

Abstract
Sulfur is an essential element for a variety of cellular constituents in all living organisms and adds considerable functionality to a wide range of biomolecules. The pathways for incorporating sulfur into central metabolites of the cell such as cysteine, methionine, cystathionine, and homocysteine have long been established. Furthermore, the importance of persulfide intermediates during the biosynthesis of thionucleotide-containing tRNAs, iron-sulfur clusters, thiamin diphosphate, and the molybdenum cofactor are well known. This review briefly surveys these topics while emphasizing more recent aspects of sulfur metabolism that involve unconventional biosynthetic pathways. Sacrificial sulfur transfers from protein cysteinyl side chains to precursors of thiamin and the nickel-pincer nucleotide (NPN) cofactor are described. Newer aspects of synthesis for lipoic acid, biotin, and other compounds are summarized, focusing on the requisite iron-sulfur cluster destruction. Sulfur transfers by using a non-core sulfide ligand bound to a [4Fe-4S] cluster are highlighted for generating certain thioamides and for alternative biosynthetic pathways of thionucleotides and the NPN cofactor. Thioamide formation by activating an amide oxygen atom via phosphorylation also is illustrated. The discussion of these topics stresses the chemical reaction mechanisms of the transformations and generally avoids comments on the gene/protein nomenclature or the sources of the enzymes. This work sets the stage for future efforts to decipher the diverse mechanisms of sulfur incorporation into biological molecules.
Keywords: sulfur metabolism, persulfide, thionucleotide, iron-sulfur cluster, thiocarboxylate, sacrificial sulfur transfer, thioamide, biosynthesis
Introduction
This review highlights selected research findings from the past decade that reveal novel mechanisms of sulfur incorporation into biological molecules. After summarizing general aspects of central sulfur metabolism in cells (Kessler 2006; Black and Dos Santos 2015), the formation of persulfides and their use as intermediates in well-established biosynthetic pathways are briefly described. The focus then turns to recently identified alternative mechanisms used to incorporate sulfur into biomolecules. For example, sacrificial sulfur donation from protein cysteinyl side chains and the transfer of core or non-core sulfide atoms associated with [4Fe-4S] clusters are explained. Throughout the text, the emphasis concerns the chemical mechanisms of the transformations, rather than dwelling on the diverse gene and gene product nomenclature, the functions of the sulfur-containing chemical species, or the disparate properties of the enzymes from different sources. This discussion of sulfur-containing biomolecules is not intended to be comprehensive; other reviews describe the incorporation of sulfur into various sugars, metabolites from marine organisms, biomolecules in plants, and other natural products along with the properties of these sulfur-containing compounds (Jiang et al. 2011; Lin et al. 2013; Dunbar et al. 2017; Francioso et al. 2020; Nakai and Maruyama-Nakashita 2020; Wang N et al. 2020; Hill et al. 2022).
Central sulfur metabolism
Figure 1 provides an overview of the pathways for incorporating sulfide into central metabolites of cells, emphasizing the production of L-cysteine, L-cystathionine, L-homocysteine, and L-methionine.
Figure 1.

Selected aspects of central sulfur metabolism. The reactions are catalyzed by: 1, L-serine O-acetyltransferase [EC:2.3.1.30]; 2, O-acetyl-L-serine sulfhydrylase or cysteine synthase [EC:2.5.1.47]; 3, L-homoserine O-acetyltransferase [EC:2.3.1.31] or L-homoserine O-succinyltransferase [EC:2.3.1.46]; 4, ATP:L-homoserine O-phosphotransferase or L-homoserine kinase [EC:2.7.1.39]; 5, cystathionine γ-synthase [EC:2.5.1.48]; 6, cystathionine γ-lyase [EC:4.4.1.1]; 7, L-cysteine-S-conjugate β-lyase or cystathionine β-lyase [EC:4.4.1.13]; 8, cystathionine β-synthase [EC:4.2.1.22]; 9, O-acetyl-L-homoserine:H2S S-(3-amino-3-carboxypropyl)transferase [EC:2.5.1.49] or O-succinyl-L-homoserine:H2S S-(3-amino-3-carboxypropyl)transferase [EC:2.5.1.-]; 10, O-acetyl-L-homoserine acetate-lyase (adding methanethiol) [EC:2.5.1.49]; 11, 5-methyltetrahydrofolate:L-homocysteine methyltransferase or L-methionine synthase [EC:2.1.1.13]; 12, adenosyl-S-methionine synthetase [EC:2.5.1.6]; 13, L-cysteine:2-oxoglutarate aminotransferase [EC:2.6.1.3].
L-Cysteine biosynthesis
In plants and bacteria, L-serine O-acetyltransferase uses acetyl-coenzyme A (CoA) to convert L-serine into O-acetyl-L-serine. This compound is then used as a substrate of cysteine synthase, a pyridoxal phosphate (PLP)-dependent enzyme that incorporates hydrogen sulfide with release of acetate. In animals, however, this de novo pathway for synthesis of L-cysteine does not exist. Rather, L-cysteine is formed by a two-step transsulfuration pathway in which (i) cystathionine β-synthase combines L-serine with L-homocysteine to form L-cystathionine, and (ii) cystathionine γ-lyase (also known as γ-cystathionase) acts on this intermediate to generate L-cysteine along with 2-oxobutyrate and ammonia (Aiken et al. 2011). Of interest, both of these enzymes also possess PLP and are structurally related to cysteine synthase (Christen and Mehta 2001).
L-Cystathionine biosynthesis
In addition to its synthesis from L-homoserine, the central sulfur-containing metabolite L-cystathionine is made by PLP-dependent cystathionine γ-synthase using L-cysteine and either O-succinyl-L-homoserine, O-acetyl-L-homoserine, or O-phospho-L-homoserine (depending on the enzyme source). These activated L-homoserine substrates are generated from L-homoserine using the appropriate acyl-CoA and acyltransferase or, in plants, by using ATP and the designated kinase.
L-homocysteine biosynthesis
In many cells, O-acyl-L-homoserine undergoes direct sulfurylation with sulfide to form L-homocysteine via 3-amino-3-carboxypropyltransferase reactions (Brewster et al. 2021). In other organisms, L-homocysteine is generated from L-cystathionine by action of the PLP-dependent enzyme cystathionine β-lyase with release of ammonia and pyruvate.
L-Methionine biosynthesis
In some cells, L-methionine is generated directly from O-acyl-L-homoserine by enzymes that utilize methane thiol. More commonly, the vitamin B12-dependent methionine synthase forms L-methionine by methyl transfer from 5-methyl-tetrahydrofolate to L-homocysteine.
Further general metabolism using the central sulfur-containing compounds
L-Cysteine and L-methionine are substrates for aminoacyl tRNA synthetases, which generate aminoacylated tRNAs that are used by ribosomes to make proteins. These amino acids also serve as precursors of a vast number other sulfur-containing compounds in cells; as two examples, L-cysteine is transformed into 3-mercaptopyruvate by a PLP-dependent aminotransferase and L-methionine is converted to S-adenosyl-L-methionine (SAM) by a vitamin B12-dependent methyltransferase (Figure 1).
Persulfide biosynthesis and utilization
Persulfides are widely used sulfur donors as well as being cellular signaling molecules (Mueller 2006; Park et al. 2015; Cuevasanta et al. 2017; Filipovic et al. 2018; Benchoam et al. 2020). The posttranslational modification of a protein thiol to a persulfide is known as sulfhydration, with aberrant incorporation being associated with pathological conditions (Paul and Snyder 2015). This section describes several reactions that generate persulfide compounds in cells and then summarizes the intensively studied biosynthetic pathways that utilize persulfides as sulfur donors.
Persulfide synthesis
One widespread and extensively studied pathway for persulfide synthesis involves cysteine desulfurase (Figure 2A), a PLP-dependent enzyme that converts L-cysteine to alanine and an enzyme-bound persulfide (Mihara and Esaki 2002). This reaction was discovered when characterizing the biosynthesis of the metallocluster of nitrogenase (Zheng L et al. 1993). The terminal or sulfane sulfur atom of the enzyme persulfide can then be transferred to cysteine residues of sulfur-transfer proteins or to small molecule thiols such as L-cysteine, CoA, and glutathione to form the corresponding persulfides. A similar two-step sequence occurs for the cofactor-free 3-mercaptopyruvate sulfurtransferase (Figure 2B) that converts its substrate into pyruvate and an enzyme-bound persulfide (Lec et al. 2018), with the sulfane sulfur subsequently transferred to make other persulfides (Kimura et al. 2017; Pedre and Dick 2021). Furthermore, thiosulfate sulfurtransferase or rhodanese (Figure 2C) can metabolize thiosulfate to form sulfite and an enzyme-persulfide that, again, undergoes sulfane sulfur transfer such as that associated with glutathione conversion to glutathione persulfide (Melideo et al. 2014). Rhodanese plays several other functions in the cell including its first identified role involving the detoxification of cyanide by conversion to thiocyanide (Kruithof et al. 2020). The PLP-containing enzymes cystathionine γ-lyase and cystathionine β-synthase catalyze side reactions that directly convert the oxidized form of L-cysteine (i.e., cystine) to L-cysteine persulfide, pyruvate, and ammonium ion without the intermediacy of a protein associated cysteinyl persulfide (Figure 2D) (Ida et al. 2014). The latter enzyme also transforms homocystine into L-homocysteine persulfide, 2-oxobutyrate, and ammonium ion. The slow, non-enzymatic transformation of disulfides (or trisulfides) into persulfides as well as the conversion of sulfenic acid to persulfides by sulfide addition also have long been known (Figure 2E) (Rao and Gorin 1959; Francoleon et al. 2011; Cuevasanta et al. 2015). Of medical interest, a variety of small molecule prodrugs have been designed for the controlled generation of sulfide that could participate in such reactions (Bora et al. 2018). Once formed, the sulfane sulfur atom of persulfides can be distributed to other thiol species by transpersulfidation reactions.
