previously (6). A total of 2X104 MES83, 5X104 GBM12, 105 GBM6/GBM6 Vector control/GBM6 SSR3 overexpressing cells and 2×105 TS543 cells were used to develop orthotropic tumors for the listed cell lines (6). Typically for every intracranial injection, 2.5 μl of cell suspension was prepared in sterile PBS and was loaded into a 29G Hamilton Syringe. Injection was done slowly over a period of three minutes into the left hemisphere of the mouse brain at 3 mm depth through the transcranial burr hole created at 3mm lateral and 2 mm caudal relative to midline and bregma sutures. Following injection, incision was closed using 9 mm stainless steel wound clips and mouse was placed into a clean cage placed onto a heating pad until recovery from anesthesia. Abraxane (ABX) treatment was started five days after intracranial tumor implantation. ABX (24 mg/kg) was injected intravenously through the retro-orbital route while the control animals were injected with PBS. Mice were monitored over the period of study and were euthanized when they approached the end point as described in the IACUC protocol.
Mammary fat pad injections
The mammary fat pad injection protocol was approved by the IACUC at Northwestern University. For the mammary fat pad injections, 2 million MDA-MB-468 NTC (vector control) and SSR3 knockout (KO) cells were diluted 1:1 with Matrigel Matrix (BD Biosciences) at a final volume of 50 μl and injected in the inguinal mammary fat pad of nude female mice. Four weeks post tumor cell injection, when the tumors reached 62.5 mm3, the mice were randomized to the treatment groups. Nanoparticle albumin-bound PTX formulation nab-paclitaxel or Abraxane® (ABX) was administered by intraperitoneal injection as a single dose of 10 mg/kg and the control groups were administered equal volume of PBS (vehicle control).
PTX CRISPR screen
To perform the whole genome KO CRISPR screen, we used the Brunello Library that contains 70,000 sgRNA at the coverage of 3–4 gRNA/gene plus 10,000 gRNA non-targeting controls. The library preparation, virus production and multiplicity of infection (MOI) determination was done as described in (13). We used 50 million of selected cells for the extraction of genomic DNA (Day 0). 50 million cells each were treated with PTX (0.025 μM PTX in DMSO) or DMSO (vehicle control). On the 14th day and 21st day respectively the cells were harvested, the gDNA was extracted with the Zymo Research Quick-DNA midiprep plus kit, and the gRNA was amplified with the unique barcode primer.
For the next generation sequencing, the gRNAs were pooled together and sequenced in a Next generation sequencer (Next Seq). Along with Day 0 (post-puromycin selection), 20% of the cells were collected on day 14 (D14) from both PTX and DMSO groups as an intermediate time point. The terminal time point was day 21 (D21), where there was an expansion of resistant clones in the PTX-treated group. The samples were sequenced according to the Illumina user manual with 80 cycles of read 1 (forward) and 8 cycles of index 1. 20% PhiX were added on the Nextseq to improve library diversity and aiming for a coverage of >1000 reads per SgRNA in the library. The CRISPR screen data analysis. was performed with the bioinformatics tool CRISPR Analyzer45 (14). Deseq and sgRSEA algorithms were used to identify enriched genes. Guides with raw read counts <40 were excluded from the analysis. For gene ontology analysis DAVID42, 43 were used (15). The CRISPR screen raw data and analysis are available as separate spread sheets (supplemental information).
Generation of single gene knockouts and SSR3 overexpression clones
Single gene knockout clones for SSR3 and IRE1α were generated in lentiCRISPRv2 (one vector system). sgRNA sequences for SSR3 and IRE1α knockout are listed in (Supplementary Table S2). The vector backbone was purchased from Addgene and the protocol for guide cloning and generation of virus was as described in (16). For SSR3 overexpression, plasmid containing SSR3 open reading frame (Catalog ID:OHS6085–213574251) was purchased from Horizon (a PerkinElmer company).
Cell Viability Assay
GBM cells were seeded at a density of 5000 cells per well in a 96 well plate. One day after seeding, cells were checked for attachment and confluency (60–70%). PTX ranging in concentrations from 0.0005 μM to 0.5 μM was added to the wells. After 72 hours of treatment cell viability was determined by CellTiter Glo (Promega).
Immunofluorescence staining
H4 SSR3 knockout and non-targeting control cells were grown on glass coverslips. After 48 h of treatment with PTX or DMSO the coverslips were fixed in methanol for 5 minutes. The cells were then stained with α–tubulin and SSR3 antibodies at 1:100 dilution and the images were acquired on the Nikon A1 (C) confocal microscope. Number of microtubule bundles per cell cluster were counted for the analysis.
Immunohistochemistry (IHC) staining
Immunohistochemistry and H&E staining were performed using standard immunoperoxidase staining on formalin-fixed paraffin-embedded tissue sections of 5 μm thick from the mouse tumors. Mouse sections were stained with anti-SSR3 monoclonal antibodies. The procedure was performed on a DAKO Autostainer Link 48 slide stainer (Agilent Technologies). Sections were counterstained with hematoxylin, dehydrated, and mounted with coverslips. The slides were scanned and digitalized with the Hamamatsu K.K. Nanozoomer 2.0 HT and were visualized with the NDP.view2 Viewing software. HistoQuest version 6.0 software (TissueGnostics) was used to generate quantitative measurements of SSR3 expression, Ki67 expression and Cleaved caspase-3 (CC3) expression from the tumor region (cells/mm2).
Statistical analyses
Cox Proportional-Hazards Model (Cox) was used to identify genes that predict favorable survival in taxane treated group in the TCGA breast cancer dataset from among the 51 CRISPR shortlisted genes. Independent variables used included gene expression, treatment indicator (none, other or taxane) and interaction effect. The 3 patient groups were compared in pairs of 2- Taxane treated patients compared to untreated patients and second group as Taxane treated patients compared to patients treated with non-taxane drugs. All the patients in the TCGA breast cancer dataset that had annotation for chemotherapy administered, gene expression and survival data, were included in this analysis. Missing data for gene expression, overall survival, relapse-free survival with values equal to 0 or NA, were excluded from the analysis. Some of the patients did not have data on relapse-free survival hence the patient number in the relapse-free survival dataset is less than that of the patient number on the overall survival dataset. The analysis was done using Cox Proportional-Hazards Model (Cox) in latex table generated in R 3.5.2 by xtable 1.8–3 package. The analysis was performed on the TCGA dataset downloaded from Xena browser (https://xena.ucsc.edu/) on 13th April, 2019. All other statistical analyses were performed using Graphpad Software (Prism). In-vitro dose response curves were generated by fitting experimental cell viability data to a sigmoidal curve. One-way ANOVA with Dunnett’s multiple comparison test was used-to compute significance for the dose response curves. For the densitometry analysis-Unpaired two-tailed t test was used. Statistical analysis of animal survival was performed using a log-rank test. Pearson correlation coefficient r was determined to measure the strength of correlation. Image J analysis (17) was used to perform densitometry analysis on the western blots. Statistically significant p-values were considered as < 0.05. The graphs summarize significance using asterisks, as follows: *p< 0.05; **p< 0.01, ***p< 0.001, ****p<0.0001.
Data availability
All the data including PTX screen raw guide counts and analysis of PTX screen by DESeq and sgRSEA, that support the findings of this study, are available as part of the main text or as supplementary data.
Results
PTX is a potent drug against several cancers but exhibits a variable susceptibility across individual tumor cell lines
PTX was found to be one of the most potent drugs among the commonly used chemotherapeutics across multiple cancer cell lines (Fig. 1A) from the Genomics of Drug Sensitivity in Cancer Project (GDSC) database (18). Further, we found a comparable susceptibility range for human glioma cell lines as observed for lung and breast cancers in which PTX is clinically used (Fig. 1B).
Fig. 1. Whole genome CRISPR screen reveals genes that confer susceptibility to PTX treatment.
