

Abstract
In the past decade, single-cell technologies have proliferated and improved from their technically challenging beginnings to become common laboratory methods capable of determining the expression of thousands of genes in thousands of cells simultaneously. The field has progressed by taking the CNS as a primary research subject — the cellular complexity and multiplicity of neuronal cell types provide fertile ground for the increasing power of single-cell methods. Current single-cell RNA sequencing methods can quantify gene expression with sufficient accuracy to finely resolve even subtle differences between cell types and states, thus providing a great tool for studying the molecular and cellular repertoire of the CNS and its disorders. However, single-cell RNA sequencing requires the dissociation of tissue samples, which means that the interrelationships between cells are lost. Spatial transcriptomic methods bypass tissue dissociation and retain this spatial information, thereby allowing gene expression to be assessed across thousands of cells within the context of tissue structural organization. Here, we discuss how single-cell and spatially resolved transcriptomics have been contributing to unravelling the pathomechanisms underlying brain disorders. We focus on three areas where we feel these new technologies have provided particularly useful insights: selective neuronal vulnerability, neuroimmune dysfunction and cell-type-specific treatment response. We also discuss the limitations and future directions of single-cell and spatial RNA sequencing technologies.
Subject terms: Diseases of the nervous system, Amyotrophic lateral sclerosis, Neurological disorders
In this Review, the authors discuss the ways in which single-cell and spatially resolved transcriptomics are contributing to our understanding of the pathophysiology of neurological conditions. They also discuss the limitations and possible future directions of these technologies.
Key points
High-throughput single-cell technologies enable multiple layers of molecular biology to be probed at the single-cell level.
Single-cell transcriptomic atlases spanning multiple developmental stages have been generated with the aim of dissecting the cellular complexity of the human brain.
Single-cell RNA sequencing technologies increase our understanding of pathomechanisms of brain disorders, providing information on selective neuronal vulnerability, neuroimmune aspects and cell-type-specific treatment responses.
Spatially resolved transcriptomics provides information about the relationships between spatial tissue organization and dysregulated molecular networks in the vicinity of pathogenic hallmarks.
Single-cell transcriptomic methods have proven useful in the biomedical field and are emerging as valuable future tools for diagnostics and the development of precision treatments in clinical practice.
Introduction
The first attempts to combine brain histology with gene expression aimed to detect multiple transcripts in situ in brain sections from individuals with tuberous sclerosis. In the mid-1990s, Eberwine and co-workers1 developed a multistep protocol consisting of Nestin immunostaining, in situ reverse transcription, in situ cDNA synthesis, aspiration of labelled cells and detection of mRNAs by reverse northern blotting. That elegant yet technically complex procedure enabled detection of over 20 mRNAs in individual nestin-positive giant cells and neurons within cortical tubers1. However, high-throughput determination of gene expression profiles in individual neural cells has become accessible only in the past decade. This advance was the result of next-generation DNA sequencing technologies, the adaptation of molecular biology techniques to subnanomolar amounts of starting material and scaling-up of computational analyses to accommodate large numbers of samples. To date, the most mature single-cell investigation technique merged with genome-scale analysis is single-cell RNA sequencing (scRNA-seq). Although scRNA-seq has limitations, including data sparsity and low detection efficiency, it enables the measurement of thousands of transcripts in thousands of cells in a single experiment.
The rapid evolution of scRNA-seq methods has led to manifold discoveries over a short time, including the identification of novel cell types and subtypes2–4, description of rare cell populations5,6, insights into the evolution of the human brain6,7, revisions to established differentiation hierarchies8 and characterization of cell-state changes during development and in response to external stimuli9,10. In addition to providing a static snapshot of cell states within a given tissue or organ, rapid advances in computational methods have enabled the use of single-nucleus RNA sequencing (snRNA-seq) and scRNA-seq to characterize dynamic processes in cells (Figs. 1 and 2). Furthermore, novel spatial transcriptomic methods are now providing a structural layer of information. This spatially resolved RNA sequencing can provide information about the relationships between spatial organization and function, which is crucial for studying dysregulated cellular networks in the vicinity of pathogenic hallmarks.
Fig. 1. Experimental pipeline of a droplet-based single-cell RNA sequencing in a nutshell.

a, Generation of single-cell or single-nuclei suspensions from a tissue. b, Use of microfluidic device to encapsulate the individual cells or nuclei in nanolitre droplets with barcoded beads. Next steps include: cell lysis, capture of polyadenylated RNA, reverse transcription combined with the introduction of unique molecular identifiers and cell barcodes, and amplification and fragmentation of cDNA. c, Next-generation sequencing of the obtained cDNA library is performed on a standard platform, typically by solid-phase amplification.
Fig. 2. Analysis of single-cell RNA sequencing data set can provide multiple types of information on cell types, states and their activation and enables inference of dynamic cellular processes.

a, High-dimensional single-cell RNA sequencing data can be visualized by using dimensionality reduction algorithms to reveal cell clusters. b, Cell clusters are assigned to specific cell types and subtypes on the basis of marker genes. c, Deeper analysis of single-cell transcriptomes can provide information about cell states and activation or uncover rare or new cell subtypes. d, Advanced algorithms and computational tools are designed to infer differentiation trajectories and transition states or compare phenotypes at the cellular level. AD, Alzheimer disease; ALS, amyotrophic lateral sclerosis; CSC, cancer stem cell; DAM, disease-associated microglia; MS, multiple sclerosis; RA, reactive astrocyte; t-SNE, t-distributed stochastic neighbour embedding; WT, wild type. Part d reprinted with permission from ref. 81, Elsevier.
In this Review, we describe how single-cell technologies have advanced our understanding of several aspects related to brain diseases: selective neuronal vulnerability, neuroimmune dysfunction and heterogeneity of brain tumour cells. We comment on the current limitations and challenges of single-cell techniques and provide our perspective on future applications of these technologies to neurology research.
Single-cell technologies
scRNA-seq and snRNA-seq
The first description of single-cell transcriptome analysis based on next-generation sequencing was published in 2009, wherein single mouse blastomeres and oocytes were manually picked under the microscope and subjected to mRNA sequencing11. Since then, multiple efforts have been made to improve and scale up the technology. Important milestones include the introduction of unique molecular identifiers for accurate RNA molecule counting12, cell barcoding13, adapting microfluidics for cell capture14,15, optimization of experimental workflows and streamlining of computational analysis. As the degree of automation and the throughput have increased, the cost per cell has substantially reduced. This progress was accelerated with the introduction, in 2015, of droplet-based microfluidic methods for scRNA-seq (Drop-seq16 and inDrop17). Droplet-based capture has also been successfully adapted for snRNA-seq. snRNA-seq has been shown to provide an effective alternative to scRNA-seq for the analysis of cellular diversity18,19. The biggest advantages of snRNA-seq over scRNA-seq are that nuclei isolation can be easily applied to cells that are difficult to dissociate intact, such as neurons, and that it can be performed from cryopreserved tissues (Supplementary information). The latter is particularly important in the context of clinical tissue samples. snRNA-seq has been widely used in brain research, allowing the analysis of archived clinical material, which is stored through brain banks as flash-frozen or fixed tissue19–22. Other available approaches for sequencing multiple cells or nuclei at a time include combinatorial indexing (as in SPLiT-seq23 or sci-RNA-seq24) as well as Microwell-seq, which uses an agarose-constructed microwell array and barcoded beads25. Microwell-seq has been used for the single-cell dissection of over 50 fetal and adult human tissues within the human cell landscape atlas26. The most commonly used platforms for single-cell, single-nucleus and spatial RNA sequencing are summarized in Table 1.
Table 1.
Comparison of different single-cell and single-nucleus RNA sequencing methods
| Method | Advantages | Disadvantages | Platforms |
|---|---|---|---|
| Single-cell RNA sequencing | Dissociated cells can be stored fixed in methanol |
Requires cells dissociation from fresh tissue Not applicable to adult brain tissue Transcriptomes perturbed during isolation No spatial information preserved |
Smart-Seq147 Smart-Seq2 (ref. 148) Fluidigm C1 (ref. 149) Drop-seq16 10× Chromium150 CEL-seq2 (ref. 151) inDrop-seq17 MATQ-seq152 ScNaUMi-seq153 |
| Single-nucleus RNA sequencing |
Fresh, fixed or frozen tissue can be used Applicable to adult or sensitive tissue Reduced perturbation of transcriptomes during isolation High throughput |
Dissociated nuclei cannot be stored No spatial information preserved Mostly nuclear transcripts measured |
sNuc-Seq154 Div-Seq154 DroNc-seq19 |
| Spatial RNA sequencing |
Fresh and fixed tissue can be used Spatial information preserved |
Low rate or limited number of mRNA captures Low throughput |
Seq-FISH155 MERFISH156 Slide-seq157 GeoMx32 CosMx33 |
The main steps of an scRNA-seq experiment are: isolation of live single cells, cell lysis, capture of polyadenylated RNAs, reverse transcription, cDNA amplification, library preparation and sequencing (Fig. 1). Computational data analysis is an integral part of single-cell RNA sequencing. In brief, computational workflows consist of several steps including read alignment, generation of gene and unique molecular identifier (UMI) counts, cell quality control, data normalization and downstream analyses such as clustering of cells, differential gene expression and, in the case of cycling and differentiating cells, pseudotemporal ordering. A common practice for clustering and interpreting sequencing results is to preprocess the data by selecting highly variable genes and performing dimensionality reduction using principal component analysis, t-distributed stochastic neighbour embedding or uniform manifold approximation and projection27. An overview of the most commonly used computational tools is provided in Box 1. Several scRNA-seq analysis pipelines have been developed that guide the user from raw data to defined single-cell populations28, and a detailed step-by-step scRNA-seq data processing and analysis outline can be found in ref. 29.
Box 1 Summary of the most commonly used single-cell computational tools.
Read alignment
Quality control
Normalization
Batch correction and merging of datasets
Imputation
Visualization
UMAP27
t-SNE
Gene expression quantification
Pseudotime and trajectory interference
Spatially resolved RNA-seq
Spatially resolved RNA sequencing, or spatial transcriptomics, generates quantitative transcriptome-wide RNA-seq data from tissue sections. This result is achieved by capturing polyadenylated RNA on arrays of spatially barcoded DNA capture probes and introducing positional molecular barcodes in the cDNA synthesis reaction30 (Fig. 3). These positional barcodes enable each transcript to be mapped back to its original spot on the tissue section. Spatial transcriptomics techniques have been commercialized; they include the Visium Spatial Gene Expression platform30,31 (10× Genomics), GeoMx32 (NanoString Technologies), CosMx33 (NanoString Technologies) and Molecular Cartography34 (Resolve Biosciences), and the latter three are in situ hybridization-based spatial profiling platforms (Supplementary information). Although spatial transcriptomics methodologies are still in their infancy, they hold promise for gleaning essential insights into the molecular mechanisms that trigger and maintain disease phenotypes. Of note, computational strategies that couple single-cell and spatially resolved transcriptomics have been introduced, and tools such as cell2location35 are designed to perform a joint analysis of multiple scRNA-seq and spatial transcriptomic datasets.
Fig. 3. Spatial transcriptomics: the principle and a workflow.

a, The tissue is subjected to cryosectioning on an mRNA capture slide, fixation and permeabilization to release RNA. The poly-A tail of the mRNA binds to an oligo(dT) ending fragment (single-stranded sequence of deoxythymines) on the capture DNA probes, which also contain embedded positional barcodes. b, After library preparation and sequencing, the computational analysis includes retrieval of the positional barcodes and tissue coordinates to reconstruct the relationship between transcripts and their locations.
