

Abstract
Stem cell differentiation methods have been developed to produce cells capable of insulin secretion which are showing promise in clinical trials for treatment of type-1 diabetes. Nevertheless, opportunities remain to improve cell maturation and function. Three-dimensional (3D) culture has demonstrated improved differentiation and metabolic function in organoid systems, with biomaterial scaffolds employed to direct cell assembly and facilitate cell-cell contacts. Herein, we investigate 3D culture of human stem cell-derived islet organoids, with 3D culture initiated at the pancreatic progenitor, endocrine progenitor, or immature β-cell stage. Clusters formed by reaggregation of immature β-cells could be readily seeded into the microporous poly(lactide-co-glycolide) scaffold, with control over cell number. Culture of islet organoids on scaffolds at the early to mid-stage beta cell progenitors had improved in vitro glucose stimulated insulin secretion relative to organoids formed at the pancreatic progenitor stage. Reaggregated islet organoids were transplanted into the peritoneal fat of streptozotocin-induced diabetic mice, which resulted in reduced blood glucose levels and the presence of systemic human C-peptide. In conclusion, 3D cell culture supports development of islet organoids as indicated by insulin secretion in vitro and supports transplantation to extrahepatic sites that leads to a reduction of hyperglycemia in vivo.
Graphical Abstract
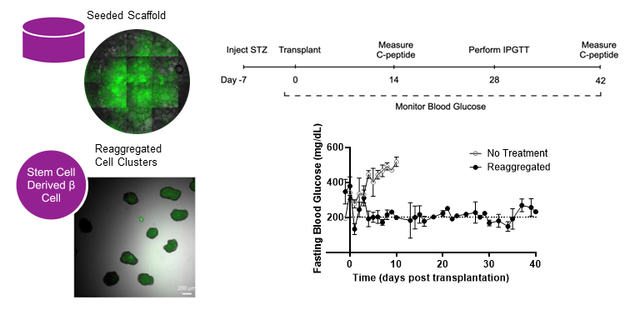
Microporous scaffolds facilitates 3D cultures of stem cell-derived islet organoids and allows for transplantation to extrahepatic sites.
1. Introduction
Type-1 diabetes (T1D) is a chronic autoimmune disease predominantly caused by the death or dysregulation of pancreatic β-cells, the insulin-secreting cells of the pancreas.[1,2] T1D is currently treated through administration of exogenous insulin, either manually via injection or through the use of an insulin pump. While automated insulin pumps and continuous glucose monitoring devices improve the management of blood glucose levels, the risk of complications, such as tissue or organ damage, from hyperglycemic and hypoglycemic events remains.[3] Complications can be minimized with islet transplantation, which was recently developed as a treatment to restore glycemic control in patients with brittle T1D.[4,5] The therapeutic benefits of this protocol are hindered by the scarcity of cadaveric human islets relative to the number of patients with T1D and poor islet survival.[6]
Stem cells are being investigated for their potential to provide an unlimited source of β-cells or islet organoids that are capable of restoring normoglycemia.[7] Various protocols have implemented suspension culture[8], air-liquid interfaces[9], planar culture[10], or combinations. Refinement of these protocols over the past decade have led to the generation of immature β-cells in vitro that are able to complete their maturation and become functional after transplantation[8–12], which have led to ongoing clinical trials.[13–16] Initial data released by Vertex Pharmaceuticals indicate high safety and tolerability of the therapy, with the first two patients demonstrating lower insulin requirements and improved glucose time-in-range.[13–14] Nevertheless, opportunities remain to improve differentiation, cell yield, viability, maturation, and metabolic function that may further improve their performance following transplantation.[17,18]
3D culture has been proposed to enable assembly of differentiating stem cells into islet organoids, consisting of multiple cell types normally found within an islet, that can enable improved function following transplantation. Improved differentiation and metabolic function have been obtained with the 3D culture of other organoid systems, such as cardiomyocytes[19,20], and assembly of cells can be driven by cell-to-cell contacts.[21,22] Reaggregation of stem cell-derived islet organoids into 3D cultures has been done in the immature beta-cell stage with orbital shakers[12] and microwell culture plates[23], yet few studies have analyzed 3D culture in earlier stages of stem cell differentiation to islet organoids. Prior studies have employed scaffolds with defined pore sizes for the culture of pancreatic progenitors, which had been seeded as single cells and ultimately formed cell aggregates of directed sizes within the pores of the scaffold.[24] Initiation of 3D culture at different stages of beta cell development may inform on time points that enhance maturation of cells.
Clinical trials with islets and stem cell derived beta cells have involved transplantation to the hepatic portal vein, for which immunosuppression may have detrimental effects on transplanted cells and has motivated studies to seek alternative sites. Much of the pre-clinical work, particularly with transplantation of stem cell derived beta cells or islet organoids, have been performed in the kidney capsule, which is generally not considered a translational site clinically. An extrahepatic site, such as the peritoneal fat, has been used for clinical islet transplantation [25] and offers the opportunity to retrieve the graft or engineer the site to reduce or eliminate immune suppression.[26,27] Microporous scaffolds have been successfully employed for the transplantation of islets or islet organoids into the peritoneal fat [28–32]. The culturing or seeding of islet organoids on microporous scaffolds for transplantation to the peritoneal fat is a translatable approach that may contribute to the ongoing efforts to treat type-1 diabetes.
In this report, we investigate the transplantation of 3D cultured stem cell-derived islet organoids to the extrahepatic intraperitoneal fat using the microporous scaffold. Scaffolds were seeded with reaggregated clusters on the day of transplantation or as single cells at the beginning of the immature β-cell or endocrine progenitor stages of differentiation. Through reaggregation or seeding into the microporous scaffold, cell differentiation into islet organoids was promoted in a 3D setting. The maturation and function of the organoids were assessed using qPCR and glucose stimulated insulin secretion (GSIS). Scaffolds seeded with reaggregated cells, which allowed for delivery of sufficient numbers of cells, were transplanted and organoid function was monitored in vivo according to fasting blood glucose levels, intraperitoneal glucose tolerance testing, and measurement of circulating human C-peptide. Collectively, these demonstrate the differentiation, maturation, and function of islet organoids on scaffolds in vitro, and following transplantation at extrahepatic sites.
2. Experimental section
2.1. Microporous scaffold fabrication
Poly(lactide-co-glycolide) (PLG) microporous scaffolds were fabricated as previously described.[24] Briefly, PLG microporous scaffolds were fabricated by compression molding PLG microspheres (75:25 mole ratio D,L-lactide to glycolide) and 250 to 425 μm salt crystals in a 1:30 ratio of PLG microspheres to salt. The mixture was humidified in an incubator for 7 min. Scaffolds were compression molded with 77.5 mg of polymer–salt mixture into cylinders 5 mm in diameter by 2 mm in height using a 5 mm KBr die (International Crystal Laboratories, Garfield, NJ) at 1500 psi for 30 seconds. Molded constructs were gas foamed in 800 psi carbon dioxide for 16 hours in a pressure vessel. The vessel was depressurized at a controlled rate for 30 min. On the day of cell seeding, scaffolds were leached in water for 1.5 hours, changing the water once after 1 hour. Scaffolds were disinfected by submersion in 70% ethanol for 30 seconds and rinsed multiple times with phosphate buffer solution (PBS).
2.2. Designing sfGFP-Cpep fluorescent insulin reporter hPSC line
Briefly, HEK293FT cells were co-transfected using lentiviral packaging vectors (pMDL-GagPol, pRSV-Rev, pIVS-VSV-G, with a CpepSfGFP construct using Lipofectamine 2000 (Life Technologies, Grand Island, NY,) for 48 hours.[23,33] The CpepSfGFP construct (i.e., with the Superfolder-GFP cDNA ligated into the XhoI site of the human C-peptide coding sequence)[29–31] driven by the upstream 2.2-kb rat Ins1 promoter included a phosphoglycerol kinase (PGK)-promoter-mCherry selection marker. Using PEG-it (System Biosciences, Mountain View, CA), supernatant was concentrated for 24 hours. Then it was precipitated using ultracentrifugation, resuspended in PBS and stored at −80 °C until use. Through viral transfection and FACS sorting, clones can be identified in which the vector was correctly integrated into the cell line. The H1-sfGFP-Cpep cell line was then expanded into a subclonal population and characterized.