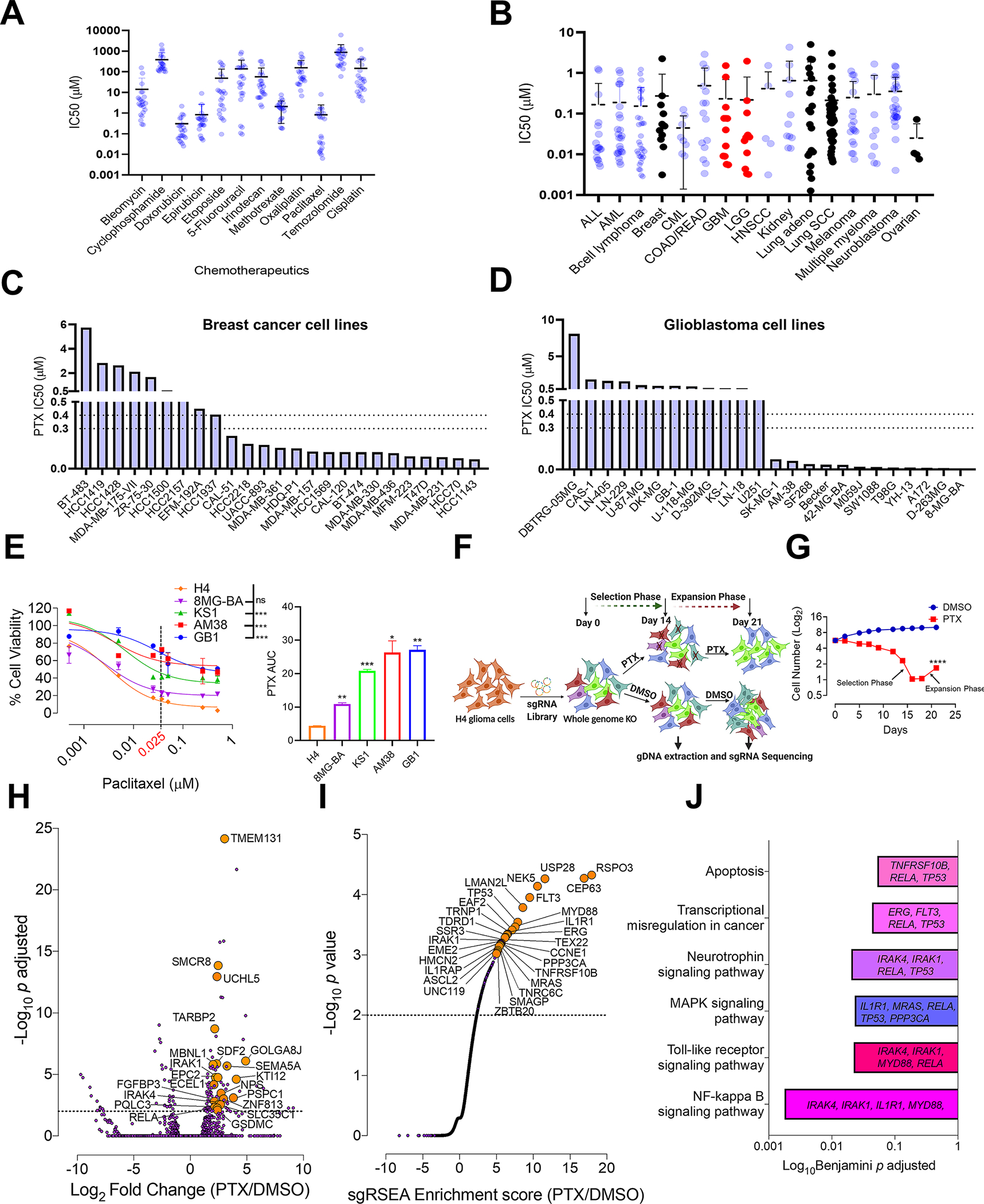
A, Comparison of IC50 drug concentrations of the commonly used chemotherapeutics against glioma cell lines from Sanger database (n=24). B, Comparison of PTX IC50 concentrations against all the available cancer cell lines (n=268) from Sanger database. On the graph-ALL (acute lymphoblastic leukemia), AML (acute myeloid leukemia), CML (chronic myelogenous leukemia), COAD (colon adenocarcinoma), READ (rectum adenocarcinoma), GBM (glioblastoma), LGG (low grade glioma), HNSCC (head and neck squamous cell carcinoma) and Lung SCC (lung squamous cell carcinoma). Colored in black are the IC50 drug concentrations of PTX for the cancers commonly treated with PTX. Colored in red are LGG’s and GBM’s. Histogram showing PTX IC50 drug concentrations for C, breast cancer and D, glioma cell lines from Sanger database. The dotted line indicates the brain PTX concentration (0.3–0.4 μM) that can be achieved by US-mediated delivery of PTX in the mouse brain (6). Graphs from A, B, C and D are generated using data from Sanger database (https://www.cancerrxgene.org/) (18). E, PTX dose response curve using glioma cell lines. One-way ANOVA with Dunnett’s multiple comparison test was used- ***p < 0.001, ns=non-significant. The histogram alongside shows the Area under curve (AUC) for each of these cell lines. Unpaired two-tailed t test was used for the analysis comparing most sensitive H4 cell line with other cell lines- *p< 0.05; **p< 0.01, ***p< 0.001. The experiment was repeated twice in triplicates. H4 cell line showed the highest degree of sensitivity to PTX hence was selected for the PTX CRISPR screen. F, Schematic representation of CRISPR screen. H4 cells were transduced with a genome-scale gRNA library (four gRNAs/gene). The edited cells were called as (Day 0). These cells were subjected to PTX (0.025 μM) or vehicle (DMSO) treatment. Cells from both these groups were sampled at D14 and D21 for Illumina sequencing of gRNA. G, Graphical representation depicting number of live cells (Y) during the CRISPR screen in PTX treated and DMSO control groups. Unpaired two-tailed t test was used for the analysis- ****p< 0.0001. H, Deseq analysis shows top genes (orange) for enriched gRNAs from PTX screen. Dashed line indicates p<0.05. Grey dots are all the genes with p>0.05. I, sgRSEA analysis shows top genes (orange) for enriched gRNAs from PTX screen. Dashed line indicates a p<0.05. Grey continuous line like dots are all the genes with p>0.05. J, Significantly enriched pathways along with their corresponding genes, selected in the PTX CRISPR screen. DAVID was used for gene ontology analysis (15).
Given the known molecular heterogeneity of human cancer, and its characteristic variable susceptibility to therapies, we investigated the susceptibility of individual breast cancer and glioma cell lines to PTX using GDSC (18). This analysis revealed that cell lines from these tumors exhibit a wide range of susceptibility to PTX (Fig. 1C and D). This suggests that the molecular characteristics of individual tumors may be more important to predict susceptibility to PTX than the overall cancer entity.
Genome-wide CRISPR knock-out screen reveals genes that confer susceptibility to PTX
To identify genes that influence PTX susceptibility in glioma, we first characterized different glioma cell lines for susceptibility to PTX (Fig. 1E). This analysis revealed the human glioma cell line H4 as the most sensitive amongst those tested. Therefore, H4 cells were used to perform PTX- genome-wide CRISPR knock-out screen. H4 cells transduced with a genome-scale gRNA library were subjected to treatment with 0.025 μM PTX or DMSO. The PTX concentration used was the lowest that was sufficient to kill most of the sensitive cell lines, but had minimal effect on the resistant cell lines (Fig. 1E). Cells were continuously treated with PTX or DMSO for 21 days. Over the course of the screen, we observed a selection phase in which PTX-treated cells declined in number, followed by an expansion phase in which resistant clones increased in number (Figs. 1F and G). The gRNAs enriched in the PTX group were determined by comparing gRNA from PTX-treated samples (D14 and D21) against DMSO-treated samples (D14 and D21) using DESeq and sgRSEA R algorithms (Figs. 1H and I). Using an adjusted p<0.01 for DESeq and p<0.001 for sgRSEA R, we obtained 51 genes enriched in PTX compared to the DMSO group (Supplementary Table S3 and S4). Gene ontology analysis of these 51 genes revealed that PTX treatment led to the selection of KO clones for genes involved in pathways like NF-kappa B signaling (Benjamini-Hochberg (BH) adjusted p=0.0017), toll-like receptor signaling (BH p=0.022), MAPK signaling (BH p=0.023), neurotrophin signaling (BH p=0.020), transcriptional misregulation in cancer (BH p=0.043) and apoptosis (BH p=0.053) (Fig. 1J). Along with previously reported genes involved in PTX susceptibility like P53 (19), apoptosis markers like FADD, caspase 8, and BAX (20–22), the screen also identified several novel genes that had not been previously implicated in susceptibility to PTX.
Identification of putative predictive biomarkers for PTX susceptibility by combining CRISPR screen results with outcomes from taxane-treated breast cancer patients
The functional implication by the CRISPR KO screen does not necessarily mean that the baseline expression of a gene hit on this experiment is a good predictive biomarker for susceptibility to a drug, as genes that are involved in susceptibility might be induced by drug treatment. Therefore, to identify a subset of genes functionally implicated and whose baseline expression predicts susceptibility to PTX, we refined our candidate gene list through Cox Proportional-Hazards Model (Cox) analysis using the TCGA breast cancer dataset (23). This included patients that had annotation for chemotherapy administered, RNA-seq-based expression data for tumor tissue, and overall survival. We chose breast cancer since PTX is commonly used in this setting, allowing robust correlation with patient outcomes for biomarker discovery. On the other hand, discovery of a predictive biomarker for breast cancer would allow immediate validation and application of this discovery for patients that are already routinely being treated this drug. We divided the patients into 1. Patients treated with taxanes 2. Patients who did not receive any chemotherapy and 3. Patients treated with drugs other than taxanes. We queried the expression of 51 genes identified as CRISPR screen hits. Correlation analysis of expression of these genes with chemotherapy used (taxanes, non-taxane drugs or no chemotherapy) with overall survival was analyzed in a continuous fashion by Cox. 9/51 genes were found to have a significant interaction coefficient between the drug used and gene expression on overall survival, as the interaction between gene expression and survival was only observed in the taxane-treated patients and not in the untreated group (Supplementary Table S5). 5/51 genes were found to have significant interaction coefficient between the drug used and gene expression on overall survival as the interaction between gene expression and survival was only observed in the taxane-treated group and not in the non-taxane chemotherapy group (Supplementary Table S6).
Baseline expression of SSR3 predicts susceptibility to PTX in breast cancer and GBM
Baseline expression of SSR3 (also known as translocon-associated protein gamma-TRAPγ) was found to be predictive for susceptibility to taxanes in two independent breast cancer datasets, among the final list of putative biomarkers significantly enriched from the CRISPR screen. SSR3 showed favorable overall survival in TCGA breast cancer patients treated with taxanes (n=123, p=0.003) and not in patients who did not receive any chemotherapy (n=358, p=0.391) or who received non-taxane chemotherapies (n=722, p=0.349, Fig. 2A). Similarly, SSR3 showed significant correlation with relapse-free survival in TCGA breast cancer patients treated with taxanes (n=115, p=0.037) and not in patients who did not receive any chemotherapy (n=254, p=0.622) or who received non-taxane chemotherapies (n=659, p=0.719, Fig. 2B). Indeed, whereas 9/51 putative biomarkers were identified using the TCGA breast cancer dataset as the discovery dataset, only 1/9 (SSR3), was found to be predictive in an independent validation cohort. In the validation cohort (GSE25066 dataset) which includes only taxane-treated patients (24,25), high SSR3 expression showed significant association with relapse-free survival (n=205, p=0.024). In contrast, no association between relapse-free survival and SSR3 expression was observed in the combined GEO datasets (GSE16716, GSE19615, GSE31519, GSE37946, GSE45255 and GSE65194) which is composed of patients who have not received hormonal therapy or chemotherapy (n=1023, p=0.947, Fig. 2C) (26–28). For the validation cohort, among the 9 putative biomarker genes, 6 were not significantly predictive of relapse-free survival (CEP63, IRAK4, TMEM131, MBNL1, ZBTB20, and TDRD1) and 2 of these genes (EPC2 and ZNF813) did not have gene expression data available for the analysis. Thus, SSR3 was predictive of relapse-free survival as a measure of susceptibility to taxanes in two independent breast cancer datasets, and predictive of OS in the context of taxne-treatment in the TCGA dataset. Although, SSR3 RNA expression was predictive of susceptibility to taxanes, SSR3 RNA expression alone (not in the context of taxane treatment) was not predictive of favorable outcome in either GBM (n=167, p=0.55) or breast cancers (n=1260, p=0.20) (Supplementary Figs. S1A and B).
