

Abstract
Objective
Identification of genetic risk factors for Parkinson’s disease (PD) has to date been primarily limited to the study of single nucleotide variants, which only represent a small fraction of the genetic variation in the human genome. Consequently, causal variants for most PD risk are not known. Here we focused on structural variants (SVs), which represent a major source of genetic variation in the human genome. We aimed to discover SVs associated with PD risk by performing the first large-scale characterization of SVs in PD.
Methods
We leveraged a recently developed computational pipeline to detect and genotype SVs from 7,772 Illumina short-read whole genome sequencing samples. Using this set of SV variants, we performed a genome-wide association study using 2,585 cases and 2,779 controls and identified SVs associated with PD risk. Furthermore, to validate the presence of these variants, we generated a subset of matched whole-genome long-read sequencing data.
Results
We genotyped and tested 3,154 common SVs, representing over 412 million nucleotides of previously uncatalogued genetic variation. Using long-read sequencing data, we validated the presence of three novel deletion SVs that are associated with risk of PD from our initial association analysis, including a 2kb intronic deletion within the gene LRRN4.
Interpretation
We identify three SVs associated with genetic risk of PD. This study represents the most comprehensive assessment of the contribution of SVs to the genetic risk of PD to date.
Summary for Social Media If Published
1. If you and/or a co-author has a Twitter handle that you would like to be tagged, please enter it here. (format: @AUTHORSHANDLE).
@kimberleybill10
2. What is the current knowledge on the topic?
Identification of genetic risk factors for Parkinson’s disease (PD) has to date been primarily limited to the study of single nucleotide variants, which only represent a small fraction of the genetic variation in the human genome. Consequently, causal variants for most PD risk are not known. Here we focused on structural variants, which represent a major source of genetic variation in the human genome that are yet to be explored in PD on a genome-wide scale.
3. What question did this study address?
We aimed to discover new variants associated with Parkinson’s disease risk by performing the first large-scale characterization of Structural variants in Parkinson’s disease.
4. What does this study add to our knowledge?
We identify three structural variants associated with genetic risk of Parkinson’s disease. This study represents the most comprehensive assessment of the contribution of Structural variants to the genetic risk of Parkinson’s disease to date.
5. How might this potentially impact on the practice of neurology?
These results demonstrate that Structural variants may contribute to disease risk in Parkinson’s disease and highlight the need for such variants to be considered in future surveys of the genetics of neurodegenerative diseases.
Introduction
There is substantial evidence that genetic factors contribute to the risk of developing Parkinson’s disease (PD). The most recent genome-wide association study (GWAS) which included approximately 40,000 cases, 20,000 first degree relatives of PD cases and 1.4 million controls, identified 90 independent risk signals across 78 regions of the genome 1. Despite these large-scale efforts, we have a limited understanding of which variants and genes are driving the signal at the known risk loci as GWAS inherently nominates chromosomal regions not individual variants. Furthermore, these loci cumulatively explain 16–30% of the heritable component of PD, meaning that most of the common genetic variation that contributes to disease risk is yet to be discovered 1.
Previous genetic studies have focused on single-nucleotide variants (SNVs), which represent only a fraction of the genetic variation in the human genome. Structural variants (SVs), which are duplications, deletions or inversions of stretches of DNA, represent over ten times more genetic variation than SNVs2. However, SVs are more difficult to identify and accurately genotype compared to SNVs due to common sequencing and alignment artifacts. As a result, SVs have been largely understudied, but recent advances in whole genome sequencing (WGS) technology and improved SV detection algorithms, may now allow for assessment of the contribution of SVs to disease risk in large cohorts.
SVs can have a substantial phenotypic impact by disrupting gene function and regulation or modifying gene dosage. Further, recent studies have shown that SVs drive functional changes across populations and cell and tissue types3,4. Although the role of SVs is yet to be comprehensively assessed in the context of risk of sporadic PD, several SVs are causative of monogenic forms of Parkinsonism. Examples include partial deletions in the gene PARK2 that causes autosomal recessive PD5 and a SV encompassing the gene SNCA that causes autosomal dominant PD6. Since then, causative SVs have been reported in other familial PD genes, such as the genes PINK1 7 and PARK7 (DJ-1) 8.
In this study, we performed the first genome-wide characterization of SVs in sporadic PD. We detected a total of 227,357 SVs in 7,772 individual samples, and validated several new variants associated with PD risk. These results demonstrate that SVs may contribute to disease risk in PD and highlight the need for such variants to be considered in future surveys of the genetics of neurodegenerative diseases.
Methods
Short-Read WGS Samples
The following ten cohorts were used in this study: Biofind (https://biofind.loni.usc.edu/), Harvard Biomarkers Study (HBS) (https://amp-pd.org/unified-cohorts/hbs), North American Brain Expression Consortium (NABEC)9, Laboratory of Neurogenetics pathologically confirmed collection, the NINDS Parkinson’s Disease Biomarker Program (PDBP) (https://pdbp.ninds.nih.gov/), samples from the NIH Parkinson’s Disease Clinic (NIH PD CLINIC), the Parkinson’s Progression Markers Initiative (PPMI) (https://www.ppmi-info.org/), Wellderly (controls), and the United Kingdom Brain Expression Consortium (UKBEC). Clinical and demographic characteristics of the cohorts under study are shown in (Supplementary Table 1). Participants included PD cases clinically diagnosed by experienced neurologists. All PD cases met criteria defined by the UK Parkinson’s Disease Society Brain Bank10. Each cohort abided by the ethics guidelines set out by their institutional review boards, and all participants gave informed consent for inclusion in both their initial cohorts and subsequent studies. The research using data from the NIH Parkinson’s Disease clinic cohort was approved by the NIH Intramural IRB under protocol number 01-N-0206. The overall study, working with genetic information, is deemed “not human subjects research” by the NIH Office of IRB Operations, waiving IRB approval.
Short-read WGS data generation through AMP-PD has been reported in detail previously by Iwaki et al11. Briefly, DNA sequencing was performed using two providers, Macrogen or Uniformed Services University of the Health Sciences (USUHS). Paired-end read sequences were processed in accordance with the pipeline standard developed by the Centers for Common Disease Genomics12. The GRCh38DH reference genome was used for alignment as specified in the standardized functional equivalence (FE) pipeline13. The Broad Institute’s implementation of this FE standardized pipeline, which incorporates the GATK (2016) Best Practices14, is publicly available and used for WGS processing. SNVs and indels were called from the processed WGS data using the GATK (2016) Best Practices 14 using the Broad Institute’s workflow for joint discovery and Variant Quality Score Recalibration (VQSR) 15. For quality control, each sample was checked using common methods for genotypes and sequence related metrics.
