

Abstract
Epilepsy is one of the most common chronic neurologic diseases, with a prevalence of 1% in the U.S. population. Many people with epilepsy live normal lives, but are at risk of sudden unexpected death in epilepsy (SUDEP). This mysterious co-morbidity of epilepsy causes premature death in 17 to 50% of those with epilepsy. Most SUDEP occurs after a generalized seizure, and patients are typically found in bed in the prone position. Until recently it was thought that SUDEP was due to cardiovascular failure, but patients who died while being monitored in hospital epilepsy units revealed that most SUDEP is due to postictal central apnea. Some cases may occur when seizures invade the amygdala and activate projections to the brainstem. Evidence suggests that the pathophysiology is linked to defects in the serotonin system and central CO2 chemoreception, and that there is considerable overlap with mechanisms thought to be involved in sudden infant death syndrome (SIDS). Future work is needed to identify biomarkers for patients at highest risk, improve ascertainment, develop methods to alert caregivers when SUDEP is imminent, and find effective approaches to prevent these fatal events.
Keywords: SUDEP, breathing, ventilation, apnea, CO2, chemoreception, SIDS, serotonin
1). Introduction
Seizures can have powerful inhibitory effects on breathing, and in some cases this can be fatal. Recent data from epilepsy patients and animal models suggest that seizure-induced central apnea may be a common cause of sudden unexpected death in epilepsy (SUDEP). In this chapter we will discuss data from SUDEP cases monitored at the time of death, from human peri-ictal respiratory physiology during non-fatal seizures, and from animal models. We will then focus on the mechanisms of respiratory dysfunction, including the role that impaired serotonin neuronal function and an abnormal response to hypercapnia may play in the pathophysiology of SUDEP.
2). Epidemiology of epilepsy and SUDEP
Epilepsy affects more than 65 million people worldwide, making it one of the most common chronic neurologic disorders (Thurman et al., 2011). In the United States alone, 3.4 million people have active epilepsy and 150,000 new cases are diagnosed every year (Sirven, 2015). Around 1 in 26 people in the United States will develop epilepsy at some point in their lifetime (England et al., 2012). Although many antiepileptic drugs (AEDs) are available, one third of people with epilepsy – or about 20 million people worldwide – fail to achieve seizure control with pharmacotherapy (Chen et al., 2018). Over time, convulsive seizures can progressively impair cognitive abilities and alter brain structure (Hermann et al., 2002). Moreover, patients with epilepsy, many of whom are young, have a risk of sudden death approximately 27-fold higher than control populations (Holst et al., 2013). The lifetime risk of SUDEP is estimated to be ~17% in patients with epilepsy (Thurman et al., 2014), a number that can drastically increase to 50% in patients with severe uncontrolled epilepsy, making it the leading cause of death in this patient population (Shorvon and Tomson, 2011).
SUDEP is defined as sudden, unexpected, non-traumatic and non-drowning death in a person with epilepsy in whom postmortem examination does not reveal another cause for death (Nashef et al., 2012). In terms of “potential years of life lost,” SUDEP is second only to stroke among neurologic diseases (Thurman et al., 2014), with the highest risk of SUDEP in persons aged 20–40 years. In Denmark during 2007–2009, persons with epilepsy younger than 50 years had a 34-fold increased risk of sudden death compared to the general population (Kløvgaard et al., 2021). In 2017, the American Academy of Neurology (AAN) estimated the overall global incidence of SUDEP to be 0.58/1000 patient-years, with a lower incidence in children compared to adults (0.22 and 1.2/1000 patient-years, respectively) (Harden et al., 2017). However, more recent data from discrete population-based studies suggest that the incidence is comparable across age groups, and in young individuals the incidence of SUDEP is much more common among males (Figure 1) (Sveinsson et al., 2017, Keller et al., 2018). The reported incidence of SUDEP varies depending on the study population and the study setting, and can range from 0.9 – 2.3 per 1000 person-years in the general epilepsy population (Shorvon and Tomson, 2011). However, studies of SUDEP incidence are strongly influenced by ascertainment bias, since it is a diagnosis of exclusion and there is no definitive way to identify a SUDEP case. Many cases of sudden death in epilepsy patients are determined to be of unknown cause rather than being identified as SUDEP (Schraeder et al., 2009, Devinsky et al., 2016).
Figure 1.
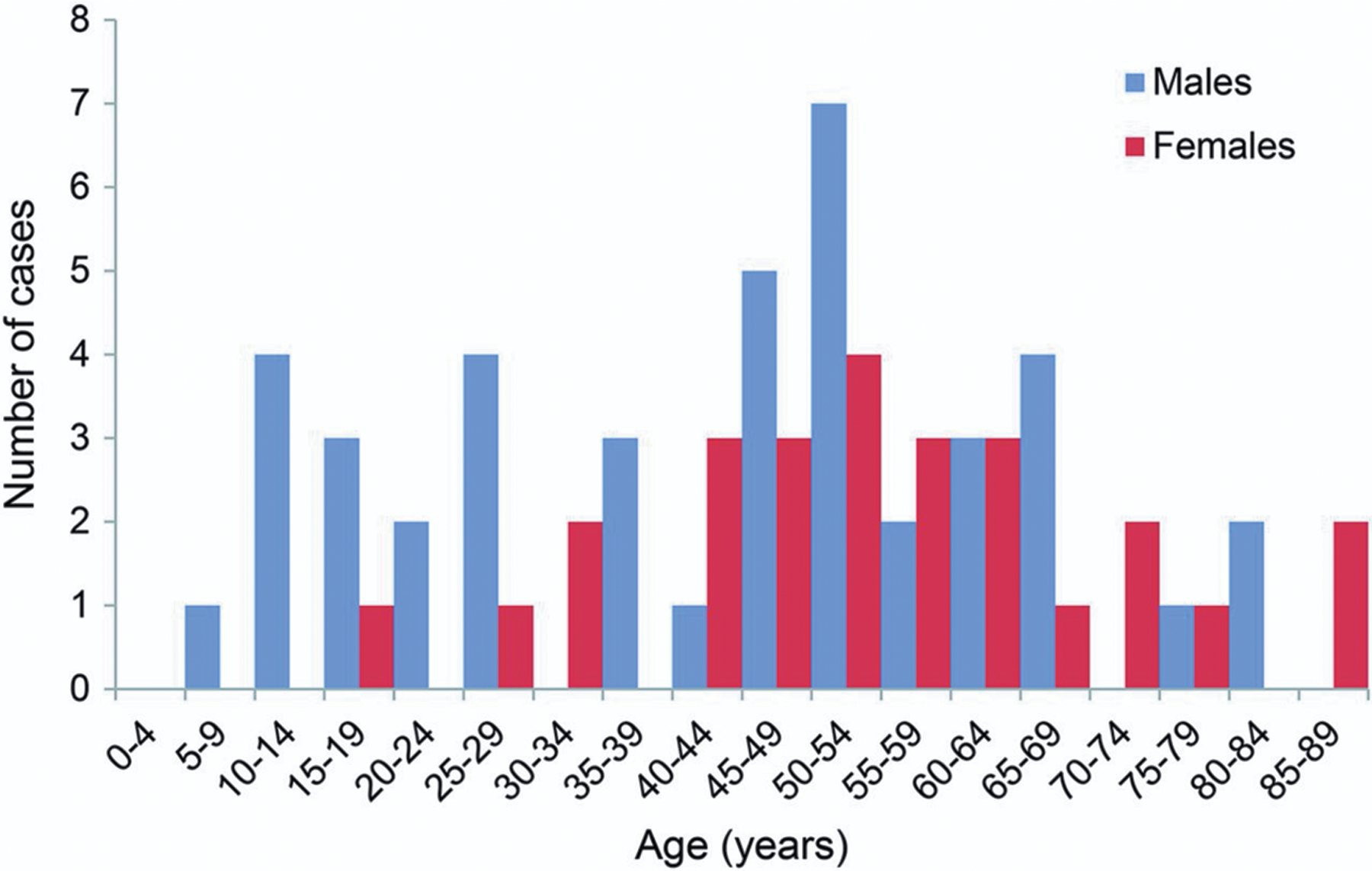
Age and sex distribution of definite and probable sudden unexpected death occurring in epilepsy cases in Sweden in 2008. Reproduced with permission from Sveinsson et al (2017).
3). Clinical risk factors for SUDEP
The unpredictable and often unwitnessed occurrence of SUDEP, which is rarely captured during physiological monitoring, presents a challenge to research studies. Fortunately, case-control studies in recent decades have provided insight into circumstances leading to SUDEP (Hesdorffer et al., 2011, Tomson et al., 2016). This has helped physicians and family members identify high-risk situations. Most SUDEP cases are preceded by a generalized tonic-clonic seizure (GTCS) (Nashef et al., 1998, Ryvlin et al., 2013), and more often occur unsupervised and during nighttime (Hajek and Buchanan, 2016). The presence and frequency of GTCSs is the strongest predictor of risk of SUDEP (Harden et al., 2017). Compared with epilepsy patients with no known GTCS, patients having three or more GTCS per month are 15 times more likely to die from SUDEP (Hesdorffer et al., 2012b). Other factors such as male sex, early age of seizure onset (<16 years), and duration of epilepsy have also been proposed as predictors of risk for SUDEP (Hesdorffer et al., 2012b, Hesdorffer et al., 2011), with male sex being a strong predictor in young patients (Sveinsson et al., 2017). Nocturnal GTCSs in patients sleeping alone represent both the primary risk factor and the most frequent triggering event of SUDEP (Sveinsson et al., 2020). Despite this, there is limited, low-certainty evidence that supervision at night reduces the risk of SUDEP (Maguire et al., 2020).
Genetics
Several genes have been associated with documented cases of SUDEP. Genetic conditions such as Dravet syndrome (DS) harbor mutations that lead to frequent seizures and a high risk of SUDEP. DS, also known as severe myoclonic epilepsy of infancy, typically manifests within the first year of life with febrile seizures that progress to polymorphous seizures that are drug-resistant (Kalume et al., 2013, Dravet, 2011). Within the second to fifth years of life, DS patients develop comorbidities such as cognitive impairment, hyperactivity, ataxia, and autistic-like behaviors. In patients with DS, the risk of SUDEP is estimated to be 15 times higher than in other pediatric epilepsies (Skluzacek et al., 2011, Kearney, 2013). By age 25 years, overall mortality in DS is about 21%, with nearly 50% of deaths due to SUDEP (Shmuely et al., 2016). More than 80% of DS cases are caused by de novo loss-of-function mutations in the SCN1A gene due to premature stop codons, deletions, or inactivating residue mutations (Claes et al., 2001, Dravet, 2011). SCN1A is expressed in both the brain and the heart, and encodes NaV1.1. In the heart, mutations of SCN1A are thought to predispose to fatal arrhythmias. In the cortex and hippocampus, NaV1.1 is more abundant in interneurons, so loss-of-function leads to hyperexcitability (Catterall, 2000).
Another channelopathy is early infantile epileptic encephalopathy type 13 (EIEE13), which is a recently recognized syndrome characterized by intractable seizures and significant developmental delay (Larsen et al., 2015). This severe form of epilepsy is caused by de novo gain-of-function mutations in the SCN8A gene, which encodes NaV1.6. Such mutations prevent complete channel inactivation, and cause a large increase in persistent sodium current, leaving neurons in a hyperexcitable state (Veeramah et al., 2012). NaV1.6 is expressed at highest levels in excitatory neurons of the forebrain, hence a gain-of-function mutation leads to brain hyperexcitability. There is an increased risk of SUDEP in patients with EIEE13 (Veeramah et al., 2012), and ictal cyanosis and respiratory dysfunction are common (Gardella et al., 2016, Johannesen et al., 2018), suggesting that seizure-induced respiratory failure is the underlying mechanism contributing to premature mortality. SCN8A epileptic encephalopathy displays many features seen in patients with DS: infantile-onset seizures that ultimately become refractory to pharmacotherapy and cause epileptic encephalopathy as a result of regression of developmental abilities. However, there are important distinctions: 1) SCN8A patients do not usually manifest prolonged GTCS, myoclonic seizures or febrile seizures in the first year of life; and 2) SCN8A patients typically also present with muscle spasms and movement disorders (Gardella et al., 2018).
Mutations in a number of other genes are also linked to SUDEP in patients, including SCN5A, KCNQ1, KCNH2, NOS1AP, RYR2 and DEPDC5 (Goldman, 2015, Yuskaitis et al., 2018). Although all of these genes are associated with cardiac arrhythmias, they are all also expressed in the brain. Nevertheless, the mechanisms of death in SUDEP in patients with mutations in such genes are unknown, and some are likely to be cardiac.
4). Pathophysiology
The underlying mechanisms of SUDEP remain elusive, but case reports of SUDEP and nearSUDEP in epilepsy monitoring units (EMUs) have refined our understanding of the final events occurring prior to death (Ryvlin et al., 2013). These data underscore the conclusion that in most cases GTCSs trigger the cascade of events leading to SUDEP. A variety of mechanisms may cause or contribute to SUDEP, including cardiac arrhythmias (Auerbach et al., 2013, Catterall et al., 2010, Anderson et al., 2014, Surges et al., 2009), autonomic dysfunction (Surges et al., 2009, Glasscock et al., 2010, Delogu et al., 2011), hypoventilation (Bateman et al., 2008, Massey et al., 2014, Kim et al., 2018, Dlouhy et al., 2015, Vilella et al., 2019b), airway obstruction (Lacuey et al., 2018, Stewart, 2018), brainstem spreading depolarization (BSD) (Aiba and Noebels, 2015), and postictal generalized EEG suppression (PGES) (Lhatoo et al., 2010). However, a growing body of evidence now strongly indicates that a substantial subset of SUDEP is due to seizure-induced respiratory arrest (Ryvlin et al., 2013, Massey et al., 2014).
Seizures and respiratory dysfunction
Until recently, it was widely believed that SUDEP was nearly always due to cardiac mechanisms (Lathers and Schraeder, 1990). This was probably because sudden cardiac death is very common in the general population, and it was assumed that cardiac death could be induced by the stress of a seizure. Despite the challenges of collecting real-time physiologic data from an event that occurs so unpredictably, cases of SUDEP that occurred while patients were being monitored in an EMU and data collected from animal models indicate that SUDEP is due to seizure-induced respiratory dysfunction more often than previously thought, and may represent the majority of cases.
Hughlings Jackson noted over a century ago that his human patients and monkeys turned blue during seizures (Jackson, 1899). Since then, others have reported apnea and oxygen desaturation during and after non-fatal seizures (Figure 2) (Bateman et al., 2008, Vilella et al., 2019a, Watanabe et al., 1982, Nashef et al., 1996). Although ictal and postictal apnea was known to commonly occur, there was no compelling evidence that it was ever fatal until publication of the Mortality in Epilepsy Monitoring Unit Study (MORTEMUS), the most extensive compilation of SUDEP cases that occurred during EMU surveillance (Ryvlin et al., 2013). In all nine patients with adequate cardiorespiratory monitoring, SUDEP occurred after a GTCS, and terminal central apnea preceded terminal asystole, implicating a primary respiratory mechanism of death (Figure 3). However, the mechanisms and triggering factors that lead from seizures to peri-ictal respiratory dysfunction to death remain largely unknown.
Figure 2.
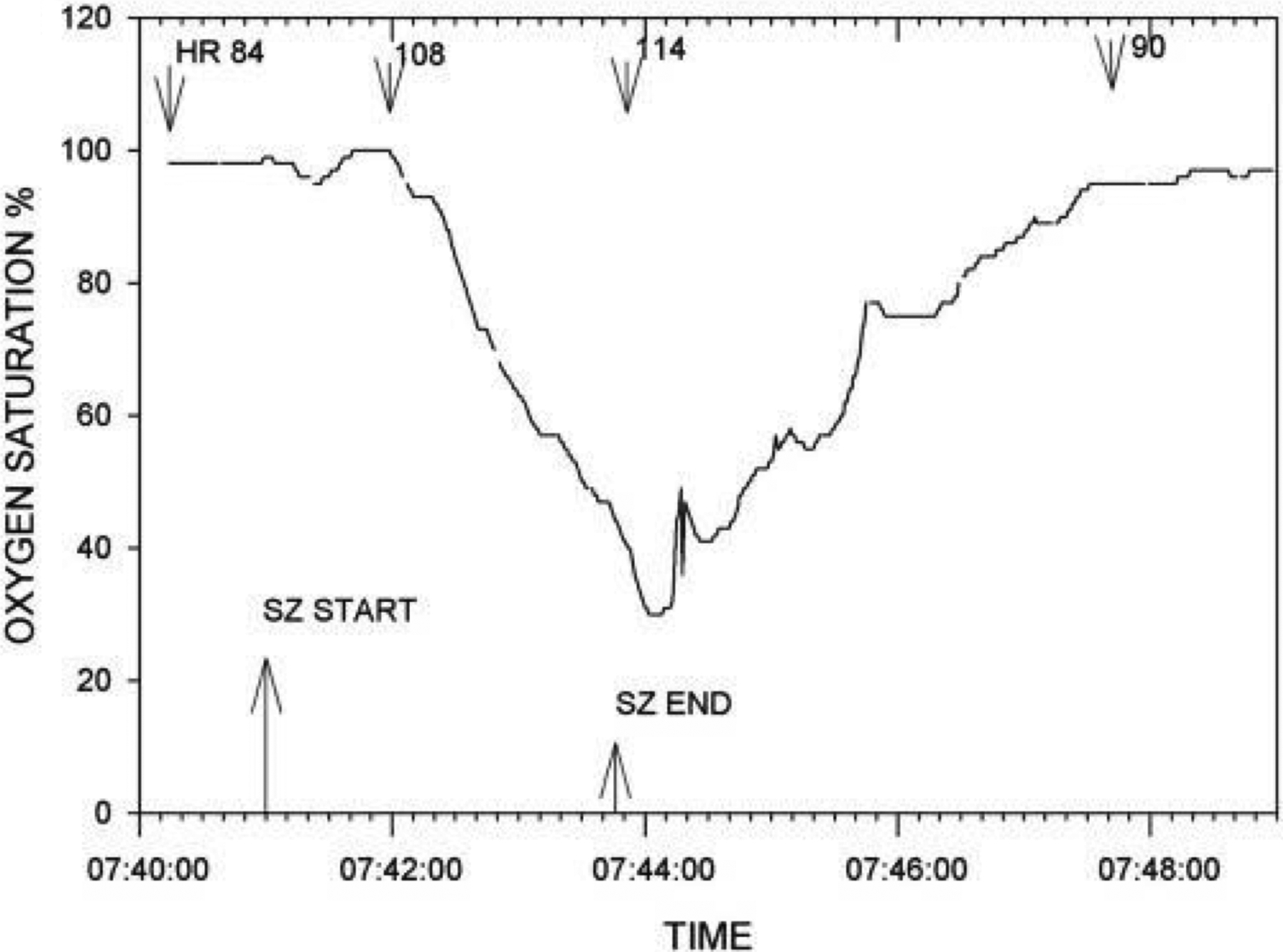
Pronounced oxygen desaturation with a complex partial left temporal onset seizure without secondary generalization. Patient was a 19-year-old male with a body mass index of 19.9. Seizure onset occurred with the patient awake and sitting in bed. He became unresponsive with lip smacking, a slight head turn to the left followed by forceful head turning to the right. He remained sitting for the duration of the seizure. The heart rate (beats per minute.) at various times is shown. Two other complex partial seizures in this patient (one left and one right temporal onset) were accompanied by oxygen desaturations below 50%. Oxygen saturation percent is shown on the ordinate. Reproduced with permission from Bateman et al (2008).