Fig. 2. SSR3 mRNA levels correlate with susceptibility to taxanes in breast cancer patients.
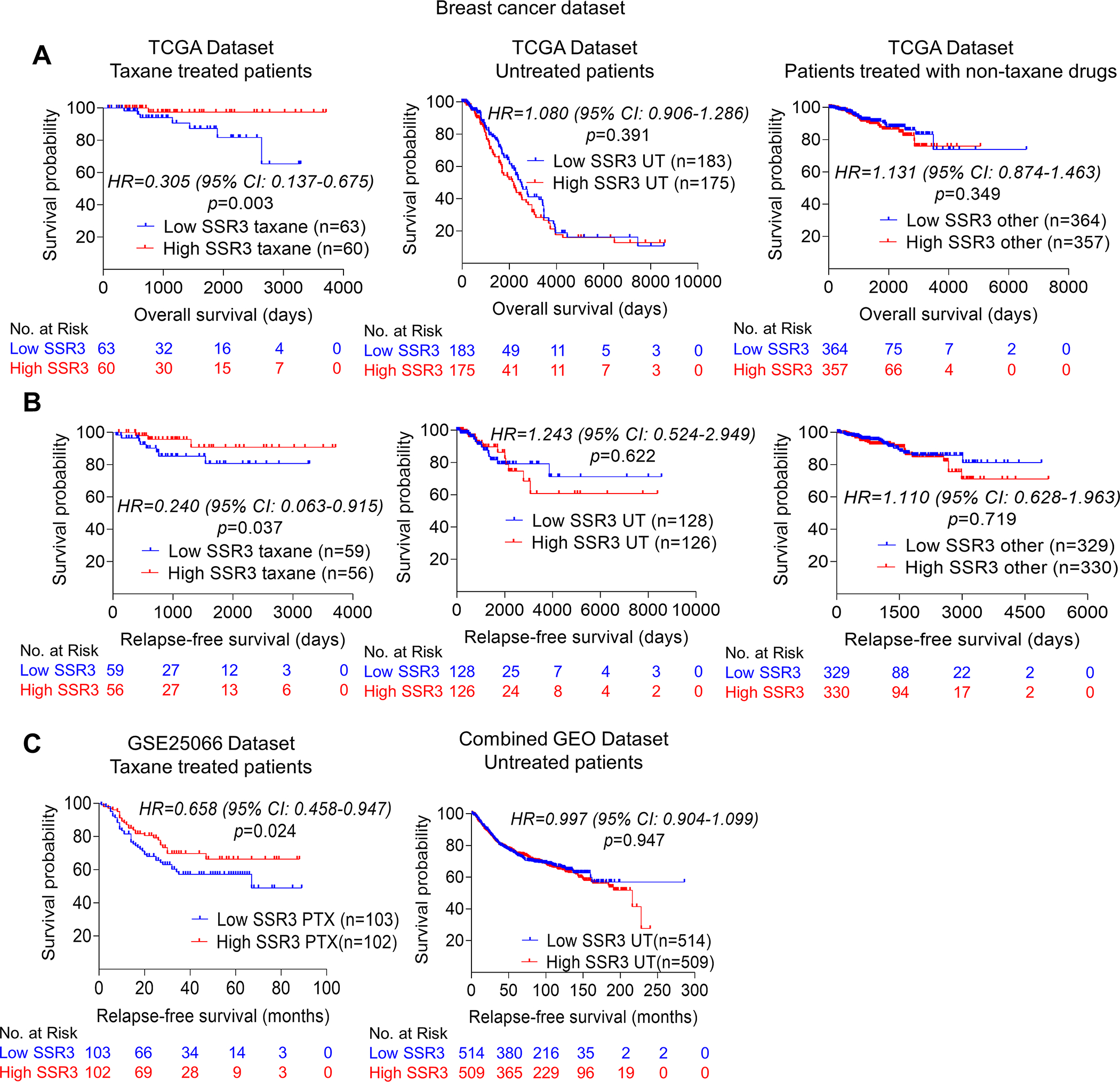
A, Kaplan-Meier survival analysis showing the association of high SSR3 mRNA levels with favorable overall survival in TCGA taxane treated breast cancer patients (n=123) and not in patients who did not receive any chemotherapy (n=358) or who received non-taxane chemotherapies (n=722) (https://xena.ucsc.edu/) (23). B, Kaplan-Meier survival analysis showing the association of high SSR3 mRNA levels with relapse-free survival in TCGA taxane treated breast cancer patients (n=115) and not in patients who did not receive any chemotherapy (n=254) or who received non-taxane chemotherapies (n=659) (https://xena.ucsc.edu/) (23). Taxane treated patients included patients who have received taxol, paclitaxel, docetaxel, ABX and taxotere as indicated in TCGA breast cancer dataset. C, Kaplan-Meier survival analysis showing the association of high SSR3 expression with relapse-free survival in the GSE25066 dataset which includes only taxane-treated patients who have not received hormonal therapy (n=205) (24,25). Kaplan-Meier survival analysis showing no such association in combined GEO datasets (GSE16716, GSE19615, GSE31519, GSE37946, GSE45255 and GSE65194) which includes patients who have not received hormonal therapy or chemotherapy (n=1023) (26–28). For A, B and C median was used as a cutoff to separate high and low SSR3 expression. p value, Hazard ratio (HR) and 95% confidence interval (CI) were computed using Cox proportional hazards analysis.
Further, we characterized breast cancer cell lines and glioma patient-derived xenografts (PDX) cells for PTX susceptibility. Area under the curve (AUC) value was determined for each of these lines as a measure of susceptibility to PTX treatment. Correspondingly, SSR3 protein levels were determined for these cell lines by western blot. A negative correlation was observed between SSR3 protein levels (image J quantification of both the bands of SSR3) and resistance to PTX (AUC values) in the case of glioma PDX cell lines (Pearson r= −0.70, p=0.03, n=9) (Fig. 3A–C). Similarly, analysis of breast cancer cell lines revealed a negative correlation between SSR3 protein levels and resistance to PTX albeit non-significant (Pearson r=−71, p=0.17, n=5) (Fig. 3D–F). Next, we investigated whether baseline expression of SSR3 is predictive of susceptibility to PTX in in-vivo setting across intracranial glioma models. Four human intracranial glioma xenograft models with different SSR3 protein expression determined by western blot (Fig. 3G), were treated with ABX. Survival curves for MES83 and GBM12 were adopted from (6). MES83 (median survival extended from 20 days to 31 days, p=0.0041) and GBM12 (median survival extended from 24 days to 38 days p<0.0001), which express higher levels of SSR3 demonstrated better susceptibility to PTX. On the other hand GBM6 (median survival extended from 29 days to 30 days, p=0.77) and TS543 (median survival extended from 38.5 days to 43 days, p=0.0050) which expresses relatively lower levels of SSR3, demonstrated poor susceptibility to PTX (Fig. 3H and I). The data on the titration of SSR3 antibody and quantification of staining is shown in Supplementary Fig. S2 and S3. The IHC analysis of the tumors developed from PTX responsive MES83 and GBM12 and PTX non-responsive GBM6 and TS543 models showed significant differences in the baseline expression of SSR3 (Fig. 3J). The baseline expression of SSR3 in these models strongly correlated with the percent increase in median survival (Pearson r= 0.97, p=0.02) (Fig. 3K). No significant difference was seen in SSR3 expression (Supplementary Fig. S4A and B) or Cleaved caspase-3 (CC3) expression (Supplementary Fig. S5A and B) in tumor cells at the time of death between ABX treated group and control group as measured in SSR3 high PTX responsive GBM12 and SSR3 low PTX non-responsive GBM6 tumors. Moreover, difference was seen between ABX treated group and control group in Ki67 expression (albeit non-significant) for GBM12 tumors and significant for GBM6 tumors (Supplementary Fig. S6A and B).
Fig. 3. SSR3 expression correlates with susceptibility to PTX in glioma, breast cancer cells and in intracranial glioma models.
