

Abstract
Head and neck squamous cell carcinoma (HNSCC) includes a subset of cancers driven by human papillomavirus (HPV). Here, we use single-cell RNA-seq to profile both HPV-positive and HPV-negative oropharyngeal tumors, uncovering a high level of cellular diversity within and between tumors. First, we detect diverse chromosomal aberrations within individual tumors, suggesting remarkable genomic instability, enabling the identification of malignant cells even at pathologically-negative margins. Second, we uncover diversity with respect to HNSCC subtypes and other cellular states such as the cell cycle, senescence, and epithelial-mesenchymal transitions. Third, we find heterogeneity in viral gene expression within HPV-positive tumors. HPV expression is lost or repressed in a subset of cells, which are associated with a decrease in HPV-associated cell cycle phenotypes, decreased response to treatment, increased invasion, and poor prognosis. These findings suggest that HPV expression diversity must be considered during diagnosis and treatment of HPV-positive tumors, with important prognostic ramifications.
Introduction
HNSCC tumors of the oral cavity and larynx are typically linked to alcohol and tobacco exposure, while oropharyngeal squamous cell carcinoma (OPSCC) is more commonly associated with infection by human papillomavirus (HPV)1. HPV-associated OPSCCs have better prognosis than other forms of HNSCC, calling for treatment de-escalation to reduce side effects while maintaining an excellent prognosis. However, a subset of HPV-related OPSCCs respond poorly to treatment and recur2, underscoring the need for a deeper understanding of these tumors and the development of new therapeutic approaches.
HPV is a sexually transmitted DNA virus, accounting for more than 5% of all cancer cases worldwide including all cervical cancers, most OPSCCs, and a majority of vaginal and anal cancers3. The HPV oncogenic proteins, E6 and E7, inhibit the tumor suppressor proteins p53 and Rb, respectively, thereby activating the proliferation of infected epithelial cells4,5. While these initial effects of HPV are largely understood, the subsequent events leading to tumorigenesis6, the biology of the resulting tumors, and their vulnerabilities still remain poorly characterized.
Previously, we used single cell RNA sequencing (scRNA-seq) to interrogate patient samples of HPV-negative oral cavity tumors7–10. Here, we turn our focus to OPSCC, profiling both HPV-positive and HPV-negative tumors. We uncover unanticipated diversity of chromosomal aberrations and of HPV expression patterns. Strikingly, each HPV-positive tumor harbors a subset of malignant cells in which HPV expression is not detected, and HPV-related phenotypes are decreased. These cells may influence prognosis and therapy response, highlighting their significance and opportunities for new interventions.
Results
Single cell RNA-seq analysis of OPSCC
We profiled 16 treatment-naïve OPSCC primary tumor samples using the 10x chromium platform (Fig. 1a, Supplementary Table 1–2). After removing low-quality cells and potential doublets (see Methods), we retained 70,970 cells, which were used to describe four distinct layers of cellular diversity (Fig. 1a): cell types, genetic clones, cellular states, and HPV expression patterns.
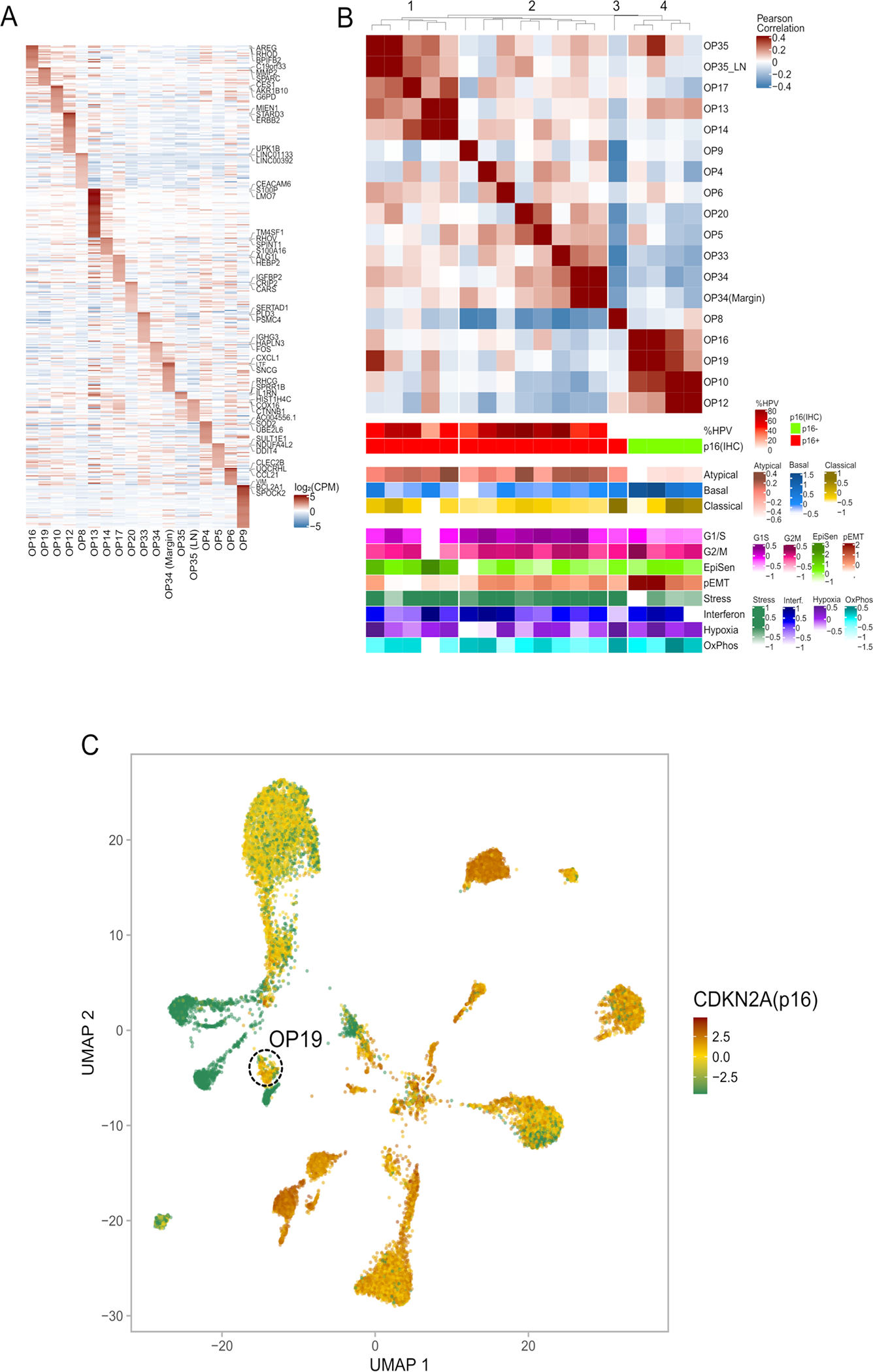
Figure 1. ScRNA-seq analysis of 16 OPSCC tumors.
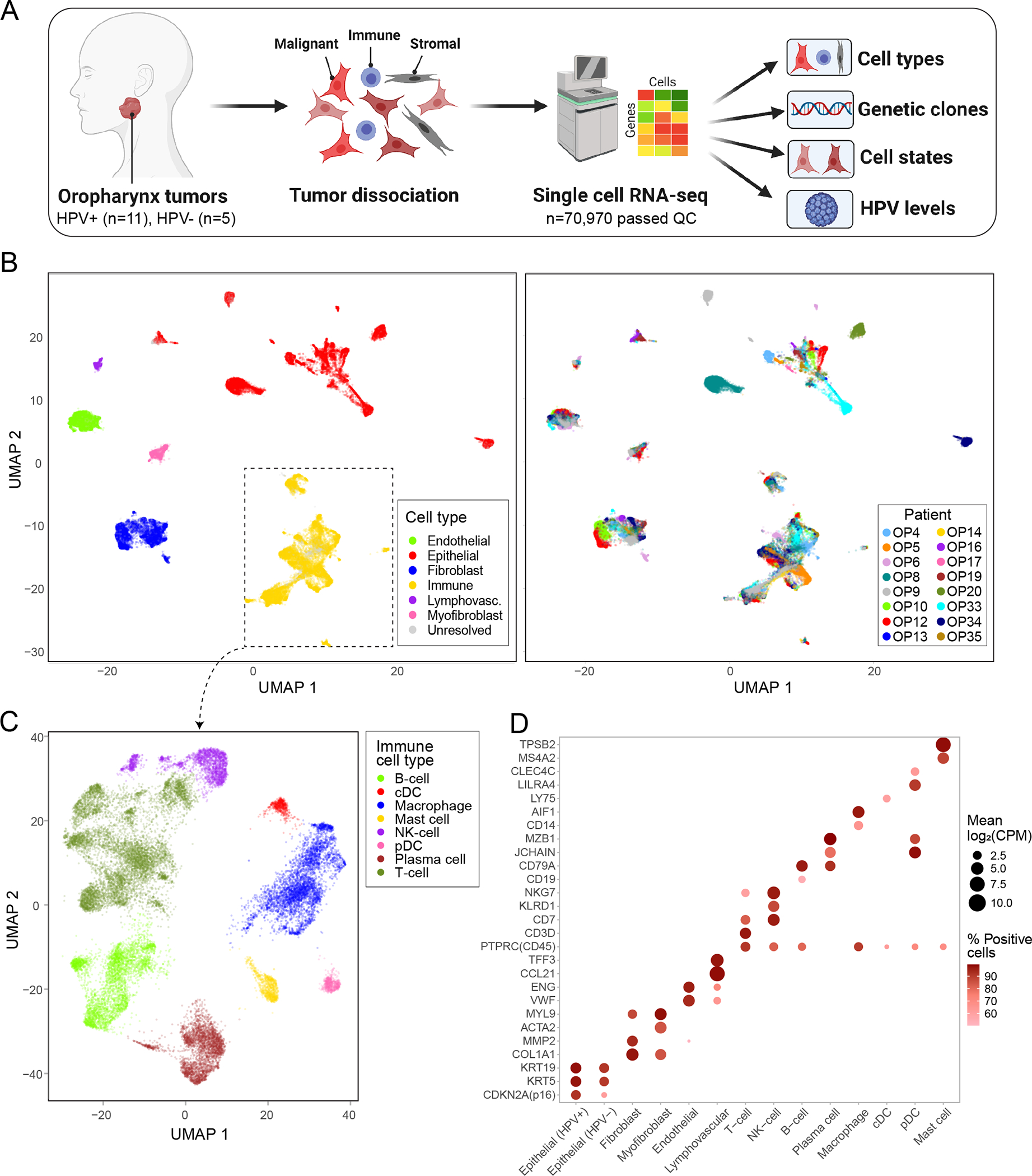
(A) Scheme of the workflow for OPSCC profiling and subsequent analysis. (B) UMAP plot of all cells that passed QC (n=70,970), colored by cell type and patient. (C) UMAP plot of all immune cells (n=22,818), colored by immune cell type. (D) Dot plot showing expression of selected marker genes (Y-axis) by all cells assigned to each cell type (X-axis). Dot size represents average expression, and dot color represents the fraction of cells with non-zero expression.
We first clustered all cells and annotated the clusters by differentially expressed marker genes (Fig. 1b–c, Extended Data Fig. 1a–b; Supplementary Table 3–4). Epithelial cells clustered primarily by patient identity, while non-epithelial cell types clustered together regardless of patient identity. We defined 12 non-epithelial clusters, including typical tumor components (e.g. T-cells and fibroblasts) as well as less common components (e.g. myofibroblasts and lymphovascular cells), each of which contained cells from multiple patients and expressed characteristic marker genes (Fig. 1d).
Based on standard p16 staining, twelve of the OPSCC tumors were clinically defined as HPV-positive and four as HPV-negative (Extended Data Fig. 1c). We mapped the scRNA-seq reads of all epithelial cells to the five most common high-risk HPV genotypes11, and identified transcripts of the most common HPV genotype (HPV16) in 11 of the 12 tumors clinically defined as HPV-positive and in none of the tumors clinically defined as HPV-negative (Extended Data Fig. 1d–e). In these 11 HPV-positive tumors, HPV16 transcripts were identified in an average of 53% (20–78% range) of epithelial cells (Supplementary Table 4). In one exceptional tumor (OP8) we did not identify any HPV transcripts despite clinical diagnosis as HPV-positive. Further testing and sequence analysis failed to identify evidence for any other HPV genotypes (see Methods), although we cannot formally exclude the possibility of a rare undetected genotype. These results suggest either a false positive clinical diagnosis by p16, clearance of the virus, or a limitation in detecting HPV transcripts, which seems unlikely based on the other tumors.
The HPV16 genome contains eight genes, and these were detected at variable frequencies, with E5 being the most commonly detected, followed by E1. The pattern of HPV gene expression varied between tumors, with seven tumors expressing E5 at particularly high levels and others with a relative enrichment of E7 (Extended Data Fig. 1e). These distinct patterns of HPV expression, rather than a uniform expression of viral proteins, are consistent with previous findings12.
Chromosomal aberrations identify malignant cells and clonality
We classified the epithelial cells into malignant and non-malignant cells based on inference of chromosomal copy-number aberrations (CNAs)8,13–15. At each chromosomal locus, an estimated copy number was calculated by averaging the normalized expression levels of the hundred adjacent genes compared to their expression in a reference set of fibroblasts and endothelial cells (see Methods). Most epithelial cells had multiple CNAs, including characteristic CNAs of OPSCC (e.g. 3p loss, 3q and 8q gain) (Fig. 2a, Extended Data Fig. 2a). We classified epithelial cells into malignant and non-malignant cells, by the combined evidence for CNAs across all chromosomes (i.e. CNA signal) and the similarity of the CNA pattern to that of other cells from the same tumor (i.e. CNA correlation), thereby defining a robust separation (Fig. 2b).
Figure 2. Inference of chromosomal aberrations for identification of malignant cells, genetic subclones and invasive cells.
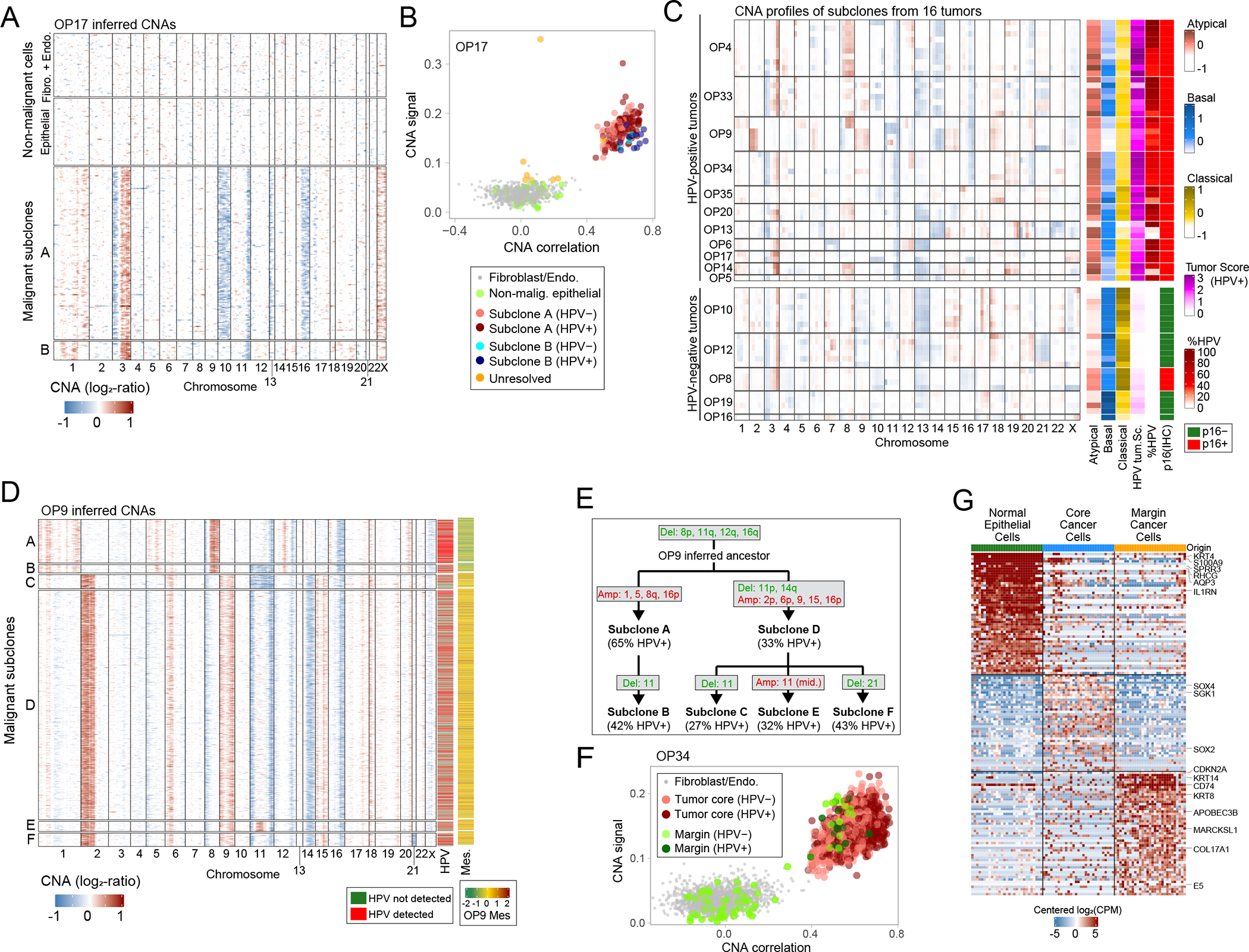
(A) CNA plot of OP17, inferred through taking a 100-gene moving average of relative expression values across the transcriptome (Methods). Rows represent cells, arranged by genetic subclones, and columns genes, arranged by chromosomal position. Fibroblasts and endothelial cells, used as reference for CNA inference, as well as cells classified as non-malignant epithelial cells, are shown above the malignant cells. (B) Scatter plot of two CNA metrics used for classification of cells as malignant, CNA signal (Y-axis) and CNA correlation (X-axis). All epithelial and stromal cells of OP17 are shown, colored by their cell type, subclone assignment and HPV expression. (C) Left: average CNA profiles for all identified genetic subclones; rows represent subclones, ordered by patient, and columns represent chromosomal positions (with five bins per chromosome). Right: scores of subclones (arranged as in left panel) for the TCGA subtypes and the HPV+ tumor signatures, the percentage of cells with HPV reads, and the HPV clinical classification of the corresponding tumor based on p16 staining. Subclone scores reflect average scores of the cells in each subclone. (D) CNA plot of malignant cells in OP9 as in (A). Columns on the right show detection of HPV reads and average expression of a mesenchymal signature found in OP9. (E) Inferred phylogenetic tree of genetic subclones in OP9. The percentage of cells with detection of HPV reads is noted for each observed subclone; chromosomal deletions (green) and amplifications (red) are noted for each observed subclone as well as for the inferred ancestral clone. (F) CNA signal and correlation scatter plot for OP34 as in (B). Cells are colored by their origin (tumor core or margin sample) and by HPV expression. (G) Heatmap of differentially expressed genes between the three subsets of epithelial cell in OP34 – normal epithelial cells, invasive malignant cells and malignant cells from the tumor core. Rows represent genes, columns represent cells. An equal number of cells is shown from each subset (to that end, cells from the normal and tumor core subsets were randomly sampled).
