

Abstract
Background:
Calcific aortic valve disease (CAVD) is characterized by a phenotypic switch of valvular interstitial cells (VICs) to bone-forming cells. Toll-like receptors (TLRs) are evolutionary conserved pattern recognition receptors at the interface between innate immunity and tissue repair. Type I interferons (IFN) are not only crucial for an adequate antiviral response, but also implicated in bone formation. We hypothesized that the accumulation of endogenous TLR3 ligands in the valvular leaflets may promote the generation of osteoblast-like cells through enhanced type I IFN signaling.
Methods:
Human VICs isolated from aortic valves were challenged with mechanical strain or synthetic TLR3 agonists, and analyzed for bone formation, gene expression profiles and IFN signaling pathways. Different inhibitors were employed to delineate the engaged signaling pathways. Moreover, we screened a variety of potential lipids as well as proteoglycans known to accumulate in CAVD lesions as potential TLR3 ligands. Ligand - receptor interactions were characterized by in-silico modeling and verified through immunoprecipitation experiments. Biglycan (Bgn), Tlr3, and Interferon-alpha/beta receptor alpha chain (Ifnar1) deficient mice, and a specific zebrafish model were used to study the implication of the BGN-TLR3-IFN axis in both CAVD and bone formation in vivo. Two large-scale cohorts (GERA, n = 55,192 with 3,469 aortic stenosis cases; UK Biobank, n = 257,231 with 2,213 aortic stenosis cases) were examined for genetic variation at genes implicated in BGN-TLR3-IFN signaling associating with CAVD in humans.
Results:
Here, we identify TLR3 as a central molecular regulator of calcification in VICs, and unravel BGN as a new endogenous agonist of TLR3. Posttranslational BGN maturation by xylosyltransferase 1 (XYLT1) is required for TLR3 activation. Moreover, BGN induces the trans-differentiation of VICs into bone-forming osteoblasts via the TLR3-dependent induction of type I IFNs. Intriguingly, Bgn−/−, Tlr3−/−, and Ifnar1−/− mice are protected against CAVD and display impaired bone formation. Meta-analysis of two large-scale cohorts with >300,000 individuals reveals that genetic variation at loci relevant to the XYLT1-BGN-TLR3-IFNAR1 pathway is associated with CAVD in humans.
Conclusion:
This study identifies the BGN-TLR3-IFNAR1 axis as an evolutionarily conserved pathway governing calcification of the aortic valve and reveals a potential therapeutic target to prevent CAVD.
Keywords: Toll-like receptor 3, calcific aortic valve disease, skeletal development, extracellular matrix, biglycan, protein maturation
Introduction
Calcific aortic valve disease (CAVD) is the third leading cause of cardiovascular related disease, constituting a major socioeconomic burden in the Western world1. Age is the principal risk factor for CAVD, and the aging of the population predicts an increased prevalence of CAVD 2. Valvular interstitial cells (VICs), the predominant cell type within the aortic valve, are specialized fibroblasts responsible for maintaining valvular function3. Progressive aortic calcification occurs when VICs acquire the phenotype of bone-forming osteoblasts4. There are currently no pharmacological options for slowing or preventing the progression of CAVD.
The aortic valve is relentlessly subjected to high levels of mechanical strain and cellular injury, in turn promoting inflammation and the formation of calcific lesions5. Endogenous alarm signals, including the release of RNA and protein from injured cells, activate Toll-Like Receptors (TLRs)6,7, which are typically involved in detecting pathogens8. The first described TLRs expressed on VICs were TLR2 and TLR4. Their activation by bacterial membrane components such as lipopolysaccharide (LPS) or peptidoglycan induces expression of pro-osteogenic factors, including BMP-2 and Runx2 via NOTCH1 and NFKB9. VICs from stenotic valves express higher levels of TLR2 and 4 and are more responsive to inflammatory stimuli, resulting in osteogenic activity10. IL-37 suppresses the osteogenic response upon TLR2 and TLR4 activation. TLR2 and TLR4 deficient mice are protected from high-fat diet induced changes of the aortic valve11. Besides TLR2 and TLR4, aortic valves express TLR912. TLR9 recognizes CpG motifs in nucleic acids, but also bind endogenous ligands including ECM components and lipids and is possibly involved in sustained inflammation and subsequent valvular calcification13. Here, we show that TLR3 is abundantly expressed in aortic valves, exhibiting a potential distinct role in the calcification process of aortic valves due to its unique signaling features.