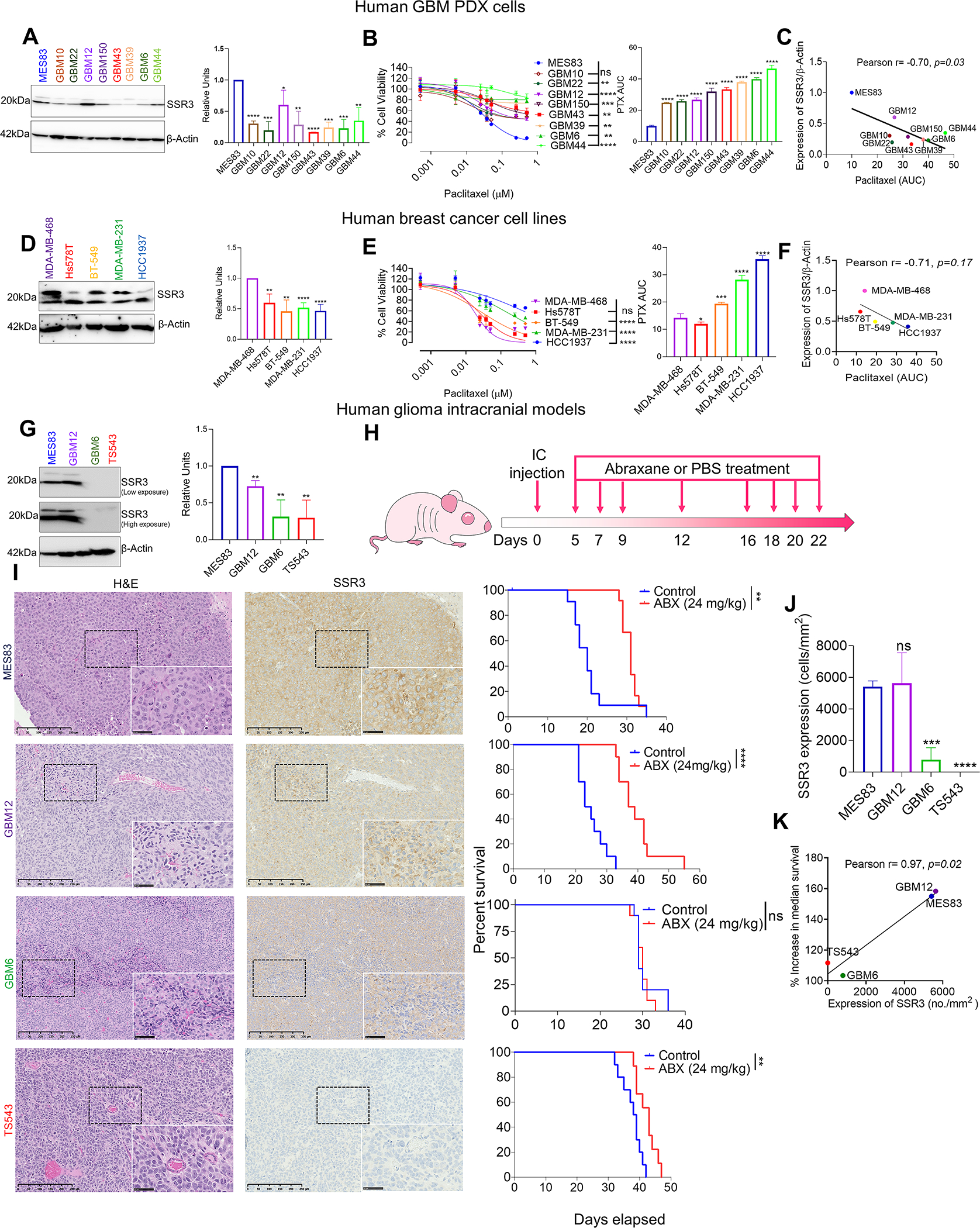
A, Western blot showing SSR3 protein levels for the 9 glioma PDX cell lines. The histogram alongside western blot represents densitometry analysis (using Image J). B, PTX dose response curves for the glioma PDX cell lines. The histogram alongside shows the AUC for each of these cell lines. C, Scatter plot showing correlation between susceptibility to PTX (AUC) and protein levels of SSR3 as quantified by Image J. Pearson correlation coefficient r= −0.70, p=0.03. D, Western blot showing SSR3 protein levels for the indicated breast cancer cell lines. Histogram alongside western blot represents densitometry analysis. E, PTX dose response curves for the breast cancer cell lines. The histogram alongside shows the AUC for each of these cell lines. F, Scatter plot showing correlation between susceptibility to PTX (AUC) and protein levels of SSR3 as quantified by Image J. Pearson correlation coefficient r= −0.71, p=0.17. G, Western blot showing baseline protein levels of SSR3 in the MES83, GBM12, GBM6 and TS543 cell lines. The histogram alongside western blot represents densitometry analysis. H, Schematic representation of the dosing scheme followed to treat mice having intracranial tumors with ABX. Three PDX lines (MES83, GBM12, GBM6 and one glioma cell line (TS543) were implanted intracranially. I, Representative H and E staining and immunostaining of SSR3 on the tumor tissues from the xenografts developed from these cell lines. Scale bar in black=250 μm. Scale bar in the inset=50 μm. The survival curve alongside, shows the survival of the mice injected with these cell lines in ABX treated and control treated groups. Log-rank analysis was used to determine the survival differences. For MES83, median survival was 20 for control vs. 31 days for ABX treated group, p=0.0041, (n=12). For GBM12, median survival was 24 for control vs. 38 days for ABX treated group, p<0.0001, (n=10). For GBM6, median survival was 29 for control vs. 30 days for ABX treated group, p=0.77, n=10. For TS543, median survival was 38.5 for control vs. 43 days for ABX treated group, p=0.0050, (n=10) for control and (n=9) for ABX treated group. J, Histogram shows SSR3 protein expression (cells/mm2) in xenograft tumor tissues in the SSR3 high PTX sensitive MES83 and GBM12 and SSR3 low PTX resistant GBM6 and TS543 models. K, Scatter plot showing correlation between baseline SSR3 protein expression (cells /mm2) in xenograft tumor tissues and percent increase in median survival. Pearson correlation coefficient r= 0.97, p=0.02. Histoquest software was used to quantitate SSR3 tumor expression by setting 25 as threshold for SSR3 mean intensity using positive and negative control slides. The data represents n=3 tumors/group. For B and E PTX dose response curves-One-way ANOVA with Dunnett’s multiple comparison test was used-ns=non-significant, **p< 0.01, ***p< 0.001, ****p<0.0001. The comparison was made between the most sensitive cell lines and all the other cell lines. For the densitometry and AUC histogram in A, B, D, E, G and J-.Unpaired two-tailed t test was used for the analysis comparing most sensitive cell line with other cell lines- *p< 0.05; **p< 0.01, ***p< 0.001, ****p<0.0001. For J and K MES83, GBM12 and GBM6 (n=3 tumors/group) and for TS543 (n=2 tumors/group). All the experiments in A, B, D, E, G and J were done in triplicates. Β-actin was used as a loading control for western blots.
SSR3 expression contributes to the susceptibility to PTX in breast cancer and glioma cells
SSR3 is overexpressed in GBM as compared to the normal brain tissue, whereas in breast cancer SSR3 shows a spectrum of expression as compared to the non-tumor breast tissue (Fig. 4A). In the PTX CRISPR screen, gRNAs for SSR3 were 2-fold more enriched in PTX group as compared to its DMSO control group (Fig. 4B). KO of SSR3 in PTX sensitive glioma cell line H4 and breast cancer cell line MDA-MB-468, rendered these cells resistant to PTX (Figs. 4C–F). For MDA-MB-468 cells, SSR3 KO 1 clone and for H4, SSR3 KO 2 clone were used for mechanistic studies. Correspondingly, overexpression of SSR3 in PTX resistant-SSR3 low-GBM6 cells led to significant increase in susceptibility of these cells to PTX (Fig. 4G and H). In line with the increased in-vitro susceptibility to PTX, intracranial tumors developed with GBM6 overexpressing SSR3 protein, showed significant improvement in the susceptibility to ABX (median survival extended from 29 days to 38 days, p<0.0016) as compared to the GBM6 vector control cells (median survival extended from 47 days to 50 days, p<0.01) (Figs. 5A–D). Similarly, mammary fat pad tumors developed from MDA-MB-468 SSR3 KO cells showed poorer susceptibility to ABX as compared to its vector control cells (p<0.018) (Figs. 5E–G). These experiments confirm that SSR3 not only correlates but also contributes to the susceptibility to paclitaxel in the intracranial setting using patient derived xenograft model.
Fig. 4. SSR3 protein levels determine susceptibility to PTX in breast and GBM cell lines.
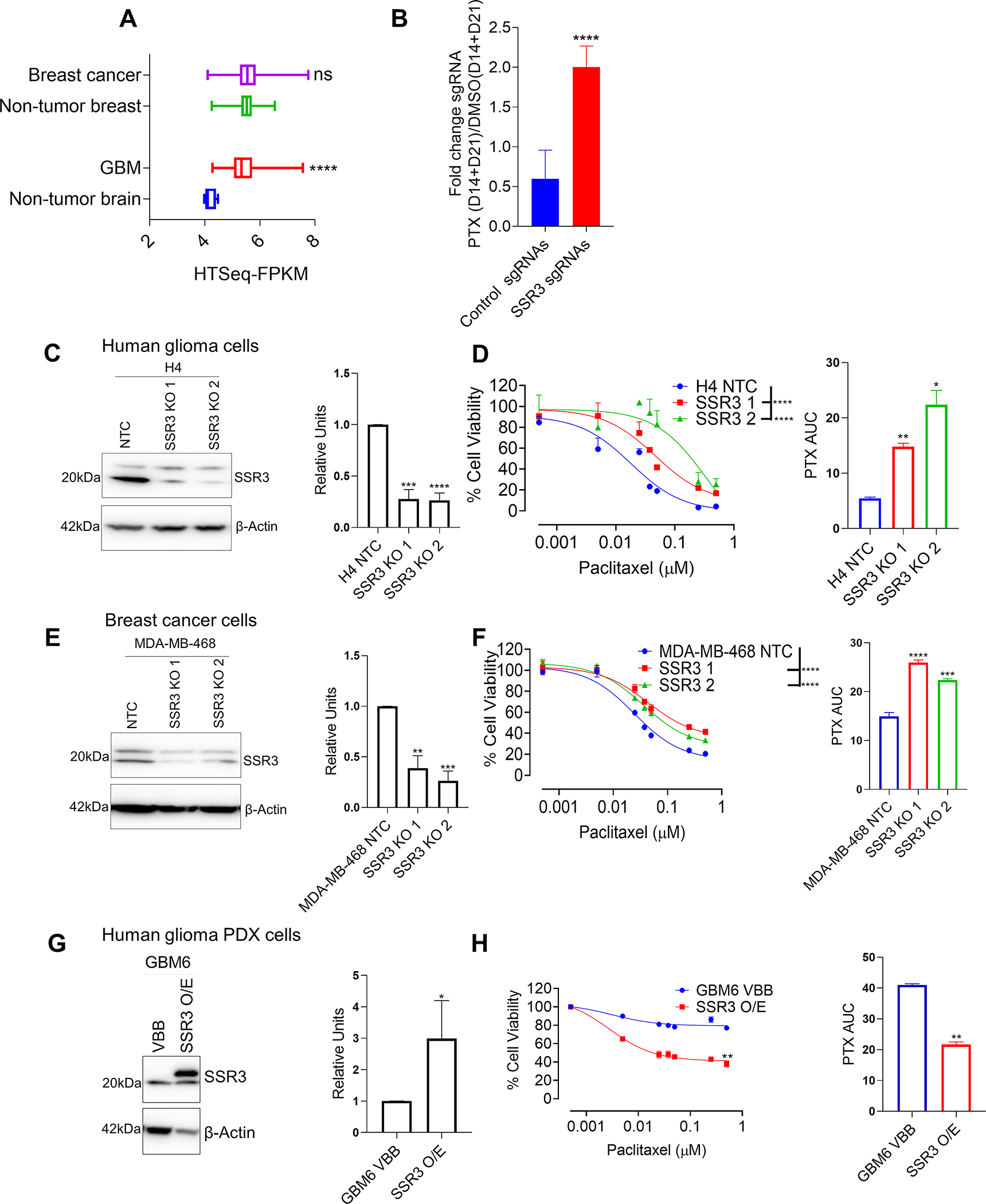
A, The graph shows SSR3 mRNA expression in GBM and breast cancers in relation to its non-tumor regions from TCGA GBM and breast cancer datasets respectively. B, The graph shows the fold enrichment of SSR3 gRNA’s (n=2) and non-targeting control gRNA’s (n=1000) in PTX treated group (D14 and D21) over DMSO control group (D14 and D21) in the CRISPR screen. Unpaired two-tailed t test was used for the analysis- ****p<0.0001. C, Western blot for SSR3 gene KO clones derived from H4 glioma cells. The histogram alongside western blot represents densitometry analysis. D, PTX dose response curve for SSR3 gene KO clones derived from H4 glioma cells. The histogram alongside shows the AUC for each of these clones. E, Western blot for SSR3 gene KO clones derived from MDA-MB 468 breast cancer cells. The histogram alongside western blot represents densitometry analysis. F, PTX dose response curve for SSR3 gene KO clones derived from MDA-MB 468 breast cancer cells. The histogram alongside shows the AUC for each of these clones. G, Western blot for SSR3 overexpressing clones derived from GBM PDX line GBM6. The histogram alongside western blot represents densitometry analysis H, PTX dose response curve for SSR3 overexpressing cells derived from GBM PDX line GBM6. The histogram alongside shows the AUC for these clones. For the densitometry and AUC histogram in C, D, E, F, G and H-Unpaired two-tailed t test was used for the analysis comparing control clones with the edited clones- *p< 0.05; **p< 0.01, ***p< 0.001, ****p<0.0001. For D and F PTX dose response curves-One-way ANOVA with Dunnett’s multiple comparison test was used- ****p<0.0001. The comparison was made between the control clones and the edited clones. For H, PTX dose response curve-Unpaired two-tailed t test was used for the analysis-**p<0.01. All the experiments in C-H were done in triplicates.