Overall, we classified 20,323 (85%) of epithelial cells as malignant cells, while 2,625 (11%) cells were classified as non-malignant (“normal”) epithelial cells (Extended Data Fig. 2a). The remainder were defined as unresolved and excluded from further analysis, likely reflecting doublets or low-quality cells. To validate the CNA-based classification, we compared epithelial cells from HPV-positive tumor samples to those from normal adjacent tissue of the same patients to derive a gene expression signature of HPV-related malignancy (Supplementary Table 3). Scoring the epithelial cells by this signature showed remarkable congruence with the CNA-based classification (Extended Data Fig. 2b–c). In HPV-positive tumors, CNA classification was largely consistent with the identification of HPV transcripts, although we also detected a small subset of non-malignant cells with HPV transcripts (Extended Data Fig. 3f–g; Supplementary Note 1).
The CNA analyses also uncovered distinct genetic subclones within individual tumors. For example, in OP17, the malignant cells were separated into two genetic subclones with both shared and clone-specific gains and losses (Fig. 2a–b). Overall, multiple CNA subclones were identified within 14 of 16 tumors (Fig. 2c). OP9 displayed particularly extensive subclonal diversity, with six different genetic subclones (Fig. 2d), for which we inferred a phylogenetic tree (Fig. 2e). The tree defined two major branches (subclones A-B and subclones C-F) that also differed in expression, with one branch expressing a unique program with many mesenchymal genes (e.g. collagens) (Fig. 2d, right-most column).
The two branches of OP9 also differed by the frequencies of HPV detection (62% vs. 33%, p<2.2*10−16, chi-square test, Fig. 2d). Similarly, a significant difference in HPV detection between subclones was found for eight of the ten HPV-positive tumors that had multiple subclones (Fig. 2c). For example, in OP13, three subclones had HPV detected in 90%, 4%, and 0% of cells. In some cases, subclones differed not only in the frequency of HPV detection, but also in the relative detection of distinct HPV genes (Extended Data Fig. 2e). Thus, the overall abundance and the relative expression patterns of HPV genes appear to be modified during tumor evolution and to vary both between and within HPV-positive tumors.
Malignant cells found in the histologically negative tumor margin
In three cases, we were also able to profile histologically-negative margin tissues (“adjacent normal”). Most epithelial cells from these samples were classified by CNA analysis as non-malignant, as expected. However, in one negative margin sample (OP34; Extended Data Fig. 3a), 29 out of 80 epithelial cells were classified as malignant by CNAs and by the malignancy expression signature (Fig. 2f, Extended Data Fig. 3b). A subset of these cells expressed HPV genes, further supporting their malignant classification (Fig. 2F). These malignant cells harbored all of the CNAs shared across OP34 subclones, and also unique CNAs (in chromosomes 4q, 9 and 22), thus representing a separate genetic subclone (Extended Data Fig. 3c). These results suggest further evolution of an invasive subclone beyond the leading front (histological edge) of the tumor. Notably, OP34 had clear resection margins on frozen and permanent histopathologic analysis (see Methods), indicating that malignant cells were not expected in the margin sample by traditional pathologic techniques. However, in a subset of OPSCCs, tumor recurrence occurs despite surgery with widely clear margins, suggesting that individual malignant cells likely remain undetected in these cases, as might be the case in OP3416. We compared the expression of malignant cells from the margin to both malignant cells from the core of the tumor and to normal epithelial cells in the margin sample (Fig. 2g, Supplementary Table 2). Fifty-seven genes were significantly upregulated in the malignant cells from the margin, including cytokeratins, EMT-related genes, APOBEC genes, immune-related genes, and the HPV E5 gene, clearly distinguishing this invasive population.
To explore the generalizability of malignant cells within histologically negative margins, we searched for other scRNA-seq datasets containing matched tumor and negative margin samples. We identified two lung adenocarcinoma samples17 meeting these criteria and classified the cells from these samples by inferred CNAs (Extended Data Fig. 3d–e). One lung tumor did not contain malignant cells in the margin, while another tumor had malignant cells in the negative margin sample, representing a distinct subclone by CNAs and upregulating 47 genes (Extended Data Fig. 3d; Supplementary Table 3). These data highlight the presence of malignant cells in histologically negative margin biopsies that may drive adverse clinical outcomes.
OPSCC tumor diversity highlights three cellular subtypes
Overall, the diversity among malignant cells is linked to patient identity, to HPV status and to three TCGA subtypes - atypical, basal and classical18 (Fig. 3a, Extended Data Fig. 4; Supplementary Table 5). Each tumor had a dominant subtype, on average covering 96% of the cells in the tumor that are confidently assigned to any subtype (Fig. 3a–b). All HPV-positive tumors had a dominant atypical subtype. In contrast, HPV-negative tumors included two with a dominant basal subtype, two with a dominant classical subtype, and OP8, with a majority of intermediate cells (not confidently assigned to any TCGA subtype), possibly reflecting its unique pattern as p16-positive but HPV-negative tumor.
Figure 3. Diversity of OPSCC malignant cells.
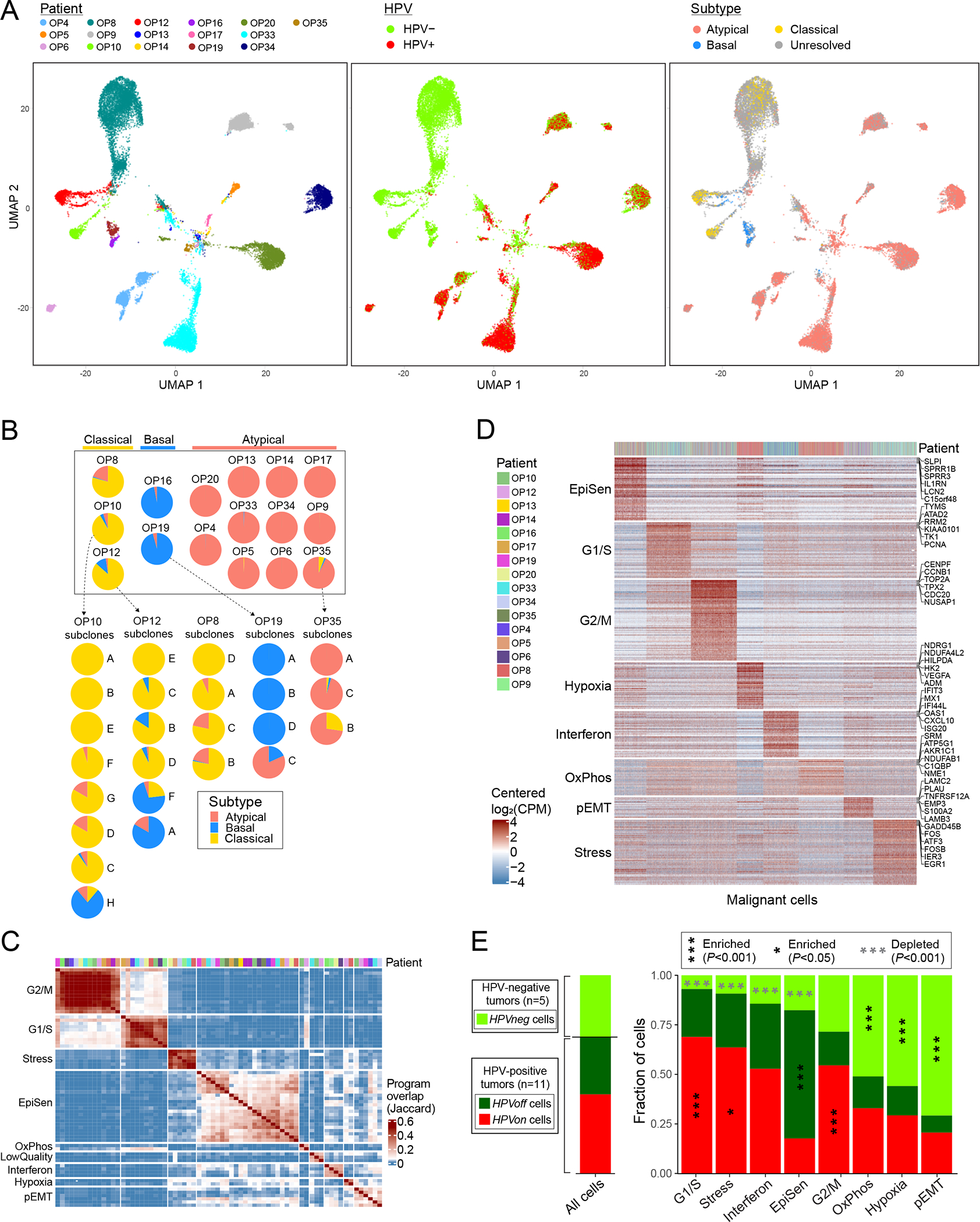
(A) UMAPs of all malignant cells (n=20,323) colored by patient (left panel), HPV expression (middle panel) and TCGA subtype (right panel). Cells with smaller than 1.5 fold-change between the top and the second highest subtype scores were defined as unresolved and marked in grey. (B) Pie charts representing the fraction of cells assigned to each TCGA subtype (excluding unresolved cells), per patient (above) and per subclone for patients with multiple subtypes and multiple subclones (below). (C) Hierarchical clustering of 69 NNMF-derived program signatures from 16 patients (see Methods). Signatures are clustered by Jaccard overlap. Groups of signatures, from which meta-programs are derived, are annotated on the left. Top panel shows the patient origin for each program using the same color map as in (D). (D) Expression of meta-program genes (rows) in all malignant cells (columns). Top panel indicates the patient origin for every cell. (E) For each meta-program, bar-plot shows the fraction of cells, out of those assigned to that meta-program, in three HPV-related classes: cells from HPV-negative tumors (HPVneg, light green) and cells from HPV-positive tumors in which HPV reads are detected (HPVon, red) or undetected (HPVoff, dark green). Asterisks denote enrichment (black and vertical) or depletion (grey and horizontal); asterisks within the HPVneg area denote enrichment/depletion in HPVneg vs. HPV-positive tumors (HPVon and HPVoff), and asterisks within the HPVon or HPVoff area denote enrichment in comparison between those two classes. Significance of enrichment/depletion was calculated using a hypergeometric test, corrected for multiple-testing. Bar-plot at the left shows the same analysis for all malignant cells. When calculating all fractions, 100 cells per patient and subset were randomly sampled 100 times to avoid patients with more cells skewing the results.
Despite the dominant subtype of each tumor, subsets of cells from five tumors were confidently assigned to a secondary subtype. In all of these cases, subtype heterogeneity was linked to genetic subclones (Fig. 3b). For example, while OP19 is dominated by the basal subtype, 4% of its malignant cells are classified as atypical and these primarily derive from subclone C. Interestingly, while OP19 is HPV-negative, it has high mRNA expression of CDKN2A (p16) (Extended Data Fig. 4c), perhaps relating to its secondary atypical subtype.
Intra-tumor heterogeneity and epithelial senescence
To systematically search for additional patterns of intra-tumor heterogeneity, malignant cells from each tumor were analyzed by non-negative matrix factorization. The expression programs identified as variable within tumors were compared across tumors to define eight groups of recurrent expression programs (Fig. 3c). For each of the eight groups, we defined a consensus “meta-program”, annotated them by functional enrichments, and scored all malignant cells for these meta-programs (Fig. 3d; Supplementary Table 6). Similar analysis was also performed for six common non-malignant cell types (Extended Data Fig. 5; Supplementary Table 7).
The malignant meta-programs included cell cycle (G1/S and G2/M phases), stress and hypoxia responses, oxidative phosphorylation, interferon response, hybrid, partial EMT (p-EMT), and an epithelial senescence-associated (EpiSen) program. Notably, the latter two meta-programs appear to be enriched in HNSCC and associated with metastasis and drug responses, respectively19,20. EpiSen was the most common pattern of heterogeneity in OPSCC, detected in 14 tumors (Fig. 3c), with high similarity to previously identified programs in oral cavity tumors8, cell lines19, and other squamous cancers21,22. The fraction of EpiSen-high cells varied markedly between tumors, from less than 5% to more than 50% of malignant cells. Consequently, pseudo-bulk tumor profiles segregated the HPV-positive tumors primarily by the frequency of EpiSen-high cells (Extended Data Fig. 4b).
Undetectable HPV expression in a subset of malignant cells from HPV-driven tumors
Except for the G2/M meta-program, all other meta-programs differed significantly in their abundance between HPV-positive and HPV-negative tumors (Fig. 3e). Interestingly, we also noticed differences in meta-program abundances within the HPV-positive tumors when distinguishing between 66% of the malignant cells in which we detected HPV reads (denoted as HPVon cells) and the remaining 34% in which we did not detect any HPV reads (denoted as HPVoff cells) (Fig. 3e). For example, expression of the G1/S meta-program was enriched in HPVon cells, not only relative to cells from HPV-negative tumors (denoted in Fig. 3e by asterisks within HPVneg) but also relative to the HPVoff cells from HPV-positive tumors (denoted in Fig. 3e by asterisks within HPVon). Conversely, the EpiSen meta-program was specifically enriched in HPVoff relative to HPVon cells among the HPV-positive tumors.
These results raise the possibility that lack of HPV detection in HPVoff cells may not merely reflect limited scRNA-seq sensitivity, but may also correspond to unique cellular states associated with genetic or epigenetic repression of HPV genes. Such repression is consistent with the variability in HPV expression profiles (Extended Data Fig. 1e and 2e) and in the fraction of cells with detected HPV that we observed between tumors and subclones (Fig. 2c–d). Thus, HPV status may define not only two types of tumors (HPV-positive and HPV-negative), but at least three types of malignant cells (HPVneg, HPVon and HPVoff). Notably, no tumors were entirely HPVoff, rather just subsets of malignant cells within each of the HPV-positive tumors.