2.3. Differentiation of hPSCs and scaffold seeding
hPSC differentiations were performed by a planar method that has been previously described.[8,10] consisting of cells seeded on polystyrene plates coated with Matrigel. H1 human embryonic stem cells were cultured in a humidified incubator set at 5% CO2 and 37 °C. Briefly, undifferentiated cells were single-cell dispersed and seeded at 5 × 106 cells/mL in 6-well cell culture treated plates. Cells were cultured for 72 hours in mTeSR1 and then cultured in the differentiation media [10,12] for 6 stages. Differentiation to pancreatic progenitors and stem-cell derived β-cells was assessed via flow cytometry for stage-specific transcription factor expression (Supplementary Figure 1). To initiate scaffold culture differentiations from planar culture, cells were single cell dispersed using TrypLE and seeded on scaffolds at a density of 125 × 106 cells/cm3. Prior to seeding, scaffolds were washed in cell media solution then briefly dried on sterile gauze to improve the absorption of the cell solution into the scaffold. Cells were distributed across both faces of the scaffold and then incubated for 2 hours to allow cell solution to be further absorbed into the scaffold before differentiation media was added. For reaggregation from planar culture, cells were single cell dispersed using TrypLE and seeded onto suspension culture 6-well plates at 8 × 106 cells/well. Y27632 was included in the media for the first 24 hours of culture, then media was changed every other day for the duration of culture. Plates were placed on an orbital shaker set to 100 rpm inside a humidified incubator. After 3–4 days in culture, reaggregated cell clusters were collected and seeded on the scaffold in the same manner described above.
2.4. qRT-PCR analysis
Gene expression from scaffold cultures was obtained by homogenizing the scaffold in Trizol with ethanol, followed by RNA isolation using the DirectZol RNA MiniPrep kit (Zymo) according to manufacturer instructions. RNA concentration was determined using a NanoDrop spectrophotometer. The Applied Biosystems High-Capacity RNA-to-cDNA kit was used to transcribe RNA into cDNA. Universal RT microRNA PCR assays were performed using SYBR Green MasterMix Universal RT (Exiqon), according to the manufacturer’s instructions. The amplification profile was assessed using a LightCycler® 480 (Roche, Germany). Gene expression was quantified using the ΔΔCt method and fold change was calculated using the formula 2−ΔΔCt. Values for the genes of interest were normalized to the housekeeping gene (GAPDH) followed by normalization to marker expression in pluripotent hPSCs. Primers used for qPCR analysis are listed in Tables 1 and Supplementary Table 1.
Table 1.
qRT-PCR primers used during late stages of β-cell differentiation
| Gene | Sequence (5’ to 3’) | Function |
|---|---|---|
| GAPDH | Forward: AAGGTGAAGGTCGGAGTCAA Reverse: AATGAAGGGGTCATTGATGG |
Housekeeping gene used to normalize gene expression |
| PDX1 | Forward: CCTTTCCCATGGATGAAGTC Reverse: CGTCCGCTTGTTCTCCTC |
Pancreatic and duodenal homeobox 1; expression begins during posterior foregut stage and remains high5,6,36 |
| NKX6.1 | Forward: GGGGATGACAGAGAGTCAGG Reverse: CGAGTCCTGCTTCTTCTTGG |
Homeobox 1 protein NK6; expression begins during pancreatic progenitor stage and indicates differentiation into monohormonal cells6,37 |
| NEUROD1 | Forward: GCCCCAGGGTTATGAGACTAT Reverse: ATCAGCCCACTCTCGCTGTA |
Basic helix-loop-helix transcription factor; endocrine precursor marker involved in β-cell differentiation and expansion33 |
| NGN3 | Forward: CTATTCTTTTGCGCCGGTAG Reverse: CTTCGTCTTCCGAGGCTCT |
Neurogenin 3, member of basic helix-loop-helix transcription factor family; proendocrine gene critical for determination of endocrine cell specification36 |
| PCSK1 | Forward: CTCTGGCTGCTGGCATCT Reverse: CGGGTCATACTCAGAGGTCC |
Gene encoding proprotein convertase 1 involved in pro-insulin processing29 |
| MafA | Forward: GAGAGCGAGAAGTGCCAACT Reverse: TTCTCCTTGTACAGGTCCCG |
V-maf musculoaponeurotic fibrosarcoma oncogene homolog A; maturation marker expressed late in β-cell maturation36,38 |
| ECAD | Forward: TTGACGCCGAGAGCTACAC Reverse: GACCGGTGCAATCTTCAAA |
E-cadherin; cell surface adhesion protein involved in intercellular communication between β-cells25 |
| Insulin | Forward: TTCTACACACCCAAGACCCG Reverse: CAATGCCACGCTTCTGC |
β-cell hormone |
| Glucagon | Forward: TGCTCTCTCTTCACCTGCTCT Reverse: AGCTGCCTTGTACCAGCATT |
α-cell hormone |
2.5. Static glucose-stimulated insulin secretion assay
GSIS was initiated by washing planar wells, reaggregated cells, and scaffold cultures with KRB buffer three times (128 mM NaCl, 5 mM KCl, 2.7 mM CaCl2, 1.2 mM MgSO4, 1mM NaH2PO4, 1.2 mM KH2PO4, 5 mM NaHCO3, 10 mM HEPES (Gibco, 15630–080), and 0.1% BSA (Proliant). Cells and scaffolds were exposed to basal (2mM) levels of glucose for one hour then washed with KRB and exposed again to basal glucose levels for one hour. Samples from this glucose exposure period were saved from cells and scaffolds. After a KRB wash, cells and scaffolds were exposed to high (20mM) levels of glucose were then added for one hour. Supernatant samples were taken, and insulin levels were measured by ELISA (Alpco, NC0038324). Cells were dispersed with TrypLE and counted to normalize insulin secretion values to cell number.
2.6. Cell number analysis
Cell number analysis was performed using PicoGreen (Invitrogen™ Quant-iT™ PicoGreen™ dsDNA Assay Kits and dsDNA Reagents). A standard curve was developed using known quantities of stage 6 stem cell β-cell progenitors. Scaffolds were lysed in Trizol (Invitrogen) and incubated at 37° C overnight. Samples and standards were diluted in TE buffer (10mM Tris-HCl, 1mM EDTA, pH 7.5) and mixed with PicoGreen working solution (200-fold dilution in TE buffer). Total Radiant Efficiency [p s−1] [μW cm2]−1 was quantified with 465 nm excitation and 520 nm emission wavelengths and compared to a standard curve to estimate cell quantities after scaffold culture.
2.7. Intracellular flow cytometry
Single cell dissociations were fixed in 4% paraformaldehyde and blocked with 5% normal donkey serum in 0.1% Triton-X PBS. Cells were incubated overnight with primary antibodies (CHGA, C-pep, NKX6.1, PDX1, OCT3/4), washed and subsequently incubated with appropriate secondary antibodies. Stained, fixed cells were analyzed by ZE5 cell analyzer (Bio-Rad). Primary and secondary antibodies used: mouse anti-OCT-3/4 (Santa Cruz Biotechnology, cat. no. sc-5279, clone C-10; RRID: AB_628051), chromogranin A Rabbit anti-Human, Invitrogen (catalog #: PIMA533052), rat anti-C-peptide (Developmental Studies Hybridoma Bank, University of Iowa, cat. no. GN-ID4-S), mouse anti-NKX6–1 (Developmental Studies Hybridoma Bank, University of Iowa, cat. no. F55A12-S), goat anti-PDX1 (R&D Systems, cat. no. AF2419), anti-rat AlexaFluor 488 (Invitrogen, cat. no. A21208), anti-mouse AlexaFluor 488 (Invitrogen, cat. no. A21202), anti-mouse AlexaFluor 647 (Invitrogen, cat. no. A31571), anti-goat AlexaFluor 647 (Invitrogen, cat. no. A21447), anti-rabbit AlexaFluor 647 (Invitrogen, cat. no. A31573).
2.8. Transplantation of Reaggregated cells in Diabetic Mice
All animal studies were approved by the University of Michigan Animal Care and Use Committee. An intraperitoneal injection of 150 mg kg−1 streptozotocin (STZ) was given to male NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG )mice (Jackson Laboratories) 7–10 days prior to transplant. In the days leading up to surgery, blood glucose was monitored using a tail vein prick (Accu-chek). Glucose readings above 300 mg dL−1 on two consecutive days were considered diabetic. Mice with weight loss less than 20% of body weight and sufficient glucose readings were considered eligible for transplantation. On the day of transplantation, the abdomen was shaved and sterilized. A small incision was made in the peritoneal wall and the epididymal fat pads were unwrapped outside the body. As described in section 2.3, reaggregated cell clusters were collected and seeded on both faces of the scaffold and incubated at least 2 hours before transplantation. Reaggregated cell-laden scaffolds were placed on the fat pads, wrapped in fat pad tissue, and placed back into the peritoneal cavity. Each mouse received approximately 5 million cells in clusters on each scaffold, equaling a total of 10 million cells transplanted per mouse. Mice were sutured, surgically stapled, and received carprofen (0.5 mg/mL) the day of and after surgery. Blood glucose and weights were measured 3 times a week.
