

Abstract
Objective:
In gout, hyperuricemia promotes urate crystal deposition that stimulates the NLRP3 inflammasome and IL-1β-mediated arthritis. Incident gout without background hyperuricemia is rarely reported. To identify hyperuricemia-independent mechanisms driving gout incidence and progression, we characterized erosive urate crystalline inflammatory arthritis meeting ACR/EULAR gout classification criteria in a normouricemic young adult female.
Methods:
Whole genome sequencing, quantitative proteomics, whole blood RNA-seq, and IL-1β-induced murine knee synovitis characterized proband candidate genes, biomarkers, and pathogenic mechanisms.
Results:
Lubricin was attenuated in proband serum, associated with elevated acute phase reactants and inflammatory whole blood transcripts and transcriptional pathways. The proband had predicted damaging gene variants of NLRP3 and of Inter-Alpha-Trypsin Inhibitor Heavy Chain 3, an inhibitor of lubricin-degrading Cathepsin G. Proband serum protein interactome network changes supported enhanced lubricin degradation, with Cathepsin G activity increased relative to its inhibitors SERPINB6 and Thrombospondin1. TLR2 activation suppressed cultured human synovial fibroblast lubricin mRNA and release (p<0.01). Lubricin blunted urate crystal precipitation, and IL-1β induction of xanthine oxidase and urate in cultured macrophages (p<0.001). In lubricin-deficient mice, IL-1β knee injection increased xanthine oxidase positive synovial resident M1 macrophages (p<0.05).
Conclusion:
We linked normouricemic erosive gout to attenuated lubricin, with impaired control of Cathepsin G activity, compounded by deleterious NLRP3 variants. Lubricin suppressed monosodium urate crystallization, and blunted IL-1β-induced increases in macrophage xanthine oxidase and urate. Collective activities of articular lubricin that could limit incident and erosive gouty arthritis independently of hyperuricemia are subject to disruption by inflammation, activated Cathepsin G, and synovial fibroblast TLR2 signaling.
Keywords: PRG4, xanthine oxidase, NLRP3, IL-1β, macrophage, synovium, Cathepsin G, SERPINB3, SERPINB6, Thrombospondin1, ITIH3, TLR2, tophus
Graphical Abstract
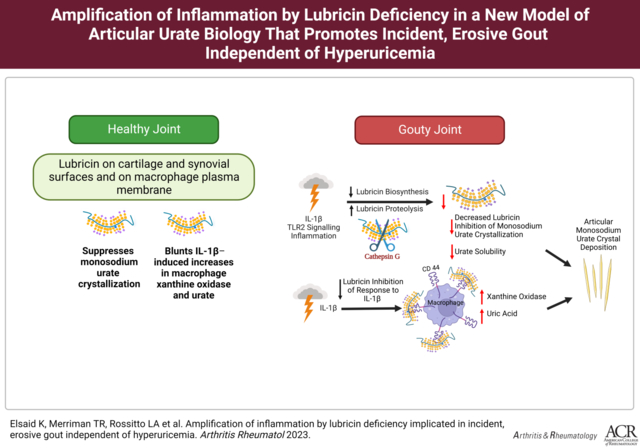
Introduction:
In gout, hyperuricemia drives tissue supersaturation with urate, with consequent deposition of monosodium urate crystals in tophi, on the articular cartilage surface, and in other joint tissues (1). Within tophi, urate crystal aggregates are mixed with foci of chronic synovitis, granulomatous and multicellular pannus-like inflammation, and fibrosis (2). Urate crystal ingestion by monocytes and macrophages stimulates NLRP3 inflammasome activation and IL-1β release (1,3). Consequent endothelial and phagocyte activation amplify inflammatory arthritis (1,3) and promote NETosis that can augment tissue urate levels (4), and modulate urate crystal aggregation, tophus development, and chronicity of gouty arthritis (5).
Major effects of gene variants on urate transport, purine metabolism, and serum urate predisposing to hyperuricemia, gout, and tophaceous disease include population-specific loss of function polymorphisms of the urate transporter ABCG2 (6). Disease duration, older age, and CKD are among other risk factors for tophi (7). However, major gout GWAS findings have principally been from hyperuricemia studies (8). Consequently, genetic factors promoting progression of hyperuricemia to incident gout, and development of tophaceous and erosive gout phenotypes, are poorly understood (8). Notably, asymptomatic hyperuricemia is over 5 times as prevalent as gout (9). Furthermore, gouty synovial fluid urate levels have been reported to be enriched relative to serum urate (10). Hence, passive filtering of excess circulating urate into joints cannot be the sole factor stimulating articular urate crystal deposition (11). Moreover, GWAS signals for urate levels are enriched for genes expressed in joint tissues including the synovium (12). To identify novel gene variants and constitutively active mediators that might limit development of gout to a minority of those with hyperuricemia, and additionally suppress disease progression, we systematically analyzed a normouricemic young female adult proband who met sufficient and weighted 2015 ACR/EULAR classification criteria for gout (12) and developed erosive arthritis. We identified proband attenuation of PRG4 gene product lubricin, linked to genetic variants and other changes particularly permissive for NLRP3/IL-1β-mediated autoinflammation, proteolysis of lubricin, and fibrosis.
Methods
The Supplementary Materials and Methods and results, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi covers approaches, and in some cases, results for WGS genetics and proteomics studies and whole blood RNA-seq, and details on antibodies, and cytokines used. Also described there are approaches for quantitative real time RT-PCR (qPCR) and primers used, urate crystal precipitation analyses, and mouse macrophage analyses. WGS and transcriptome profiling data will be uploaded to NCBI Gene Expression Omnibus database. All patients and healthy volunteers signed a consent form in a protocol approved by each local institutional review board involved in human subjects studies. All animal procedures were approved by Chapman University and San Diego VA Institutional Animal Care and Use Committees. For statistical analyses, P < 0.05 was considered statistically significant.
Results
Erosive Urate Crystalline Arthritis without Hyperuricemia:
A 22-year-old White female, previously in excellent health, and without personal or family history of gout or hyperuricemia, reported sudden onset non-traumatic left hip pain in the left hip, and had antalgic gait and positive impingement testing, but negative plain radiography for fracture. Rheumatoid factor, anti-CCP antibody, and ANA were negative. Serum ESR and CRP were elevated. Renal function was normal. Left hip MRI showed inflammatory synovitis, joint effusion, and cartilage loss (Figure 1A). Left hip aspiration yielded bacterial culture negative synovial fluid loaded with urate crystals and neutrophils. Serum urate was normal (5 mg/dL). Progressive left hip pain required total hip arthroplasty at age 25, with erosive joint damage with severe cartilage loss documented on the operative report. In 2012, at age 27, monthly episodes of self-limiting joint pain and swelling developed, affecting the right wrist, both ankles and feet. Foot radiographs revealed great toe joint erosions with well-defined margins (Figure 1B, arrows), consistent with tophaceous, erosive gout. First metatarsophalangeal joint ultrasound showed subchondral bone erosions and urate crystal deposits (Figure 1C, arrows). Ultrasound and dual energy CT results confirmed urate crystalline macroaggregates in numerous foot joints (Figure 1C–D). In 2014, after treatment with 400 mg allopurinol daily lowered serum urate to ~2 mg/dL, acute arthritis flares markedly lessened. Research studies on her genome, serum acute phase reactants and other serum constituents (proteome, lubricin, proteases) were done in 2018. In 2019, the proband reported diarrhea, which increased in 2020. Crohn’s disease was diagnosed in early 2021 by terminal ileal biopsy revealing moderate to severe chronic active enteritis, and mucosal ulceration and mucopurulent membranes. She was treated with low dose budesonide, and her symptoms improved. RNA-seq studies were then performed in 2021. None of the research samples were collected at the time of an acute gouty arthritis flare.
Figure 1. Proband skeletal imaging confirms urate crystal deposition in multiple joints and erosive disease.