To examine if HPV genes are repressed in HPVoff cells, or whether their expression is not detected due to technical limitations, we compared the frequency of HPVoff cells to the frequency in which other sets of genes are not detected. The fraction of HPVoff was significantly higher than expected based on detection of other sets of control genes sampled so that each gene in the control gene set had similar average expression levels to one HPV gene (34% vs. 9%, p<2.2*10−16, z-test) (Fig. 4a). RNAscope in situ hybridization (ISH) (Fig. 4b, Extended Data Fig. 6a) as well as immunohistochemistry (IHC) (Extended Data Fig. 6b) further supported the presence of HPVoff cells, by demonstrating the absence of E6 and E7 RNA and E6 protein in several tumor areas marked by p16. Together, these data uncover a subset of malignant cells (HPVoff) in which HPV expression is lost or reduced.
Figure 4. HPVoff cells and their association with cell cycle and senescence.
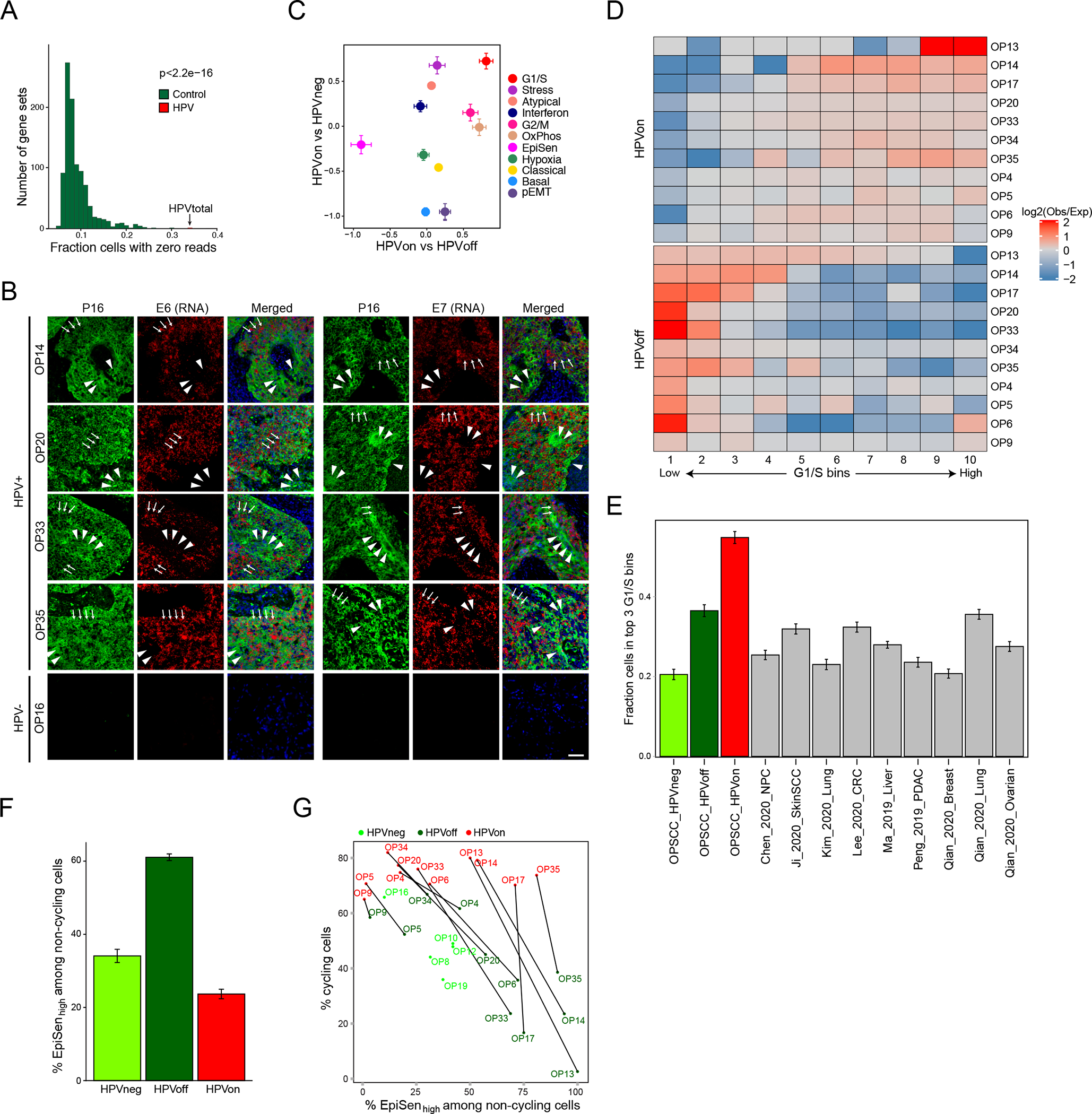
(A) Fraction of cells with zero reads for 1000 control gene-sets and for the set of five detected HPV genes (E1, E2, E5, E6 and E7). Each control gene-set includes one non-HPV gene as the matched control for each of the five HPV genes. Control genes were randomly sampled among the 100 genes closest to the respective HPV gene, based on average expression across all cancer cells from HPV-positive patients (p<2.2e-16, z-test). (B) RNA ISH (RNAScope) of representative HPV-positive (OP14, OP20, OP33, and OP35) and HPV-negative (OP16) tumors for viral E6 (left) and E7 (right) RNA (red) with immunofluorescence co-staining for regions of tumor as marked by p16 protein (green) and nuclei by DAPI (blue). HPV-positive tumors display regions of p16 positivity with absence of E6 and E7 RNA signal, consistent with an HPVoff state (arrowheads), while other regions have p16 along with E6 and E7 expression (HPVon; arrows). HPV-negative tumors do not have signal for p16 protein or E6 or E7 RNA. Scale bar = 1000 μm. (C) Scatter plot of differences in program expression between cells from different HPV classes. The X-axis shows mean difference, for all genes in each metaprogram, between HPVon and HPVoff cells within the same patient (n=11 patients). The Y-axis shows mean difference between HPVon cells, averaged across all HPV-positive patients (n=11), and HPVneg cells, averaged across HPV-negative patients (n=5). Error bars represent the standard error of the mean for each geneset. (D) Log2-ratio of observed to expected number of cells in each bin of G1/S scores ranked from low (left) to high (right), for each HPV-positive tumor (rows). Top and bottom rows correspond to the HPVon and HPVoff cells, respectively. I Mean fraction of malignant cells with high G1/S expression, as defined by the top 3 bins of G1/S scores, in each HPV class from this work and in multiple external datasets (n=9). Error bars show standard error of the mean fraction from 100 repetitions with sampling of 1000 cells per dataset. (F) Mean proportions of EpiSen-high noncycling cells across HPV subsets in n=5 (HPVneg) and n=11 (HPVon, HPVoff) patients. The top 20% of all malignant cells by average expression of the EpiSen program genes were defined as EpiSen-high. The y-axis shows, per subset, the mean proportion of EpiSen-high noncycling cells among all noncycling cells. Error bars represent standard error after resampling 100 times, each time sampling 200 cells per patient and subset. (G) Proportions of cycling cells and EpiSen-high noncycling cells for each patient and HPV subset.
HPVoff cells are associated with HPV-negative phenotypes
We next identified genes that were differentially expressed between HPVon and HPVoff cells across multiple tumors (Supplementary Table 8). EpiSen genes were enriched in HPVoff cells, while G1/S cell cycle genes were enriched in HPVon cells (Fig. 4c, Extended Data Fig. 6c–d). To further characterize cell cycle differences between HPVon and HPVoff cells, we divided all malignant cells into 10 bins by expression of the G1/S program. In all HPV-positive tumors, HPVon and HPVoff cells were significantly enriched (p<0.01 for every patient, chi-square test) in higher and lower G1/S bins, respectively (Fig. 4d, Extended Data Fig. 6e). This consistent association of HPVon with cell cycle is also seen across subclones (Extended Data Fig. 6f).
While all cancers are associated with increased proliferation, HPVon cells have higher expression of the G1/S program than other cancer types, based on reanalysis of multiple 10x scRNA-seq datasets. Most HPVon cells (54%) were among the G1/S-high cells, compared to significantly lower fractions (defined in a similar manner) for the HPV-negative OPSCCs (from this work) as well as for nine other tumor cohorts (Fig. 4e). For HPVoff cell, 36% were G1/S-high, which is comparable to the other tumor cohorts, although still higher than most of them. Thus, HPV is associated with an aberrant activation of the G1/S expression program, which is reduced in HPVoff cells, consistent with the possibility that HPV expression is suppressed in those cells. Loss of pRb repression by the HPV-E7 oncogene4,5 may thus decrease the proportion of HPVoff cells in G1/S phase.
We further speculated that the second major difference between HPVon and HPVoff cells – the enrichment of EpiSen in HPVoff cells (Fig. 4c) – reflects the inactivation of p53 by HPV-E6, as absence of HPV-E6 may enable cells to induce senescence. Multiple observations link the EpiSen meta-program to senescence: It is induced in senescent keratinocytes and bronchial cells19 and is enriched in non-cycling HNSCC cells, both in the oral cavity8 and in oropharynx tumors (Extended Data Fig. 6g). Notably, the enrichment of EpiSen in HPVoff cells remained significant when restricting the analysis to non-cycling cells, in order to decouple the differences in induction of a senescence program from the differences in proliferation (Fig. 4f). In summary, HPVon and HPVoff cells from the same tumor differ in the fraction of cycling cells and in the induction of EpiSen among the non-cycling cells, presumably reflecting the reduced activity of the two major HPV oncogenes (E6 and E7) in HPVoff cells. Importantly, this observation is consistent across all 11 HPV-positive tumors (Fig. 4g).
TCGA data and cell lines support a lower proliferation of HPVoff cells
To explore the functional significance of HPVoff cells, we examined the TCGA dataset of HPV-positive OPSCC tumors. We reasoned that bulk expression levels of HPV transcripts (normalized for tumor purity)could serve as an approximation for the fraction of HPVon versus HPVoff malignant cells. Consistent with our scRNA-seq analysis, normalized HPV expression correlates with G1/S scores across HPV-positive TCGA tumors (Fig. 5a). Similar results were obtained in analysis of TCGA specimens for cervical cancer, suggesting that HPV expression levels are associated with G1/S induction across distinct contexts (Extended Data Fig. 7a).
Figure 5. Regulation and function of HPVoff cells.
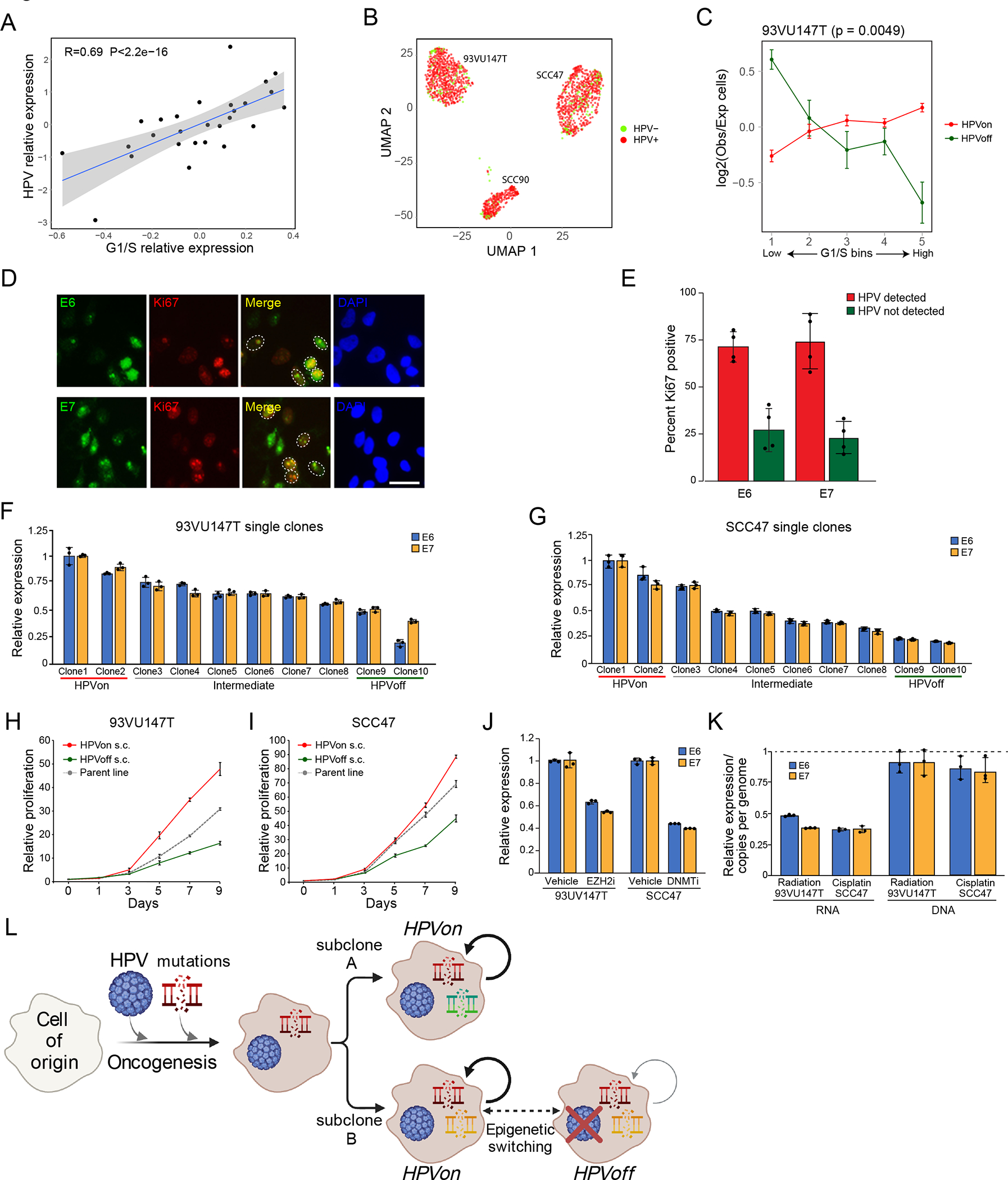
(A) Scatter plot of all HPV-positive OPSCC samples in the TCGA cohort, showing correlation (two-sided Pearson correlation test) between the relative expression of HPV and of the genes in the G1/S program. Relative expression values reflect residuals, after normalizing each sample for malignant cell content (using the epithelial signature from Supplementary Table 3). (B) UMAP of 1,422 cells from three HPV-positive cell lines colored by HPV expression. Cells with at least one read from an HPV16 gene were considered HPV+. (C) Differences in expression of the G1/S genes between HPV subsets in the HPV-positive cell line 93VU147T. Cells were divided into 5 bins of equal size, ranked by average expression. The Y-axis shows mean ratio of cells belonging to an HPV subset in a bin versus the expected number of cells, assuming random distribution across bins. Error bars are standard error after 100 resampling runs, where 100 cells per subset were randomly selected. P-value based on chi-square test, comparing the distribution of cells per bin between the groups. (D) Immunocytochemistry images of 93VU147T cells probed with Ki67 (red) and E6 (green, top) or E7 (green, bottom). Nuclei were stained and visualized with DAPI (blue). Scale bar = 100 μm. (E) Bar plot (mean +/− SEM) shows percentage of Ki67 positive cells among E6 and E7 positive cells (HPV detected; red) and E6 and E7 negative cells (HPV not detected; green). 50 cells were counted across four fields (p<0.00001, chi-square). (F and G) Bar plot (mean +/− SEM) shows relative expression of E6 and E7 among single clones (n=10) derived from 93VU147T (F) and SCC47 (G) revealing diversity in HPV expression (p<0.00001, ANOVA).(H and I) Line graph (mean +/− SEM) shows relative proliferation of HPVon and HPVoff single clones derived from 93VU147T (H) and SCC47 (I) compared to parent line. HPVon single clones displayed substantially more relative proliferation than HPVoff single clones (n=3; p<0.00001, two-sided t-test). (J) Left: Bar plot (mean +/− SEM) shows relative expression of E6 and E7 in 93VU147T cells treated with vehicle or tazemetostat (EZH2 inhibitor) (left). Right: Bar plot (mean +/− SEM) shows relative expression of E6 and E7 in SCC47 cells treated with vehicle or decitabine (DNMT inhibitor). Tazemetostat and decitabine significantly reduced relative E6 and E7 expression compared to vehicle in 93VU147T cells and SCC47 cells, respectively (n=3; p<0.001 and p<0.00001, ANOVA). (K) Left: Bar plot (mean +/− SEM) shows relative expression of E6 and E7 in 93VU147T and SCC47 cells treated with radiation or cisplatin, respectively, normalized to control cells (dashed line) (n=3; p<0.00005, two-sided t-test). Right: Bar plot (mean +/− SEM) depicts HPV copies per genome of E6 and E7 (normalized to albumin) for 93VU147T and SCC47 cells treated with radiation or cisplatin, respectively, normalized to control cells (dashed line). There were no significant differences in HPV copies in genomic DNA in radiation or cisplatin treated cells compared to control (n=3). (L) Model of genomic and viral heterogeneity in HPV-related OPSCC. A combination of HPV infection and associated genetic mutations trigger oncogenesis. Some genetic subclones continue to express HPV (HPVon), while others may undergo epigenetic switching with repression of HPV expression (HPVoff) and an associated decrease in cell cycle (circled arrows).