Figure 2.

Persulfide generating mechanisms. (A) Cysteine desulfurase [EC:2.8.1.7] forms an enzyme cysteinyl persulfide from L-cysteine using its PLP cofactor (blue) that initially is bound to an enzyme lysyl side chain. (B) 3-Mercaptopyruvate sulfurtransferase [EC:2.8.1.2] transfers the sulfur atom of its substrate to an enzyme cysteinyl residue while producing pyruvate. (C) Thiosulfate sulfurtransferase (rhodanese) [EC:2.8.1.1] reacts with thiosulfate to generate an enzyme cysteinyl persulfide and sulfite. The enzyme-associated persulfides generated in reactions A-C donate their sulfane sulfur to protein or small molecule thiols. (D) Cystathionine γ-lyase [EC:4.4.1.1] and cystathionine β-synthase [EC:4.2.1.22] use their PLP cofactors to transform cystine to cysteine persulfide, pyruvate, and ammonium ion. (E) Non-enzymatic persulfide synthesis by reacting sulfide with a disulfide (or trisulfide) or with a sulfenic acid.
Thionucleotide biosynthesis using persulfides
In the best-studied example, bacteria synthesize 4-thiouridine (s4U) at position 8 of tRNA using an enzyme-associated persulfide (Mueller 2006; Cavuzic and Liu 2017; Leimkühler et al. 2017; Shigi 2018; Bimai et al. 2020) (Figure 3A). The nucleotide base is first activated by adenylylation with the release of pyrophosphate. A nucleophilic persulfide then attacks this electrophile to form a covalent C-S bond while releasing AMP. Finally, a catalytic cysteinyl residue generates an enzyme disulfide while releasing s4U. Reduction of the disulfide subsequently regenerates the starting form of the enzyme. Analogous reactions form 2-thiouridine (s2U) at position 34 in many organisms along with other tRNA modifications (Tanabe et al. 2019; Bimai et al. 2020; Shigi 2021), and a similar process is used to synthesize 6-thioguanosine, 6-thio-GMP, 6-thio-GDP, and 6-thio-GTP (Coyne et al. 2013; Litomska et al. 2018; Mahanta et al. 2019). As will be described in a later section, persulfides also may participate in thionucleotide biosynthesis with the intermediacy of an iron-sulfur cluster (Shigi 2021).
Figure 3.

Well-known biosynthetic pathways using persulfides. (A) 4-Thiouridine in tRNA is formed by initial activation of the precursor nucleotide using an adenylyltransferase [EC:2.7.7.73], persulfide attack on the intermediate with release of AMP catalyzed by tRNA uracil 4-sulfurtransferase [EC:2.8.1.4], and addition by an enzyme thiol to generate the product. (B) Persulfides also provide the sulfur atoms, with other sources donating iron and electrons, for generating various forms of iron-sulfur clusters. Scaffold proteins are utilized for assembly of [2Fe-2S] and [4Fe-4S] clusters prior to transfer to the target proteins. (C) The synthesis of thiamin diphosphate and the molybdenum cofactor (Moco) both initiate by adenylylation of the carboxyl terminus of proteins, attack by an enzyme persulfide, and reduction to generate protein thiocarboxylates. This key species is used by thiazole synthase [EC:2.8.1.10] to provide the sulfur atom for synthesis of thiazole phosphate, which incorporates into the vitamin B1 product, or by molybdopterin synthase [EC:2.8.1.12] catalyzing successive steps to transfer two sulfur atoms into cyclic pyranopterin monophosphate to form molybdopterin, which is converted to Moco.
Iron-sulfur cluster biosynthesis using persulfides
Another large and essential group of persulfide sulfur acceptors are proteins containing iron-sulfur clusters. The complex and highly orchestrated synthesis of iron-sulfur clusters has been extensively reviewed recently (Pérard and Ollagnier de Choudens 2018; Zheng C and Dos Santos 2018; Blahut et al. 2020; Lill and Freibert 2020; Braymer et al. 2021; Esquilin-Lebron et al. 2021; Talib and Outten 2021) and will not be repeated here. A key feature of this process is the transfer of sulfane sulfur atoms from enzymatically produced persulfides to scaffold proteins that also accept iron atoms and reducing equivalents to generate iron-sulfur clusters (Figure 3B). The transfer of sulfur from cysteine desulfurase to a scaffold protein may occur directly or it may be mediated by a dedicated zinc-dependent sulfur transferase that exhibits homology to the scaffold (Selbach et al. 2013). The initial metalloclusters are sometimes modified as they localize to their ultimate destination as [2Fe-2S], [4Fe-4S], or more complex cluster types.
Persulfide-dependent biosynthesis of thiocarboxylates
Persulfides also sometimes are used to generate thiocarboxylates (Figure 3C), which can then function as sulfur donors during the biosynthesis of other molecules (Cavuzic and Liu 2017; Leimkühler et al. 2017). In these systems, the carboxyl-terminal residue of a protein is first activated by adenylylation, then a persulfide attacks the acyl adenylylate to release AMP. The resulting acyl persulfide covalent adduct is reduced to generate the carboxyl-terminal thiocarboxylate. These steps are reminiscent of those used in thionucleotide biosynthesis. Alternatively, a cysteinyl residue may attack the adenylylated protein to form a thioester that then reacts with the persulfide to form the acyl persulfide (Termathe and Leidel 2018). The figure depicts two types of reactivity using thiocarboxylates. First, a thiocarboxylate combines with 1-deoxy-D-xylulose-5-phosphate and dehydroglycine to form thiazole phosphate during the synthesis of thiamin diphosphate (Jurgenson et al. 2009). Second, two thiocarboxylates sequentially react with cyclic pyranopterin monophosphate to generate thiopyranopterin then molybdopterin during synthesis of the molybdenum cofactor (Moco) (Mayr et al. 2021). Thiocarboxylate-dependent sulfur donation also is associated with the biosynthesis of other molecules (not shown). Eukaryotes and archaea use thiocarboxylates during the synthesis of s2U, 5-methyl-s2U, 5-methoxycarbonylmethyl-s2U tRNAs (Cavuzic and Liu 2017; Leimkühler et al. 2017). Furthermore, thiocarboxylates are utilized when making several small molecules including the bacterial siderophores thioquinolobactin (Godert et al. 2007) and pyridine dithiocarboxylic acid (Krishnamoorthy and Begley 2010) and, in particular microorganisms, the amino acids L-cysteine (Burns et al. 2005) and L-methionine (Krishnamoorthy and Begley 2011). The thiocarboxylates for some of these systems derive by processes not involving persulfides. Genes encoding proteins with a thiocarboxylate at their carboxyl termini are suggested to be widespread in bacteria where they are used to generate thiocarboxylic acid-containing natural products, e.g., thioplatensimycin and thioplatencin are generated by reacting the corresponding CoA-activated carboxylates with a thiocarboxylate-containing protein, followed by hydrolysis (Dong et al. 2018).
Other persulfide-dependent synthetic reactions
A variety of other sulfur-containing molecules are generated by reactions that utilize persulfides. Whereas the best characterized biosynthetic pathway for ergothioneine utilizes L-cysteine as a sulfur source (Cordell and Lamahewage 2022), an anaerobe generates this thiohistidine derivative using a protein persulfide to supply the sulfur atom (Figure 4A) (Cheng et al. 2020). In this case, the protein persulfide is formed by using polysulfide. An example of sulfur incorporation into a large molecule (Figure 4B) is the replacement of a nonbridging oxygen atom of phosphate in DNA, a process known as phosphorothioation. In this incompletely detailed process, a protein persulfide is generated and donates the sulfane sulfur atom to DNA with the participation of a protein containing a [4Fe-4S], an ATP pyrophosphatase (hinting at substrate activation by adenylylation), and an ATPase (Wang L et al. 2019; Liu L et al. 2020; Pu et al. 2020). A final example is the sulfhydrylation of tRNA-bound O-phosphoserine (Sep) to generate cysteinyl-tRNA (Figure 4C). This process occurs by initial PLP-catalyzed β-elimination of phosphate while forming tRNA-bound dehydroalanine (Dha) followed by attack using a sulfide generated from a protein persulfide (Liu Y et al. 2012).