Latest developments
Single-cell technologies are now moving in two main directions. The first is to perform integrated, multiple measurements from individual single cells. A great example is Patch-seq, a method in which, after electrophysiological recording in a brain slice, the neurons are aspirated and subjected to an scRNA-seq procedure to profile their transcriptomes36,37. The second direction is the combination of measurements from multiple molecular layers in single cells; that is, overlaying transcriptomic data with proteomic, metabolomic or chromatin accessibility data in an approach termed single-cell multi-omics. For example, cellular indexing of transcriptomes and epitopes by sequencing integrates protein and transcriptome measurements into an efficient, single-cell readout38. It uses oligonucleotide-conjugated antibodies to combine multiplexed protein-marker detection with unbiased transcriptome profiling of single cells. The possible applications of multiomics approaches in the fields of neuroscience and neurology are discussed in more detail in this Review.
Exploring brain complexity
Surveying the mouse CNS provided a showcase for single-cell RNA transcriptomics. In 2015, Zeisel et al.2 analysed the transcriptomes of more than 3,000 individual cells from the mouse somatosensory cortex and hippocampal CA1 region. The next year, Tasic et al.3 reported transcriptomic characteristics of more than 1,600 single cells from the visual cortex. Both studies provided information on the transcriptomic diversity of neurons and non-neuronal brain cell types and highlighted the exploratory power of scRNA-seq. Since these initial publications, both embryonic and adult mouse brains have been exhaustively studied using single-cell RNA transcriptomic methods (Box 2). Various transcriptomic cell atlases have been generated for many regions of the nervous system and developmental stages in mouse39–41, and increasing numbers are available for human and non-human primates4,42–46. Jointly, these atlases reveal highly complex transcription factor network topology and excitability-related gene expression for neurons within numerous identified types and subtypes, depending on the brain region. These findings have markedly expanded the repertoire of morphology-based and connectivity-based definitions of neuronal types and subtypes. Non-neuronal cells generally display less diversity than neurons; however, substantial regional specificity has been reported for astrocytes47 and microglia48. Additionally, many non-neuronal cell types can exhibit a wide range of cell states under different physiological or diseased conditions49.
The growing amount of single-cell transcriptomic data has contributed to a paradigm shift in brain cell taxonomy, and the new gene-expression-based taxonomies3,42,50,51 have been compared and combined with traditional morphological, physiological and connectomic taxonomies52. For example, by combining biocytin staining, Patch-seq and multiplexed error-robust fluorescence in situ hybridization of primary motor cortex, researchers from the BRAIN Initiative Cell Census Network connected transcriptome-based cell-type taxonomies with physiology, morphology and circuit-based brain function53. This pilot study revealed good correspondence among molecular, anatomical and physiological datasets, thereby reinforcing the transcriptomic classification of neuronal types and subtypes. Another study combined single-projection-level analysis of neuron morphology with single-cell transcriptomic profiles and reported overall good concordance between the transcriptome and the major projection types defined by morphology54. However, worse correlation was observed between the fine-grained subtypes defined with the two different methods. Cortical GABAergic interneurons have also been classified using a combination of transcriptomic and physiological properties, showing robust mutual predictability of both properties55. Such cross-modality studies are necessary to achieve a proper description of complete single-cell anatomy and cell-type classification in healthy and pathological tissues, including the brain. Importantly, emerging transcriptome-based cell-type taxonomies of the homeostatic, healthy mammalian brain provide crucial references for studies focusing on pathologies, neurodevelopment and evolution4,25,39,46,53,56.
Indeed, single-cell transcriptomic approaches have now also been applied to the study of diseased CNS tissue, and in the subsequent sections we provide an overview of how scRNA-seq and spatial transcriptomics have contributed to our current understanding of pathomechanisms of brain disorders, including neurodegenerative diseases, neurodevelopmental disorders, psychiatric diseases and brain tumours (Fig. 4). We focus on three topics in which we feel the most progress has been made: selective neuronal vulnerability, neuroimmune responses and cell-type sensitivity to treatment.
Fig. 4. Single-cell RNA-sequencing, single-nuclei RNA-sequencing and spatial transcriptomics studies reveal cellular and molecular heterogeneity in human neurological disorders.

Samples collected from the brain, spinal cord, cerebrospinal fluid (CSF) and peripheral blood have been used to analyse transcriptomes of thousands of cells and/or nuclei from individuals with multiple sclerosis (MS)87,158–160, Rett syndrome (Rett S)161, Alzheimer disease (AD)60,82,84,162,163, Huntington disease (HD)164, COVID-19 (refs. 98–100), amyotrophic lateral sclerosis (ALS)83,165, major depressive disorder (MDD)65, schizophrenia (SCZ)166 and autism spectrum disorders (ASD)71,167 and to compare them with healthy controls. Such an approach has also been used to compare different neurodevelopmental (Dev) stages168,169. GM, grey matter; WM, white matter.
Box 2 Expanding landscape of the brain cell-type diversity.
Neuronal and non-neuronal cell types have been characterized with single-cell transcriptomics in multiple regions of the mouse brain, including the striatum50, midbrain189,190, hypothalamus191–194, suprachiasmatic nucleus195, lateral geniculate nucleus of the thalamus196, dorsal and median raphe nuclei197, cerebellum198–200, olfactory bulb201, prefrontal cortex202, visual cortex3,113,203, motor cortex42,203, somatosensory cortex2, developing neocortex114, amygdala112, subventricular zone204,205 and dentate gyrus206,207. Single-cell transcriptomics have provided information on specific cell populations; for example, interneuron diversity within CA1 hippocampus208 and cortex209; dopamine neurons194,210; serotonin neurons197; oligodendrocyte progenitor cells, their transcriptome profiles within the spinal cord and the forebrain10 and stages in the maturation path to mature oligodendrocytes189; mouse brain macrophages211; microglia throughout mouse lifespan49; and across different brain regions9.
In humans, the characterization of single cells or nuclei has been performed in adult visual cortex, frontal cortex and cerebellum212, developing midbrain213 and early stages of human brain development (gestational weeks 6–10: telencephalon, diencephalon, midbrain, hindbrain, cerebellum, ganglionic eminences, thalamus, hypothalamus and cortex)4. Most recently, snRNA-seq of over 3 million nuclei from the human post-mortem brain across the forebrain, midbrain and hindbrain has been reported, revealing 461 clusters and 3,313 subclusters of distinct cell types or subtypes190. These data, which have been posted on a preprint server, are a long-awaited and comprehensive resource that provides new evidence for the molecular complexity of human neural cells and constitutes a valuable reference dataset for comparative studies, for example, those focusing on brain disorders.
Selective neuronal vulnerability
In the majority of neurodegenerative conditions, there are specific groups of cells in the CNS that seem to be particularly vulnerable to degeneration. This selective neuronal vulnerability is a widely appreciated but poorly understood phenomenon. Indeed, our lack of an understanding of the molecular mechanisms underlying selective neuronal vulnerability is recognized as one of the major obstacles to treating neurodegenerative conditions57. However, single-cell technologies have emerged as useful methods to identify intrinsic properties of vulnerable cell types and subtypes; subsequently, we discuss selected studies that we feel illustrate this application of single-cell methods particularly well.
Neurodegenerative disease
Alzheimer disease (AD) is a slowly progressing neurodegenerative disorder and the pathological process begins years, if not decades, before clinical symptoms occur58. In the early stages of AD, neurons from several areas of the brain — for example, principal neurons of the entorhinal cortex layer II, hippocampal CA1 pyramidal cells and pyramidal neurons in neocortex association areas — are particularly susceptible to degeneration. By contrast, other brain regions, such as the primary sensory cortices, are relatively resistant to degeneration59. However, the molecular basis underlying this selective vulnerability remains unclear. In a study published in 2021, a combination of snRNA-seq and quantitative immunofluorescence for different markers of excitatory neurons was used to analyse post-mortem human brain samples, with the aim of defining the vulnerable neuronal populations in the entorhinal cortex of individuals with AD60. In the caudal entorhinal cortex, specific excitatory neuron subpopulations expressing RORB were depleted during disease progression in AD. These RORB-positive excitatory neurons included both large multipolar neurons and pyramidal neurons and preferentially accumulated tau inclusions, which are a key neuropathological hallmark of AD. No substantial changes in the abundance of inhibitory neuron subpopulations were observed, and there was no evidence of selective vulnerability among any subpopulation of inhibitory neurons in the entorhinal cortex60.
Parkinson disease (PD) is defined by the neurodegeneration of nigrostriatal dopaminergic neurons. α-Synuclein aggregates and mitochondrial dysfunction have been implicated in PD pathogenesis, but the mechanisms underlying vulnerability of dopaminergic neurons to neurodegeneration are not yet well understood61. In 2022, snRNA-seq and spatial transcriptomics were applied to the definition of molecular features associated with vulnerability of dopamine neurons within the substantia nigra pars compacta to neurodegeneration in PD62. On the basis of a published scRNA-seq data set of mouse midbrain39, the researchers selected the Nr4a2 gene as a marker of mammalian midbrain dopamine neurons and used fluorescence-activated nuclei sorting to enrich human dopamine neuron samples for NR4A2-expressing cells. In-depth snRNA-seq analysis of dopamine neurons from neurotypical donors revealed 10 subtypes that were then analysed with spatial transcriptomics in macaque brain. The authors identified a subpopulation of dopaminergic neurons characterized by SOX6 and AGTR1 expression that was in a location suggestive of PD susceptibility. Indeed, when samples from individuals with PD were compared with those from neurotypical individuals, the largest decline in dopamine cell number was observed within the SOX6+ AGTR1+ subpopulations, indicating selective vulnerability of these neurons. In another study from the same year, midbrains from individuals with idiopathic PD and age-matched control individuals were analysed post-mortem with snRNA-seq. The researchers identified a small subpopulation of neurons that was proportionally increased in individuals with PD compared with control individuals63. These neurons had a dopaminergic origin; were characterized by high expression of CADPS2, low expression of TH and elevated levels of TIAM1; and, according to validation analyses in laser-microdissected neurons, probably represented degenerating dopamine neurons63.
Neuropsychiatric disorders
In neuropsychiatric disorders, consistent evidence of gross brain malformations or a characteristic neuropathology is lacking. In the case of schizophrenia or major depressive disorder (MDD), a prevalent view is that the underlying pathophysiology involves the dorsolateral prefrontal cortex (DLPFC) and hippocampal areas and their associated circuitries. That view is based on converging evidence from neurophysiological, functional neuroimaging and cognitive studies64. An approach that has been used for disentangling the neuronal types and subtypes involved in neuropsychiatric disorders is to combine scRNA-seq data with information on gene sets known to be perturbed in neuropsychiatric disorders. snRNA-seq data obtained from the DLPFC of individuals with MDD and psychiatrically healthy individuals revealed sets of dysregulated genes that were mainly assigned to two types of cell: immature oligodendrocyte precursors and deep-layer excitatory neurons65. Of the 96 dysregulated genes, 26 were linked to mental illnesses in data from genome-wide association studies (GWASs) provided by the PsyGeNET consortium. Similarly, another study linked genetic associations from a schizophrenia GWAS to specific cell types66. The common genomic variants for schizophrenia consistently mapped to pyramidal cells, medium spiny neurons and specific types of interneurons, but far less consistently to embryonic, progenitor or glial cells. The genetic risk associated with medium spiny neurons did not overlap with that of glutamatergic pyramidal cells and interneurons, suggesting that different cell types have biologically distinct roles in schizophrenia.
Finally, a spatial topography of gene expression in human DLPFC has been published and analysed with respect to gene sets that are perturbed in neuropsychiatric disorders67. The brain sections used for the study encompassed all six grey matter layers of the DLPFC and were collected from three neurotypical adults. To enhance gene annotation, the spatial sequencing was overlaid with snRNA-seq data. Differential gene expression analysis across the laminar organization of the DLPFC revealed sets of layer-enriched genes. Laminar enrichment of genes in the DLPFC has been linked to common genetic variation associated with schizophrenia68, autism spectrum disorder (ASD)69, bipolar disorder and MDD70. The authors identified an overlap between L2-enriched and L5-enriched genes and risk for schizophrenia, with additional overlap between L2-enriched genes and risk for bipolar disorder67. This proof-of-principle spatial transcriptomics study in DLPFC highlighted a preferential layer-enriched expression of the genes implicated in neuropsychiatric disorders and showed the power of spatial transcriptomics for studying cell-specific basis of neuropathologies.