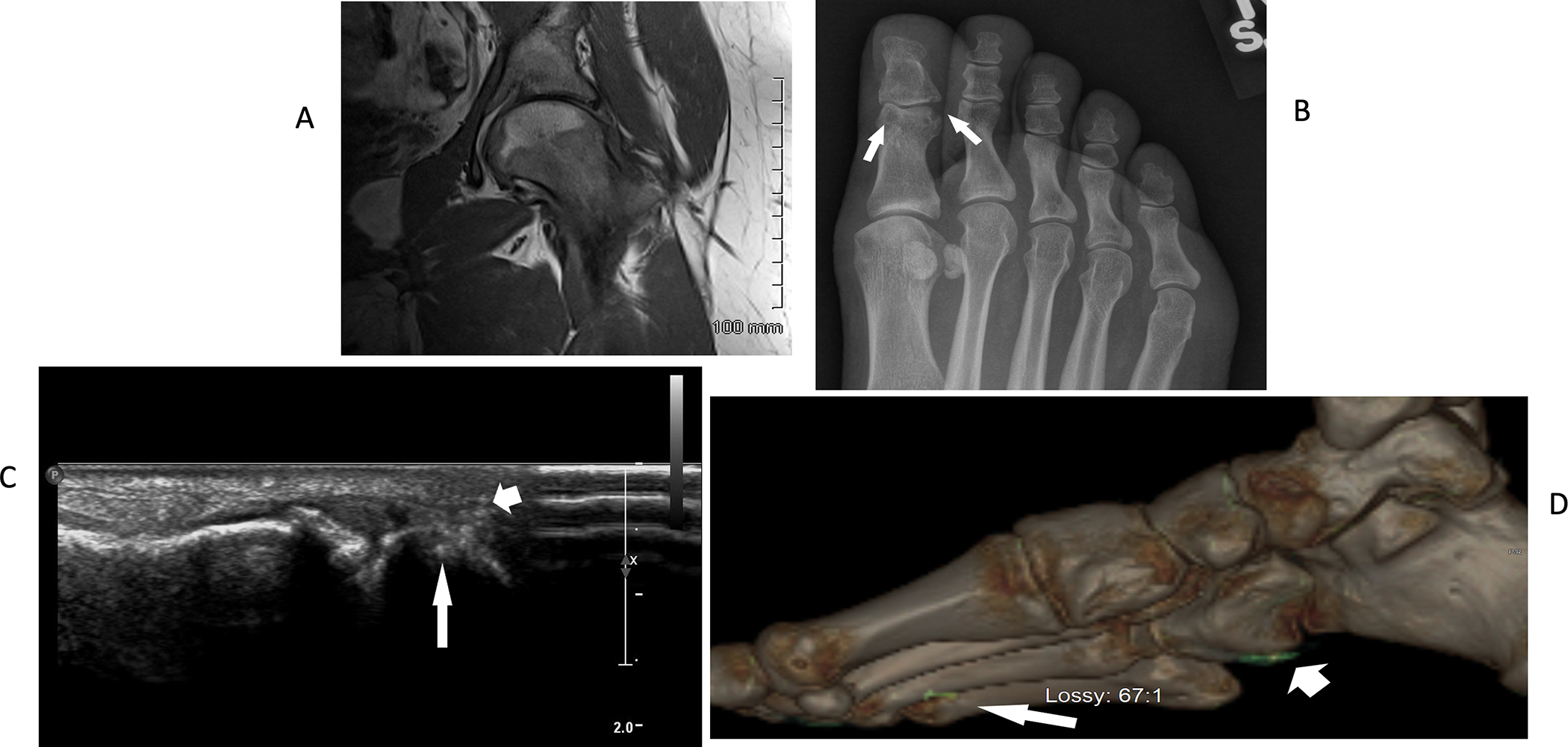
A. Left hip MRI revealed inflammatory synovitis, joint effusion, and articular cartilage loss. B. Foot radiographs revealed classic punched out great toe joint interphalangeal joint erosions, with well-defined margins (arrows), features characteristic of tophaceous, erosive gout. C. First MTP joint ultrasound showed hyperechoic areas consistent with urate crystal deposits (arrowhead), accompanied by cortical erosion (arrow). D. Dual energy CT exams of the foot confirmed urate crystalline macroaggregates affecting foot joints and surrounding soft tissues (arrow and arrowhead).
Characterization of Proband Inflammatory State:
Multiple acute phase reactants, including CRP and fibrinogens, were increased in proband serum, in serum of proband’s father (who also bore variant NLRP3 V198M), and to a lesser degree in serum of proband’s mother (Figure 2A). Whole blood RNA-seq, on globin-depleted RNA, when gout had long been symptomatically controlled, and inflammatory bowel disease symptoms had improved, showed multiple changes supporting inflammatory disease diathesis. The proband had 769 differentially expressed genes (DEGs) compared to healthy controls; 120 DEGs were upregulated, and 649 downregulated. Pathway enrichment analysis of upregulated genes (Figure 2B) showed predominance for interferon and cytokine signaling in proband compared to healthy controls. Conversely, we observed decreased IL-4 and IL-13, which partially share cell signaling pathways and anti-inflammatory and immune-modulating effects. Proband RNA-seq also showed downregulated chaperone-mediated autophagy and DNA damage and repair response pathways, suggesting impaired cell homeostasis. Further details on RNA-seq data are in the Supplementary Materials and Methods and Results, Supplementary Figures b–o.
Figure 2. A. Serum quantitative proteome of a panel of acute phase reactants in the proband.

A. Relative protein abundances were measured by quantitative multiplexed proteomics. Bars represent mean protein relative abundance between replicates (if protein detected in both replicates). Error bars represent SEM. Serum quantitative proteomics showed multiple acute phase reactants increased in the proband, with many also elevated in the proband’s father (who also bore the NLRP3 V198M variant), and some increased in the proband’s mother. Non-gout healthy controls (n=3); controls with “common gout” n=3, none with palpable tophi. B. Key results for whole blood RNA-seq in proband compared to 3 healthy non-gout controls (using prior depletion of globin RNA, with 2 replicates for each sample). Data analysis shown is detailed further in the Supplementary Materials. STRING application-Protein Query generated a node network enriched in differentially expressed genes (DEGs), further separated into 8 clusters. Reactome Pathway queries identified functional pathways in each cluster.
Proband lubricin deficiency without decreased whole blood lubricin mRNA expression:
To efficiently screen for erosive gout without hyperuricemia candidate mechanisms, we first examined proband sera by label-free proteomics (described in Supplementary Methods). Thirteen serum proteins, including lead candidate PRG4/lubricin, were at least one order of magnitude decreased in proband compared to either parent, and to 4 pooled healthy controls (Figure 3A). Multiplex serum quantitative proteomics, with binary comparisons (Figure 3B)(Supplementary Methods), allowed classification of proteins altered “only in proband,” “only in family member” (changed vs. healthy controls), “only in common gout” (changed in proband and “common gout” vs. healthy controls) and “nonspecifically” (changed in all binary comparisons). Since significance testing could not be performed for n=1 proband, log2 fold-change cutoff of >0.5 or <0.5 (~40%change) was used to classify proteins as “up” or “down,” respectively (Supplementary Methods). In the proband, 191 proteins were increased or decreased relative to non-gout controls, with 119 of these present in the interactome connecting known, physical protein-protein interactions (Figure 3B). Proband and “common gout” patient sera had increased neutrophil-secreted activation biomarkers myeloperoxidase (MPO) and calprotectin (S100A8/S100A9)(14)(Figure 3B). Significantly, “pin-dropping” NLRP3 and SERPINB3, via described approach (15), pointed to NLRP3 and SERPINB3 involvement in the proband interactome.
Figure 3. Serum proteomics identifies lubricin attenuation and proband interactome network changes suggesting increased lubricin degradation potential.