Figure 4.

Other persulfide-dependent sulfur incorporation reactions. (A) Anaerobic ergothioneine biosynthesis. (B) Phosphorothioation of DNA. (C) Synthesis of Cys-tRNA from O-phosphoserine-bound tRNA.
Sacrificial sulfur transfer pathways
The following subsections describe recent examples in which sulfur is provided for incorporation into biomolecules by the sacrifice of protein cysteinyl residues or the destruction of iron-sulfur clusters.
Cysteinyl side chains as sulfur donors
Two cases illustrate the transfer of a sulfur atom from a protein side chain into a metabolite. In the first example, yeast thiamin thiazole synthase catalyzes a remarkable sequence of reactions (Figure 5A) including the release of nicotinamide from NAD+, ribose ring opening, tautomerization, glycine addition, tautomerization, and dehydration. A sulfur atom then adds from Cys205 of the enzyme in a ferrous ion-dependent reaction, resulting in an inactive enzyme containing Dha at this position (Chatterjee A et al. 2011). Such a result is consistent with attack on the electrophilic carbon atom by cysteine, with sulfur transfer being accompanied by deprotonation of the α-carbon backbone. Further dehydration and tautomerization of the intermediate yields adenylated thiazole (ADT), which is further converted by other enzymes into thiamin diphosphate. The gene encoding this enzyme with its key cysteine residue is conserved among eukaryotes where the protein appears to be used as a reagent rather than an enzyme. Curiously, the ortholog from a methanogenic archaeon catalyzes an essentially identical series of reactions, but it catalytically incorporates inorganic sulfide into the product rather than being limited to a single turnover (Eser et al. 2016). Notably, this microorganism lives in sulfide-rich environments, and it possesses a histidine residue at the position corresponding to Cys205 in the yeast protein.
Figure 5.
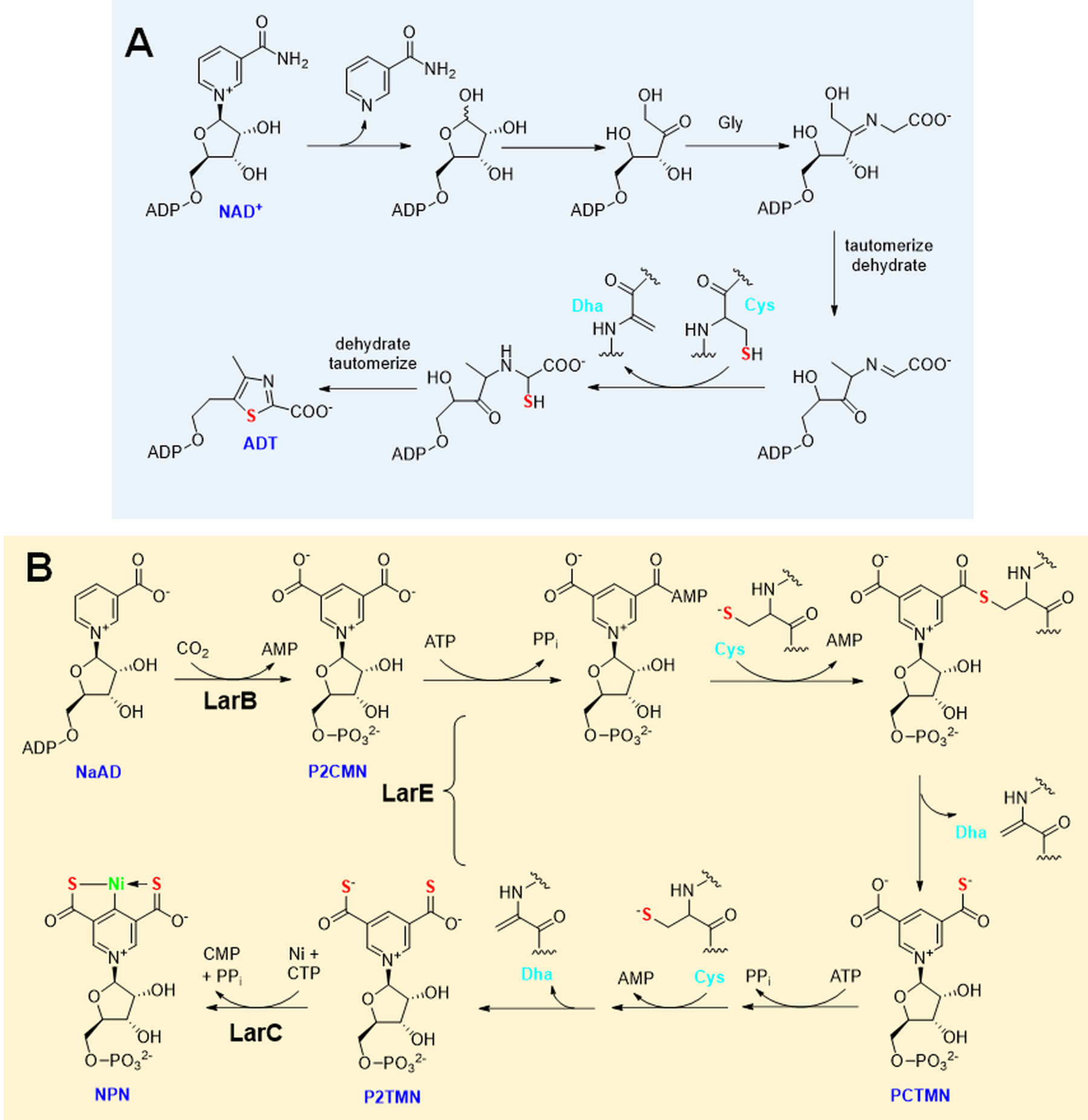
Sulfur donation from a protein cysteinyl residue. (A) Thiamin thiazole synthase [EC:2.4.2.60] converts NAD+ plus glycine into adenylated thiazole (ADT) by a complex series of steps including the transfer of a sulfur atom from one of its cysteinyl residues, yielding an inactive protein containing dehydroalanine (Dha). (B) Nickel-pincer nucleotide (NPN) cofactor biosynthesis is catalyzed by the sequential actions of pyridinium-3,5-biscarboxylic acid mononucleotide synthase or LarB [EC:2.5.1.143], pyridinium-3,5-bisthiocarboxylic acid mononucleotide synthase or LarE [EC:4.4.1.37], and pyridinium-3,5-bisthiocarboxylic acid mononucleotide nickel chelatase or LarC [EC:4.99.1.12], with the cysteinyl sulfur atoms from two molecules of LarE used to convert P2CMN to P2TMN in ATP-dependent reactions that yield Dha residues in the protein molecules.
The second example of a protein side chain being used for sacrificial sulfur incorporation involves one class of LarE sulfur insertase that participates in the biosynthesis of the nickel-pincer nucleotide (NPN) cofactor (Nevarez et al. 2020). The synthesis of NPN requires three enzymes (Figure 5B), with LarB first converting nicotinic acid dinucleotide (NaAD) to pyridinium-3,5-biscarboxylic acid mononucleotide (P2CMN) in a reaction that incorporates carbon dioxide and releases AMP (Rankin et al. 2021). Two molecules of LarE are needed to sequentially transform P2CMN first into pyridinium-3-carboxy-5-thiocarboxylic acid mononucleotide (PCTMN) and then into pyridinium-3,5-bisthiocarboxylic acid mononucleotide (P2TMN) by catalyzing two cycles of a process involving substrate adenylylation, protein cysteinyl-substrate thioester formation, and sulfur transfer with formation of a Dha residue (Fellner et al. 2017). The resulting Dha-containing form of LarE is inactive, but the native protein can be recovered by incubation with coenzyme A persulfide followed by reduction (Fellner et al. 2018); however, it remains uncertain whether this recycling reaction occurs in vivo. Insertion of nickel into P2TMN to obtain NPN is catalyzed by LarC, a CTP-dependent enzyme (Desguin et al. 2018; Turmo et al. 2022).