Structural Variant discovery
For SV discovery and downstream filtering, the Broad Institute GATK-SV pipeline was run in cohort mode https://github.com/broadinstitute/gatk-sv16. All computations were finished on the Google Cloud Platform (https://cloud.google.com). We filtered the dataset using 10 QC measurements (median sequencing coverage in 100bp bins, dosage bias score δ, autosomal ploidy spread, Z-score of outlier 1Mb bins, chimera rate, pairwise alignment rate, read length, library contamination, ambiguous sex genotypes, and discordant inferred and reported sex) and excluded 164 samples (2.03%) for failing at least one criterion. We kept samples with non-canonical sex chromosome configurations in their batches and manually removed all raw SV calls on X/Y from their raw VCF files. We followed the batch scheme designed by Collins et al to subdivide all samples that passed QC into 20 batches with ~400 samples per batch16. The ratio of female samples and male samples in each batch is around 1:1.19, balanced across all batches.
The complexity of calling SVs from short-read sequencing data requires the use of multiple SV calling tools in order to accurately and completely capture the different types of SVs. For example, calling copy number variants requires a different algorithm than calling mobile element insertions, hence separate SV tools are needed. Once the individual tools are run separately a multi-algorithm pipeline such as GATK-SV is then required to merge the overlapping SV calls from the multiple callers into one final call set and perform downstream filtering. In this present study, the SV evidence was collected from three different SV algorithms (Manta v1.417, MELT v2.2.0 18, and Wham v1.7 19) and CNV calls using cn.MOPS v1.20.1 20 and GATK gCNV21. The GATK-SV pipeline integrates the SV calls from the three algorithms and the CNV calls of each sample and standardizes the calls to meet specifications required for the SV discovery pipeline.
For filtering, we ran all four downstream filtering steps included in the GATK-SV pipeline: minGQ filtering, FilterOutlierSamples, BatchEffect and FilterCleanupQualRecalibration. The final filtering step usually requires trio data, however since the PD cohort lacks family structures, we ran minGQ filtering using the table pre-trained with 1000 genomes samples at the 1% FDR thresholds. 142 outliers were removed in the FilterOutlierSamples step, executed the procedures of BatchEffect, and ran FilterCleanupQualRecalibration on the remaining cleaned 7772 samples. The final SV callset included 366,555 SV calls.
Genetic Analysis
Filtering
Initial sample inclusion criteria included: age at disease onset or last examination at 18 years of age or older, no genetically ascertained relation to other samples (proportional sharing at a maximum of 12.5%) at the cousin level or closer, and majority European ancestry confirmed through principal-components determined by HapMap3. All individuals recruited as part of a biased and/or genetic cohort, such as GBA and LRRK2 rare variant carriers within a specific effort of PPMI cohort, were also excluded. After sample QC, a total of 2,585 PD cases and 2,779 neurologically healthy controls were included. PD cases ranged from 19 to 92 years of age of onset. Control subjects ranged from 19 to 110 years of age. For the association analyzes in order to assess the impact of high-quality variants, SVs with the filter label “PASS” were extracted from the final SV callset leaving a total of 227,357 biallelic autosomal SVs.
Genome-wide Association Study
We performed a SV PD GWAS (n= 2,585 cases, 2,779 controls) using logistic regression in PLINK (v2.0) with a minor allele frequency threshold of >1%. Principal components (PCs) were generated in PLINK (v1.9) for the common SNV datasets and common SV dataset separately. The step function in R MASS package was used to identify the minimum number of PCs required to correct for population substructure22 using both sets of PCs. Based on this analysis sex, age and 18 PCs were incorporated in the model. Overall genomic inflation was minimal with a lambda estimate of 1.011 and a lambda scaled to 1,000 cases and 1,000 controls at 1.004. Multiple test correction was handled using standard Bonferroni correction in PLINKv1.9 under default settings.
Linkage Disequilibrium Analysis
We next integrated the new SV dataset with the corresponding SNV data to identify if any SV tags any of the 90 PD risk SNVs. LD between SNVs and SVs was computed with the “--r2 inter-chr dprime” parameter in Plink v1.923.
Rare variants within PD genes
We wanted to identify variants within PD causal genes that were only present in PD cases. To do this we included 21 genes that have been reported to carry mutations that cause PD. The following genes were included: SNCA, PRKN, UCHL1, PARK7, LRRK2, PINK1, POLG, HTRA2, ATP13A2, FBX07, GIGYF2, GBA, PLA2G6, EIF4G1, VPS35, DNAJC6, SYNJ1, DNAJC13, TMEM230, VPS13C and LRP10.
Long-Read Structural Variant Confirmation
Matched Oxford Nanopore Technologies (ONT) long-read sequencing data was generated for 8 PPMI samples from blood to in-silico validate the SV of interest that were discovered from the short-read sequencing data. The samples were processed and sequenced using a protocol optimized for population scale long-read sequencing from frozen human blood (https://dx.doi.org/10.17504/protocols.io.ewov1n93ygr2/v1).
Fast5 files containing raw signal data were obtained from sequencing performed using minKNOW v21.05.13. All fast5 files were used to perform “super accuracy” basecalling on each sample with Guppy v6.0.1. Fastq files that passed quality control filters in the basecalling step were then mapped to the GRChg38 reference genome.The resulting sam files were sorted, converted to bams and indexed using samtools24 and one final bam file per sample was created. Chimera rate was calculated using the Liger2LiGer tool 25.
To detect and genotype SVs in the matched long-read sequencing data Sniffles2 26 v2.0.3 was run using default parameters. To improve SV calling in repetitive regions, the “–tandem-repeats” option was used. To filter out possible false positive SV calls Survivor27 v1.0.7 was used with the “ –filter” option to remove SV below 50bp.
Next to calculate the overall in silico confirmation rate of the short-read GATK-SV calls and validate the PD associated short-read SV of interest, Truvari28 v3.1.2 was run using the default parameters along with the --pctsim=0 parameter to turn off sequence comparison. To note, Truvari only classifies SVs as “confirmed” if the SV type (e.g., INS, DEL, DUP) is an exact match between the two callsets being compared. Because the naming of SVs was different between the two tools (i.e., with GATK-SV the type is named SVTYPE=DUP and with Sniffles2 SVTYPE=INS) before we ran Truvari all SVTYPE=DUPs in the GATK-SV callset were converted to SVTYPE=INS.
Results
Structural Variant discovery and distribution
Using the largest PD WGS short-read sequencing dataset available, we surveyed 7772 individuals at a mean genome coverage of 30X. For SV discovery and genotyping we used the GATK-SV pipeline to capture the main classes of SVs. Using this approach, we genotyped a total of 366,555 SVs and then filtered for high-quality variants, leaving a total callset of 227,357 SVs. In line with recent population studies that estimate that 401–10,884 SVs can be detected per short-read genome 16,29–32, our final callset contained on average 5,626 SVs per genome with a median of 1361 insertions, 2991 deletions, 1194 duplications, 115 complex SVs and 11 inversions (Figure 2). Also, in line with previous population-scale SV studies, the majority of the SVs were small (median 329 bp in size) with 21.40% < 100bp and 62.12% < 1kb in size (Figure 3). As expected, we observed three main peaks of insertion size at around 300bp, 2.5kb and 6kb, corresponding to Alu, SVA and LINE mobile elements insertions16. The majority of SVs are only discovered in one individual, or rare (46.69 % minor allele frequency > 0.01%). Overall, these results demonstrate that our dataset of SV discovery in this PD series contains variants consistent with expectations from other surveys of the human genome.