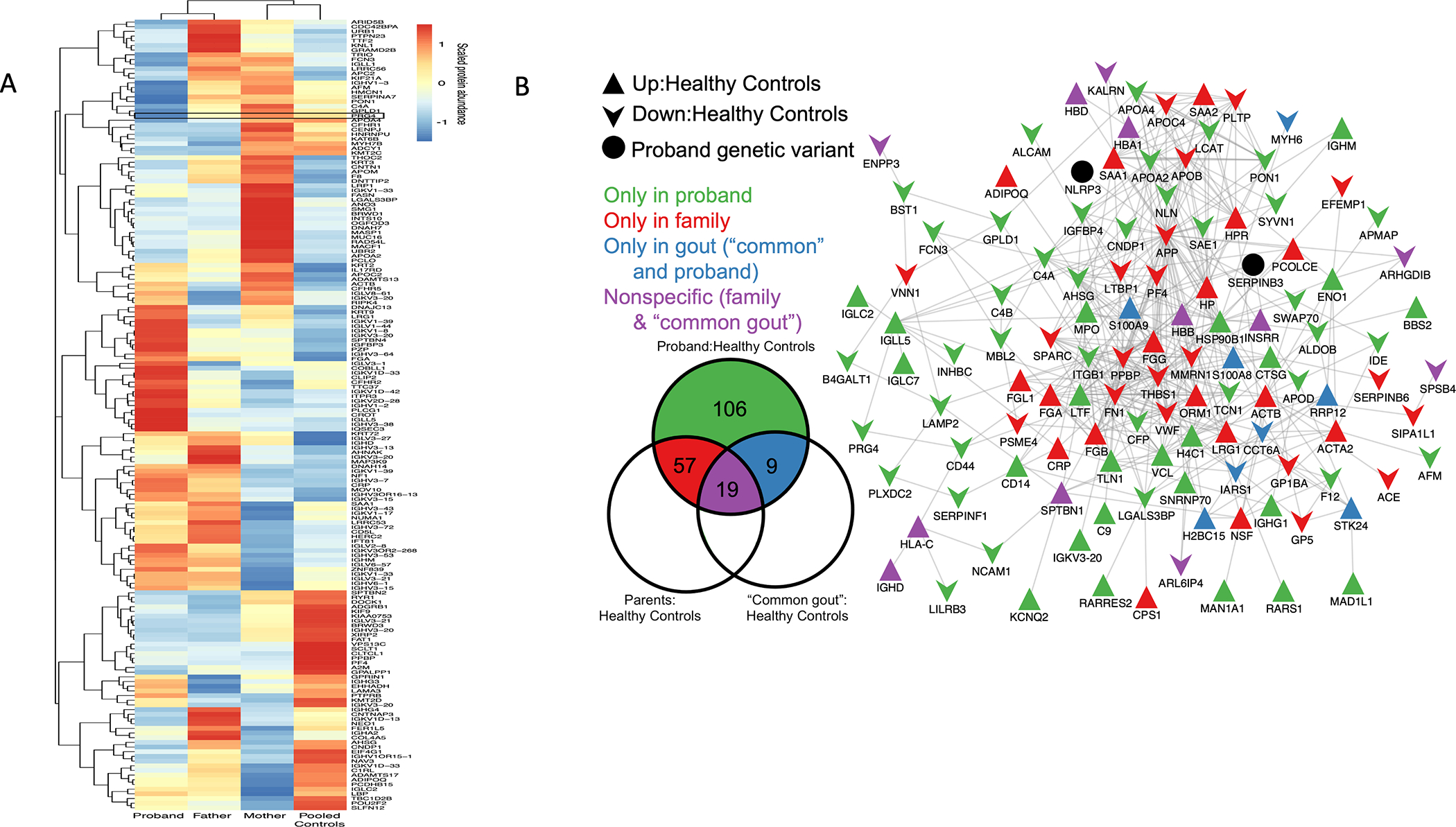
A. Label-free proteomics heatmap showing serum proteins with ~1 order magnitude reduction in proband vs. father, mother, pooled (n=4) healthy non-gout controls; PRG4/lubricin lead candidate. B. Quantitative multiplexed proteomics-defined binary comparisons for proteins with cutoff ~≥40% difference between groups). (Left) proteins of interest specific to or overlapping between groups, without directionality (full lists in Supplementary Data). (Right) overall proteomics changes in Metascape-generated protein-protein networks; background, edges (known protein-protein interactions), and “pin-dropping” NLRP3 and SERPINB3 into network. Network presence of 119 of 191 interacting proteins of interest (Venn diagram) suggested central dysregulated pathways.
We analyzed Figure 3B for other altered core pathways, beyond those involving NLRP3 and SERPINB3, that may have promoted the proband’s gout phenotype. PRG4/lubricin connected to the soluble form of its receptor CD44, was in the proteome network of interacting proteins changed in the proband. Furthermore, the most centrally connected proteins in the interactome were the Cathepsin G (CTSG) substrate (16) amyloid precursor protein (APP), which was relatively low in each kindred member, and had 51 network connections, the CTSG inhibitor Thrombospondin 1 (TSP1, THBS1) with 20 connections, lactoferrin (LTF) with 13 connections, and CTSG with 7 connections (Figure 3B). The CTSG inhibitor SERPINB6 also was in the interactome (17). Results linked proband lubricin depletion to potentially enhanced lubricin-degrading capacity.
We confirmed attenuated serum lubricin in the proband compared to parents and controls (Supplementary Figure 1A), using Dissociation-Enhanced Lanthanide Fluorescent Immunoassay (DELFIA), with catching antibody 1E12 (described in Supplementary Methods). In immunoassays of lubricin lectin binding, kindred members had slightly elongated O-linked oligosaccharides with increased sialylation in the mucin domain (Supplementary Figure 1A). This suggested disproportionate dominance of more protease-resistant sialic acid-capped lubricin in kindred member sera. Competition ELISA, using lubricin monoclonal antibody 9G3 (18) further validated markedly low proband serum lubricin, which was significantly decreased relative to either parent (neither of them with gout), and to 18 control patients with “common gout” and uncontrolled hyperuricemia at the baseline before entering the VA STOP GOUT urate-lowering therapy clinical trial (19)(Supplementary Figure 1B). The over 30-fold range of serum PRG4/lubricin seen in “common gout” patients was far wider than previously seen for RA, where serum lubricin levels did not differ significantly from healthy controls (18). Moreover, 5 of the 18 “common gout” subjects had very low serum lubricin.
PRG4 is induced by Wnt signaling in chondrocytes (20). RNA-seq pathway enrichment analysis revealed three Wnt signaling pathways among those downregulated in proband whole blood relative to healthy non-gout controls (Figure 2B). Despite this, and blunted proband serum lubricin levels, proband whole blood PRG4/Lubricin mRNA was not decreased compared to either parent or to healthy non-gout controls (Supplementary Figure 1C).
Imbalance between lubricin-degrading proteases and their inhibitors in the proband:
Serum activity of the lubricin-degrading protease Cathepsin G (CTSG) was increased in the proband, both her parents, and in “common gout” patient sera, though with substantial variability; by contrast, it was undetectable in unrelated, healthy, non-gout controls (Figure 4C). By ELISA, “common gout” patient sera also had increased neutrophil-secreted CTSG co-activator LTF compared to matched, non-gout healthy controls (Figure 4B). Multiplex serum proteomics revealed decreased levels of the normally abundant CTSG and elastase inhibitor TSP1 (21), and of the predominantly intracellular myeloid leukocyte CTSG inhibitor SERPINB6 in the proband and her parents (17)(Figure 4A). There were no substantial differences between proband, proband father and mother, and non-gout and gout control sera in other CTSG inhibitors detected by proteomics (α1 antitrypsin (AAT, SERPINA1); α1-antichymotrypsin (SERPINA3), α-1 microglobulin/bikunin precursor (AMBP), and Inter-α trypsin inhibitor (ITI).
Figure 4. Serum quantitative multiplex proteomics for proteins relevant to proteolysis of lubricin.

Shown here are the proteomics-detectable species from serum multiplex quantitative proteomics, studying the proband, father, and mother of the kindred with gout without hyperuricemia. Controls were serum from small panels of “common gout” and non-gout healthy subjects. Bars represent the mean protein relative abundance between technical replicates (if protein detected in both replicates) and error bars represent standard error of the mean. Relevance of individual findings is discussed in the text. A. Bars represent the mean protein relative abundance between technical replicates (if the protein was detected in both replicates). Error bars represent SEM. B. By ELISA, serum LTF was increased in “common gout” patients vs. non-gout healthy controls matched for age, sex, and presence or absence of obesity. Statistics by 1 way ANOVA. C. Serum CTSG activity (assayed in triplicate) was undetectable in healthy non-gout, but was detected in the proband, her parents, and multiple gout patients. 1: Healthy non-gout controls (n=6); 2: Proband; 3: Proband’s Mother; 4: Proband’s Father; 5: Poorly controlled gout with hyperuricemia at time of sampling (n=10); 6: Well-controlled gout without hyperuricemia (n=6). *P<0.05, ** P<0.01 by 1 way ANOVA.