As a complementary approach, we analyzed scRNA-seq data from three HPV-positive cell lines19. Although HPV expression was identified in most cells, we found HPVoff subpopulations in each of the cell lines (Fig. 5b, Extended Data Fig. 7b). In two cell lines, HPVoff cells were also associated with decreased G1/S scores (Fig. 5c, Extended Data Fig. 7c). Immunocytochemistry confirmed that expression of HPV proteins E6 and E7 (but not of p16) correlates with proliferation (Fig. 5d–e, Extended Data Fig. 7d–e). Moreover, knockdown of E6 and E7 in these lines did not affect p16 expression but reduced proliferation (Extended Data Fig. 7f–g; Supplementary Fig. 1).
Single cell clones from these two cell lines showed a spectrum of HPV expression, which we denoted as HPVon, HPVoff, and intermediate clones (Fig. 5f–g). HPVon and HPVoff clones largely maintained their relative HPV expression levels over multiple passages (Extended Data Fig. 7h–i), demonstrating the heritability of these states. HPVon clones were enriched with cycling cells and were more proliferative (Fig. 5h–i, Extended Data Fig. 7j). Notably, serum starvation of HPVon clones suppressed their proliferation with little to no effect on HPV expression (Extended Data Fig. 7k–l), suggesting that HPV expression does not merely reflect that a cell is cycling but rather directly promotes the cell cycle through the function of E6 and E74,5. Taken together, these results highlight an association between heterogeneity of HPV expression levels and of G1/S cell cycle activity.
HPVoff cells are epigenetically regulated and may be associated with invasion and drug resistance
In contrast to the observed expression differences of HPV genes between HPVon and HPVoff clones, the genomic copy numbers of HPV genes were comparable between HPVon and HPVoff clones (Extended Data Fig. 8a). Moreover, DNAScope ISH experiments did not show substantial differences in E6 and E7 between and within different tumors (Extended Data Fig. 8b–c). These observations support the possibility of epigenetic regulation of HPV expression. We therefore treated HPV-positive cell lines with inhibitors of two epigenetic regulators, EZH2 and DNA methyltransferases (DNMT). Inhibition of EZH2 significantly reduced HPV expression in 93VU147T with limited effect on SCC47, while DNMT inhibition reduced HPV expression in SCC47 but not in 93VU147T (Fig. 5j, Extended Data Fig. 8d–e). These effects were largely specific to HPVon clones (Extended Data Fig. 8f). Thus, epigenetic regulators may direct HPV expression and heterogeneity.
Next, we turned to examine the impact of heterogeneity in HPV expression on cancer phenotypes. Aberrant cell cycle activity of HPVon cells might render them susceptible to standard cancer treatments, while the less proliferative HPVoff cells may have reduced susceptibility to such treatments. Indeed, treatment of SCC47 cells with cisplatin and of 93UV147T cells with radiation, reduced the expression of HPV genes (Fig. 5k, left) without affecting HPV genomic copy numbers (Fig. 5k, right), consistent with the possibility that HPVon cells were preferentially eliminated. As expected, treatment of individual HPVon and HPVoff clones revealed that HPVon clones are more susceptible to these cytotoxic agents (Extended Data Fig. 8g). The aberrant cell cycle of HPVon cells might also diminish their migration and invasive capacity, due to potential migration-proliferation tradeoffs23,24. Indeed, we found increased invasiveness of HPVoff clones compared to HPVon clones in both cell lines (Extended Data Fig. 8h–i).
The association of HPVoff cells with increased invasion and resistance to treatments suggests that the fraction of HPVoff cells in a tumor might have clinical implications. Accordingly, HPV-positive tumors with low normalized HPV expression tend to have reduced recurrence-free survival compared to those with higher HPV expression (Extended Data Fig. 8j). This analysis was hindered by small sample size, and the effect on survival had borderline statistical significance (p=0.05), highlighting the need for further analysis with a larger patient cohort, while raising the intriguing possibility that loss or reduction of HPV expression in subsets of cells may have a negative effect on patient survival.
Discussion
Our comprehensive scRNA-seq analysis reveals unappreciated diversity, both in genomic CNA profiles (Fig. 2) and in HPV gene expression (Fig. 4), within individual OPSCC tumors. The observed genomic diversity may reflect an HPV-driven genomic instability, consistent with previous studies25–27. This instability allowed us to robustly detect invasive malignant cells in pathologically-normal tissue, which needs to be examined further in larger cohorts but may ultimately guide improved analyses of tumor margins.
The diversity of HPV expression is observed at three levels. First, different HPV genes are expressed at distinct levels in each tumor and cell line examined. Overall, E5 is the most highly expressed HPV gene in tumors, but not in cell lines, highlighting the need to better understand its regulation and function. Second, these HPV expression patterns vary among tumors, among cell lines, and even among genetic subclones of the same tumor. Thus, HPV integration and/or expression patterns may be modulated during tumor initiation and clonal evolution. Third, we do not detect HPV expression in a subset of cells, and the number of such cells is significantly higher than would be expected by the technical limitations of scRNA-seq. While we cannot distinguish between partial and complete repression of HPV genes, cells with undetected HPV mRNA (HPVoff) are associated with a decrease in the phenotypes that are driven by HPV oncogenes, namely aberrant cell cycle (through E7) and avoidance of senescence (through E6). These results suggest that reduced HPV levels persist for a sufficient degree and time to invoke phenotypes that partially resemble HPV-negative cells. HPVoff cells present a paradigm of heterogeneity in HPV expression within each HPV-positive tumor that is associated with unique cell states and clinical implications.
Given the strong evolutionary pressures to suppress viruses, and the multitude of viral-protective mechanisms, it is tempting to speculate that even in a successful viral infection and a resulting tumor, the virus may still be suppressed in a subset of cells, thereby leading to the observed HPVoff cells. In cell lines, such suppression appears to be driven by epigenetic mechanisms: reduced HPV expression in HPVoff clones is not mirrored by reduced copy numbers at the DNA level, and can be achieved by inhibition of epigenetic regulators. However, we also found significant variability in the fraction of HPVoff cells between tumor subclones, suggesting that genetic evolution further modulates the transition towards HPVoff cells. We therefore speculate that multiple mechanisms, both genetic and epigenetic, regulate HPV expression levels and the emergence of HPVoff cells (see model in Fig. 5l).
The potential clinical significance of HPVoff cells is hinted by their decreased response to treatments, increased invasion in vitro, and by the trend to worse disease-free survival in HPV-positive patients with a larger proportion of HPVoff cells. We speculate that aberrant HPV-driven cell cycle activity facilitates responses to chemotherapy and radiation, partially accounting for the improved prognosis of HPV-positive tumors. HPVoff cells could resume their growth after treatment, possibly even switching their HPV expression back on, and may provide the basis for recurrent HPV-positive tumors.
While the cell cycle behavior of HPVoff cells is reminiscent of HPV-negative cells, the levels of CDKN2A (encoding p16) are indistinguishable between HPVoff and HPVon cells based on scRNA-seq, RNAscope ISH and IHC (Fig. 4b, Extended Data Fig. 6d and 8k). Moreover, knockdown of E6 and E7 decreased the proliferation of cells but not their levels of p16 (Extended Data Fig. 1 and 7f–g). CDKN2A was, however, significantly upregulated in the few non-malignant epithelial cells in which we detected HPV reads (Extended Data Fig. 3g), highlighting the robust and early effect of HPV infection on CDKN2A. These observations raise the possibility that CDKN2A activation is more stable than other HPV-driven effects and may persist for a long time through unknown mechanisms.
If HPV expression varies after infection while CDKN2A (p16) expression remains constitutively high long after infection, then CDKN2A could theoretically become a more sensitive readout for latent or past HPV infection than HPV itself, potentially explaining the common and reliable use of p16 as a clinical marker of HPV infection. Interestingly, CDKN2A is highly expressed in several OPSCC tumors and cell lines in which we do not detect any HPV reads (Extended Data Fig. 4c). It is conceivable that such tumors had initially been driven by HPV, but that during tumor progression one/few of the clones lost HPV or its expression (as appears to be the case in OP13, see Fig. 2c), and these clones could have taken over the tumor via clonal evolution. Such a scenario could explain our observation that OP8 is p16-positive, yet HPV-negative, and has a mixture of transcriptional TCGA subtypes associated with HPV-negative and HPV-positive tumors (Fig. 3a–b). Similarly, the subset of atypical cells in OP19 (a tumor with undetected HPV transcripts but high CDKN2A expression) may be a remnant of latent or past HPV infection, although CDKN2A expression could also reflect non-viral mechanisms of regulation.
In summary, our single cell atlas of OPSCC shows that genes encoded by an oncovirus (in this case, HPV) may cease to be expressed in a subset of cells, thereby reducing the oncogenic properties of cells, but also relieving their associated vulnerabilities, which may allow malignant cells to survive anti-tumor treatments and then potentially re-express the virally encoded oncogene and resume growth. This model is conceptually similar to that of reversible drug-tolerant persister cells28, except that the source of drug tolerance is directly connected to repression of the oncogene. This model may be particularly relevant for virally-induced cancers, in which anti-viral mechanisms may drive such oncogenic diversity. Future studies will determine if this model is relevant in additional contexts and might reveal new opportunities to eradicate the elusive persister cells in OPSCC and other cancers.
Methods
Human Tumor Specimens
Patients with OPSCC at the Washington University School of Medicine gave informed consent preoperatively to take part in the study following Institutional Review Board approval (#201911095 and 201102323), complying with all relevant ethical regulations. A total of 16 patients were included in the study, and received no compensation for providing tissue samples. Twelve patients were initially clinically classified as HPV-positive based on p16-staining performed in a CLIA-certified clinical laboratory and interpreted by a dedicated head and neck pathologist, while four were classified as HPV-negative. After further analysis of HPV-related reads, one of the HPV-positive samples (OP8) was reclassified as HPV-negative due to the absence of any detectable HPV reads (see ‘Detection of rare HPV genotypes’). Age, gender, and other demographic characteristics of human subjects providing samples are summarized in Supplementary Table 1, while pathologic features are summarized in Supplementary Table 2.
Sample Processing and Sequencing
Fresh biopsies of OPSCC were collected from the primary tumor at the time of surgical resection, and in some cases, additional tissue was obtained from metastatic lymph node (LN) tissue. For histologically negative margins, the surgeon thoroughly irrigated the primary tumor defect site and then obtained a separate biopsy just beyond the intra-operative, frozen section margin sent to pathology. In all cases, the intra-operative, frozen margin analysis returned clear and final permanent margin status was also confirmed to be negative. A small fragment was snap frozen for bulk whole exome sequencing, and the remainder of the provided tissue was processed for scRNA-seq. Fresh samples were minced, washed with phosphate buffered saline (PBS) (Thermo Fisher Scientific, Waltham, MA), and dissociated using a Human Tumor Dissociation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) per manufacturer guidelines. Red blood cell lysis was performed with ACK lysis buffer per manufacturer protocol (Thermo Fisher Scientific), followed by dead cell removal using a dead cell removal kit to improve the viability if needed (Miltenyi Biotec, Bergisch Gladbach, Germany). Viability was confirmed to be >80% in all samples based on trypan blue analysis (ThermoFisher Scientific). Cell suspensions were filtered using a 40 μm filter (ThermoFisher Scientific) and dissociated cells were pelleted and re-suspended in AutoMACS Rinsing Solution with 0.5% bovine serum albumin (BSA; Miltenyi Biotech). The single-cell suspension was sorted using human CD45 magnetic MicroBeads (Miltenyi Biotec) to enrich for CD45+ cells. Briefly, 20μl of CD45 MicroBeads per 107 total cells was added to the cell suspension and incubated for 15 minutes at 2–8°C. After incubation, CD45+ and CD45− cells were collected after the cells passed through the magnetic column. The CD45+ and CD45− cell pellets were then obtained after centrifuging at 450g for 5 minutes at 4°C. Samples were processed using the Chromium Single Cell 3′ (v2 Chemistry), and in two cases 5’, platform with the target of ~10,000 cells (10x Genomics, Pleasanton, CA) following manufacturer’s instructions. Briefly, cells were added onto a chip to form Gel Bead-in-Emulsion (GEMs) in the Chromium instrument followed by cell lysis, barcoding, fragmentation, adaptor ligation and addition of sample index to the libraries before sequencing. scRNA-seq libraries were sequenced on Illumina NovaSeq 6000 machines with a minimal target read count of 0.5 billion per sample. In 4 patients (OP4, OP6, OP9 and OP14), including the two (OP9 and OP14) who underwent 5’ sequencing, CD45+ and CD45− cell fractions were sequenced separately. In the remaining cases, cells from the two fractions were mixed at a CD45+:CD45− ratio of 1:2 (twice as many CD45− cells) before single cell barcoding. After sequencing, the resulting FASTQ files were aligned to a custom genome, combining the human genome (grch38) with genomes of the main high-risk HPV genotypes – HPV16, 18, 31, 33 and 35 – using Cell Ranger v4.0. Assemblies for the high-risk HPV types were downloaded from NCBI with the following GenBank accession numbers: HPV16 (GCA_000863945.2), HPV18 (GCA_000865665.1), HPV31 (GCA_003179095.1), HPV33 (GCA_003179955.1), HPV35 (GCA_003180695.1). All viral FASTA files were concatenated onto the grch38 FASTA. All viral gtf files were adapted for Cell Ranger usage with the mkgtf function in Cell Ranger and concatenated onto the grch38 gtf. Thereafter, cellranger mkref was used on the new FASTA and gtf to create the custom reference. All reads that aligned to HPV genes were aligned exclusively to HPV16.
Detection of rare HPV genotypes
One sample classified as HPV-positive by p16 staining turned out not to have any reads for the genotypes used in our alignment (16, 18, 31, 33, 35). For this particular patient, single-cell alignment was also done against HPV45, without any aligned reads. In order to make sure that this sample actually was HPV-negative, we also tested this patient for 13 HPV genotypes (16, 18, 31, 33, 35, 39, 45, 52, 56, 58, 59, 66, and 68), all of which were negative (Supplementary Table 9), using a previously validated RT-PCR method29.
Furthermore, to exclude the possibility that a rare HPV genotype might still be present, we used the HPV-EM tool30, which detects all known HPV genotypes in human sequencing data, to reanalyze a number of known HPV-positive samples as positive controls in addition to OP12 (known HPV-negative, negative control) and OP8 (patient in question). The FASTQ files from each patient were treated as a pseudobulk sample and aligned to the human genome, whereafter unmapped reads were mapped, allowing for mismatches, to all known HPV genotypes. In the HPV-positive patients, HPVon and HPVoff cells were analyzed separately. While we did find HPV16 reads in HPVon cells from all analyzed HPV-positive patients in this cohort, as expected, we did not have any reads mapped to any HPV genotype in OP12 nor in OP8 nor in the HPVoff cells. No HPV genotypes other than HPV16 were detected in any sample (Supplementary Fig. 2).
Cell Lines
HNSCC HPV positive cell lines SCC47, 93VU147T, and SCC90 were generously provided by Dr. James Rocco and colleagues. They were cultured in 3:1 Ham’s F12 (ThermoFisher Scientific): DMEM (ThermoFisher Scientific) supplemented with 10% fetal bovine serum (FBS) (Peak Serum, Fort Collins, CO) and 1X penicillin-streptomycin-glutamine (PSG) (ThermoFisher Scientific). Cells were maintained at or below a confluency of 90% to optimize growth conditions.
Filtering and Preprocessing
For each sample, cells with fewer than 1000 detected genes were removed as low-quality. Doublets were identified by combining the results of three alternative methods that were published recently and implemented in R packages – scdDblFinder31, the hybrid score from scds32 and the doubletCells algorithm from scran33. For each method, we set the expected doublet rate at 0.6%, per 500 cells per sample. Cells classified as doublets by at least two methods, totaling 2,744 of all 73,714 cells (3.7%), were removed as probable doublets.
The UMI matrix was transformed to CPM (counts per million) by normalizing every gene by the total number of UMIs per sample. The CPM-matrix was then log2-transformed, as log2(CPM/10+1). Then, the data was mean-centered by subtracting the average expression of each gene from all values of that gene. We further filtered out the data, keeping only genes with either an average expression of >4 log2(CPM) across all cells or genes with >5 UMI counts in >20 cells. After filtering and doublet removal, the resulting matrix had 10,034 genes and 70,970 cells.