Sacrificial iron-sulfur clusters
A few metabolites obtain their sulfur atoms by destruction of iron-sulfur clusters (Jarrett 2015; Lanz and Booker 2015; Mulliez et al. 2017). One such example is the coenzyme biotin produced by biotin synthase, an enzyme that uses a [4Fe-4S] cluster as a SAM-binding site for initiating radical-based chemistry and an auxiliary [2Fe-2S] cluster that provides the sulfur atom for cofactor biosynthesis (Bui et al. 1998; Ugulava et al. 2001; Jameson et al. 2004). Reductive cleavage of SAM yields methionine and a 5’-deoxyadenosine radical that abstracts a hydrogen atom from C9 of desthiobiotin to yield 5’deoxyadenosine (5’-dAH) (Figure 6A). The resulting C9 radical reacts with a μ-sulfur atom in the [2Fe-2S] cluster yielding cluster-associated 9-mercaptodesthiobiotin that also accepts an electron from a donor. The resulting paramagnetic intermediate has been extensively characterized by pulse EPR spectroscopy and quantum chemical modeling (Fugate et al. 2012; Tao et al. 2018). Consumption of a second molecule of SAM results in release of Met and 5’-dAH along with abstraction of the C6 hydrogen atom with the resulting radical reacting with the same μ-sulfur, forming the thiophane ring and a defective [2Fe-1S] cluster. Dissociation of this aberrant cluster followed by reassembly of the [2Fe-2S] cluster is required for reconstitution of activity.
Figure 6.
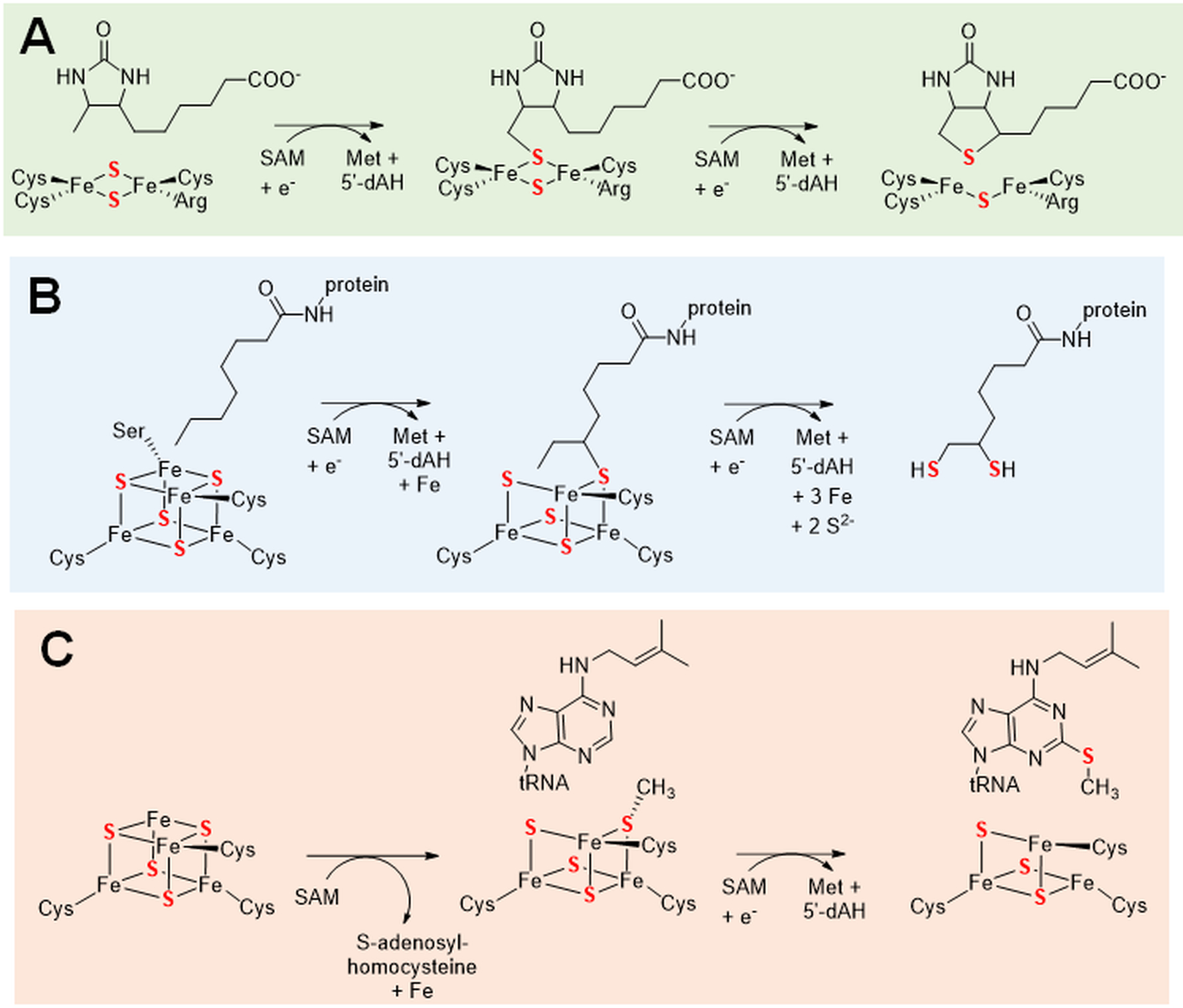
Sulfur donation accompanying destruction of an iron-sulfur cluster. The reactions are shown for (A) biotin synthase or BioB [EC:2.8.1.6], lipoyl synthase or LipA [EC:2.8.1.8], and tRNA-2-methylthio-N6-dimethylallyladenosine synthase or MiaB [EC:2.8.4.3]. Each of these enzymes also possesses a SAM-binding [4Fe-4S] cluster bound by three cysteine residues.
A second example of coenzyme production accompanied by iron-sulfur cluster destruction occurs during the biosynthesis of the protein-bound lipoyl group (Cronan 2016). This process begins by LipB-catalyzed covalent attachment of lipoic acid via amide linkage to a lysyl residue of the lipoyl transfer protein. The derivatized protein is then used as the substrate for lipoyl synthase, LipA, containing two [4Fe-4S] clusters. The structure of LipA revealed a channel separating the two clusters allowing for the close juxtapositioning of SAM and the octanoyl sidechain located between the clusters (Harmer et al. 2014). One of these clusters sequentially binds two molecules of SAM and facilitates their reductive cleavage, whereas the second [4Fe-4S] cluster provides the source of the two sulfur atoms (Figure 6B). The initially formed 5’-deoxyadenosine radical abstracts a C6 hydrogen atom of the substrate (Lanz et al. 2014), with the carbon centered radical adding to one sulfur atom of the auxiliary cluster, resulting in the loss of an iron atom (specifically the one possessing an unusual Ser ligand). The crystal structure of this intermediate state and that of the resting enzyme have been described in a different LipA (McLaughlin et al. 2016). A second round of SAM cleavage leads to abstraction of the C8 hydrogen atom, which adds to a second sulfur atom of the auxiliary cluster. Release of the dithiol product is accompanied by disassembly of the cluster. The cluster is reformed using the cluster carrier proteins NfuA or IscU (McCarthy and Booker 2017). The overall process of single turnover product formation followed by cluster reconstitution is analogous to what was described for biotin synthase.
Three related proteins, RimO, MiaB, and MtaB, are methylthiotransferases containing SAM-binding [4Fe-4S] and auxiliary [4Fe-4S] clusters, each with three cysteinyl ligands, where the latter clusters are cannibalized to provide the sulfur atom. Destruction of the auxiliary clusters might be expected to allow for only a single turnover reaction in vitro; however, product ratios greater than one have been reported for RimO and MiaB (Forouhar et al. 2013). These proteins transfer a methylthio group to a particular aspartyl residue on protein S12 of the ribosome 23 S subunit, the C2 position of 6-isopentenyladenosine in tRNA, and the C2 atom of 6-threonyladenosine in tRNA, respectively (Jarrett 2015). Biochemical and structural studies of RimO and MtaB are consistent with a mechanism in which (i) SAM binds to cluster-1 and alkylates a sulfur atom of cluster-2 while releasing S-adenosylhomocysteine and (ii) a second molecule of SAM binds to cluster-1, undergoes reductive cleavage to generate methionine and the 5’-deoxyadenosyl radical, the latter abstracts a hydrogen atom from substrate, and the methylthio group is transferred to form the product (Forouhar et al. 2013; Landgraf et al. 2013; Maiocco et al. 2016; Molle et al. 2016). Of particular interest, recent studies of MiaB revealed that methylation of the auxiliary cluster leads to loss of an iron atom, converting it to a [3Fe-4S] cluster with unusual Mössbauer spectroscopic properties that are consistent with methylation of a core sulfide (Zhang et al. 2020). Subsequent structural studies identified additional intermediates in the enzymatic process (Esakova et al. 2021). A simplified view of the reaction mechanism proposed for methylthiotransferases is illustrated for MiaB (Figure 6C).