Whole Genome Sequencing (WGS) of Kindred Members:
Proband DNA whole genome sequencing (WGS) (key results summarized in Supplementary Table 1) revealed no obvious functional mutations in PRG4. However, proband and father shared the NLRP3 rs121908147 low penetrance variant that encodes NLRP3 V198M, whose allele frequency is between 0.0038 and 0.0074 in healthy White controls (22,23). NLRP3 V198M, linked to several-fold higher rates of stimulated monocyte IL-1β release in vitro, is occasionally associated with IL-1β-driven autoinflammatory diseases and periodic fever syndromes (23). The proband also was heterozygous for other potentially disease-promoting NLRP3 variants (rs4612666, rs10754558, rs4353135, rs6672995) (Supplementary Table 1, and Supplementary Materials Genetics Section B: Supplementary Table 4). To examine PRG4 gene further, we extracted all variants in PRG4 +/−100KB: 1:186165405–186383694. We analyzed 1318 variants, including those filtered as missense (n=25), frameshift (n=5), LOF (n=5) and splice (n=6). Nothing of interest was discovered (Supplementary Materials Genetics Section B: Supplementary Tables 2–3).
The proband was heterozygous for the rs74320783(G523R) variant (Supplementary Materials Genetics Section B: Supplementary Table 5) of Inter-Alpha-Trypsin Inhibitor Heavy Chain 3 (ITIH3), a multifunctional chondrocyte-expressed Kunitz family inhibitor of multiple serine proteases, including leucocyte elastase and CTSG (24). ITIH3 binds hyaluronan, protects the cartilage surface, and exerts anti-inflammatory effects (25). The rs74320783 ITIH3 variant minor allele frequency is low in EUR (MAF = 0.012), and SIFT, PolyPhen and CADD scores predicted the variant to be deleterious, probably damaging, and likely deleterious, respectively. Moreover, the variant is located at a 11-zinc finger protein or CCCTC-binding factor transcriptional repressor site (CTFC binding site).
The proband also was homozygous for SERPINB3 variant rs3180227, which encodes a cysteine protease inhibitory gain-of-function G351V substitution. SERPINB3 is anti-apoptotic, pro-inflammatory, and pro-fibrotic, and promotes epithelial to mesenchymal transition (25,26). SERPINB3 rs3180227 is linked with susceptibility to cirrhosis (27). Deeper analyses of WGS results for selected gene candidates focused on two other inhibitors of lubricin-degrading proteases (SERPINB6, THBS1) that were relatively low in proband serum. No obvious variants of interest were identified for SERPINB6 and THBS1 (Supplementary Materials Genetics Section B: Supplementary Tables 5–7). Last, no knockout mutations, or other known rare disease-causing mutations were seen by WGS analysis for any other candidate genes.
Lubricin blunted urate crystal formation and suppressed IL-1β-induced XO activity and uric acid generation by cultured macrophages:
IL-1β and TNF are known to suppress osteoarthritis fibroblast-like synoviocyte (FLS) lubricin mRNA levels, and both cytokines promote release of TLR2 ligands (28,29). Here, we examined additional mechanisms by which deficient intra-articular lubricin release could arise in gouty arthritis. We observed that a TLR2-selective ligand suppressed PRG4 mRNA and lubricin secretion by human FLS, as did selective TLR2 ligand activated macrophage co-culture in a Transwell system (Supplementary Figure 2). Under the same conditions, the TLR2 ligand induced human FLS mRNA for COX-2, and multiple inflammatory cytokines and matrix-degrading enzymes.
Articular cartilage homogenates and certain cartilage constituents promote urate crystallization (30,31). Screening proband serum, we did not detect any evidence for “hyper-precipitation” of urate crystals in vitro (Figure 5A). Since normal serum lubricin (~1 μg/ml) is much lower than in normal joint fluid(~200 μg/ml)(18), we tested effects of rhPRG4 on urate crystal formation in vitro (Figure 5B). As a control, we used the large, heavily O-glycosylated molecule bovine submaxillary mucin (BSM), which fails to inhibit urate crystal phagocytosis and cytokine release by macrophages under conditions where lubricin is inhibitory (32). BSM did not significantly affect urate crystallization, whereas lubricin blunted urate crystal formation in vitro (Figure 5B). Lubricin did so dose-dependently, starting at 25 μg/ml, well below the ~200 μg/ml lubricin concentration of normal synovial fluid
Figure 5. Effects of proband serum, and of lubricin on monosodium urate crystallization, and effects of lubricin on IL-1b-induced XO and uric acid level in macrophages in vitro.
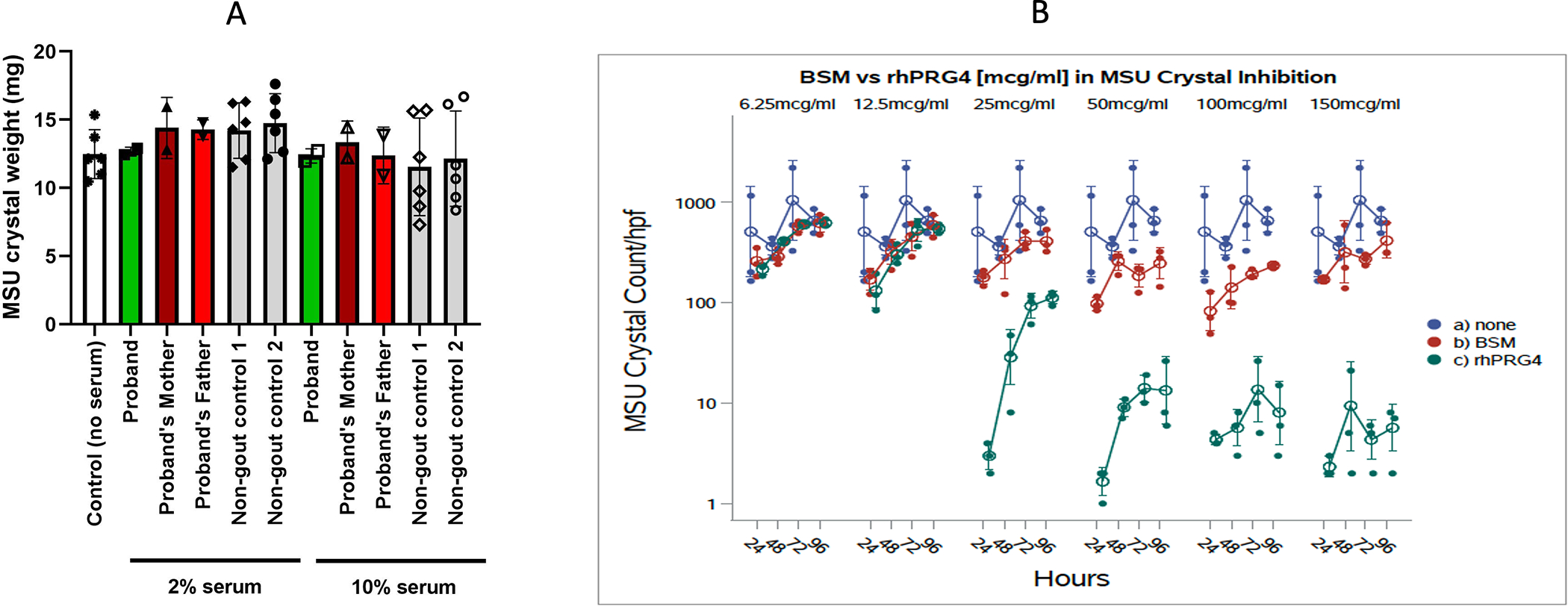
A. Urate concentration in 2% and 10% serum-sodium urate solutions was assessed for reduction of monosodium urate crystallization over time. Starting serum urate concentrations were 0.31–0.38 mmol/L in proband, father and mother, and 2 non-gout controls. Results were validated by assessing samples using compensated polarized light microscopy. Data shown as mean (SD) with each symbol showing data from an experimental replicate. B. rhPRG4/Lubricin alone inhibited the number of urate crystals formed between 25–150 mg/ml concentration, compared to buffer or the bovine submaxillary mucin (BSM) control (p<0.001 for 25–150 mg/ml rhPRG4/Lubricin at each dose).
