

Abstract
Objective:
The Transatlantic Australasian Retroperitoneal Sarcoma Working Group (TARPSWG) conducted a retrospective study on the disease course and clinical management of ganglioneuromas.
Background:
Ganglioneuromas are rare tumors derived from neural crest cells. Data on these tumors remain limited to case reports and single-institution case series.
Methods:
Patients of all ages with pathologically confirmed primary retroperitoneal, intra-abdominal, and pelvic ganglioneuromas between January 1, 2000 and January 1, 2020 were included. We examined demographic, clinicopathologic, radiologic characteristics as well as clinical management.
Results:
Overall, 328 patients from 29 institutions were included. The median age at diagnosis was 37 years with 59.1% of patients being female. Symptomatic presentation comprised 40.9% of cases, and tumors were often located in the extra-adrenal retroperitoneum (67.1%). At baseline, the median maximum tumor diameter was 7.2 cm. One hundred sixteen (35.4%) patients underwent active surveillance while 212 (64.6%) patients underwent resection with 74.5% of operative cases achieving an R0/R1 resection. Serial tumor evaluations showed that malignant transformation to neuroblastoma was rare (0.9%, N=3). Tumors undergoing surveillance had a median follow-up of 1.9 years, with 92.2% of ganglioneuromas stable in size. With a median follow-up of 3.0 years for resected tumors, 84.4% of patients were disease-free following resections while recurrences were observed in 4 (1.9%) patients.
Conclusion:
Most ganglioneuromas have indolent disease courses and rarely transform to neuroblastoma. Thus, active surveillance may be appropriate for benign and asymptomatic tumors particularly when the risks of surgery outweigh the benefits. For symptomatic or growing tumors, resection may be curative.
Introduction:
Ganglioneuromas are rare tumors composed of mature ganglion cells embedded in a Schwannian stroma that arise along the sympathetic plexus and within the adrenal medulla1. They originate from primordial neural crest cells and develop as primary neoplasms or from differentiation of immature neurogenic tumors such as neuroblastomas1, 2.
Ganglioneuromas are usually hormonally silent, although some have been reported to secrete vasoactive intestinal peptide (VIP), cortisol, catecholamines, and/or testosterone3-10. Moreover, ganglioneuromas have been occasionally found in the setting of familial syndromes, including Neurofibromatosis type-1 (NF-1), Multiple Endocrine Neoplasia Type 2A (MEN 2A), Multiple Endocrine Neoplasia 2B (MEN 2B), Turner’s Syndrome, Cowden Syndrome, or PTEN Hamartomatous Tumor Syndrome11-19. The clinical significance of these associations has yet to be determined.
Pre-operative diagnosis of ganglioneuromas is challenging because these tumors have no defined clinical signs and symptoms, laboratory findings, or imaging characteristics; this may lead to misdiagnoses that result in inappropriate treatment courses6, 20, 21. As benign tumors, ganglioneuromas are often discovered incidentally or during work-up for non-specific symptoms caused by mass effect10, 22, 23 . An additional challenge to management, their potential for recurrence and malignant transformation to neuroblastoma remains unclear24-27.
Although these lesions develop in children and adults, little data are available for these tumors beyond case reports and small single-institution case series. Herein, we present an international, multi-institutional, retrospective study of ganglioneuromas conducted by the Transatlantic Australasian Retroperitoneal Sarcoma Working Group (TARPSWG). We examine patient demographics, clinicopathologic features, radiological findings, approaches to treatment, and outcomes with the goal of expanding our understanding of the disease course and clinical management of ganglioneuromas.
Methods:
Collaborative database
Institutions across North America, South America, Europe, Asia, and Australia collaborated to create a ganglioneuroma database through TARPSWG. Institutional review board approvals were obtained by all institutions and data-sharing agreements were completed according to local institutional policies. Patient data abstracted from electronic medical records was de-identified and submitted to the coordinating institution (University of California, San Diego) for central collation and analysis. Clinical features examined were patient demographics, tumor presentation, and histopathology. Mass size and growth characteristics were assessed using CT scans, MRI, or ultrasound. Surveillance approaches and outcomes of tumor interventions (operative and non-operative), as well as their follow-up were investigated.
Patient parameters
Patients of all ages presenting with primary ganglioneuroma between January 1, 2000 and January 1, 2020 were included in the analysis. Only biopsy or histopathology confirmed ganglioneuromas located in the retroperitoneum, abdomen, or pelvis were included in the study. Head and neck, mediastinal, and gastrointestinal tract tumors were excluded to ensure a relatively homogenous population of ganglioneuromas without outliers and potentially variable biology of other tumor sites.
Tumor measurements
Imaging scans provided tumor width, length, and height measurements in centimeters. Tumor maximum diameter was determined using the largest measurement amongst width, length, or height from each scan. Percentage changes per year in tumor maximum diameter from baseline were assessed over a series of follow-up scans, with a minimum interval of one year between baseline to the first follow-up scan. Percentage changes per year from baseline to the last follow-up scan were visualized using a waterfall plot created with Prism GraphPad9 (GraphPad Software, La Jolla, CA). Adopting the modified RECIST 1.1 criteria, ganglioneuromas displaying changes greater than 20% were designated as progressive, between 20% and −30% as stable disease, and less than −30% as regressive28.