2.9. Circulating Serum C-peptide and Intraperitoneal Glucose Tolerance Test
On 2-week intervals, blood was collected following a 4-hour fast and a 20% intraperitoneal glucose injection. Approximately 100–200 μL of blood was collected from the saphenous vein 30 minutes after injection. The serum was separated by centrifugation at 2000g for 20 minutes. Circulating human C-peptide was measured by ELISA. ELISAs were done by the University of Michigan Cancer Center Immunology Core. Intraperitoneal glucose tolerance test was done 4 weeks after transplantation. Blood glucose measurements were taken at baseline and after a 4-hour fast and a 20% intraperitoneal glucose injection. Measurements were taken at 0, 15, 30, 45, 60, 90, and 120 minutes after injection.
2.10. Immunostaining
Explanted scaffolds were flash frozen in isopentane cooled on dry ice and then embedded within OCT embedding medium and cryosectioned. Sections were stained using human insulin (Dako) and human glucagon (Sigma, cat. no. G2654) primaries, followed by AlexaFluor donkey anti-mouse 488nm (Invitrogen, cat. no. A21202) and AlexaFluor 647nm AffiniPure donkey anti-guinea pig (Jackson Immuno Research Labs, cat. no. 706605148) secondaries. Fluoromount with DAPI was applied before placing coverslips. Digital images were acquired with a ZEISS Axio Observer Z1 inverted microscope.
2.11. Statistical Methods
All statistical analyses were conducted using Prism graphing and data analysis software (GraphPad Software, Inc., La Jolla, CA, United States). Statistical differences were determined using a two-tailed Student t test, one-way ANOVA with Dunnett test for multiple comparisons, or Kruskal Wallis one-way analysis of variance. Values were reported as the mean ± standard error of mean (SEM). n indicates the total number of biological replicates.
3. Results and discussion
3.1. Reaggregation step in stage 6 produces high quality stem cell-derived islet organoids
We developed a fluorescent C-peptide reporter for the H1 hESC cell line using Superfolder-GFP cDNA ligated into the XhoI site of the human C-peptide coding sequence (Figure 1A). The sfGFP-Cpep reporter is a fusion protein in which sfGFP is inserted within the C-peptide region of pro-insulin, such that cleavage of C-peptide leaves a functional insulin protein. The H1-sfGFP-Cpep line was differentiated according to previously published adherent planar protocols (Figure 1B).[10] As the cells mature, the cells begin to fluoresce due to expression of the superfolder-GFP, indicating production of insulin (Figure 1C). A 3D assembly of these cells can be obtained in the middle of stage 6 (approximately stage 6 day 7) by disassociation and subsequent reaggregation (as described in section 2.3) to form clusters with a diameter of approximately 200 μm.[12] The reaggregation process resulted in cell loss of approximately 50% which is consistent with previous studies [10] that indicate a loss of non-endocrine cells upon lifting from planar culture. Fluorescence imaging indicates that cells in planar culture and following reaggregation are GFP positive in stage 6 (Figure 1C). Co-positive populations of C-peptide and NKX6.1 indicated the presence of mono-hormonal β-cells. The number of cells in the reaggregated cultures that are co-positive for C-pep+/NKX6.1+ averaged 43.5% (Figure 1D), whereas the planar cells averaged 34% C-pep+/NKX6.1+ co-positive (Figure 1E). Chromogranin A (CHGA), which is expressed in endocrine cells, was also analyzed between planar and reaggregated cells. CHGA+/NKX6.1+ co-positive populations were not significantly different between planar and reaggregated cells (Figure 1F). The H1-sfGFP-Cpep engineered cell line produced islet organoids containing β-cells that could readily be seeded within the pores of a microporous PLG scaffold.
Figure 1.
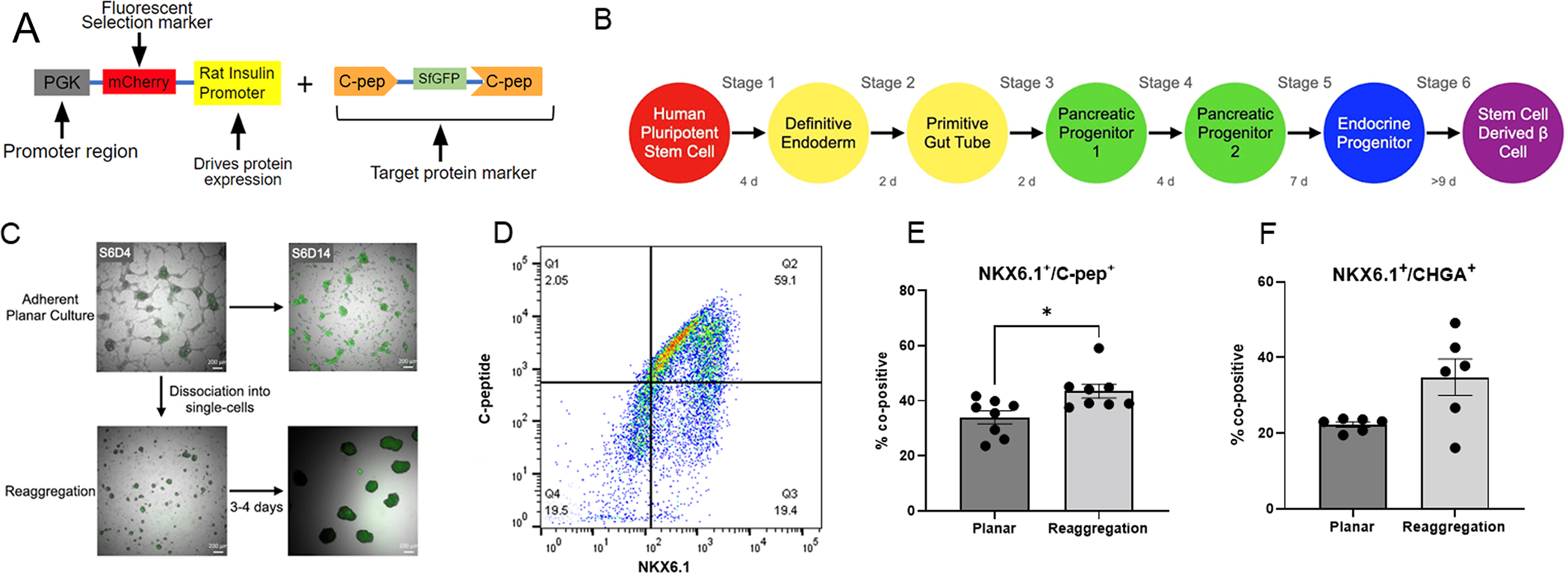
(A) Schematic of the sfGFP-Cpep Fluorescent Insulin Reporter. (B) Schematic of culture timeline. (C) Representative brightfield images of cells in planar culture and after reaggregation during the stem cell-derived β-cell stage. 488 channel overlaid with brightfield images indicates insulin secretion of reaggregated and planar cells. (D) Representative image of flow analysis of Cpep+/NKX6.1+ after reaggregation. (E) Comparison of Cpep+/NKX6.1+ populations analyzed with flow cytometry before and after reaggregation (n= 9 Planar, n=9 Reaggregation). (F) Comparison of CHGA+/NKX6.1+ populations analyzed with flow cytometry before and after reaggregation (n=6 Planar, n=6 Reaggregation).
3.2. Maturation of endocrine progenitor cells on the scaffold
We subsequently investigated formation of islet organoids following aggregation of cells within the pores of the scaffold at earlier stages, with subsequent culture through Day 10 of Stage 6. Cells were isolated on the first day of stage 6 (S6D1), which are generally regarded as endocrine progenitors, and seeded onto scaffolds at densities to promote aggregation within the pores (Figure 2A). The expression dynamics of sfGFP on the scaffold were similar to the dynamics observed with planar culture, indicating that the maturation of the cells to insulin-expressing organoids was supported by seeding and culture on the scaffold (Figure 2B). Similarly, no significant differences in maturation between cells seeded onto the scaffold at S6D1 were observed relative to planar culture (Figure 2C), as assessed by qRT-PCR (Table 1). Similar results have also been obtained with the HUES8 cell line (Supplementary Figure 2B).