Neurodevelopmental conditions
Similar to neuropsychiatric disorders, neurodevelopmental conditions have a strong and very complex genetic basis, involving many independent loci containing common and rare variants69. scRNA-seq or snRNA-seq of in vivo and/or in vitro models of ASD has been extremely helpful to define how the presence of high-confidence risk genes affects brain development. In one of the first applications of this technology to the study of ASD, Kriegstein and co-workers used snRNA-seq to examine post-mortem tissue samples from prefrontal cortex and anterior cingulate cortex of individuals with ASD, individuals with sporadic epilepsy and control individuals71. They identified cell-type specific dysregulation of gene expression in individuals with ASD compared with the control individuals. The neuronal genes that were most downregulated in ASD were expressed in layer 2 and layer 3 excitatory neurons and vasoactive intestinal polypeptide-expressing interneurons. The most upregulated genes were expressed in non-neuronal cell types: protoplasmic astrocytes and microglia. The dysregulated genes overlapped substantially with high-confidence ASD-associated genetic risk factors71. Another group used in vivo Perturb-seq to functionally evaluate selected de novo ASD or neurodevelopmental delay risk genes72. Using CRISPR–Cas9, they introduced frameshift mutations in 35 risk genes in the developing mouse brain in utero and analysed the transcriptomes in the perturbed cells at the postnatal stages. These experiments helped to identify cell-type-specific and evolutionarily conserved gene modules from both neuronal and glial cell classes and determine how individual mutations affected cell types diversification and activation in the developing brain72.
Next steps
From the studies described earlier, it is clear that single-cell technologies are becoming extremely helpful for the identification of the molecular mechanisms involved in selective neuronal vulnerability. The next step is to perform mechanistic studies with the aim of designing and testing therapeutic strategies to alter the features that make neurons vulnerable to death. In theory, such therapies could slow down or stop the progression of PD, AD or other neurodegenerative disorders. However, it is also very important to investigate whether the identified vulnerable cell types or structures are disease-prone owing to primary (that is, heritable) features or whether their vulnerability is secondary to other influences, for example, from the surrounding neuroimmune compartment (as discussed subsequently). Both possibilities could be assessed with modern genomics and multi-omics approaches, especially if applied to unbiased single-cell or single-nuclei suspensions from a given brain structure. One approach would be to use machine learning to integrate neuron-type-specific molecular profiles from mouse models with post-mortem human functional genomics and quantitative genetics data to pinpoint the functional gene modules that underlie selective vulnerability — this has been achieved for AD59. Pharmacological or genetic (for example, CRISPR-based) perturbations in healthy or diseased conditions, in combination with single-cell profiling (for example, Perturb-seq), could also be used to understand how mutations affect the adaptation of disruptive or restorative cell states and whether these states affect cell-type function or dysfunction.
Neuroimmune dysfunction
Neurodegeneration
An immune contribution to neurodegeneration was regarded as little more than a curiosity a decade ago, but neuroinflammation is now considered to have a role in AD, PD, amyotrophic lateral sclerosis (ALS), Huntington disease and multiple sclerosis (MS). Microglia are the resident immune cells of the CNS and, when activated, they secret pro-inflammatory mediators such as tumour necrosis factor-α, IL-1b and reactive oxygen species, which are detrimental to neurons73. The brain also hosts several other myeloid populations including perivascular cells, meningeal macrophages and choroid plexus macrophages74. Moreover, astrocytes can produce immune factors when activated by neuronal dysfunction and microglial activation75. Single-cell sequencing technologies have become a key tool for studying the landscape and heterogeneity of the neuroimmune compartment. Resource studies in mice and humans revealed higher than expected complexity of microglial cellular states in the healthy brain throughout the lifespan49, as well as during disease9,76. Single-cell transcriptomics of the CNS have also revealed the great plasticity and diversity of other myeloid cell types and reactive astrocytes77–79.
The innate immune response
In a recent AD GWAS, risk loci and disease-associated genes were enriched for immune-related features and immune cell types80. The authors concluded that the data indicate a causal role of the immune system in AD, as opposed to an immune response to AD pathology. Single-cell approaches have been used to characterize the microglial population during neurodegeneration. In a study by Keren-Shaul et al.81, single-microglia transcriptomes were generated from 5XFAD mice — a transgenic model of AD that expresses five human familial AD gene mutations — and a microglial type associated with neurodegenerative disease was identified. These disease-associated microglia (DAM) were characterized by a substantial decrease in expression of homeostatic microglia genes (such as those encoding the purinergic receptors P2RY12/P2RY13, CX3CR1 and TMEM11) and upregulation of known AD risk genes (Apoe, Ctsd, Lpl, Tyrobp and Trem2)81. In another study, researchers examined microglia from the hippocampus of the CK-p25 inducible mouse model, which develops AD-like pathology82. Analysis of four timepoints during the course of neurodegeneration revealed different microglial cell states discriminating early and late responses. In the late stage of neurodegeneration, the researchers identified a subset of microglia typified by strongly upregulated expression of antiviral and interferon response genes and major histocompatibility complex (MHC) class II components. They noted a substantial similarity between the expression profiles of these late-response microglia and the DAMs identified by Keren-Shaul et al.81.
In the study by Keren-Shaul et al.81, the spatial distribution of DAMs was analysed using immunohistochemistry and single-molecule fluorescence in situ hybridization for marker genes — DAMs were located close to Aβ plaques in CK-p25 mouse brains and post-mortem brain samples from individuals with AD and were present in the spinal cords of mSOD1 mice (a model of ALS). In a more recent study, Maniatis et al.83 applied spatial transcriptomics to generate a more detailed profile of gene expression in spinal cord from wild-type mice and the SOD1-G93A mouse model of ALS at several stages of disease progression, as well as from humans with the disease. The spatiotemporal gene expression analysis recapitulated the perturbed expression pattern of several known ALS-associated genes. Furthermore, the results pointed towards a microglial dysfunction preceding astroglial dysfunction in ALS. The microglial dysfunction was observed proximal to motor neurons and occurred well before the onset of disease symptoms83. In another study, induction of LPS-induced neuroinflammation in the mouse brain resulted in distinct reactive astrocyte states and subtypes, revealing spatial (that is, depending on the anatomical localization in the brain) and subtype-specific variation in response, including ‘super-responding’ subpopulations79.
One key question regarding neuroimmune dysfunction in AD is whether reactive microglia and reactive astrocytes have common gene-expression patterns. To test that, and also to uncover novel therapeutic targets for AD, Xu et al.84 combined single-cell and single-nucleus transcriptome data from transgenic mouse models and brains of individuals with AD and compared them with datasets on drug-target networks, metabolite–enzyme associations, human protein–protein interactomes and longitudinally collected patient information. The molecular networks shared between DAMs and disease-associated astrocytes were significantly enriched in neuro-inflammatory pathways and genetic variants associated with an increased risk of AD (that is, BIN1)84.
snRNA-seq has also been used to investigate the cell-type-specific contribution of the pro-inflammatory IL-12 and IL-23 signalling pathways to AD-driven neuroinflammation in the APPS1 mouse model85. The results, which have been posted on a preprint server but not yet published in a peer-reviewed journal, indicated that IL-12, but not IL-23, is the main driver of AD-specific neuroinflammation and that it alters neuronal and oligodendrocyte functions. Interestingly, genetic ablation of IL-12 and IL-23 signalling did not affect the inflammatory gene expression profiles of DAMs, but reversed the loss of mature myelin-producing oligodendrocytes and alterations in neuronal homeostasis in APPPS1 mice. Perhaps targeting of ‘an immune component’, specifically the IL-12 signalling cascade, could be a promising interventional approach in the future treatment of AD.
Application of single-cell technologies has provided a new, exciting chapter for studying MS, a chronic inflammatory, demyelinating and disabling neurodegenerative disorder. Much effort has been directed towards disentangling activated microglial subtypes in demyelinating injury in mice9,49 as well as human MS lesions86. In the experimental autoimmune encephalomyelitis (EAE) mouse model of autoimmune demyelination, single-cell RNA profiles were generated for immune cells isolated from several CNS compartments — including leptomeninges, perivascular space, parenchyma and choroid plexus — and a high level of myeloid cell diversity was observed among compartments77. One study analysed RNA profiles from cells isolated from the cerebrospinal fluid (CSF) and blood of individuals with MS and compared them with those of control individuals. The results indicated that MS is associated with diverse transcriptional changes within cell populations in the blood, without changes in cell-type ratios. By contrast, CSF from individuals with MS showed altered cell-type ratios and an overall increase in cell-type diversity, which could be attributed to compartmentalized mechanisms driving human autoimmunity in the brain87. In another study, Masuda et al.9 performed scRNA-seq on microglia isolated from the brains of individuals with early-active MS and control individuals. The researchers identified an increase in the expression of genes encoding MHC class II-related molecules, as well as SPP1, PADI2 and LPL, in the individuals with MS; this transcriptional profile was similar to that of microglia associated with demyelination in mice9. Interestingly, in mice, the microglial response differed between those with neurodegenerative pathology (facial nerve axotomy) and those with demyelinating pathology (cuprizone)9. Another study profiled microglia in mice during neurodevelopment, ageing and brain injury and observed Spp1+ microglia in the subcortical axon tracts of the corpus callosum in the forebrain, as well as in the axon tracts of the cerebellum49. This specialized subpopulation of cells has been termed axon tract-associated microglia.
More recently, Schirmer et al.86 performed snRNA-seq on samples from cortical and subcortical lesion and non-lesion areas in individuals with MS. The results indicated that most dysregulated genes in MS mapped to vulnerable upper-cortical-layer neurons and reactive glia at the borders of subcortical MS lesions. Activated microglia were enriched for transcripts encoding activation markers, complement factors, MHC class II-associated proteins and lipid degradation proteins (in line with the findings of Masuda et al.9) and were localized to the chronic active boundaries of subcortical MS lesions. Additionally, the authors detected a subpopulation of microglial cells involved in myelin phagocytosis. Together, the studies discussed here indicate the presence of context-dependent transcriptomic subtypes of microglia in MS, raising the possibility that one or more of these subtypes could be targeted therapeutically.
The adaptive immune response
Single-cell transcriptomic approaches have also proven useful for understanding the role of the adaptive immune response, that is, T cells and B cells, in neurodegeneration. scRNA-seq performed on CSF from individuals with MS and healthy individuals identified altered cell-type ratios — an expansion of cytotoxic CD4+ T cell and late-stage B lineage cell populations — in individuals with MS87. The transcriptomic profile of the different cell types did not differ significantly between individuals with MS and healthy individuals. Using a method called the cell-set enrichment analysis, the researchers observed an expansion of the T follicular helper cell population in the CSF of individuals with MS and also in the EAE mouse model. The results of reverse translation experiments in mice indicated that these T follicular helper cells drive local B cell responses in the CNS to promote MS-like autoimmunity87.
More recently, another group of researchers performed single-cell transcriptome and T-cell receptor sequencing on blood and CSF from individuals with PD and healthy individuals; the aim of the study was to decode the composition, function and lineage relationship of T cells in these individuals88. In blood and CSF from individuals with PD, they identified a large population of CD8+ T cells showing continuous progression from central-memory to terminal-effector T cells. Two specific groups of cells — terminal-effector CD8+ T cells and cytotoxic CD4+ T cells — were expanded in individuals with PD compared with healthy individuals. Similar to the MS study described earlier, the results of this study indicate an influence of peripheral T cells on neuronal degeneration and identify specific cell populations likely to be involved. Indeed, lymphocyte infiltration has been observed in post-mortem brain samples from individuals with PD89.