Figure 3.
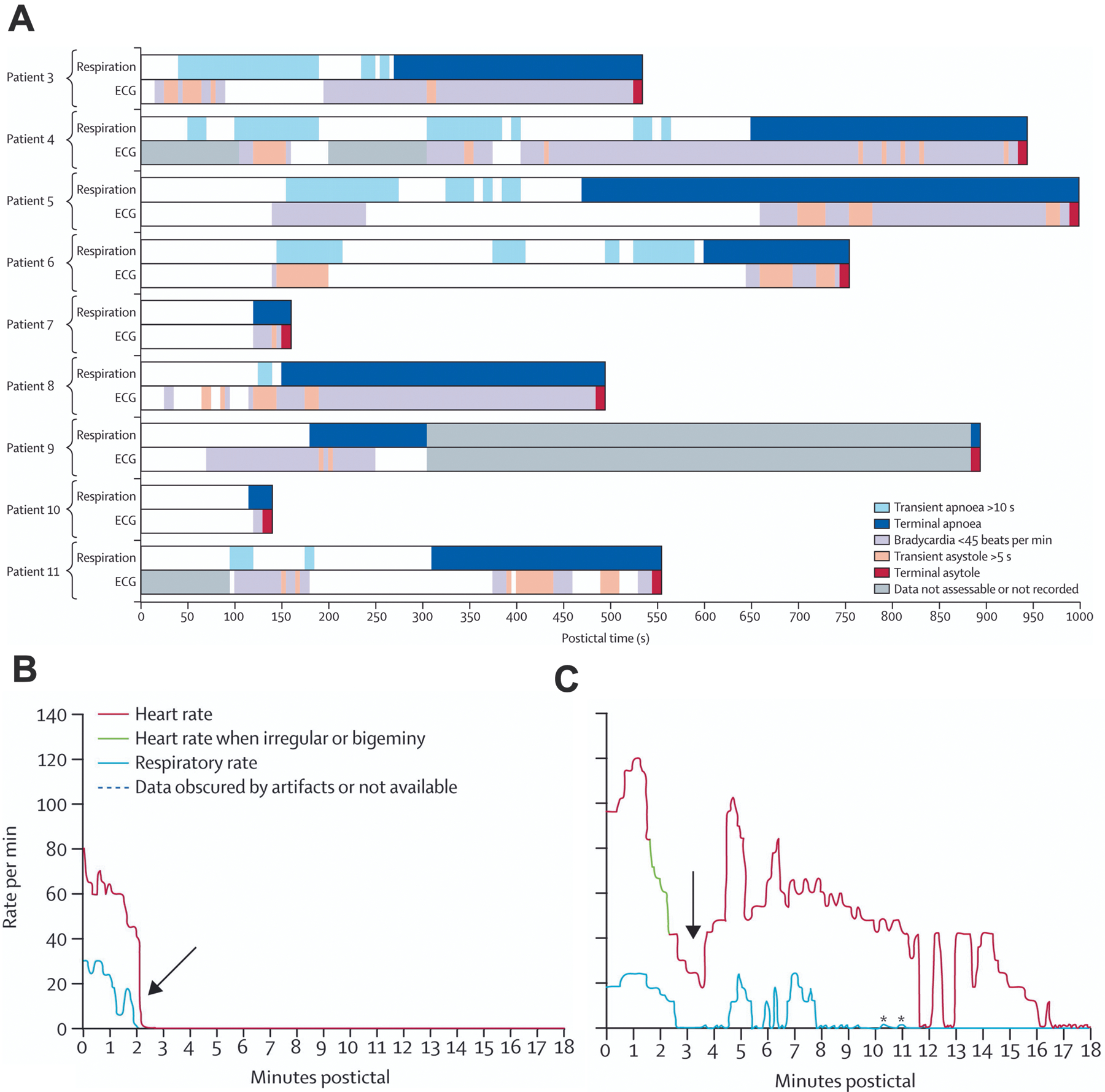
SUDEP cases monitored in epilepsy units at the time of death always occurred after a generalized tonic-clonic seizure and had terminal apnea that preceded terminal asystole. A) Shown are data from nine separate patients with bar graphs colored to show time of breathing patterns plotted above those of cardiac patterns. Time = 0 marks the end of each electrographic seizure. B) Pattern of postictal cardiorespiratory function from an individual patient (Patient 10) with near-simultaneous cessation of respiratory and cardiac activity. C) Postictal cardiorespiratory activity from an individual patient (Patient 5) with severe disruption of respiratory activity 14 minutes before cessation of cardiac activity. Breathing was measured by observation of respiratory movements on video. Breathing could not be determined during seizures due to movement artifact. Reproduced with permission from Ryvlin et al, (2013).
Interest in forebrain effects on respiratory motor output and its implication in seizures has its origins in early reports (Smith, 1938, Kaada and Jasper, 1952). W. G. Spencer first showed that breathing can be slowed and arrested with electrical stimulation of specific brain regions in animals (Spencer and Horsley, 1894). Primary centers for automatic respiratory rhythm generation lie in the lower brainstem, including the pre-Bötzinger complex (pre-BötC) cells of the ventrolateral medulla (Smith et al., 1991) and parafacial nucleus (Onimaru et al., 2009). However, a number of suprapontine regions commonly affected by epilepsy have also been implicated in respiratory control (Kaada and Jasper, 1952, Zelano et al., 2016, Nobis et al., 2018, Dlouhy et al., 2015, Lacuey et al., 2019). Apnea in temporal lobe seizures has been reported to be more common when seizures spread to the contralateral temporal pole (Seyal and Bateman, 2009), but others have not found this to be true (Nadkarni et al., 2012).
The view that many cases of SUDEP are due to respiratory dysfunction is a major shift from previous dogma, which held that cardiovascular dysfunction was the primary cause of SUDEP (Lathers and Schraeder, 1990). It is likely that cardiac mechanisms are the primary cause of death in a subset of SUDEP, but it is not clear how commonly that occurs, because there have been no cases of SUDEP in which breathing and EKG were both recorded and death was documented to be initiated by cardiovascular failure. In other cases, cardiac mechanisms may make death be more likely to occur even though respiratory arrest may be the initiating event.
5). Insights from animal models of SUDEP
Animal models have been instrumental in the study of SUDEP because they readily allow spontaneous death to be captured while recording cardiac and respiratory activity, and in some animal models SUDEP can be induced by various stimuli. Under these conditions the pathophysiological mechanisms of death have been defined for several models of epilepsy.
SCN1A-Related Epilepsy
The development of DS mouse models has proven to be a valuable research tool for understanding the pathophysiology of SUDEP, as they recapitulate many aspects of DS in patients: seizures occur spontaneously and can also be reliably provoked by elevating body temperature, and they have a high incidence of seizure-induced death (Kim et al., 2018, Ogiwara et al., 2007, Kalume et al., 2013). Several studies in heterozygous Scn1a knockout (Scn1a−/+) mice have revealed reduced sodium currents and decreased excitability in GABAergic interneurons (Goff and Goldberg, 2019, Favero et al., 2018, Catterall et al., 2010). These findings have led to the hypothesis that impaired excitability of GABAergic inhibitory neurons is the cause of epilepsy and premature death in DS. This is supported by findings in which selective deletion of NaV1.1 in GABAergic interneurons in the forebrain is sufficient to replicate the severe spontaneous convulsive seizures and premature death observed in DS (Cheah et al., 2012).
NaV1.1 is also expressed in pyramidal neurons, albeit at lower levels (Duflocq et al., 2008, Trimmer and Rhodes, 2004, Westenbroek et al., 1989). Glutamatergic neurons derived from induced pluripotent stem cells from DS patients have increased sodium currents (Jiao et al., 2013, Liu et al., 2013). This was also observed in acutely dissociated hippocampal pyramidal neurons from Scn1a−/+ mice (Mistry et al., 2014). The increased sodium current and hyperexcitability seen in Scn1a−/+pyramidal neurons appear to be agedependent. Multiple studies have reported greater spontaneous firing frequencies and excitability to injected current in dissociated pyramidal neurons and in hippocampal and prefrontal cortex slices from postnatal day (P)21-P25 Scn1a−/+ mice (Mistry et al., 2014, Han et al., 2012), presumably due to compensation for loss of NaV1.1. This age correlates with the onset of seizures and accelerated mortality (Kalume et al., 2013). Increased sodium current density in pyramidal neurons combined with reduced sodium current in GABAergic interneurons likely results in a significant inhibitory-excitatory imbalance, promoting a hyperexcitable state and frequent convulsive seizures.
Another DS model is a knock-in mouse with a heterozygous (Scn1aR1407X/+) loss-of-function mutation seen in some DS patients that results in a truncated NaV1.1 protein (Ogiwara et al., 2007). Similar to Scn1a−/− mice, homozygous (Scn1aR1407X/R1407X) mice show recurrent seizures in the second postnatal week and invariably die before P20, whereas heterozygotes (Scn1aR1407X/+) develop recurrent seizures at P18 and a high rate of sudden death between P21–25. Importantly, a study in Scn1aR1407X/+ mice demonstrated that seizure-induced respiratory arrest (S-IRA) invariably precedes terminal asystole and SUDEP (Figure 4), indicating that the primary cause of death is central apnea (Kim et al., 2018).
Figure 4.
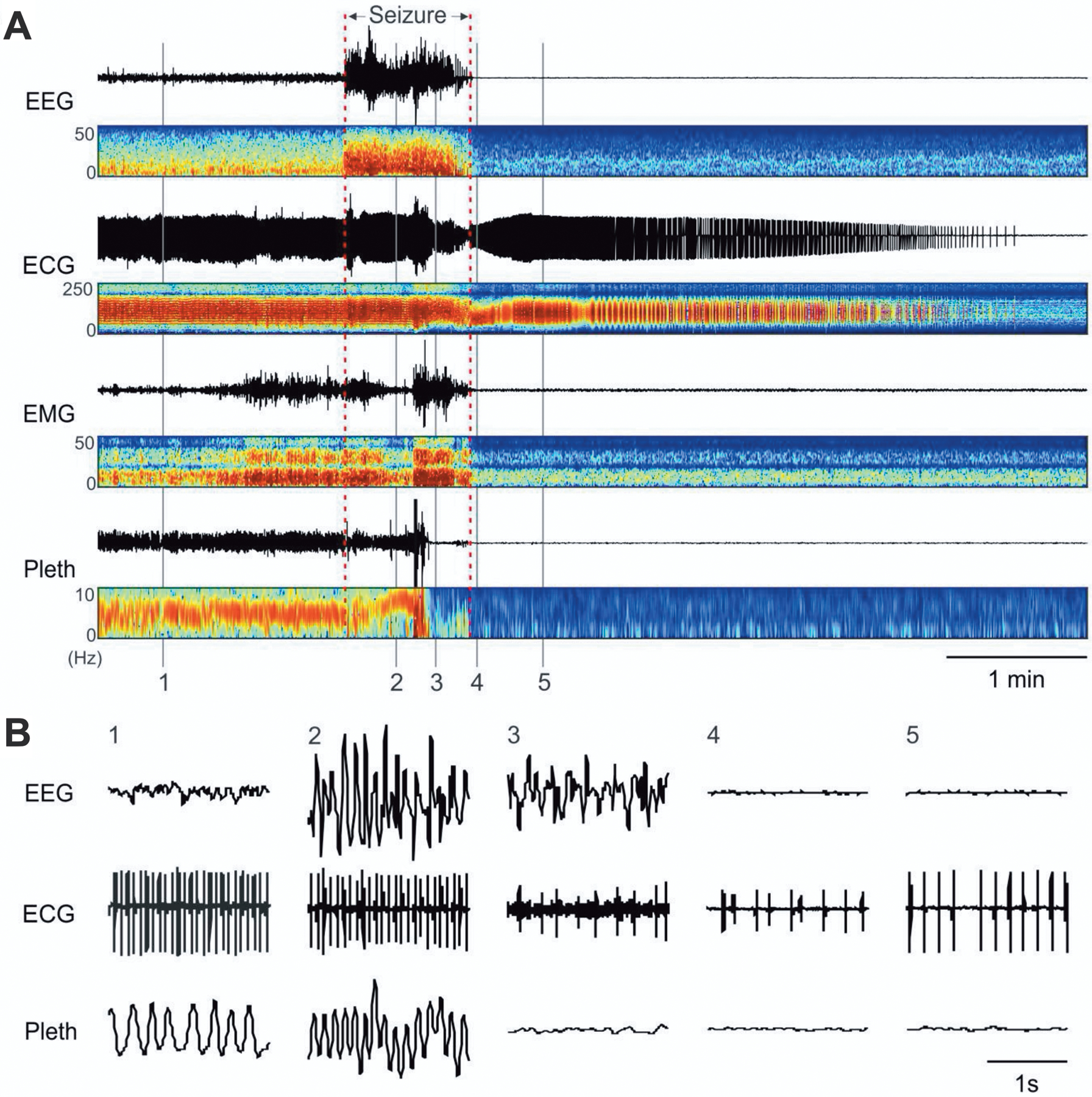
Death after spontaneous seizures in Scn1aR1407X/+ mice is initiated by respiratory arrest. A) Events leading to death in an Scn1aR1407X/+ mouse. Traces from top down are EEG, ECG, EMG, and whole-animal plethysmography (Pleth), in each case with paired power spectrum heatmaps shown below. A spontaneous seizure occurred at the time indicated, and this was followed by electrocerebral silence in the EEG. Shortly after the onset of the seizure, breathing became disrupted. Respiratory activity ceased halfway through the seizure. In contrast, the ECG did not change until near the end of the seizure, after which the frequency and amplitude slowly decreased over the next 4 minutes. B) Expanded traces of EEG, ECG, and plethysmography at the times labeled in (A) as 1–5. Reproduced with permission from Kim et al (2018).
SCN8A-Related Epilepsy
Mutations in the SCN8A gene cause a spectrum of neurologic conditions, including epilepsy, developmental delay, autism spectrum disorder, intellectual disability and ataxia (Hammer et al., 1993). The severity of epilepsy in children with SCN8A mutations is highly variable. Some pathogenic mutations can result in developmental and epileptic encephalopathy (DEE), a severe form of epilepsy characterized both by seizures, which are often drug-resistant, as well as significant developmental delay or regression of developmental abilities (Larsen et al., 2015). Early infantile epileptic encephalopathy type 13 (EIEE13) is one such syndrome that was recently recognized. This SCN8A epileptic encephalopathy is characterized by developmental delay, seizure onset within the first 18 months of life (mean 4 months), and intractable epilepsy. The prevalence of SCN8A-related epilepsy with encephalopathy is not known. To date, approximately 50 cases have been documented in the literature, of which SUDEP has been reported in about 10–12% of published cases (Hammer et al., 1993).
SCN8A epileptic encephalopathy is caused by de novo, gain-of-function mutations in the gene that encodes the α subunit of the voltage-gated sodium channel NaV1.6, which is widely expressed in the brain. NaV1.6 is located at the axon initial segment and nodes of Ranvier of myelinated axons, and provides the molecular basis for initiation and propagation of neuronal action potentials in most neuronal populations (Tzoumaka et al., 2000, Trudeau et al., 2006, Gardella et al., 2016). The first known mutation in humans was discovered in 2012, where the genome of a child demonstrating epileptic encephalopathy was sequenced and revealed a de novo missense mutation, p.Asn1768Asp. This mutation increases channel function by increasing the duration of the persistent sodium current and preventing complete inactivation following hyperpolarization, leaving neurons in a hyperexcitable state (Veeramah et al., 2012).
Several mouse models harboring SCN8A mutations identified in patients with epileptic encephalopathy have been generated. Scn8aN1768D/+ (D/+) mice have a germline knock-in mutation that is expressed globally (Wagnon et al., 2016), whereas Scn8aR1872W/+ (W/+Emx-Cre) mice have a conditional knock-in mutation expressed primarily in forebrain glutamatergic neurons (Bunton-Stasyshyn et al., 2019). Phenotypically, expression of either mutation recapitulates the clinical features seen in patients with SCN8A epileptic encephalopathy, including frequent spontaneous tonic-clonic seizures and seizure-induced death (Bunton-Stasyshyn et al., 2019, Ottolini et al., 2017, Wagnon et al., 2015). W/+Emx-Cre mice die from central apnea after spontaneous convulsive seizures (Wenker et al., 2021).