TLR3 can be activated by extracellular dsRNA intermediates or by-products of viral infection, and by unknown endogenous agonists. Its activation on VICs was previously shown to induce osteogenic response through the NF-KB and ERK 1/2 pathways9. Besides NFKB, TLR3 activation appears to be a major pathway resulting in an IRF3-dependent induction of type I IFNs 8, therefore aiding in the control of physiological type I IFN levels14.
Type I IFNs have been implicated not only in the host response to viruses, but also in CAVD, physiological calcification and bone formation. Consistent with this role, Ifnb−/− and Ifnar1−/− mice, which have compromised type I IFN signaling, display osteoporosis 15,16.
Human Mendelian disorders associated with increases in the production of type I IFNs are known as ‘type I interferonopathies’ 17. The causal genes and the cells displaying enhanced type I IFN production differ between disorders, accounting for their clinical heterogeneity. Gain-of-function (GOF) mutations in the human IFIH1/MDA5 gene, which regulates the antiviral response, cause pathological vascular and valvular calcification with osteopenia in children with Singleton-Merton syndrome 18. Aicardi-Goutières syndrome, which is genetically heterogeneous, is characterized by pathological calcifications of the basal ganglia in the brain 19. Finally, ADAR-related type I interferonopathy has been linked to valvular calcifications in children 20. The impact of human inherited deficiencies of type I IFNs on calcification in physiology and disease is unknown.
Here, we show that the activation of TLR3 by endogenous ligands leads to type I IFN production, promoting bone formation in young mice but also triggering the phenotypic switch to osteoblast-like cells and contributing to the pathogenesis of CAVD in aged mice. We identified biglycan, as endogenous TLR3 ligand. Posttranslational BGN maturation by xylosyltransferase 1 (XYLT1) is required for TLR3 activation. We propose that the evolutionary advantage of TLRs for improved defense against infections might come at the cost of increased risk for cardiovascular disease, representing a model of pleiotropic antagonism at later age.
Methods
Institutional approval
Ethics committee approval for the use of human material was obtained from the Medical University of Innsbruck (IRB number AN2014–026 340/4.34), all subjects gave informed consent. Ethics committee approval was obtained for all animal experiments (BMWFW 66.011/0152-WF/V/3b/2014 and BMWF-66.011/0101-V/3b/2018). Experiments were performed in accordance with the ‘Guide for the Care and Use of Laboratory Animals’ published by the US National Institutes of Health (NIH Publication No. 85–23, 1996, revised 2011; available from: www.nap.edu/catalog/5140.html). All genetic analyses in GERA were approved by the appropriate review boards at Kaiser Permanente Northern California and the McGill University Health Centre (2015–1292). The UK Biobank was approved by the North West Multi-Center Research Ethics Committee (11/NW/0382) as a research tissue bank. Analyses performed on the UK Biobank data as part of application 41025 were approved by the internal review board at the McGill University Health Centre (2015–1292).
Availability of data and materials
All high-throughput data are available via GEO. Accession numbers are GSE138360 and GSE223543. The datasets generated and/or analyzed during this study are available from the corresponding author on reasonable request except for GERA and UK Biobank data, which are available directly from those cohorts upon application.
Cell culture
Valvular interstitial cells from patients undergoing aortic valve replacement or heart transplantation were isolated via collagen digestion, as previously described 21. WT and TLR3−/− human dermal fibroblasts were kindly provided by Jean-Laurent Casanova.
Mouse models
Experiments were performed blinded on C57BL/6N mice (Charles River Laboratories, Wilmington, MA) and Tlr3−/− mice (C57BL/6N background). Tlr3−/− mice were crossed with ApoE−/− mice obtained from Jackson Laboratories (Bar Harbor, ME). C57BL/6N, Tlr3−/−, Bgn−/− (C3.129S4(B6)-Bgntm1Mfy/Mmmh; MMRRC) and Ifnar1−/− (B6.129S2-Ifnar1tm1Agt/Mmjax; Jackson Laboratory, Bar Harbor, ME) mice were used. For the induction of aortic valve stenosis, mice were switched onto a proatherogenic high-fat/high-carbohydrate (HF/HC) diet without added cholesterol (#F3282, BioServ, Frenchland, NJ) as previously described.