Figure 2: Properties of SVs detected in the average genome.
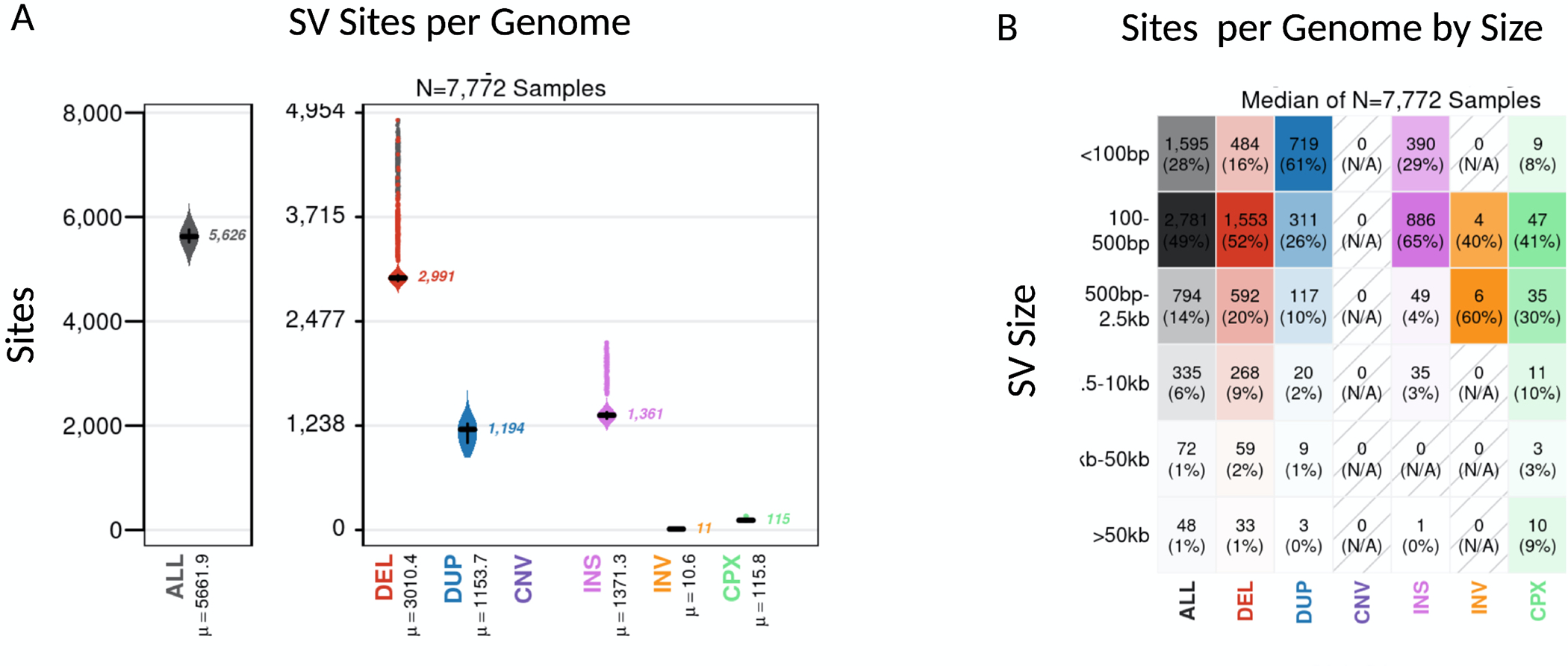
We analyzed a total of 7772 short-read genomes after quality control. The plots show the breakdown across SV class and size. a) Overall, on average each genome carried 5,626 SV, with a median of 1361 insertions, 2991 deletions, 1194 duplications, 115 complex SVs and 11 inversions. b) The majority of SVs were small with a medium size of 329 bp. Overall only a total of 8% of SV per genome were larger than 2.5kb and 1% of SVs per genome were >50kb.
Figure 3: Size and allele frequency distribution of “PASS” SVs in the short-read data.
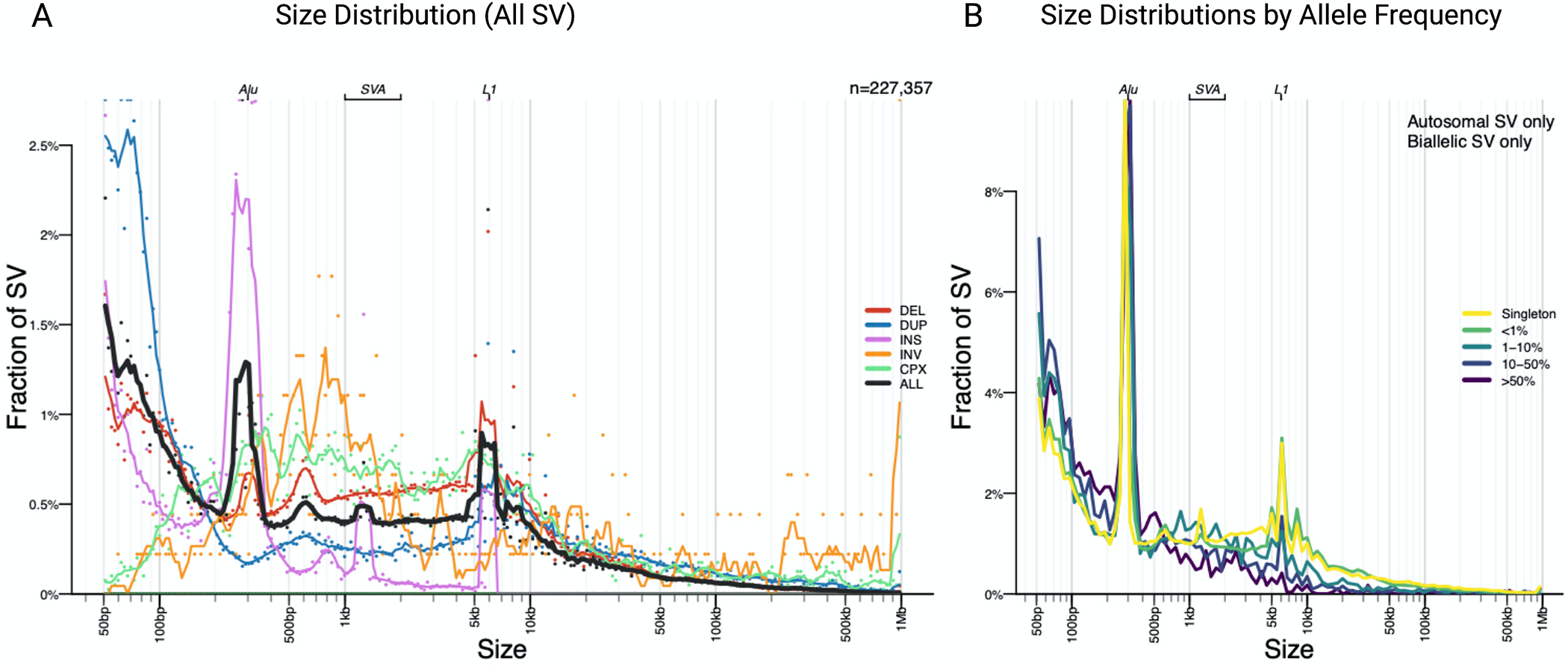
a) The majority of SVs are small and rare. As previously reported in other large-scale short-read studies three peaks are observed at 300bp, 2kb and 6kb, representing Alu, SVA and LINE1 mobile element insertions respectively. b) Most SVs were singleton variants (46.87%) or rare (AF<1%) (46.69 %).