Fig. 5. SSR3 protein levels determine susceptibility to PTX in in-vivo glioma and breast cancer models.
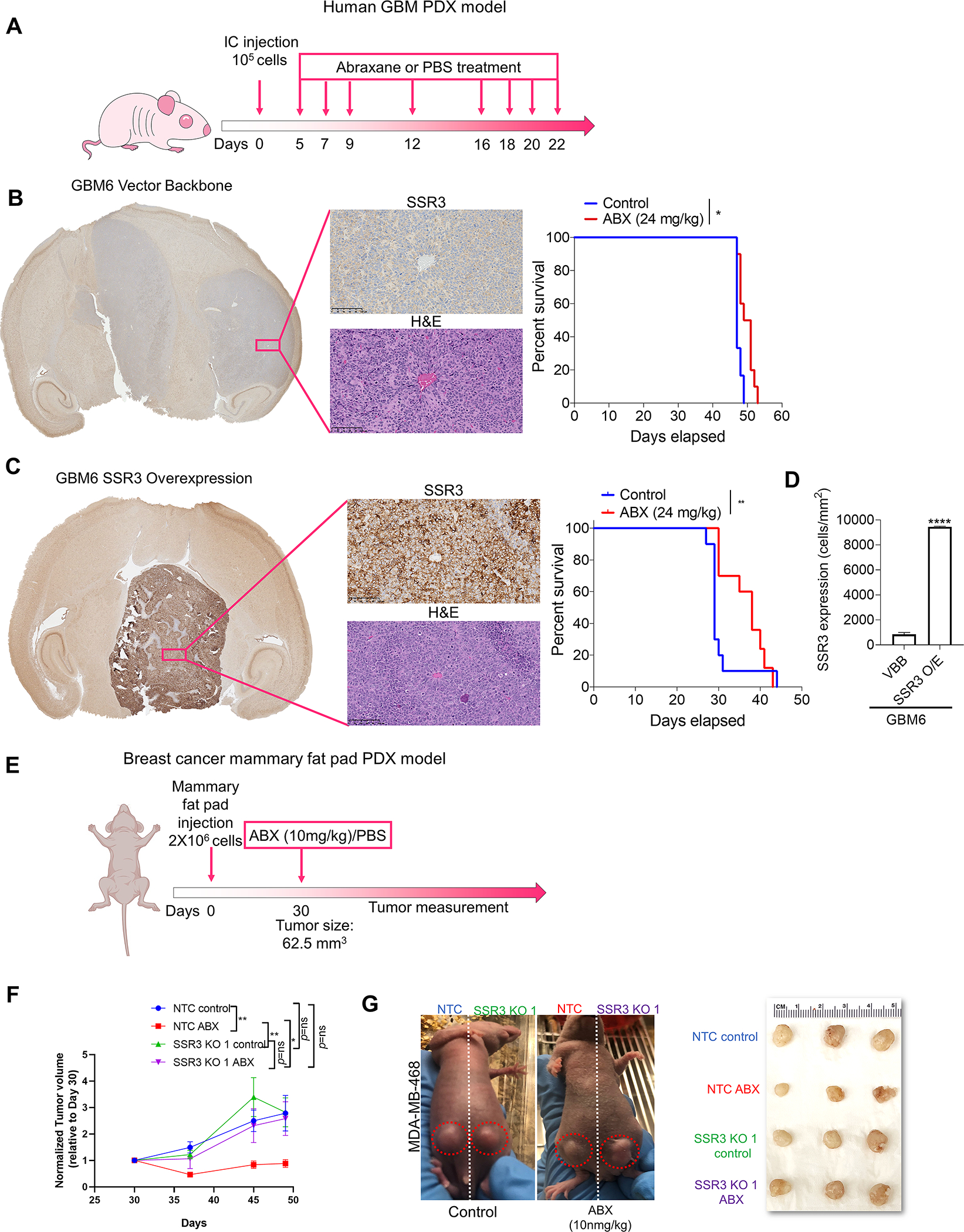
A, Schematic representation of the dosing scheme followed to treat mice having intracranial tumors with ABX. B and C, Representative H and E staining and immunostaining of SSR3 on the tumor tissues from the xenografts developed from GBM6 vector control and SSR3 overexpressing cells respectively. Scale bar in black=100 μm. Kaplan–Meier survival curves for mice injected with vector control (GBM6 VBB) and GBM6 SSR3 overexpressing (GBM6 SSR3 O/E) clones-with and without the treatment with ABX. Log-rank analysis was used to determine the survival differences. For GBM6 VBB, median survival was 47 days for control vs. 50 days for ABX treated group, p<0.01, number of mice (n=10/group). For GBM6 SSR3 O/E, median survival was 29 for control vs. 38 days for ABX treated group, p<0.0016 (n=10/group). **p<0.01, *p<0.05. D, Histogram shows baseline SSR3 protein expression (cells/mm2) in tumor tissues from the xenografts developed from GBM6 VBB (n=2 tumors/group) and GBM6 SSR3 O/E (n=3 tumors/group) in the control group. E, Schematic representation of the dosing scheme followed to treat mice bearing mammary fat pad tumors with ABX. F, The mice were randomized on day 30 when the tumors measured 62.5 mm3. Single dose of ABX (10mg/kg) was administered to the mice in the experimental group and PBS was administered to the mice in the control group, after randomization on day 30. The tumors were measured once every week after the drug treatment (n=5/group). The data was analyzed using two-way ANOVA with Tukey’s multiple comparison test-**p<0.01, *p<0.05. G, Representative images of the mice bearing mammary fat pad tumors-injected on one side with MDA-MB-468 vector control (NTC) and on contralateral side with SSR3 KO (SSR3 KO1) clones and treated with ABX or PBS. Alongside is a photograph of the tumors from all the four groups at the endpoint.
Phosphorylation of IRE1α contributes to SSR3-mediated susceptibility to PTX in glioma cells
Basal levels of SSR3 were found to determine susceptibility to PTX. In line with this, SSR3 was not induced by 48 h of treatment with PTX in PTX sensitive cell lines H4 NTC and MES83 and in PTX resistant GBM6 cells (Supplementary Fig. S7A). SSR3 is a subunit of translocon-associated protein (TRAP) complex, localized in the endoplasmic reticulum (ER) lumen and is primarily involved in the folding and transport of proteins destined to ER (29–31). Inhibition of TRAP complex function by depletion of SSR3 was found to influence ER stress related unfolded protein response (UPR pathway) (32). ER stress response and ER transport machineries are shown to interact with each other via UPR gene IRE1α (33). Moreover, SSR3 KO mice and IRE1α KO mice have shown to confer similar phenotypes on the development of placenta (34,35). With this in mind, we investigated IRE1α and other ER stress markers in the context of susceptibility to PTX. We observed a positive correlation between SSR3 and IRE1α levels (Pearson r=0.88, p=0.001, n=9) but not with other ER stress markers like BIP and PERK in glioma PDX cell lines (Supplementary Fig. S7B–E). A positive correlation was also observed between SSR3 protein levels and pIRE1α in glioma PDX cell lines (Pearson r= 0.97, p=0.0001, n=9, Fig. 6A). In H4 glioma cells, SSR3 KO led to reduction in phosphorylation of IRE1α in presence of PTX as showed at 2 h and 5 h time points (Fig. 6B). Further, knockout of IRE1α in PTX sensitive H4 glioma cells rendered them resistant to PTX (H4 PTX AUC- NTC vs IRE1α KO 1 and 2, p<0.01) (Fig. 6C and D). We performed a rescue experiment in GBM6 SSR3-overexpressing cells that had acquired susceptibility to PTX, to investigate whether IRE1α loss would restore the PTX resistance phenotype found in GBM6 wild-type cells. We found that knockout of IRE1α in SSR3-overexpression background rendered the cells more resistant to PTX (GBM6 SSR3 O/E PTX AUC- NTC vs IRE1α KO, p<0.01). (Fig. 6E–G). On the other hand, the knockout of IRE1α in PTX-resistant-wild-type GBM6 cells (SSR3 low), showed a modest increase in resistance to PTX (GBM6 PTX AUC- NTC vs IRE1α KO 1, p=ns and NTC vs IRE1α KO 2, p<0.05). (Fig. 6H and I).
Fig. 6. SSR3 mediated susceptibility to PTX seems to be regulated through phosphorylation of IRE1α in glioma cells.