In summary, studies using single-cell transcriptomics have provided evidence that the immune system — specifically, CNS-resident immune cells and infiltrating peripheral immune cells — has an important role in the onset and progression of neurodegenerative disease. Many of these discoveries would not have been possible with bulk RNA sequencing. We expect that this line of research will continue to unravel new aspects of immune system biology that might be translated into clinical practice. In theory, therapeutic strategies that target disease-specific and CNS region-specific myeloid cell subsets could have fewer adverse effects than the global immune suppressive therapies currently administered to individuals with neuroinflammatory disorders. Indeed, in a publication from 2022, researchers describe an approach in mice that induced expression of the immune-modulating cytokine IL-2 specifically in astrocytes, with the aim of targeted delivery of IL-2 to regions of inflammation90. Mice with IL-2 delivery were effectively protected from neuroinflammation during brain injury, stroke or EAE.
Glioma
Glioma is a type of CNS tumour that originates from glial cells and, depending on the cellular origin, it can be classified as oligodendroglioma, astrocytoma, ependymoma or glioblastoma91. Glioma tumour cells, along with the tumour microenvironment, create a complex milieu that ultimately promotes tumour cell adaptability and disease progression. scRNA-seq studies have broadened our view of the tumour microenvironment, especially of the immune cells that occupy the tumour and peritumoural regions. Knowledge afforded by these technologies is providing substantially more granularity to the concept of intratumoural heterogeneity and complex interactions between cancer cells and immune cells. For example, comparison of scRNA-seq profiles from isocitrate dehydrogenase-mutant astrocytomas and oligodendrogliomas, and mutual analysis of 165 bulk RNA profiles from The Cancer Genome Atlas, revealed that isocitrate dehydrogenase-mutant astrocytomas and oligodendrogliomas differed in their tumour microenvironment, in particular, in the abundance of microglia and tumour-associated macrophage cells5. In another study, distinct myeloid cell populations were found in the glioblastoma core when compared with the peritumoural tissue92; that is, tumour-infiltrating macrophages and microglia preferentially occupied the tumour and peritumoural spaces, respectively92. Analysis of resident myeloid subpopulations from naive and glioma-bearing mice suggested that sex-specific gene expression in glioma-activated microglia could be relevant to the incidence and outcomes of glioma93.
In addition to revealing heterogeneity in the immune component of the tumour microenvironment, single-cell analyses have also provided information on how the immune microenvironment alters the physiology and fate of cancer cells. In a study that used a high-throughput single-cell transcriptomic analysis of human tumours and mouse models in combination with functional assays, macrophages were found to promote the transition of cancer cells into a mesenchymal-like state94. Mesenchymal-like cell states were also associated with increased abundance and cytotoxicity of tumour-infiltrating T cells, and targeting these T cells might represent a therapeutic opportunity for mesenchymal-like glioblastomas94. A spatial transcriptomics study published in 2022 identified enhanced immunosuppressive interactions between tumour cells and myeloid cells in segregated niches within glioblastoma tumours95. A combination of spatial transcriptomics and high-throughput proteomics was used to characterize these ‘reactive-immune regions’. It revealed enrichment in tumour-associated myeloid cells, T cells and cells with mesenchymal-like and astrocytic-like transcriptional signatures, described ‘MES–AC-hybrid’states. Overall, these findings could have implications for testing next-generation molecular targets for therapeutic interventions, especially designing immunotherapies for treating glioblastomas.
COVID-19
Although SARS-CoV-2 primarily targets the respiratory system, patients and survivors of COVID-19 often have neurological manifestations, for example, headache, anosmia, aheusia, acute encephalopathy, coma and stroke96. The pathomechanism of neurological abnormalities in COVID-19 is currently not well understood97–99, but single-cell approaches have provided some useful information. In snRNA-seq studies of post-mortem brain tissue from individuals with COVID-19 and control individuals, perturbation of gene expression was observed in the brains of individuals with COVID-19. This perturbation was observed mainly in microglia and astrocytes and was characterized by inflammatory and dysregulated homeostatic pathways, including activation of antiviral defence genes99 and activation of the innate immune response98. Interestingly, some of the detected activation states shared features with pathological cell states that have been reported in human neurodegeneration99. Moreover, peripheral T cells were detected in the brain parenchyma of individuals with COVID-19 but not control individuals99. Analyses of choroid plexus helped to identify an increased proportion of stromal cells, monocytes and macrophages in individuals with COVID-19 (ref. 98) and broad cellular perturbations including upregulation of inflammatory genes across choroid plexus cell types99. Overall, these observations suggest that barrier cells forming choroid plexus sense and relay peripheral inflammation into the brain in COVID-19.
In another study, CSF samples from individuals with COVID-19 who developed neuro-COVID were compared with those from individuals with non-inflammatory and autoimmune neurological diseases or with viral encephalitis100. This comparison revealed an expansion of dedifferentiated monocytes and of exhausted CD4+ T cells that was specific to individuals with COVID-19. Interestingly, individuals with severe neuro-COVID had broader clonal T cell expansion and a lower interferon response than individuals with mild neuro-COVID, which might be the reason for impaired antiviral responses. Leukocytes exhibited disease-specific signs of local immune overactivation, despite the absence of SARS-CoV-2 in the CSF. The findings support the hypothesis that immune-mediated mechanisms contribute to neurological sequelae in individuals with COVID-19 (ref. 100).
Drug-sensitive cell types
Single-cell technologies are becoming recognized as powerful tools to investigate and monitor response to drugs and treatments, a fact that is already well appreciated in cancer research. Elimination of the cancer stem cells that drive tumour progression is considered a promising treatment strategy, and a few early clinical trials of cancer stem cell-targeting therapies are underway101. In a study published in 2021, scRNA-seq revealed multiple subtypes of glioblastoma stem cells within individuals and even greater heterogeneity among individuals102. The common feature of these subtypes was upregulation of cell cycling programmes, with overexpression of genes known to promote self-renewal and progenitor expansion in the neocortex. Furthermore, the gene expression profiles of glioblastoma stem cells correlated strongly with the gene expression profiles of reactive astrocytes, which suggest active inflammatory processes in glioblastoma stem cells102. These findings highlight the heterogeneity of glioblastoma stem cells and raise the possibility of developing future therapies to simultaneously target the developmental and inflammatory processes observed in glioblastoma stem cells.
Another paper from 2021 describes an approach for rapid screening of cell-type-specific drug sensitivities in glioma103. In this approach, an acute slice culture from a freshly resected tumour is subjected to multiplexed drug perturbation followed by scRNA-seq to profile transcriptome-wide drug responses. Such personalized drug screening was performed for six individuals and provided information on cell-type-specific responses to the chemotherapy drugs etoposide and panobinostat. In this study, screening and analysis were completed less than 1 week after surgery, which highlights the potential for this approach to inform the administration of personalized therapies.
Medulloblastoma is the most prevalent malignant (WHO grade 4) brain tumour in children104 and is classified into four major tumour subgroups: wingless, sonic hedgehog (SHH), group 3 and group 4 (refs. 105,106). In 2019, three independent scRNA-seq studies performed on clinical medulloblastoma samples or mouse models were published. The aim of these studies was to address the gap in our understanding of the cells of origin for different medulloblastoma subgroups107 and to define treatment-resistant populations for SHH medulloblastoma108,109. In one of these studies, OLIG2-positive stem-like cells were identified as a key cellular component of SHH medulloblastomas during both the initiation and a recurrence of tumour108. In another study, scRNA-seq was used to investigate the responses to the SHH-pathway inhibitor vismodegib109. Cells expressing the Notch pathway transcription factor HES1 were found to be sensitive to the drug treatment, and MYOD1 expression was observed in vismodegib-resistant tumour cells109.
Challenges and limitations
Sample collection
The major limitations of single-cell transcriptomic approaches stem from technical challenges in obtaining good quality single cells or nuclei from complex brain tissue. Defining the proper conditions of harvesting, handling, freezing and monitoring, the quality of brain samples for scRNA-seq and spatial transcriptomics is crucial, but not yet adopted in standard clinical practice. Also, sample handling time (that is, the time between sample collection and analysis) has a tremendous impact on the quality of single-cell transcriptome data and needs to be reduced to minimum to preserve the quality of RNA; however, this timing is difficult to monitor during surgery110. These initial steps of tissue handling need to be monitored, further optimized and standardized for clinical use to ensure high-quality data and reproducibility.
Isolation of single cells
From the methodological side, isolating intact and live cells from the adult brain without losing their integrity, viability and compromising their RNA content is difficult (see Supplementary information). Transcriptomic analyses of single-cell suspensions are limited to freshly isolated neurosurgical tissues and rely on harsh enzymatic dissociation111. The isolation process might bias the proportions of recovered cell types and subtypes39 or affect gene expression profiles of cells, for example, by induction of stress-dependent transcription112. Solutions to this problem have been proposed, and they are mostly used to minimize the activation profiles in brain and tumour cells that can arise from single-cell dissociation procedures20,112–114. Nuclei are less susceptible to isolation artefacts than whole cells, are easier to isolate intact and can be obtained from cryopreserved material115,116; therefore, for frozen or fixed post-mortem brain tissues, snRNA-seq is the method of choice117,118. However, with snRNA-seq, all the information encrypted in neuronal processes is lost. A growing body of evidence indicates that many genes act locally at the synapse119, so that single-nucleus data might not always capture the full picture of neuronal transcriptomes. Given these limitations, spatial transcriptomics emerges as an attractive method to capture cellular RNA in intact brain tissue, regardless of its low resolution and low throughput. However, it is difficult to perform spatial transcriptomics in a large-scale or high-throughput manner. Spatial transcriptomics experiments are also expensive — in our experience, it costs a few thousand US dollars for library preparation and sequencing of a 6 mm × 6 mm × 10 µm section. Currently, the expense limits access to spatial transcriptomics to well-funded research laboratories and makes the use of the technology in the standard clinical context unlikely unless the cost drops.
Data analysis and interpretation
Owing to high dimensionality and increasing scalability, single-cell datasets are highly complex and their analysis and interpretation are a challenge. First, datasets generated from scRNA-seq or snRNA-seq have technical limitations, mainly associated with a low mRNA capture rate. Because of low capture efficiency, many transcripts seem to be lost during reverse transcription, which generates sparse data with common occurrences of so-called dropout events, when a transcript is detected in one cell but missing in another cell120. To address this shortcoming, imputation methods have been designed to estimate the gene expression of the dropout genes using other cells from the dataset as a guide; these methods include MAGIC121, SAVER122, scImpute123 and RESCUE124. The relatively low mRNA capture rate and high cDNA amplification bias can also lead to distortion in gene expression profiles and can inflate the estimates of cell-to-cell variability125. The technical noise in single-cell data may also contribute to other factors such as batch effects, which are generated when cells from the same group are captured or sequenced separately, and cell-to-cell variability, for example, variability that might be result from differences in cell cycle state125,126. Several of these issues can be corrected using computational methods, and selection of these is summarized in Box 1.
There is a growing need to integrate multiple datasets generated across samples from different timepoints, treatment groups or individuals, as well as samples with different sequencing depths or performed on different platforms. Eventually, integration across different types of single-cell measurement (DNA, RNA and protein) might be required and beneficial. This integration presents another major challenge127, which is particularly important for drawing proper conclusions from comparisons between healthy and diseased tissues. More information on the challenges of computational analysis of scRNA-seq datasets can be found elsewhere120.