Scoring Cells for Gene Expression Signatures
Cells were scored for expression of various gene expression signatures, following our previously used approach13.For each cell, a relative expression score was defined by subtracting the average expression of the gene signature in a cell by that of a control gene set. The control gene set was defined by dividing all analyzed genes into 30 bins by average expression level, and for each gene in the gene signature randomly sampling 100 genes from the same bin.
Cell Type Assignment
The gene-cell matrix underwent dimension reduction using UMAP and Louvain clustering (k=200), with each cluster assigned as either epithelial, stromal or immune based on the clusters’ top 50 differentially expressed genes. All cells were also individually assigned to cell types by scoring for the expression of cell type signature genes (Supplementary Table 3). Cells fulfilling either of the three following conditions: 1) HPV-positive cell in non-epithelial cluster, 2) highest cell signature score discordant with cluster assignment or 3) highest cell signature score less than 1.15*second highest signature score AND second highest signature scoring cell type discordant with cluster assignment, were set as unresolved and removed from further analysis; 3,342 cells were filtered out using this approach. Assignment to any non-immune subtype in an immune cluster was defined as discordant, and likewise for stromal and epithelial clusters. Cells classified as fibroblasts in epithelial clusters were kept, so as not to miss malignant cells undergoing EMT.
Cells assigned to any of the immune cell types were reclassified separately. A matrix was formed from just the immune cells and batch correction applied to samples from four patients – OP4, OP6, OP9 and OP14, since these samples had their CD45+ fractions sequenced separately, and showed batch effects. Batch correction was performed through assigning every cell to an immune cell type by individual scoring as described above, followed by centering the expression values of cells from the four above-mentioned patients to the expression values of all other cells from the same cell type. Final assignments were achieved through dimension reduction and clustering of the corrected matrix.
Differential Gene Expression Analysis
Whenever differential gene expression analysis was performed, a new UMI matrix was created containing only the relevant cells. It was then log2(CPM/10+1)-transformed, filtered to only keep highly expressed genes, and mean-centered as described above. P-values were corrected using the Benjamini-Hochberg method.
Non-negative Matrix Factorization
Diversity within cell types was studied through non-negative matrix factorization (NNMF). For every cell type, patients with at least 30 cells belonging to that cell type were selected. For each patient and the cells of that cell type, a new matrix was created by gene filtering and centering as described above. All negative values were set to zero, and NNMF was performed using the snmf/r factorization algorithm from the NMF R package34. For each sample and cell type, the algorithm was run 100 times and the factorization yielding the lowest approximation error was kept.
Every matrix was split into ten factors, each represented by 100 factor-genes. The cells were then assigned to the factor with highest average factor-genes expression. Factors for which fewer than 10 cells were assigned were removed. The factor-gene lists from all patients were then compared, and Jaccard similarities were calculated between every pair of gene lists. Factors that did not have Jaccard similarities >= 0.2 with any other factor were removed, since these did not represent recurrent programs. For the malignant cells, a total of 69 factors were kept for downstream analysis. The remaining factors were then hierarchically clustered, using Euclidean (1-Jaccard similarity) distance as distance metric, with average linkage.
Clusters of factors (metaclusters) were then used to define subtypes. For each of the metaclusters representing at least two patients, all genes present in >50% of patients included in that metacluster were defined as a subtype signature. Cells from every cell type were then assigned to subtypes by creating matrices consisting just of cells from that cell type, and scoring every cell for the subtype signatures as described in the section “Scoring Cells for Gene Expression Signatures”. Annotations for fibroblast and endothelial cell subtypes were aided by subtype data35,36.
HPV-positive Tumor Signature
An HPV-positive malignant signature score was defined from the three patients where adjacent as well as tumor tissue was provided. Firstly, all HPV-positive epithelial cells in adjacent tissue samples were excluded. Thereafter, differential expression analysis between epithelial cells from tumor samples and epithelial cells from adjacent tissue samples was performed for each patient separately. Genes that ranked among the top 50 overexpressed genes in either the tumor sample (“up in cancer” genes) or the adjacent sample (“down in cancer” genes), consistently in all three comparisons, were kept. The signature score was then defined as the average normalized expression of the former genes minus the average normalized expression of the latter genes.
CNA Inference
To define malignant cells, we firstly inferred copy number aberrations from single cell data using the method earlier published in15. For each patient, a matrix was created from all epithelial and stromal (endothelial/fibroblasts) cells from that patient. The matrix was filtered, normalized and centered as described above. The genes were then reordered according to their chromosomal position, and extreme values of normalized expression were limited by setting the extremes at −3 and 3. For each chromosome separately, a moving average was calculated at every chromosomal position by using a 100-gene window. As a baseline reference for normalization, an average CNA value was calculated for each stromal cell type, defining multiple potential reference profiles that represent cells with normal karyotype. Then, for each positive CNA value we subtracted the maximum value of these potential references and for each negative CNA value we subtracted the minimum value of these potential references. Finally, all values between −0.15 and 0.15, which we consider as likely reflecting noise rather than a genuine CNA signal, were set to zero. This resulted in a final matrix of CNA signal by cell.
Subclone Assignments
The epithelial cells in the matrix of CNA values, derived as described above, were clustered to identify genetic subclones. Given the difficulty in selecting optimal clustering parameters, our approach was to initially use parameters that define a relatively large number of cluster (over-clustering) and subsequently merge clusters that have the same set of inferred aberrations. Specifically, the matrix was first filtered, keeping only the top 2/3 of genes by absolute value of their CNA signal. The matrix was then subjected to dimension reduction through UMAP, followed by over-clustering of the UMAP coordinate matrix through Louvain clustering with k set at 15. Clusters containing fewer than 10 cells were merged into the most similar larger cluster by KNN distance, with k=ln(number of cells). For each cluster, an average CNA value for all cells in that cluster was calculated for every chromosome arm. Clusters were assigned as deleted or amplified at a chromosome arm if the average CNA value across the cells in the cluster over the genes in the chromosome arm was < −0.15 or > 0.15, respectively. Clusters were then merged if they satisfied two requirements: (i) equal assignments across all chromosome arms, and (ii) maximum difference between clusters (across all chromosome arm) smaller than 0.15. This merging process was repeated, with new average values calculated, until all remaining clusters differed by at least one chromosome arm.
Malignant Cell Definitions
To separate malignant cells from non-malignant epithelial cells, two metrics – CNA signal and CNA correlation – were calculated for each epithelial cell. CNA signal was defined as the average of absolute CNA values across the top 2/3 of genes by CNA value. CNA correlation was defined as the correlation between the CNA values of every cell and the average CNA profile of the top 25% epithelial cells by CNA signal. For every subclone analyzed, cutoffs were set for both CNA signal and CNA correlation so that <1% of cells passing each threshold were stromal reference cells. Cells passing both thresholds were classified as malignant cells, cells passing neither were classified as non-malignant epithelial cells, and those passing only one threshold were classified as unresolved.
Signature Score Comparisons by Binning
The expression of gene-set signatures between HPV-related subsets of malignant cells were compared through a binning approach. First, all cells received a signature score as described above. Cells were then ranked by their expression score and divided into 10 bins of equal size. For each subset of cells, 100 cells per patient within the subset were randomly sampled in order to control for uneven number of cells across patients. Then, for each bin and subset, the actual number of cells in that bin was compared to the expected number, provided an even distribution across all bins. This process was repeated 100 times, and a mean value of observed/expected was calculated across all runs.
Comparison of G1/S Scores Across Datasets
Nine external scRNA-seq cancer datasets, acquired through 10x sequencing and comprising a total of 60056 cancer cells21,37–42 were used for this analysis. For each dataset, the cells annotated as malignant by the authors were selected, and the UMI matrix log2(CPM/10+1)-transformed as described above. Genes were not filtered nor were expression values normalized by centering. Each dataset was then separately scored for the genes in our G1/S signature as described above. The HPVon, HPVoff and HPVneg cells from our study were considered as three separate datasets for this analysis and processed in the same way.
From each dataset, 1000 cells were randomly sampled and all cells divided into 10 bins of equal size, ranked by G1/S score. We then looked at what proportion of cells per dataset were in the top 3 bins, representing a high G1/S score. This sampling was repeated 100 times, and the mean fraction of cells in the top bins, as well as standard error across the 100 runs, used for comparison of datasets.
TCGA Analyses
An earlier study43 provided data on HPV reads in ppm for each sample in a subset of the TCGA HNSCC cohort. Forty patients with available HPV data, of which 28 were HPV-positive, were oropharyngeal (ICD codes C01, C01.9, C09.9, C10.9), and were used for our analysis. HPV read counts in ppm were log2(ppm)-transformed, and mRNA expression data was log2(TPM)-transformed. To account for differences in sample composition, we scored each sample for expression of epithelial genes (Supplementary Table 3) and created a linear model where we regressed all gene expression scores of interest, including HPV expression, against the epithelial score. Model residuals were used as final scores for downstream analysis. Grouping of patients into HPV-high and HPV-low groups for survival analysis was done using the maxstat algorithm44.
Cell Cycle Assignment
To set cutoffs for defining which epithelial cells express the cell cycle programs of the G1/S or G2/M phases and can thus be defined as cycling, we reasoned that: (i) the vast majority of non-malignant epithelial cells would not be cycling; and (ii) by permuting the expression of each cell cycle gene across the non-malignant epithelial cells we could reduce the signal of the potential few cycling cells, thereby defining reference profiles representing non-cycling epithelial cells. We thus scored all the epithelial cells, as well as the permuted non-malignant cells, for the G1/S and G2/M signatures. We then used the maximal observed scores of the permuted non-malignant cells as cutoffs for the G1/S and G2/M signature scores.
Flow Cytometry and Single-cell Clone Expansion
Upon reaching 80% confluence, SCC47 and 93VU147T cells were trypsinized from the plate and filtered through 40 μm filters (ThermoFisher Scientific). Filtered cells were washed with PBS and suspended in Hank’s Balanced Salt Solution supplemented with 2 mM EDTA. To obtain a single cell from the bulk suspension, we performed cell sorting at the Siteman Flow Cytometry Core (Washington University School of Medicine). Briefly, standard forward scatter height versus area criteria were used to discard doublets and capture singlets. Cells were sorted into a 96-well plate containing 100 μL of complete growth medium with 1X penicillin-streptomycin (ThermoFisher Scientific). Plates containing sorted single cells were spun down at 200g for 5 minutes and incubated at 37°C and 5% CO2. Plates were scanned for single cell colonies via microscope as soon as small aggregates of cells were visible by microscope, with single clone-derived colonies usually appreciable two weeks later. The confirmed single clones were transferred to 12-well plates and incubated for an additional two weeks to expand the clonal populations. To characterize the HPV genetic and transcriptomic heterogeneity in SCC47 and 93VU147T cell lines, we selected 10 single-cell clones from each cell line.
For flow cytometric cell cycle analyses, cells were fixed with 70% ice cold ethanol and stained with propidium iodide (30 μg/ml of PI (Sigma) with 200 μg/ml of RNase (Sigma) in 0.1% of Triton-X-100 (Sigma) in PBS) for one hour at room temperature. Cell cycle analysis was completed with at least 10,000 cells using CytoFLEX Flow Cytometer and data were analyzed using FlowJo v9.0 software. The gating strategy is shown in Supplementary Fig. 3.
Cell Treatment
SCC47 was treated with 0.2 μM of cisplatin (Sigma) or equal volume of DMSO (vehicle) for 72 hours. HPV positive 93VU147T cells were irradiated with a dose of 8 GY and the controls were untreated, then collected 24 hours later. SCC47 and 93VU147T cells were treated with varying dosages of the DNMT inhibitor decitabine (generously provided by Dr. Ting Wang) and the H3K27 histone methyltransferase EZH2 inhibitor tazemetostat (MedKoo), respectively, with DMSO (vehicle) treatment as a control. Cells were treated for 72 hours before they were collected for expression analysis of HPV genes.
CRISPRi Knockdown
Golden-Gate cloning protocol was used to clone the HPV-16 E6 and E7 targeting sgRNA oligos (Supplementary Table 10) into the sgOpti lentiviral vector backbone (Addgene). Lentivirus was generated via co-transfection of CRISPR plasmids into 293T cells with psPAX2 packaging plasmid (containing the GAG/POL genes) and pMD2.G plasmid supplying the VSVG envelope gene (Addgene) using PEI (2ug/ml). dCAS9-KRAB (lenti-dCAS9-dKRAB-blast, Addgene) SCC47 and 93VU147T cells expressing catalytically inactive dead Cas9 fused to transcriptional repressor KRAB were transduced and then selected with puromycin and blastocidin (Life Technologies). Knockdown of genes was confirmed by qPCR.
Nucleic Acid Extraction and Reverse Transcription
The genomic DNA and total RNA of the HPV positive cell lines (SCC47 and 93VU147T) were extracted by DNeasy Blood & Tissue Kit (QIAGEN, Germany) and RNeasy Plus Mini Kit (QIAGEN, Germany), respectively, according to manufacturer’s instructions. We performed first strand synthesis with Maxima H Minus Reverse Transcriptase (Thermo Fisher Scientific) per manufacturer protocols using a dT18VN primer (Supplementary Table 10). Both genomic DNA and cDNA were stored at −20°C.
qPCR Analysis
qPCR was used to quantify relative HPV gene copies and expression. Primer sequences are listed in Supplementary Table 10. For the quantification of relative viral gene copies, we used the single copy gene (albumin) as an internal reference. The amplification efficiency of the viral genes (E6 and E7) was compared to that of albumin in order to estimate the relative viral gene copies45. We also generated a standard curve with a 10-fold dilution series of plasmids containing the albumin gene (spanning 5 logs from 105 to 109 copies) to estimate the albumin gene copies. To quantify the relative HPV expression, we used GAPDH as an internal reference. We performed the qPCR with either 50 ng of the genomic DNA for gene copy analysis or the cDNA for gene expression analysis per manufacturer protocols using the ABI QuantStudio Q3 (Applied Biosystems). We generated melting curves after each PCR and all samples yielded a single peak.
Matrigel Invasion Assay
Matrigel invasion assay was performed following an established protocol46. Briefly, preformed matrigel invasion chambers (Corning) were prepared per manufacturer protocol. Serum-containing media was placed below the invasion chambers and 2.5 × 104 cells suspended in 500 μL serum-free media were placed above the invasion chambers and incubated for 24 hours. Cells on the lower surface of the membrane were fixed with methanol, stained with crystal violet, and counted in a blinded manner. Cells in serum-containing media were used as a negative control.
Immunocytochemistry
Cells were fixed in freshly prepared 4% paraformaldehyde for 20 minutes at room temperature, washed with PBS and subsequently blocked and permeabilized with 0.1% Triton X in 10% goat serum containing PBS in room temperature for 1 hour. Cells were then probed with primary antibodies, Ki-67 1:500 dilution (D2H0 rabbit mAb, Cell Signaling), E6 1:100 dilution (mouse anti-virus, clone C1P5, Invitrogen), E7 1:100 dilution (mouse anti-virus, clone TVG701Y, Invitrogen) diluted in 10% goat-serum PBS and incubated overnight at 4 degree Celsius. Cells were washed with PBS and then probed with secondary antibody, goat anti-rabbit IgG (H+L), Fab2 Alexa Fluor 594, goat anti-mouse IgG (H+L), or Fab2 Alexa Fluor 488 (Cell Signaling) at 1:400 dilution in 2% goat serum containing PBS for 1 hour at room temperature followed by PBS washes and mounted with DAPI (Fluoroshield, Sigma). Imaging was completed using Fluorescence Eclipse Ti2 Inverted microscope (Nikon).
Molecular Fluorescent In Situ Hybridization (MFISH)
Viral RNA ISH (RNAScope) and DNA ISH (DNAScope) were performed using RNAscope 2.5 HD Reagent Kit Red assay combined with Immunohistochemistry (Advanced Cell Diagnostics) according to manufacturer’s instructions. Briefly, slides were baked in dry air oven for one hour at 60°C, deparaffinized (Xylene for five minutes twice followed by 100% ethanol for two minutes twice), hydrogen peroxide was applied for 10 minutes at room temperature, and co-detection target retrieval was done using Steamer (BELLA) for twenty minutes and washed with PBS-T. Tissue slides were then incubated overnight with p16-INK4a antibody (LSBio) in HybEz Slide Rack in the Humidity Control Tray with damp humidifying paper and incubated overnight at 4°C. The next day, post-primary fixation was done by washing slides with PBS-T and submerging slides in 10% NBF for 30 minutes at room temperature. Slides are washed with PBS-T and Protease Plus was added to each slide for 30 minutes at 40°C then washed with distilled water. RNAscope antisense probes were utilized to target RNA of specified viral genes, while DNAScope sense probes were utilized targeting DNA of specified viral genes47. Selected probes were warmed at 40°C and hybridized with specific oligonucleotide probes for 2 hours at 40°C in HybEZ Humidifying System. Details of antibodies, probes, and sequences are in Supplementary Table 11. RNA/DNA was then serial amplified and stained with Fast Red solution. Slides were blocked with co-detection blocker for 15 minutes at 40°C and washed in PBS-T. Secondary Alexa Flour 488 antibody (Abcam) was applied for one hour at room temperature in the dark. Finally, slides were washed with PBS-T and counter stained with DAPI (Sigma) and mounted with ProLong Gold Antifade Reagent (Invitrogen). RNAScope was optimized with a PPIB probe as a positive control, while a DapB probe and no secondary antibody served as negative controls. All slides were imaged on the EVOS M5000 Imaging System (Invitrogen) for scoring.