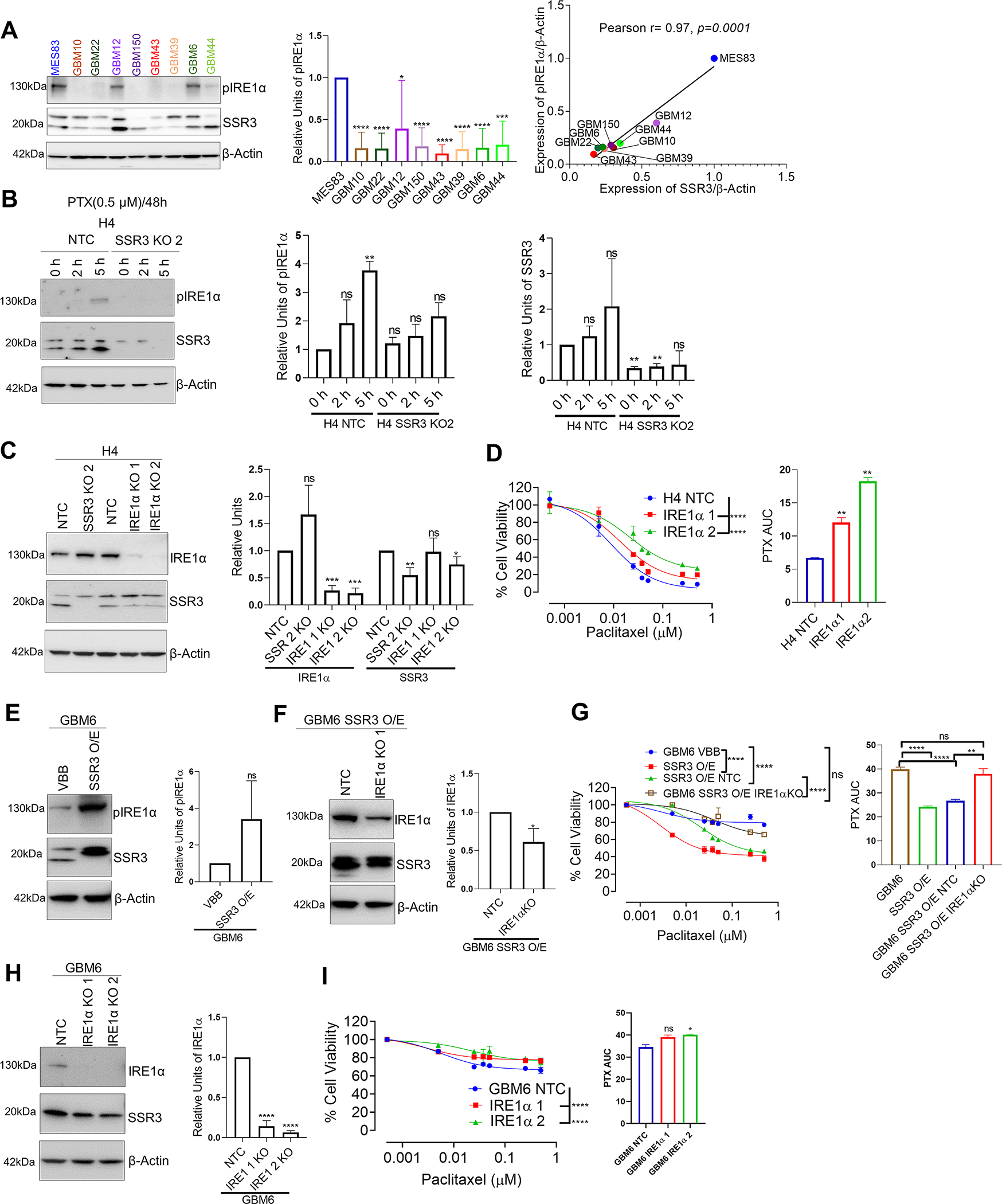
A, Western blot showing pIRE1α and SSR3 protein levels in glioma PDX cell lines. The histogram alongside western blot represents densitometry analysis. The Scatter plot shows correlation between SSR3 (from Figure 3) and pIRE1α protein levels, as quantified by Image J using data from three independent experiments. Pearson correlation coefficient r= 0.97, p=0.0001. B, Western blot showing pIRE1α levels in 0.5 μM PTX treated H4 SSR3 KO and vector control clones at 2 h and 5 h time points. The histogram alongside western blot represents densitometry analysis of 2 independent experiments for pIRE1α and SSR3. C, Western blot for IRE1α gene KO clones derived from H4 glioma cells. The histogram alongside western blot represents densitometry analysis for IRE1α and SSR3. D, PTX dose response curve for IRE1α gene KO clones derived from H4 glioma cells. The histogram alongside shows the AUC for each of these clones. E, Western blot showing pIRE1α levels in GBM6 SSR3 overexpressing and vector control clones. The histogram alongside western blot represents densitometry analysis. F, Western blot of IRE1α gene KO clones and its vector control cells derived from GBM6 SSR3 overexpressing cells. The histogram alongside western blot represents densitometry analysis for IRE1α. G, PTX dose response curve of IRE1α gene KO clones. Curves for GBM6 SSR3 overexpressing clones and GBM6 parental cells are plotted for comparison. The histogram alongside shows the AUC for each of these clones. H, Western blot of IRE1α gene KO clones and its vector control cells derived from GBM6 wild type cells. The histogram alongside western blot represents densitometry analysis for IRE1α. I, PTX dose response curve of IRE1α gene KO clones derived from GBM6 wild type cells. The histogram alongside shows the AUC for each of these clones. For the densitometry and AUC histogram in A-I -Unpaired two-tailed t test was used for the analysis comparing control clones with the edited clones- *p< 0.05; **p< 0.01, ***p< 0.001, ****p<0.0001, ns=non-significant. For D, G and I, PTX dose response curves-One-way ANOVA with Dunnett’s multiple comparison test was used- ****p<0.0001, ns=non-significant. The comparison was made between the control clones and the edited clones. All the experiments in A-I were done in triplicates.
Next, we investigated the effect of SSR3 KO on microtubule polymerization following PTX treatment, as this is the canonical mechanism of action of PTX. As previously reported, PTX led to stabilization of microtubule polymerization, yet no significant difference was seen on the microtubule polymerization in the presence of PTX between control and SSR3 KO cells (Supplementary Fig. S8). Furthermore, overexpression of SSR3 in GBM6 conferred increased proliferation potential while the reduction in proliferation potential was observed upon knockout of SSR3 in H4 and MDA-MB-468 cells (Supplementary Fig. S9A–C). On the similar lines, animals injected with GBM6 overexpressing SSR3 cells (median survival=29 days) reached the endpoint earlier as compared to their vector control group (median 47 days, p<0.0001) in the absence of any treatment (Supplementary Fig. S9D). Concurrently, no correlation was seen between doubling time and susceptibility to PTX in both glioma PDXs and breast cancer cell lines (Supplementary Fig. S9E and F).
Discussion
The exquisitely low concentration of PTX necessary to achieve tumor cell death, makes this agent among the most potent drugs for GBM. Numerous approaches are being pursued including the use of convection-enhanced delivery, biodegradable wafers that release PTX, peptide-drug conjugates, nanoparticle-drug coupling- to enhance the PTX penetration across the BBB, and most recently the use of ultrasound for transient BBB opening (6,8,12). In this context, we are conducting a first-in-human Phase 1–2 clinical trial in recurrent GBM where skull implantable ultrasound for BBB opening is used to deliver PTX across the BBB (NCT04528680).
In mice, we showed that following systemic delivery of ABX, ultrasound-based BBB opening can enhance the brain penetration of PTX by 3–5 fold, achieving parenchymal drug concentrations of approximately 0.3–0.4 μM (6). The concentrations achieved by these means should be interpreted carefully as there are limitations to comparing the bio-distribution and dosing between mice and humans, and there might be differences between susceptibility of GBM to PTX in patients vs that of PTX susceptibility in vitro. Yet, these concentrations serve as an initial parameter to designate glioma cells with a PTX IC50 greater than 0.3–0.4 uM as resistant and those with IC50 lower than this as sensitive to this drug.
Whereas taxanes are highly potent, there is a known variability in susceptibility across tumors (7,36). Indeed, it is estimated that approximately 50% of patients do not experience clinical benefit to taxane-based treatments (37). Our study supports these previous observations as approximately half of the glioma and breast cancer cell lines were susceptible to PTX. Based on this, we estimate that roughly half of the patients enrolled in our clinical trial and other cohorts, will have tumors that are resistant to PTX. For this reason, the overall survival of our Phase 1–2 study and other clinical trials evaluating means of enhancing delivery of PTX across the BBB for treatment of malignant gliomas (8,12), is heavily influenced by the composition of susceptible vs resistant tumors, a variable that we cannot control or measure at this time.
Given the need for personalizing the use of PTX, a predictive biomarker for this drug is being investigated by various groups. As the canonical mechanism of action for PTX is stabilization of microtubule polymerization, some studies have explored genes that relate to microtubule assembly and related cytoskeleton biology. Rouzier et al. and colleagues identified expression of microtubule-associated protein tau in conferring resistance to PTX treatment in breast cancer using U133A chips. They showed that knockdown of tau protein rendered the cells susceptible to PTX (38), However, in the NSABP B28 trial, tau expression correlated with improved outcome independent of whether patients received taxane-containing chemotherapy regimen or not (39). Another study by Swanton et al. and colleagues using a kinome and ceramidome siRNA screen, identified the role of ceramide transport protein, COL4A3BP in conferring resistance to PTX in ovarian cancer setting (40). The PTX resistant gene signature identified from this study was successfully validated in patients with triple-negative breast cancer (41). Whole genome siRNA screen was performed by Whitehurst and colleagues to identify chemosensitizer for PTX i.e. to identify gene targets that specifically reduce cell viability in the presence of sublethal concentrations of PTX. They demonstrated that inhibition of proteasomal subunits chemosensitizes lung cancer cells to PTX (42). In an independent study MCL1 expression was shown to confer resistance to anti-tubulin chemotherapeutics like PTX and vincristine (43). Another group has reported the role of solute carrier transporters in conferring resistance to PTX by mediating efflux of the drug (44). Cyclin G1 levels were determined to be resistant to PTX and ovarian cancer patients with cyclin G1 amplification showing poor post-surgery survival in a taxane treated group (45). Gargini et al. showed that overexpression of tau can confer resistance to taxanes since both tau and taxane compete for the same microtubule binding site (46). A biomarker signature consisting of three gene products-ELF5, SCUBE2 and NFIB, was shown to predict susceptibility to taxane-based neoadjuvant chemotherapy in breast cancer (47). Qualitative transcriptional signature consisting of four gene pairs (FUS>THBS2, GUCY2C>RCVRN, PCSK6>ZNF365, PASK>DNAJB14) was shown to predict recurrence-free survival in high-grade serous ovarian cancer patients treated with platinum-taxane adjuvant chemotherapy (48). Taxol score consisting of combination of six genes (CLIP2, LAX1, PCK2, THOC1, LEPR, GSTZ1) was used to predict the sensitivity to taxol in ovarian cancer patients who underwent taxol chemotherapy (49). Protein and gene expression profiling on pre-treatment biopsies of breast tumors showed α-defensins and MAP2 (a microtubule-associated protein) as tumor markers associated with susceptibility to neoadjuvant taxane–based therapy (50). Several of these efforts identified genes whose expression is associated with resistance to PTX, yet none of these studies has led to a clinically-valid biomarker that predicts susceptibility to PTX treatment, and none of these have been explored in GBM.