Figure 2.
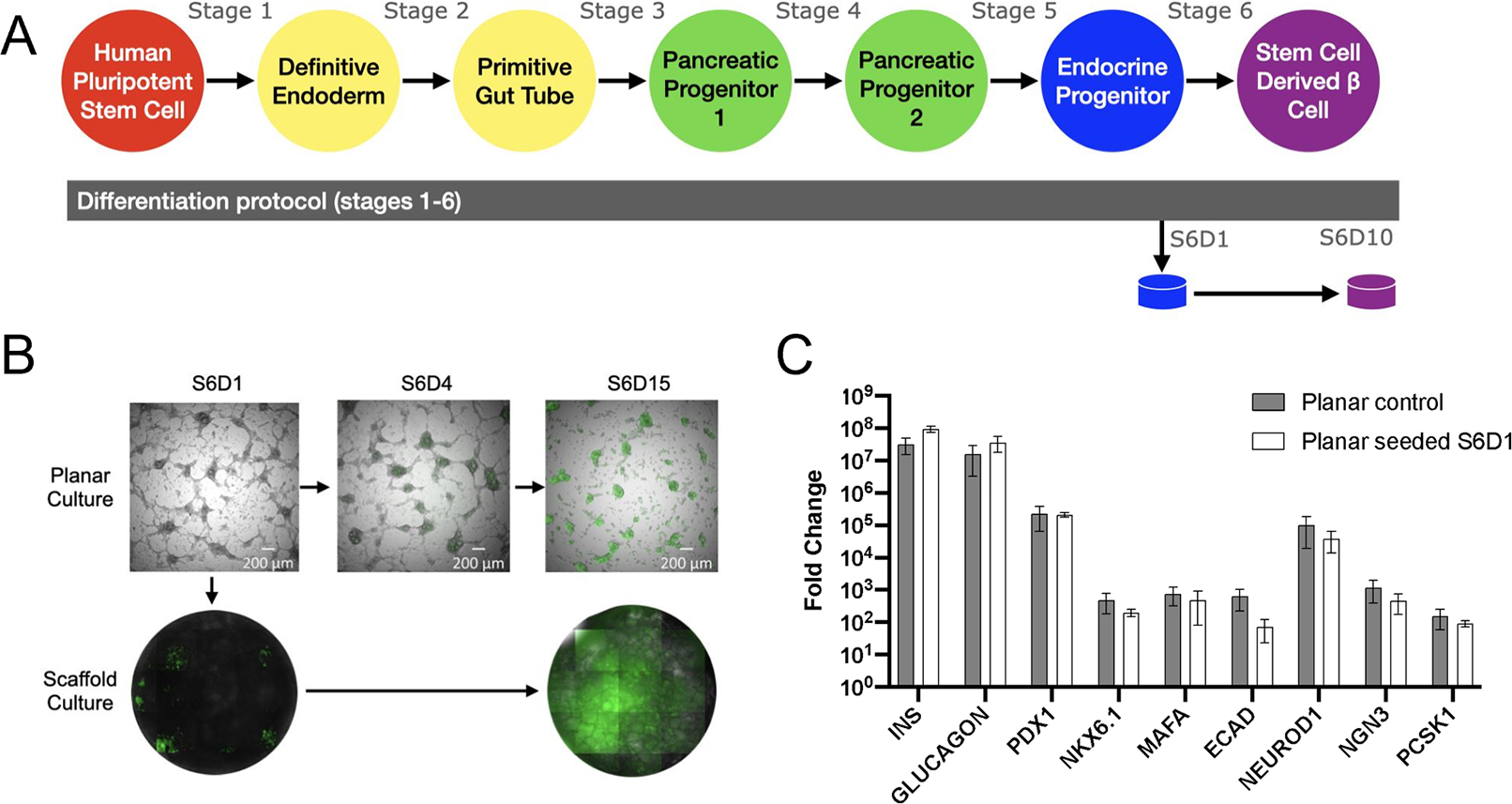
(A) Schematic of differentiation protocol including scaffold seeding at Stage 6 Day 1 (S6D1). (B) Fluorescent images of planar culture cells (top) and scaffold culture (bottom) throughout stage 6 of differentiation. 488 channel overlaid with brightfield images to demonstrate sfGFP expression aligned with cells. (C) Gene expression at Stage 6 Day 10 of planar cultured cells compared to planar cells seeded onto a scaffold at Stage 6 Day 1 (n=6 Planar control, n=16 Planar seeded S6D1).
3.3. Pancreatic progenitor cell differentiation to β-cell progenitors
We subsequently investigated seeding cells from the beginning of stage 5 (S5D1) into the scaffold (Figure 3A), which is the stage at which cells begin to transition from pancreatic progenitors to endocrine progenitors. Scaffolds were collected for analysis on stage 6 day 10, which totals 17 days of culture on the scaffold. From initiation of scaffold culture to maturation in stage 6, the sfGFP reporter indicates that insulin production is occurring on the scaffold early into Stage 6 (Figure 3B). No significant differences in the expression of genes associated with maturation were detected between planar culture cells and scaffolds seeded with planar culture cells (Figure 3C). Similar results have also been obtained with the HUES8 cell line (Supplementary Figure 2A).
Figure 3.
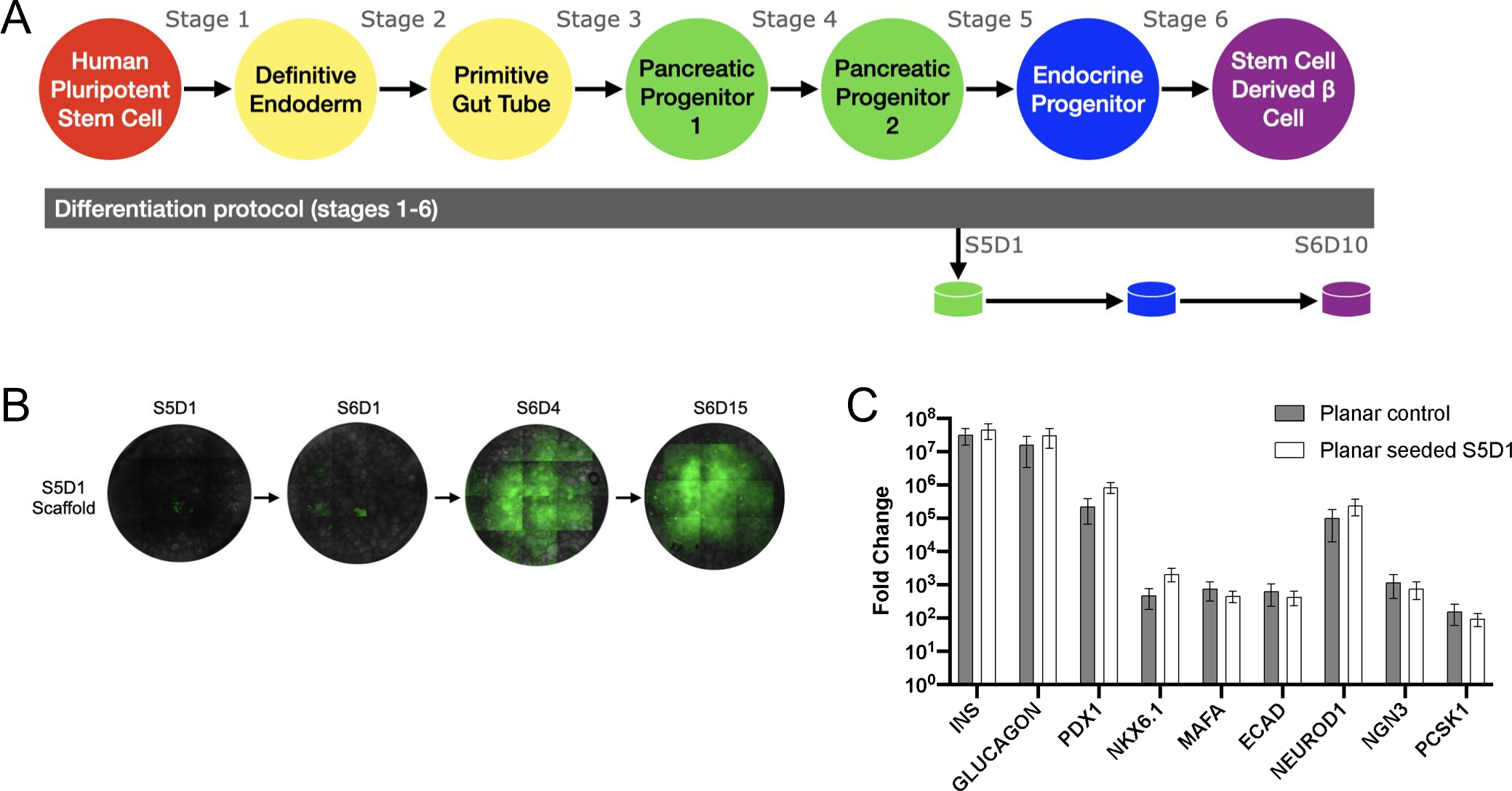
(A) Schematic of differentiation protocol including scaffold seeding at Stage 5 Day 1 (S5D1). (B) Fluorescent images throughout stage 5 and 6 of differentiation. (C) Gene expression at Stage 6 Day 10 of planar cultured cells compared to planar cells seeded onto a scaffold at Stage 5 Day 1 (n=6 Planar control, n=12 Planar seeded S5D1).