XO promotes macrophage differentiation to an activated state (33). Hence, we tested if lubricin could limit the potential capacity of macrophage lineage cells within the joint to increase XO activity and generate uric acid in response to IL-1β (Supplementary Figure 3). IL-1β induced increased XO activity and uric acid in wild type mouse bone marrow macrophages. Since myeloid cells do not make lubricin, we added rhPRG4 to macrophages at 200 μg/ml. Lubricin blunted IL-1β induction of XO and the associated increase in uric acid in cultured bone marrow macrophages. Last, we assessed synovial resident macrophages (SRMs) via harvesting of mouse knee joint capsules, and flow cytometry, done as previously reported (34). At baseline, XO positivity was enriched in M1 relative to M2 SRMs in all mouse knee joints studied in vivo (Figure 6A). Baseline XO+ SRMs were higher in Prg4GT/GT synovia compared to PRG4-reconstituted Prg4GTR/GTR synovia, associated with an increase in M1 SRMs. IL-1β injection into the mouse knee joint significantly increased XO in total SRMs, and in M1 SRMs, in Prg4GT/GT vs. Prg4GTR/GTR mice. Collective results supported a model (Figure 6B) in which IL-1β-mediated “inflammatory drive” and phagocyte activation can promote decreased lubricin mRNA in FLS and increased lubricin degradation in the gouty joint. Results suggested that this pathway to lubricin depletion could facilitate IL-1β-induced macrophage XO activity and uric acid generation in the joint, and further lead to intra-articular urate crystal formation in vitro, with these effects independent of hyperuricemia.
Figure 6. intra-articular modulation of XO in synovium-resident macrophages (SRMs), and model for the lubricin and IL-1β circuit regulating gouty arthritis incidence and progression.
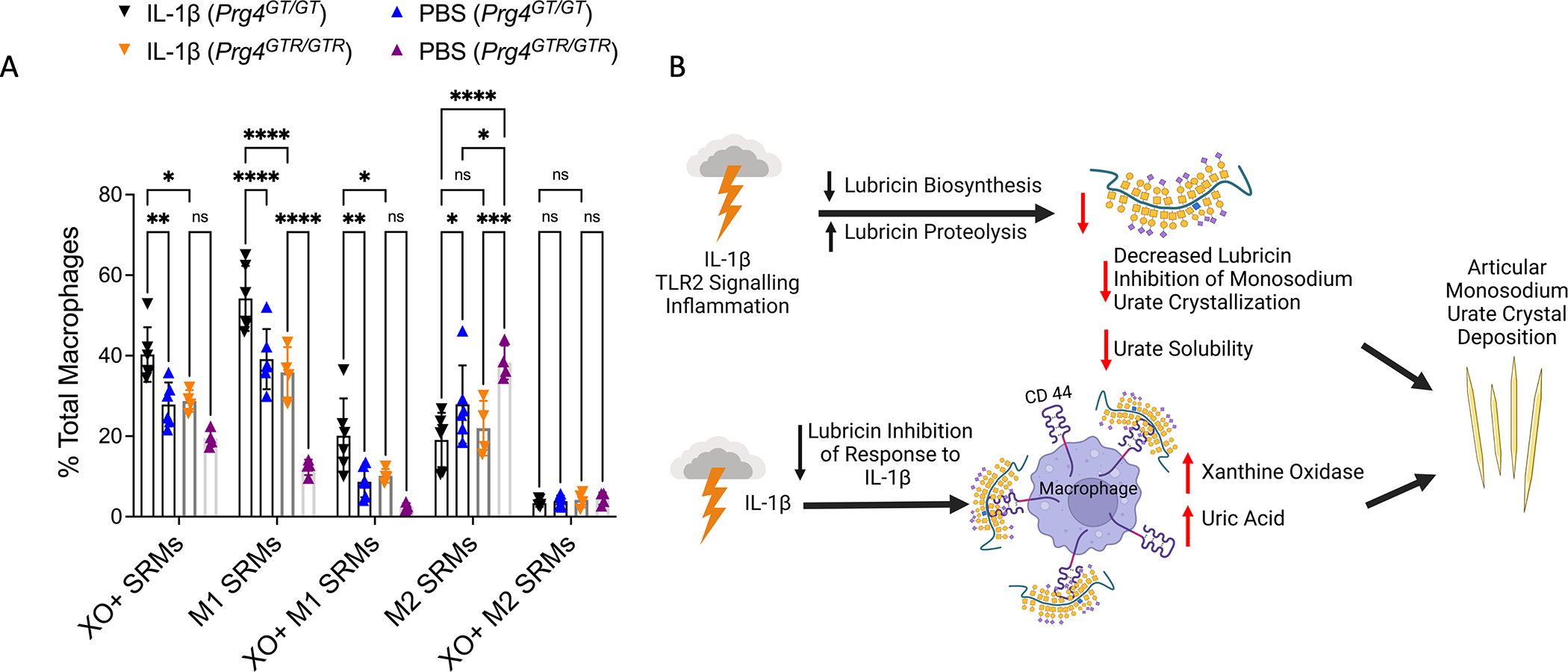
A. Intra-articular IL-1β (10 ng) injection induced XO in (SRMs) knees of 2–3 months old Prg4GT/GT mice (n=6) and Prg4GTR/GTR mice (n=4). Prg4 reconstitution via recombination of Prg4GTR mice was done, with all mice receiving intraperitoneal tamoxifen at 3 weeks of age. Flow cytometry results for M1 and M2 SRMs and XO+ M1 and XO+ M2. 2-way ANOVA with Tukey’s pairwise comparisons. ns: non-significant; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. B. Model for dysfunction of a lubricin and IL-1β regulatory circuit that promotes gout development and progression independent of hyperuricemia, derived from collective results.
Discussion:
We characterized a normouricemic premenopausal female with widespread urate crystal deposition, acute inflammatory arthritis flares, destructive unilateral hip arthropathy, and great toe erosive joint disease with classic radiographic features of gout. The collective findings shed new light on dysregulated articular cell functions, inflammatory responses, and pathways that can impact gout incidence and disease progression independently of hyperuricemia. The proband’s arthritis phenotype, compounded later by Crohn’s disease, could be classified as an unusual autoinflammatory NLRP3 variant syndrome, compounded by associated lubricin deficiency and urate crystal deposition that amplified the arthritis. In this context, destructive arthritis first affecting the hip joint is a highly unusual presentation for gout. On the other hand, the proband’s arthritis met both sufficient and weighted scale 2015 ACR/EULAR classification criteria for gout (13). The self-limited peripheral arthritis flares and great toe erosions were classical for gout (1,13). Furthermore, the arthritis markedly responded to long-term allopurinol treatment.
Proband WGS revealed heterozygosity of NLRP3 V198M, linked to increased potential for CAPS and certain other autoinflammatory diseases, and the proband had other potentially deleterious NLRP3 variants (22,23). Myeloperoxidase (MPO), LTF, and calprotectin (S100A8/A9) elevation in proband and gout patient sera buttressed evidence of inflammation linked with heightened neutrophil activation, which can trigger and amplify acute gout (3,35,36). Furthermore, several proband whole blood transcript and transcriptional pathway changes supported more severe inflammation but also increased macrophage M1 differentiation, which is mediated by XO activity (33). Specifically, IL-4, decreased in proband whole blood mRNA, promotes macrophage polarization to M2 cells and it inhibits macrophage differentiation from M0 to M1 cells. Increase in M2 macrophages is associated with greater release of IL-10 and TGFβ, which may help self-limit gouty inflammation (35,37). The proband also had downregulation in a transcriptional pathway for autophagy, which self-limits gouty inflammation (35), whereas there was an upregulated cytokine signaling pathway. Hence, the proband appeared predisposed to mount inefficiently resolving inflammatory responses, likely contributing to her erosive hip and toe arthritis, and later development of Crohn’s Disease.
The PRG4 gene product lubricin was attenuated in proband serum. Lubricin, a large, mucinous boundary lubricant O-glycoprotein, is released in the joint principally by fibroblast like synoviocytes (FLS), complemented by chondrocyte release, in the superficial zone, of the alternative PRG4 gene product superficial zone protein (SZP)(28,29,38,39). Lubricin regulates synovial macrophage polarization and inflammatory macrophage infiltration (34). Furthermore, Prg4-deficient mice demonstrate elevated urate crystal phagocytosis, inflammatory signaling, urate crystal-induced IL-1β release and inflammation in vitro and in vivo, and impaired resolution of model acute gouty inflammation (40,41). These effects are mediated by urate crystal binding to the cognate lubricin transmembrane receptor CD44 (42). Since both CD44 knockout and lubricin limit urate crystal-induced inflammation in vivo (40–42), lubricin “cloaking” of CD44 may constitutively suppress synovial proliferation and inflammation in gout.