Statistical analysis
Categorical data were reported as percentages, while medians and interquartile ranges were used to describe quantitative values. With an α level of 5% (p < 0.05), statistical significance of continuous variables was evaluated with Mann–Whitney U or Kruskal–Wallis test, while chi-square or Fisher's exact test was used for categorical variables. Statistical analyses were performed using Prism GraphPad9.
Results
Patient demographics and presentation
Overall, 328 patients from 29 institutions across 5 continents met the inclusion criteria for analysis, with 66.5% of patients presenting from 2011 to 2020 (Table 1, Supplemental Figure 1, Supplemental Digital Content 1, http://links.lww.com/SLA/E77). The median age at diagnosis was 37 years (range, 4 – 79) and 59.1% of patients were females. Ganglioneuromas were most often found in the retroperitoneum (67.1%), followed by the adrenal glands (18.9%), pelvis (11.0%), and the peritoneal cavity (3.0%). The majority of ganglioneuromas presented sporadically (97.6%) in the absence of known familial syndromes. Less than half (N=134, 40.9%) of patients were symptomatic while symptoms were often non-specific, with the most frequent being pain/discomfort (N=108), emesis (N=6), weight loss (N=5), and hematuria (N=5).
Table 1:
Patient Demographics and Clinical Characteristics of Ganglioneuromas.
| Characteristics | Number (%) |
|---|---|
| Total Number of Patients | 328 |
| Median Age at Diagnosis | 37 |
| Sex | |
| Male | 134 (40.9) |
| Female | 194 (59.1) |
| Location of Ganglioneuroma | |
| Retroperitoneal | 220 (67.1) |
| Adrenal | 62 (18.9) |
| Pelvic | 36 (11.0) |
| Intra-abdominal | 10 (3.0) |
| Presentation | |
| Symptomatic | 134 (40.9) |
| Incidental | 192 (58.5) |
| Unknown status | 2 (0.6) |
| ASA Grade at Referral | |
| I | 149 (45.4) |
| II | 102 (31.1) |
| III | 21 (6.4) |
| IV | 1 (0.3) |
| Unknown status | 55 (16.8) |
| Ganglioneuroma Suspected on Imaging | |
| Yes | 79 (24.1) |
| No | 201 (61.3) |
| Unknown status | 48 (14.6) |
| Vascular Involvement | 56 (17.1) |
| Retroperitoneal | 43 (76.8) |
| Adrenal | 4 (7.1) |
| Pelvic | 5 (8.9) |
| Intra-abdominal | 4 (7.1) |
| Major Nerve Involvement | 26 (7.9) |
| Retroperitoneal | 17 (65.4) |
| Pelvic | 9 (34.6) |
| History of Peripheral Nerve Tumors | 8 (2.4) |
| Neurofibromatosis Type 1 | 6 |
| Schwannamotosis | 1 |
| Men 2A | 1 |
Frequencies are reported in parentheses.
Amongst patients with hereditary syndromes (2.4%), NF-1 (N=6) was the most common association followed by MEN 2A (N=1) and Schwannomatosis (N=1). These patients presented with solitary ganglioneuromas. In addition, five adrenal ganglioneuromas co-occurred with pheochromocytomas.
Tumor size, involvement, and growth
Ganglioneuromas were suspected on only 24.1% of initial imaging studies based on the differential diagnoses in radiology reports (Table 1). Tumors abutted or encased major blood vessels (arterial and/or venous) in 17.1% (N=56) of patients with the majority of involvement located in the retroperitoneum (76.8%). Major nerve involvement occurred in 7.9% (N=26) of cases and again these were primarily in the retroperitoneum (65.4%).
At baseline, the median maximum tumor diameter was 7.2 cm (IQR [5.0, 9.9]) (Table 2). At presentation, tumors with vascular involvement and those located in the pelvis had the largest diameters of 9.4 cm (IQR [7.2, 12.0]) and of 9.1 cm (IQR [6.9, 10.5]), respectively. Those with vascular involvement were larger than those without (p < 0.001), and pelvic tumors were the largest amongst the various locations (p < 0.001). In addition, non-resected tumors had diameters of 8.0 cm (IQR [6.1, 10.0]) and were larger than resected ones (p = 0.003) (Table 2) but were not more likely to have vascular involvement.
Table 2:
Maximum Tumor Diameter on Presentation.