Positive results in the pancreatic progenitor stage led attempts to culture pluripotent cells (Stage 0) on scaffolds and culture to the end of either stage 1 or stage 4. Pluripotent cells seeded on the scaffold prior to stage 1 had reduced expression of CER and CXCR4 compared to cells at the end of stage 1 culture (Supplementary Figure 3B). Scaffold cultures initiated prior to stage 1 and cultured until the end of stage 4 had reduced expression of NKX6.1, MAFA, ECAD, NEUROD1, and NGN3 compared to control cells (Supplementary Figure 3C), indicating planar culture is beneficial in the early stages of β-cell differentiation.
3.4. Cell number decline on the scaffold is observed early after seeding
We next quantified the number of cells on scaffolds following the seeding of reaggregated clusters or scaffold culture. At S6D1, 5 million cells were seeded on the scaffold and continued in scaffold culture. Cell number was then determined at multiple time points (t =2, 24, 48, 96, and 240 hours) (Figure 4A), which indicated the greatest cell number immediately after seeding and corresponded to a seeding efficiency of approximately 98%. The cell number subsequently decreased through 24 hours, and no significant difference was observed between 24 hours and 240 hours, suggesting that the majority of cells were lost in the first 24 hours after seeding. Cell number was also quantified at the conclusion of differentiation with scaffolds seeded with cells at S5D1 and S6D1, with similar cell densities at the end of culture. For the seeding of reaggregated clusters in the middle of stage 6 (S6D7), the number of cells on the scaffold was high, with an average of 4.9 million cells present 2 hours after seeding. The cell numbers on the scaffold were significantly greater with the seeding of cells on the day of transplant relative to the S5D1 or S6D1 seeding (Figure 4B). Cells are lost with the formation of reaggregated clusters, with the number of cells lost during reaggregation similar to the decline in cell number following the seeding of scaffolds at S5D1 or S6D1. Given our studies of cell loss during on-scaffold culture, we determined that seeding of reaggregated clusters on the scaffold achieved greater cell numbers for the duration of the studies. This evidence promoted the use of reaggregated cell-seeding onto the scaffold for transplant studies, where cell number is critically important to blood glucose control.
Figure 4.
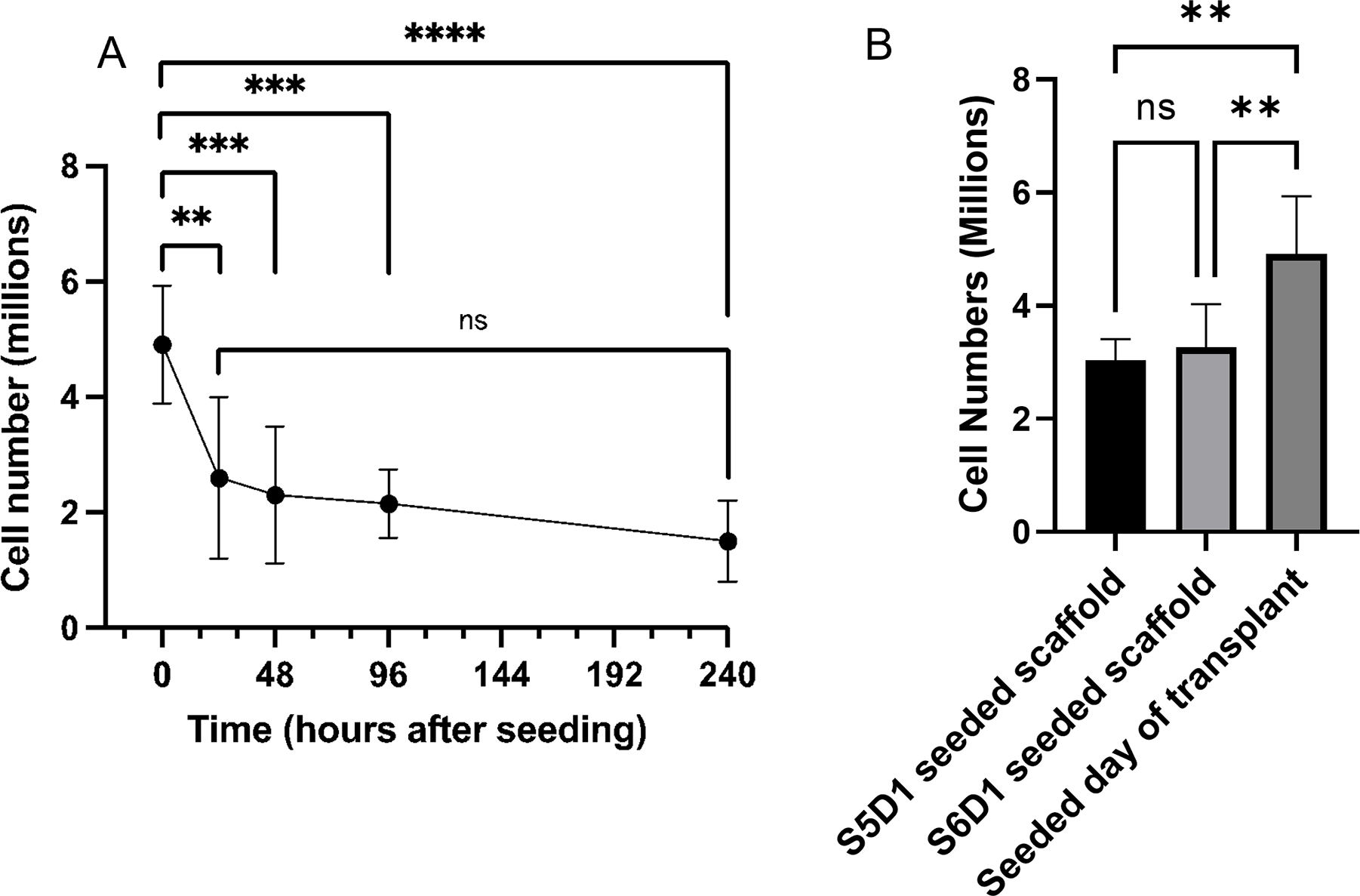
(A) Cell number at various time points after seeding cells S6D1 on the scaffold and continuing cell culture on the scaffold. Scaffolds were analyzed right after seeding, at 24, 48, 96, and 240 hours. Stage media was changed on the scaffold similarly to planar culture. (n=5: 2 hours, n=8: 24 hours, n=8: 48 hours, n=8: 96 hours, n=13: 240 hours). **P<0.01, ***P<0.001, ****P<0.0001. (B) Cell number at the end of differentiation (S6D10) when comparing different seeding starting times. Cells seeded at S5D1 are on the scaffold for about 17 days and 10 days for cells seeded at S6D1. Samples taken the day of transplant were after a brief (2 hour) incubation. (n=5 S5D1 seeded scaffold, n=10 S6D1 seeded scaffold, n=5 Seeded day of transplant). **P<0.01.
3.5. Glucose-stimulated insulin secretion maintained by scaffold culture, higher from reaggregated clusters
Glucose-stimulated insulin secretion assays (GSIS) were performed to assess cell function following 3D culture or reaggregation. At high glucose, S6D7 reaggregated cells secreted more insulin than S5D1 seeded cells, while there was no significant difference between reaggregated cells and S6D1 seeded cells. Cells seeded at S6D1 was the only condition that secreted significantly more insulin at high glucose than at low glucose across all samples (Figure 5A). Insulin secretion at high glucose was normalized to low glucose to compare the insulin secretion index across conditions (Figure 5B). No significant differences were consistently observed between conditions, though some samples had high insulin secretion indexes indicating batch to batch variability. These results indicate greater insulin secretion with S6D1 and S6D7 reaggregated cells compared to S5D1 scaffolds. The combination of high cell numbers on the scaffold (Figure 4B) and enhanced insulin secretion led us to investigate scaffolds seeded with S6D7 reaggregated clusters for the analysis of in vivo function.