Structural Variants are candidate causal variants at Parkinson’s Disease risk loci
Previous studies have shown that SVs are often localized to GWAS loci and are strong candidate causal variants for hundreds of human traits 16,33,34. To identify SVs that may drive signals at PD risk loci we integrated our SV data set with prior, SNV, based GWAS for PD risk. After filtering for SVs that are co-inherited with the lead GWAS risk variant at each locus, we nominated eight SVs that may explain PD risk at eight distinct genomic regions.
SV detection from short-read sequencing data can lead to false positive identification of SVs, especially in repetitive regions35, and hence it is crucial to validate nominated events. We therefore performed extensive SV validation by generating matched long-read sequencing data on a subset of individuals from the discovery cohort. We first optimized a protocol to yield high-quality long-read sequencing data from frozen human blood samples (see materials and methods). A SV was considered confirmed if there was evidence of the SV in the corresponding long-read sequencing data and high genotype concordance across samples (defined here as > 60% of genotypes matching between the two datasets). Of the eight variants tested from the short read sequencing data, three SVs were confirmed with the matched long-read sequencing data with high confidence (Supplementary Table 2).
Two of the validated SVs; PD_DEL_chr4_14749 and PD_DEL_chr6_2338 are in moderate LD with the PD risk SNVs rs62333164(r2=0.33, D’=0.82) (Nalls 2019 PD GWAS, p value of SNV= 2×10 −10, OR= 0.94) and rs4140646 (r2=0.20, D’=0.65) (Nalls 2019 PD GWAS, p value of SNV= 5.62×10 −12, OR= 1.09) respectively. PD_DEL_chr4_14749 is a 0.42kb intragenic deletion 35kb downstream of the gene NEK1. PD_DEL_chr6_2338 SV is a 0.33kb intragenic deletion 2.5kb upstream of the gene ZSCAN9. Both SVs are deletions of a reference Alu mobile element. In addition, the validated SV, PD_DEL_chr20_597 (Figure 4.A) is in strong LD with the PD risk SNV rs77351827 (r2=0.89, D’=0.95) (Nalls 2019 PD GWAS, p value of SNV= 8.87×10 −9, OR= 1.08) (Figure 4.B). PD_DEL_chr20_597 is a 1.95kb intronic deletion within intron three of the gene LRRN4 (Figure 4.C) that spans two reference Alu mobile elements.
Figure 4: A 2kb deletion within intron 3 of LRRN4 is a strong candidate for causal variant at the chr20 rs77351827 locus.
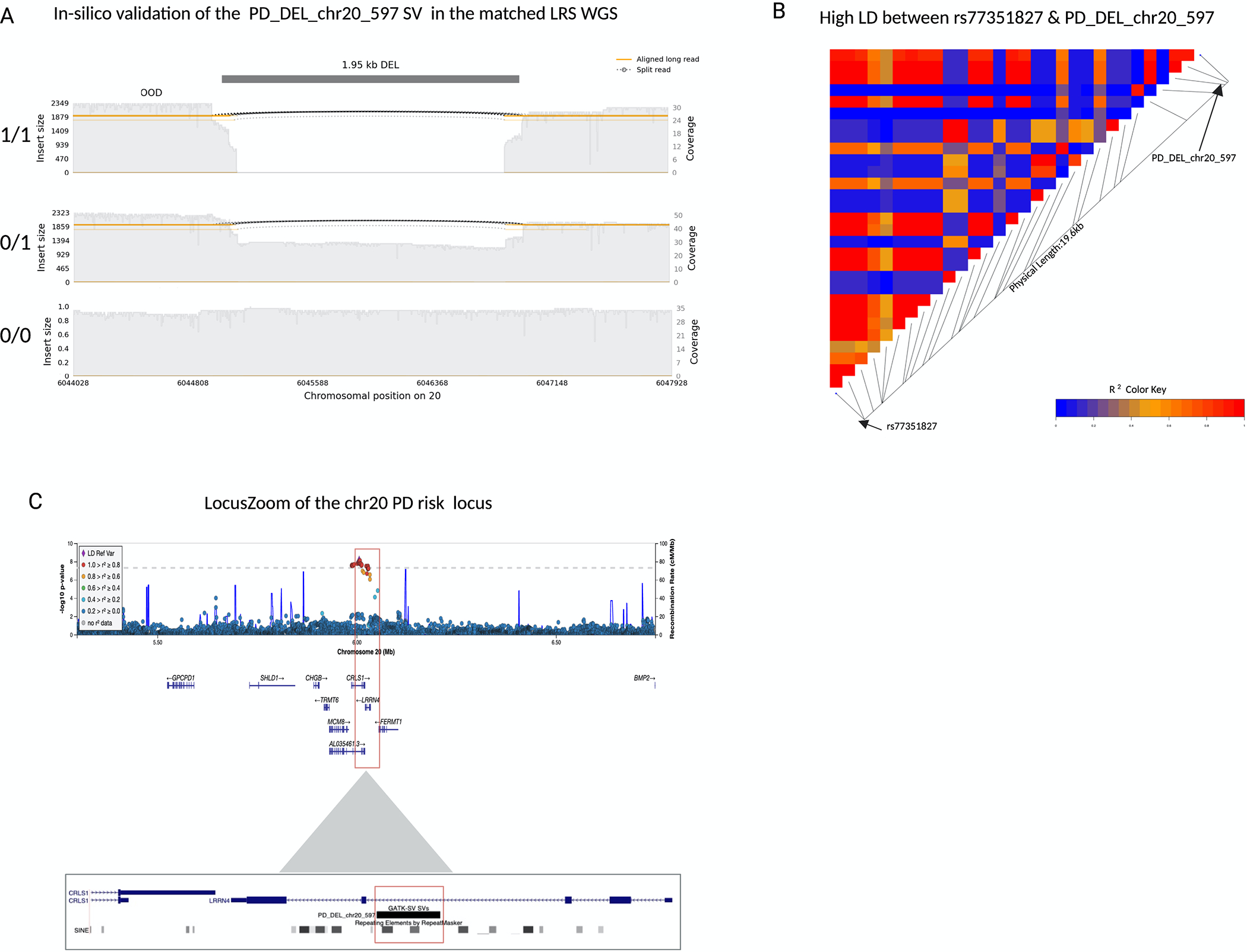
a) A samplot image showing the ~2kb deletion at chr20. Aligned regions are marked in orange and the gap represents the deletion coded in black. The height of the alignment is based on the size of its largest gap. The three sequence alignment tracks follow, each alignment file plotted as a separate track in the image. The coverage for the region is shown with the gray-filled background. The SV genotypes (homozygous deletion, heterozygous deletion, and homozygous reference allele/no deletion) that were predicted by GATK-SV from the short-read sequencing data are annotated on the left of the corresponding tracks. Each genotype was confirmed in-silico by the matched long-read sequencing data. b) A LDheatmap showing pairwise LD measurements measured in R2 between the 2kb PD_DEL_chr20_597 deletion and rs77351827. High R2 values are shown in red and low R2 values in blue. PD_DEL_chr20_597 is in high LD with the lead PD risk SNV of this locus rs77351827(r2=0.89, D’=0.95). c) Locuszoom plot of the association signal at the chr20 rs77351827 PD risk locus from the Nalls 2019 PD SNV meta-analysis. The gene LRRN4 lies directly under the risk signal and the schematic below shows the location of the deletion within intron 3 of the gene.