PRG4 expression is induced by Wnt signaling in chondrocytes (20). Three Wnt signaling transcriptional pathways were decreased in the proband. Moreover, not only IL-1β, but also TNF inhibit osteoarthritis fibroblast-like synoviocyte (FLS) lubricin mRNA level, and both cytokines promote release of TLR2 ligands (28,29). Here, TLR2-selective ligand decreased lubricin mRNA and lubricin release in human FLS. Though proband whole blood PRG4 mRNA was not decreased, lubricin mRNA in the joint space was potentially low in the proband.
Joint inflammation promotes lubricin degradation (28,29). The phagocyte-derived protease CTSG cleaves the lubricin mucin, N- and C-terminal domains to generate over 20 lubricin polypeptides (43). Here, results supported a pathogenic role in the proband of insufficient inhibition of CTSG, and potentially of lubricin-degrading neutrophil elastase. First, an ITIH3 variant, predicted to be deleterious by 3 different algorithms, was identified in the proband by WGS. ITIH3 protects articular surfaces from proteolytic damage, and acts by binding hyaluronan, inhibiting both CTSG and neutrophil elastase, and limiting complement activation, hyaluronidase activity, and induction of chemokine release (24). Second, the proband, kindred members, and gout patients tested here had increased serum CTSG activity, and associated increases in neutrophil-secreted LTF, a co-activator of the abundant, constitutively latent CTSG in azurophil granules. Third, proband serum had decreases in the CTSG inhibitor SERPINB6, and in the tight CTSG and neutrophil elastase inhibitor TSP1, whose biologic effects include promoting activation from latency of the constitutive gouty inflammation pro-resolving mediator TGFβ (44).
This study identified novel effects of lubricin on intra-articular urate homeostasis and crystallization. Specifically, lubricin suppressed not only urate crystal deposition in solution, but also IL-1β-induced XO activity and urate levels in mouse bone marrow macrophages in vitro and in IL-1β-induced XO in murine SRMs in vivo. FLS lubricin release is significantly inhibited by IL-1β, and this response is limited by lubricin supplementation in vitro (39,45,46). Hence, not only the NLRP3/IL-1β pathway, but also modulation of this pathway by lubricin could alter XO activity and urate homeostasis within the joint. Moreover, lubricin dose-dependently decreased urate crystal precipitation in vitro at 25–150 μg/ml. We tested these lubricin concentrations because synovial fluid lubricin in normal human joints is ~200 μg/ml. Since urate crystallization in vitro is promoted by cartilage homogenates (30,31), these results were unexpected. Notably, failure of the large mucinous O-glycoprotein BSM to suppress urate crystallization in vitro pointed to selectivity of the lubricin effect.
NETosis is a major consequence of neutrophil activation by urate crystals (5), and it is increased in peripheral blood neutrophils of gout patients (47). NETosis increases tissue urate levels in a DNAase-suppressed manner, potentially mediated by extracellular neutrophil DNA accumulation (4), which colocalizes with lubricin-degrading proteases (eg, neutrophil elastase, CTSG released from granules) and LTF. Since NETosis is linked to formation of tophus-like urate crystal aggregation in experimental gout (47,48), our findings could be mechanistically pertinent to gout.
Serum lubricin is mainly sourced from the liver, and it is not expressed by leukocytes (49). However, lubricin binds several leukocyte adhesion proteins, physiologically coats blood and joint fluid neutrophils, and inhibits both their rolling on endothelium and attachment to articular cartilage (49). As such, elevated phagocyte-derived lubricin-degrading protease activity in serum might be a biomarker for the capacity of phagocytes to degrade lubricin in the joint. Moreover, serum CTSG activity is a potential biomarker in gout, since CTSG promotes inflammation, transduced by effects on MMP activation, phagocyte movement, and cytokine production under conditions of high neutrophil cell death (50).
Study limitations included that lubricin expression is modulated in a tissue-specific manner; we did not study proband joint tissues or pathology specimens. Directly quantifying relative effects of the ITIH3 variant rs74320783 G523R substitution, and deficiencies of TSP1 and of intracellular SERPINB6 on lubricin-degrading CTSG and elastase activity, as well as studies on NETosis on lubricin homeostasis, were beyond the scope of the work. Since articular chondrocytes express NLRP3, XO, and the PRG4 gene product superficial zone protein (SZP), macrophages may not be the sole articular cell type subject to NLRP3, IL-1β and PRG4 regulatory effects on urate homeostasis. We have not detected XO activity in human OA FLS (Miner M, Hammaker D, Firestein GS, Terkeltaub R, unpublished observations). However, deeper analyses of articular lubricin and urate homeostasis in cartilage and phagocytes may be of interest. The proband was homozygous for a pro-inflammatory SERPINB3 gain-of-function variant, whose net effects in tophus fibrosis, impairment of autophagy, and inflammatory disease in gout remain to be determined. The proteomics studies used trypsin treatment in all samples, which fragments lubricin and other substrates for trypsin and trypsin-like serine proteases. As such, reliably distinguishing biological proteolysis in sera from experimental proteolysis that used trypsin, was not feasible using our quantitative proteomics approach. Last, this study combined in vitro bench studies, and in vivo mouse studies with proband characterization that included proteomics and transcriptomics. Causal inferences from the bench to the proband were extrapolations that do not conclusively prove the basis for the proband’s condition.
In conclusion, lubricin not only suppresses how urate crystals activate macrophages but also inhibit multiple processes central to gout development and progression, such as urate crystal precipitation, and IL-1β-induced upregulation of macrophage XO activity and urate levels. The essential roles of PRG4/lubricin in articular boundary lubrication and joint homeostasis have been illuminated via linkage of severe congenital PRG4/lubricin deficiency to synovial proliferation and joint failure in Camptodactyly-Arthropathy-Coxa Vara-Pericarditis (45,46). Lubricin-deficient FLS are more inflammatory, pro-catabolic, and proliferative (46), and these effects can promote OA progression. Hence, articular lubricin dysregulation in OA and gout could modulate the frequent coexistence of both diseases. Lubricin homeostasis and its biomarkers, and potential measures to maintain and increase articular lubricin, warrant further investigation for their potential in limiting incidence and progression of gouty arthritis.
Supplementary Material
Acknowledgements:
Ron Booth and Alicia Storey (The Ottawa Hospital, Ottawa, Canada) were instrumental in organizing study of the proband and parents in the gout without hyperuricemia kindred. Input on NLRP3 gene variants was provided by Dr. Hal Hoffman (UC San Diego). Information on potential lubricin-modulating genes expressed in synovium was shared by Dr. Inka Brockhausen (Queens University, Kingston, Ontario, Canada), and by Drs. Brian Pedersen and Gary Firestein (UC San Diego). Tracy Guo (San Diego VA Healthcare System) provided technical assistance with macrophage and mRNA sample analyses.
The author(s) wish to acknowledge the use of New Zealand eScience Infrastructure (NeSI) high performance computing facilities, consulting support and/or training services as part of this research. New Zealand’s national facilities are provided by NeSI and funded jointly by NeSI’s collaborator institutions and through the Ministry of Business, Innovation & Employment’s Research Infrastructure programme. https://www.nesi.org.nz.
The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from https://gtexportal.org/home/.
Grant support:
LAR: NIH T32 GM007752
TRM: NIH AR075990 and Health Research Council of New Zealand (14–527)
RLB: VA Research Service I01 BX002234, Rheumatology Research Foundation Innovation Research Award
ND: Health Research Council of New Zealand (19–232)
ACh: Royal Society of New Zealand Rutherford Foundation Post-Doctoral Research Fellowship
AC: NIH T32 AR064194
NK: Grants from the Swedish state under the agreement between the Swedish government and the county council, the ALF-agreement (ALFGBG-722391), the Swedish Research Council (621-2013-5895), Petrus and Augusta Hedlund’s foundation (M-2016-0353) and AFA insurance research fund (dnr 150150)
KE, GDJ: NIH AR067748
DJG: UCSD Collaborative Center of Multiplexed Proteomics
RT: VA Research Service (I01 BX005927), NIH (AR075990, PAG007996)
Footnotes
Disclosures:
KE: Patent on the use of rhPRG4 for treatment of acute gout
ND: consultant for AstraZeneca, Dyve Biosciences, Horizon, Amgen, Selecta, Arthrosi, JW Pharmaceutical Corporation, PK Med, PTC Therapeutics, Protalix (all <$10,000).