Figure 5.
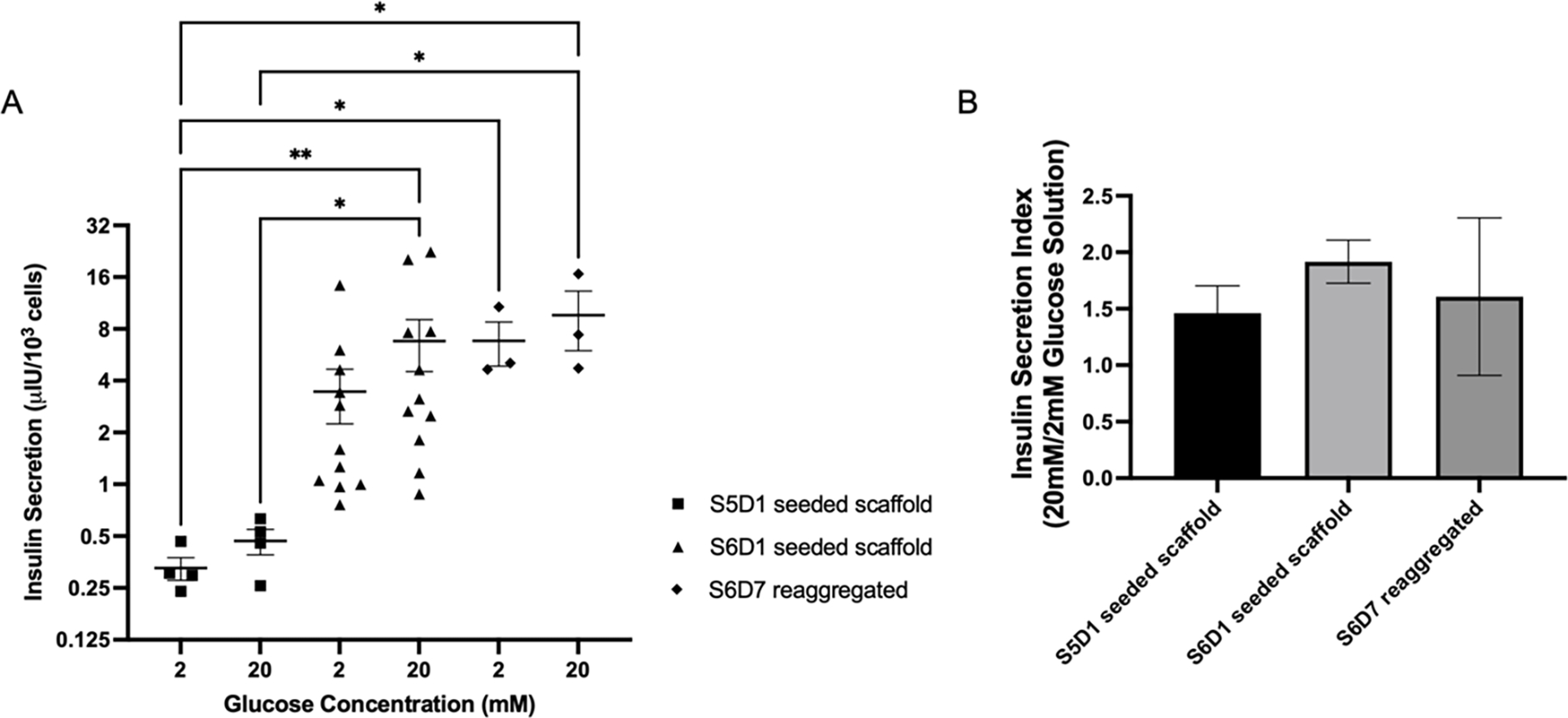
(A) Insulin secretion normalized to cell number during glucose-stimulated insulin secretion assays performed at Stage 6 Day 10 from scaffolds seeded at Stage 5 Day 1 with planar cells (squares), scaffolds seeded at Stage 6 Day 1 with planar cells (triangles), and planar cells reaggregated at Stage 6 Day 7 (diamonds). Cell number determined by PicoGreen for scaffold conditions and reaggregated. Planar cell number counted using hemocytometer after dissociation. Kruskal-Wallis test with Dunn’s multiple comparisons test performed. Significance level of α=0.05 used (n=4 S5D1 seeded scaffold, n=11 S6D1 seeded scaffold, n=3 S6D7 reaggregated), *P<0.05, **P<0.01. (B) Insulin secretion indexes for each condition.
3.6. Transplanted cells reduce blood glucose levels in STZ-induced diabetic mice
Reaggregated cells (S6D7) were seeded onto the scaffold and transplanted into the peritoneal fat of STZ-induced diabetic NSG mice to assess their in vivo function (Figure 6A). Scaffolds explanted after 14 days in vivo and stained for human insulin and human glucagon provided evidence of cell survival following transplantation (Figure 6B). Mice that received reaggregated cells transplanted on the scaffold had lower blood glucose levels than mice that received no intervention (Figure 6C), with blood glucose levels reduced to approximately 200 mg/dL. Blood glucose readings during intraperitoneal glucose tolerance testing demonstrate glycemic control by mice that received reaggregated cells, with the reduction of elevated glucose on a similar time scale as compared to healthy (non-diabetic) controls (Figure 6D). Circulating human C-peptide levels were subsequently measured in serum at 2- and 6-weeks post transplantation, demonstrating measurable human C-peptide in mice transplanted with reaggregated cell, and that their levels of C-peptide increased over time (Figure 6E). The presence and increase in C-peptide levels indicates survival and function of the transplanted cells and suggests improved function of transplanted cells with time. Together, these results indicate that the reaggregated cells transplanted on the microporous PLG scaffold secrete insulin and reduce blood glucose levels in STZ induced diabetic mice.
Figure 6.