Echocardiography
Transthoracic echocardiography was performed as previously described 22,23.
Cohorts for genetic associations with aortic stenosis in humans
After excluding individuals with congenital valvular heart disease (International Classification of Diseases, Ninth Revision [ICD-9] 746–747), aortic stenosis cases in the GERA cohort 24 were defined as participants with ICD-9 424.1 or a procedure code for aortic valve replacement in their electronic health records between January 1996 and December 2015, inclusive; all other participants were deemed controls. Analyses in the GERA cohort were limited to unrelated individuals of self-reported European ancestry, aged 55 years or older (n = 55,192 [3,469 aortic stenosis cases]).
In the UK Biobank, following the exclusion of participants with congenital valvular heart disease (ICD-9 746–747 or International Classification of Diseases, Tenth Revision [ICD-10] Q20-Q23), aortic stenosis cases were defined as participants with ICD-9 424.1 or ICD-10 I35.0 in their hospital inpatient records or death records, or OPCS Classification of Surgical Operations and Procedures, Fourth Revision codes K26.1, K26.2, K26.3, K26.4, K31.2, K32.2, or K35.2 in their hospital inpatient records 25. The remaining UKB participants were classified as controls. Analyses were restricted to unrelated participants with genetically confirmed White British ancestry (n = 257,231 [2,213 aortic stenosis cases]).
Statistical analysis
Results are presented as the mean ± standard error of the mean. Statistical comparisons between two groups were performed by Student’s t-tests or Mann-Whitney tests, whereas comparisons between multiple groups were performed by one-way ANOVA with Tukey post hoc analysis for statistical significance. P-values < 0.05 were considered statistically significant.
A detailed description of the Methods is included in the Supplementary Appendix.
Results
Tlr3 deficiency protects against age-related CAVD
To investigate a potential role for TLR3 in CAVD in vivo, we assessed its expression in the aorta and aortic valve. TLRs display a vessel-specific pattern of expression within the human cardiovascular system, depending on their anatomical site 26. In mice, Tlr3 was expressed at high levels in the ascending aorta and aortic valve, and at much lower levels in the peripheral arteries (Figure 1A). TLR3 expression levels were also high in human aortic valves (Figure 1A), which have the same embryonic origin as the ascending aorta 27. VICs isolated from human aortic valves maintained high levels of TLR3 expression in culture (Figure 1A), consistent with previous findings of TLR3 expression by other fibroblasts, including dermal fibroblasts 14. TLR3 and IFN-β levels were higher in the aortic valves from CAVD patients compared to healthy controls (Figures 1B and 1C), suggesting a role of the TLR3-IFN axis in disease. Age is the principal risk factor for CAVD, the prevalence of which increases significantly over the age of 80 years 28. As a proxy for age, we passaged human VICs in vitro, and found that TLR3 protein levels increased markedly with the number of passages (Figure 1D). To confirm our results, we have studied a second ageing model. For this purpose, we treated human valvular interstitial cells with hydroxyurea (HU) and measured senescence markers. Not only was the senescence marker p21 found upregulated 3-fold, but also did we observe a higher amount of ß-galactosidase positive cells upon treatment, indicating induction of senescence. Whereas TLR3 mRNA expression was not affected in this model, ageing with HU was associated with markedly increased IFN-ß levels (Figure S1A–D).
Figure 1: TLR3 deficiency protects against age-related CAVD.