Sulfur incorporation using non-core sulfides of iron-sulfur clusters
Selected sulfur insertion enzymes bind [4Fe-4S] clusters using three cysteine residues and possess a transferable sulfide on the iron atom with an open coordination site. This process was first unveiled and is best characterized for selected enzymes participating in tRNA thionucleotide synthesis (Bimai et al. 2020; Shigi 2021), a process that is distinct from previously described persulfide-based mechanisms (Cavuzic and Liu 2017; Leimkühler et al. 2017; Shigi 2018; Esakova et al. 2021). An early study revealed that E. coli catalyzes C2 thiolation of cytosine in tRNA using an enzyme that contains an essential oxygen-labile cluster (Bouvier et al. 2014). A mechanism involving sulfur transfer of a noncubane sulfide ligand was postulated, but not demonstrated. A report describing enzymes used for synthesis of tRNA containing s2U or s4U in a methanogen and of s2U in eukaryotes suggested they possessed [3Fe-4S] clusters (Liu Y et al. 2016), but a case was made later that these enzymes contained damaged a metallocenter derived from oxygen-labile [4Fe-4S] clusters (Bimai et al. 2020). The s4U tRNA synthase from a methanogen was able to use exogenous sulfide as a sulfur source (Liu Y et al. 2016), consistent with an extra-cubane association of the sulfide with the cluster. Synthesis of 5-methyl-s2U containing tRNA in a thermophile involves two proteins: TtuA contains a PP-loop domain for activating the substrate and an oxygen-labile [4Fe-4S] cluster, while TtuB is a sulfur transfer protein with a thiocarboxylate C-terminus (Chen et al. 2017). The structure of the TtuA holoprotein in the presence of an ATP analog confirmed the presence of a [4Fe-4S] cluster bound by three cysteine residues and demonstrated the nucleotide was positioned nearby. Furthermore, the structure of a TtuA∙TtuB complex revealed the proximity of the TtuB thiocarboxylate with the open coordination site on the [4Fe-4S] cluster (Chen et al. 2020). These authors proposed a model in which the TtuB thiocarboxylate donates its sulfide to the [4Fe-4S] cluster, with the non-cuboidal sulfide then attacking the activated substrate (Figure 7A). Direct structural evidence for an extra-cubane sulfide bound to a [4Fe-4S] cluster was obtained for TtuA from a thermophilic archaeon (Arragain et al. 2017). That enzyme uses inorganic sulfide (abundant in the microorganism’s environment) as the sulfur source, rather than thiocarboxylated TtuB, and the authors proposed a more simplified sulfur insertion mechanism (Figure 7B). Similar reactions are also likely to be used by bacterial TtuA (Arragain et al. 2017), s2U synthases (Shigi et al. 2020; Zhou, Lénon, et al. 2021), and probably by a broad range of other related enzymes (Bimai et al. 2020; Shigi et al. 2020).
Figure 7.

Sulfur transfer by proteins with non-core sulfides on [4Fe-4S] clusters. (A) A tRNA-5-methyluridine 2-sulfurtransferase [EC:2.8.1.15] uses TtuA to activate the substrate by adenylylation and has a [4Fe-4S] cluster that accepts sulfide from thiocarboxylated TtuB. (B) Another TtuA enzyme uses sulfide as the sulfur source and does not require a TtuB component. (C) The sulfuration of P2CMN to the mono-thiocarboxylated species and then to P2TMN by a class of LarE [EC:4.4.1.37] uses a sulfur atom from a non-core position of a [4Fe-4S] cluster. (D) Iterative thioamidation during synthesis of closthioamide is carried out by a [4Fe-4S] cluster-containing enzyme (CtaC) using a substrate bound to CtaE and requires ATP.
Still involving thiolated bases, but operating in the reversed direction, a [4Fe-4S] cluster with an extra noncore sulfide ligand was structurally characterized for a desulfidase that uses 2- or 4-thiouracil, rather than thionucleotide-containing tRNA (Zhou, Pecqueur, et al. 2021). The mechanism postulated for this enzyme invokes (i) attack of water on the thiocarbonyl carbon atom accompanied by formation of a sulfur bond to the cluster, (ii) generation of the product carbonyl group while forming a [4Fe-4S] cluster with a noncubane sulfide, and (iii) release of the sulfide into solution. An L-cysteine desulfidase from a methanogenic archaeon likely operates by a related reaction: the substrate thiolate coordinates the available iron atom of the [4Fe-4S] cluster, the C2 proton is abstracted, aminoacrylate forms (later decomposing to pyruvate and ammonium ion) as the sulfide transfers to a non-core position of the cluster, and the sulfide is released into solution (Tchong et al. 2005).
Another example where a non-core sulfide of a [4Fe-4S] cluster is used for sulfur assimilation occurs during NPN biosynthesis in some microorganisms. As described earlier, one class of LarE sacrifices a cysteinyl sulfur atom while converting P2CMN first into PCTMN and then into P2TMN (Desguin et al. 2016; Fellner et al. 2017), thus requiring two copies of the protein. A second class of LarE avoids this energetically costly reaction by using a noncore sulfide bound to a [4Fe-4S] cluster (coordinated by three cysteinyl ligands) to insert into the substrates after they are activated by adenylylation (Chatterjee S et al. 2022) (Figure 7C). L-cysteine provides the extra sulfide to this class of LarE via cysteine desulfurase.
A final illustration of this [4Fe-4S]-S− cluster mechanism is associated with the formation of closthioamide, a symmetrical antibiotic containing six thioamide bonds (Dunbar et al. 2019; Dunbar et al. 2020). The multi-step non-ribosomal biosynthesis pathway includes the CtaC-catalyzed iterative thioamidation of a precursor molecule bound to CteE (Figure 7D). Mutational studies allowed the authors to identify three likely cluster-binding cysteinyl residues in CteC. Additional investigations demonstrated the necessity of ATP, but both AMP and ADP were detected as products with neither correlating in amount to the concentration of processed substrate. Sulfide was used as the substrate in the reaction, but the native sulfur source was not established.
Thioamide formation by amide oxygen phosphorylation
A distinct mechanism to form thioamides is used during the synthesis of a number of ribosomally-encoded antibiotics (Schwalen et al. 2018; Liu J et al. 2019; Santos-Aberturas et al. 2019) and for the creation of a thioglycine residue in the archaeal enzyme methyl-coenzyme M reductase (MCR) (Nayak et al. 2017; Mahanta et al. 2018). The most dramatic antibiotic example is thioviridamide, a twelve-residue peptide containing five thioamides along with other modifications (Izawa et al. 2013), but the chemistry is best understood for MCR, a methane-generating metalloenzyme that is the only thioamide-containing protein. The generation of these thioamides universally requires a member of the YcaO protein family, which is best known for catalyzing cyclodehydration reactions to form azoline-containing peptides. Sulfide attack on the amide carbonyl is accompanied by O-phosphorylation using ATP, with thioamide formation coupled to release of inorganic phosphate that, by 18O-labeling studies, possesses the original amide oxygen atom (Figure 8) (Burkhart et al. 2017; Mahanta et al. 2019). In most cases, a second protein, TfuA, also is required for thioamide formation. The TfuA of MCR-modifying systems has been shown to hydrolyze the thiocarboxylate of another protein, ThiS, to provide the sulfide used in this reaction.
Figure 8.

Thioamide formation during thioglycine formation by YcaO [EC: 6.2.2.1] involving sulfide attack on the amide carbonyl group, O-phosphorylation, and elimination of inorganic phosphate.
Conclusion
Our understanding of the diverse biochemistry associated with sulfur incorporation into biological molecules has advanced significantly in recent years, but the biosynthetic pathways associated with many sulfur-containing molecules remain enigmatic. To further confound the situation, alternative biosynthetic systems exist for the same compound in different organisms. For example, thionucleotides may be synthesized by pathways that provide sulfur from persulfides, but in other cases the sulfur derives from a non-core sulfide coordinated to a [4Fe-4S] cluster. Similarly, the sulfur in thiamin diphosphate may arise from a protein thiocarboxylate (generated in turn by use of a protein persulfide) or it may derive from sacrificial sulfur transfer of a protein cysteinyl side chain. The sulfur atoms in the NPN cofactor also may originate in a cysteine residue of a single-turnover protein or they may be transferred catalytically from a [4Fe-4S]-S− cluster with the noncore sulfide ultimately provided from a persulfide. Iron-sulfur clusters are destroyed to provide sulfide for biotin, lipoate, and a small number of other compounds. Also of importance, several methods are utilized for activating a substrate prior to sulfur transfer, e.g., adenylylation, phosphorylation, and use of SAM to promote radical-based chemistry. The direct reaction of a substrate with sulfide does occur in some cases such as for archaea that grow in high sulfide concentrations, but this molecule is toxic and alternative sulfur donors are common in other cells. It is hoped that the sulfur insertion themes summarized here will help guide further investigations of this topic and accelerate the identification of new pathways for sulfur incorporation.