Regulatory non-polyadenylated transcripts
scRNA-seq and snRNA-seq techniques are based on the capture and analysis of polyadenylated transcripts, including the majority of mRNA and some of long non-coding RNAs. However, a substantial part of our transcriptome is not polyadenylated and not covered by currently available scRNA-seq or snRNA-seq pipelines. This part of the transcriptome includes transcripts often jointly referred to as ‘regulatory’ RNA molecules, for example, ribosomal RNA, transfer RNA and short RNAs such as microRNAs or piwi-interacting RNAs, small nuclear RNAs, small nucleolar RNAs and non-polyadenylated long non-coding RNAs as well as circular RNAs (circRNAs). Many of these regulatory transcripts (for example, circRNAs) are tissue-specific and regionally enriched in the CNS128,129. In the future, it would be helpful to expand single-cell methodologies in the direction of capturing and analysing these non-polyadenylated RNA classes, which are responsible for numerous transcriptional and post-transcriptional gene expression processes.
Future directions
Single-cell multi-omics
As mentioned earlier, to gain a deeper insight into brain function and human pathologies, we need experimental and computational tools to combine data from multiple single-cell approaches and then to integrate them with clinical data. Single-cell multi-omics technologies aim to overlay gene expression profiles in single cells with information on the proteome, the metabolome, chromatin accessibility and/or DNA methylation. With the aim of performing simultaneous generation of multimodal data, several experimental methods have been developed that do not require the physical separation of RNA and DNA from a single cell38,130,131. These methods have already been applied to post-mortem human frontal cortex tissue and have enabled joint analyses of the methylome, transcriptome, chromatin accessibility and conformation of over 60 human cortical cell types (results posted on preprint server)132. Such approaches might be useful for the development of new fluid biomarkers of immune dysregulation or the identification of novel therapeutic targets, especially when coupled to machine learning-based predictive algorithms133. A particularly active area of research focuses on the use of machine learning models to enable early diagnosis on the basis of molecular and genetic data134, including multi-omics data133.
A key goal of future single-cell transcriptomics research is to generate treatment recommendations on a per-patient basis that are useful for the physician and beneficial to the patients. Spatial transcriptomics is the most likely candidate for achieving this goal, as it allows spatially resolved gene-expression measurement directly in pathology-derived patient samples. Indeed, spatially resolved transcriptomics was crowned the ‘Method of the Year’ for 2020 by Nature Methods135. Given the ongoing rapid technological developments, we expect that new methods will combine single-cell multi-omics with spatial transcriptomics to capture cellular changes with both high sensitivity and high resolution. Also, the advances in applying spatial transcriptomics to formalin-fixed paraffin-embedded tissues136 are emerging as crucial development for future clinical and biomedical research.
Single-cell analysis of brain organoids
Human brain organoids are emerging as a potent experimental system that is genetically tractable and can be coupled to physiological and molecular measurements at the single-cell level. Single-cell transcriptomics and epigenomics have been already applied to address the questions of cellular composition and reproducibility of cerebral organoids, and those data served as an invaluable toolbox for benchmarking the fidelity of organoids for studies on the pathomechanisms of human brain diseases137–139. Detecting the earliest molecular alterations in brain organoids or defining pathology-initiating cell types could pave the way for a workflow to test treatments that aim to prevent the pathogenic event140. To this end, the reconstruction of regulatory lineages for cortical cell populations has already been performed in organoids, yielding insights into genetic risk for neuropsychiatric traits and enabling prediction of cell types vulnerable to disease4,46,132,140,141.
Brain organoids are also considered an effective future platform for testing personalized therapies. For example, single-cell analysis of brain tumours and tumour-derived organoids demonstrated that organoids preserve many key features of glioblastomas and can be rapidly deployed to investigate patient-specific treatment strategies142,143.
Gathering more clinical data
The current picture emerging from systems-level single-cell transcriptomics points towards cell diversity and biological pathways related to synaptic function, inflammation, proteostasis, cell death, oxidative stress and myelination as molecular drives of pathological changes in neurodegenration82. It seems crucial to implement cell transcriptomics in larger human cohorts and, importantly, also develop a standardized protocol to generate comparable data sets for reliable assessment of disease-associated cellular phenotypes. Tracking immune changes at the molecular and cellular levels both from the CSF and bloodstream with single-cell technologies is currently achievable and provides new opportunities for researchers to screen individuals with different neurological disorders, as adaptive immune activation is increasingly implicated in neurodegenerative conditions.
Conclusions
In conclusion, we are witnessing an explosion of single-cell sequencing technologies, which over the past 6–7 years have greatly matured from both the experimental and data analysis perspectives. Our understanding of the human brain and its pathologies is now progressing from descriptive histological examinations to detailed single-cell characterizations. In particular, scRNA-seq and snRNA-seq are becoming invaluable tools for investigating neuroimmune dysfunction, selective neuronal vulnerability to disease and cell-type susceptibility or resilience to treatment. Moreover, accumulating evidence from single-cell studies highlights glial cells as essential contributors to CNS homeostasis and neurological pathologies, especially their ability to acquire a wide range of activation states. The application of single-cell technologies is bringing us closer to illuminating the cellular states on the verge of homeostatic and pathological physiology, at least at the gene expression level.
Over the past few years, several international initiatives have been launched that aim to define human cell types in terms of distinctive molecular profiles. One such initiative is the Human Cell Atlas Project, which aims to bring together different groups within the biomedical community to build a comprehensive atlas comprising two branches — a cellular branch, focused on the properties of individual cells, and a spatial branch, focused on the histological organization of cells in tissues144. The pan-European LifeTime consortium aims to study human cells at single-cell resolution during the onset and progression of complex diseases and during their response to therapy, with the ultimate aim of creating a framework for cell-based interceptive medicine145. The NIH BRAIN initiative in the USA is entering a new phase and aims to achieve three main goals by 2026 — a comprehensive human brain cell atlas, a whole mammalian brain microconnectivity map and tools for precision access to brain cell types146.
We can now sequence the RNA of a single cell or a single nucleus, examine the state of the chromatin, start to explore the proteome of individual cells and even spatially resolve these data. We envision that advanced integrative technologies for measuring transcriptomes and other modalities from single cells will enable us to investigate features of the human healthy and diseased brain with cell-type, and perhaps subcellular resolution and will provide invaluable information for designing new treatments for individuals with neurological conditions. In our opinion, emerging single-cell multi-omics technologies and machine learning models have tremendous potential for clinical application, to study disease mechanisms and develop precision treatments (Fig. 5).
Fig. 5. Future directions for the application of single-cell and spatial transcriptomics in clinical use.
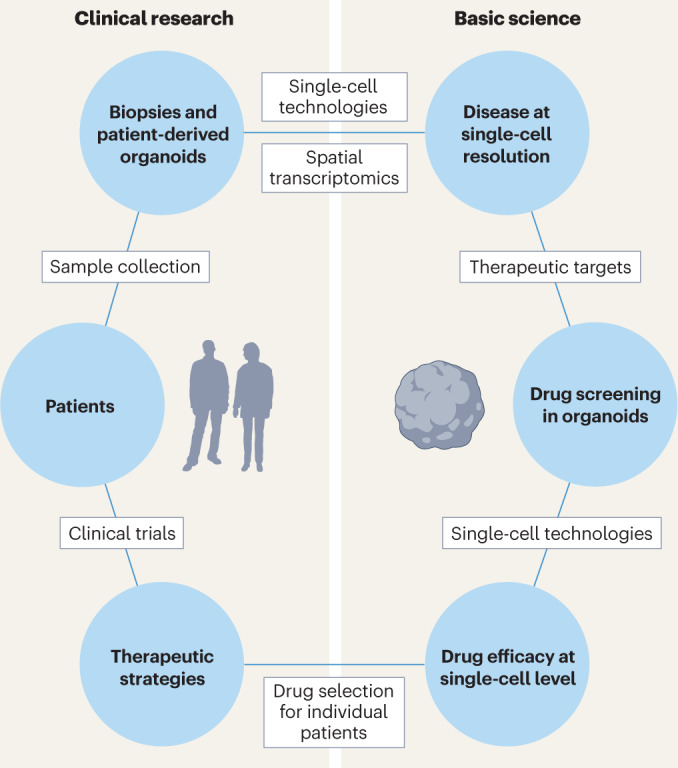
In the future, we expect that analysis of single cells from patients or patient-derived in vitro models will help to explore molecular mechanisms of diseases and define the spatial localization of rare cell types and cellular subpopulations emerging during disease. Furthermore, single-cell technologies will contribute to the discovery of new therapeutic targets. The efficacy of newly discovered drugs will then be tested in patient-derived in vitro models and monitored using single-cell technologies to define the cell‐type-specific responses of the patient to treatment, which can then be used to specify the best therapeutic strategy for the individual patient.
Supplementary information
Acknowledgements
We thank M. Jens, N. Karaiskos and D. Koppstein for a critical reading and the feedback on the manuscript. Funding: M.P. was supported by The Polish National Agency for Academic Exchange (Polish Returns grant no. PPN/PPO/2019/1/00035/U/0001) and The National Science Centre (grant no. 2018/30/E/NZ3/00624). A.R.-W. was supported by MDC and BIH funding.
Glossary
- CD4+ T cell
Subpopulation of major histocompatibility complex class II-restricted T helper cells, which are a type of T cell that has an important role in the adaptive immune system; they support the activity of other immune cells by releasing cytokines.
- CD8+ T cells
Subpopulation of major histocompatibility complex class I-restricted T helper cells, which are a type of T cell that has an important role in the adaptive immune system.
- cDNA
A DNA that is complementary to a given RNA; synthetized in an enzymatic reaction called reverse transcription.
- cDNA amplification bias
In the context of scRNA-seq, this term refers to the stochastic capture of a subset of polyadenylated transcripts from a cell and the consequent failure to detect some of the transcripts, leading to over-representation of captured RNAs during amplification of a cDNA library.
- Cell barcoding
The use of a short DNA sequence as a ‘tag’ to identify reads that originate from the same cell.
- Dimensionality reduction
Computational procedure that aims to reduce the number of separate dimensions in the high-dimensional space of scRNA-seq data; enables the comparison of cells on the basis of their expression values across multiple genes.
- Mesenchymal-like state
A cellular state that is characterized by increased expression of a mesenchymal programme (transition to mesenchymal lineages). It is also one of the four recurrent cellular states described for glioblastoma.
- Neuro-COVID
A term describing conditions related to neurological sequelae, such as headache and neuroinflammatory or cerebrovascular disease, developed by individuals with COVID-19.
- Next-generation DNA sequencing
A massively parallel sequencing technology that offers high throughput and scalability to determine the order of nucleotides in entire genomes, targeted regions of DNA, or RNA that is reverse-transcribed into cDNA libraries.
- Read alignment
A bioinformatic procedure that enables observation of the differences between the sequencing read and the reference genome.
- Reverse transcription
Enzyme-mediated synthesis of a DNA molecule (cDNA) from the RNA template.
- Single-cell multi-omics
Single-cell technologies designed to measure multiple types of molecules and modalities from an individual cell in high-throughput manner, that is, across multiple cells at the same time.
- Unique molecular identifiers
A randomized nucleotide sequence incorporated into the cDNA in the initial steps of the scRNA-seq protocol. It is used to recognize multiple sequencing reads originating from the same mRNA molecule in a given cell.
Author contributions
A.R.-W. and M.P. researched data for the article. A.R-W. and M.P. contributed substantially to discussion of the content. M.P. and A.R-F. wrote the article. All authors reviewed and/or edited the manuscript before submission.
Peer review
Peer review information
Nature Reviews Neurology thanks M. Johnson and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41582-023-00809-y.