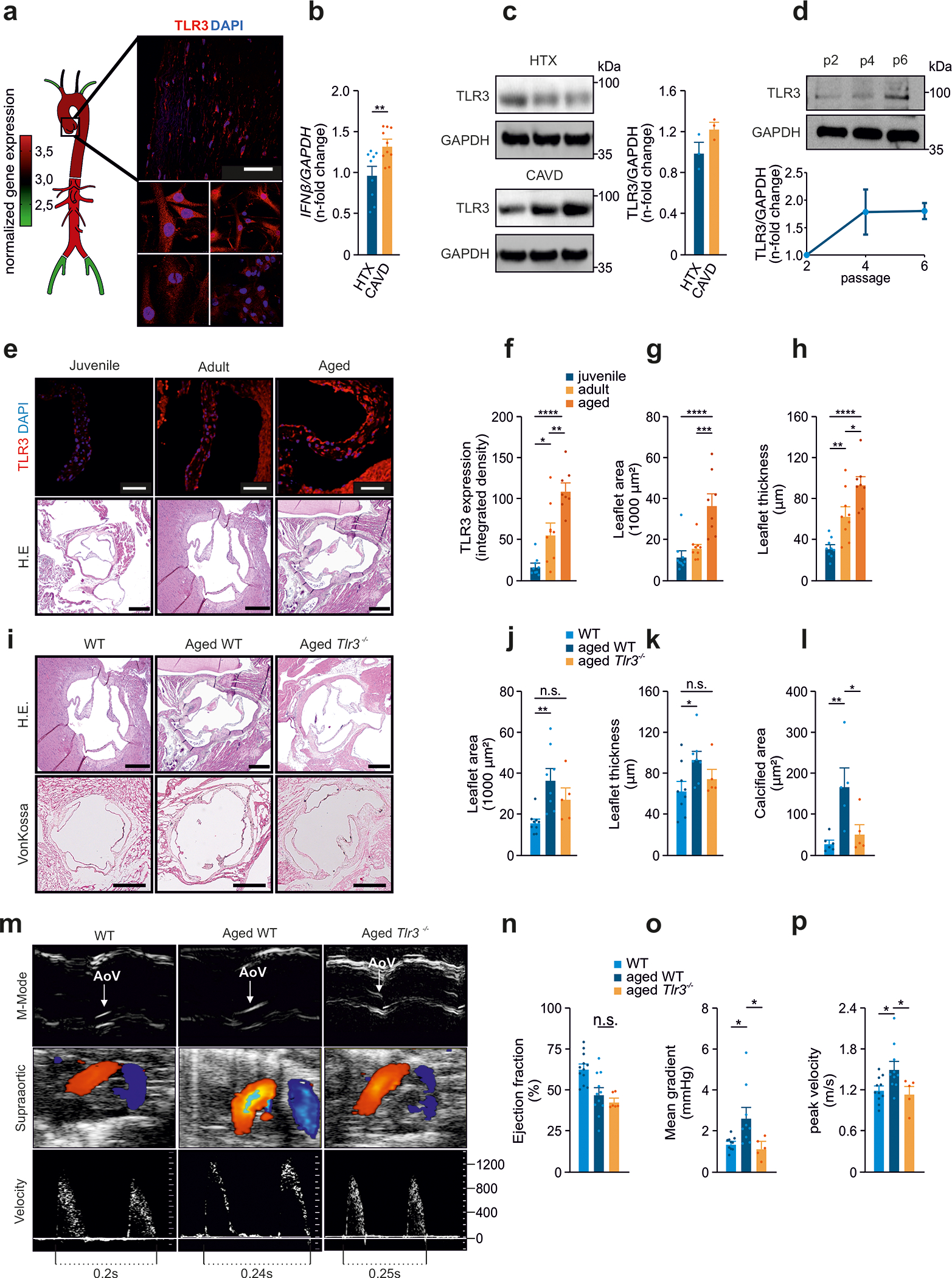
(a) Analysis of Tlr3 expression in murine ascending aortas, descending aortas, subclavian arteries, carotid arteries and femoral arteries. Analysis of TLR3 levels in healthy human aortic valves (upper panel) and valvular interstitial cells from human aortic valves (lower panel); red: TLR3, blue: DAPI, scale=50 μm; n=3.
(b) IFN-β expression, assessed by RT-PCR for CAVD samples and controls; n=9–10 per group.
(c) Immunoblot analysis of TLR3 levels in human aortic valve specimens from CAVD patients and healthy controls; n=3.
(d) Increasing levels of TLR3 with increasing passage number, for VICs analyzed by immunoblotting; n=2 independent western blotting experiments, representative western blot shown.
(e) Representative images of murine aortic valves from juvenile (3 weeks), adult (12 weeks) and aged (>18 months) mice (red: TLR3, blue: DAPI, IF-scale=50 μm, H.E.-scale=250 μm) analyzed for
(f) Tlr3 expression (n=8–9) and
(g) leaflet area (n=8–9) and
(h) leaflet thickness (n=8–9).
(i) Aortic valves from adult wild-type mice (12 weeks), aged wild-type mice (>18 months) and aged Tlr3−/− mice (>18 months) analyzed by H.E staining for morphological analysis, and by vonKossa staining.