Further, to assess whether the SVs could be driving the PD risk signals at these loci, we attempted to run conditional analyses, however due to the current sample size of our dataset no signal existed at the three loci (Supplementary Figure 1). In summary here we identify three structural variants that are strong candidates for causal variants for future follow-up functional studies.
Structural Variant Genome-wide association analysis
Using a GWAS approach with 2585 cases and 2779 controls, we identified a total of nine genome-wide significant SV association signals (Supplementary Figure 2) with a genome-wide inflation factor λ1000 of 1.004 (Supplementary Figure 3). However, in-silico confirmation using the matched long-read sequencing data indicated that the 9 “hits” were potentially false positive signals because either 1) there was no evidence of the SV in the matched long-read data or 2) the SV genotyping accuracy was low across the 8 tested long-read samples. To note, for the majority of loci, although a non-reference SV was present in that region, the genotype concordance was low across samples, so we could not confirm the association signal. Detailed summary statistics from the SV GWAS can be found in (Supplementary Table 4).
Of interest are two large deletions (PD_DEL_ch17_4739 and PD_DEL_chr17_4744) at the 17q.21.31 locus containing the MAPT gene that were significant hits in our GWAS analysis. This locus lies within a 1.5Mb inversion region with two distinct haplotypes, H1 and the inverted H2 haplotype. The major haplotype H1 has been associated with risk of many neurodegenerative diseases including Alzheimer’s Disease36, Progressive supranuclear palsy37 and PD 38. While these deletions do not validate in the long-read sequencing data the variants are in high LD with the H1/H2 tagging SNV rs8070723 (PD_DEL_ch17_4739; r2=0.98, D’=0.99 and PD_DEL_chr17_4744 r2=0.98, D’=0.99) suggesting that they are artifacts that likely represent the known large inversion in this region with correct genotyping accuracy but inaccurate SV detection.
Further analysis of the 17q21.31 locus identified that a large 675kb inversion (PD_CPX_chr17_84) is present in the non-filtered short-read GATK-SV callset, however it was removed from downstream analyses due to the Hardy Weinberg filtering. If this inversion is the known large inversion in the 17q21.31 locus it should be in complete LD with rs8070723 the H1/H2 proxy SNV. However, the LD was r2=0.65 and D’=0.90, suggesting that the genotyping accuracy of this large inversion is moderate in the short-read callset and also inaccurate with SV detection given the difference in size (i.e., our long-read sequencing data reported 675kb compared to the 900kb reported in the literature). We next aimed to investigate carriers of the inversion in the long-read data. Two of the long-read samples were predicted to carry the inversion based on rs8070723 and PD_CPX_chr17_84 genotypes. Analysis of this region in the long-read genomes identified that the two predicted carriers in fact carried a 150kb duplication as called by Sniffles2. This suggests an artifact-based detection on the large inversion with inaccurate SV detection but with correct genotype like that of the two deletion SVs called from the short-read sequencing data in the MAPT region. So, for this large inversion that is associated with many neurodegenerative diseases, we highlight that both the short and long-read datasets were unable to both accurately detect and genotype the SV using current algorithms.
Overall, we highlight that short-read sequencing SV studies can result in a high number of false positives even when very stringent QC filtering is used. Therefore, it is important to perform experiments to validate these hits so false associations are not reported.
Long-read Sequencing is required to capture most Structural Variants in the genome
We sought to characterize the genome-wide distribution of SV in long-read datasets. In line with recent population studies that report ~25,000 SV per long-read genome 39,40, each genome carried a mean of 27,277 SVs. Unlike the short-read callset, which contains predominantly deletions, over half of the long-read SVs were insertions. Overall a median of 14,481 insertions, 12,532 deletions, 43 duplications, 98 inversions and 123 translocations were discovered per genome.
To assess the sensitivity and specificity of the short-read SV callset we performed a detailed comparison of the short-read and matched long-read sequencing data. It is important to note here that although recent benchmarking using 30X ONT data reported high accuracy with Sniffles226, the following confirmation rates assume that the long-read SV calls represent the absolute ground truth, which will not be the case for 100% of the long-read SVs.
To benchmark our dataset against other large scale SV studies, we compared our confirmation rates to those from Collins et al 16. In line with this prior approach, we only focused on “high-confidence” SVs that had breakpoint-level read support (‘split-read’ evidence) and that did not span annotated simple repeats or segmental duplications. When we focused on this restricted list of variants the overall confirmation rate was ~80%, lower than the 94% reported by Collins et al. The final filtering step in GATK-SV removes variants based on the genotype quality across populations. Usually this step requires trio families to build the minGQ filtering model. However we used a model pre-trained with the 1000 Genomes samples as there were no trios present in our short-read WGS dataset. Although we implemented a very stringent final FDR cut off (1%) for our calls, as expected this likely leads to a higher false-positive rate compared to a filtering model based on family data.
If we expand the comparison and focus on all the “PASS” short-read SVs that were used in the genetic analysis here, on average 72% of the tested SVs were confirmed in the long-read sequencing data (Figure 5). A SV was classed as confirmed if there was evidence of that variant in the matched long-read data. For the confirmed SVs, on average the genotype concordance was 78% per genome. As expected, the majority of confirmed SVs were deletions (85% of the tested short-read deletions were confirmed). In line with recent studies that highlight the difficulty of calling SV in repetitive regions with short-read sequencing data35, duplications represented 40% of the false positives in the short-read callset. When we assessed the overlap between the long-read and short-read sequencing data, 84% of the SV in the long-read data were not present in the short-read callset and the majority (58%) of the SV detected solely by long-read sequencing were insertions (Figure 5).
Figure 5: Comparison of SVs called with short-read and long-read in eight matched PPMI blood samples -.