Funding
The studies from the author’s laboratory described in review were supported by the National Science Foundation (CHE1807073) and the National Institutes of Health (GM128959).
Footnotes
Disclosure statement
The authors report no conflicts of interest.
References
- Aiken SM, Lodha PH, Morneau DJK. 2011. The enzymes of the transsulfuration pathways: Active-site characterizations. Biochim Biophys Acta. 1814:1511–1517. [DOI] [PubMed] [Google Scholar]
- Arragain S, Bimai O, Legrand P, Caillat S, Ravanat J-L, Touati N, Binet L, Atta M, Fontecave M, Golinelli-Pimpaneau B. 2017. Nonredox thiolation in tRNA occurring via sulfur activation by a [4Fe-4S] cluster. Proc Natl Acad Sci USA. 114:7355–7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benchoam D, Cuevasanta E, Möller MN, Alvarez B. 2020. Persulfides, at the crossroads between hydrogen sulfide and thiols. Essays in Biochemistry. 64:155–168. [DOI] [PubMed] [Google Scholar]
- Bimai O, Arragain S, Golinelli-Pimpaneau B. 2020. Structure-based mechanistic insights into catalysis by tRNA thiolation enzymes. Curr Opin Struct Biol. 65:69–78. [DOI] [PubMed] [Google Scholar]
- Black KA, Dos Santos PC. 2015. Shared-intermediates in the biosynthesis of thio-cofactors: Mechanism and functions of cysteine desulfurases and sulfur acceptors. Biochim Biophys Acta. 1853:1470–1480. [DOI] [PubMed] [Google Scholar]
- Blahut M, Sanchez E, Fisher CE, Outten FW. 2020. Fe-S cluster biogenesis by the bacterial Suf pathway. Biochim Biophys Acta. 1867:118829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bora P, Chauhan P, Pardeshi KA, Chakrapani H. 2018. Small molecule generators of biologically reactive sulfur species. RSC Adv. 8:27359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvier D, Labessan N, Clémancey M, Latour J-M, Ravanat J-L, Fontecave M, Atta M. 2014. TtcA a new tRNA thioltransferase with an Fe-S cluster. Nucl Acids Res. 42:7960–7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braymer JJ, Freibert SA, Rakwalska-Bange M, Lill R. 2021. Mechanistic concepts of iron-sulfur protein biogenesis in biology. Biochim Biophys Acta. 1868:118863. [DOI] [PubMed] [Google Scholar]
- Brewster JL, Pachl P, McKellar JLO, Selmer M, Squire CJ, Patrick WM. 2021. Structures and kinetics of Thermotoga maritima MetY reveal new insights into the predominant sulfurylation enzyme of bacterial methionine biosynthesis. J Biol Chem. 296:100797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui BTS, Florentin D, Fournier F, Ploux O, Mejean A, Marquet A. 1998. Biotin synthase mechanism: On the origin of sulphur. FEBS Lett. 440:226–230. [DOI] [PubMed] [Google Scholar]
- Burkhart BJ, Schwalen CJ, Mann G, Naismith JH, Mitchell DA. 2017. YcaO-dependent posttranslational amide activation: Biosynthesis, structure, and function. Chem Rev. 117:5389–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns KE, Baumgart S, Dorrestein PC, Zhai H, McLafferty FW, Begley TP. 2005. Reconstitution of a new cysteine biosynthetic pathway in Mycobacterium tuberculosis. J Am Chem Soc. 127:11602–11603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavuzic M, Liu Y. 2017. Biosynthesis of sulfur-containing tRNA modifications: A comparison of bacterial, archael, and eukaryotic pathways. Biomolecules. 7:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A, Abeydeera ND, Bale S, Pai P-J, Dorrestein PC, Russell DH, Ealick SE, Begley TP. 2011. Saccharomyces cerevisiae THI4p is a suicide thiamin thiazole synthase. Nature. 478:542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Parson KF, Ruotolo BT, McCracken J, Hu J, Hausinger RP. 2022. Characterization of a [4Fe-4S]-dependent LarE sulfur insertase that facilitates nickel-pincer nucleotide cofactor biosynthesis in Thermotoga maritima. J Biol Chem. 298:102131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Asai S-i, Narai S, Nambu S, Omura N, Sakaguchi Y, Suzuki T, Ikeda-Saito M, Watanabe K, Yao M et al. 2017. Biochemical and structural characterization of oxygen-sensitive 2-thiouridine synthesis catalyzed by an iron-sulfur protein TtuA. Proc Natl Acad Sci USA. 114:4954–4959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Ishizaka M, Narai S, Horitani M, Shigi N, Yao M, Tanaka Y. 2020. The [4Fe-4S] cluster of sulfurtransferase TtuA desulfurizes TtuB during tRNA modification in Thermus thermophilus. Commun Biol. 3:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng R, Wu L, Lai R-Y, Peng C, Naowarojna N, Hu W, Li X, Whelan SA, Lee N, Lopez J et al. 2020. Single-step replacement of an unreactive C-H bond by a C-S bond using polysulfide as the direct sulfur source in the anaerobic ergothioneine biosynthesis. ACS Catalysis. 10:8981–8994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen P, Mehta PK. 2001. From cofactor to enzymes. The molecular evolution of pyridoxal-5’-phosphate-dependent enzymes. Chem Rec. 1:436–447. [DOI] [PubMed] [Google Scholar]
- Cordell GA, Lamahewage SNS. 2022. Ergothioneine, ovothiol A, and selenoneine--histidine-derived, biologically significant, trace global alkaloids. Molecules. 27:2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne S, Chizzali C, Khalil MNA, Litomska A, Richter K, Beerhues L, Hertweck C. 2013. Biosynthesis of the antimetabolite 6-thioguanine in Erwinia amylovora plays a key role in fire blight pathogenesis. Angew Chem Int Ed. 52:10564–10568. [DOI] [PubMed] [Google Scholar]
- Cronan JE. 2016. Assembly of lipoic acid on its cognate enzymes: An extraordinary and essential biosynthetic pathway. Microbiological and Molecular Biological Reviews. 80:429–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevasanta E, Lange M, Bonanata J, Coitino EL, Ferrer-Sueta G, Filipovic MR, Alvarez B. 2015. Reaction of hydrogen sulfide with disulfide and sulfenic acid to form the strongly nucleophilic persulfide. J Biol Chem. 290:26866–26880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevasanta E, Möller MN, Alvarez B. 2017. Biological chemistry of hydrogen sulfide and persulfides. Arch Biochem Biophys. 617:9–25. [DOI] [PubMed] [Google Scholar]
- Desguin B, Fellner M, Riant O, Hu J, Hausinger RP, Hols P, Soumillion P. 2018. Biosynthesis of the nickel-pincer nucleotide cofactor of lactate racemase requires a CTP-dependent cyclometallase. J Biol Chem. 293:12303–12317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desguin B, Soumillion P, Hols P, Hausinger RP. 2016. Nickel-pincer cofactor biosynthesis involves LarB-catalyzed pyridinium carboxylation and LarE-dependent sacrificial sulfur insertion. Proc Natl Acad Sci USA. 113:5598–5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L-B, Rudolf JD, Kang D, Wang N, He CQ, Deng Y, Huang Y, Houk KN, Duan Y, Shen B. 2018. Biosynthesis of thiocarboxylic acid-containing natural products. Nat Commun. 9:2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbar KL, Dell M, Gude F, Hertweck C. 2020. Reconstitution of polythioamide antibiotic backbone formation reveals unusual thiotemplated assembly strategy. Proc Natl Acad Sci USA. 117:8850–8858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbar KL, Dell M, Molloy EM, Kloss F, Hertweck C. 2019. Reconstitution of iterative thioamidation in closthioamide biosynthesis reveals tailoring strategy for nonribosomal peptide backbones. Angew Chem Int Ed. 58:13014–13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbar KL, Scharf DH, Litomska A, Hertweck C. 2017. Enzymatic carbon-sulfur bond formation in natural product biosynthesis. Chem Rev. 117:5521–5577. [DOI] [PubMed] [Google Scholar]
- Esakova OA, Grove TL, Yennawar NH, Arcinas AJ, Wang B, Krebs C, Almo SC, Booker SJ. 2021. Structural basis for tRNA methylthiolation by the radical SAM enzyme MiaB. Nature. 597:566–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eser BE, Zhang X, Chanani PK, Ealick SE, Begley TP. 2016. From suicide enzyme to catalyst: The iron-dependent sulfide transfer in Methanococcus jannaschii thiamin thiazole biosynthesis. J Am Chem Soc. 138:3639–3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esquilin-Lebron K, Dubrac S, Barras F, Boyd JM. 2021. Bacterial approaches for assembling iron-sulfur proteins. mBio. 12:e02325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellner M, Desguin B, Hausinger RP, Hu J. 2017. Structural insights into the catalytic mechanism of a sacrificial sulfur insertase of the N-type ATP pyrophosphatase family, LarE. Proc Natl Acad Sci USA. 114:9074–9079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellner M, Rankin JA, Desguin B, Hu J, Hausinger RP. 2018. Analysis of the active site cysteine residue of the sacrificial sulfur insertase LarE from Lactobacillus plantarum. Biochemistry. 57:5513–5523. [DOI] [PubMed] [Google Scholar]