(j) Aortic valve leaflet area measured on H.E. sections (n=5–9).
(k) Aortic valve leaflet thickness measured on H.E. sections (n=5–9).
(l) Calcified area of the aortic valve leaflets analyzed by vonKossa staining (n=5–6).
(m) Echocardiographic assessment of aortic valves (AoV) from adult wild-type mice, aged wild-type mice and aged Tlr3−/− mice for the measurement of:
(n) Ejection fraction (n=8–12)
(o) Mean pressure gradients (n=8–12) and
(p) Mean transvalvular velocity (n=8–12).
All data are presented as the mean ± SEM. Statistical comparisons: (b) Two-tailed unpaired t-test, **P<0.01. (f-p) One-way ANOVA with Tukey post hoc tests: n.s. = non-significant, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Moreover, aortic valve leaflet thickness and area, as well as the levels of valvular TLR3 increased with age in wild-type mice (Figures 1E–H). Our data indicate that aging, TLR3 levels in the aortic valve, and the occurrence of CAVD associate in humans and mice. To prove a direct causality in disease development, we studied CAVD development in Tlr3−/− mice. It is well documented that TLR2/4 und TLR3 signaling may converge through TRIF and TRAF adapter molecules, therefore enabling the cell to respond (besides NF-kB) with an adequate IFN signaling, and ultimately calcification. Interestingly, it appears in our experiments that a lack of TLR3 in mice is compensated with an increase of TLR2/TLR4 throughout the murine aorta (Fig. S1E). Intriguingly, aged wild-type mice displayed aortic valve thickening and calcification, whereas age-matched Tlr3−/− mice did not display these CAVD phenotypes (Figures 1I–L). Transthoracic echocardiography confirmed increased leaflet thickness, and revealed high-pressure gradients and transvalvular velocities in aged wild-type animals, but not in aged Tlr3−/− mice (Figures 1M–P).
TLR3 induces osteoblast-like phenotype via IFN-β
We tested the hypothesis that high-pressure gradients in heart valves, and the associated mechanical strain, may lead to increased TLR3-IFN signaling in VICs. Cultured human VICs subjected to mechanical strain had increased levels of TLR3 and of its downstream signaling components TRIF and IRF3 (Figures S1F and S1G). Intriguingly, VICs subjected to mechanical strain also displayed increased levels of both BMP2 and RUNX2 (Figure S1G), which are known to promote osteoblast differentiation 29,30. Moreover, the supernatants of mechanically stretched VICs activated TLR3 signaling in HEK293 reporter cells that express a Renilla luciferase reporter under the transcriptional control of the interferon stimulated response element (ISRE) and TLR3 (Figure S1H).
To investigate whether TLR3-RUNX2 signaling in VICs induces an osteoblast expression program, we performed RNA-seq analyses in untreated VICs, VICs treated with the specific TLR3 agonist Poly(I:C), and osteoblasts. Compared to untreated VICs, the Poly(I:C)-treated VICs and human osteoblasts shared 118 upregulated genes and 199 downregulated genes (Fig. 2A+B). Besides TLR3, human VICs expressed TLR1, 4 and 6 and treatment with Poly(I:C) caused upregulation of TLR1, 2, 3, 4 and to a smaller extent TLR 5–7 and 9 (Figure S2A).
Figure 2: TLR3 is a conserved inducer of calcification via IFN I signaling.