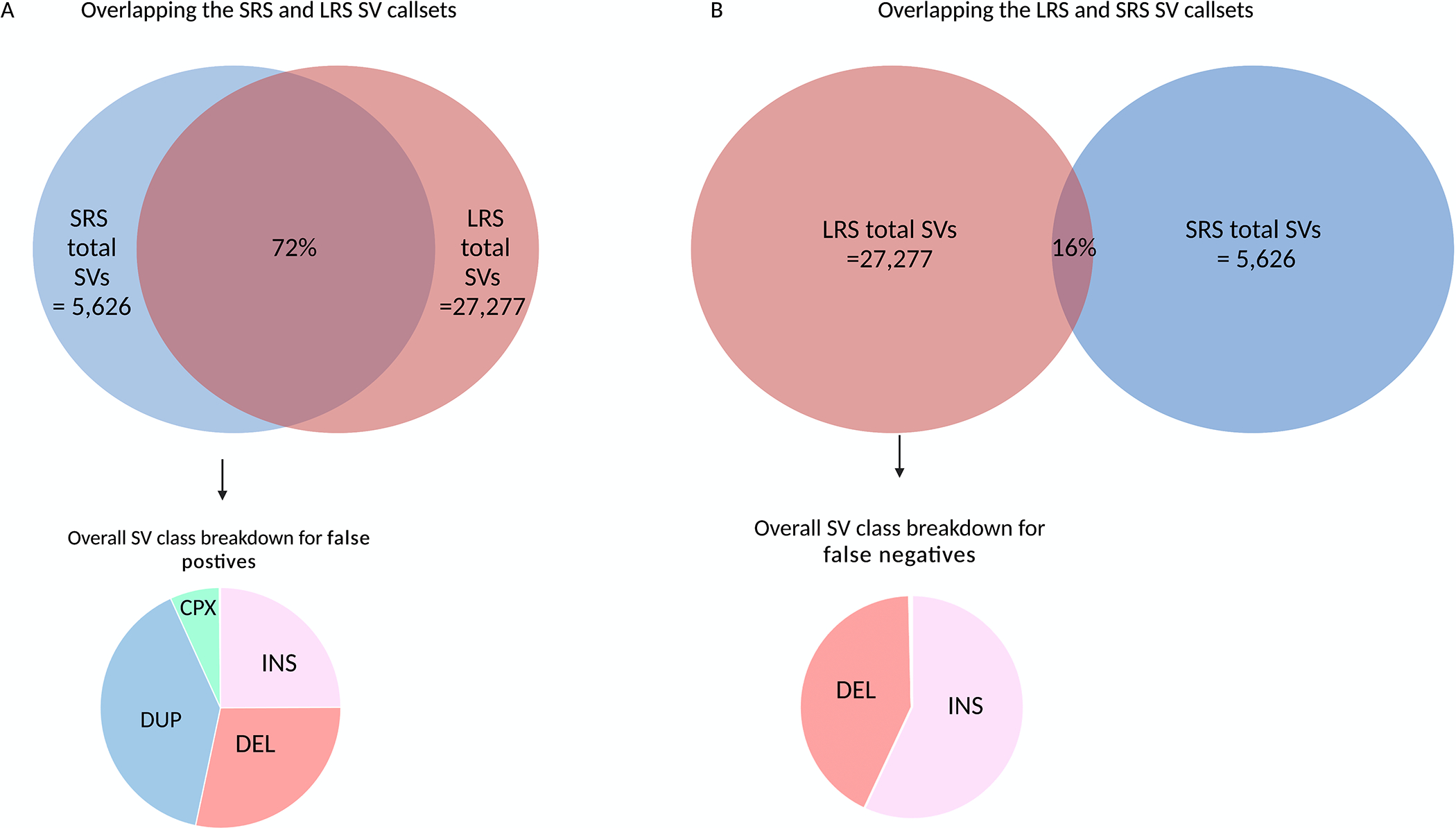
ONT long-read sequencing detects significantly more SV than the short-read sequencing on average per genome. a) On average 5,626 SVs were detected per short-read genome compared to 27,277 with long-read sequencing. Of the 5,626 SV discovered in the short-read sequencing data, 72% of the SV were confirmed in-silico with long-read sequencing. As expected, duplications drove the false positive rate. b) The majority of the SV in the genome cannot be detected with sequencing data alone. Of the 27,277 SVs detected with long-read sequencing, only 14% of the SVs were present in the short-read callset. Most of these false negative calls, i.e SVs that were detected by long-read sequences but not present in the short-read callset were insertions.
Rare variants within reported causal or high-risk genes for Parkinson’s Disease
Although the primary focus of this present study was to characterize the role of common variants in PD given the utility of this new SV dataset, we aimed to report rare SV of potential interest. To date, rare variants in more than 20 genes have been reported to cause PD 41. We explored this in our SV callset by extracting SVs within these genes that were only present in PD cases. A total of 106 rare variants lay within these genes in cases only (Supplementary Table 5). It’s important to stress here that this list of SVs were not validated and based on the rate of validation between our short-read and long-read SV callsets some of the variants may be false positives.
Discussion
Gaining a full understanding of the genetic architecture underpinning PD is critical for the development and application of therapeutic treatments that could slow or stop disease. Despite this, most of the common variants driving disease are unknown and even for well-characterized loci, the identity of the functional effector variant remains unknown. One reason for this is that previous genetic studies have focused solely on SNVs which represent only a fraction of the genetic variation in the human genome. SVs are a major source of genetic diversity, however this type of variability, while widespread and of significant functional consequence, has been difficult to assay accurately, even with the evolution of short read whole genome sequencing42. Here we report the first comprehensive genome-wide analysis of SVs in PD to date. We characterized SVs in 7772 individuals, representing over 412 million nucleotides of unexplored genetic variation and validated three new variants associated with PD risk.
A major bottleneck in genetics is determining the true causal variant(s) and functionally affected gene(s) within the associated risk loci. Consequently, only a few PD risk loci have been functionally validated and these mainly consist of genes that are known to cause monogenic forms of PD. One of the main motivations of this present study was to integrate new SV data at these loci with the hope that a more complete understanding of the genetic variation at these regions would provide insight into the biology driving risk of disease. Although this initial work is the largest and most complete assessment of SV in PD to date, it is still of relatively modest size for genetic discovery, hence no genome-wide significant variants were identified in the GWAS. However, we identified three SVs that are located at a known risk loci in strong (PD_DEL_chr20_597)or moderate LD (PD_DEL_chr4_14749 and PD_DEL_chr6_2338) with the lead risk SNV at each locus. Of interest, all three variants were deletions of one or more reference Alu mobile elements. Alu mobile elements are usually ~ 300 bp in length and constitute ~11% of the human reference genome with over one million copies43. Recent studies have shown that presence/absence variation within reference Alu elements can have a profound functional impact 44,45.
This study suggests that a 2kb deletion within intron 3 of the gene LRRN4 (Leucine-rich repeat neuronal protein-4) is a strong candidate for causal variant at the PD risk locus at chromosome 20 (Locus 77 - https://pdgenetics.shinyapps.io/GWASBrowser/). LRRN4 is a type I transmembrane protein that is a member of the LRRN family. Previous studies report that it is expressed in the lung, heart, ovary, hippocampus and cortex and suggest it may be involved in hippocampus dependent memory retention in mice46. Clearly, it will be important to further understand the importance of these and other yet to be identified SVs on risk loci, and this will likely require a combination of higher powered genetic investigation, along with the integration of functional modalities to include genomic and transcriptomic regulatory assays.