| Median Baseline Tumor Maximum Diameter (cm, IQR range) |
P value |
|
|---|---|---|
| Patients* (N=265) | 7.2 (5.0 – 9.9) | |
| Sex (N=265) | 0.21 | |
| Male (N=115) | 7.0 (4.6 – 9.4) | |
| Female (N=150) | 7.5 (5.7 – 10.0) | |
| Tumor Location (N=265) | <0.001 | |
| Retroperitoneal (N=177) | 7.6 (5.8 – 10.0) | |
| Adrenal (N=55) | 5.5 (3.7 – 7.0) | |
| Pelvic (N=28) | 9.1 (6.9 – 10.5) | |
| Intra-abdominal (N=5) | 5.2 (4.8 – 5.9) | |
| Presentation (N=264) | 0.95 | |
| Symptomatic (N=111) | 7.5 (5.1 – 10.0) | |
| Incidental (N=153) | 7.0 (5.0 – 9.7) | |
| Operative vs. Non-Operative Management (N=265) | 0.003 | |
| Resected Tumors (N=162) | 6.5 (4.5 – 9.6) | |
| Non-resected Tumors (N=103) | 8.0 (6.1 – 10.0) | |
| Resection Margin (N=146) | 0.29 | |
| R0/R1 (N=126) | 6.3 (4.4 – 8.5) | |
| R2 (N=20) | 7.0 (4.5 – 12.0) | |
| ASA Grade (N=219) | 0.01 | |
| I (N=120) | 7.4 (5.0 – 10.3) | |
| II (N=82) | 7.1 (4.7 – 9.4) | |
| III (N=16) | 4.2 (3.1 – 6.3) | |
| IV (N=1) | 3.2 | |
| Vascular Involvement (N=196) | <0.001 | |
| Yes (N=43) | 9.4 (7.2 – 12.0) | |
| No (N=153) | 6.3 (4.4 – 9.0) | |
| Nerve Involvement (N=189) | 0.60 | |
| Yes (N=21) | 7.0 (4.6 – 9.3) | |
| No (N=168) | 7.0 (4.6 – 9.3) |
The largest measurement among length, width, and height from the first scan were used to determine the maximum tumor diameter. Values are medians with interquartile ranges in parentheses. Mann–Whitney U test or Kruskal–Wallis test were used to calculate significance.
Patients with maximum tumor diameter data upon presentation.
Among the 66 patients with available measurements for percentage changes in maximum tumor diameter per year, only 4 (6.1%) cases had progression in tumor size (Figure 1). Three out of the four patients were female, and all four were adults with an ASA grade of II or higher. All of the progressive tumors were located in the retroperitoneum (one adrenal), and two co-occurred with lung cancers. Furthermore, for the lesion that progressed with associated symptoms, final pathology did not reveal malignant transformation to neuroblastoma following resection. Maximum tumor diameters were found to be relatively stable over time for 62 (93.9%) patients undergoing active surveillance. Among the stable lesions, four initially displayed progression, but stabilized in subsequent scans. Overall, 34 (51.5%) of the stable tumors had decreases in size (changes less than 0%).
Figure 1:
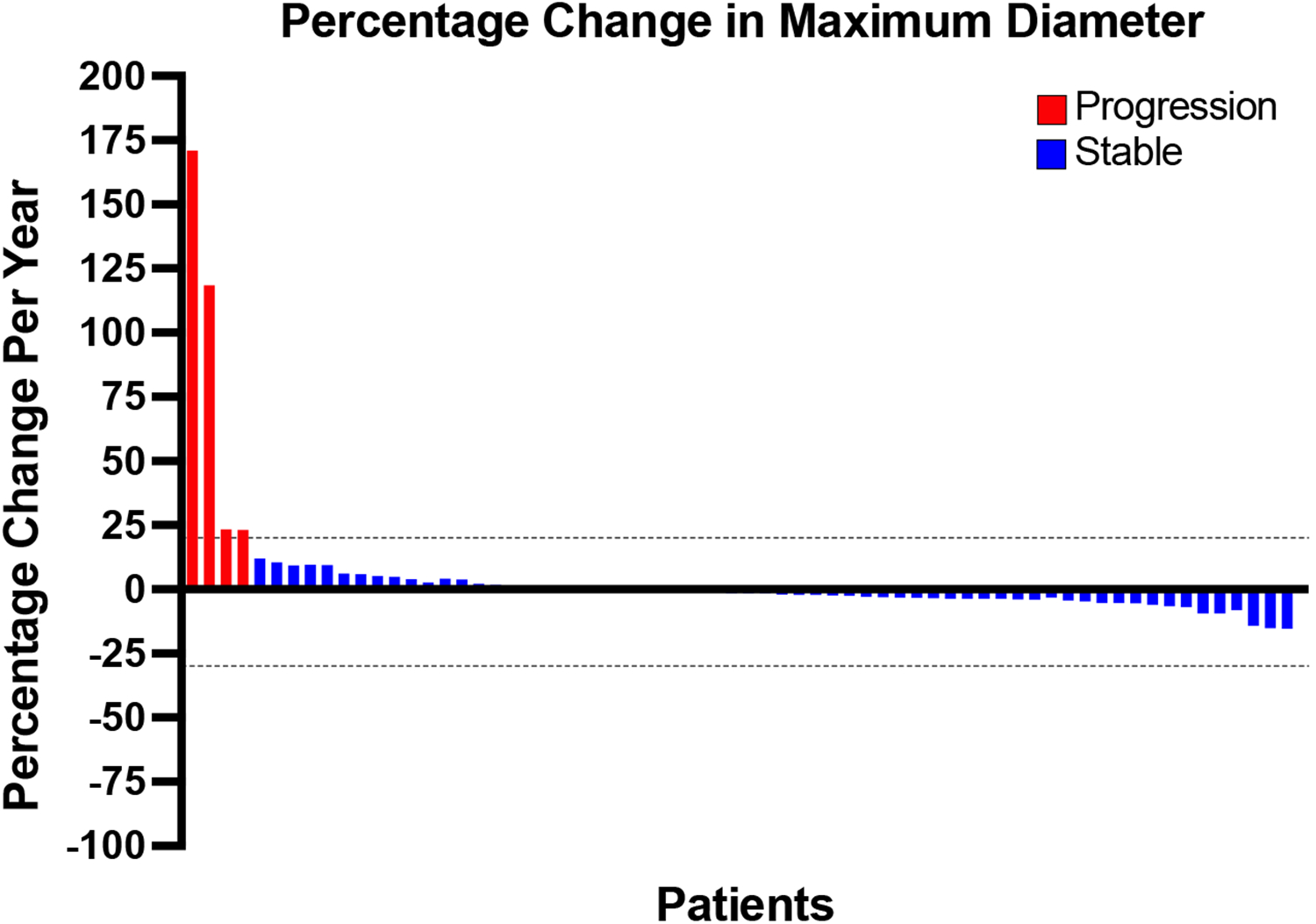
Percent change in maximum tumor diameter over time. Waterfall plot showing the percent change per year in maximum diameter from baseline to the terminal follow-up scan for each patient. Ganglioneuromas displaying changes greater than 20% were designated as progressive (red) and between 20% and −30% as stable disease (blue).