TS: authored patents on rhPRG4, is a paid consultant for Lubris LLC, MA, USA and holds equity in Lubris LLC, MA, USA.
GDJ: Authored patents on lubricin and holds equity in Lubris LLC, MA, USA.
TRM, AC, LAR, RLB, AP-G, MC, SE, MQ, MM, MG, NM, JH, TJD, DJG: None
NGK: Authored a patent on lubricin as biomarker and holds equity in Lynxon AB, Gothenburg, Sweden
RT: has served as a consultant for Dyve, Fortress Bio, Allena, and Acquist (all less than $10,000).
Contributor Information
Khaled Elsaid, Chapman University School of Pharmacy, 401 Jeronimo Rd. Irvine, CA 92618..
Tony R. Merriman, Division of Clinical Immunology and Rheumatology, University of Alabama at Birmingham, United States; Professor of Biochemistry, Department of Biochemistry, University of Otago, New Zealand..
Leigh-Ana Rossitto, Dept of Pharmacology, School of Medicine, and Skaggs School of Pharmacy and Pharmaceutical Sciences, UC San Diego..
Ru Liu-Bryan, VA San Diego Healthcare System, San Diego, CA, Dept. of Medicine, UC San Diego, La Jolla, CA..
Jacob Karsh, The Ottawa Hospital, Division of Rheumatology, University of Ottawa, Canada..
Amanda Phipps-Green, Department of Biochemistry, University of Otago, New Zealand..
Gregory D. Jay, Department of Emergency Medicine, Alpert School of Medicine, Brown University & Division of Biomedical Engineering, School of Engineering, Brown University, RI..
Sandy Elsayed, Sandy Elsayed, MSc. Chapman University School of Pharmacy 9401 Jeronimo Rd. Irvine, CA 92618..
Marwa Qadri, Assistant Professor of Pharmacology, Jazan University, Jazan, Saudi Arabia..
Marin Miner, VA San Diego Healthcare System, San Diego, CA..
Murray Cadzow, Department of Biochemistry, University of Otago, New Zealand..
Talia J. Dambruoso, Division of Biomedical Engineering, School of Engineering, Brown University, RI..
Tannin A Schmidt, Biomedical Engineering Department, School of Dental Medicine, UConn Health, Farmington, CT, USA..
Nicola Dalbeth, Department of Medicine, University of Auckland, Auckland, New Zealand..
Ashika Chhana, Department of Medicine, University of Auckland, Auckland, New Zealand..
Jennifer Höglund, Department of Medical Biochemistry, Institute for Biomedicine, University of Gothenburg, Gothenburg, Sweden..
Majid Ghassemian, Biomolecular & Proteomics Mass Spectrometry Facility, Department of Chemistry/Biochemistry, UC San Diego, La Jolla, CA..
Anaamika Campeau, Dept of Pharmacology, School of Medicine, and Skaggs School of Pharmacy and Pharmaceutical Sciences, UC San Diego..
Nancy Maltez, The Ottawa Hospital, Division of Rheumatology, University of Ottawa, Canada..
Niclas G. Karlsson, Faculty of Health Sciences, Oslo Metropolitan University, 0130 Oslo, Norway, and Department of Medical Biochemistry, Institute for Biomedicine, University of Gothenburg, Gothenburg, Sweden..
David J. Gonzalez, Dept of Pharmacology, School of Medicine, and Skaggs School of Pharmacy and Pharmaceutical Sciences, Collaborative Center for Multiplexed Proteomics, Program for Integrative Omics and Data Science in Disease Prevention and Therapeutics, UC San Diego, La Jolla, CA..
Robert Terkeltaub, VA San Diego Healthcare System, San Diego, CA, Dept. of Medicine, UC San Diego, La Jolla, CA..
References:
- 1.Dalbeth N, Merriman TR, Stamp LK. Gout. Lancet. 2016;388:2039–2052. [DOI] [PubMed] [Google Scholar]
- 2.Chhana A, Dalbeth N. The gouty tophus: a review. Curr Rheumatol Rep. 2015;17:19. [DOI] [PubMed] [Google Scholar]
- 3.So A, Dumusc A, Nasi S. The role of IL-1 in gout: from bench to bedside. Rheumatology (Oxford). 2018;57(suppl_1):i12–i19. [DOI] [PubMed] [Google Scholar]
- 4.Liu C, Zhou M, Jiang W, Ye S, Tian S, Jiang C, Hao K, et al. GPR105-Targeted Therapy Promotes Gout Resolution as a Switch Between NETosis and Apoptosis of Neutrophils. Front Immunol. 2022;13:870183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suzuki K, Tsuchiya M, Yoshida S, Ogawa K, Chen W, Kanzaki M, et al. Tissue accumulation of neutrophil extracellular traps mediates muscle hyperalgesia in a mouse model. Sci Rep. 2022;12:4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He W, Phipps-Green A, Stamp LK, Merriman TR, Dalbeth N. Population-specific association between ABCG2 variants and tophaceous disease in people with gout. Arthritis Res Ther. 2017;19:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu B, Lu Q, Huang B, Li C, Zheng F, Wang P. Risk factors of ultrasound-detected tophi in patients with gout. Clin Rheumatol. 2020;39:1953–1960. [DOI] [PubMed] [Google Scholar]
- 8.Major TJ, Dalbeth N, Stahl EA, Merriman TR. An update on the genetics of hyperuricaemia and gout. Nat Rev Rheumatol. 2018;14:341–353. [DOI] [PubMed] [Google Scholar]
- 9.Chen-Xu M, Yokose C, Rai SK, Pillinger MH, Choi HK. Contemporary Prevalence of Gout and Hyperuricemia in the United States and Decadal Trends: The National Health and Nutrition Examination Survey, 2007–2016. Arthritis Rheumatol. 2019;71:991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaidya B, Bhochhibhoya M, Nakarmi S. Synovial fluid uric acid level aids diagnosis of gout. Biomed Rep. 2018;9:60–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Towiwat P, Chhana A, Dalbeth N. The anatomical pathology of gout: a systematic literature review. BMC Musculoskelet Disord. 2019;20:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tin A, Marten J, Halperin Kuhns VL, Li Y, Wuttke M, Kirsten H, et al. Target genes, variants, tissues and transcriptional pathways influencing human serum urate levels. Nat Genet. 2019;51:1459–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neogi T, Jansen TL, Dalbeth N, Fransen J, Schumacher HR, Berendsen D, et al. 2015 Gout Classification Criteria. Arthritis Rheum. 2015;67:2557–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wozniak JM, Mills RH, Olson J, Caldera JR, Sepich-Poore GD, et al. Mortality Risk Profiling of Staphylococcus aureus Bacteremia by Multi-omic Serum Analysis Reveals Early Predictive and Pathogenic Signatures. Cell. 2020;182:1311–1327.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shrivastava S, Chelluboina S, Jedge P, Doke P, Palkar S, Mishra AC, et al. Elevated Levels of Neutrophil Activated Proteins, Alpha-Defensins (DEFA1), Calprotectin (S100A8/A9) and Myeloperoxidase (MPO) Are Associated With Disease Severity in COVID-19 Patients. Front Cell Infect Microbiol. 2021;11:751232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Savage MJ, Iqbal M, Loh T, Trusko SP, Scott R, Siman R. Cathepsin G: localization in human cerebral cortex and generation of amyloidogenic fragments from the beta-amyloid precursor protein. Neuroscience. 1994;60:607–19. [DOI] [PubMed] [Google Scholar]
- 17.Scott FL, Hirst CE, Sun J, Bird CH, Bottomley SP, Bird PI. The intracellular serpin proteinase inhibitor 6 is expressed in monocytes and granulocytes and is a potent inhibitor of the azurophilic granule protease, cathepsin G. Blood. 1999;93:2089–97. [PubMed] [Google Scholar]
- 18.Ai M, Cui Y, Sy MS, Lee DM, Zhang LX, Larson KM, et al. Anti-lubricin monoclonal antibodies created using lubricin-knockout mice immunodetect lubricin in several species and in patients with healthy and diseased joints. PLoS One. 2015;10:e0116237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Dell JR, Brophy MT, Pillinger MH, Neogi T, Palevsky PM, Wu H, et al. Comparative Effectiveness of Allopurinol and Febuxostat in Gout Management. NEJM Evid. 2022; 1: 10.1056/evidoa2100028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xuan F, Yano F, Mori D, Chijimatsu R, Maenohara Y, Nakamoto H, et al. Wnt/β-catenin signaling contributes to articular cartilage homeostasis through lubricin induction in the superficial zone. Arthritis Res Ther. 2019;21:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hogg PJ, Owensby DA, Chesterman CN. Thrombospondin 1 is a tight-binding competitive inhibitor of neutrophil cathepsin G. Determination of the kinetic mechanism of inhibition and localization of cathepsin G binding to the thrombospondin 1 type 3 repeats. J Biol Chem. 1993;268:21811–8. [PubMed] [Google Scholar]