The focus of our study was to identify a predictive biomarker for PTX susceptibility that would be causally and correlatively implicated, and that would be applicable for GBM and breast cancer. We used a genome-wide CRISPR KO approach to identify genes that confer susceptibility to PTX. Our approach of combining CRISPR screen with the publicly available databases led to the identification of a biomarker with causal as well as correlative properties.
Our study suggests that SSR3 expression leads to PTX susceptibility by a mechanism that is either independent or downstream of the effect that PTX has on microtubule polymerization. This conclusion is supported by the fact that the microtubule polymerization pattern following PTX treatment was not influenced by SSR3 KO. Whereas microtubule polymerization has been considered the canonical mechanism of action for PTX cytotoxicity, emerging studies are reporting that PTX can elicit deleterious effects by other mechanisms. For example, one study has described the release of apoptosis-inducing cytokine TNFα while other has described phosphorylation of anti-apoptotic Bcl-2 protein to induce PTX mediated cell death (19,51). Our experiments show that the interaction between SSR3 and IRE1α contributes to PTX susceptibility and in the resistant cell line GBM6, where IRE1α levels are already low, further reducing the IRE1α levels exerts minimal difference in susceptibility to PTX.
Our approach has several limitations. We have performed CRISPR KO screen in glioma cells and have harnessed breast cancer dataset to determine correlation between baseline expression of gene and susceptibility to taxane. Although, this approach can lead to a biomarker that is more generalizable to both the cancers it may not be able to capture cancer specific differences. Other potential weaknesses include validation only on publicly available breast cancer databases with limited curation/annotation, where information on previous treatment/s administered, and the small number of samples/subjects tested might not be optimal. More importantly, this analysis is retrospective, and relies on RNA-based expression as opposed to protein levels of SSR3. Given this, our study serves as a hypothesis-generating report, and the predictive property of SSR3 expression for PTX susceptibility will require prospective investigation and validation. Moreover, whereas our over-expression and KO experiments in breast cancer and glioma cell lines show that SSR3 contributes to susceptibility to PTX, the effect is modest, and is clear that there are other biological factors that we did not control, that can also contribute to the susceptibility phenotype. Indeed, a single gene like SSR3 might have a partial contribution towards susceptibility to taxanes and the interaction of multiple putative biomarker candidates may more closely predict and determine susceptibility to taxanes. Although, SSR3 expression appears to be predictive of susceptibility to PTX for both GBM and breast cancer, we have only investigated the mechanism by which SSR3 is modulating PTX susceptibility in gliomas. Important molecular differences between these two cancers warrant disease-specific validation of this biomarker and the mechanisms by which SSR3 modulates this susceptibility.
Identification of susceptible GBM tumors might allow early elucidation of an efficacy signal on our study and other ongoing trials, and eventual refinement on the inclusion criteria to enrich for patients with susceptible tumors in a future trial design. On a broader perspective, the application of personalized medicine for commonly used chemotherapeutic drugs such as taxanes might have a direct impact on the management of a lot of patients that routinely get these drugs, including a large number of patients that do not benefit from this therapy. On the other hand, personalized use of PTX could also open therapeutic possibilities for patients who have tumors that are susceptible to this drug, that currently do not get this treatment as is not part of the standard chemotherapeutic regimen for their cancer diagnosis.
Supplementary Material
Translational Relevance:
Despite the widespread use of paclitaxel for breast cancer, there are no predictive biomarkers to inform which patients will benefit from this therapy. Approximately half of the patients who get taxanes do not enjoy a therapeutic benefit, yet these patients remain exposed to the toxicity associated with these drugs. Moreover, there are scenarios where tumors might be sensitive to paclitaxel, yet this drug is not indicated. A predictive biomarker might allow identification of an efficacy signal and refine patient selection for ongoing paclitaxel-related trials in gliomas. Therefore, to identify predictive biomarkers for susceptibility to PTX, we used an unbiased-CRISPR screen approach to first identify the causally implicated biomarkers. This biomarker list was refined based on correlation with survival in the breast cancer patient cohort treated with PTX. This led to the identification of SSR3 as a putative biomarker for susceptibility to PTX.
Acknowledgments:
This work was funded by 1R01CA245969–01A1 (A.M.S & R.S), funding support from the Lou and Jean Malnati Brain Tumor Institute (A.M.S), philanthropic support from the Moceri Family Foundation and Developmental funds from The Robert H Lurie NCI Cancer Center Support Grant #P30CA060553 (A.M.S.). P50CA221747 SPORE for Translational Approaches to Brain Cancer (M.L.), B.G. was supported by the Higher Education Institutional Excellence Programme (2020–4.1.1.-TKP2020) of the Ministry for Innovation and Technology in Hungary. Imaging work was performed at the Northwestern University Center for Advanced Microscopy generously supported by NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center. Histology services were provided by the Northwestern University Research Histology and Phenotyping Laboratory which is supported by NCI P30-CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center. We sincerely thank our deceased colleague Joshua Robert Kane for his contributions towards performing of experiments and data analyses.
Financial Support:
This work was funded by 5DP5OD021356–05 (A. M. S), 1R01CA245969–01A1 (A.M.S & R.S), P50CA221747 SPORE for Translational Approaches to Brain Cancer (A.M.S. & R.S.), funding support from the Lou and Jean Malnati Brain Tumor Institute (A.M.S), philanthropic support from Dan and Sharon Moceri and Developmental funds from The Robert H Lurie NCI Cancer Center Support Grant #P30CA060553 (A.M.S.).
Footnotes
Declaration of Interest: C.D., R.S., A.S. and A.M.S. have submitted a patent application on the proposed biomarker through Northwestern University.
References
- 1.Gradishar WJ, Tjulandin S, Davidson N, Shaw H, Desai N, Bhar P, et al. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J Clin Oncol 2005;23(31):7794–803 doi 10.1200/JCO.2005.04.937. [DOI] [PubMed] [Google Scholar]
- 2.Markman M, Liu PY, Wilczynski S, Monk B, Copeland LJ, Alvarez RD, et al. Phase III randomized trial of 12 versus 3 months of maintenance paclitaxel in patients with advanced ovarian cancer after complete response to platinum and paclitaxel-based chemotherapy: a Southwest Oncology Group and Gynecologic Oncology Group trial. J Clin Oncol 2003;21(13):2460–5 doi 10.1200/JCO.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 3.Rosell R, Gatzemeier U, Betticher DC, Keppler U, Macha HN, Pirker R, et al. Phase III randomised trial comparing paclitaxel/carboplatin with paclitaxel/cisplatin in patients with advanced non-small-cell lung cancer: a cooperative multinational trial. Ann Oncol 2002;13(10):1539–49 doi 10.1093/annonc/mdf332. [DOI] [PubMed] [Google Scholar]
- 4.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369(18):1691–703 doi 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017;318(23):2306–16 doi 10.1001/jama.2017.18718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang DY, Dmello C, Chen L, Arrieta VA, Gonzalez-Buendia E, Kane JR, et al. Ultrasound-mediated Delivery of Paclitaxel for Glioma: A Comparative Study of Distribution, Toxicity, and Efficacy of Albumin-bound Versus Cremophor Formulations. Clin Cancer Res 2020;26(2):477–86 doi 10.1158/1078-0432.CCR-19-2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chamberlain MC, Kormanik P. Salvage chemotherapy with paclitaxel for recurrent primary brain tumors. J Clin Oncol 1995;13(8):2066–71 doi 10.1200/JCO.1995.13.8.2066. [DOI] [PubMed] [Google Scholar]
- 8.Drappatz J, Brenner A, Wong ET, Eichler A, Schiff D, Groves MD, et al. Phase I study of GRN1005 in recurrent malignant glioma. Clin Cancer Res 2013;19(6):1567–76 doi 10.1158/1078-0432.CCR-12-2481. [DOI] [PubMed] [Google Scholar]
- 9.Gabikian P, Tyler BM, Zhang I, Li KW, Brem H, Walter KA. Radiosensitization of malignant gliomas following intracranial delivery of paclitaxel biodegradable polymer microspheres. J Neurosurg 2014;120(5):1078–85 doi 10.3171/2014.1.JNS13235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koziara JM, Lockman PR, Allen DD, Mumper RJ. Paclitaxel nanoparticles for the potential treatment of brain tumors. J Control Release 2004;99(2):259–69 doi 10.1016/j.jconrel.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 11.Kumthekar P, Tang SC, Brenner AJ, Kesari S, Piccioni DE, Anders C, et al. ANG1005, a Brain-Penetrating Peptide-Drug Conjugate, Shows Activity in Patients with Breast Cancer with Leptomeningeal Carcinomatosis and Recurrent Brain Metastases. Clin Cancer Res 2020;26(12):2789–99 doi 10.1158/1078-0432.CCR-19-3258. [DOI] [PubMed] [Google Scholar]
- 12.Lidar Z, Mardor Y, Jonas T, Pfeffer R, Faibel M, Nass D, et al. Convection-enhanced delivery of paclitaxel for the treatment of recurrent malignant glioma: a phase I/II clinical study. J Neurosurg 2004;100(3):472–9 doi 10.3171/jns.2004.100.3.0472. [DOI] [PubMed] [Google Scholar]