Based upon histopathology, malignant transformation to neuroblastoma occurred in 3 (0.9%) cases among all patients with all cases occurring in children (Table 3). No ganglioneuromas in adults that lacked atypia, significant growth, or symptoms transformed into neuroblastoma.
Table 3:
Approaches to Clinical Management.
| Number (%) | |
|---|---|
| Total Patients | 328 |
| Non-Operative Management | 116 (35.4) |
| Surgery | 212 (64.6) |
| Vascular Resection | 32 (15.1) |
| Vessel Ligation (N=32) | 28 (87.5) |
| Vessel Reconstruction (N=32) | 1 (3.1) |
| Vessel Ligation and Reconstruction (N=32) | 3 (9.4) |
| Major Nerve Resection (N=212) | 7 (3.3) |
| Nerve Reconstruction (N=7) | 0 (0.0) |
| Bowel Resection/Stoma Formation (N=212) | 1 (0.5) |
| Resection Margin (N=212) | |
| R0/R1 | 158 (74.5) |
| R2 | 24 (11.3) |
| Not applicable | 21 (9.9) |
| Unknown status | 9 (4.2) |
| Histopathology (N=212) | |
| Malignant | 12 (5.7) |
| Transformation to Neuroblastoma in all patients | 3 (0.9) |
Frequencies are reported in parentheses.
Approaches to tumor management
One hundred sixteen (35.4%) patients underwent active surveillance (Table 3). Common reasons for non-operative management were benign nature of the lesion (N=49), lack of symptoms (N=41), risks of surgery (N=25), and stable tumor size (N=19).
Resection was carried out in 212 (64.6%) patients, of which 74.5% cases achieved an R0/R1 resection (Table 3). Common reasons for operative management were: the presence of symptoms (N=52), curative chance (N=25), diagnostic uncertainty (N=21), surgeon preference (N=20), suspicion of other diseases (N=19), concerns of malignancy (N=14), patient choice (N=13), and mass effect (N=11).
Named vessel and nerve resections
Overall, 32 (15.1%) patients underwent resection of named arteries and/or veins (Table 3, Supplemental Table 1, Supplemental Digital Content 1, http://links.lww.com/SLA/E77). Among those patients, 28 (87.5%) underwent arterial and/or venous ligations only, 3 (9.4%) underwent arterial and/or venous ligations with reconstructions, and one (3.1%) underwent venous resection with reconstruction only. Common reasons for resection in this subset of patients included symptoms (16.2%), surgeon recommendation (16.2%), suspicion of other tumors (16.2%), patient decision (10.8%), and large tumor size (10.8%).
Seven patients (3.3%) underwent resection of named nerves (Table 3, Supplemental Table 2, Supplemental Digital Content 1, http://links.lww.com/SLA/E77). There were no reported reconstructions or nerve grafts. The most common reasons for operative intervention in context of nerve resection were due to symptoms (28.6%) and patient decision (28.6%).
Postoperative complications
Among the 212 patients who underwent resection, 40 (18.9%) developed post-operative complications (Table 4). Of those, 8 (20.0%) were Clavien-Dindo grade III or higher, which included massive bleeding, pneumonia, portal vein thrombosis, organ dysfunction (liver and renal), and/or fistula formation. Resection of tumors in the retroperitoneum was more likely to result in post-operative complications than resection in any other location (p = 0.003). Six of the 32 (18.8%) patients with named artery and/or vein resections had post-operative complications, although vascular resections were not associated with an increased overall perioperative complication rate (p = 0.81). There were 3 (42.9%) post-operative complications among those with named nerve resections. No significant association between nerve resection with post-operative complications were found (p = 0.19). There were no surgery-related deaths.