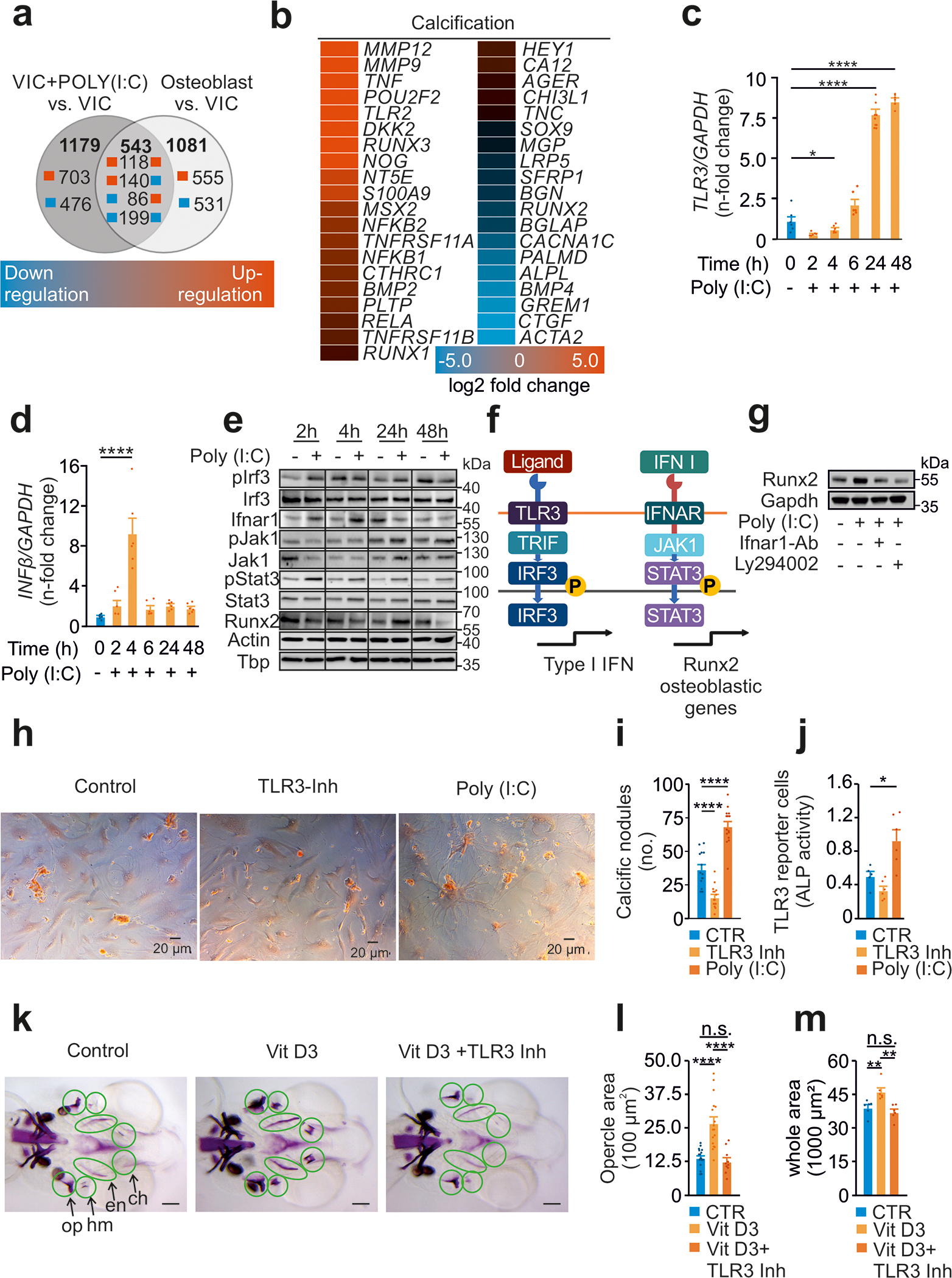
(a) Numbers of at least four-fold change differentially expressed genes in human VICs with or without Poly(I:C) treatment (20 μg/mL) for 72h and human osteoblasts.
(b) Expression profile of genes involved in calcification.
(c) RT-PCR analysis of VICs treated with Poly(I:C) for TLR3 (n=6) and
(d) IFN-β (n=6), measured in duplicates.
(e) Immunoblot analysis of VICs treated with Poly(I:C) assessing the TLR3-RUNX2 pathway (n=2 independent western blot experiments, representative western blot shown).
(f) Schematic illustration of the involved TLR3-RUNX2 pathway.
(g) Expression of the osteoblastic transcription factor RUNX2 after Poly(I:C) treatment (20 μg/mL) in the presence of either LY294002 (10μM), an IRF3 inhibitor, or a specific IFNAR1 blocking antibody (4μg/ml). GAPDH served as loading control (n=2 independent western blot experiments, representative western blot shown).
(h) VICs cultured with osteoblastic medium in the presence of TLR3 agonist Poly(I:C) or inhibitor (scale=40μm).
(i) Quantification of calcific nodules upon treatment applying Alizarin Red staining (n=9).
(j) Activity of alkaline phosphatase measured in the supernatant of treated VICs (n=9).