- 22.Rowczenio DM, Trojer H, Russell T, Baginska A, Lane T, Stewart NM, et al. Clinical characteristics in subjects with NLRP3 V198M diagnosed at a single UK center and a review of the literature. Arthritis Res Ther. 2013;15:R30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuemmerle-Deschner JB, Verma D, Endres T, Broderick L, de Jesus AA, Hofer F, et al. Clinical and Molecular Phenotypes of Low-Penetrance Variants of NLRP3: Diagnostic and Therapeutic Challenges. Arthritis Rheumatol. 2017;69:2233–2240. [DOI] [PubMed] [Google Scholar]
- 24.Lord MS, Melrose J, Day AJ, Whitelock JM. The Inter-α-Trypsin Inhibitor Family: Versatile Molecules in Biology and Pathology. J Histochem Cytochem. 2020;68:907–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turato C, Biasiolo A, Pengo P, Frecer V, Quarta S, Fasolato S, et al. Increased antiprotease activity of the SERPINB3 polymorphic variant SCCA-PD. Exp Biol Med (Maywood).2011;236:281–90. [DOI] [PubMed] [Google Scholar]
- 26.Sivaprasad U, Kinker KG, Ericksen MB, Lindsey M, Gibson AM, Bass SA, et al. SERPINB3/B4 contributes to early inflammation and barrier dysfunction in an experimental murine model of atopic dermatitis. J Invest Dermatol. 2015;135:160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turato C, Ruvoletto MG, Biasiolo A, Frecer V, Quarta S, Fasolato S, et al. Squamous cell carcinoma antigen-1 (SERPINB3) polymorphism in chronic liver disease. Dig Liver Dis. 2009;41:212–6. [DOI] [PubMed] [Google Scholar]
- 28.Elsaid KA, Jay GD, Chichester CO. Reduced expression and proteolytic susceptibility of lubricin/superficial zone protein may explain early elevation in the coefficient of friction in the joints of rats with antigen-induced arthritis. Arthritis Rheum. 2007;56:108–16. [DOI] [PubMed] [Google Scholar]
- 29.Svala E, Jin C, Rüetschi U, Ekman S, Lindahl A, Karlsson NG, et al. Characterisation of lubricin in synovial fluid from horses with osteoarthritis. Equine Vet J. 2017;49:116–123. [DOI] [PubMed] [Google Scholar]
- 30.Chhana A, Pool B, Wei Y, Choi A, Gao R, Munro J, et al. Human Cartilage Homogenates Influence the Crystallization of Monosodium Urate and Inflammatory Response to Monosodium Urate Crystals: A Potential Link Between Osteoarthritis and Gout. Arthritis Rheumatol. 2019;71:2090–2099 [DOI] [PubMed] [Google Scholar]
- 31.Chhana A, Lee G, Dalbeth N. Factors influencing the crystallization of monosodium urate: a systematic literature review. BMC Musculoskelet Disord. 2015;16:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qadri M, Jay GD, Zhang LX, Wong W, Reginato AM, Sun C, et al. Recombinant human proteoglycan-4 reduces phagocytosis of urate crystals and downstream nuclear factor kappa B and inflammasome activation and production of cytokines and chemokines in human and murine macrophages. Arthritis Res Ther. 2018;20:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gibbings S, Elkins ND, Fitzgerald H, Tiao J, Weyman ME, Shibao G, et al. Xanthine oxidoreductase promotes the inflammatory state of mononuclear phagocytes through effects on chemokine expression, peroxisome proliferator-activated receptor-{gamma} sumoylation, and HIF-1a. J Biol Chem. 2011;286:961–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qadri M, Jay GD, Zhang LX, Schmidt TA, Totonchy J, Elsaid KA. Proteoglycan-4 is an essential regulator of synovial macrophage polarization and inflammatory macrophage joint infiltration. Arthritis Res Ther. 2021; 23:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Terkeltaub R What makes gouty inflammation so variable? BMC Med. 2017;15:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson JL, Ramadass M, Haimovich A, McGeough MD, Zhang J, Hoffman HM, et al. Increased Neutrophil Secretion Induced by NLRP3 Mutation Links the Inflammasome to Azurophilic Granule Exocytosis. Front Cell Infect Microbiol. 2017;7:507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen YH, Hsieh SC, Chen WY, Li KJ, Wu CH, Wu, et al. Spontaneous resolution of acute gouty arthritis is associated with rapid induction of the anti-inflammatory factors TGFβ1, IL-10 and soluble TNF receptors and the intracellular cytokine negative regulators CIS and SOCS3. Ann Rheum Dis. 2011;70:1655–63. [DOI] [PubMed] [Google Scholar]
- 38.Rhee DK, Marcelino J, Baker M, Gong Y, Smits P, Lefebvre V, et al. The secreted glycoprotein lubricin protects cartilage surfaces and inhibits synovial cell overgrowth. J Clin Invest. 2005;115:622–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alquraini A, Jamal M, Zhang L, Schmidt T, Jay GD, Elsaid KA. The autocrine role of proteoglycan-4 (PRG4) in modulating osteoarthritic synoviocyte proliferation and expression of matrix degrading enzymes. Arthritis Res Ther.2017;19:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qadri M, Jay GD, Zhang LX, Wong W, Reginato AM, Sun C, et al. Recombinant human proteoglycan-4 reduces phagocytosis of urate crystals and downstream nuclear factor kappa B and inflammasome activation and production of cytokines and chemokines in human and murine macrophages. Arthritis Res Ther. 2018;20:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.ElSayed S, Jay GD, Cabezas R, Qadri M, Schmidt TA, Elsaid KA. Recombinant Human Proteoglycan 4 Regulates Phagocytic Activation of Monocytes and Reduces IL-1β Secretion by Urate Crystal Stimulated Gout PBMCs. Front Immunol. 2021;12:771677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bousoik E, Qadri M, Elsaid KA. CD44 Receptor Mediates Urate Crystal Phagocytosis by Macrophages and Regulates Inflammation in A Murine Peritoneal Model of Acute Gout. Sci Rep. 2020;10:5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang S, Thomsson KA, Jin C, Alweddi S, Struglics A, Rolfson O, et al. Cathepsin g Degrades Both Glycosylated and Unglycosylated Regions of Lubricin, a Synovial Mucin. Sci Rep. 2020;10:4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murphy-Ullrich JE, Suto MJ. Thrombospondin-1 regulation of latent TGF-β activation: A therapeutic target for fibrotic disease. Matrix Biol. 2018;68–69:28–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jones AR, Flannery CR. Bioregulation of lubricin expression by growth factors and cytokines. Eur Cell Mater. 2007;13:40–5. [DOI] [PubMed] [Google Scholar]
- 46.Alquraini A, Jamal M, Zhang L, Schmidt T, Jay GD, Elsaid KA. The autocrine role of proteoglycan-4 (PRG4) in modulating osteoarthritic synoviocyte proliferation and expression of matrix degrading enzymes. Arthritis Res Ther. 2017;19:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vedder D, Gerritsen M, Nurmohamed MT, van Vollenhoven RF, Lood C. A neutrophil signature is strongly associated with increased cardiovascular risk in gout. Rheumatology (Oxford). 2021;60:2783–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schauer C, Janko C, Munoz LE, Zhao Y, Kienhöfer D, Frey B, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med. 2014;20:511–7. [DOI] [PubMed] [Google Scholar]
- 49.Jin C, Ekwall AK, Bylund J, Björkman L, Estrella RP, Whitelock JM, et al. Human synovial lubricin expresses sialyl Lewis × determinant and has L-selectin ligand activity. J Biol Chem. 2012;287:35922–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao S, Zhu H, Zuo X, Luo H. Cathepsin G and Its Role in Inflammation and Autoimmune Diseases. Arch Rheumatol. 2018;33:498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.