Table 4:
Post-operative Complications in 40 Patients.
| Complications | Number (%) | P value |
|---|---|---|
| Post-Operative Complications (N=212) | 40 (18.9) | |
| High Grade Complications* (N=40) | 8 (20.0%) | |
| Sex | 0.59 | |
| Male | 17 (42.5) | |
| Female | 23 (57.5) | |
| ASA | 0.64 | |
| I | 18 (45.0) | |
| II | 16 (40.0) | |
| III | 3 (7.5) | |
| Unknown status | 3 (7.5) | |
| Presentation | 0.16 | |
| Symptomatic | 23 (57.5) | |
| Incidental | 17 (42.5) | |
| Location | 0.003 | |
| Retroperitoneal | 26 (65.0) | |
| Adrenal | 3 (7.5) | |
| Pelvic | 9 (22.5) | |
| Intra-abdominal | 2 (5.0) | |
| Major Nerve Involvement | 5 (12.5) | 0.35 |
| Named Nerve Resection | 3 (7.5) | 0.19 |
| Vascular Involvement | 7 (17.5) | 0.41 |
| Named Vessel Resection | 6 (15.0) | 0.81 |
| Resection Margin | 0.69 | |
| R0/R1 | 28 (70.0) | |
| R2 | 5 (12.5) | |
| Not applicable | 6 (15.0) | |
| Unknown status | 1 (2.5) |
Frequencies are reported in parentheses. Chi Square test or Fisher’s exact test were used to calculate significance.
High grade complications are defined as a Clavien–Dindo grade of III or more.
Clinical outcomes of non-operative management
Throughout a median follow-up of 1.9 years (IQR [0.8, 4.2]) for 116 non-resected tumors, serial imaging in 92.2% showed predominantly stable disease while only 7.8% (N = 9) exhibited growth that later stabilized on subsequent cross-sectional imaging (Table 5). This indolent course continued to be seen in 52 ganglioneuromas (44.8% of the non-operative cohort) with a follow-up more than 2.0 years (median, 4.3 years; range, 2.0 – 16.8 years). Forty-nine (94.2%) of these tumors remained stable while only 3 (5.8%) displayed growth with a median increase of 5.3% in maximum tumor diameter (range, 4.7% – 20.0%). Long-term functional deficits were seen among 10 patients (8.6%) after a period of observation. Only one patient (0.9%) died due to an unknown cause.
Table 5:
Results and Follow-up of Operative and Non-operative management.
| Operative Management Number (%) |
Non-Operative Management Number (%) |
P value | |
|---|---|---|---|
| Median Follow-Up | 3.0 years (IQR 1.0 – 5.5) | 1.9 years (IQR 0.8 – 4.2) | |
| Status at Last Contact | N=212 | N=116 | |
| Recurrence | 4 (1.9) | 0 | |
| Disease Free Post Excision | 179 (84.4) | 4* (3.4) | |
| Residual disease | 18 (8.5) | 0 | |
| Ganglioneuroma still present | 0 | 103 (88.8) | |
| Unknown status | 11 (5.2) | 9 (7.8) | |
| Living Status | N=212 | N=116 | 0.46 |
| Alive | 199 (93.9) | 112 (96.5) | |
| Dead | 6 (2.8) | 1 (0.9) | |
| Unknown status | 7 (3.3) | 3 (2.6) | |
| Functional Deficits | N=212 | N=116 | 0.75 |
| Neurological sequelae | 6 (2.8) | 2 (1.7) | |
| Bladder dysfunction | 2 (0.9) | 0 | |
| Bowel dysfunction | 4 (1.9) | 1 (0.9) | |
| Bladder and Bowel Dysfunction | 1 (0.5) | 0 | |
| Lower limb edema | 1 (0.5) | 2 (1.7) | |
| Persistent pain | 8 (3.8) | 5 (4.3) |
Frequencies are reported in parentheses. Chi Square test or Fisher’s exact test were used to calculate significance.
Disease-free Post Biopsy.
Clinical outcomes of operative management
Over a median follow-up of 3.0 years (IQR [1.0, 5.5]) for resected tumors, 84.4% of patients were disease-free post-resection (Table 5). Macroscopic or microscopic residual disease was present in 18 patients (8.5%), of which 15 (83.3%) underwent R2 resections, 1 (5.6%) underwent R0/R1 resection, and 2 (11.1%) had unknown resection margins. Two of these 18 patients were later operated on for removal of residual tumors, after which they were disease-free. Recurrences were documented in 4 (1.9%) out of all operative cases. Final status of the remaining 11 (5.2%) operative patients were unknown.
Long-term functional deficits were seen among 22 (10.4%) patients after a period of observation (Table 5). For the 7 patients with named nerve resections, long-term functional deficits were reported in 2 (28.6%), including impaired ambulation due to femoral nerve resection and permanent sweat loss in the right lower extremity. The remaining 5 patients (71.4%) had no long-term functional deficits. There was a total of 6 (2.8%) deaths in patients with operative management, with 1 death due to recurrence of a malignant ganglioneuroma that progressed to the lungs. The remaining 5 patients who died had benign ganglioneuromas; 4 of these deaths were due to unknown causes and 1 was due to a metachronous lung cancer. There were no deaths reported in patients with ganglioneuromas that transformed into neuroblastoma.
Recurrence
In our study, 4 (1.9%) recurrences from sporadic ganglioneuromas were observed, 3 of which occurred following R0/R1 resections (resection margin data was unavailable for 1 patient). Based upon histopathology, 2 out of the 4 original tumors were malignant, with one displaying transformation to neuroblastoma. Malignancy occurred more frequently with recurrent tumors than with non-recurrent lesions (p = 0.02). Moreover, the time to recurrence varied among cases, ranging from 1.3 to 5.6 years. Two of the recurrences were identified on imaging within 1.6 years following resection, while the remaining two recurred after 3.0 and 5.6 years. Only one patient underwent a second operation and subsequently displayed no evidence of disease.