Although this study marks a significant step forward for cataloging structural variation in PD, indicative of any SV analysis from short-read sequencing data, it has several limitations. Firstly, very stringent variant QC parameters were used to reduce the false positive rate of the short-read SV calls. Therefore, it is possible that disease relevant SVs detectable via short-read sequencing may have been filtered out from the downstream analyses. Second, as shown by our long-read and short-read comparison, most SVs are not detectable using short-read sequencing data alone, suggesting that we have only been able to assess a small fraction of the SVs present in each genome. Taken together these factors highlight that this study likely represents a massive underestimate of the contribution of SV to risk of PD.
Our study emphasizes that long-read sequencing data is needed to resolve much of the genetic variation in the human genome. Generating population-scale long-read WGS datasets to capture SVs that are currently hidden from traditional methods is an essential step towards solving the architecture of complex genetic disorders47. For neurodegenerative diseases specifically, there are two large-scale initiatives underway to generate such datasets. The first is a Global Parkinson’s Genetics Program (GP2, www.gp2.org) led initiative. GP2 is the first supported resource project of the Aligning Science Across Parkinson’s48. Through this endeavor, GP2 will long-read sequence ~1000 PD cases and control blood samples. The second initiative is led by the NIH Center of Alzheimer’s Dementias and Related Dementias (CARD, https://card.nih.gov), whereby CARD is generating long-read WGS in a total of ~ 4000 brain samples to catalog SVs in Alzheimer’s disease, Lewy Body Dementia, Frontotemporal Dementia and neurologically healthy controls.
Conclusion:
In conclusion, this present study represents a step forward in understanding the genetic factors contributing to PD risk. We performed the first SV GWAS of PD and ran comprehensive validation analyses, identifying three structural variants associated with PD risk. These variants are strong candidates for causal variants at these loci, therefore an essential next step will be to run follow-up functional studies to identify the true gene or genes driving the risk of disease at these regions. Without a complete understanding of the gene(s) truly involved in disease, identifying viable therapeutic targets is extremely challenging. Through this study we also show the limitations of using short-read sequencing data for calling SVs and report that even with very stringent QC a high number of false positive associations are observed, thus extensive experimental validation studies are required. Finally, we show the benefits of using long-read sequencing data and present a workflow for generating high quality long-read sequencing data. With this data we are powered to detect many new variants once invisible with previous methods.
Supplementary Material
Figure 1: SV analysis workflow.
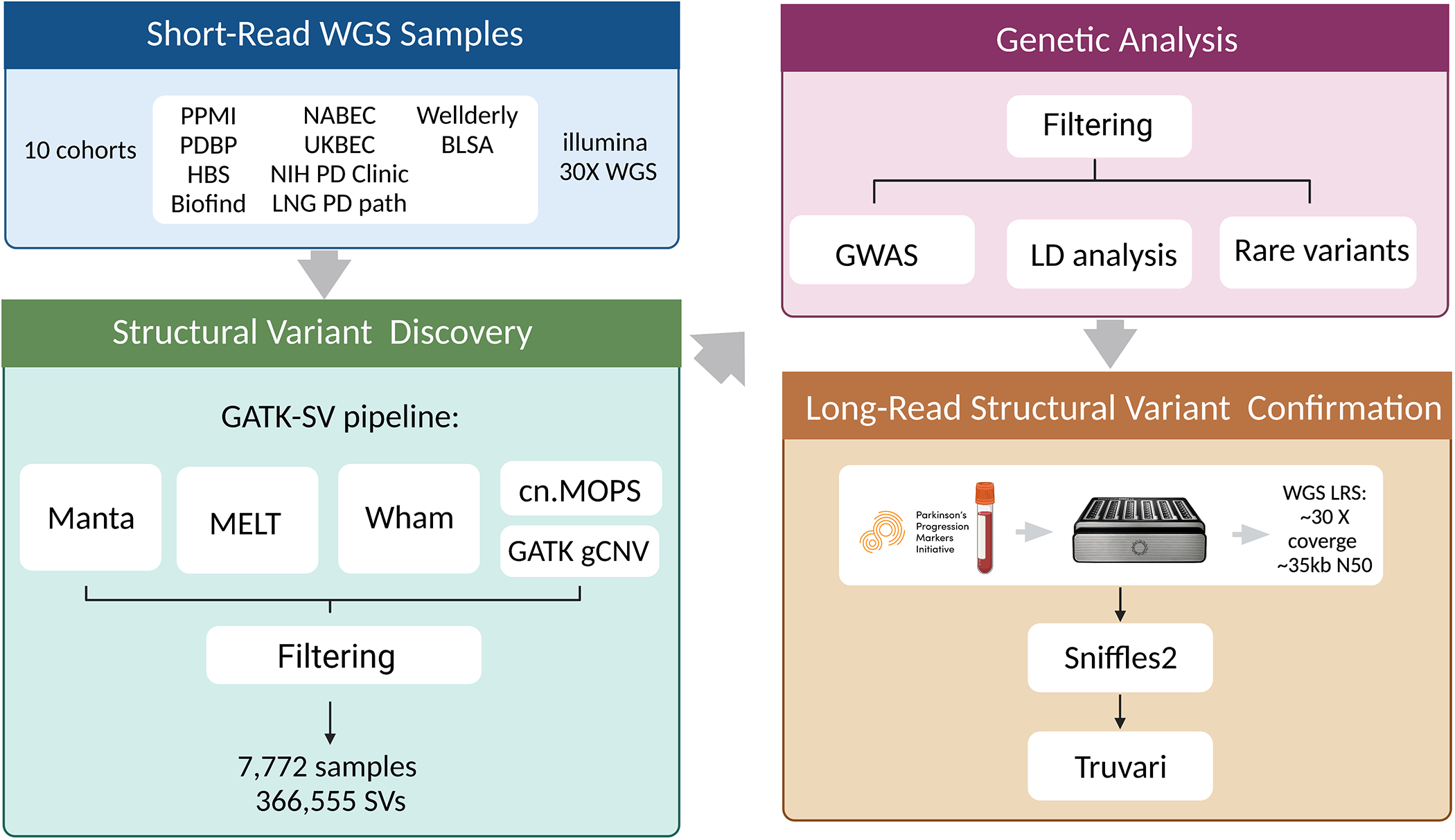
This figure describes the study design behind the analyses included in this report.
Acknowledgements
We would like to thank all the participants who donated their time and biological samples to be a part of this study. This work was supported in part by the Intramural Research Programs of the National Institute on Aging (NIA) and the National Institute of Neurological Disorders and Stroke (NINDS), part of the National Institutes of Health, Department of Health and Human Services; project numbers Z01-AG000949, 1ZIANS003154. A full list of acknowledgements for each of cohort used in this study can be found in the Supplementary material.