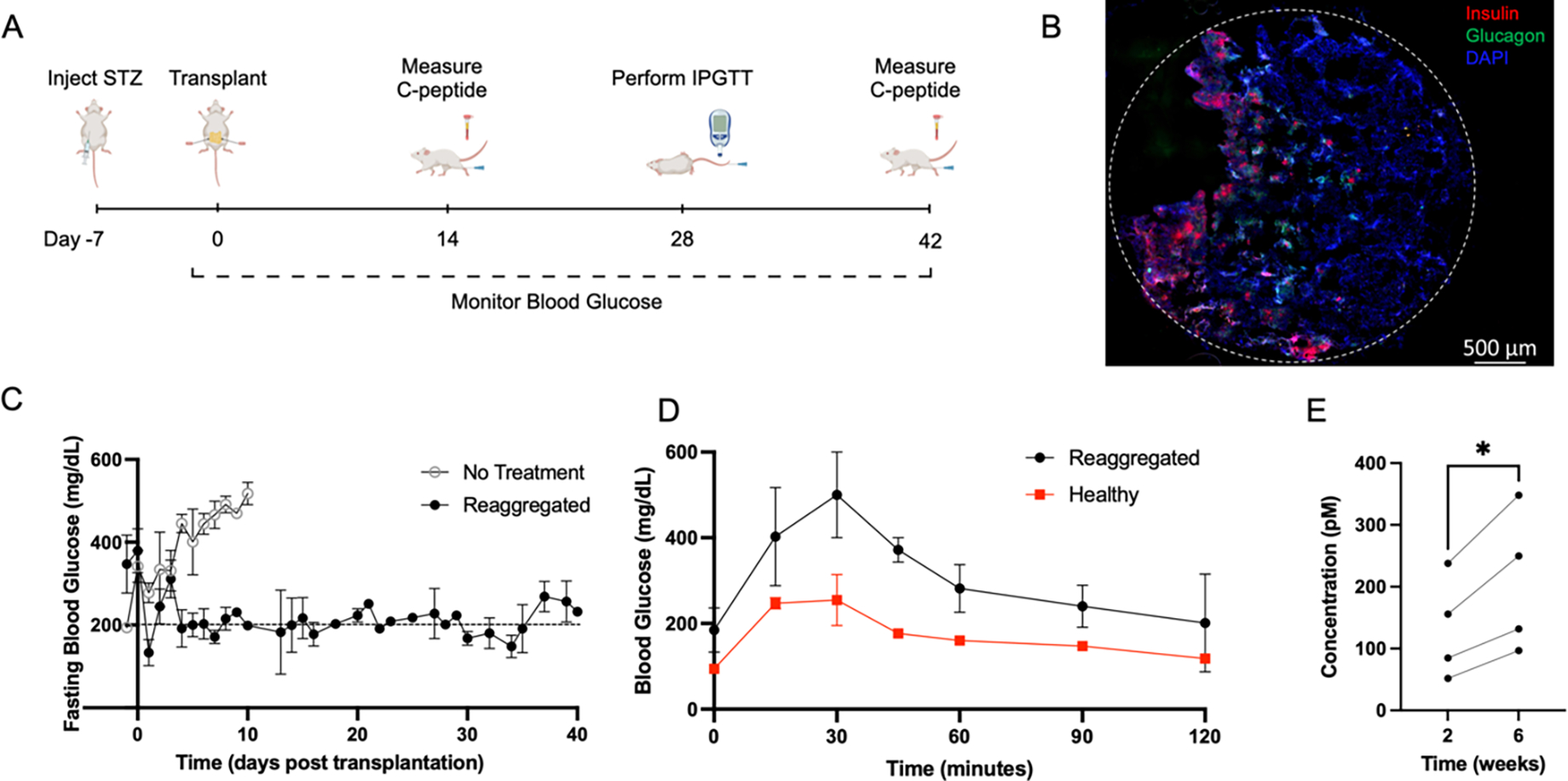
(A) Timeline of in vivo studies, including timepoints induction of diabetes, transplantation of scaffolds to the peritoneal fat, collection of blood serum, and intraperitoneal testing of glucose tolerance. (B) Representative image of a scaffold section explanted after 14 days in vivo indicating survival of transplanted islet organoids. Scaffolds were cryosectioned and stained for human insulin (red), human glucagon (green) and DAPI (blue). The dashed line indicates the circumference of the scaffold. (C) Fasting blood glucose measurements from STZ-induced diabetic mice that received 10M reaggregated cells transplanted on two PLG scaffolds (5M cells/scaffold) (n=3) or no treatment (n=4). Mice were fasted for 4–6 hours prior to blood glucose readings. Blood glucose was measured daily for at least 10 days following transplantation, then 3 times per week for the duration of the study. (D) Blood glucose measurements taken during intraperitoneal glucose tolerance testing of mice that received reaggregated cells (S6D7) transplanted on PLG scaffolds (n=2) and healthy mice (n=2). Mice were fasted for 4–6 hours, then received an intraperitoneal injection of glucose at 2g/kg body weight at t=0. Average area under the curve for treated mice was 37,042 and for healthy mice was 20,655. (E) Circulating C-peptide levels at 2- and 6-weeks post transplantation, measured from serum collected through saphenous vein blood draw. Paired t-test performed using repeated measures from four mice at both time points. Significance level of α=0.05 used.
This report investigated the 3D culture or aggregation of hPSC into islet organoids that sense glucose and secrete insulin, with the potential to reduce hyperglycemia in diabetic mice. Planar culture of the hPSCs are not technically considered islet organoids because the clusters lack the cellular organization of the native islets.[34–36] The 3D culture environment for organoids allows for the development of cellular heterogeneity and their structural and cellular organization may mimic features of the native tissue. Herein, we investigated the stage of culture for hPSCs at which they are aggregated to 3D culture. Cells at the latter stage (S6D7) are able to spontaneously form into 3D aggregates, whereas earlier stages were placed on a microporous scaffold to support a 3D organization.
Cells seeded and aggregated on scaffolds at S5D1 or S6D1 allowed for maturation of the cells through S6D10 culture. Cultures with cells seeded prior to the definitive endoderm stage (Stage 0) did not effectively develop on the scaffold. The early stages in the differentiation protocol, definitive endoderm (stage 1), primitive gut tube (stage 2), and pancreatic progenitor 1 (stage 3) are accompanied by rapid proliferation and changes in cluster morphology that may not be supported within the scaffold. Fluorescence imaging of the sfGFP-Cpep reporter allowed visualization of the maturation dynamics on the scaffold, which demonstrated initial fluorescence around S6D1 that gradually increased to S6D7 for cultures beginning on either S6D1 or S5D1. Assessment of function for scaffold cultures beginning at S5D1 or S6D1 demonstrated glucose responsiveness as indicated by an insulin secretion index greater than 1; however, scaffold cultures beginning on S6D1 had significantly more insulin secretion relative to S5D1. A cellular analysis of the organoids within the scaffold was not possible as single cell dispersions could not be retrieved for flow cytometry analysis.
Reaggregation of cells at S6D7 of culture is common for multiple laboratory studies and serves to increase the percentage of C-peptide and NKX6.1 positive cells, which are thought to represent functional β-cells. The reaggregation process leads to a loss of approximately 50% of the cells, which are believed to mostly consist of off-target or undifferentiated cells.[10] Cell loss during reaggregation is similar to cell numbers observed when comparing seeding the day of transplant (4.92 ± 0.46M) to 24 hours (2.60 ± 0.50M) after seeding (Figure 4A). Planar cells seeded on the scaffold are single-cell dispersed in the same manner done during reaggregation, which could initiate similar cell loss. Cells lost during planar seeding may be progenitor cells while cells that are maintained through culture become fully differentiated and functional. The reaggregated cell clusters fit within the pores of the scaffold and are of similar size to islets. (Supplementary Figure 4) [37] Clusters can be readily seeded onto the scaffold with retention of cell-cell connections for subsequent culture or transplantation. Interestingly, reaggregated cells seeded on the scaffold had greater insulin secretion compared to cultures from S5D1. Collectively, the 3D culture of cells as reaggregated clusters or on scaffolds can support their maturation towards functional organoids, though the timing of cell seeding and duration of culture can influence cell number and function.
Reaggregated cell clusters from S6D7 were able to reverse hyperglycemia, though the blood glucose levels were not fully normalized. Transplantation studies were performed with reaggregated cells and not with scaffold cultured cells primarily due to the ability to control the cell number, which has been implicated as a major factor impacting transplantation outcomes. While the kidney capsule and hepatic portal vein are prominent sites for transplantation, the scaffold transplants were performed into the peritoneal fat,[28–30]. Most studies with the transplantation of hPSC-derived β-cells are performed in the kidney capsule, which is not considered a translatable site. We employed the peritoneal fat, an extrahepatic site that is similar to the omentum in humans, as this site has been investigated clinically and can support the transplantation of large numbers of cells necessary for treating a patient.[25,38] Extrahepatic sites may also enable alternative immune modulation strategies that may ultimately reduce or eliminate the use of immune suppression.[39] The biomaterial scaffold functions as a support for cell delivery, distributes the cells across the surface and allows integration with the host vasculature, and can be functionalized to enhance engraftment and modulate immune responses.[27,40] Cells from S6D1 culture had a strong GSIS response that would support transplantation, yet additional studies are needed to refine the initial seeding and culture approach to attain higher cell numbers at the end of culture. Untreated mice exhibited elevated levels of blood glucose (>500 mg dL−1 at day 10), whereas mice receiving cell-scaffold constructs had blood glucose remaining around 200 mg dL−1, which is not a level that indicates full function. The inability to attain fully normalized blood glucose levels likely results from a combination of factors, such as the site of transplantation that may impact cell survival and function post-transplantation as well as the intrinsic cell capabilities. These studies were performed using the H1 cell line, which has been reported to produce less insulin than other cell lines commonly used for transplantation (e.g., HUES8).[12,41] Nevertheless, human C-peptide levels were substantial and indicative of insulin secretion, with an increased production of C-peptide over time. IPGTT results are also consistent with insulin secretion and the ability to modulate blood glucose.
4. Conclusion
In conclusion, the 3D culture of stem cells to form islet organoids can support their maturation and function in vitro, with the capacity to reduce hyperglycemia in vivo. Endocrine progenitor cells (S6D1) seeded onto scaffolds and cultured secreted more insulin than seeded and cultured pancreatic progenitor cells (S5D1), and similar levels to reaggregated clusters. Collectively, the support of 3D architectures was most effective for Stage 6 cells, with reaggregated clusters from S6D7 able to obtain the highest cell numbers and reduce hyperglycemia with transplantation at extrahepatic sites. Culture of islet organoids on the scaffold may reduce the manipulation of the cells and the associated disruption of the cellular microenvironment when collecting cultures for transplantation, and the design of the biomaterial scaffold provides opportunities to further enhance the maturation and function of islet organoids, or to provide local immunomodulation [25,26].