Discussion
This is the first international, multi-institutional large series examining the disease course and clinical management of ganglioneuromas. Through collaborative efforts of TARPSWG, we examine patient demographics, clinicopathologic features, radiological findings, and management approaches of ganglioneuromas. Based upon this series of 328 patients, we found that ganglioneuromas predominantly develop sporadically, seldom occur in the setting of familial syndromes, frequently present asymptomatically, and tend to have indolent growth patterns. Moreover, they have low rates of recurrence and rarely have malignant transformation to neuroblastoma except in young children.
By examining growth patterns of ganglioneuromas, we found that most lesions displayed stable disease and that only a minority of tumors showed disease progression during surveillance. But the minority that progress in size can become symptomatic and prompt the need for resection. Although the indolent growth rate of ganglioneuroma is well-identified, the imaging characteristics of these tumors are variable. Pre-operative diagnosis based on CT, MRI, or ultrasonography remain challenging. The morphology of ganglioneuromas vary from a well-circumscribed tumor to a poly-lobulated lesion, to a mass with cystic or cavitating components. CT scans display hypodense or isodense tumors. On MRI, T1 often shows hypointense to isointense lesions, while T2 exhibits mostly heterogeneously hyperintense masses with some examples of hypointensity and isointensity. Calcifications and multifocality are also observed in some cases. This range of signal intensity on CT or MRI is attributable to the varied composition of myxoid stroma, collagen fibers, and ganglion and Schwann cells9, 21, 29. In addition, the 12 malignant tumors identified in this study demonstrate no hallmark imaging findings indicating malignant transformation. Overall, there are no imaging characteristics pathognomonic for ganglioneuromas nor their evolution into cancer. It is thus of no surprise that ganglioneuromas were suspected on imaging in only 24.1% of patients, and that surgery was pursued due to uncertainty of the lesion or due to concerns for a more aggressive, malignant entity. Thus, it is worthwhile to consider a biopsy to establish a diagnosis when patients present with a mass, rather than undergoing surgical resection without a definitive preoperative diagnosis.
Approximately two-thirds of the cases that we identified underwent operative management. Resection was often pursued due to symptoms, or due to concerns of other tumors, such as pheochromocytoma, renal cell carcinoma, and adrenocortical carcinoma. The prognoses following resection were like those in other case series, as most patients remained disease free after index surgery at a median follow-up of 3 years (84.4%)1, 10, 24, 30. Tumor progression in incompletely resected lesions was occasionally observed in other series, but residual macroscopic tumors (8.5%) in our study did not demonstrate any evidence of progressive disease24, 30. Overall, the post-operative complication rate was 18.9%, which was comparable to those in other studies24, 26. However, the complications were predominantly minor (80.0%), and there were no surgery-related deaths. With no significant differences in long-term functional deficits between operative and non-operative management (p = 0.75), resection is the treatment of choice for symptomatic, rapidly growing, malignant, and diagnostically uncertain tumors based upon our findings.
In contrast, one-third of patients underwent non-operative management with imaging surveillance. This was in stark contrast to the clinical approach of other case series in which surgery was pursued in nearly all patients1, 10, 24, 30. In our study, imaging over time demonstrated predominantly stable tumor sizes, with only 7.8% of non-resected ganglioneuromas displaying slight growth that later stabilized in subsequent scans. Although surgery is often curative, non-operative management with active surveillance may be more suitable in select cases, such as for asymptomatic or indolent tumors which have been pathologically confirmed as benign or for lesions that are deemed non-resectable or could result in morbid operations due to involvement of multiple viscera. Certainly, aggressive resections can lead to fatal complications24, 30. Furthermore, our findings show that biopsy proven benign ganglioneuromas that remain stable during serial imaging for at least 2 years are unlikely to undergo malignant transformation or significantly grow. Thus, treating physicians may consider ceasing follow-up scans after 24 months in these patients or perform cross-sectional imaging with either MRI or CT annually or biannually depending on other clinical considerations or concerns.
The differentiation of neuroblastoma to a secondary ganglioneuroma has been well established1, 2. Conversely, occurrence of a neuroblastoma from a ganglioneuroma is extremely rare. Kulkarni et al. reported a case of a retroperitoneal ganglioneuroma that had transformed into a spinal neuroblastoma 11 years after resection25. Based on the histopathology in our study, there were 3 (0.9%) ganglioneuromas with transformation to neuroblastoma (one pelvic, one adrenal, and one retroperitoneal). All 3 were initially identified as pure ganglioneuromas on biopsy, and subsequent resection showed transformation to neuroblastoma. It is plausible that the biopsy was performed in a region without neuroblastoma, but that cancer was already present. Those that presented initially with biopsy-proven ganglioneuroblastoma intermixed were excluded from our study. In contrast to the transformation observed 11 years following resection in Kulkarni et al.’s report, the 3 cases in our study demonstrated a more rapid transformation. That is, resections showing neuroblastomas were performed within 6 months of the initial presentation and biopsy of pure ganglioneuromas. With a median age of diagnosis at 10 years (range, 9 – 11), these patients presented at an earlier age than those with pure ganglioneuromas at 37 years (p < 0.001). One of the lesions displayed malignant features, and recurrence was observed on MRI 3.0 years following R0/R1 resection with no evidence of disease after a second operation. Furthermore, one patient with vascular involvement demonstrated a positive MIBG, prompting surgical removal of the tumor that ended up transforming into a neuroblastoma despite initial biopsy showing a pure ganglioneuroma. The incorporation of MIBG and PET scans at initial or follow-up imaging may have utility in clinical management in cases with suspicious growth 31, 32. The genetic and biochemical development of a neuroblastoma from a ganglioneuroma still remains unclear. However, based upon our study, as younger patients appear to be at higher risk for transformation to neuroblastoma, patient age may be an important factor in determining the need for resection.