- Filipovic MR, Zivanovic J, Alvarez B, Banerjee R. 2018. Chemical biology of H2S signaling through persulfidation. Chem Rev. 118:1253–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forouhar F, Arragain S, Atta M, Gambarelli S, Mouesca J-M, Hussain M, Xiao R, Kieffer-Jaquinod S, Seetharaman J, Acton TB et al. 2013. Two Fe-S clusters catalyze sulfur insertion by radical SAM methylthiotransferases. Nat Chem Biol. 9:333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francioso A, Conrado AB, Mosca L, Fontana M. 2020. Chemistry and biochemistry of sulfur natural compounds: Key intermediates of metabolism and redox biology. Ox Med Cell Long.8294158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francoleon NE, Carrington SJ, Fukuto JM. 2011. The reaction of H2S with oxidized thiols: Generation of persulfides and implications to H2S biology. Arch Biochem Biophys. 516:146–153. [DOI] [PubMed] [Google Scholar]
- Fugate CJ, Stich TA, Kim EG, Myers WK, Britt RD, Jarrett JT. 2012. 9-Mercaptodethiobiotin is generated as a ligand to the [2Fe-2S]+ cluster during the reaction catalyzed by biotin synthase from Escherichia coli. J Am Chem Soc. 134:9042–9045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godert AM, Jin M, McLafferty FW, Begley TJ. 2007. Biosynthesis of the thioquinolobactin siderophore: An interesting variation on sulfur transfer. J Bacteriol. 189:2941–2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmer JE, Hiscox MJ, Dinis PC, Fox SJ, Iliopoulos A, Hussey JE, Sandy J, Van Beek FT, Essex JW, Roach PL. 2014. Structures of lipoyl synthase reveal a compact active site for controlling sequential sulfur insertion reactions. Biochem J. 464:123–133. [DOI] [PubMed] [Google Scholar]
- Hill CR, Shafaei A, Balmer L, Lewis JR, Hodgson JM, Millar AH, Blekkenhorst LC. 2022. Sulfur compounds: From plants to humans and their role in chronic disease prevention. Crit Rev Food Sci Nutr. 5:1–23. [DOI] [PubMed] [Google Scholar]
- Ida T, Sawa T, Ihara H, Tsuchiya Y, Watanabe Y, Kumagai Y, Suematsu M, Motohashi H, Fujii S, Matsunaga T et al. 2014. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc Natl Acad Sci USA. 111:7606–7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa M, Kawasaki T, Hayakawa Y. 2013. Cloning and heterologous expression of the thioviridamide biosynthesis gene cluster from Streptomyces olivoviridis. Appl Environ Microbiol. 79:7110–7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jameson GNL, Cosper MM, Hernandez HL, Johnson MK, Huynh BH. 2004. Role of the [2Fe-2S] cluster in recombinant Escherichia coli biotin synthase. Biochemistry. 43:2022–2031. [DOI] [PubMed] [Google Scholar]
- Jarrett JT. 2015. The biosynthesis of thiol- and thioether-containing cofactors and secondary metabolites catalyzed by radical S-adenosylmethionine enzymes. J Biol Chem. 290:3972–3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C-S, Müller WEG, Schröder HC, Guo Y-W. 2011. Disulfide- and multisulfide-containing metabolites from marine organisms. Chem Rev. 112:2179–2307. [DOI] [PubMed] [Google Scholar]
- Jurgenson CT, Begley TJ, Ealick SE. 2009. The structural and biochemical foundations of thiamin biosynthesis. Annu Rev Biochem. 78:569–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler D 2006. Enzymatic activation of sulfur for incorporation into biomolecules in prokaryotes. FEMS Microbiol Rev. 30:825–840. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Koike S, Shibuya N, Lefer D, Ogasawara Y, Kimura H. 2017. 3-Mercaptopyruvate sulfurtransferase produces potential redox regulators cysteine- and glutathione-persulfide (Cys-SSH and GSSH) together with signaling molecules H2S2, H2S3 and H2S. Sci Rep. 7:10459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy K, Begley TP. 2010. Reagent for the detection of protein thiocarboxylase in the bacterial proteome: Lissamine rhodamine B sulfonyl azide. J Am Chem Soc. 132:11608–11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy K, Begley TP. 2011. Protein thiocarboxylate-dependent methionine biosynthesis in Wolinella succinogenes. J Am Chem Soc. 133:379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruithof PD, Lunev S, Aguilar Lozano SP, de Assis Batista F, Al-dahmani ZM, Joles JA, Dolga AM, Groves MR, van Goor H. 2020. Unraveling the role of thiosulfate sulfurtransferase in metabolic diseases. Biochim Biophys Acta. 1866:165716. [DOI] [PubMed] [Google Scholar]
- Landgraf BJ, Arcinas AJ, Lee K-H, Booker SJ. 2013. Identification of an intermediate methyl carrier in the radical S-adenosylmethionine methyltransferases RimO and MiaB. J Am Chem Soc. 135:15404–15416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz ND, Booker SJ. 2015. Auxiliary iron-sulfur cofactors in radical SAM enzymes. Biochim Biophys Acta. 1853:1316–1334. [DOI] [PubMed] [Google Scholar]
- Lanz ND, Rectenwald JM, Wang B, Kakar ES, Laremore TN, Booker SJ, Silakov A. 2014. Characterization of a radical intermediate in lipoyl cofactor biosynthesis. J Am Chem Soc. 137:13216–13219. [DOI] [PubMed] [Google Scholar]
- Lec J-C, Boutserin S, Mazon H, Mulliert G, Boschi-Muller S, Talfournier F. 2018. Unraveling the mechanism of cysteine persulfide formation catalyzed by 3-mercaptopyruvate sulfurtransferase. ACS Catalysis. 8:2049–2059. [Google Scholar]
- Leimkühler S, Bühning M, Beilschmidt L. 2017. Shared sulfur mobilization routes for tRNA thiolation and molybdenum cofactor biosynthesis in prokaryotes adn eukaryotes. Biomolecules. 7:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lill R, Freibert SA. 2020. Mechanisms of mitochondrial iron-sulfur protein biogenesis. Annu Rev Biochem. 89:471–499. [DOI] [PubMed] [Google Scholar]
- Lin C-I, McCarty RM, Liu H-w. 2013. The biosynthesis of nitrogen-, sulfur-, and high-carbon chain-containing sugars. Chemical Society Reviews. 42:4377–4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litomska A, Ishida K, Dunbar KL, Boettger M, Coyne S, Hertweck C. 2018. Enzymatic thioamide formation in a bacterial antimetabolite pathway. Angew Chem Int Ed. 57:11574–11578. [DOI] [PubMed] [Google Scholar]
- Liu J, Lin Z, Li Y, Zheng Q, Chen D, Liu W. 2019. Insights into the thioamidation of thiopeptins to enhance the understanding of the biosynthetic logic of thioamide-containing thiopeptides. Org Biomolec Chem. 17:3727–3731. [DOI] [PubMed] [Google Scholar]
- Liu L, Jiang S, Xing M, Chen CD, Lai C, Liu G, Wu D, Gao H, Hong L, Tan P et al. 2020. Structural analysis of an L-cysteine desulfurase from an Ssp DNA phosphorothioation system. mBio. 11:e00488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Dos Santos PC, Zhu X, Orlando R, Dean DR, Söll D, Yuan J. 2012. Catalytic mechanism of Sep-tRNA:Cys-tRNA synthase. Sulfur transfer is mediated by a disulfide and persulfide. J Biol Chem. 287:5426–5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Vinyard DJ, Reesbeck ME, Suzuki T, Manakongtreecheep K, Holland PL, Brudvig GW, Soll D. 2016. A [3Fe-4S] cluster is required for tRNA thiolation in archaea and eukaryotes. Proc Natl Acad Sci USA. 113:12703–12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanta N, Liu A, Dong S, Nair SK, Mitchell DA. 2018. Enzymatic reconstitution of ribosomal peptide backbone thioamidation. Proc Natl Acad Sci USA. 115:3030–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanta N, Szantai-Kis DM, Petersson EJ, Mitchell DA. 2019. Biosynthesis and chamical applications of thioamides. ACS Chem Biol. 14:142–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiocco SJ, Arcinas AJ, Landgraf BJ, Lee K-H, Booker SJ, Elliott SJ. 2016. Transformations of the FeS clusters of the methylthiotransferases MiaB and RimO, detected by direct electrochemistry. Biochemistry. 55:5531–5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr S, Mendel RR, Schwarz G. 2021. Molybdenum cofactor biology, evolution and deficiency. Biochim Biophys Acta. 1868:118883. [DOI] [PubMed] [Google Scholar]