Supplementary Material
Acknowledgements
Funding for this work was provided by NIH R01DK121462, and the Juvenile Diabetes Research Foundation, and the JDRF Center of Excellence at the University of Michigan. We are grateful to Dr. Jeff Millman and those in his laboratory for guidance with the culture protocols. The authors would like to acknowledge Zoe Beekman, Namit Padgaonkar, and the Shea Lab.
Funding Statement:
Funding for this work was provided by NIH R01DK121462, and the Juvenile Diabetes Research Foundation, and the JDRF Center of Excellence at the University of Michigan.
Footnotes
Conflicts of Interest: There are no conflicts to declare.
Ethics Statement: All animal procedures studies were performed at the University of Michigan (Protocol Number PRO00009714) in compliance with the Institutional Animal Care & Use Committee (IACUC) guidelines.
References
- [1].Cryer PE, Diabetes 2008, 57, 3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pambianco G, Costacou T, Ellis D, Becker DJ, Klein R, Orchard TJ, Diabetes 2006, 55, 1463. [DOI] [PubMed] [Google Scholar]
- [3].Kalra S, Mukherjee J, Venkataraman S, Bantwal G, Shaikh S, Saboo B, Das A, Ramachandran A, Indian J Endocr Metab 2013, 17, 819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Shapiro AMJ, Lakey JRT, Ryan EA, Korbutt GS, Toth E, Warnock GL, Kneteman NM, Rajotte RV, N Engl J Med 2000, 343, 230. [DOI] [PubMed] [Google Scholar]
- [5].Shapiro AMJ, Auchincloss H, Secchi A, Brennan DC, Ryan EA, Polonsky KS, Bertuzzi F, Sutherland DER, Segal M, Barton FB, et al. , n engl j med 2006. [Google Scholar]
- [6].Emamaullee JA, Shapiro AMJ, Cell Transplant 2007, 16, 1. [DOI] [PubMed] [Google Scholar]
- [7].Helman A, Melton DA, Cold Spring Harb Perspect Biol 2021, 13, a035741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Velazco-Cruz L, Song J, Maxwell KG, Goedegebuure MM, Augsornworawat P, Hogrebe NJ, Millman JR, Stem Cell Reports 2019, 12, 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, O’Dwyer S, Quiskamp N, Mojibian M, Albrecht T, et al. , Nat Biotechnol 2014, 32, 1121. [DOI] [PubMed] [Google Scholar]
- [10].Hogrebe NJ, Augsornworawat P, Maxwell KG, Velazco-Cruz L, Millman JR, Nat Biotechnol 2020, 38, 460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rezania A, Bruin JE, Riedel MJ, Mojibian M, Asadi A, Xu J, Gauvin R, Narayan K, Karanu F, O’Neil JJ, et al. , Diabetes 2012, 61, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hogrebe NJ, Maxwell KG, Augsornworawat P, Millman JR, Nat Protoc 2021, 16, 4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Vertex Pharmaceuticals Incorporated, A Phase 1/2 Study to Evaluate the Safety, Tolerability, and Efficacy of VX-880 in Subjects Who Have Type 1 Diabetes Mellitus With Impaired Hypoglycemic Awareness and Severe Hypoglycemia, Clinicaltrials.Gov, 2022. [Google Scholar]
- [14].“Vertex Presents New Data from VX-880 Phase 1/2 Clinical Trial at the American Diabetes Association 82nd Scientific Sessions | Vertex Pharmaceuticals,” can be found under https://investors.vrtx.com/news-releases/news-release-details/vertex-presents-new-data-vx-880-phase-12-clinical-trial-american, n.d.
- [15].A Safety, Tolerability, and Efficacy Study of VC-02™ Combination Product in Subjects With Type 1 Diabetes Mellitus and Hypoglycemia Unawareness, Clinicaltrials.Gov, 2022. [Google Scholar]
- [16].De Klerk E, Hebrok M, Front. Endocrinol. 2021, 12, 631463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pagliuca FW, Millman JR, Gürtler M, Segel M, Van Dervort A, Ryu JH, Peterson QP, Greiner D, Melton DA, Cell 2014, 159, 428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hrvatin S, O’Donnell CW, Deng F, Millman JR, Pagliuca FW, DiIorio P, Rezania A, Gifford DK, Melton DA, Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jha R, Wu Q, Singh M, Preininger MK, Han P, Ding G, Cho HC, Jo H, Maher KO, Wagner MB, et al. , Sci Rep 2016, 6, 30956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Correia C, Koshkin A, Duarte P, Hu D, Carido M, Sebastião MJ, Gomes-Alves P, Elliott DA, Domian IJ, Teixeira AP, et al. , Biotechnol. Bioeng. 2018, 115, 630. [DOI] [PubMed] [Google Scholar]
- [21].Lecomte M-J, Pechberty S, Machado C, Da Barroca S, Ravassard P, Scharfmann R, Czernichow P, Duvillié B, Cell Med 2016, 8, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Toyoda T, Mae S-I, Tanaka H, Kondo Y, Funato M, Hosokawa Y, Sudo T, Kawaguchi Y, Osafune K, Stem Cell Research 2015, 14, 185. [DOI] [PubMed] [Google Scholar]
- [23].Li X, Yang KY, Chan VW, Leung KT, Zhang X-B, Wong AS, Chong CCN, Wang CC, Ku M, Lui KO, Stem Cell Reports 2020, 15, 1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Youngblood RL, Sampson JP, Lebioda KR, Shea LD, Acta Biomaterialia 2019, 96, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Baidal DA, Ricordi C, Berman DM, Alvarez A, Padilla N, Ciancio G, Linetsky E, Pileggi A, Alejandro R, N Engl J Med 2017, 376, 1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Headen DM, Woodward KB, Coronel MM, Shrestha P, Weaver JD, Zhao H, Tan M, Hunckler MD, Bowen WS, Johnson CT, et al. , Nature Mater 2018, 17, 732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Skoumal M, Woodward KB, Zhao H, Wang F, Yolcu ES, Pearson RM, Hughes KR, García AJ, Shea LD, Shirwan H, Biomaterials 2019, 192, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kasputis T, Clough D, Noto F, Rychel K, Dye B, Shea LD, ACS Biomater. Sci. Eng. 2018, acsbiomaterials.7b00912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gibly RF, Zhang X, Graham ML, Hering BJ, Kaufman DB, Lowe WL, Shea LD, Biomaterials 2011, 32, 9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gibly RF, Zhang X, Lowe WL, Shea LD, Cell Transplant 2013, 22, 811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hlavaty KA, Gibly RF, Zhang X, Rives CB, Graham JG, Luo X, Lowe WL, Shea LD, Am J Transplanation 2014, 14, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rives CB, des Rieux A, Zelivyanskaya M, Stock S, Lowe WL, Shea LD, Biomaterials 2009, 30, 394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhu S, Larkin D, Lu S, Inouye C, Haataja L, Anjum A, Kennedy R, Castle D, Arvan P, Diabetes 2016, 65, 699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kim Y, Kim H, Ko UH, Oh Y, Lim A, Sohn J-W, Shin JH, Kim H, Han Y-M, Sci Rep 2016, 6, 35145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Vegas AJ, Veiseh O, Gürtler M, Millman JR, Pagliuca FW, Bader AR, Doloff JC, Li J, Chen M, Olejnik K, et al. , Nat Med 2016, 22, 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Velazco-Cruz L, Goedegebuure MM, Millman JR, Front. Bioeng. Biotechnol. 2020, 8, 786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bosco D, Rouiller DG, Halban PA, Journal of Endocrinology 2007, 194, 21. [DOI] [PubMed] [Google Scholar]
- [38].Stice MJ, Dunn TB, Bellin MD, Skube ME, Beilman GJ, Cell Transplant 2018, 27, 1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Clough DW, King JL, Li F, Shea LD, Endocrinology 2020, 161, bqaa156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Li F, Crumley K, Bealer E, King JL, Saito E, Grimany-Nuno O, Yolcu ES, Shirwan H, Shea LD, ACS Appl. Mater. Interfaces 2022, acsami.2c12939. [DOI] [PubMed] [Google Scholar]
- [41].Rezania A, Bruin JE, Xu J, Narayan K, Fox JK, O’Neil JJ, Kieffer TJ, Stem Cells 2013, 31, 2432. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.