Other animal models
Several other mouse models have been proposed as tools to study the pathophysiology of SUDEP. One such model is DBA/2 mice, as these mice often die from respiratory arrest following audiogenic seizures (Venit et al., 2004, Marincovich et al., 2019, Tupal and Faingold, 2006). However, the usefulness of the DBA/2 mouse as a model of SUDEP is temporally limited, as consistent susceptibility to S-IRA in these animals lasts approximately one week. DBA/1 mice, another closely-related strain, are also susceptible to fatal audiogenic seizures, as first reported more than 50 years ago (Huff and Fuller, 1964). Faingold and colleagues later confirmed that audiogenic seizures in DBA/1 mice culminate in respiratory arrest (Faingold et al., 2010). Importantly, they showed that once DBA/1 mice exhibit susceptibility to S-IRA when tested between P21–30, susceptibility lasts up to at least P100, which is the oldest age DBA/1 mice were tested. Although DBA/1 and DBA/2 mice do not meet the criteria for SUDEP, because they do not have spontaneous seizures, they are still useful models to study the disruptive effects of seizures on breathing, as death from S-IRA can be prevented with mechanical ventilation allowing multiple trials in individual mice.
Another model is homozygous knockout Kcna1-null (KV1.1−/−) mice, in which the α subunit of the delayed rectifier potassium channel is deleted. KV1.1 subunits are widely expressed in the brain, where they regulate action potential propagation and neurotransmitter release (Wang et al., 1994). Kcna1-null mice develop spontaneous seizures including myoclonic seizures and GTCS by their third week of life (Smart et al., 1998, Chun et al., 2018, Wenzel et al., 2007) and progress in frequency until seizure-induced death occurs between postnatal weeks 5 and 7 (Simeone et al., 2018, Simeone et al., 2016). Of note, mutations in KCNA1 are primarily associated with episodic ataxia in human patients, but partial epilepsy has also been reported in a few cases (Eunson et al., 2000, Zuberi et al., 1999, Paulhus et al., 2020).
6). Hypotheses of postictal respiratory depression in SUDEP
There are no data available from patients that provide direct evidence for the mechanisms by which seizures cause fatal respiratory arrest. Therefore, current hypotheses for how this occurs derive from human data from nonfatal seizures, and from animal models. These hypotheses must explain how seizures in the forebrain propagate to the brainstem, what part of the respiratory network forebrain projections target, and the mechanisms by which descending projections disrupt respiratory output. There are a variety of plausible explanations for these mechanisms. For example, it has been proposed that laryngospasm or the laryngeal chemoreflex can lead to airway obstruction after seizures (Mandal et al., 2021, Stewart, 2018). This might explain a subset of SUDEP cases, although data from the MORTEMUS study and from mouse models indicate that most cases of postictal death are due to central rather than obstructive apnea (Kim et al., 2018, Ryvlin et al., 2013, Wenker et al., 2021). Similarly, during the tonic phase of convulsive seizures in Scn8a mutant mice, apnea may be worsened by the inability to contract the already fully contracted diaphragm (Wenker et al., 2021). This might contribute to some deaths, but would only partially explain apnea since central apnea occurs in some of these mice prior to tonic activation of the diaphragm, and they do not recover breathing after the end of relatively short episodes of tonic activity (Wenker et al., 2021). These observations suggest that the brainstem becomes incapable of generating normal respiratory activity during and/or after seizures.
There are other possible targets of descending activity that, if inhibited, could make the brainstem incapable of respiratory output. These include neurons that generate rhythmic respiratory output, such as those in the pre-Bötz (Smith et al., 1991), or neurons that provide modulatory input to the respiratory network such as in the parabrachial nucleus. Apnea might also result if seizures inactivated neurons that detect changes in hypoxia and/or hypercapnia (chemoreceptors), effectively shutting down chemoreceptor drive for breathing. The main chemoreceptors for hypoxia are in the carotid body and enter the brain in the nucleus tractus solitarius. Chemoreceptors for CO2 have been proposed to be located in the raphe nuclei (Richerson, 2004, Teran et al., 2014), retrotrapezoid nucleus (Guyenet et al., 2019), locus coeruleus (Pineda and Aghajanian, 1997), nucleus tractus solitarius (Dean et al., 1990) and other brainstem sites. It is possible that seizures inhibit activity at one or more of these sites.
It is also not known how descending projections inhibit respiratory output. An obvious explanation is that GABAergic input inhibits the respiratory network. Alternatively, it has been proposed that adenosine is released locally in the medulla, causing the respiratory network to become unstable (Purnell et al., 2021). Finally, it has been found that spreading depolarization can occur after seizures in some mouse models, and it has been proposed that this prevents normal function of the respiratory network (Aiba and Noebels, 2015).
Each of the possibilities described above has its proponents, but none of them are uniformly supported. It is possible that more than one of them are involved in combination. In the remainder of this chapter, we will focus on a working hypothesis for which there is considerable evidence, but for which some components remain speculative, namely that seizures project to the medulla via the amygdala, and cause a defect in CO2 chemoreception by 5-HT neurons. The resulting loss of tonic chemoreceptor drive leads to central apnea. This hypothesis is not necessarily exclusive of other potential mechanisms, and in fact other mechanisms (release of adenosine, BSD and inhibition of the pre-Bötz) may act in combination.
7). The 5-HT system and SUDEP
The existing clinical and experimental evidence indicates that S-IRA is the primary cause of death in most cases of SUDEP. The cellular and network basis of this dysfunction has not been defined, but serotonin dysfunction is a probable contributing factor in part due to its role in control of breathing.
The 5-HT system and respiratory control
Serotonin (5-hydroxytryptamine, 5-HT) is a monoamine neurotransmitter first identified in mammalian brains in 1953 (Twarog and Page, 1953) that plays a role in many physiologic processes. Although 5-HT neurons represent less than 1% of all neurons in the brain, they exhibit a wide array of effects mediated by seven classes of receptors. Cell bodies of 5-HT neurons are located in the brainstem raphe nuclei and can be divided into a caudal group in the medulla and a rostral group in the midbrain (Jacobs and Azmitia, 1992, Jensen et al., 2008). 5-HT neurons in the medulla are an important subset of central respiratory chemoreceptors that detect changes in blood CO2 via changes in pH (Richerson, 2004, Wu et al., 2019, Richerson, 1995, Wang et al., 2001, Teran et al., 2014). A subset of medullary 5-HT neurons is located immediately adjacent to the basilar artery and its large midline branches, where they can reliably monitor arterial blood CO2 levels (Figure 5A) (Bradley et al., 2002). Studies in unanesthetized cats in vivo have shown that around 20% of 5-HT neurons in the raphe obscurus and dorsal raphe increase their firing rate in response to hypercapnia (Veasey et al., 1997, Veasey et al., 1995). Mature 5-HT neurons are highly responsive to acidosis, and when they are isolated from synaptic input they increase their firing rate by ~300% in response to a decrease in pH from 7.4 to 7.2 (Wang et al., 2002, Wang et al., 2001, Wang et al., 1998). Hypercapnia has been shown to result in release of 5-HT using in vivo microdialysis in unanesthetized mice (Kanamaru and Homma, 2007). 5-HT neurons in the medullary raphe project to all major respiratory nuclei, including the nucleus ambiguus, nucleus tractus solitarius (NTS), phrenic motor nucleus, hypoglossal motor nucleus (XII), pre-Bötz and retrotrapezoid nucleus (Figure 5B) (Jacobs and Azmitia, 1992) where they enhance chemosensitivity of other chemoreceptor neurons (Wu et al., 2019) and stimulate respiratory output (Teran et al., 2014, Ptak et al., 2009), increasing breathing to keep blood PCO2 within the normal physiologic range (Richerson, 2004, Teran et al., 2014). Optogenetic stimulation of medullary 5-HT neurons in vivo induces an increase in ventilation in rats (Depuy et al., 2011). A separate subset of 5-HT neurons in the midbrain are also chemosensitive to increased blood PCO2 and are located close to large arteries (Severson et al., 2003). These neurons stimulate cortical arousal (Buchanan and Richerson, 2010, Smith et al., 2018), which is also a crucial component of the protective response to hypercapnia.
Figure 5.
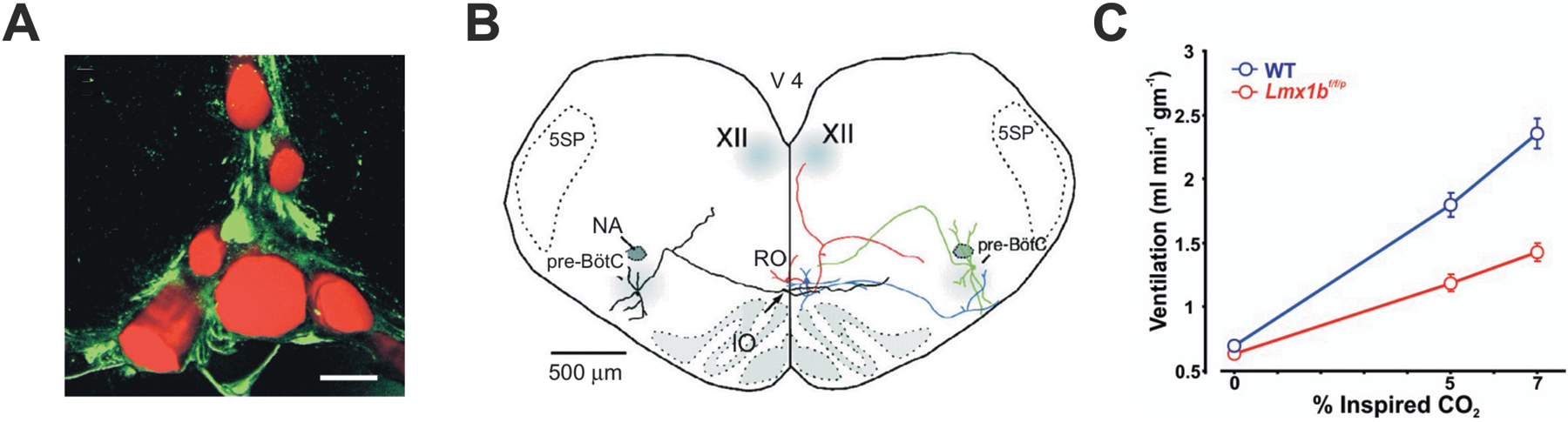
The 5-HT system is involved in the ventilatory response to hypercapnia. A) Medullary raphe neurons are located close to the basilar artery and its large penetrating branches. Shown is a transverse section of the midline rostral medulla (dorsal to the basilar artery). 5-HT neurons in the raphe are immunostained with an antibody against tryptophan hydroxylase (green). Blood vessels are filled with fluorescently-tagged albumin (red). Scale bar: 50 μm. Reproduced with permission from Fiske (2002). B). 5-HT neurons project to neurons in the major respiratory nuclei. Reconstruction of two biocytin-filled raphe obscurus 5-HT neurons (red, blue) and two pre-Bötzinger complex neurons (green, black) in a brain slice illustrating dendrites and axonal projections. Reproduced from Ptak et al (2009). C) Awake mice with genetic deletion of 5-HT neurons (Lmx1bf/f/p) have a smaller ventilatory response to an increase in ambient CO2 compared to WT littermates. Adapted from Hodges et al (2008).
The generation of Lmx1bf/f/p mice, in which more than 99% of central 5-HT neurons are deleted embryonically, has facilitated the study of the serotonergic system (Zhao et al., 2006). These mice exhibit prolonged apneas and high mortality in the neonatal period (Hodges et al., 2009), and those that survive display an attenuated ventilatory response to hypercapnia as adults (Figure 5C) (Hodges et al., 2008). Moreover, Lmx1bf/f/p mice fail to arouse from sleep in response to high levels of CO2 (Buchanan and Richerson, 2010). Hypercapnic arousal is a critical response that assists resumption of breathing after a seizure, especially one that occurs during sleep.
5-HT and seizures
A relationship between 5-HT and seizure inhibition was first proposed in 1957 by Bonnycastle et al. (1957) based on the observation that many AEDs increase the concentration of brain 5-HT in rodents. For example, sodium valproate increases 5-HT levels in rats (Baf et al., 1994) and in audiogenic seizure-prone mice (Maciejak et al., 2014, Vriend and Alexiuk, 1996). Similarly, lamotrigine, carbamazepine, phenytoin and zonisamide all elevate basal 5-HT levels and/or 5-HT release as part of their anticonvulsant action in rodents (Ahmad et al., 2005, Dailey et al., 1997, Dailey et al., 1996, Okada et al., 1992). Subsequent studies have demonstrated that other agents that increase extracellular 5-HT such as the precursor 5-hydroxytryptophan (5-HTP) (Truscott, 1975) or selective serotonin reuptake inhibitors (SSRIs) such as fluoxetine inhibit both focal and generalized seizures in rodents (Pasini et al., 1992, Hernandez et al., 2002, Wada et al., 1995) and in humans (Albano et al., 2006). In contrast, decreasing 5-HT appears to increase seizure susceptibility. Woolley first noted in 1954 that injecting a serotonin antagonist into mice ‘calls forth convulsions much resembling those in human epilepsy’ (Shaw and Woolley, 1954). Similarly, depletion of 5-HT following administration of parachlorophenylalanine (PCPA) to block tryptophan hydroxylase, the rate-limiting enzyme in 5-HT synthesis, increases seizure susceptibility in mice (Buchanan et al., 2014). Lmx1bf/f/p mice, which lack central 5-HT neurons, also have a lower seizure threshold and more convulsions than littermate controls when subjected to maximal electroshock or pilocarpine-induced seizures (Buchanan et al., 2014).
Seizures may also influence 5-HT metabolism. Lower levels of the 5-HT metabolite 5-hydroxyindoleacetic acid (5-HIAA) have been reported in the cerebrospinal fluid (CSF) of pediatric epilepsy patients (Shaywitz et al., 1975, Giroud et al., 1990) and adults with progressive myoclonic epilepsy (Pranzatelli et al., 1995). Pediatric epilepsy patients also exhibit lower plasma and CSF concentrations of L-tryptophan, the amino acid precursor to 5-HT (Marion et al., 1985, Ko et al., 1993). Interestingly, increased serum 5-HT levels relative to baseline have been noted immediately after seizures in patients, and the magnitude of 5-HT increase negatively correlates with the duration of the tonic seizure phase, suggesting an association between more severe seizures and lower 5-HT in the postictal state (Murugesan et al., 2018).
Studying the link between epilepsy and certain comorbidities such as depression has the potential to inform our understanding of the underlying pathology. Depression and epilepsy are thought to be related based on the increased risk of occurrence and severity of one in the presence of the other (Kanner, 2011, Hesdorffer et al., 2012a, Gilliam et al., 2004). A history of depression is associated with an increased risk for developing epilepsy (Hesdorffer et al., 2012a, Hesdorffer et al., 2000, Hesdorffer et al., 2007), which suggests that depression and epilepsy may share a common pathophysiology involving serotonergic dysfunction, rather than depression resulting from a reaction to carrying the diagnosis of epilepsy. Drevets and colleagues first identified consistent widespread reduction in 5-HT1A receptor binding in the raphe and mesial temporal cortex of depressed patients (Drevets et al., 1999). Additional neuroimaging studies in epilepsy patients with depression have reported similar abnormalities in limbic regions, such as the hippocampus, insula, and cingulate gyrus (Savic et al., 2004, Merlet et al., 2004, Toczek et al., 2003, Theodore et al., 2007, Giovacchini et al., 2005, Lothe et al., 2008), further supporting involvement of 5-HT system dysfunction. In placebo-controlled studies of psychopharmacologic drugs to treat depression, patients treated with a 5-HT-modulating agent had a 50% reduction in the occurrence of seizures, whereas those on placebo showed a 19-fold increased risk for developing seizures compared to the general population (Alper et al., 2007). Collectively, the existing clinical and experimental evidence supports that 5-HT helps maintain a higher seizure threshold and defects in the 5-HT system can increase susceptibility to seizures.
Evidence of 5-HT mechanisms in SUDEP
Although the definitive causes of SUDEP are not known, serotonergic dysfunction may be involved (Massey et al., 2014, Petrucci et al., 2020). The first evidence that 5-HT defects can increase the risk of SUDEP came from mice in which the 5-HT2C receptor was genetically deleted. These mice are prone to spontaneous GTCS that are followed by respiratory arrest and death (Tecott et al., 1995). Similarly, Lmx1bf/f/p mice show a higher incidence of death after seizures induced by maximal electroshock or pilocarpine. DBA/1 and DBA/2 mice have aberrant 5-HT receptor expression in some brainstem regions (Faingold et al., 2011a, Uteshev et al., 2010, Feng and Faingold, 2017). S-IRA in DBA/1 and DBA/2 mice can be prevented with immediate mechanical ventilation (Venit et al., 2004). Interestingly, 5-HT augmentation via pretreatment with an SSRI (Zeng et al., 2015, Faingold et al., 2011b, Tupal and Faingold, 2006), fenfluramine (Tupal and Faingold, 2019), or optogenetic activation of 5-HT neurons in the dorsal raphe (Zhang et al., 2018) can also prevent S-IRA. These findings are translatable to humans, as SSRIs have been reported to decrease the likelihood of ictal hypoxemia in patients with refractory epilepsy (Bateman et al., 2008). Several studies have evaluated the impact of SSRIs on seizure frequency in patients with epilepsy and found them to usually be effective (Richerson et al., 2016, Albano et al., 2006, Favale et al., 2003, Favale et al., 1995). Moreover, data from recent randomized-controlled trials investigating the effect of fenfluramine as an adjunctive therapy in children with DS showed a significant decrease in convulsive seizure frequency compared with placebo (Nabbout et al., 2020, Lagae et al., 2019). Nevertheless, it remains unknown whether SSRIs or other serotonergic agents can prevent seizure-induced respiratory depression in epilepsy patients, and if there is a decrease in the incidence of SUDEP in patients on these medications.