Quantification was performed with CellProfiler using the ISH pipeline by Erben et al48. Adjustments were made to accommodate cell size as well as green versus red staining. Dot staining was identified based on intensity and distinct pixel ranges for DAPI (nucleus, 20–50 pixels), green (p16, 10–30 pixels), and red (E6 or E7, 3–12 pixels). Cell size was identified using a 5-pixel radius from nucleus and images were overlaid to count dots per cell. A positive stain scoring of p16 was determined as greater than 1, while a negative stain score was determined as less than 1. Red (E6 and E7) RNA ISH signal was detected within individual cells and scored using ACD scoring bins. Bin scoring ranged from < 1 dot/cell designated as bin 0, 1–3 dots/cell designated as bin 1, 4–9 dots/cell designated as bin 2, 10–15 dots/cell designated as bin 3, and > 15 dots/cell designated as bin 4. Stain scoring remained the same for all genes of interest with positive staining determined as >0 dots/cell and negative staining as 0 dots/cell per ACD guidelines. DNA ISH signal was detected and scored in a similar fashion. RNAse treatment was used to confirm that signal was specific to DNA. All RNA and DNA ISH images were reviewed with a dedicated head and neck pathologist, who confirmed the heterogeneous pattern of HPV RNA expression compared to a more homogeneous pattern of HPV DNA detected.
Dual-Stain Immunohistochemistry
FFPE blocks of the patient tumors were sectioned onto slides at 4 μm. Slides were baked at 60 degrees for 30 minutes followed by deparaffinization with xylene and graded ethanol. Diva Decloaker (Biocare Medical) was used for heat mediated antigen retrieval for all stains. Blocking was performed with Dako Dual Endogenous Enzyme Block (5 minutes). HPV TYPE 16/18 E6 Mouse Monoclonal antibody (1:50 dilution, Thermofisher, cat# MA1–46057) was applied first and incubated for 30 minutes. Secondary antibody incubation was performed with the Dako EnVision+ Dual Link System-HRP for 30 minutes, followed by DAB staining for 5 minutes. Blocking with Dako Dual Endogenous Enzyme Block was then repeated for 5 minutes. Staining with P16-INK4A polyclonal antibody (1:75 dilution, Thermofisher, cat# 10883–1-AP) was then performed with a 30 minute incubation time. Dako Powervision Poly-AP was used for secondary antibody staining (30 minutes), followed by incubation with AP Red substrate for 5 minutes. Sections were then mounted with a coverslip with Glycergel (Dako).
Cell Proliferation
CellTitre-Glo (CTG) proliferation assays (Promega) were completed according to manufacturer protocols. Briefly, 2000 cells were seeded per 96 wells in technical replicates of 5. Cells were lysed on day 0 (one hour after seeding of cells), 1, 3, 5, 7, and 9 for HPV single clones and day 0, day 2, day 4, day 6, day 8, day 10 and day 12 for E6 and E7 HNSCC knockdown cell lines by addition of the CTG reagent followed by measurement of luminescence using the Biotek Cytation 5 (BioTek, Winooski, VT). Background luminescence was removed. Luminescence values were adjusting based on 2μM Adenosine triphosphate (ATP) luminescence measured on the same plate for each day.
Statistics
All functional experiments were performed with at least three independent biological replicates, with number of replicates indicated in figure legends. Statistical analyses for functional experiments were performed with GraphPad Prism 4.0. All histograms are presented as mean + s.e.m. Student’s t-test was used for comparisons in experiments with two sample groups. In experiments with more than two sample groups, ANOVA was performed followed by Bonferroni’s post hoc test.
Software Packages
Data analysis was performed in R (version 4.1.0) with the following packages used: caTools version 1.18.2 for calculating moving averages, circlize version 0.4.13 for creating colour palettes, class version 7.3–19 for classifying cells by kNN, clusterProfiler version 4.0.2 for enrichment analysis, ComplexHeatmap version 2.8.0 for plotting heatmaps, dplyr version 1.0.7 for data handling, FNN version 1.1.3 for creating kNN graphs, ggplot2 version 3.3.4 for creating plots, ggrepel version 0.9.1 for separating text labels in plots, gtools version 3.9.2 for random permutation of values, igraph version 1.2.6 for Louvain clustering, Matrix version 1.3–4 and Matrix.utils version 0.9.8 for working with sparse matrices, msigdbr version 7.4.1 for enrichment analysis, NMF version 0.23.0 for performing NMF, parallel version 4.1.0 for parallellising computation, reshape2 version 1.4.4 for data handling, scDBlFinder version 1.7.1, scds version 1.8.0 and scran version 1.22.1 for doublet detection, SingleCellExperiment version 1.14.1 for data handling, stringdist version 0.9.6.3 for calculating similarity between character strings and uwot version 0.1.10 for creating UMAP plots.
Extended Data
Extended Data Fig. 1. Expression of marker genes and HPV genes, related to Figure 1.
(A) UMAP of all cells (n=70,970) colored by expression of selected marker genes.
(B) UMAP of immune cells (n=22,818) colored by expression of selected marker genes.
(C) Histologic sections of two representative HPV+ (p16+) and HPV− (p16−) oropharynx tumors (OP34 and OP12), stained by H&E (top) and p16 (bottom). Staining was repeated three independent times with similar results. Scale bar = 100 μm.
(D) UMAP of all cells colored by detection of at least one read from HPV16 genes.
(E) Dot plot showing variability in expression of HPV genes (rows) across patients (columns). The last column summarizes all HPV-negative tumors. The top row shows the sum of HPV gene expression per patient (HPVtotal). The size of each dot represents the fraction of cells with at least one read for that gene in each patient, while the color represents the fraction of HPV reads in one patient that reflect the corresponding gene. For the latter metric, HPVtotal is set to 1.
Extended Data Fig. 2. CNA patterns and controls, related to Figure 2.
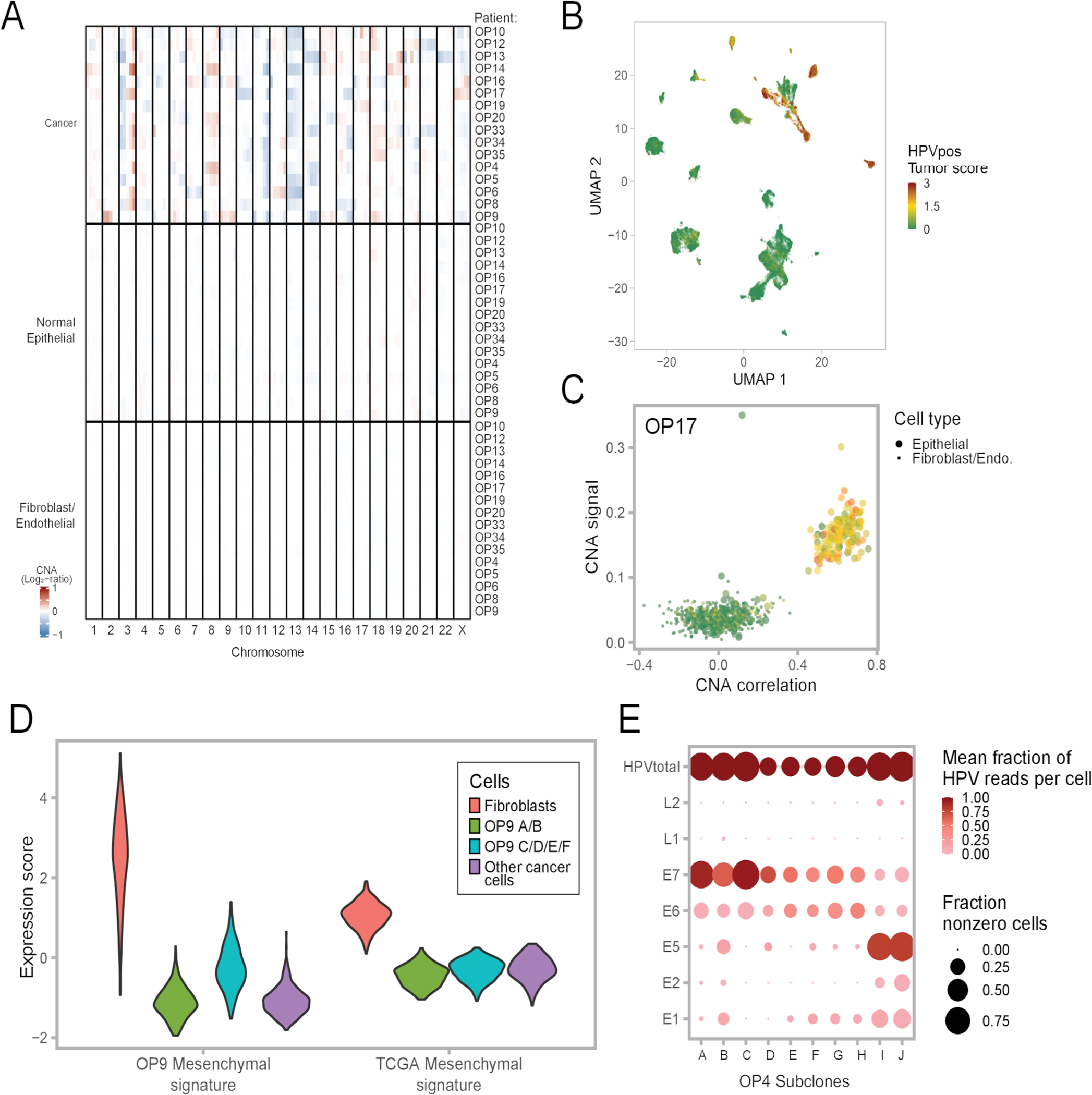
(A) Average CNA profiles of malignant cells, normal epithelial cells and fibroblasts/endothelial cells used as reference for each patient. Each row is a cell subset within a patient. Rows are ordered by cell subset and patient ID. Columns are chromosomal positions. For each row and chromosome, the chromosome was split into five bins.
(B) UMAP of all cells colored by HPV-positive tumor score.
(C) CNA signal and correlation scatter plot of OP17. Cells are colored by their expression of the HPV-positive tumor score.
(D) Violin plots showing expression of the OP9 mesenchymal signature (left panel) and the TCGA HNSCC mesenchymal signature (right panel) in four subsets of cells; 300 cells were randomly sampled from each subset to ensure equal-sized groups.
(E) Dot plot showing variability in HPV gene expression between subclones in one patient, OP4. The size of each dot represents the fraction of cells with at least one read for that gene in each subclone, while the color represents the fraction of HPV reads in one subclone that reflect the corresponding gene. For the latter metric, HPVtotal is set to 1.
Extended Data Fig. 3. CNA-based detection of invasive malignant cells, related to Figure 2.
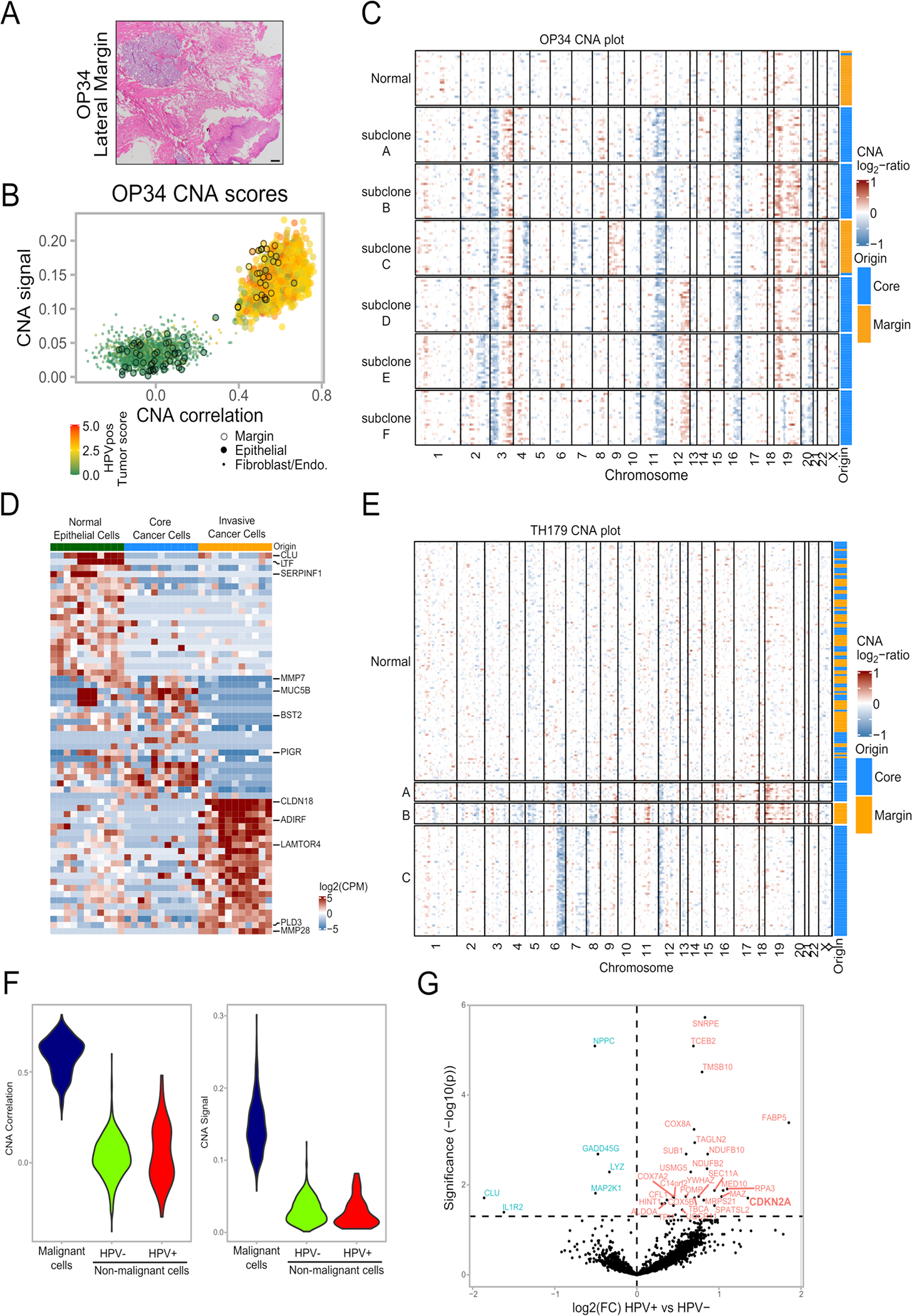
(A) Histologic section of the lateral margin from OP34, stained by H&E. A piece of mucosa was taken beyond this histologically clear (pathologically negative) margin for scRNA-seq (labeled ‘margin’). Staining was repeated three independent times with similar results. Scale bar = 1000 μm.
(B) CNA signal and correlation scatter plot of OP34. Cells are colored by their expression of the HPV-positive tumor score. Epithelial cells from the margin sample are circled.
(C) CNA plot of OP34. Cells were randomly sampled from all subclones in equal numbers to ensure equal-sized groups. Column at the right shows the origin of cells from the tumor core and margin samples.
(D) Heatmap of differentially expressed genes in the three epithelial cell subsets of lung adenocarcinoma sample TH179 – normal epithelial cells, invasive malignant cells and malignant cells from the tumor core. Rows are genes, columns are cells. Cells were randomly sampled from the normal and core subsets to ensure equal-sized groups.
(E) CNA plot of lung adenocarcinoma sample TH179. Column at the right shows the origin of cells from the tumor core and margin samples.
(F) HPV expression in normal epithelial cells. Violin plots showing values for CNA signal and CNA correlation for the 51 HPV-positive and 779 HPV-negative negative nonmalignant epithelial cells from HPV-positive patients, as well as for 830 randomly sampled cancer cells from the same patients, one cancer cell per patient sampled per nonmalignant epithelial cell.
(G) Volcano plot of differentially expressed genes between nonmalignant epithelial cells (defined by lack of CNAs) with or without HPV expression. P-value derived from two-sided t-test adjusted for multiple comparisons.