- 13.Joung J, Konermann S, Gootenberg JS, Abudayyeh OO, Platt RJ, Brigham MD, et al. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc 2017;12(4):828–63 doi 10.1038/nprot.2017.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winter J, Schwering M, Pelz O, Rauscher B, Zhan T, Heigwer F, et al. CRISPRAnalyzeR: Interactive analysis, annotation and documentation of pooled CRISPR screens. 2017:109967 doi 10.1101/109967 %J bioRxiv. [Google Scholar]
- 15.Dennis G Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 2003;4(5):P3. [PubMed] [Google Scholar]
- 16.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 2014;11(8):783–4 doi 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012;9(7):671–5 doi 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res 2013;41(Database issue):D955–61 doi 10.1093/nar/gks1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lanni JS, Lowe SW, Licitra EJ, Liu JO, Jacks T. p53-independent apoptosis induced by paclitaxel through an indirect mechanism. Proc Natl Acad Sci U S A 1997;94(18):9679–83 doi 10.1073/pnas.94.18.9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janssen K, Pohlmann S, Janicke RU, Schulze-Osthoff K, Fischer U. Apaf-1 and caspase-9 deficiency prevents apoptosis in a Bax-controlled pathway and promotes clonogenic survival during paclitaxel treatment. Blood 2007;110(10):3662–72 doi 10.1182/blood-2007-02-073213. [DOI] [PubMed] [Google Scholar]
- 21.Mielgo A, Torres VA, Clair K, Barbero S, Stupack DG. Paclitaxel promotes a caspase 8-mediated apoptosis through death effector domain association with microtubules. Oncogene 2009;28(40):3551–62 doi 10.1038/onc.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shimada K, Matsuyoshi S, Nakamura M, Ishida E, Kishi M, Konishi N. Phosphorylation of FADD is critical for sensitivity to anticancer drug-induced apoptosis. Carcinogenesis 2004;25(7):1089–97 doi 10.1093/carcin/bgh130. [DOI] [PubMed] [Google Scholar]
- 23.Goldman MJ, Craft B, Hastie M, Repecka K, McDade F, Kamath A, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol 2020;38(6):675–8 doi 10.1038/s41587-020-0546-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hatzis C, Pusztai L, Valero V, Booser DJ, Esserman L, Lluch A, et al. A genomic predictor of response and survival following taxane-anthracycline chemotherapy for invasive breast cancer. JAMA 2011;305(18):1873–81 doi 10.1001/jama.2011.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Itoh M, Iwamoto T, Matsuoka J, Nogami T, Motoki T, Shien T, et al. Estrogen receptor (ER) mRNA expression and molecular subtype distribution in ER-negative/progesterone receptor-positive breast cancers. Breast Cancer Res Treat 2014;143(2):403–9 doi 10.1007/s10549-013-2763-z. [DOI] [PubMed] [Google Scholar]
- 26.Maire V, Baldeyron C, Richardson M, Tesson B, Vincent-Salomon A, Gravier E, et al. TTK/hMPS1 is an attractive therapeutic target for triple-negative breast cancer. PLoS One 2013;8(5):e63712 doi 10.1371/journal.pone.0063712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maire V, Nemati F, Richardson M, Vincent-Salomon A, Tesson B, Rigaill G, et al. Polo-like kinase 1: a potential therapeutic option in combination with conventional chemotherapy for the management of patients with triple-negative breast cancer. Cancer Res 2013;73(2):813–23 doi 10.1158/0008-5472.CAN-12-2633. [DOI] [PubMed] [Google Scholar]
- 28.Maubant S, Tesson B, Maire V, Ye M, Rigaill G, Gentien D, et al. Transcriptome analysis of Wnt3a-treated triple-negative breast cancer cells. PLoS One 2015;10(4):e0122333 doi 10.1371/journal.pone.0122333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fons RD, Bogert BA, Hegde RS. Substrate-specific function of the translocon-associated protein complex during translocation across the ER membrane. J Cell Biol 2003;160(4):529–39 doi 10.1083/jcb.200210095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gorlich D, Hartmann E, Prehn S, Rapoport TA. A protein of the endoplasmic reticulum involved early in polypeptide translocation. Nature 1992;357(6373):47–52 doi 10.1038/357047a0. [DOI] [PubMed] [Google Scholar]
- 31.Hartmann E, Gorlich D, Kostka S, Otto A, Kraft R, Knespel S, et al. A tetrameric complex of membrane proteins in the endoplasmic reticulum. Eur J Biochem 1993;214(2):375–81 doi 10.1111/j.1432-1033.1993.tb17933.x. [DOI] [PubMed] [Google Scholar]
- 32.Nagasawa K, Higashi T, Hosokawa N, Kaufman RJ, Nagata K. Simultaneous induction of the four subunits of the TRAP complex by ER stress accelerates ER degradation. EMBO Rep 2007;8(5):483–9 doi 10.1038/sj.embor.7400933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Acosta-Alvear D, Karagoz GE, Frohlich F, Li H, Walther TC, Walter P. The unfolded protein response and endoplasmic reticulum protein targeting machineries converge on the stress sensor IRE1. Elife 2018;7 doi 10.7554/eLife.43036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iwawaki T, Akai R, Yamanaka S, Kohno K. Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc Natl Acad Sci U S A 2009;106(39):16657–62 doi 10.1073/pnas.0903775106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamaguchi YL, Tanaka SS, Oshima N, Kiyonari H, Asashima M, Nishinakamura R. Translocon-associated protein subunit Trap-gamma/Ssr3 is required for vascular network formation in the mouse placenta. Dev Dyn 2011;240(2):394–403 doi 10.1002/dvdy.22528. [DOI] [PubMed] [Google Scholar]
- 36.Fetell MR, Grossman SA, Fisher JD, Erlanger B, Rowinsky E, Stockel J, et al. Preirradiation paclitaxel in glioblastoma multiforme: efficacy, pharmacology, and drug interactions. New Approaches to Brain Tumor Therapy Central Nervous System Consortium. J Clin Oncol 1997;15(9):3121–8 doi 10.1200/JCO.1997.15.9.3121. [DOI] [PubMed] [Google Scholar]
- 37.Weaver BA. How Taxol/paclitaxel kills cancer cells. Mol Biol Cell 2014;25(18):2677–81 doi 10.1091/mbc.E14-04-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rouzier R, Rajan R, Wagner P, Hess KR, Gold DL, Stec J, et al. Microtubule-associated protein tau: a marker of paclitaxel sensitivity in breast cancer. Proc Natl Acad Sci U S A 2005;102(23):8315–20 doi 10.1073/pnas.0408974102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pusztai L, Jeong JH, Gong Y, Ross JS, Kim C, Paik S, et al. Evaluation of microtubule-associated protein-Tau expression as a prognostic and predictive marker in the NSABP-B 28 randomized clinical trial. J Clin Oncol 2009;27(26):4287–92 doi 10.1200/JCO.2008.21.6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swanton C, Marani M, Pardo O, Warne PH, Kelly G, Sahai E, et al. Regulators of mitotic arrest and ceramide metabolism are determinants of sensitivity to paclitaxel and other chemotherapeutic drugs. Cancer Cell 2007;11(6):498–512 doi 10.1016/j.ccr.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 41.Juul N, Szallasi Z, Eklund AC, Li Q, Burrell RA, Gerlinger M, et al. Assessment of an RNA interference screen-derived mitotic and ceramide pathway metagene as a predictor of response to neoadjuvant paclitaxel for primary triple-negative breast cancer: a retrospective analysis of five clinical trials. Lancet Oncol 2010;11(4):358–65 doi 10.1016/S1470-2045(10)70018-8. [DOI] [PubMed] [Google Scholar]
- 42.Whitehurst AW, Bodemann BO, Cardenas J, Ferguson D, Girard L, Peyton M, et al. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature 2007;446(7137):815–9 doi 10.1038/nature05697. [DOI] [PubMed] [Google Scholar]
- 43.Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011;471(7336):110–4 doi 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- 44.Njiaju UO, Gamazon ER, Gorsic LK, Delaney SM, Wheeler HE, Im HK, et al. Whole-genome studies identify solute carrier transporters in cellular susceptibility to paclitaxel. Pharmacogenet Genomics 2012;22(7):498–507 doi 10.1097/FPC.0b013e328352f436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Russell P, Hennessy BT, Li J, Carey MS, Bast RC, Freeman T, et al. Cyclin G1 regulates the outcome of taxane-induced mitotic checkpoint arrest. Oncogene 2012;31(19):2450–60 doi 10.1038/onc.2011.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gargini R, Segura-Collar B, Herranz B, Garcia-Escudero V, Romero-Bravo A, Nunez FJ, et al. The IDH-TAU-EGFR triad defines the neovascular landscape of diffuse gliomas. Sci Transl Med 2020;12(527) doi 10.1126/scitranslmed.aax1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kallarackal J, Burger F, Bianco S, Romualdi A, Schad M. A 3-gene biomarker signature to predict response to taxane-based neoadjuvant chemotherapy in breast cancer. PLoS One 2020;15(3):e0230313 doi 10.1371/journal.pone.0230313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Y, Zhang Z, Li T, Li X, Zhang S, Li Y, et al. A Qualitative Transcriptional Signature for Predicting Recurrence Risk for High-Grade Serous Ovarian Cancer Patients Treated With Platinum-Taxane Adjuvant Chemotherapy. Front Oncol 2019;9:1094 doi 10.3389/fonc.2019.01094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hou S, Dai J. Transcriptome-based signature predicts the effect of taxol in serous ovarian cancer. PLoS One 2018;13(3):e0192812 doi 10.1371/journal.pone.0192812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bauer JA, Chakravarthy AB, Rosenbluth JM, Mi D, Seeley EH, De Matos Granja-Ingram N, et al. Identification of markers of taxane sensitivity using proteomic and genomic analyses of breast tumors from patients receiving neoadjuvant paclitaxel and radiation. Clin Cancer Res 2010;16(2):681–90 doi 10.1158/1078-0432.CCR-09-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Husemann LC, Reese A, Radine C, Piekorz RP, Budach W, Sohn D, et al. The microtubule targeting agents eribulin and paclitaxel activate similar signaling pathways and induce cell death predominantly in a caspase-independent manner. Cell Cycle 2020;19(4):464–78 doi 10.1080/15384101.2020.1716144. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data including PTX screen raw guide counts and analysis of PTX screen by DESeq and sgRSEA, that support the findings of this study, are available as part of the main text or as supplementary data.