Activation by seizures of projections to brainstem regions responsible for respiratory output could explain breathing disruption and may underlie seizure-induced respiratory failure. For example, suppression of breathing during and after seizures in rats is associated with suppressed activity of medullary 5-HT neurons (Zhan et al., 2016) (Figure 6). It is possible that increasing 5-HT signaling could prevent respiratory dysfunction during and after seizures, as greater seizure-induced increases in serum 5-HT levels are associated with a lower incidence of ictal and postictal apnea in epilepsy patients (Murugesan et al., 2019) (Figure 7). Since peripheral (serum) 5-HT does not readily cross the blood-brain barrier (BBB), an increase in 5-HT levels in the serum does not normally cause an increase in 5-HT in the brain parenchyma. However, BBB permeability increases during intense seizure activity (van Vliet et al., 2015) and BBB dysfunction as a consequence of seizures has been reported (Ruber et al., 2018). The degree of this BBB breakdown and the extent to which it allows exchange of 5-HT between the peripheral circulation and the CNS remain unclear, but if breakdown does occur the increase in serum 5-HT would add to the stimulatory effects of synaptically-released 5-HT on respiration and arousal. Alternatively, if BBB breakdown does not occur, factors influencing brain 5-HT (metabolism, 5-HT transporter function, etc) may be paralleled by similar factors operative in the periphery, and blood levels may act as a biomarker of brain levels rather than directly altering them.
Figure 6.
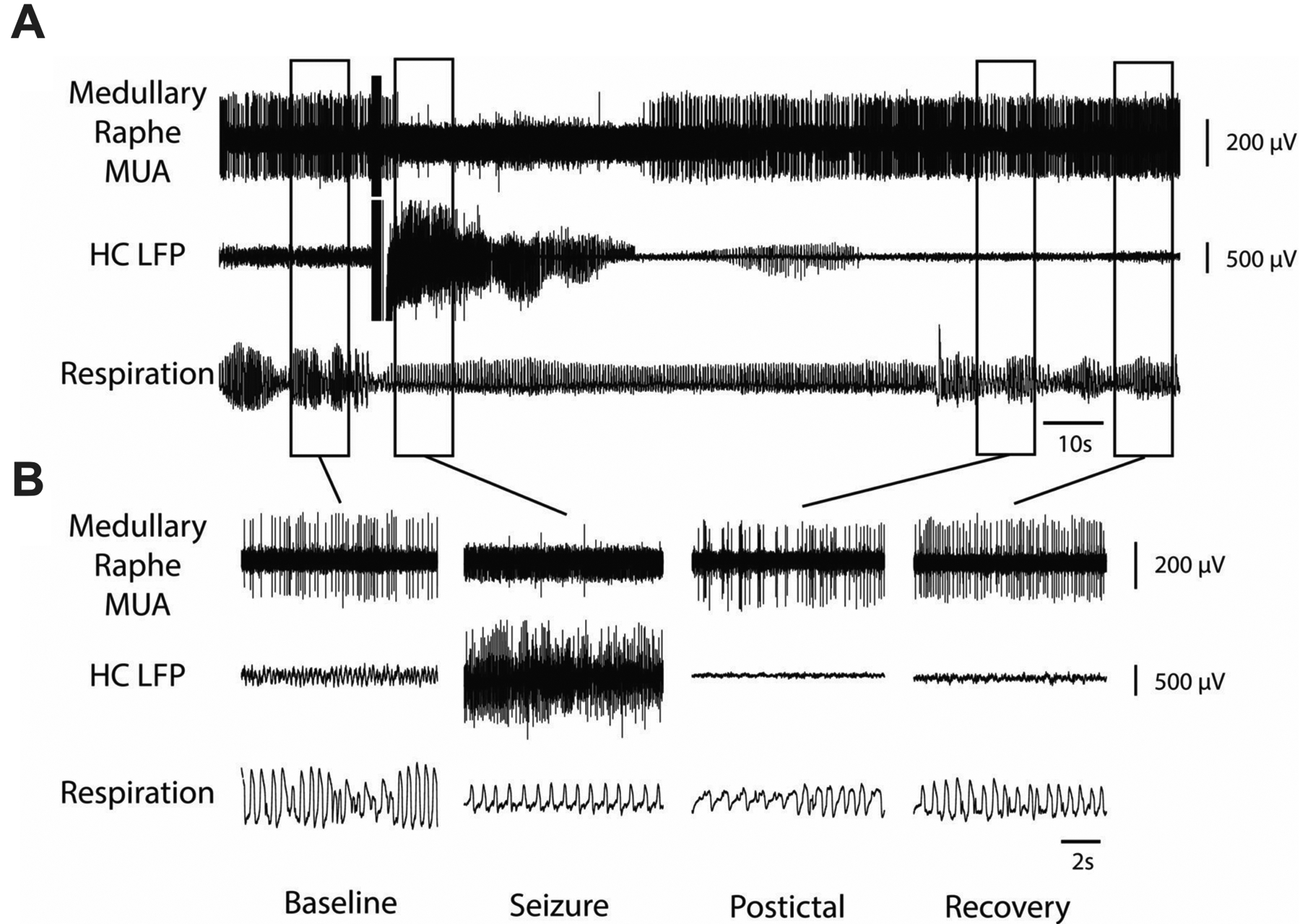
Decreased multiunit activity (MUA) in medullary raphe and reduced breathing during a seizure. A) Seizure induced by 2 s stimulation of hippocampus (HC). After the stimulus, fast polyspike activity is seen in the HC local field potential (LFP). MUA from the medullary raphe shows marked suppression of neuronal firing during the seizure with slow recovery in the post-ictal period. Respiratory trace shows airflow amplitude (proportional to volume) and respiratory rate both decreased markedly during the ictal and post-ictal periods. B) Expanded segments of data from baseline, seizure, post-ictal, and recovery periods from the boxed regions in (A). Reproduced from Zhan et al (2016).
Figure 7.
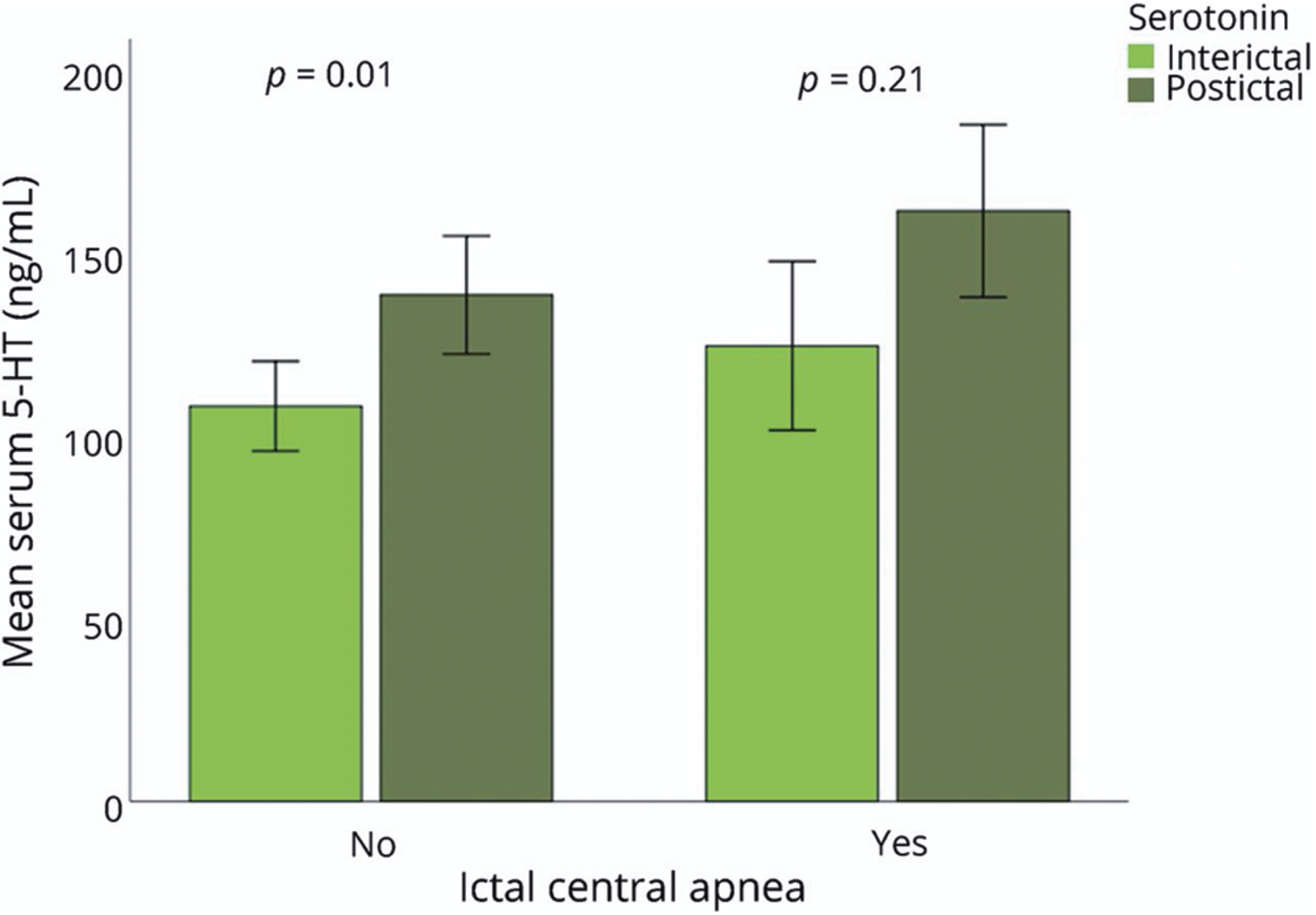
Serum 5-HT levels and postconvulsive central apnea (PCCA) after generalized convulsive seizures. Elevated levels of postictal 5-HT in generalized convulsive seizures (GCS). The mean serum interictal 5-HT levels are shown in light green bars and postictal 5-HT levels (ng/mL) are shown in dark green bars for the 2 seizure groups: PCCA (n = 8) and non-PCCA (n = 19). The levels of postictal serum 5-HT in the absence of PCCA were higher when compared to interictal levels (p < 0.001), but not when PCCA occurred (p = 0.22), suggesting the elevated 5-HT may be protective against PCCA. Reproduced with permission from Murugesan et al (2019).
Most patients who die of SUDEP are found prone in bed (Langan et al., 2000), and monitored cases have shown that patients remain motionless after a seizure and do not attempt to change position before death (Ryvlin et al., 2013). Postictal unconsciousness is due to impairment of many neural systems in the brain, but the lack of arousal after prolonged apnea suggests that there is loss of the normal arousal response to hypercapnia. Hypercapnia would be expected to occur as a result of seizure-induced hypoventilation (Buchanan and Richerson, 2010). In support of the hypothesis that there is loss of hypercapnic arousal, suppressed activity of midbrain 5-HT neurons, which are chemosensitive and are required for the arousal response to hypercapnia (Severson et al., 2003, Buchanan and Richerson, 2010), has been documented in mice during and after seizures (Zhan et al., 2016). It should be noted that other possibilities exist for the lack of arousal and impaired restoration of breathing, instead of or in addition to 5-HT neuron inhibition, such as dysfunction of chemosensitivity of nonserotonergic neurons, or impairment of effectors downstream of chemoreceptors. It is also possible that dysfunction of a subset of 5-HT neurons that are not chemosensitive plays a role. However, existing evidence suggests that postictal depression of chemosensitive serotonergic neurons in the medullary and midbrain raphe could help to explain the impaired respiratory function and decreased arousal after seizures.
Impaired chemoreception and the pathophysiology of SUDEP
Under awake, resting conditions, ventilation is primarily regulated by central CO2 chemoreceptor drive (Teran et al., 2014, Richerson, 2004, Richerson and Boron, 2003) and wakefulness drive (Dubois et al., 2016, Fink, 1961). There is increasing evidence that defects in CO2 homeostasis may play a major role in SUDEP. Peri-ictal hypoventilation has been documented in both focal seizures and GTCS (Bateman et al., 2008, Vilella et al., 2019a, Nashef et al., 1996), and in some DS patients long after a GTCS despite increased transcutaneous CO2 (tcCO2) readings (Kim et al., 2018). Video recordings from 7 patients with DS revealed peri-ictal respiratory abnormalities that can extend long into the postictal period, such as paradoxical breathing and apnea lasting longer than 5 seconds. These changes were transient in nonfatal episodes, but severe defects in breathing in a patient who later died of SUDEP suggest that respiratory changes may be biomarkers of patients at high risk. One of the patients was a 9-year-old girl with DS and an associated de novo mutation in the SCN1A gene. Her case was remarkable for hypoventilation and oxygen desaturation during and after seizures despite supplementary oxygen, suggesting impaired central CO2 chemoreception (Figure 8). Unfortunately, three years after the patient was monitored, she was found prone in bed, cyanotic and pulseless, and could not be resuscitated. The cause of death was determined to be SUDEP upon autopsy.
Figure 8.
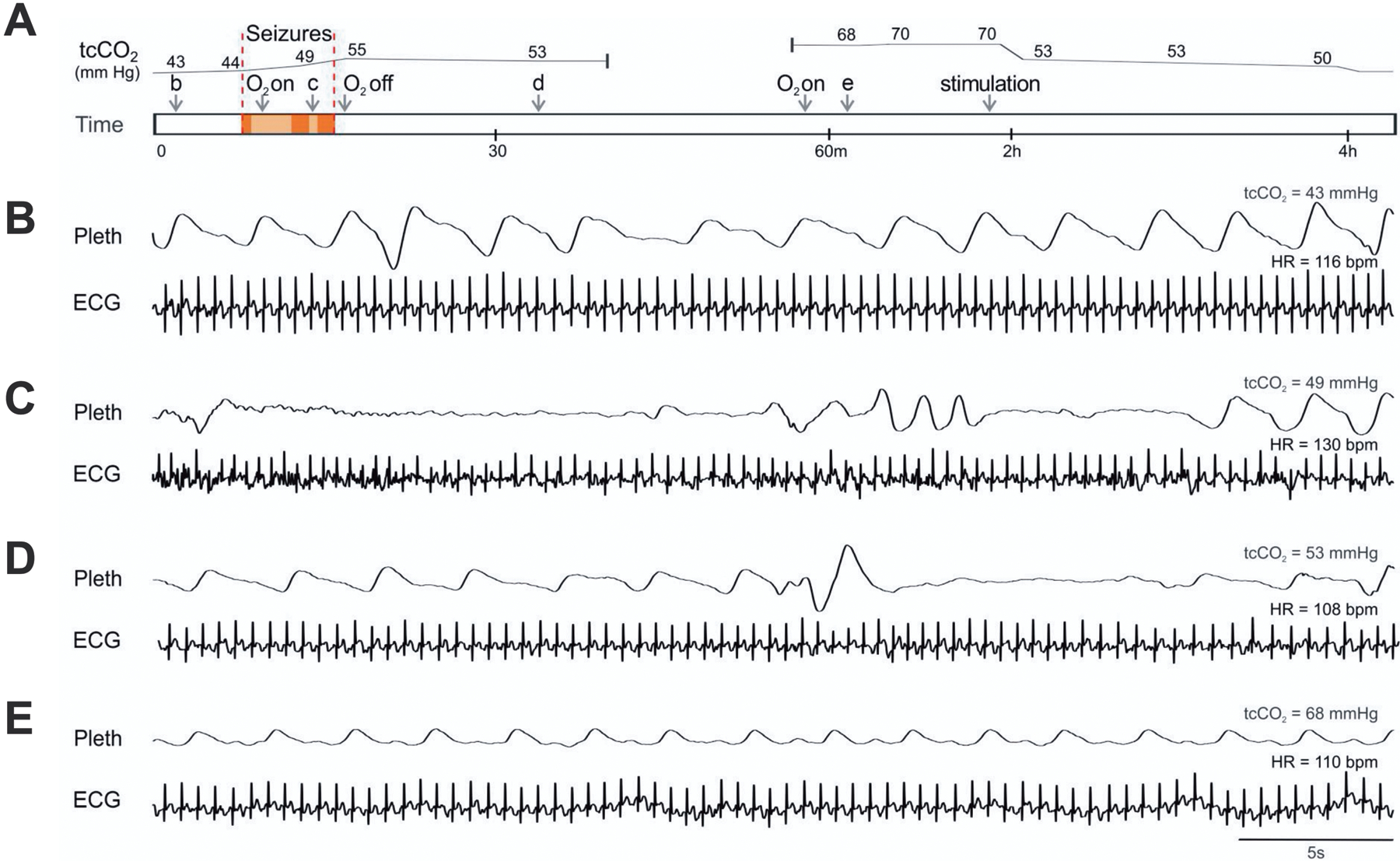
Patients with DS have abnormal breathing after seizures. A) Schematic of events while recording cardiorespiratory activity during and after a seizure in a 9-year-old girl with DS. Arrows labeled “b-e” denote the time of recordings shown in parts B–E, respectively. Area within red bars denotes convulsive seizures. B–E) Respiratory impedance plethysmography (Pleth) and ECG during normal breathing when tcCO2 was 43 mmHg (B), between convulsive seizures when there was apnea (C), after the seizures when apnea continued (D), and 44 minutes after the seizures when tcCO2 had risen to 68 mmHg (E). At 2 hours, tcCO2 decreased when the patient was stimulated to arouse, but did not return to baseline until 4 hours. Reproduced with permission from Kim et al (2018).
Recent data also suggest that epilepsy patients with blunted interictal respiratory chemoreception may be at greater risk of SUDEP. Central respiratory CO2 chemosensitivity can be quantified using the hypercapnic ventilatory response (HCVR), which is calculated by measuring the increase in minute ventilation (VE) induced by an increase in end-tidal CO2 (ETCO2) (Mohan and Duffin, 1997, Fan et al., 2010). A recent study found that epilepsy patients with a depressed HCVR exhibit more severe respiratory depression following GTCS (Figure 9A–B) (Sainju et al., 2019). One patient with a particularly low HCVR later died from SUDEP (Figure 9C–E), suggesting that the HCVR may be a biomarker for high risk for SUDEP.
Figure 9.