References
- 1.Crino PB, Trojanowski JQ, Dichter MA, Eberwine J. Embryonic neuronal markers in tuberous sclerosis: single-cell molecular pathology. Proc. Natl Acad. Sci. USA. 1996;93:14152–14157. doi: 10.1073/pnas.93.24.14152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zeisel A, et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347:1138–1142. doi: 10.1126/science.aaa1934. [DOI] [PubMed] [Google Scholar]
- 3.Tasic B, et al. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci. 2016;19:335–346. doi: 10.1038/nn.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eze UC, Bhaduri A, Haeussler M, Nowakowski TJ, Kriegstein AR. Single-cell atlas of early human brain development highlights heterogeneity of human neuroepithelial cells and early radial glia. Nat. Neurosci. 2021;24:584–594. doi: 10.1038/s41593-020-00794-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tirosh I, et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 2016;539:309–313. doi: 10.1038/nature20123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krienen FM, et al. Innovations present in the primate interneuron repertoire. Nature. 2020;586:262–269. doi: 10.1038/s41586-020-2781-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geirsdottir L, et al. Cross-species single-cell analysis reveals divergence of the primate microglia program. Cell. 2019;179:1609–1622. doi: 10.1016/j.cell.2019.11.010. [DOI] [PubMed] [Google Scholar]
- 8.Durante MA, et al. Single-cell analysis of olfactory neurogenesis and differentiation in adult humans. Nat. Neurosci. 2020;23:323–326. doi: 10.1038/s41593-020-0587-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masuda T, et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature. 2019;566:388–392. doi: 10.1038/s41586-019-0924-x. [DOI] [PubMed] [Google Scholar]
- 10.Marques S, et al. Transcriptional convergence of oligodendrocyte lineage progenitors during development. Dev. Cell. 2018;46:504–517. doi: 10.1016/j.devcel.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang F, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods. 2009;6:377–382. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- 12.Islam S, et al. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat. Methods. 2014;11:163–166. doi: 10.1038/nmeth.2772. [DOI] [PubMed] [Google Scholar]
- 13.Jaitin DA, et al. Massively parallel single-cell RNA-Seq for marker-free decomposition of tissues into cell types. Science. 2014;343:776–779. doi: 10.1126/science.1247651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pollen AA, et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat. Biotechnol. 2014;32:1053–1058. doi: 10.1038/nbt.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Treutlein B, et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014;509:371–375. doi: 10.1038/nature13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Macosko EZ, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klein AM, et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015;161:1187–1201. doi: 10.1016/j.cell.2015.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lake BB, et al. A comparative strategy for single-nucleus and single-cell transcriptomes confirms accuracy in predicted cell-type expression from nuclear RNA. Sci. Rep. 2017;7:6031. doi: 10.1038/s41598-017-04426-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Habib N, et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods. 2017;14:955–958. doi: 10.1038/nmeth.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Slyper M, et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat. Med. 2020;26:792–802. doi: 10.1038/s41591-020-0844-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rousselle TV, et al. An optimized protocol for single nuclei isolation from clinical biopsies for RNA-seq. Sci. Rep. 2022;12:9851. doi: 10.1038/s41598-022-14099-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amamoto R, et al. FIN-Seq: transcriptional profiling of specific cell types from frozen archived tissue of the human central nervous system. Nucleic Acids Res. 2019;48:e4. doi: 10.1093/nar/gkz968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosenberg AB, et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science. 2018;360:176–182. doi: 10.1126/science.aam8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao J, et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science. 2017;357:661–667. doi: 10.1126/science.aam8940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han X, et al. Mapping the mouse cell atlas by Microwell-Seq. Cell. 2018;172:1091–1107. doi: 10.1016/j.cell.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 26.Han X, et al. Construction of a human cell landscape at single-cell level. Nature. 2020;581:303–309. doi: 10.1038/s41586-020-2157-4. [DOI] [PubMed] [Google Scholar]
- 27.Becht E, et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2019;37:38–44. doi: 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- 28.Vieth B, Parekh S, Ziegenhain C, Enard W, Hellmann I. A systematic evaluation of single cell RNA-seq analysis pipelines. Nat. Commun. 2019;10:4667. doi: 10.1038/s41467-019-12266-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luecken MD, Theis FJ. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol. 2019;15:e8746. doi: 10.15252/msb.20188746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ståhl PL, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353:78–82. doi: 10.1126/science.aaf2403. [DOI] [PubMed] [Google Scholar]
- 31.Vickovic S, et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods. 2019;16:987–990. doi: 10.1038/s41592-019-0548-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Merritt CR, et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol. 2020;38:586–599. doi: 10.1038/s41587-020-0472-9. [DOI] [PubMed] [Google Scholar]
- 33.He, S. et al. High-plex multiomic analysis in FFPE tissue at single-cellular and subcellular resolution by spatial molecular imaging. Preprint at bioRxiv10.1101/2021.11.03.467020 (2021).
- 34.D’Gama PP, et al. Diversity and function of motile ciliated cell types within ependymal lineages of the zebrafish brain. Cell Rep. 2021;37:109775. doi: 10.1016/j.celrep.2021.109775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kleshchevnikov V, et al. Cell2location maps fine-grained cell types in spatial transcriptomics. Nat. Biotechnol. 2022;40:661–671. doi: 10.1038/s41587-021-01139-4. [DOI] [PubMed] [Google Scholar]
- 36.Cadwell CR, et al. Electrophysiological, transcriptomic and morphologic profiling of single neurons using Patch-seq. Nat. Biotechnol. 2016;34:199–203. doi: 10.1038/nbt.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cadwell CR, et al. Multimodal profiling of single-cell morphology, electrophysiology, and gene expression using Patch-seq. Nat. Protoc. 2017;12:2531–2553. doi: 10.1038/nprot.2017.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stoeckius M, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods. 2017;14:865–868. doi: 10.1038/nmeth.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saunders A, et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell. 2018;174:1015–1030. doi: 10.1016/j.cell.2018.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zeisel A, et al. Molecular architecture of the mouse nervous system. Cell. 2018;174:999–1014. doi: 10.1016/j.cell.2018.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.La Manno G, et al. Molecular architecture of the developing mouse brain. Nature. 2021;596:92–96. doi: 10.1038/s41586-021-03775-x. [DOI] [PubMed] [Google Scholar]
- 42.Bakken TE, et al. Comparative cellular analysis of motor cortex in human, marmoset and mouse. Nature. 2021;598:111–119. doi: 10.1038/s41586-021-03465-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khrameeva E, et al. Single-cell-resolution transcriptome map of human, chimpanzee, bonobo, and macaque brains. Genome Res. 2020;30:776–789. doi: 10.1101/gr.256958.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hardwick SA, et al. Single-nuclei isoform RNA sequencing unlocks barcoded exon connectivity in frozen brain tissue. Nat. Biotechnol. 2022;40:1082–1092. doi: 10.1038/s41587-022-01231-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhong S, et al. A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature. 2018;555:524–528. doi: 10.1038/nature25980. [DOI] [PubMed] [Google Scholar]
- 46.Polioudakis D, et al. A single-cell transcriptomic atlas of human neocortical development during mid-gestation. Neuron. 2019;103:785–801.e8. doi: 10.1016/j.neuron.2019.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Batiuk MY, et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat. Commun. 2020;11:1220. doi: 10.1038/s41467-019-14198-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tan Y-L, Yuan Y, Tian L. Microglial regional heterogeneity and its role in the brain. Mol. Psychiatry. 2020;25:351–367. doi: 10.1038/s41380-019-0609-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hammond TR, et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity. 2019;50:253–271. doi: 10.1016/j.immuni.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gokce O, et al. Cellular taxonomy of the mouse striatum as revealed by single-cell RNA-Seq. Cell Rep. 2016;16:1126–1137. doi: 10.1016/j.celrep.2016.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yao Z, et al. A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation. Cell. 2021;184:3222–3241. doi: 10.1016/j.cell.2021.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miller JA, et al. Common cell type nomenclature for the mammalian brain. eLife. 2020;9:e59928. doi: 10.7554/eLife.59928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Callaway EM, et al. A multimodal cell census and atlas of the mammalian primary motor cortex. Nature. 2021;598:86–102. doi: 10.1038/s41586-021-03950-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peng H, et al. Morphological diversity of single neurons in molecularly defined cell types. Nature. 2021;598:174–181. doi: 10.1038/s41586-021-03941-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gouwens NW, et al. Integrated morphoelectric and transcriptomic classification of cortical GABAergic. Cells Cell. 2020;183:935–953. doi: 10.1016/j.cell.2020.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Darmanis S, et al. A survey of human brain transcriptome diversity at the single cell level. Proc. Natl Acad. Sci. USA. 2015;112:7285–7290. doi: 10.1073/pnas.1507125112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fu H, Hardy J, Duff KE. Selective vulnerability in neurodegenerative diseases. Nat. Neurosci. 2018;21:1350–1358. doi: 10.1038/s41593-018-0221-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sperling RA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roussarie J-P, et al. Selective neuronal vulnerability in Alzheimer’s disease: a network-based analysis. Neuron. 2020;107:821–835. doi: 10.1016/j.neuron.2020.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leng K, et al. Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat. Neurosci. 2021;24:276–287. doi: 10.1038/s41593-020-00764-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Surmeier DJ. Determinants of dopaminergic neuron loss in Parkinson’s disease. FEBS J. 2018;285:3657–3668. doi: 10.1111/febs.14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kamath T, et al. Single-cell genomic profiling of human dopamine neurons identifies a population that selectively degenerates in Parkinson’s disease. Nat. Neurosci. 2022;25:588–595. doi: 10.1038/s41593-022-01061-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Smajić S, et al. Single-cell sequencing of human midbrain reveals glial activation and a Parkinson-specific neuronal state. Brain J. Neurol. 2022;145:964–978. doi: 10.1093/brain/awab446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Birnbaum R, Weinberger DR. Genetic insights into the neurodevelopmental origins of schizophrenia. Nat. Rev. Neurosci. 2017;18:727–740. doi: 10.1038/nrn.2017.125. [DOI] [PubMed] [Google Scholar]
- 65.Nagy C, et al. Single-nucleus transcriptomics of the prefrontal cortex in major depressive disorder implicates oligodendrocyte precursor cells and excitatory neurons. Nat. Neurosci. 2020;23:771–781. doi: 10.1038/s41593-020-0621-y. [DOI] [PubMed] [Google Scholar]
- 66.Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium et al. Genetic identification of brain cell types underlying schizophrenia. Nat. Genet. 2018;50:825–833. doi: 10.1038/s41588-018-0129-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maynard KR, et al. Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. Nat. Neurosci. 2021;24:425–436. doi: 10.1038/s41593-020-00787-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pardiñas AF, et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 2018;50:381–389. doi: 10.1038/s41588-018-0059-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grove J, et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019;51:431–444. doi: 10.1038/s41588-019-0344-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wray NR, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 2018;50:668–681. doi: 10.1038/s41588-018-0090-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Velmeshev D, et al. Single-cell genomics identifies cell type-specific molecular changes in autism. Science. 2019;364:685–689. doi: 10.1126/science.aav8130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jin X, et al. In vivo Perturb-Seq reveals neuronal and glial abnormalities associated with autism risk genes. Science. 2020;370:eaaz6063. doi: 10.1126/science.aaz6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baidya F, et al. Neuroimmune crosstalk and evolving pharmacotherapies in neurodegenerative diseases. Immunology. 2021;162:160–178. doi: 10.1111/imm.13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014;15:300–312. doi: 10.1038/nrn3722. [DOI] [PubMed] [Google Scholar]