Extended Data Fig. 4. Diversity of malignant cells across tumors, related to Figure 3.
(A) Heatmap showing relative expression of differentially expressed genes (rows) across all tumor samples (columns). Selected genes include the top 50 preferentially expressed genes from each tumor.
(B) Hierarchical clustering of “pseudobulk” tumor profiles (defined by averaging all malignant cells per sample). Shown are Pearson correlations, ordered by the clustering of samples. Bottom panels show additional tumor characteristics with the same tumor ordering as in the heatmap, including (from top to bottom): the percentage of cells with detected HPV reads, the clinical HPV status (defined by p16 staining), three TCGA subtype scores, and scores for all meta-programs defined in Fig. 3c–d.
(C) UMAP of all malignant cells, colored by mRNA expression of CDKN2A (encoding for p16). OP19 is circled.
Extended Data Fig. 5. Heterogeneity among common cell types in the OPSCC microenvironment, related to Figure 3.
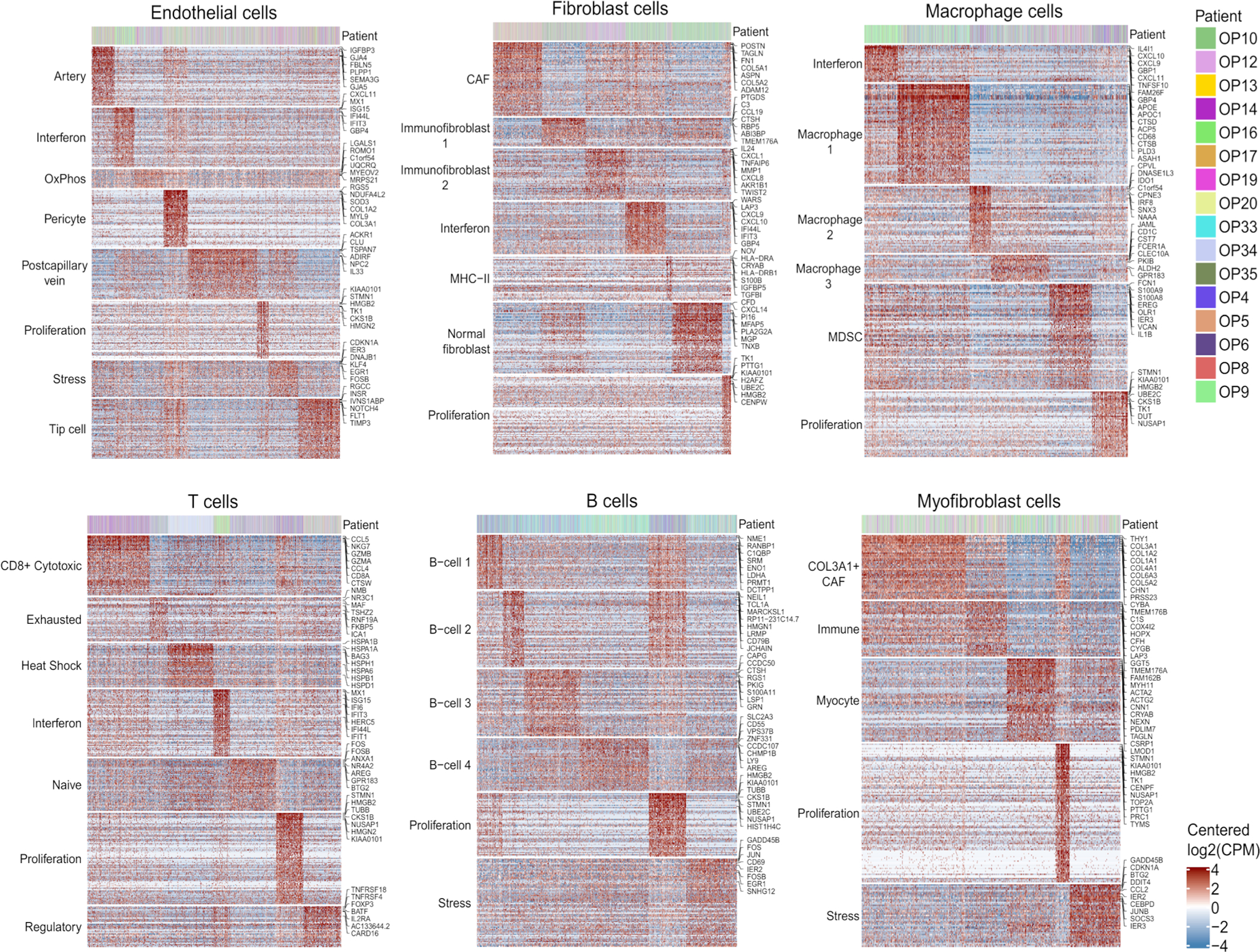
For each of the common cell types in the OPSCC microenvironment (endothelial cells, fibroblasts, macrophages, T cells, B cells, and myofibroblasts), the corresponding panel shows meta-programs, as defined using the same approach as performed for malignant cells and shown in Fig. 3d. Shown are the relative expression levels of meta-program genes (rows) in all cells of the corresponding cell types (columns). Top panels indicate the patient of origin for all cells.
Extended Data Fig. 6. Characteristics of HPVoff cells, related to Figure 4.
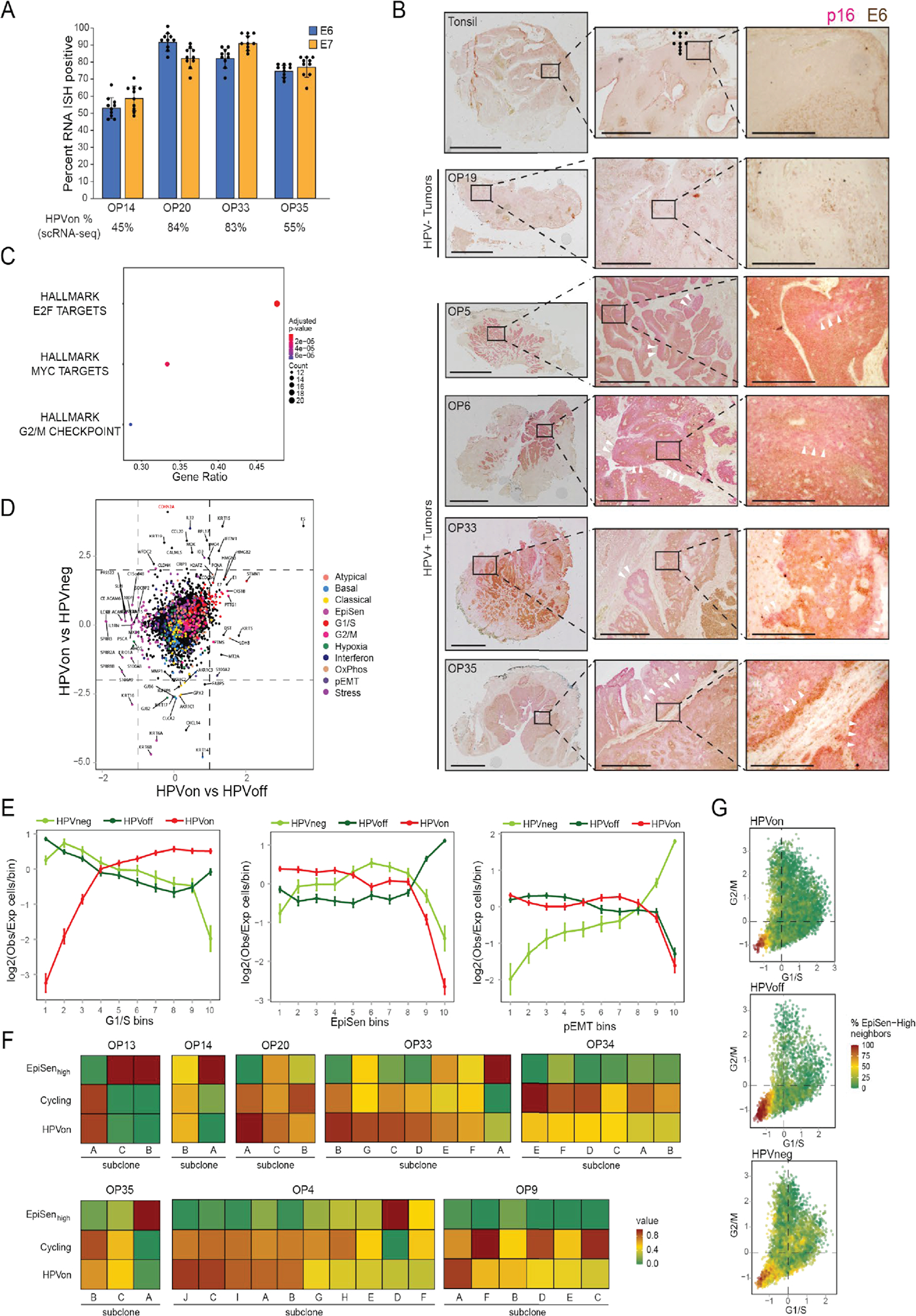
(A) Percentage of cells positive for E6 or E7 in RNA ISH analyses (n=4 tumors, shown are mean and standard errora cross nine regions per tumor). Percentage of HPVon cells by scRNA-seq (bottom) correlates with RNA ISH values (p<0.01, ANOVA).
(B) IHC of representative HPV-positive (OP5, OP6, OP33, and OP35) and HPV-negative (OP19) tumors and normal tonsil stained for malignant-cell specific marker p16 (pink) and viral E6 protein (brown). Similar results were obtained in three independent experiments. White arrowheads denote p16 positivity without E6 expression. Scale bars: Low magnification = 10 mm (tonsil, OP5, OP6), 5 mm (OP19), 7.5mm (OP35); intermediate magnification = 1000 μm; highest magnification = 250 μm.
(C) Enriched MSigDB Hallmark gene-sets among genes significantly overexpressed in HPVon versus HPVoff cells. X-axis: fraction of significantly upregulated genes in the gene set.
(D) Differential expression of all analyzed genes between HPV-related classes of malignant cells. X-axis: difference between HPVon and HPVneg cells; Y-axis: difference between HPVon and HPVoff cells, averaged across all HPV-positive patients. Genes are colored by their assignment to meta-program(right legend). CDKN2A (p16, highlighted in red) was not significantly different between HPVon versus HPVoff cells, but was the most overexpressed gene in HPVon cells compared to HPVneg cells.
(E) For three meta-programs (panels), cells were divided into 10 bins of equal size, ranked by average expression from low (left) to high (right). Y-axis: mean ratio of cells belonging to an HPV subset versus the expected number assuming random distribution across bins. Error bars reflect SEM based on 100 re-sampling runs (n=5 patients for HPVneg, n=11 patients for HPVon and HPVoff). P-values are based on chi-square test.
(F) Fractions of cycling cells, EpiSen-high cells and HPVon cells across genetic subclones. Subclones with a high fraction of HPVon cells tend to also have higher proliferation (p<0.05 for correlations in OP13, OP33 and OP35).
(G) G1/S (X-axis) and G2/M (Y-axis) scores of all malignant cells, colored by the percentage of cycling cells among their neighbors (20 closest cells in this plot).
Extended Data Fig. 7. Regulation and function of HPVoff cells, related to Figure 5.
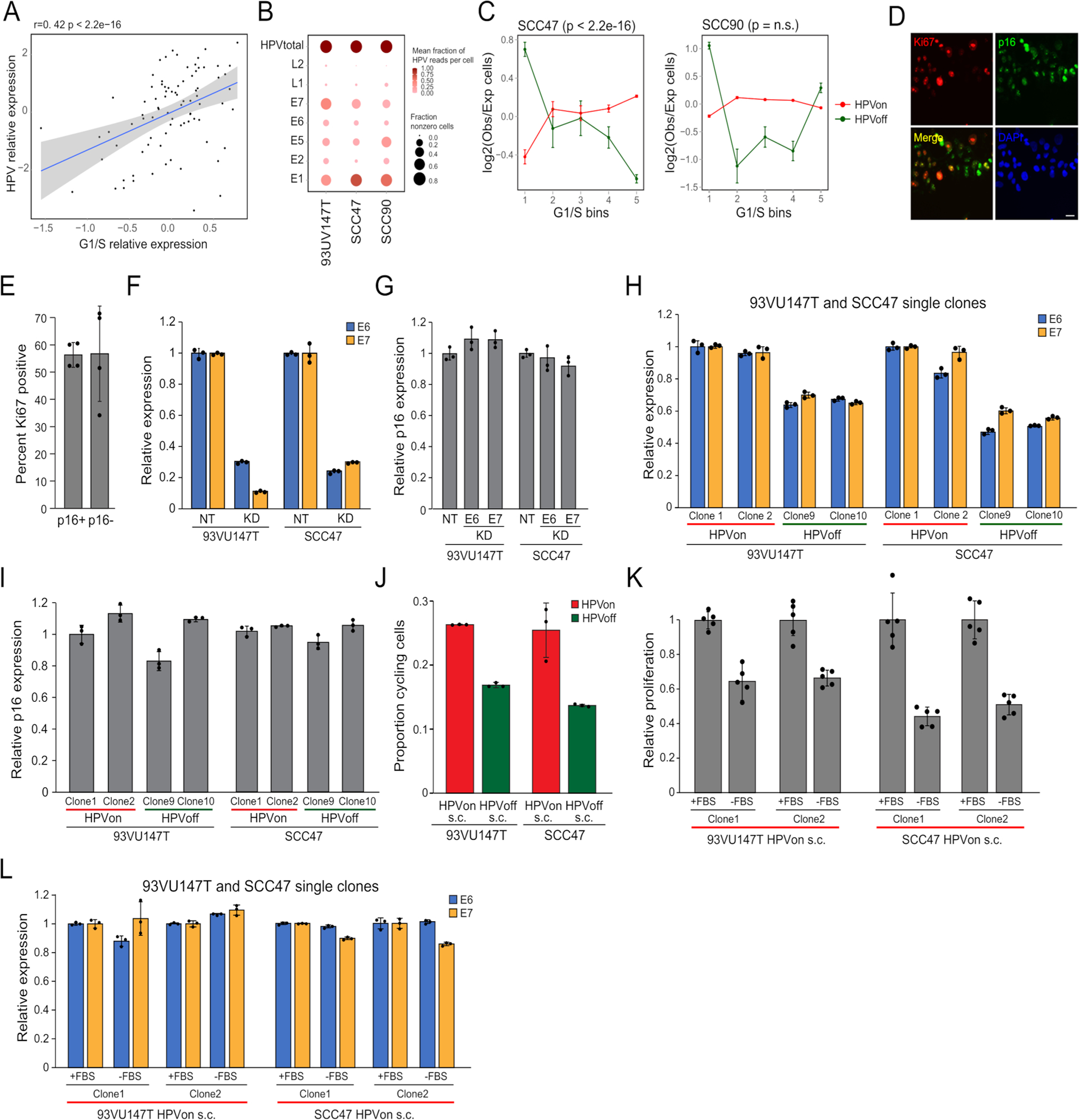
(A) HPV expression and G1/S gene expression across cervical squamous cell carcinoma TCGA samples. Shown are residuals after regression (Supplementary Table 3).
(B) Variability in HPV expression between cell lines. Dot size and color represent fraction of cells with at least one read and fraction of HPV reads that reflect the corresponding gene, respectively.
(C) Cells were divided into 5 bins by average G1/S expression from low (left) to high (right). Y-axis: mean ratio of cells in an HPV subset versus expected number assuming random distribution. Error bars are SEM by 100 resampling runs. P-value based on chi-square test.
(D) Immunocytochemistry of 93VU147T cells probed with Ki67 (red), p16 (green), and DAPI (blue). Scale bar = 100 μm.
(E) Percentage of Ki67 positive cells among p16 positive and negative cells. 50 cells were counted across four fields (n=4).
(F) Relative expression of E6 and E7 in non-target, control (NT) compared to E6 or E7 CRISPRi knockdown (KD) 93VU147T (left) or SCC47 (right) lines (n=3; p<0.0001, t-test).
(G) Relative expression of p16 in same lines as in (F). Data are presented as mean +/− SEM. There was no change in p16 upon E6 or E7 knockdown (n=3).
(H) Relative expression of E6 and E7 among HPVon and HPVoff single clones derived from 93VU147T (left) and SCC47 (right) after three weeks of culture and numerous passages. HPVon and HPVoff clones maintained relatively high and low expression states (n=3; p<0.005, t-test).
(I) Relative expression of p16 in same clones as in (H).
(J) Proportion of cycling cells in HPVon and HPVoff single clones in 93VU147T (left) and SCC47 (right) by flow cytometry (n=3; p<0.05, t-test).