There are several limitations arising from the retrospective nature of this study. Due to the rarity ganglioneuromas, not all data fields were completed for each patient, leading to absence of some information in the database and hence measurement bias. Frequencies of variables were consistently reported with common denominators, with gaps in percentages indicating unavailable data. There also may be a limitation of a relatively short follow-up period in this study with an overall median follow-up of 2.3 years (IQR [0.9, 5.0]). Despite this, an indolent disease course continues to be seen for ganglioneuromas with a follow-up greater than 2.0 years (median, 4.3 years; range, 2.0 – 16.8 years). Because there is a potential of sampling bias with any tumor biopsy, there is a possibility that the findings of neuroblastoma from an initially pure ganglioneuroma may be due to sampling bias as opposed to a true transformation. However, various sampling methods were performed via either an incisional or core biopsy, and the stable disease course of one of the neuroblastomas was like that of the rest of the ganglioneuromas (percentage change over time data unavailable for the other two neuroblastomas). Nonetheless, resection remains the therapy of choice for ganglioneuromas with malignant transformation to neuroblastoma. There is also selection bias due to inclusion of cases only from participating sarcoma institutions through TARPSWG. Despite these limitations, the global nature of this collaboration across 29 institutions and 5 continents provides a comprehensive and large dataset that is valuable to the knowledge of ganglioneuromas occurring worldwide.
Conclusion
To our knowledge, this is the largest study to date on ganglioneuromas describing their demographics, clinicopathologic features, radiologic characteristics, and management. Although more than 40% of cases presented symptomatically, most ganglioneuromas have indolent disease courses and rarely transform to neuroblastoma except in children. Non-operative management with serial imaging evaluations may be appropriate and sufficient for biopsy-proven benign, asymptomatic, and/or stable tumors particularly when the risks outweigh benefits of surgery. If safe and technically feasible, definitive operative management with R0/R1 resection may be recommended in symptomatic, rapidly growing, biopsy-proven malignant, or diagnostically uncertain tumors. Overall, this study provides a broad, global analysis for informing ganglioneuroma surveillance and clinical management guidelines.
Supplementary Material
Funding
We appreciate the funding support from the Summer Student’s Research Fellowship Program by the Western University of Health Sciences (SN), as well as the James IV Association of Surgeons Traveling Fellowship (JKS), the UC San Diego LMS and Soft Tissue Sarcoma Research Fund (JKS).
Footnotes
Conflict of Interest
The authors declare no competing interests relevant to this study.
References
- 1.Geoerger B, Hero B, Harms D, et al. Metabolic activity and clinical features of primary ganglioneuromas. Cancer 2001; 91(10):1905–13. [DOI] [PubMed] [Google Scholar]
- 2.Ambros IM, Zellner A, Roald B, et al. Role of ploidy, chromosome 1p, and Schwann cells in the maturation of neuroblastoma. N Engl J Med 1996; 334(23):1505–11. [DOI] [PubMed] [Google Scholar]
- 3.Diab DL, Faiman C, Siperstein AE, et al. Virilizing adrenal ganglioneuroma in a woman with subclinical Cushing syndrome. Endocr Pract 2008; 14(5):584–7. [DOI] [PubMed] [Google Scholar]
- 4.Erem C, Fidan M, Civan N, et al. Hormone-secreting large adrenal ganglioneuroma in an adult patient: a case report and review of literature. Blood Press 2014; 23(1):64–9. [DOI] [PubMed] [Google Scholar]
- 5.Koch CA, Brouwers FM, Rosenblatt K, et al. Adrenal ganglioneuroma in a patient presenting with severe hypertension and diarrhea. Endocr Relat Cancer 2003; 10(1):99–107. [DOI] [PubMed] [Google Scholar]
- 6.Linos D, Tsirlis T, Kapralou A, et al. Adrenal ganglioneuromas: incidentalomas with misleading clinical and imaging features. Surgery 2011; 149(1):99–105. [DOI] [PubMed] [Google Scholar]
- 7.Lucas K, Gula MJ, Knisely AS, et al. Catecholamine metabolites in ganglioneuroma. Med Pediatr Oncol 1994; 22(4):240–3. [DOI] [PubMed] [Google Scholar]
- 8.Mendelsohn G, Eggleston JC, Olson JL, et al. Vasoactive intestinal peptide and its relationship to ganglion cell differentiation in neuroblastic tumors. Lab Invest 1979; 41(2):144–9. [PubMed] [Google Scholar]
- 9.Rondeau G, Nolet S, Latour M, et al. Clinical and biochemical features of seven adult adrenal ganglioneuromas. J Clin EndocrinolMetab 2010; 95(7):3118–25. [DOI] [PubMed] [Google Scholar]