- 75.Han RT, Kim RD, Molofsky AV, Liddelow SA. Astrocyte–immune cell interactions in physiology and pathology. Immunity. 2021;54:211–224. doi: 10.1016/j.immuni.2021.01.013. [DOI] [PubMed] [Google Scholar]
- 76.Mathys H, et al. Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep. 2017;21:366–380. doi: 10.1016/j.celrep.2017.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jordão MJC, et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science. 2019;363:eaat7554. doi: 10.1126/science.aat7554. [DOI] [PubMed] [Google Scholar]
- 78.Wheeler MA, et al. MAFG-driven astrocytes promote CNS inflammation. Nature. 2020;578:593–599. doi: 10.1038/s41586-020-1999-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hasel P, Rose IVL, Sadick JS, Kim RD, Liddelow SA. Neuroinflammatory astrocyte subtypes in the mouse brain. Nat. Neurosci. 2021;24:1475–1487. doi: 10.1038/s41593-021-00905-6. [DOI] [PubMed] [Google Scholar]
- 80.Jansen IE, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019;51:404–413. doi: 10.1038/s41588-018-0311-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Keren-Shaul H, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169:1276–1290.e17. doi: 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 82.Mathys H, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 2019;570:332–337. doi: 10.1038/s41586-019-1195-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maniatis S, et al. Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science. 2019;364:89–93. doi: 10.1126/science.aav9776. [DOI] [PubMed] [Google Scholar]
- 84.Xu J, et al. Multimodal single-cell/nucleus RNA sequencing data analysis uncovers molecular networks between disease-associated microglia and astrocytes with implications for drug repurposing in Alzheimer’s disease. Genome Res. 2021;31:1900–1912. doi: 10.1101/gr.272484.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schneeberger S, et al. The neuroinflammatory interleukin-12 signaling pathway drives Alzheimer’s disease-like pathology by perturbing oligodendrocyte survival and neuronal homeostasis. bioRxiv. 2021 doi: 10.1101/2021.04.25.441313. [DOI] [Google Scholar]
- 86.Schirmer L, et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature. 2019;573:75–82. doi: 10.1038/s41586-019-1404-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schafflick D, et al. Integrated single cell analysis of blood and cerebrospinal fluid leukocytes in multiple sclerosis. Nat. Commun. 2020;11:247. doi: 10.1038/s41467-019-14118-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang P, et al. Single-cell transcriptome and TCR profiling reveal activated and expanded T cell populations in Parkinson’s disease. Cell Discov. 2021;7:52. doi: 10.1038/s41421-021-00280-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brochard V, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Invest. 2009;119:182–192. doi: 10.1172/JCI36470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yshii L, et al. Astrocyte-targeted gene delivery of interleukin 2 specifically increases brain-resident regulatory T cell numbers and protects against pathological neuroinflammation. Nat. Immunol. 2022;23:878–891. doi: 10.1038/s41590-022-01208-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Louis DN, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 2021;23:1231–1251. doi: 10.1093/neuonc/noab106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Darmanis S, et al. Single-cell RNA-Seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Rep. 2017;21:1399–1410. doi: 10.1016/j.celrep.2017.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ochocka N, et al. Single-cell RNA sequencing reveals functional heterogeneity of glioma-associated brain macrophages. Nat. Commun. 2021;12:1151. doi: 10.1038/s41467-021-21407-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hara T, et al. Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma. Cancer Cell. 2021;39:779–792. doi: 10.1016/j.ccell.2021.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ravi VM, et al. Spatially resolved multi-omics deciphers bidirectional tumor–host interdependence in glioblastoma. Cancer Cell. 2022;40:639–655. doi: 10.1016/j.ccell.2022.05.009. [DOI] [PubMed] [Google Scholar]
- 96.Chou SH-Y, et al. Global incidence of neurological manifestations among patients hospitalized with COVID-19 — a report for the GCS-NeuroCOVID Consortium and the ENERGY Consortium. JAMA Netw. Open. 2021;4:e2112131. doi: 10.1001/jamanetworkopen.2021.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ellul MA, et al. Neurological associations of COVID-19. Lancet Neurol. 2020;19:767–783. doi: 10.1016/S1474-4422(20)30221-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fullard JF, et al. Single-nucleus transcriptome analysis of human brain immune response in patients with severe COVID-19. Genome Med. 2021;13:118. doi: 10.1186/s13073-021-00933-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yang AC, et al. Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature. 2021;595:565–571. doi: 10.1038/s41586-021-03710-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Heming M, et al. Neurological manifestations of COVID-19 feature T cell exhaustion and dedifferentiated monocytes in cerebrospinal fluid. Immunity. 2021;54:164–175. doi: 10.1016/j.immuni.2020.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Caras IW. Two cancer stem cell-targeted therapies in clinical trials as viewed from the standpoint of the cancer stem cell model. Stem Cells Transl. Med. 2020;9:821–826. doi: 10.1002/sctm.19-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Richards LM, et al. Gradient of developmental and injury response transcriptional states defines functional vulnerabilities underpinning glioblastoma heterogeneity. Nat. Cancer. 2021;2:157–173. doi: 10.1038/s43018-020-00154-9. [DOI] [PubMed] [Google Scholar]
- 103.Zhao W, et al. Deconvolution of cell type-specific drug responses in human tumor tissue with single-cell RNA-seq. Genome Med. 2021;13:82. doi: 10.1186/s13073-021-00894-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ostrom QT, et al. CBTRUS Statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2012–2016. Neuro Oncol. 2019;21:v1–v100. doi: 10.1093/neuonc/noz150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gibson P, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010;468:1095–1099. doi: 10.1038/nature09587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Menyhárt O, Giangaspero F, Győrffy B. Molecular markers and potential therapeutic targets in non-WNT/non-SHH (group 3 and group 4) medulloblastomas. J. Hematol. Oncol. 2019;12:29. doi: 10.1186/s13045-019-0712-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hovestadt V, et al. Resolving medulloblastoma cellular architecture by single-cell genomics. Nature. 2019;572:74–79. doi: 10.1038/s41586-019-1434-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang L, et al. Single-cell transcriptomics in medulloblastoma reveals tumor-initiating progenitors and oncogenic cascades during tumorigenesis and relapse. Cancer Cell. 2019;36:302–318. doi: 10.1016/j.ccell.2019.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ocasio J, et al. scRNA-seq in medulloblastoma shows cellular heterogeneity and lineage expansion support resistance to SHH inhibitor therapy. Nat. Commun. 2019;10:5829. doi: 10.1038/s41467-019-13657-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liu X, et al. Clinical challenges of tissue preparation for spatial transcriptome. Clin. Transl. Med. 2022;12:e669. doi: 10.1002/ctm2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kulkarni A, Anderson AG, Merullo DP, Konopka G. Beyond bulk: a review of single cell transcriptomics methodologies and applications. Curr. Opin. Biotechnol. 2019;58:129–136. doi: 10.1016/j.copbio.2019.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wu YE, Pan L, Zuo Y, Li X, Hong W. Detecting activated cell populations using single-cell RNA-Seq. Neuron. 2017;96:313–329. doi: 10.1016/j.neuron.2017.09.026. [DOI] [PubMed] [Google Scholar]
- 113.Hrvatin S, et al. Single-cell analysis of experience-dependent transcriptomic states in the mouse visual cortex. Nat. Neurosci. 2018;21:120–129. doi: 10.1038/s41593-017-0029-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Loo L, et al. Single-cell transcriptomic analysis of mouse neocortical development. Nat. Commun. 2019;10:134. doi: 10.1038/s41467-018-08079-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bakken TE, et al. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS ONE. 2018;13:e0209648. doi: 10.1371/journal.pone.0209648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nadelmann ER, et al. Isolation of nuclei from mammalian cells and tissues for single-nucleus molecular profiling. Curr. Protoc. 2021;1:e132. doi: 10.1002/cpz1.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lake BB, et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science. 2016;352:1586–1590. doi: 10.1126/science.aaf1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lacar B, et al. Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nat. Commun. 2016;7:11022. doi: 10.1038/ncomms11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Perez JD, et al. Subcellular sequencing of single neurons reveals the dendritic transcriptome of GABAergic interneurons. eLife. 2021;10:e63092. doi: 10.7554/eLife.63092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kharchenko PV. The triumphs and limitations of computational methods for scRNA-seq. Nat. Methods. 2021;18:723–732. doi: 10.1038/s41592-021-01171-x. [DOI] [PubMed] [Google Scholar]
- 121.van Dijk D, et al. Recovering gene interactions from single-cell data using data diffusion. Cell. 2018;174:716–729. doi: 10.1016/j.cell.2018.05.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Huang M, et al. SAVER: gene expression recovery for single-cell RNA sequencing. Nat. Methods. 2018;15:539–542. doi: 10.1038/s41592-018-0033-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li WV, Li JJ. An accurate and robust imputation method scImpute for single-cell RNA-seq data. Nat. Commun. 2018;9:997. doi: 10.1038/s41467-018-03405-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tracy S, Yuan G-C, Dries R. RESCUE: imputing dropout events in single-cell RNA-sequencing data. BMC Bioinforma. 2019;20:388. doi: 10.1186/s12859-019-2977-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lähnemann D, et al. Eleven grand challenges in single-cell data science. Genome Biol. 2020;21:31. doi: 10.1186/s13059-020-1926-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Haghverdi L, Lun ATL, Morgan MD, Marioni JC. Batch effects in single-cell RNA sequencing data are corrected by matching mutual nearest neighbours. Nat. Biotechnol. 2018;36:421–427. doi: 10.1038/nbt.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Luecken MD, et al. Benchmarking atlas-level data integration in single-cell genomics. Nat. Methods. 2022;19:41–50. doi: 10.1038/s41592-021-01336-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.A R-W, et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol. Cell. 2015;58:870–885. doi: 10.1016/j.molcel.2015.03.027. [DOI] [PubMed] [Google Scholar]
- 129.Piwecka M, et al. Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science. 2017;357:eaam8526. doi: 10.1126/science.aam8526. [DOI] [PubMed] [Google Scholar]
- 130.Luo C, et al. Robust single-cell DNA methylome profiling with snmC-seq2. Nat. Commun. 2018;9:3824. doi: 10.1038/s41467-018-06355-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pott S. Simultaneous measurement of chromatin accessibility, DNA methylation, and nucleosome phasing in single cells. eLife. 2017;6:e23203. doi: 10.7554/eLife.23203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Luo C, et al. Single nucleus multi-omics identifies human cortical cell regulatory genome diversity. Cell Genomics. 2022;2:100107. doi: 10.1016/j.xgen.2022.100107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Reel PS, Reel S, Pearson E, Trucco E, Jefferson E. Using machine learning approaches for multi-omics data analysis: a review. Biotechnol. Adv. 2021;49:107739. doi: 10.1016/j.biotechadv.2021.107739. [DOI] [PubMed] [Google Scholar]
- 134.Myszczynska MA, et al. Applications of machine learning to diagnosis and treatment of neurodegenerative diseases. Nat. Rev. Neurol. 2020;16:440–456. doi: 10.1038/s41582-020-0377-8. [DOI] [PubMed] [Google Scholar]
- 135.Marx V. Method of the year: spatially resolved transcriptomics. Nat. Methods. 2021;18:9–14. doi: 10.1038/s41592-020-01033-y. [DOI] [PubMed] [Google Scholar]
- 136.Gracia Villacampa E, et al. Genome-wide spatial expression profiling in formalin-fixed tissues. Cell Genomics. 2021;1:100065. doi: 10.1016/j.xgen.2021.100065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Klaus J, et al. Altered neuronal migratory trajectories in human cerebral organoids derived from individuals with neuronal heterotopia. Nat. Med. 2019;25:561–568. doi: 10.1038/s41591-019-0371-0. [DOI] [PubMed] [Google Scholar]