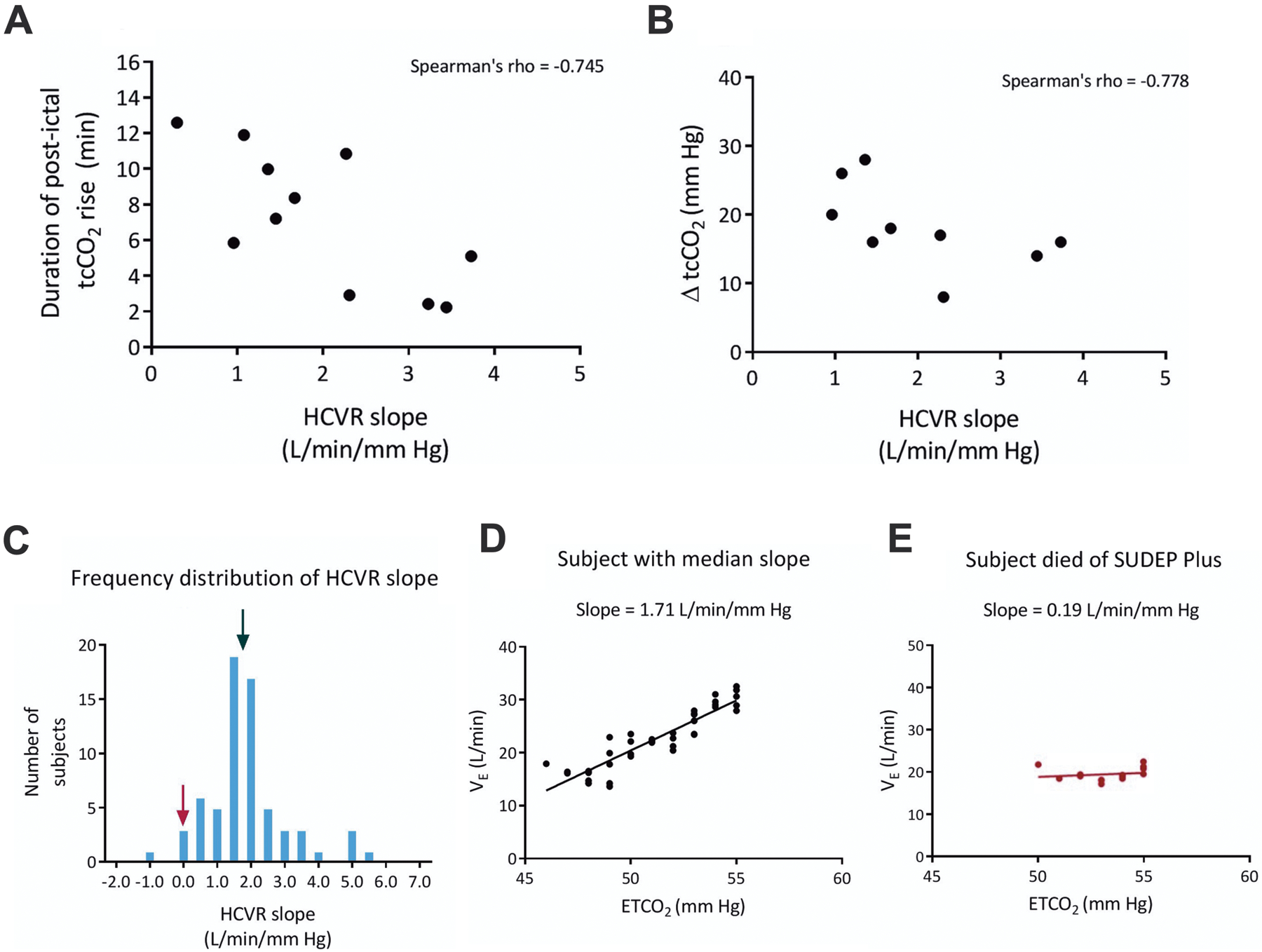
A low interictal HCVR slope is associated with more severe postictal apnea, and may increase the risk of SUDEP in epilepsy patients. A) Scatterplot depicting the correlation between HCVR slope and duration of postictal tcCO2 rise and, B) HCVR and magnitude of postictal tcCO2 rise (ΔtcCO2). C) Frequency distribution of HCVR slopes in epilepsy patients. The black arrow points to the median slope for all patients and the maroon arrow indicates a patient who later died of SUDEP plus. D) Linear regression of VE and ETCO2 for a patient with the median response indicated in (C). E) Linear regression for the patient in (C) shown with the maroon arrow. Adapted with permission from Sainju et al (2019).
Similarities between SUDEP and SIDS
Sudden infant death syndrome (SIDS) is the leading cause of postneonatal infant mortality in the United States and the third leading cause of infant mortality overall (Kinney and Thach, 2009). SUDEP shares many similarities with SIDS (Table 1) (Richerson and Buchanan, 2011). In both, there is an increased risk in males, and death is often unwitnessed, occurring more commonly at night, with the individual usually found dead in bed/crib and in the prone position (Kloster and Engelskjon, 1999). A shared hypothesis has also been proposed: a defect in response to CO2 and dysfunctional arousal responses create an intrinsic vulnerability that leads to death in response to a challenge to homeostasis (Richerson and Buchanan, 2011, Buchanan and Richerson, 2009, Sowers et al., 2013). Indeed, studies in monitored infants who subsequently died of SIDS have revealed cardiorespiratory abnormalities (Kahn and Blum, 1982, Kelly et al., 1986, Meny et al., 1994, Poets et al., 1999, Southall et al., 1980) and arousal impairments (Horne et al., 2002).
Table 1.
There is considerable overlap in the features of SIDS and SUDEP, suggesting that there may be shared pathophysiologic mechanisms.
| SIDS | SUDEP | |
|---|---|---|
| Diagnostic method | Diagnosis of exclusion | Diagnosis of exclusion |
| Baseline health | Normal | Normal, except seizures |
| Routine autopsy findings | Normal | Normal |
| Incidence | 0.6 per 1000 live births | 1.1–1.3 per 1000 persons with epilepsy |
| Proposed mechanism of death | Respiratory | Respiratory |
| Cardiac | Cardiac | |
| Arousal impairment | Arousal impairment | |
| Thermoregulatory | ? | |
| Circumstance of death | Often found prone in crib | Often found prone in bed |
| Link to 5-HT | Yes | Yes |
| Sex dependence | Male | Male |
| Time of day | Sleep period | Night or sleep period |
| Hippocampal pathology | Present in 40% of cases | Common |
Dysregulation of the 5-HT system has also been proposed to play a role in SIDS. Multiple defects in the medullary 5-HT system have been identified in several independent datasets of pathologic specimens from SIDS cases (Kinney et al., 2009). These include increased immature forms of 5-HT neurons (Paterson et al., 2006), reduced levels of the enzyme tryptophan hydroxylase (Duncan et al., 2010), reduced 5-HT1A receptor ligand and transporter binding in the raphe nuclei (Paterson et al., 2006), and reduced overall 5-HT content in the brainstem (Duncan et al., 2010, Kinney et al., 2003). The many similarities between the two entities reinforce the evidence that 5-HT dysfunction plays a role in SUDEP.
It has recently been proposed that some cases of SIDS may in fact be due to unrecognized seizures (Kinney et al., 2013, Rodriguez et al., 2012). There are case reports of witnessed seizures in infants who would have died if unattended (Kinney et al., 2013). The case for this hypothesis has gained momentum in the past decade, as recent studies reported multiple anatomic abnormalities in the hippocampus and temporal lobe classically associated with temporal lobe epilepsy in approximately 40% of SIDS cases (Brownstein et al., 2018, Kinney et al., 2016, Kinney et al., 2015). Genetic variants seen in SUDEP have also been identified in SIDS cases. A recent study identified loss-of-function mutations in the SCN1A gene, which is implicated in febrile seizures and DS, in 2 of 10 infants who died of SIDS (Brownstein et al., 2018). Both of these cases also displayed hippocampal abnormalities, namely dentate gyrus bilamination. It is possible that these two infants were too young (<2 months of age) to have exhibited convulsive seizures and other features of DS, and were therefore not yet diagnosed. These cases and the many similarities between the two syndromes suggest that some cases of SIDS may in fact be SUDEP in infants who had epilepsy, but who had not yet been diagnosed.
Overall, 5-HT is an attractive candidate to be involved in the pathophysiology of SIDS and SUDEP because it plays a role in modulating many brain functions implicated in these diseases, including breathing, cardiovascular control, sleep and wakefulness, arousal, and seizures.
8). Forebrain inhibition of breathing: Role of the amygdala
Since respiratory motor output can be generated automatically by the pons and medulla, an important question is how a seizure arising from the forebrain influences neurons in the lower brainstem to inhibit respiratory motor output. Descending projections could inhibit brainstem neurons involved in generation and shaping of respiratory motor output or its modulation, including those in the preBötz (Smith et al., 1991), the NTS, the retrotrapezoid nucleus (Guyenet et al., 2019) or 5-HT neurons in the raphe nuclei (Zhan et al., 2016). Recent data indicate that the amygdala is a critical node in the pathway for seizures in the forebrain to project to and inhibit respiratory motor output.
Forebrain effects on breathing in patients
Under most conditions, breathing is automatic, and is stimulated by tonic drive from CO2 chemoreceptors (Richerson and Boron, 2003) and wakefulness drive (Dubois et al., 2016, Fink, 1961). Voluntary control of breathing is required for activities such as eating, singing, voluntary breath holding, meditation, laughing, and other situations using the respiratory apparatus for non-ventilatory functions (Butler et al., 2014, Provine, 2016). Changes in breathing during these voluntary behaviors is mediated by projections from the forebrain. Neuronal activity observed with depth electrode recordings in some forebrain nuclei has been associated with changes in breathing in epilepsy patients. Such structures include the piriform cortex, hippocampus, amygdala, and other regions (Zelano et al., 2016), indicating a possible role in voluntary breathing modulation. Pathways from these forebrain regions to the brainstem might also be activated by seizures, inducing peri-ictal breathing abnormalities.
Seizure spread into the limbic system is known to induce apnea. This phenomenon was first described by John Hughlings Jackson in 1899 (Jackson, 1899). In his work, he described seizure discharges in the uncinate gyrus that occurred coincident with asphyxia. In the last decade, it has been shown that seizure spread to different nuclei of the limbic system, such as the hippocampus (Nobis et al., 2018, Lacuey et al., 2017) and amygdala complex (Rhone et al., 2020, Park et al., 2020, Nobis et al., 2018, Lacuey et al., 2017, Dlouhy et al., 2015) can also occur coincident with breathing abnormalities, including oxygen desaturation and central apnea (Figure 10A). Of these recent studies, the amygdala has been identified as having a particularly powerful effect in causing ictal apnea in adult (Dlouhy et al., 2015, Lacuey et al., 2017, Nobis et al., 2019) and pediatric epilepsy patients (Rhone et al., 2020). These and other findings suggest a functional connection between the amygdala and medullary respiratory networks in humans.
Figure 10.
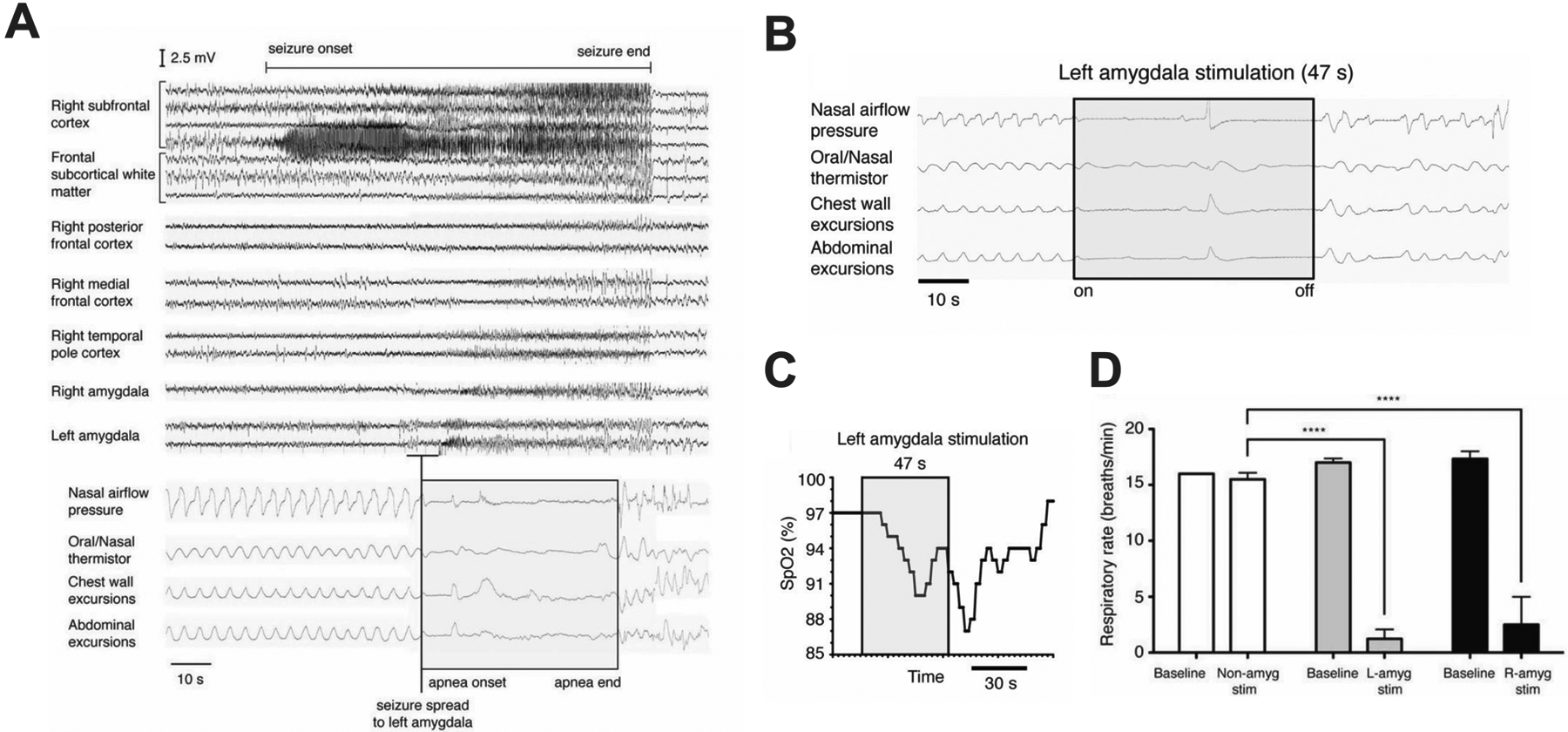
Central apnea occurs in patients when seizures spread to the left amygdala, and can be induced by electrical stimulation. A) EEG of complex partial seizure with onset in the right subfrontal cortex. Breathing pattern is normal before and after seizure initiation. Apnea occurred when the seizure spread to the left amygdala and before it spread to the right amygdala, right temporal pole, right medial frontal cortex, and right posterior frontal cortex. B) Apnea could be induced by electrical stimulation at the electrode contact in (A) coinciding with apnea. C) Apnea was severe enough to cause O2 desaturation below 90%. D) Apnea was induced by stimulation of either the left or right amygdala. Adapted from Dlouhy et al (2015).
Electrical stimulation of the amygdala and breathing abnormalities
Electric stimulation of the lateral amygdala in adolescent (Nelson and Ray, 1968) and basolateral (BLA) or basomedial (BMA) amygdala in pediatric patients (Rhone et al., 2020) can induce apnea. Central apnea and oxygen desaturation also occur in adult epilepsy patients when the amygdala is stimulated (Figure 10B–D) (Nobis et al., 2018, Dlouhy et al., 2015, Lacuey et al., 2017). Interestingly, patients were unaware that they were apneic for as long as 48 seconds, and did not develop any dyspnea despite having significant oxygen desaturation. Patients were able to voluntarily resume breathing during stimulation when prompted, or during talking, suggesting apnea was due to loss of involuntary ventilatory drive rather than dysfunction of respiratory motor output pathways or musculature (Dlouhy et al., 2015). The effect of amygdala stimulation was strong enough to induce breath holding for longer than patients could hold their breath voluntarily.
There have been contradictory results obtained with stimulation of limbic regions other than the amygdala in patients, with some investigators able to induce apnea with stimulation of the hippocampus (Lacuey et al., 2017), and others unable to induce this response (Dlouhy et al., 2015). There are a variety of possible reasons for this difference, including the use of different stimulation parameters and electrode configurations. Dlouhy et al (2015) reported that they were only able to induce apnea with stimulation of a discrete region of the amygdala. This may have been due to their use of low levels of current injection to restrict the region stimulated, and high-resolution imaging to verify the anatomic localization. This approach is important because injection of too much current can activate neuronal cell bodies or axonal tracts in neighboring regions, or can induce seizures that spread widely to distant brain regions. It is of most interest to only activate local neurons to identify those with oligosynaptic projections to the brainstem.
Seizure propagation and ventilatory arrest in animal models
The amygdala is a structure that is frequently involved early during spread of seizures (Baram et al., 1997, Sharma et al., 2007), likely facilitated by hippocampal commissural fibers (Toyoda et al., 2013). The amygdala has been described as critical for propagation of seizures from the brainstem to the forebrain in rat and mouse audiogenic seizure models (Hirsch et al., 1997, Klein et al., 2004), but its role in propagation of seizures from the cortex to the brainstem is still unknown.
Experiments in DBA/1 mice have shown that the amygdala is a required node in the pathway from the forebrain to the respiratory network. Seizures induce apnea in DBA/1 mice that is normally fatal (Faingold et al., 2010). However, these apneas and death are prevented in DBA/1 mice if the amygdala is ablated (Marincovich et al., 2019), indicating that the amygdala is key in propagation of the seizure from the forebrain to the brainstem. It remains to be determined which specific neurons in the amygdala form this pathway, what neurotransmitter(s) they contain, and to what neurons they project in the brainstem. One possible candidate is serotonin neurons, which are inhibited during seizures (Zhan et al., 2016).
9). Summary and future directions
Figure 11 illustrates a model for our working hypothesis of the mechanisms of SUDEP. Under normal conditions hypercapnia stimulates breathing to maintain blood gas homeostasis. During a seizure, projections from the amygdala inhibit 5-HT neurons in the medulla and midbrain leading to loss of the ventilatory and arousal response to hypercapnia. Patients are obtunded postictally, unaware they are not breathing, and do not experience the dyspnea that would normally lead to protective reflexes to restore homeostasis. The resulting rise in CO2 and decrease in O2 would be fatal if severe and prolonged.