(K) Relative proliferation of HPVon single clones from 93VU147T (left) and SCC47 (right) cultured under normal growth conditions (+FBS) or serum starvation (−FBS) for 48 hours. Proliferation was reduced with serum starvation (n=5; p<0.001, t-test).Relative expression of E6 and E7 in HPVon single clones in 93VU147T (left) and SCC47 (right) under normal growth conditions (+FBS) or serum starvation (−FBS) for 48 hours (n=3).
Extended Data Fig. 8. Functional impact of HPVoff cells and p16 expression, related to Figure 5.
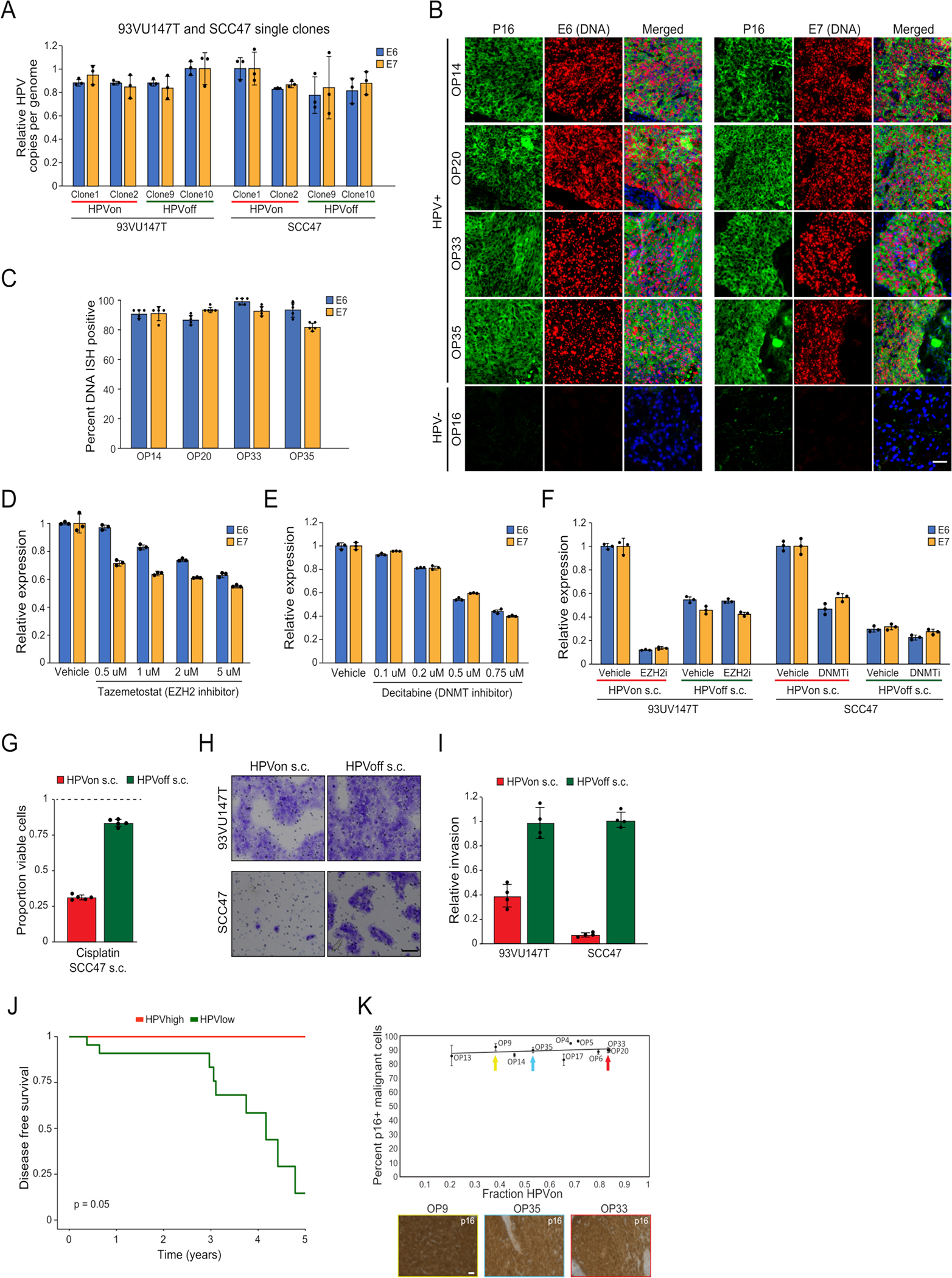
(A) HPV copies per genome of E6 and E7 (normalized to albumin) for HPVon and HPVoff single clones from 93VU147T (left) and SCC47 (right).
(B) DNA ISH (DNAScope) of representative HPV-positive (OP14, OP20, OP33, and OP35) and HPV-negative (OP16) tumors for viral E6 (left) and E7 (right) DNA (red) with immunofluorescence co-staining for regions of tumor as marked by p16 protein (green) and nuclei by DAPI (blue). HPV-positive tumors display p16 positive malignant cells with homogenous E6 and E7 DNA signal. HPV-negative tumors do not have signal for p16 protein or E6 or E7 DNA. Scale bar = 1000 μm.
(C) Percentage of cells positive for E6 or E7 DNA among p16 positive malignant cells in DNA ISH analyses (n=4 tumors, five areas per tumor). Nearly all p16 positive malignant cells demonstrated E6 or E7 DNA signal.
(D) Relative expression of E6 and E7 in 93VU147T cells treated with vehicle or tazemetostat (n=3). All doses did not significantly affect cell viability.
(E) Relative expression of E6 and E7 in SCC47 cells treated with vehicle or escalating concentrations of decitabine (n=3). All doses did not significantly affect cell viability.
(F) Relative expression of E6 and E7 in HPVon and HPVoff single clones from 93VU147T (left) and SCC47 (right) treated with tazemetostat, decitabine, or vehicle. HPVon clones show reduction in E6 and E7 expression upon tazemetostat or decitabine treatment compared to HPVoff clones (n=3; p<0.00001, t-test).
(G) Proportion of viable cells after treatment of SCC47 HPVon and HPVoff single cell clones with cisplatin, relative to cells treated with vehicle (dashed line). HPVon clones were more susceptible to cisplatin compared to HPVoff clones (n=5; p<0.00001, t-test).
(H) Invasion of HPVon and HPVoff single clones from 93VU147T (top) and SCC47 (bottom). Scale bar = 100 μm.
(I) Relative invasion of HPVon and HPVoff single clones from 93VU147T (left) and SCC47 (right) cells. HPVoff cells were more invasive than HPVon (n=4; p<0.05, t-test).
(J) Improved disease-free survival in HPVhigh compared to HPVlow samples, among TCGA p16+ oropharyngeal samples (n=28; p = 0.05).
(K) Top: percentage of p16 positive malignant cells (by IHC) and proportion of HPVon cells (by scRNA-seq). Bottom: p16 staining from tumors with low (OP9), intermediate (OP35) and high (OP20) proportions of HPVon cells (bottom). No correlation between HPVon proportion and percentage of p16 positive cells (n=10 tumors). Scale bar = 100 μm.
Supplementary Material
Acknowledgements
This work was supported by the V Foundation (S.V.P.), Cancer Research Foundation (S.V.P.), Barnes Jewish Hospital Foundation (S.V.P.), NCI 1K08CA237732 (S.V.P), Doris Duke Fund to Retain Clinician Scientists (S.V.P.), Doris Duke Clinician Scientist Development Award (S.V.P.), NIDCD T32DC000022 (T.F.B.), the Swedish Society of Medicine (M.M.), Washington University Department of Medicine Faculty Diversity Award (J.S.F.), Israel Science Foundation (I.T.), Rising Tide Foundation (S.V.P.; I.T.), Mexican Friends New Generation Grant (I.T.), Mauricio Schwarz (I.T.), Zuckerman STEM Leadership Program (I.T.), and The Dr. Celia Zwillenberg-Fridman and Dr. Lutz Zwillenberg Career Development Chair (I.T.). The funding sources had no involvement in the design, conduct, and reporting of the research.
Footnotes
Competing Interests
I.T is a member of the Scientific Advisory Board (SAB) of Immunitas Therapeutics. All other authors report no competing interests.
Code availability
R code and functions for the analysis are available at https://github.com/micmin3/HPV_OPSCC_Analysis.
Data availability
All single cell RNA-seq data produced by this study is available through the Gene Expression Omnibus with GEO accession GSE182227. TCGA bulk RNAseq and clinical data for head and neck and cervical cancer is available through the Broad Genome Data Analysis Center Firehose (https://gdac.broadinstitute.org/). Single-cell datasets reanalyzed to compare proliferation rates are available through the Gene Expression Omnibus with accession numbers GSE150430 (nasopharyngeal carcinoma), GSE131907 (lung carcinoma), GSE132465, GSE132257, GSE144735 (CRC), GSE125449 (HCC), through the Chinese National Centre for Bioinformation Genome Sequence Archive (CNCB-GSA) with accession: CRA001160 (PDAC) and through EMBL-EBI ArrayExpress with accession numbers E-MTAB-8107 (breast, ovarian, colorectal cancer), E-MTAB-6149 (lung) and E-MTAB-6653 (lung). Cell line data used for validation analysis is available through GEO with accession number GSE157220. The NSCLC dataset used to validate finding malignant cells in normal samples is deposited as an NCBI BioProject with accession number PRJNA591860. Source data are provided with this paper.
References
- 1.Gillison ML et al. Distinct risk factor profiles for human papillomavirus type 16-positive and human papillomavirus type 16-negative head and neck cancers. J. Natl. Cancer Inst. 100, 407–420 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Ang KK et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 363, 24–35 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brianti P, De Flammineis E & Mercuri SR Review of HPV-related diseases and cancers. New Microbiol. 40, 80–85 (2017). [PubMed] [Google Scholar]
- 4.Doorbar J, Egawa N, Griffin H, Kranjec C & Murakami I Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 25 Suppl 1, 2–23 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graham SV The human papillomavirus replication cycle, and its links to cancer progression: a comprehensive review. Clin. Sci. Lond. Engl. 1979 131, 2201–2221 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Litwin TR, Clarke MA, Dean M & Wentzensen N Somatic Host Cell Alterations in HPV Carcinogenesis. Viruses 9, E206 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parikh A et al. Malignant cell-specific CXCL14 promotes tumor lymphocyte infiltration in oral cavity squamous cell carcinoma. J. Immunother. Cancer 8, e001048 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Puram SV et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 171, 1611–1624.e24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qi Z, Barrett T, Parikh AS, Tirosh I & Puram SV Single-cell sequencing and its applications in head and neck cancer. Oral Oncol. 99, 104441 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qi Z et al. Single-Cell Deconvolution of Head and Neck Squamous Cell Carcinoma. Cancers 13, 1230 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castellsagué X et al. HPV Involvement in Head and Neck Cancers: Comprehensive Assessment of Biomarkers in 3680 Patients. J. Natl. Cancer Inst. 108, djv403 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Ramqvist T et al. Studies on human papillomavirus (HPV) 16 E2, E5 and E7 mRNA in HPV-positive tonsillar and base of tongue cancer in relation to clinical outcome and immunological parameters. Oral Oncol. 51, 1126–1131 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Neftel C et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 178, 835–849.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tirosh I et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tirosh I et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Slootweg PJ, Hordijk GJ, Schade Y, van Es RJJ & Koole R Treatment failure and margin status in head and neck cancer. A critical view on the potential value of molecular pathology. Oral Oncol. 38, 500–503 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Maynard A et al. Therapy-Induced Evolution of Human Lung Cancer Revealed by Single-Cell RNA Sequencing. Cell (2020) doi: 10.1016/j.cell.2020.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517, 576–582 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kinker GS et al. Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat. Genet. 52, 1208–1218 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parikh AS et al. Immunohistochemical quantification of partial-EMT in oral cavity squamous cell carcinoma primary tumors is associated with nodal metastasis. Oral Oncol. 99, 104458 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ji AL et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 182, 497–514.e22 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yao J et al. Single-cell transcriptomic analysis in a mouse model deciphers cell transition states in the multistep development of esophageal cancer. Nat. Commun. 11, 3715 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerlee P & Nelander S The impact of phenotypic switching on glioblastoma growth and invasion. PLoS Comput. Biol. 8, e1002556 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giese A et al. Dichotomy of astrocytoma migration and proliferation. Int. J. Cancer 67, 275–282 (1996). [DOI] [PubMed] [Google Scholar]
- 25.Akagi K et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 24, 185–199 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duensing S & Münger K The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 62, 7075–7082 (2002). [PubMed] [Google Scholar]
- 27.Korzeniewski N, Spardy N, Duensing A & Duensing S Genomic instability and cancer: lessons learned from human papillomaviruses. Cancer Lett. 305, 113–122 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen S, Vagner S & Robert C Persistent Cancer Cells: The Deadly Survivors. Cell 183, 860–874 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Gao G et al. A novel RT-PCR method for quantification of human papillomavirus transcripts in archived tissues and its application in oropharyngeal cancer prognosis. Int. J. Cancer 132, 882–890 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Inkman MJ et al. HPV-EM: an accurate HPV detection and genotyping EM algorithm. Sci. Rep. 10, 14340 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Germain P-L scDblFinder. R package version 1.6.0, https://github.com/plger/scDblFinder.(2021).
- 32.Bais AS & Kostka D scds: computational annotation of doublets in single-cell RNA sequencing data. Bioinforma. Oxf. Engl. 36, 1150–1158 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lun ATL, McCarthy DJ & Marioni JC A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Research 5, 2122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaujoux R & Seoighe C A flexible R package for nonnegative matrix factorization. BMC Bioinformatics 11, 367 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goveia J et al. An Integrated Gene Expression Landscape Profiling Approach to Identify Lung Tumor Endothelial Cell Heterogeneity and Angiogenic Candidates. Cancer Cell 37, 21–36.e13 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Buechler MB et al. Cross-tissue organization of the fibroblast lineage. Nature 593, 575–579 (2021). [DOI] [PubMed] [Google Scholar]
- 37.Chen Y-P et al. Single-cell transcriptomics reveals regulators underlying immune cell diversity and immune subtypes associated with prognosis in nasopharyngeal carcinoma. Cell Res. (2020) doi: 10.1038/s41422-020-0374-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim N et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat. Commun. 11, 2285 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee H-O et al. Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat. Genet. 52, 594–603 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Ma L et al. Tumor Cell Biodiversity Drives Microenvironmental Reprogramming in Liver Cancer. Cancer Cell 36, 418–430.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peng J et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 29, 725–738 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qian J et al. A pan-cancer blueprint of the heterogeneous tumor microenvironment revealed by single-cell profiling. Cell Res. 30, 745–762 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang K-W, Alaei-Mahabadi B, Samuelsson T, Lindh M & Larsson E The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat. Commun. 4, 2513 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ogłuszka M, Orzechowska M, Jędroszka D, Witas P & Bednarek AK Evaluate Cutpoints: Adaptable continuous data distribution system for determining survival in Kaplan-Meier estimator. Comput. Methods Programs Biomed. 177, 133–139 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Barczak W, Suchorska W, Rubiś B & Kulcenty K Universal real-time PCR-based assay for lentiviral titration. Mol. Biotechnol. 57, 195–200 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Puram SV et al. STAT3-iNOS Signaling Mediates EGFRvIII-Induced Glial Proliferation and Transformation. J. Neurosci. Off. J. Soc. Neurosci. 32, 7806–7818 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deleage C et al. Defining HIV and SIV Reservoirs in Lymphoid Tissues. Pathog. Immun. 1, 68–106 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Erben L, He M-X, Laeremans A, Park E & Buonanno A A Novel Ultrasensitive In Situ Hybridization Approach to Detect Short Sequences and Splice Variants with Cellular Resolution. Mol. Neurobiol. 55, 6169–6181 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All single cell RNA-seq data produced by this study is available through the Gene Expression Omnibus with GEO accession GSE182227. TCGA bulk RNAseq and clinical data for head and neck and cervical cancer is available through the Broad Genome Data Analysis Center Firehose (https://gdac.broadinstitute.org/). Single-cell datasets reanalyzed to compare proliferation rates are available through the Gene Expression Omnibus with accession numbers GSE150430 (nasopharyngeal carcinoma), GSE131907 (lung carcinoma), GSE132465, GSE132257, GSE144735 (CRC), GSE125449 (HCC), through the Chinese National Centre for Bioinformation Genome Sequence Archive (CNCB-GSA) with accession: CRA001160 (PDAC) and through EMBL-EBI ArrayExpress with accession numbers E-MTAB-8107 (breast, ovarian, colorectal cancer), E-MTAB-6149 (lung) and E-MTAB-6653 (lung). Cell line data used for validation analysis is available through GEO with accession number GSE157220. The NSCLC dataset used to validate finding malignant cells in normal samples is deposited as an NCBI BioProject with accession number PRJNA591860. Source data are provided with this paper.