- 138.Amiri A, et al. Transcriptome and epigenome landscape of human cortical development modeled in organoids. Science. 2018;362:eaat6720. doi: 10.1126/science.aat6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Paulsen B, et al. Autism genes converge on asynchronous development of shared neuron classes. Nature. 2022;602:268–273. doi: 10.1038/s41586-021-04358-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.He Z, et al. Lineage recording in human cerebral organoids. Nat. Methods. 2022;19:90–99. doi: 10.1038/s41592-021-01344-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ziffra RS, et al. Single-cell epigenomics reveals mechanisms of human cortical development. Nature. 2021;598:205–203. doi: 10.1038/s41586-021-03209-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chen C-C, et al. Patient-derived tumor organoids as a platform of precision treatment for malignant brain tumors. Sci. Rep. 2022;12:16399. doi: 10.1038/s41598-022-20487-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jacob F, et al. A patient-derived glioblastoma organoid model and biobank recapitulates inter- and intra-tumoral heterogeneity. Cell. 2020;180:188–204. doi: 10.1016/j.cell.2019.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Regev A, et al. The human cell atlas white paper. ArXiv. 2018 doi: 10.48550/arXiv.1810.05192. [DOI] [Google Scholar]
- 145.Rajewsky N, et al. LifeTime and improving European healthcare through cell-based interceptive medicine. Nature. 2020;587:377–386. doi: 10.1038/s41586-020-2715-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ngai J. BRAIN 2.0: transforming neuroscience. Cell. 2022;185:4–8. doi: 10.1016/j.cell.2021.11.037. [DOI] [PubMed] [Google Scholar]
- 147.Goetz JJ, Trimarchi JM. Transcriptome sequencing of single cells with Smart-Seq. Nat. Biotechnol. 2012;30:763–765. doi: 10.1038/nbt.2325. [DOI] [PubMed] [Google Scholar]
- 148.Picelli S, et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods. 2013;10:1096–1098. doi: 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- 149.Xin Y, et al. Use of the fluidigm C1 platform for RNA sequencing of single mouse pancreatic islet cells. Proc. Natl Acad. Sci. USA. 2016;113:3293–3298. doi: 10.1073/pnas.1602306113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zheng GXY, et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8:14049. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hashimshony T, et al. CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 2016;17:77. doi: 10.1186/s13059-016-0938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sheng K, Cao W, Niu Y, Deng Q, Zong C. Effective detection of variation in single-cell transcriptomes using MATQ-seq. Nat. Methods. 2017;14:267–270. doi: 10.1038/nmeth.4145. [DOI] [PubMed] [Google Scholar]
- 153.Lebrigand K, Magnone V, Barbry P, Waldmann R. High throughput error corrected nanopore single cell transcriptome sequencing. Nat. Commun. 2020;11:4025. doi: 10.1038/s41467-020-17800-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Habib N, et al. Div-Seq: single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science. 2016;353:925–928. doi: 10.1126/science.aad7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Eng C-HL, et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature. 2019;568:235–239. doi: 10.1038/s41586-019-1049-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Xia C, Babcock HP, Moffitt JR, Zhuang X. Multiplexed detection of RNA using MERFISH and branched DNA amplification. Sci. Rep. 2019;9:7721. doi: 10.1038/s41598-019-43943-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Rodriques SG, et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science. 2019;363:1463–1467. doi: 10.1126/science.aaw1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Beltrán E, et al. Early adaptive immune activation detected in monozygotic twins with prodromal multiple sclerosis. J. Clin. Invest. 2019;129:4758–4768. doi: 10.1172/JCI128475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Esaulova E, et al. Single-cell RNA-seq analysis of human CSF microglia and myeloid cells in neuroinflammation. Neurol. Neuroimmunol. Neuroinflamm. 2020;7:e732. doi: 10.1212/NXI.0000000000000732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kaufmann M, et al. Identifying CNS-colonizing T cells as potential therapeutic targets to prevent progression of multiple sclerosis. Med. 2021;2:296–312. doi: 10.1016/j.medj.2021.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Renthal W, et al. Characterization of human mosaic Rett syndrome brain tissue by single-nucleus RNA sequencing. Nat. Neurosci. 2018;21:1670–1679. doi: 10.1038/s41593-018-0270-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Grubman A, et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019;22:2087–2097. doi: 10.1038/s41593-019-0539-4. [DOI] [PubMed] [Google Scholar]
- 163.Del-Aguila JL, et al. A single-nuclei RNA sequencing study of Mendelian and sporadic AD in the human brain. Alzheimers Res. Ther. 2019;11:71. doi: 10.1186/s13195-019-0524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Al-Dalahmah O, et al. Single-nucleus RNA-seq identifies Huntington disease astrocyte states. Acta Neuropathol. Commun. 2020;8:19. doi: 10.1186/s40478-020-0880-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gregory JM, et al. Spatial transcriptomics identifies spatially dysregulated expression of GRM3 and USP47 in amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 2020;46:441–457. doi: 10.1111/nan.12597. [DOI] [PubMed] [Google Scholar]
- 166.Jaffe AE, et al. Profiling gene expression in the human dentate gyrus granule cell layer reveals insights into schizophrenia and its genetic risk. Nat. Neurosci. 2020;23:510–519. doi: 10.1038/s41593-020-0604-z. [DOI] [PubMed] [Google Scholar]
- 167.Herrero MJ, et al. Identification of amygdala-expressed genes associated with autism spectrum disorder. Mol. Autism. 2020;11:39. doi: 10.1186/s13229-020-00346-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sorrells SF, et al. Immature excitatory neurons develop during adolescence in the human amygdala. Nat. Commun. 2019;10:2748. doi: 10.1038/s41467-019-10765-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zhong S, et al. Decoding the development of the human hippocampus. Nature. 2020;577:531–536. doi: 10.1038/s41586-019-1917-5. [DOI] [PubMed] [Google Scholar]
- 170.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinforma. Oxf. Engl. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kim D, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.McCarthy DJ, Campbell KR, Lun ATL, Wills QF. Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinforma. Oxf. Engl. 2017;33:1179–1186. doi: 10.1093/bioinformatics/btw777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lun ATL, McCarthy DJ, Marioni JC. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with bioconductor. F1000Research. 2016;5:2122. doi: 10.12688/f1000research.9501.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Bacher R, et al. SCnorm: robust normalization of single-cell RNA-seq data. Nat. Methods. 2017;14:584–586. doi: 10.1038/nmeth.4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Tang W, et al. bayNorm: Bayesian gene expression recovery, imputation and normalization for single-cell RNA-sequencing data. Bioinformatics. 2020;36:1174–1181. doi: 10.1093/bioinformatics/btz726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Stein CK, et al. Removing batch effects from purified plasma cell gene expression microarrays with modified ComBat. BMC Bioinforma. 2015;16:63. doi: 10.1186/s12859-015-0478-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Stuart T, et al. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Pertea M, et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 185.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Wolf FA, et al. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 2019;20:59. doi: 10.1186/s13059-019-1663-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Cao J, et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019;566:496–502. doi: 10.1038/s41586-019-0969-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.La Manno G, et al. RNA velocity of single cells. Nature. 2018;560:494–498. doi: 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Marques S, et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science. 2016;352:1326–1329. doi: 10.1126/science.aaf6463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Siletti K, et al. Transcriptomic diversity of cell types across the adult human brain. bioRxiv. 2022 doi: 10.1101/2022.10.12.511898. [DOI] [PubMed] [Google Scholar]
- 191.Campbell JN, et al. A molecular census of arcuate hypothalamus and median eminence cell types. Nat. Neurosci. 2017;20:484–496. doi: 10.1038/nn.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Kim D-W, et al. Multimodal analysis of cell types in a hypothalamic node controlling social behavior. Cell. 2019;179:713–728. doi: 10.1016/j.cell.2019.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Mickelsen LE, et al. Single-cell transcriptomic analysis of the lateral hypothalamic area reveals molecularly distinct populations of inhibitory and excitatory neurons. Nat. Neurosci. 2019;22:642–656. doi: 10.1038/s41593-019-0349-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Romanov RA, et al. Molecular interrogation of hypothalamic organization reveals distinct dopamine neuronal subtypes. Nat. Neurosci. 2017;20:176–188. doi: 10.1038/nn.4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Wen S, et al. Spatiotemporal single-cell analysis of gene expression in the mouse suprachiasmatic nucleus. Nat. Neurosci. 2020;23:456–467. doi: 10.1038/s41593-020-0586-x. [DOI] [PubMed] [Google Scholar]
- 196.Kalish BT, et al. Single-cell transcriptomics of the developing lateral geniculate nucleus reveals insights into circuit assembly and refinement. Proc. Natl Acad. Sci. USA. 2018;115:E1051–E1060. doi: 10.1073/pnas.1717871115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ren J, et al. Single-cell transcriptomes and whole-brain projections of serotonin neurons in the mouse dorsal and median raphe nuclei. eLife. 2019;8:e49424. doi: 10.7554/eLife.49424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Gupta I, et al. Single-cell isoform RNA sequencing characterizes isoforms in thousands of cerebellar cells. Nat. Biotechnol. 2018;36:1197–1202. doi: 10.1038/nbt.4259. [DOI] [PubMed] [Google Scholar]
- 199.Carter RA, et al. A single-cell transcriptional atlas of the developing murine cerebellum. Curr. Biol. 2018;28:2910–2920. doi: 10.1016/j.cub.2018.07.062. [DOI] [PubMed] [Google Scholar]
- 200.Peng J, et al. Single-cell transcriptomes reveal molecular specializations of neuronal cell types in the developing cerebellum. J. Mol. Cell Biol. 2019;11:636–648. doi: 10.1093/jmcb/mjy089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Zeppilli S, et al. Molecular characterization of projection neuron subtypes in the mouse olfactory bulb. eLife. 2021;10:e65445. doi: 10.7554/eLife.65445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Bhattacherjee A, et al. Cell type-specific transcriptional programs in mouse prefrontal cortex during adolescence and addiction. Nat. Commun. 2019;10:4169. doi: 10.1038/s41467-019-12054-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Tasic B, et al. Shared and distinct transcriptomic cell types across neocortical areas. Nature. 2018;563:72–78. doi: 10.1038/s41586-018-0654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Zywitza V, Misios A, Bunatyan L, Willnow TE, Rajewsky N. Single-cell transcriptomics characterizes cell types in the subventricular zone and uncovers molecular defects impairing adult neurogenesis. Cell Rep. 2018;25:2457–2469. doi: 10.1016/j.celrep.2018.11.003. [DOI] [PubMed] [Google Scholar]
- 205.Dulken BW, et al. Single-cell analysis reveals T cell infiltration in old neurogenic niches. Nature. 2019;571:205–210. doi: 10.1038/s41586-019-1362-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Hochgerner H, Zeisel A, Lönnerberg P, Linnarsson S. Conserved properties of dentate gyrus neurogenesis across postnatal development revealed by single-cell RNA sequencing. Nat. Neurosci. 2018;21:290–299. doi: 10.1038/s41593-017-0056-2. [DOI] [PubMed] [Google Scholar]
- 207.Artegiani B, et al. A single-cell RNA sequencing study reveals cellular and molecular dynamics of the hippocampal neurogenic niche. Cell Rep. 2017;21:3271–3284. doi: 10.1016/j.celrep.2017.11.050. [DOI] [PubMed] [Google Scholar]
- 208.Harris KD, et al. Classes and continua of hippocampal CA1 inhibitory neurons revealed by single-cell transcriptomics. PLoS Biol. 2018;16:e2006387. doi: 10.1371/journal.pbio.2006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Mayer C, et al. Developmental diversification of cortical inhibitory interneurons. Nature. 2018;555:457–462. doi: 10.1038/nature25999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Tiklová K, et al. Single-cell RNA sequencing reveals midbrain dopamine neuron diversity emerging during mouse brain development. Nat. Commun. 2019;10:581. doi: 10.1038/s41467-019-08453-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Hove HV, et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci. 2019;22:1021–1035. doi: 10.1038/s41593-019-0393-4. [DOI] [PubMed] [Google Scholar]
- 212.Lake BB, et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol. 2018;36:70–80. doi: 10.1038/nbt.4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.La Manno G, et al. Molecular diversity of midbrain development in mouse, human, and stem cells. Cell. 2016;167:566–580. doi: 10.1016/j.cell.2016.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.