Figure 11.
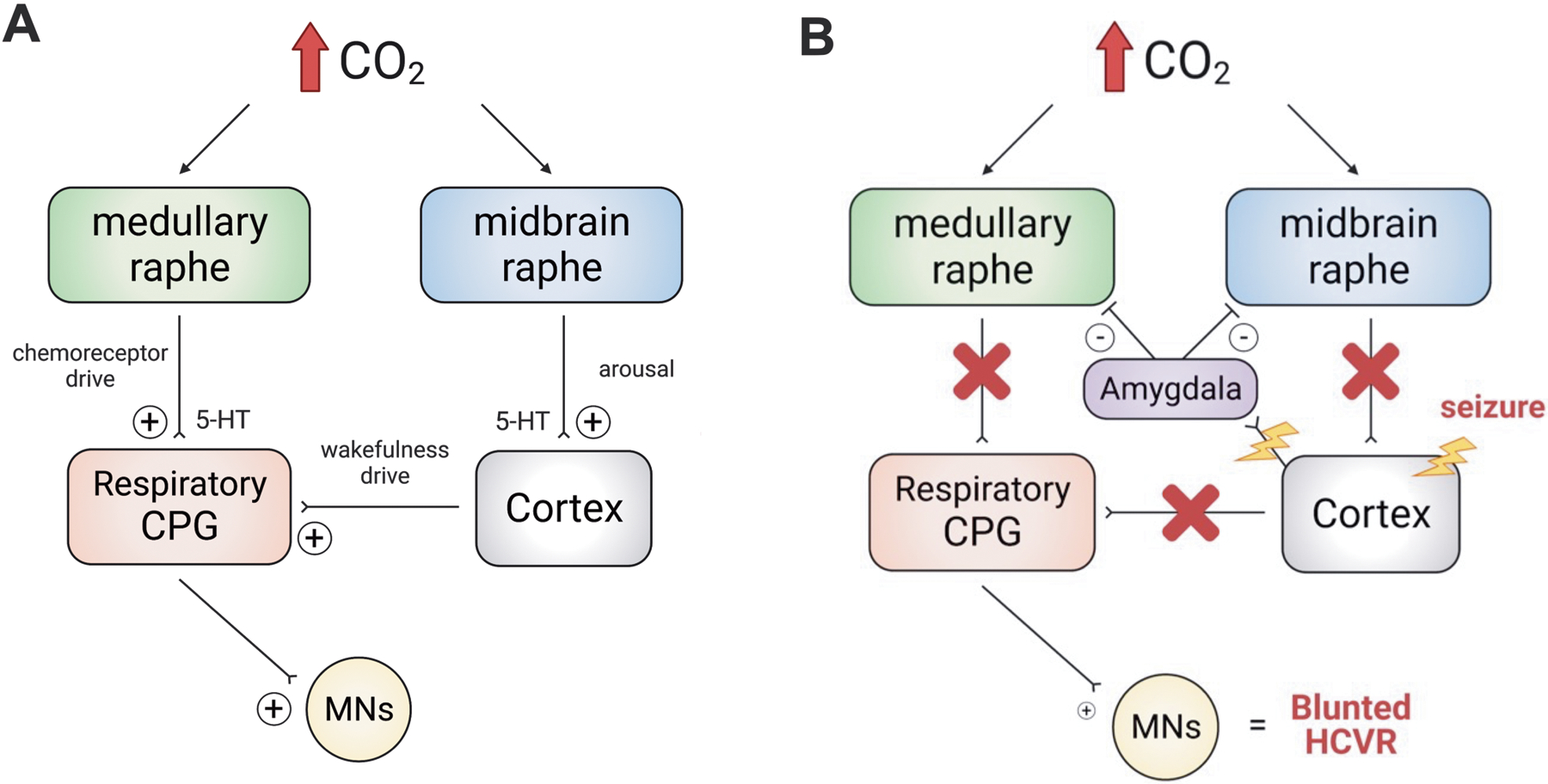
Working model for seizure-induced respiratory depression. A) There are two main sources of respiratory drive. While awake, breathing is maintained by drive from chemoreceptors (including medullary raphe neurons) and cortical “wakefulness drive.” When CO2 increases it causes arousal from sleep and stimulates chemoreceptor drive. B) When a seizure occurs and propagates from the cortex to the brainstem (including via the amygdala), both chemoreceptor and wakefulness drives are lost. This leads to decreased baseline ventilation and a blunted HCVR. This postictal HCVR depression and hypoventilation could increase the risk of respiratory arrest and SUDEP. MNs = motor neurons, CPG = central pattern generator.
As follows from the preceding discussion, there are other mechanisms that may be involved, either in addition to or instead of those illustrated here. The serotonin neurons involved may not be those that are chemosensitive. Non-serotonergic chemoreceptors or portions of the respiratory network downstream of the chemoreceptors, including the respiratory rhythm generator may be involved. Adenosine appears to play a role (Purnell et al., 2021) but it is not known how that fits into this model. Thus, future work will be needed to test the proposed hypothesis for the pathophysiology of SUDEP, to identify biomarkers of high-risk patients, to improve ascertainment of SUDEP cases, to develop devices that can warn caregivers of impending SUDEP, and to find approaches for prevention of death. Based on the rapid growth of research in this field (Figure 12), it is likely that many of these issues will be addressed in the coming years.
Figure 12.
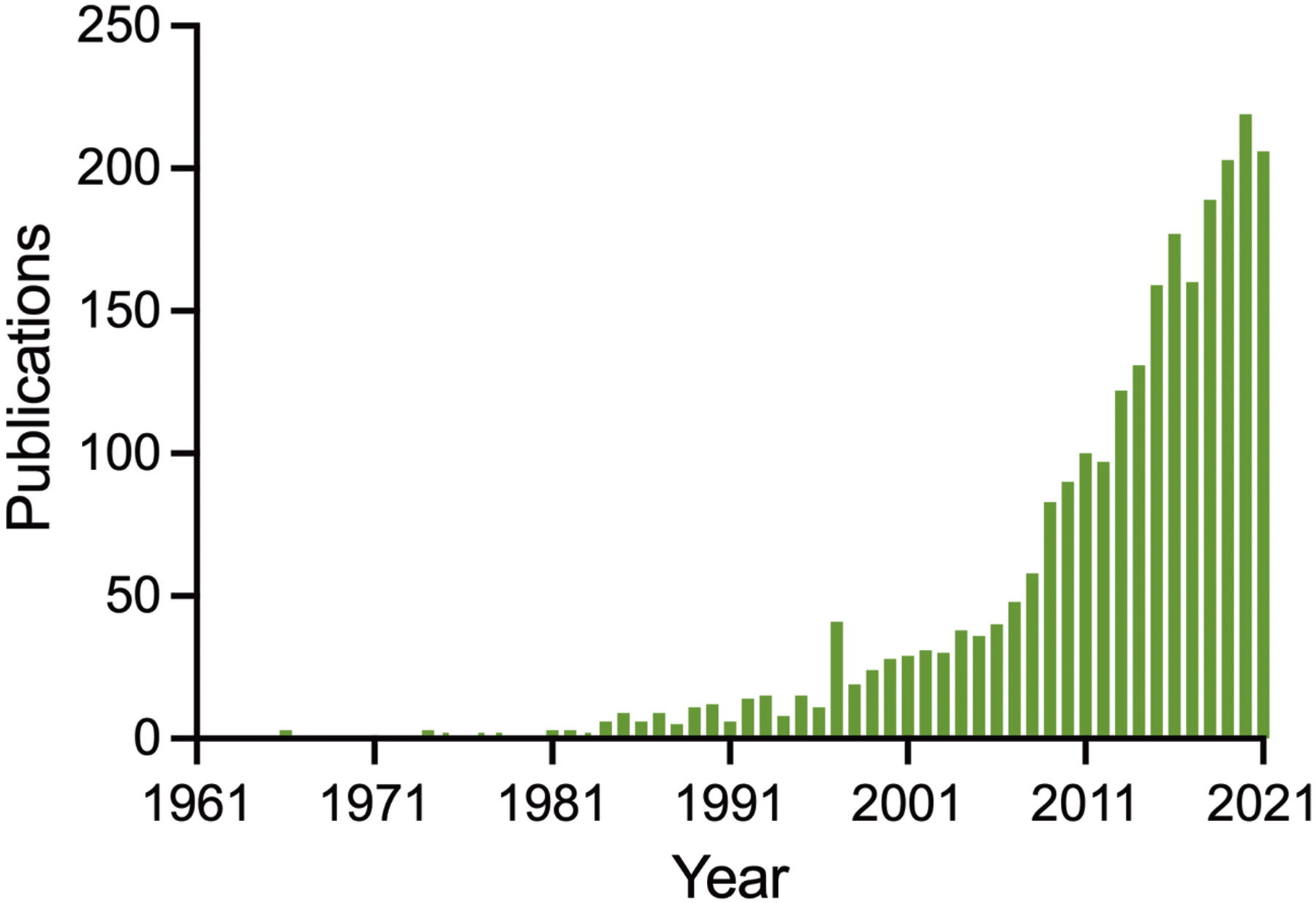
The number of published studies on SUDEP has increased dramatically from 1971 to 2021. PubMed was used to search for articles using ‘sudden unexpected death in epilepsy’ or ‘SUDEP’.
REFERENCES
- Ahmad S, Fowler LJ & Whitton PS (2005). Effects of combined lamotrigine and valproate on basal and stimulated extracellular amino acids and monoamines in the hippocampus of freely moving rats. Naunyn Schmiedebergs Arch Pharmacol, 371, 1–8. [DOI] [PubMed] [Google Scholar]
- Aiba I & Noebels JL (2015). Spreading depolarization in the brainstem mediates sudden cardiorespiratory arrest in mouse SUDEP models. Sci Transl Med, 7, 282ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albano C, Cupello A, Mainardi P, et al. (2006). Successful treatment of epilepsy with serotonin reuptake inhibitors: proposed mechanism. Neurochem Res, 31, 509–14. [DOI] [PubMed] [Google Scholar]
- Alper K, Schwartz KA, Kolts RL, et al. (2007). Seizure incidence in psychopharmacological clinical trials: an analysis of Food and Drug Administration (FDA) summary basis of approval reports. Biol Psychiatry, 62, 345–54. [DOI] [PubMed] [Google Scholar]
- Anderson LL, Thompson CH, Hawkins NA, et al. (2014). Antiepileptic activity of preferential inhibitors of persistent sodium current. Epilepsia, 55, 1274–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach DS, Jones J, Clawson BC, et al. (2013). Altered cardiac electrophysiology and SUDEP in a model of Dravet syndrome. PLoS One, 8, e77843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baf MH, Subhash MN, Lakshmana KM, et al. (1994). Sodium valproate induced alterations in monoamine levels in different regions of the rat brain. Neurochem Int, 24, 67–72. [DOI] [PubMed] [Google Scholar]
- Baram TZ, Gerth A & Schultz L (1997). Febrile seizures: an appropriate-aged model suitable for long-term studies. Brain Res Dev Brain Res, 98, 265–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman LM, Li CS & Seyal M (2008). Ictal hypoxemia in localization-related epilepsy: analysis of incidence, severity and risk factors. Brain, 131, 3239–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnycastle DD, Giarman NJ & Paasonen MK (1957). Anticonvulsant compounds and 5-hydroxytryptamine in rat brain. Br J Pharmacol Chemother, 12, 228–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley SR, Pieribone VA, Wang W, et al. (2002). Chemosensitive serotonergic neurons are closely associated with large medullary arteries. Nat Neurosci, 5, 401–2. [DOI] [PubMed] [Google Scholar]
- Brownstein CA, Goldstein RD, Thompson CH, et al. (2018). SCN1A variants associated with sudden infant death syndrome. Epilepsia, 59, e56–e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan GF, Murray NM, Hajek MA, et al. (2014). Serotonin neurones have anti-convulsant effects and reduce seizure-induced mortality. J Physiol, 592, 4395–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan GF & Richerson GB (2009). Role of chemoreceptors in mediating dyspnea. Respir Physiol Neurobiol, 167, 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan GF & Richerson GB (2010). Central serotonin neurons are required for arousal to CO2. Proc Natl Acad Sci U S A, 107, 16354–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunton-Stasyshyn RKA, Wagnon JL, Wengert ER, et al. (2019). Prominent role of forebrain excitatory neurons in SCN8A encephalopathy. Brain, 142, 362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler JE, Hudson AL & Gandevia SC (2014). The neural control of human inspiratory muscles. Prog Brain Res, 209, 295–308. [DOI] [PubMed] [Google Scholar]
- Catterall WA (2000). From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron, 26, 13–25. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Kalume F & Oakley JC (2010). Nav1.1 channels and epilepsy. J Physiol, 588, 1849–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheah CS, Yu FH, Westenbroek RE, et al. (2012). Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc Natl Acad Sci U S A, 109, 14646–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Brodie MJ, Liew D, et al. (2018). Treatment Outcomes in Patients With Newly Diagnosed Epilepsy Treated With Established and New Antiepileptic Drugs: A 30-Year Longitudinal Cohort Study. JAMA Neurol, 75, 279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun KC, Ma SC, Oh H, et al. (2018). Ketogenic diet-induced extension of longevity in epileptic Kcna1-null mice is influenced by gender and age at treatment onset. Epilepsy Res, 140, 53–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes L, Del-Favero J, Ceulemans B, et al. (2001). De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet, 68, 1327–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dailey JW, Reith ME, Yan QS, et al. (1997). Anticonvulsant doses of carbamazepine increase hippocampal extracellular serotonin in genetically epilepsy-prone rats: dose response relationships. Neurosci Lett, 227, 13–6. [DOI] [PubMed] [Google Scholar]
- Dailey JW, Yan QS, Adams-Curtis LE, et al. (1996). Neurochemical correlates of antiepileptic drugs in the genetically epilepsy-prone rat (GEPR). Life Sci, 58, 259–66. [DOI] [PubMed] [Google Scholar]
- Dean JB, Bayliss DA, Erickson JT, et al. (1990). Depolarization and stimulation of neurons in nucleus tractus solitarii by carbon dioxide does not require chemical synaptic input. Neuroscience, 36, 207–216. [DOI] [PubMed] [Google Scholar]
- Delogu AB, Spinelli A, Battaglia D, et al. (2011). Electrical and autonomic cardiac function in patients with Dravet syndrome. Epilepsia, 52 Suppl 2, 55–8. [DOI] [PubMed] [Google Scholar]
- Depuy SD, Kanbar R, Coates MB, et al. (2011). Control of breathing by raphe obscurus serotonergic neurons in mice. J Neurosci, 31, 1981–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinsky O, Hesdorffer DC, Thurman DJ, et al. (2016). Sudden unexpected death in epilepsy: epidemiology, mechanisms, and prevention. Lancet Neurol, 15, 1075–88. [DOI] [PubMed] [Google Scholar]
- Dlouhy BJ, Gehlbach BK, Kreple CJ, et al. (2015). Breathing inhibited when seizures spread to the amygdala and upon amygdala stimulation. J Neurosci, 35, 10281–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dravet C (2011). The core Dravet syndrome phenotype. Epilepsia, 52 Suppl 2, 3–9. [DOI] [PubMed] [Google Scholar]
- Drevets WC, Frank E, Price JC, et al. (1999). PET imaging of serotonin 1A receptor binding in depression. Biol Psychiatry, 46, 1375–87. [DOI] [PubMed] [Google Scholar]
- Dubois M, Chenivesse C, Raux M, et al. (2016). Neurophysiological Evidence for a Cortical Contribution to the Wakefulness-Related Drive to Breathe Explaining Hypocapnia-Resistant Ventilation in Humans. J Neurosci, 36, 10673–10682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duflocq A, Le Bras B, Bullier E, et al. (2008). Nav1.1 is predominantly expressed in nodes of Ranvier and axon initial segments. Mol Cell Neurosci, 39, 180–92. [DOI] [PubMed] [Google Scholar]
- Duncan JR, Paterson DS, Hoffman JM, et al. (2010). Brainstem serotonergic deficiency in sudden infant death syndrome. JAMA, 303, 430–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England MJ, Liverman CT, Schultz AM, et al. (2012). Epilepsy across the spectrum: promoting health and understanding. A summary of the Institute of Medicine report. Epilepsy Behav, 25, 266–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eunson LH, Rea R, Zuberi SM, et al. (2000). Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann Neurol, 48, 647–56. [PubMed] [Google Scholar]
- Faingold CL, Randall M, Mhaskar Y, et al. (2011a). Differences in serotonin receptor expression in the brainstem may explain the differential ability of a serotonin agonist to block seizure-induced sudden death in DBA/2 vs. DBA/1 mice. Brain Res, 1418, 104–10. [DOI] [PubMed] [Google Scholar]
- Faingold CL, Randall M & Tupal S (2010). DBA/1 mice exhibit chronic susceptibility to audiogenic seizures followed by sudden death associated with respiratory arrest. Epilepsy Behav, 17, 436–40. [DOI] [PubMed] [Google Scholar]
- Faingold CL, Tupal S & Randall M (2011b). Prevention of seizure-induced sudden death in a chronic SUDEP model by semichronic administration of a selective serotonin reuptake inhibitor. Epilepsy Behav, 22, 186–90. [DOI] [PubMed] [Google Scholar]
- Fan JL, Burgess KR, Basnyat R, et al. (2010). Influence of high altitude on cerebrovascular and ventilatory responsiveness to CO2. J Physiol, 588, 539–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favale E, Audenino D, Cocito L, et al. (2003). The anticonvulsant effect of citalopram as an indirect evidence of serotonergic impairment in human epileptogenesis. Seizure-European Journal of Epilepsy, 12, 316–318. [DOI] [PubMed] [Google Scholar]
- Favale E, Rubino V, Mainardi P, et al. (1995). Anticonvulsant effect of fluoxetine in humans. Neurology, 45, 1926–7. [DOI] [PubMed] [Google Scholar]
- Favero M, Sotuyo NP, Lopez E, et al. (2018). A Transient Developmental Window of Fast-Spiking Interneuron Dysfunction in a Mouse Model of Dravet Syndrome. J Neurosci, 38, 7912–7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng HJ & Faingold CL (2017). Abnormalities of serotonergic neurotransmission in animal models of SUDEP. Epilepsy Behav, 71, 174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink BR (1961). Influence of cerebral activity in wakefulness on regulation of breathing. J Appl Physiol, 16, 15–20. [DOI] [PubMed] [Google Scholar]
- Fiske B (2002). Putative chemoreceptors get close to arteries. Nature Neuroscience, 5, 396–396. [Google Scholar]
- Gardella E, Becker F, Moller RS, et al. (2016). Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation. Ann Neurol, 79, 428–36. [DOI] [PubMed] [Google Scholar]
- Gardella E, Marini C, Trivisano M, et al. (2018). The phenotype of SCN8A developmental and epileptic encephalopathy. Neurology, 91, e1112–e1124. [DOI] [PubMed] [Google Scholar]
- Gilliam FG, Santos J, Vahle V, et al. (2004). Depression in epilepsy: ignoring clinical expression of neuronal network dysfunction? Epilepsia, 45 Suppl 2, 28–33. [DOI] [PubMed] [Google Scholar]
- Giovacchini G, Toczek MT, Bonwetsch R, et al. (2005). 5-HT 1A receptors are reduced in temporal lobe epilepsy after partial-volume correction. J Nucl Med, 46, 1128–35. [PMC free article] [PubMed] [Google Scholar]
- Giroud M, Dumas R, Dauvergne M, et al. (1990). 5-Hydroxyindoleacetic acid and homovanillic acid in cerebrospinal fluid of children with febrile convulsions. Epilepsia, 31, 178–81. [DOI] [PubMed] [Google Scholar]
- Glasscock E, Yoo JW, Chen TT, et al. (2010). Kv1.1 potassium channel deficiency reveals braindriven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J Neurosci, 30, 5167–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff KM & Goldberg EM (2019). Vasoactive intestinal peptide-expressing interneurons are impaired in a mouse model of Dravet syndrome. Elife, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman AM (2015). Mechanisms of sudden unexplained death in epilepsy. Curr Opin Neurol, 28, 166–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG, Stornetta RL, Souza G, et al. (2019). The Retrotrapezoid Nucleus: Central Chemoreceptor and Regulator of Breathing Automaticity. Trends Neurosci, 42, 807–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajek MA & Buchanan GF (2016). Influence of vigilance state on physiological consequences of seizures and seizure-induced death in mice. Journal of Neurophysiology, 115, 2286–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer MF, Wagnon JL, Mefford HC, et al. (1993). SCN8A-Related Epilepsy with Encephalopathy. In: ADAM MP, ARDINGER HH, PAGON RA, WALLACE SE, BEAN LJH, MIRZAA G & AMEMIYA A (eds.) GeneReviews((R)). Seattle (WA). [Google Scholar]
- Han S, Yu FH, Schwartz MD, et al. (2012). Na(V)1.1 channels are critical for intercellular communication in the suprachiasmatic nucleus and for normal circadian rhythms. Proc Natl Acad Sci U S A, 109, E368–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harden C, Tomson T, Gloss D, et al. (2017). Practice guideline summary: Sudden unexpected death in epilepsy incidence rates and risk factors: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology, 88, 1674–1680. [DOI] [PubMed] [Google Scholar]
- Hermann BP, Seidenberg M & Bell B (2002). The neurodevelopmental impact of childhood onset temporal lobe epilepsy on brain structure and function and the risk of progressive cognitive effects. Prog Brain Res, 135, 429–38. [DOI] [PubMed] [Google Scholar]
- Hernandez EJ, Williams PA & Dudek FE (2002). Effects of fluoxetine and TFMPP on spontaneous seizures in rats with pilocarpine-induced epilepsy. Epilepsia, 43, 1337–45. [DOI] [PubMed] [Google Scholar]
- Hesdorffer DC, Hauser WA, Annegers JF, et al. (2000). Major depression is a risk factor for seizures in older adults. Annals of Neurology, 47, 246–249. [PubMed] [Google Scholar]
- Hesdorffer DC, Ishihara L, Mynepalli L, et al. (2012a). Epilepsy, suicidality, and psychiatric disorders: a bidirectional association. Ann Neurol, 72, 184–91. [DOI] [PubMed] [Google Scholar]
- Hesdorffer DC, Ludvigsson P, Hauser WA, et al. (2007). Co-occurrence of major depression or suicide attempt with migraine with aura and risk for unprovoked seizure. Epilepsy Res, 75, 220–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesdorffer DC, Tomson T, Benn E, et al. (2011). Combined analysis of risk factors for SUDEP. Epilepsia, 52, 1150–9. [DOI] [PubMed] [Google Scholar]