- 10.Xie J, Dai J, Zhou WL, et al. Adrenal Ganglioneuroma: Features and Outcomes of 42 Cases in a Chinese Population. World J Surg 2018; 42(8):2469–2475. [DOI] [PubMed] [Google Scholar]
- 11.Brady S, Lechan RM, Schwaitzberg SD, et al. Composite pheochromocytoma/ganglioneuroma of the adrenal gland associated with multiple endocrine neoplasia 2A: case report with immunohistochemical analysis. Am J Surg Pathol 1997; 21(1):102–8. [DOI] [PubMed] [Google Scholar]
- 12.Chetty R, Duhig JD. Bilateral pheochromocytoma-ganglioneuroma of the adrenal in type 1 neurofibromatosis. Am J Surg Pathol 1993; 17(8):837–41. [DOI] [PubMed] [Google Scholar]
- 13.DeParis SW, Bloomer M, Han Y, et al. Uveal Ganglioneuroma due to Germline. Ocul Oncol Pathol 2017; 3(2):122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Efared B, Atsame-Ebang G, Tahirou S, et al. Bilateral pheochromocytoma with ganglioneuroma component associated with multiple neuroendocrine neoplasia type 2A: a case report. J Med Case Rep 2017; 11(1):208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamoun M, Mnif MF, Rekik N, et al. Ganglioneuroma of adrenal gland in a patient with Turner syndrome. Ann Diagn Pathol 2010; 14(2):133–6. [DOI] [PubMed] [Google Scholar]
- 16.Lora MS, Waguespack SG, Moley JF, et al. Adrenal ganglioneuromas in children with multiple endocrine neoplasia type 2: a report of two cases. J Clin Endocrinol Metab 2005; 90(7):4383–7. [DOI] [PubMed] [Google Scholar]
- 17.Stanich PP, Pilarski R, Rock J, et al. Colonic manifestations of PTEN hamartoma tumor syndrome: case series and systematic review. World J Gastroenterol 2014; 20(7):1833–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan CY, Liu JW, Lin Y, et al. Bilateral and symmetric C1-C2 dumbbell ganglioneuromas associated with neurofibromatosis type 1: A case report. World J Clin Cases 2019; 7( 1): 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamasaki M, Sato Y, Nomura T, et al. Composite paraganglioma-ganglioneuroma concomitant with adrenal metastasis of medullary thyroid carcinoma in a patient with multiple endocrine neoplasia type 2B: A case report. Asian J Endosc Surg 2017; 10(l):66–69. [DOI] [PubMed] [Google Scholar]
- 20.Elnady B, Abdelgawaad AS, Elkhayat H. Giant intrathoracic ganglioneuroma with scoliosis treated by one-stage posterior resection and scoliosis correction: a case report. SICOT J 2020; 6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qing Y, Bin X, Jian W, et al. Adrenal ganglioneuromas: a 10-year experience in a Chinese population. Surgery 2010; 147(6):854–60. [DOI] [PubMed] [Google Scholar]
- 22.Mylonas KS, Schizas D, Economopoulos KP. Adrenal ganglioneuroma: What you need to know. World J Clin Cases 2017; 5(10):373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng X, Luo L, Han FG. Cause of postprandial vomiting - a giant retroperitoneal ganglioneuroma enclosing large blood vessels: A case report. World J Clin Cases 2019; 7(17):2617–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Bernardi B, Gambini C, Haupt R, et al. Retrospective study of childhood ganglioneuroma. J Clin Oncol 2008; 26(10):1710–6. [DOI] [PubMed] [Google Scholar]
- 25.Kulkarni AV, Bilbao JM, Cusimano MD, et al. Malignant transformation of ganglioneuroma into spinal neuroblastoma in an adult. Case report. JNeurosurg 1998; 88(2):324–7. [DOI] [PubMed] [Google Scholar]
- 26.Sánchez-Galán A, Barrena S, Vilanova-Sánchez A, et al. Ganglioneuroma: to operate or not to operate. Eur J Pediatr Surg 2014; 24(l):25–30. [DOI] [PubMed] [Google Scholar]
- 27.Fang CW, Wang JS, Wu TT, et al. Occurrence of paratesticular ganglioneuroma 18 years after concurrent adrenal ganglioneuroma and papillary thyroid carcinoma - a case report. BMC Cancer 2019; 19(1):1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010; 363(18):1693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Nishimura H, Kato S, et al. MRI of ganglioneuroma: histologic correlation study. J Comput Assist Tomogr 2001; 25(4):617–23. [DOI] [PubMed] [Google Scholar]
- 30.Decarolis B, Simon T, Krug B, et al. Treatment and outcome of Ganglioneuroma and Ganglioneuroblastoma intermixed. BMC Cancer 2016; 16:542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharp SE, Shulkin BL, Gelfand MJ, et al. 123I-MIBG scintigraphy and 18F-FDGPET in neuroblastoma. J Nucl Med 2009; 50(8): 1237–43. [DOI] [PubMed] [Google Scholar]
- 32.Bleeker G, Tytgat GA, Adam JA, et al. 123I-MIBG scintigraphy and 18F-FDG-PET imaging for diagnosing neuroblastoma. Cochrane Database SystRev 2015(9):CD009263. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.