- McCarthy EL, Booker SJ. 2017. Destruction and reformation of an iron-sulfur cluster during catalysis by lipoyl synthase. Science. 358:373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin MI, Lanz ND, Goldman PJ, Lee K-H, Booker SJ, Drennan CL. 2016. Crystallographic snapshots of sulfur insertion by lipoyl synthase. Proc Natl Acad Sci USA. 113:9446–9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melideo SL, Jackson MR, Jorns MS. 2014. Biosynthesis of a central intermediate in hydrogen sulfide metabolism by a novel human sulfurtransferase and its yeast ortholog. Biochemistry. 53:4739–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihara H, Esaki N. 2002. Bacterial cysteine desulfurases: Their function and mechanisms. Appl Microbiol Biotech. 60(1–2):12–23. [DOI] [PubMed] [Google Scholar]
- Molle T, Moreau P, Clemancey M, Forouhar F, Ravanat J-L, Duraffourg N, Fourmond V, Latour J-M, Gambarelli S, Mulliez E et al. 2016. Redox behavior of the S-adenosylmethionine (SAM)-binding Fe-S cluster in methylthiotransferase RimO, toward understanding dual SAM activity. Biochemistry. 55:5798–5808. [DOI] [PubMed] [Google Scholar]
- Mueller EG. 2006. Trafficking in persulfides: Delivering sulfur in biosynthetic pathways. Nat Chem Biol. 2:185–194. [DOI] [PubMed] [Google Scholar]
- Mulliez E, Duarte V, Arragain S, Fontecave M, Atta M. 2017. On the role of additional [4Fe-4S] clusters with a free coordination site in radical-SAM enzymes. Frontiers in Chemistry. 5:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai Y, Maruyama-Nakashita A. 2020. Biosynthesis of sulfur-containing small biomolecules in plants. Intl J Molec Sci. 21:3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak DD, Mahanta N, Mitchell DA, Metcalf WW. 2017. Post-translational thioamidation of methyl-coenzyme M reductase, a key enzyme in methanogenic and methanotrophic archaea. eLife. 6:e29218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevarez JL, Turmo A, Hu J, Hausinger RP. 2020. Biochemical pincer complexes. ChemCatChem. 12:4242–4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C-M, Weerasinghe L, Day JJ, Fukuto JM, Xian M. 2015. Persulfides: Current knowledge and challenges in chemistry and chemical biology. Molec Biosyst. 11:1775–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul BD, Snyder SH. 2015. Protein sulfhydration. Meth Enzymol. 555:79–90. [DOI] [PubMed] [Google Scholar]
- Pedre B, Dick TP. 2021. 3-Mercaptopyruvate sulfurtransferase: An enzyme at the crossroads of sulfane sulfur trafficking. Biol Chem. 402:223–237. [DOI] [PubMed] [Google Scholar]
- Pérard J, Ollagnier de Choudens S. 2018. Iron-sulfur clusters biogenesis by the SUF machinery: Close to the molecular mechanism understanding. J Biol Inorg Chem. 23:581–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu T, Mei Z, Zhang W, Liang W-J, Zhou X, Liang J, Deng Z, Wang Z. 2020. An in vitro DNA phosphorothioate modification reaction. Molec Microbiol. 113:452–463. [DOI] [PubMed] [Google Scholar]
- Rankin JA, Chatterjee S, Tariq Z, Lagishetty S, Desguin B, Hu J, Hausinger RP. 2021. The LarB carboxylase/hydrolase forms a transient cysteinyl-pyridine intermediate during nickel-pincer nucleotide cofactor biosynthesis. Proc Natl Acad Sci USA. 118:e2106202118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao GS, Gorin G. 1959. Reaction of cystine with sodium sulfide in sodium hydroxide. J Org Chem. 24:749–753. [Google Scholar]
- Santos-Aberturas J, Chandra G, Frattaruolo L, Lacret R, Phan TH, Vior NM, Eyles TH, Truman AW. 2019. Uncovering the unexplored diversity of thioamidated ribosomal peptides in Actinobacteria using the RiPPER genome mining tool. Nucl Acids Res. 47:4624–4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalen CJ, Hudson GA, Kille B, Mitchell DA. 2018. Bioinformatic expansion and discovery of thiopeptide antibiotics. J Am Chem Soc. 140:9494–9501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbach BP, Chung AH, Scott AD, George SJ, Cramer SP, Dos Santos PC. 2013. Fe-S cluster biogenesis in Gram-positive bacteria: SufU is a zinc-dependent sulfur transfer protein. Biochemistry. 53:152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigi N 2018. Recent advances in our understanding of the biosynthesis of sulfur modifications in tRNAs. Front Microbiol. 9:2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigi N 2021. Biosynthesis and degradation of sulfur modifications in tRNAs. Intl J Molec Sci. 22:11937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigi N, Horitani M, Miyauchi K, Suzuki T, Kuroki M. 2020. An ancient type of MnmA protein is an iron-sulfur cluster-dependent sulfurtransferase for tRNA anticodons. RNA. 26:240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talib EA, Outten CE. 2021. Iron-sulfur cluster biogenesis, trafficking, and signaling: Roles for CGFS glutaredoxins and BolA proteins. Biochim Biophys Acta. 1868:118847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe TS, Leimkühler S, Dahl C. 2019. The functional diversity of the prokaryotic sulfur carrier protein TusA. Adv Microbial Physiol. 75:233–277. [DOI] [PubMed] [Google Scholar]
- Tao L, Stich TA, Fugate CJ, Jarrett JT, Britt RD. 2018. EPR-derived structure of a paramagnetic intermediate generated by biotin synthase BioB. J Am Chem Soc. 140:12947–12963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchong S-I, Xu H, White RH. 2005. L-Cysteine desulfidase: An [4Fe-4S] enzyme isolated from Methanocaldococcus jannaschii that catalyzes the breakdown of L-cysteine into pyruvate, ammonia, and sulfide. Biochemistry. 44:1659–1670. [DOI] [PubMed] [Google Scholar]
- Termathe M, Leidel SA. 2018. The Uba4 domain interplay is mediated via a thioester that is critical for tRNA thiolation through Urm1 thiocarboxylation. Nucl Acids Res. 46:5171–5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turmo A, Hu J, Hausinger RP. 2022. Characterization of the nickel-inserting cyclometallase LarC from Moorella thermoacetica and identification of a cytidinylylated reaction intermediate. Metallomics. 14:mfac014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugulava NB, Sacanell CJ, Jarrett JT. 2001. Spectroscopic changes during a single turnover of biotin synthase: Destruction of a 2Fe-2S cluster accompanies sulfur insertion. Biochemistry. 40:8352–8358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Jiang S, Deng Z, Dedon PC, Chen S. 2019. DNA phosphorothioate modification--a new multi-functional epigenetic system in bacteria. FEMS Microbiol Rev. 43:109–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Saidhareddy P, Jiang X. 2020. Construction of sulfur-containing moieties in the total synthesis of natural products. Nat Prod Rep. 37:246–275. [DOI] [PubMed] [Google Scholar]
- Zhang B, Arcinas AJ, Radle MI, Silakov A, Booker SJ, Krebs C. 2020. First step in catalysis of the radical S-adenosylmethionine methylthiotransferase MiaB yields an intermediate with a [3Fe-4S]0-like auxiliary cluster. J Am Chem Soc. 142:1911–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C, Dos Santos PC. 2018. Metallocluster transactions: Dynamic protein interactions guide the biosynthesis of Fe-S clusters in bacteria. Biochem Soc Trans. 46:1593–1603. [DOI] [PubMed] [Google Scholar]
- Zheng L, White RH, Cash VL, Jack RF, Dean DR. 1993. Cysteine desulfurase activity indicates a role for NIFS in metallocluster biosynthesis. Proc Natl Acad Sci USA. 90:2754–2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Lénon M, Ravanat J-L, Touati N, Velours C, Podskoczyi K, Leszczynska G, Fontecave M, Barras F, Golinelli-Pimpaneau B. 2021. Iron-sulfur biology invades tRNA modification: The case of U34 sulfuration. Nucl Acids Res. 49:3997–4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Pecqueur L, Aucynaitè A, Fuchs J, Rutkienè R, Vaitekunas J, Meskys R, Boll M, Fontecave M, Urbonavicius J et al. 2021. Structural evidence for a [4Fe-5S] intermediate in the non-redox desulfuration of thiouracil. Angew Chem Int Ed. 60:424–431. [DOI] [PubMed] [Google Scholar]