- Hesdorffer DC, Tomson T, Benn E, et al. (2012b). Do antiepileptic drugs or generalized tonic-clonic seizure frequency increase SUDEP risk? A combined analysis. Epilepsia, 53, 249–52. [DOI] [PubMed] [Google Scholar]
- Hirsch E, Danober L, Simler S, et al. (1997). The amygdala is critical for seizure propagation from brainstem to forebrain. Neuroscience, 77, 975–84. [DOI] [PubMed] [Google Scholar]
- Hodges MR, Tattersall GJ, Harris MB, et al. (2008). Defects in breathing and thermoregulation in mice with near-complete absence of central serotonin neurons. J Neurosci, 28, 2495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges MR, Wehner M, Aungst J, et al. (2009). Transgenic mice lacking serotonin neurons have severe apnea and high mortality during development. J Neurosci, 29, 10341–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst AG, Winkel BG, Risgaard B, et al. (2013). Epilepsy and risk of death and sudden unexpected death in the young: a nationwide study. Epilepsia, 54, 1613–20. [DOI] [PubMed] [Google Scholar]
- Horne RS, Parslow PM, Ferens D, et al. (2002). Arousal responses and risk factors for sudden infant death syndrome. Sleep Med, 3 Suppl 2, S61–5. [DOI] [PubMed] [Google Scholar]
- Huff SD & Fuller JL (1964). Audiogenic Seizures, the Dilite Locus, and Phenylalanine Hydroxylase in Dba/1 Mice. Science, 144, 304–5. [DOI] [PubMed] [Google Scholar]
- Jackson JH (1899). Neurological fragments: On asphyxia in slight epileptic paroxysms. The Lancet, 153, 79–80. [Google Scholar]
- Jacobs BL & Azmitia EC (1992). Structure and function of the brain serotonin system. Physiol Rev, 72, 165–229. [DOI] [PubMed] [Google Scholar]
- Jensen P, Farago AF, Awatramani RB, et al. (2008). Redefining the serotonergic system by genetic lineage. Nat Neurosci, 11, 417–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao J, Yang Y, Shi Y, et al. (2013). Modeling Dravet syndrome using induced pluripotent stem cells (iPSCs) and directly converted neurons. Hum Mol Genet, 22, 4241–52. [DOI] [PubMed] [Google Scholar]
- Johannesen KM, Gardella E, Scheffer I, et al. (2018). Early mortality in SCN8A-related epilepsies. Epilepsy Res, 143, 79–81. [DOI] [PubMed] [Google Scholar]
- Kaada BR & Jasper H (1952). Respiratory responses to stimulation of temporal pole, insula, and hippocampal and limbic gyri in man. AMA Arch Neurol Psychiatry, 68, 609–19. [DOI] [PubMed] [Google Scholar]
- Kahn A & Blum D (1982). Home monitoring of infants considered at risk for the sudden infant death syndrome. Four years’ experience (1977–1981). Eur J Pediatr, 139, 94–100. [DOI] [PubMed] [Google Scholar]
- Kalume F, Westenbroek RE, Cheah CS, et al. (2013). Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Invest, 123, 1798–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanamaru M & Homma I (2007). Compensatory airway dilation and additive ventilatory augmentation mediated by dorsomedial medullary 5-hydroxytryptamine 2 receptor activity and hypercapnia. Am J Physiol Regul Integr Comp Physiol, 293, R854–60. [DOI] [PubMed] [Google Scholar]
- Kanner AM (2011). Depression and epilepsy: A bidirectional relation? Epilepsia, 52 Suppl 1, 21–7. [DOI] [PubMed] [Google Scholar]
- Kearney J (2013). Sudden unexpected death in dravet syndrome. Epilepsy Curr, 13, 264–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller AE, Whitney R, Li SA, et al. (2018). Incidence of sudden unexpected death in epilepsy in children is similar to adults. Neurology, 91, e107–e111. [DOI] [PubMed] [Google Scholar]
- Kelly DH, Golub H, Carley D, et al. (1986). Pneumograms in infants who subsequently died of sudden infant death syndrome. J Pediatr, 109, 249–54. [DOI] [PubMed] [Google Scholar]
- Kim Y, Bravo E, Thirnbeck CK, et al. (2018). Severe peri-ictal respiratory dysfunction is common in Dravet syndrome. J Clin Invest, 128, 1141–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney HC, Cryan JB, Haynes RL, et al. (2015). Dentate gyrus abnormalities in sudden unexplained death in infants: morphological marker of underlying brain vulnerability. Acta Neuropathol, 129, 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney HC, Mcdonald AG, Minter ME, et al. (2013). Witnessed sleep-related seizure and sudden unexpected death in infancy: a case report. Forensic Sci Med Pathol, 9, 418–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney HC, Poduri AH, Cryan JB, et al. (2016). Hippocampal Formation Maldevelopment and Sudden Unexpected Death across the Pediatric Age Spectrum. J Neuropathol Exp Neurol, 75, 981–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney HC, Randall LL, Sleeper LA, et al. (2003). Serotonergic brainstem abnormalities in Northern Plains Indians with the sudden infant death syndrome. J Neuropathol Exp Neurol, 62, 1178–91. [DOI] [PubMed] [Google Scholar]
- Kinney HC, Richerson GB, Dymecki SM, et al. (2009). The brainstem and serotonin in the sudden infant death syndrome. Annu Rev Pathol, 4, 517–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney HC & Thach BT (2009). The sudden infant death syndrome. N Engl J Med, 361, 795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein BD, Fu YH, Ptacek LJ, et al. (2004). c-Fos immunohistochemical mapping of the audiogenic seizure network and tonotopic neuronal hyperexcitability in the inferior colliculus of the Frings mouse. Epilepsy Res, 62, 13–25. [DOI] [PubMed] [Google Scholar]
- Kloster R & Engelskjon T (1999). Sudden unexpected death in epilepsy (SUDEP): a clinical perspective and a search for risk factors. J Neurol Neurosurg Psychiatry, 67, 439–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kløvgaard M, Lynge TH, Tsiropoulos I, et al. (2021). Sudden unexpected death in epilepsy in persons younger than 50 years: A retrospective nationwide cohort study in Denmark. Epilepsia. [DOI] [PubMed] [Google Scholar]
- Ko FJ, Chiang CH, Liu WJ, et al. (1993). Alteration of amino acid in plasma and cerebrospinal fluid of children with seizure disorders. Gaoxiong Yi Xue Ke Xue Za Zhi, 9, 131–42. [PubMed] [Google Scholar]
- Lacuey N, Hampson JP, Harper RM, et al. (2019). Limbic and paralimbic structures driving ictal central apnea. Neurology, 92, e655–e669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacuey N, Vilella L, Hampson JP, et al. (2018). Ictal laryngospasm monitored by video-EEG and polygraphy: a potential SUDEP mechanism. Epileptic Disord, 20, 146–150. [DOI] [PubMed] [Google Scholar]
- Lacuey N, Zonjy B, Londono L, et al. (2017). Amygdala and hippocampus are symptomatogenic zones for central apneic seizures. Neurology, 88, 701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagae L, Sullivan J, Knupp K, et al. (2019). Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: a randomised, double-blind, placebo-controlled trial. Lancet, 394, 2243–2254. [DOI] [PubMed] [Google Scholar]
- Langan Y, Nashef L & Sander JW (2000). Sudden unexpected death in epilepsy: a series of witnessed deaths. J Neurol Neurosurg Psychiatry, 68, 211–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen J, Carvill GL, Gardella E, et al. (2015). The phenotypic spectrum of SCN8A encephalopathy. Neurology, 84, 480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathers CM & Schraeder PL (1990). Epilepsy and sudden death, New York, NY, Marcel Dekker. [Google Scholar]
- Lhatoo SD, Faulkner HJ, Dembny K, et al. (2010). An electroclinical case-control study of sudden unexpected death in epilepsy. Ann Neurol, 68, 787–96. [DOI] [PubMed] [Google Scholar]
- Liu Y, Lopez-Santiago LF, Yuan Y, et al. (2013). Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism. Ann Neurol, 74, 128–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lothe A, Didelot A, Hammers A, et al. (2008). Comorbidity between temporal lobe epilepsy and depression: a [18F]MPPF PET study. Brain, 131, 2765–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejak P, Szyndler J, Kolosowska K, et al. (2014). Valproate disturbs the balance between branched and aromatic amino acids in rats. Neurotox Res, 25, 358–68. [DOI] [PubMed] [Google Scholar]
- Maguire MJ, Jackson CF, Marson AG, et al. (2020). Treatments for the prevention of Sudden Unexpected Death in Epilepsy (SUDEP). Cochrane Database Syst Rev, 4, CD011792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal R, Budde R, Lawlor GL, et al. (2021). Utilizing multimodal imaging to visualize potential mechanism for sudden death in epilepsy. Epilepsy Behav, 122, 108124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marincovich A, Bravo E, Dlouhy B, et al. (2019). Amygdala lesions reduce seizure-induced respiratory arrest in DBA/1 mice. Epilepsy Behav, 106440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion JL, Bigot JC & Goas JY (1985). [Alcoholic epilepsy. Decrease of tryptophan levels in the blood and cerebrospinal fluid]. Presse Med, 14, 681–3. [PubMed] [Google Scholar]
- Massey CA, Sowers LP, Dlouhy BJ, et al. (2014). Mechanisms of sudden unexpected death in epilepsy: the pathway to prevention. Nat Rev Neurol, 10, 271–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meny RG, Carroll JL, Carbone MT, et al. (1994). Cardiorespiratory recordings from infants dying suddenly and unexpectedly at home. Pediatrics, 93, 44–9. [PubMed] [Google Scholar]
- Merlet I, Ostrowsky K, Costes N, et al. (2004). 5-HT1A receptor binding and intracerebral activity in temporal lobe epilepsy: an [18F]MPPF-PET study. Brain, 127, 900–13. [DOI] [PubMed] [Google Scholar]
- Mistry AM, Thompson CH, Miller AR, et al. (2014). Strain- and age-dependent hippocampal neuron sodium currents correlate with epilepsy severity in Dravet syndrome mice. Neurobiol Dis, 65, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan R & Duffin J (1997). The effect of hypoxia on the ventilatory response to carbon dioxide in man. Respir Physiol, 108, 101–15. [DOI] [PubMed] [Google Scholar]
- Murugesan A, Rani MRS, Hampson J, et al. (2018). Serum serotonin levels in patients with epileptic seizures. Epilepsia, 59, e91–e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murugesan A, Rani MRS, Vilella L, et al. (2019). Postictal serotonin levels are associated with peri-ictal apnea. Neurology, 93, e1485–e1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabbout R, Mistry A, Zuberi S, et al. (2020). Fenfluramine for Treatment-Resistant Seizures in Patients With Dravet Syndrome Receiving Stiripentol-Inclusive Regimens: A Randomized Clinical Trial. JAMA Neurol, 77, 300–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadkarni MA, Friedman D & Devinsky O (2012). Central apnea at complex partial seizure onset. Seizure, 21, 555–8. [DOI] [PubMed] [Google Scholar]
- Nashef L, Garner S, Sander JW, et al. (1998). Circumstances of death in sudden death in epilepsy: interviews of bereaved relatives. J Neurol Neurosurg Psychiatry, 64, 349–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashef L, So EL, Ryvlin P, et al. (2012). Unifying the definitions of sudden unexpected death in epilepsy. Epilepsia, 53, 227–33. [DOI] [PubMed] [Google Scholar]
- Nashef L, Walker F, Allen P, et al. (1996). Apnoea and bradycardia during epileptic seizures: relation to sudden death in epilepsy. J Neurol Neurosurg Psychiatry, 60, 297–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DA & Ray CD (1968). Respiratory arrest from seizure discharges in limbic system. Report of cases. Arch Neurol, 19, 199–207. [DOI] [PubMed] [Google Scholar]
- Nobis WP, Gonzalez Otarula KA, Templer JW, et al. (2019). The effect of seizure spread to the amygdala on respiration and onset of ictal central apnea. J Neurosurg, 132, 1313–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobis WP, Schuele S, Templer JW, et al. (2018). Amygdala-stimulation-induced apnea is attention and nasal-breathing dependent. Ann Neurol, 83, 460–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, Miyamoto H, Morita N, et al. (2007). Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci, 27, 5903–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada M, Kaneko S, Hirano T, et al. (1992). Effects of zonisamide on extracellular levels of monoamine and its metabolite, and on Ca2+ ependent dopamine release. Epilepsy Research, 13, 113–119. [DOI] [PubMed] [Google Scholar]
- Onimaru H, Ikeda K & Kawakami K (2009). Phox2b, RTN/pFRG neurons and respiratory rhythmogenesis. Respir Physiol Neurobiol, 168, 13–8. [DOI] [PubMed] [Google Scholar]
- Ottolini M, Barker BS, Gaykema RP, et al. (2017). Aberrant Sodium Channel Currents and Hyperexcitability of Medial Entorhinal Cortex Neurons in a Mouse Model of SCN8A Encephalopathy. J Neurosci, 37, 7643–7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K, Kanth K, Bajwa S, et al. (2020). Seizure-related apneas have an inconsistent linkage to amygdala seizure spread. Epilepsia, 61, 1253–1260. [DOI] [PubMed] [Google Scholar]
- Pasini A, Tortorella A & Gale K (1992). Anticonvulsant effect of intranigral fluoxetine. Brain Res, 593, 287–90. [DOI] [PubMed] [Google Scholar]
- Paterson DS, Trachtenberg FL, Thompson EG, et al. (2006). Multiple serotonergic brainstem abnormalities in sudden infant death syndrome. JAMA, 296, 2124–32. [DOI] [PubMed] [Google Scholar]
- Paulhus K, Ammerman L & Glasscock E (2020). Clinical Spectrum of KCNA1 Mutations: New Insights into Episodic Ataxia and Epilepsy Comorbidity. Int J Mol Sci, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrucci AN, Joyal KG, Purnell BS, et al. (2020). Serotonin and sudden unexpected death in epilepsy. Exp Neurol, 325, 113145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda J & Aghajanian GK (1997). Carbon dioxide regulates the tonic activity of locus coeruleus neurons by modulating a proton- and polyamine-sensitive inward rectifier potassium current. Neuroscience, 77, 723–43. [DOI] [PubMed] [Google Scholar]
- Poets CF, Meny RG, Chobanian MR, et al. (1999). Gasping and other cardiorespiratory patterns during sudden infant deaths. Pediatr Res, 45, 350–4. [DOI] [PubMed] [Google Scholar]
- Pranzatelli MR, Tate E, Huang Y, et al. (1995). Neuropharmacology of progressive myoclonus epilepsy: response to 5-hydroxy-L-tryptophan. Epilepsia, 36, 783–91. [DOI] [PubMed] [Google Scholar]
- Provine RR (2016). Laughter as a scientific problem: An adventure in sidewalk neuroscience. J Comp Neurol, 524, 1532–9. [DOI] [PubMed] [Google Scholar]
- Ptak K, Yamanishi T, Aungst J, et al. (2009). Raphe neurons stimulate respiratory circuit activity by multiple mechanisms via endogenously released serotonin and substance P. J Neurosci, 29, 3720–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purnell B, Murugan M, Jani R, et al. (2021). The Good, the Bad, and the Deadly: Adenosinergic Mechanisms Underlying Sudden Unexpected Death in Epilepsy. Front Neurosci, 15, 708304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhone AE, Kovach CK, Harmata GI, et al. (2020). A human amygdala site that inhibits respiration and elicits apnea in pediatric epilepsy. JCI Insight, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richerson GB (1995). Response to CO2 of neurons in the rostral ventral medulla in vitro. J Neurophysiol, 73, 933–44. [DOI] [PubMed] [Google Scholar]