Footnotes
Potential Conflicts of Interest
D.V, K.L and M.A.N.’s participation in this project was part of a competitive contract awarded to Data Tecnica International LLC by the National Institutes of Health to support open science research. M.A.N. also currently serves on the scientific advisory board for Clover Therapeutics and is an advisor to Neuron23 Inc. BT currently serves on the Editorial board of Clinical Medicine, JNNP, NBA, is an Associate Editor for Brain and has a collaborative research agreement with Ionis Pharmaceuticals, Roche and Optimeos. FJS receives research support from Illumina, ONT and PacBio. AM works for ONT. M.E.T. receives research funding and/or reagents from Levo Therapeutics, Microsoft Inc., and Illumina Inc.
References
- 1.Nalls MA et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pang AW et al. Towards a comprehensive structural variation map of an individual human genome. Genome Biol. 11, R52 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Han L et al. Functional annotation of rare structural variation in the human brain. Nat. Commun. 11, 2990 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scott AJ, Chiang C & Hall IM Structural variants are a major source of gene expression differences in humans and often affect multiple nearby genes. Genome Res. (2021) doi: 10.1101/gr.275488.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kitada T et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature vol. 392 605–608 Preprint at 10.1038/33416 (1998). [DOI] [PubMed] [Google Scholar]
- 6.Singleton AB et al. α-Synuclein Locus Triplication Causes Parkinson’s Disease. Science vol. 302 841–841 Preprint at 10.1126/science.1090278 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Bonifati V et al. Early-onset parkinsonism associated with PINK1 mutations: Frequency, genotypes, and phenotypes. Neurology vol. 65 87–95 Preprint at 10.1212/01.wnl.0000167546.39375.82 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Bonifati V et al. DJ-1( PARK7), a novel gene for autosomal recessive, early onset parkinsonism. Neurological Sciences vol. 24 159–160 Preprint at 10.1007/s10072-003-0108-0 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Gibbs JR et al. Abundant Quantitative Trait Loci Exist for DNA Methylation and Gene Expression in Human Brain. PLoS Genetics vol. 6 e1000952 Preprint at 10.1371/journal.pgen.1000952 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gelb DJ, Oliver E & Gilman S Diagnostic Criteria for Parkinson Disease. Archives of Neurology vol. 56 33 Preprint at 10.1001/archneur.56.1.33 (1999). [DOI] [PubMed] [Google Scholar]
- 11.Iwaki H et al. Accelerating Medicines Partnership: Parkinson’s Disease. Genetic Resource. Mov. Disord. 36, 1795–1804 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Centers for Common Disease Genomics. Genome.gov https://www.genome.gov/Funded-Programs-Projects/NHGRI-Genome-Sequencing-Program/Centers-for-Common-Disease-Genomics.
- 13.Regier AA et al. Functional equivalence of genome sequencing analysis pipelines enables harmonized variant calling across human genetics projects. Nat. Commun. 9, 4038 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van der Auwera GA et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11.10.1–33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Website. https://github.com/gatk-workflows/broad-prod-wgs-germline-snps-indels.
- 16.Collins RL et al. A structural variation reference for medical and population genetics. Nature 581, 444–451 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen X et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Gardner EJ et al. The Mobile Element Locator Tool (MELT): population-scale mobile element discovery and biology. Genome Res. 27, 1916–1929 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kronenberg ZN et al. Wham: Identifying Structural Variants of Biological Consequence. PLoS Comput. Biol. 11, e1004572 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klambauer G et al. cn.MOPS: mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res. 40, e69 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van der Auwera GA & O’Connor BD Genomics in the Cloud: Using Docker, GATK, and WDL in Terra. (O’Reilly Media, 2020). [Google Scholar]
- 22.Chang CC et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Purcell S et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. The American Journal of Human Genetics vol. 81 559–575 Preprint at 10.1086/519795 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Danecek P et al. Twelve years of SAMtools and BCFtools. Gigascience 10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Website. https://github.com/rlorigro/Liger2LiGer.
- 26.Smolka M et al. Comprehensive Structural Variant Detection: From Mosaic to Population-Level. bioRxiv 2022.04.04.487055 (2022) doi: 10.1101/2022.04.04.487055. [DOI] [Google Scholar]
- 27.Jeffares DC et al. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun. 8, 14061 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.English AC, Menon VK, Gibbs R, Metcalf GA & Sedlazeck FJ Truvari: Refined Structural Variant Comparison Preserves Allelic Diversity. Preprint at 10.1101/2022.02.21.481353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sudmant PH et al. An integrated map of structural variation in 2,504 human genomes. Nature vol. 526 75–81 Preprint at 10.1038/nature15394 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Werling DM et al. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat. Genet. 50, 727–736 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiang C et al. The impact of structural variation on human gene expression. Preprint at 10.1101/055962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brandler WM et al. Paternally inherited cis-regulatory structural variants are associated with autism. Science 360, 327–331 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jakubosky D et al. Properties of structural variants and short tandem repeats associated with gene expression and complex traits. Nat. Commun. 11, 2927 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beyter D et al. Long-read sequencing of 3,622 Icelanders provides insight into the role of structural variants in human diseases and other traits. Nat. Genet. 53, 779–786 (2021). [DOI] [PubMed] [Google Scholar]
- 35.Expectations and blind spots for structural variation detection from long-read assemblies and short-read genome sequencing technologies. Am. J. Hum. Genet. 108, 919–928 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jun G et al. A novel Alzheimer disease locus located near the gene encoding tau protein. Mol. Psychiatry 21, 108–117 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pastor P et al. Novel haplotypes in 17q21 are associated with progressive supranuclear palsy. Annals of Neurology vol. 56 249–258 Preprint at 10.1002/ana.20178 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Nalls MA et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 46, 989–993 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chaisson MJP et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Preprint at 10.1101/193144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Audano PA et al. Characterizing the Major Structural Variant Alleles of the Human Genome. Cell 176, 663–675.e19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blauwendraat C, Nalls MA & Singleton AB The genetic architecture of Parkinson’s disease. Lancet Neurol. 19, 170–178 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mahmoud M et al. Structural variant calling: the long and the short of it. Genome Biology vol. 20 Preprint at 10.1186/s13059-019-1828-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Consortium IHGS & International Human Genome Sequencing Consortium. Correction: Initial sequencing and analysis of the human genome. Nature vol. 412 565–566 Preprint at 10.1038/35087627 (2001). [DOI] [Google Scholar]
- 44.Goubert C, Zevallos NA & Feschotte C Contribution of unfixed transposable element insertions to human regulatory variation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 375, 20190331 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Payer LM et al. insertion variants alter gene transcript levels. Genome Res. (2021) doi: 10.1101/gr.261305.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bando T et al. Neuronal Leucine-Rich Repeat Protein 4 Functions in Hippocampus-Dependent Long-Lasting Memory. Molecular and Cellular Biology vol. 25 4166–4175 Preprint at 10.1128/mcb.25.10.4166-4175.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Coster W, Weissensteiner MH & Sedlazeck FJ Towards population-scale long-read sequencing. Nat. Rev. Genet. 22, 572–587 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schekman R & Riley EA Coordinating a new approach to basic research into Parkinson’s disease. Elife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.