- Richerson GB (2004). Serotonergic neurons as carbon dioxide sensors that maintain pH homeostasis. Nat Rev Neurosci, 5, 449–61. [DOI] [PubMed] [Google Scholar]
- Richerson GB, Boison D, Faingold CL, et al. (2016). From unwitnessed fatality to witnessed rescue: Pharmacologic intervention in sudden unexpected death in epilepsy. Epilepsia, 57 Suppl 1, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richerson GB & Boron WF (2003). Control of ventilation. In: BOULPAEP E & BORON WF (eds.) Textbook of Medical Physiology. 3 ed. New York, NY: WB Saunders. [Google Scholar]
- Richerson GB & Buchanan GF (2011). The serotonin axis: Shared mechanisms in seizures, depression, and SUDEP. Epilepsia, 52 Suppl 1, 28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez ML, Mcmillan K, Crandall LA, et al. (2012). Hippocampal asymmetry and sudden unexpected death in infancy: a case report. Forensic Sci Med Pathol, 8, 441–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruber T, David B, Luchters G, et al. (2018). Evidence for peri-ictal blood-brain barrier dysfunction in patients with epilepsy. Brain, 141, 2952–2965. [DOI] [PubMed] [Google Scholar]
- Ryvlin P, Nashef L, Lhatoo SD, et al. (2013). Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): a retrospective study. Lancet Neurol, 12, 966–77. [DOI] [PubMed] [Google Scholar]
- Sainju RK, Dragon DN, Winnike HB, et al. (2019). Ventilatory response to CO2 in patients with epilepsy. Epilepsia, 60, 508–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savic I, Lindstrom P, Gulyas B, et al. (2004). Limbic reductions of 5-HT1A receptor binding in human temporal lobe epilepsy. Neurology, 62, 1343–51. [DOI] [PubMed] [Google Scholar]
- Schraeder PL, Delin K, Mcclelland RL, et al. (2009). A nationwide survey of the extent of autopsy in sudden unexplained death in epilepsy. Am J Forensic Med Pathol, 30, 123–6. [DOI] [PubMed] [Google Scholar]
- Severson CA, Wang W, Pieribone VA, et al. (2003). Midbrain serotonergic neurons are central pH chemoreceptors. Nat Neurosci, 6, 1139–40. [DOI] [PubMed] [Google Scholar]
- Seyal M & Bateman LM (2009). Ictal apnea linked to contralateral spread of temporal lobe seizures: Intracranial EEG recordings in refractory temporal lobe epilepsy. Epilepsia, 50, 2557–62. [DOI] [PubMed] [Google Scholar]
- Sharma V, Babu PP, Singh A, et al. (2007). Iron-induced experimental cortical seizures: electroencephalographic mapping of seizure spread in the subcortical brain areas. Seizure, 16, 680–90. [DOI] [PubMed] [Google Scholar]
- Shaw E & Woolley DW (1954). Pharmacological properties of some antimetabolites of serotonin having unusually high activity on isolated tissues. J Pharmacol Exp Ther, 111, 43–53. [PubMed] [Google Scholar]
- Shaywitz BA, Cohen DJ & Bowers MB (1975). Reduced cerebrospinal fluid 5-hydroxyindoleacetic acid and homovanillic acid in children with epilepsy. Neurology, 25, 72–9. [DOI] [PubMed] [Google Scholar]
- Shmuely S, Sisodiya SM, Gunning WB, et al. (2016). Mortality in Dravet syndrome: A review. Epilepsy Behav, 64, 69–74. [DOI] [PubMed] [Google Scholar]
- Shorvon S & Tomson T (2011). Sudden unexpected death in epilepsy. Lancet, 378, 2028–2038. [DOI] [PubMed] [Google Scholar]
- Simeone KA, Hallgren J, Bockman CS, et al. (2018). Respiratory dysfunction progresses with age in Kcna1-null mice, a model of sudden unexpected death in epilepsy. Epilepsia, 59, 345–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeone KA, Matthews SA, Rho JM, et al. (2016). Ketogenic diet treatment increases longevity in Kcna1-null mice, a model of sudden unexpected death in epilepsy. Epilepsia, 57, e178–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirven JI (2015). Epilepsy: A Spectrum Disorder. Cold Spring Harb Perspect Med, 5, a022848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skluzacek JV, Watts KP, Parsy O, et al. (2011). Dravet syndrome and parent associations: the IDEA League experience with comorbid conditions, mortality, management, adaptation, and grief. Epilepsia, 52 Suppl 2, 95–101. [DOI] [PubMed] [Google Scholar]
- Smart SL, Lopantsev V, Zhang CL, et al. (1998). Deletion of the K(V)1.1 potassium channel causes epilepsy in mice. Neuron, 20, 809–19. [DOI] [PubMed] [Google Scholar]
- Smith HR, Leibold NK, Rappoport DA, et al. (2018). Dorsal Raphe Serotonin Neurons Mediate CO2-Induced Arousal from Sleep. J Neurosci, 38, 1915–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JC, Ellenberger HH, Ballanyi K, et al. (1991). Pre-Botzinger complex: a brainstem region that may generate respiratory rhythm in mammals. Science, 254, 726–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WK (1938). The Representation of Respiratory Movements in the Cerebral Cortex. Journal of Neurophysiology, 1, 55–68. [Google Scholar]
- Southall DP, Richards J, Brown DJ, et al. (1980). 24-hour tape recordings of ECG and respiration in the newborn infant with findings related to sudden death and unexplained brain damage in infancy. Arch Dis Child, 55, 7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowers LP, Massey CA, Gehlbach BK, et al. (2013). Sudden unexpected death in epilepsy: fatal post-ictal respiratory and arousal mechanisms. Respir Physiol Neurobiol, 189, 315–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer WG & Horsley V. a. H. (1894). The effect produced upon respiration by faradic excitation of the cerebrum in the monkey, dog, cat, and rabbit. Philosophical Transactions of the Royal Society of London. (B.), 185, 609–657. [Google Scholar]
- Stewart M (2018). An explanation for sudden death in epilepsy (SUDEP). J Physiol Sci, 68, 307–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surges R, Thijs RD, Tan HL, et al. (2009). Sudden unexpected death in epilepsy: risk factors and potential pathomechanisms. Nat Rev Neurol, 5, 492–504. [DOI] [PubMed] [Google Scholar]
- Sveinsson O, Andersson T, Carlsson S, et al. (2017). The incidence of SUDEP: A nationwide population-based cohort study. Neurology, 89, 170–177. [DOI] [PubMed] [Google Scholar]
- Sveinsson O, Andersson T, Mattsson P, et al. (2020). Clinical risk factors in SUDEP: A nationwide population-based case-control study. Neurology, 94, e419–e429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecott LH, Sun LM, Akana SF, et al. (1995). Eating disorder and epilepsy in mice lacking 5-HT2C serotonin receptors. Nature, 374, 542–6. [DOI] [PubMed] [Google Scholar]
- Teran FA, Massey CA & Richerson GB (2014). Serotonin neurons and central respiratory chemoreception: where are we now? Prog Brain Res, 209, 207–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodore WH, Hasler G, Giovacchini G, et al. (2007). Reduced hippocampal 5HT1A PET receptor binding and depression in temporal lobe epilepsy. Epilepsia, 48, 1526–30. [DOI] [PubMed] [Google Scholar]
- Thurman DJ, Beghi E, Begley CE, et al. (2011). Standards for epidemiologic studies and surveillance of epilepsy. Epilepsia, 52 Suppl 7, 2–26. [DOI] [PubMed] [Google Scholar]
- Thurman DJ, Hesdorffer DC & French JA (2014). Sudden unexpected death in epilepsy: assessing the public health burden. Epilepsia, 55, 1479–85. [DOI] [PubMed] [Google Scholar]
- Toczek MT, Carson RE, Lang L, et al. (2003). PET imaging of 5-HT1A receptor binding in patients with temporal lobe epilepsy. Neurology, 60, 749–56. [DOI] [PubMed] [Google Scholar]
- Tomson T, Surges R, Delamont R, et al. (2016). Who to target in sudden unexpected death in epilepsy prevention and how? Risk factors, biomarkers, and intervention study designs. Epilepsia, 57 Suppl 1, 4–16. [DOI] [PubMed] [Google Scholar]
- Toyoda I, Bower MR, Leyva F, et al. (2013). Early activation of ventral hippocampus and subiculum during spontaneous seizures in a rat model of temporal lobe epilepsy. J Neurosci, 33, 11100–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer JS & Rhodes KJ (2004). Localization of voltage-gated ion channels in mammalian brain. Annu Rev Physiol, 66, 477–519. [DOI] [PubMed] [Google Scholar]
- Trudeau MM, Dalton JC, Day JW, et al. (2006). Heterozygosity for a protein truncation mutation of sodium channel SCN8A in a patient with cerebellar atrophy, ataxia, and mental retardation. J Med Genet, 43, 527–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truscott TC (1975). Effects of phenylalanine and 5-hydroxytryptophan on seizure severity in mice. Pharmacol Biochem Behav, 3, 939–41. [DOI] [PubMed] [Google Scholar]
- Tupal S & Faingold CL (2006). Evidence supporting a role of serotonin in modulation of sudden death induced by seizures in DBA/2 mice. Epilepsia, 47, 21–6. [DOI] [PubMed] [Google Scholar]
- Tupal S & Faingold CL (2019). Fenfluramine, a serotonin-releasing drug, prevents seizure-induced respiratory arrest and is anticonvulsant in the DBA/1 mouse model of SUDEP. Epilepsia, 60, 485–494. [DOI] [PubMed] [Google Scholar]
- Twarog BM & Page IH (1953). Serotonin content of some mammalian tissues and urine and a method for its determination. Am J Physiol, 175, 157–61. [DOI] [PubMed] [Google Scholar]
- Tzoumaka E, Tischler AC, Sangameswaran L, et al. (2000). Differential distribution of the tetrodotoxin-sensitive rPN4/NaCh6/Scn8a sodium channel in the nervous system. J Neurosci Res, 60, 37–44. [DOI] [PubMed] [Google Scholar]
- Uteshev VV, Tupal S, Mhaskar Y, et al. (2010). Abnormal serotonin receptor expression in DBA/2 mice associated with susceptibility to sudden death due to respiratory arrest. Epilepsy Res, 88, 183–8. [DOI] [PubMed] [Google Scholar]
- Van Vliet EA, Aronica E & Gorter JA (2015). Blood-brain barrier dysfunction, seizures and epilepsy. Semin Cell Dev Biol, 38, 26–34. [DOI] [PubMed] [Google Scholar]
- Veasey SC, Fornal CA, Metzler CW, et al. (1995). Response of serotonergic caudal raphe neurons in relation to specific motor activities in freely moving cats. J Neurosci, 15, 5346–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veasey SC, Fornal CA, Metzler CW, et al. (1997). Single-unit responses of serotonergic dorsal raphe neurons to specific motor challenges in freely moving cats. Neuroscience, 79, 161–9. [DOI] [PubMed] [Google Scholar]
- Veeramah KR, O’brien JE, Meisler MH, et al. (2012). De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet, 90, 502–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venit EL, Shepard BD & Seyfried TN (2004). Oxygenation prevents sudden death in seizure-prone mice. Epilepsia, 45, 993–6. [DOI] [PubMed] [Google Scholar]
- Vilella L, Lacuey N, Hampson JP, et al. (2019a). Incidence, Recurrence, and Risk Factors for Peri-ictal Central Apnea and Sudden Unexpected Death in Epilepsy. Front Neurol, 10, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilella L, Lacuey N, Hampson JP, et al. (2019b). Postconvulsive central apnea as a biomarker for sudden unexpected death in epilepsy (SUDEP). Neurology, 92, e171–e182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriend JP & Alexiuk NA (1996). Effects of valproate on amino acid and monoamine concentrations in striatum of audiogenic seizure-prone Balb/c mice. Mol Chem Neuropathol, 27, 307–24. [DOI] [PubMed] [Google Scholar]
- Wada Y, Shiraishi J, Nakamura M, et al. (1995). Prolonged but not acute fluoxetine administration produces its inhibitory effect on hippocampal seizures in rats. Psychopharmacology (Berl), 118, 305–9. [DOI] [PubMed] [Google Scholar]
- Wagnon JL, Barker BS, Hounshell JA, et al. (2016). Pathogenic mechanism of recurrent mutations of SCN8A in epileptic encephalopathy. Ann Clin Transl Neurol, 3, 114–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagnon JL, Korn MJ, Parent R, et al. (2015). Convulsive seizures and SUDEP in a mouse model of SCN8A epileptic encephalopathy. Hum Mol Genet, 24, 506–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Kunkel DD, Schwartzkroin PA, et al. (1994). Localization of Kv1.1 and Kv1.2, two K channel proteins, to synaptic terminals, somata, and dendrites in the mouse brain. J Neurosci, 14, 4588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Bradley SR & Richerson GB (2002). Quantification of the response of rat medullary raphe neurones to independent changes in pH(o) and P(CO2). J Physiol, 540, 951–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Pizzonia JH & Richerson GB (1998). Chemosensitivity of rat medullary raphe neurones in primary tissue culture. J Physiol, 511 (Pt 2), 433–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Tiwari JK, Bradley SR, et al. (2001). Acidosis-stimulated neurons of the medullary raphe are serotonergic. J Neurophysiol, 85, 2224–35. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Hara K, Hakamada S, et al. (1982). Seizures with apnea in children. Pediatrics, 70, 87–90. [PubMed] [Google Scholar]
- Wenker IC, Teran FA, Wengert ER, et al. (2021). Postictal Death Is Associated with Tonic Phase Apnea in a Mouse Model of Sudden Unexpected Death in Epilepsy. Ann Neurol, 89, 1023–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel HJ, Vacher H, Clark E, et al. (2007). Structural consequences of Kcna1 gene deletion and transfer in the mouse hippocampus. Epilepsia, 48, 2023–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Merrick DK & Catterall WA (1989). Differential subcellular localization of the RI and RII Na+ channel subtypes in central neurons. Neuron, 3, 695–704. [DOI] [PubMed] [Google Scholar]
- Wu Y, Proch KL, Teran FA, et al. (2019). Chemosensitivity of Phox2b-expressing retrotrapezoid neurons is mediated in part by input from 5-HT neurons. J Physiol, 597, 2741–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuskaitis CJ, Jones BM, Wolfson RL, et al. (2018). A mouse model of DEPDC5-related epilepsy: Neuronal loss of Depdc5 causes dysplastic and ectopic neurons, increased mTOR signaling, and seizure susceptibility. Neurobiol Dis, 111, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelano C, Jiang H, Zhou G, et al. (2016). Nasal Respiration Entrains Human Limbic Oscillations and Modulates Cognitive Function. J Neurosci, 36, 12448–12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng C, Long X, Cotten JF, et al. (2015). Fluoxetine prevents respiratory arrest without enhancing ventilation in DBA/1 mice. Epilepsy Behav, 45, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Q, Buchanan GF, Motelow JE, et al. (2016). Impaired serotonergic brainstem function during and after seizures. J Neurosci, 36, 2711–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zhao H, Zeng C, et al. (2018). Optogenetic activation of 5-HT neurons in the dorsal raphe suppresses seizure-induced respiratory arrest and produces anticonvulsant effect in the DBA/1 mouse SUDEP model. Neurobiol Dis, 110, 47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZQ, Scott M, Chiechio S, et al. (2006). Lmx1b is required for maintenance of central serotonergic neurons and mice lacking central serotonergic system exhibit normal locomotor activity. J Neurosci, 26, 12781–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuberi SM, Eunson LH, Spauschus A, et al. (1999). A novel mutation in the human voltage-gated potassium channel gene (Kv1.1) associates with episodic ataxia type 1 and sometimes with partial epilepsy. Brain, 122 (Pt 5), 817–25. [DOI] [PubMed] [Google Scholar]