(k) Zebrafish treated with VitD3 in the presence of TLR3 inhibitor with subsequent quantification of calcification of
(l) the opercle (n=10–19, scale=100μm)
(m) all bone structures (n=6, scale=100μm).
All data are presented as mean ± SEM.
Statistical comparisons: One-way ANOVA with Tukey post hoc test: *P<0.05, **P<0.01, ****P<0.0001
Gene set enrichment analysis on Poly(I:C)-treated VICs revealed that many of the upregulated genes were involved in calcification (Figures 2B), and more specifically in CAVD (Figures S2B–D). Our data clearly show activation of TLR3 in VICs to induce an osteoblastic signature.
To gain more mechanistic insights into the phenotypic switch observed in TLR3-activated VICs, we next set up experiments aiming at better delineating the involved signaling cascade. As observed for mechanical strain, treatment of VICs with Poly(I:C) resulted in increased TLR3 and IFN-β expression (Fig. 2C–D). Immunoblot analysis revealed phosphorylation of IRF3, upregulation of IFNAR1 and associated phosphorylation of JAK1/STAT3, resulting in increased levels of the essential osteoblastic transcription factor RUNX2 (Fig. 2E). To further corroborate the newly identified TLR3-RUNX2 pathway, a set of experiments was designed employing either LY294002, a compound abolishing IFN-β synthesis 31, or a specific IFNAR1 blocking antibody (Fig. 2F). As shown in Fig. 2G, both LY294002 and a specific IFNAR1 blocking antibody abolished RUNX2 upregulation in VICs induced by Poly(I:C).
Finally, stimulation of TLR3 enhanced the production of calcific nodules in VICs, which was paralleled by increased activity of the osteoblastic enzyme alkaline phosphatase (ALP) 32, whereas TLR3 inhibition decreased calcification and ALP activity (Fig. 2H–J). These findings strongly suggest that TLR3 stimulation of VICs activates an osteoblast-related pathway and promotes calcification. This finding is consistent with the established notion that osteoblasts are essentially sophisticated fibroblasts 33.
TLR3 governs bone development in vivo via a conserved mechanism of calcification
We next considered the possibility that TLR3 is involved in the maturation of fibroblasts into osteoblasts beyond the aortic valve. TLRs are highly conserved among species and have been shown to drive dorso-ventral polarity in Drosophila melanogaster34. Specifically, the TLR3 analog tollo is of central importance for the axial development in Drosophila melanogaster by activating the transcription factor runt, the analog of vertebrate RUNX2 35. To test whether TLR3 would play an evolutionary conserved role in morphogenesis, we next targeted Tlr3 in zebrafish (Danio rerio), a well-established model organism to study calcification and bone formation (Fig. 2K). In zebrafish, the first bones develop in the head including the opercle and the branchiostegal rays. It was previously shown that at this early larval stage (8–10 days post fertilization), premature calcification of these hyomandibular bones can be experimentally hyper-induced upon vitamin D3 (VitD3) supplementation 36. Accordingly, in our experiments an early and strong calcification of landmark bones in zebrafish morphogenesis was observed in VitD3-treated larvae. Co-incubation with a TLR3 inhibitor abolished VitD3-mediated calcification of the hyomandibular bones, indicating that TLR3 activation represents an evolutionary conserved mechanism in development, promoting cellular calcification in vertebrates (Fig. 2L+M).
Ifnb−/− mice have a distinctive bone phenotype, with impaired bone formation and osteoporosis 15,16. Our data suggest that TLR3 stimulation of VICs induces calcification via IFN, IFNAR1 and JAK1/STAT3 signaling, yet no specific bone phenotype has been described for Tlr3−/− mice. Here, we show that WT and Tlr3−/− mice display a similar size and skeletal development (Figures S3A and S3B). However, micro-computed tomography revealed a distinct osteoporotic phenotype with decreased bone volume and bone density in Tlr3−/− mice compared to WT (Figures 3A–C). The number of trabecles and trabecular networks were decreased, resulting in higher intertrabecular distances and in increased trabecular thickness in Tlr3−/− mice compared to WT (Figures 3D–G). Histological analysis confirmed the osteoporotic phenotype in Tlr3−/− mice, with decreased trabecular volume (Figures 3H and 3I). As incidental finding, we noted a shorter femur length in Tlr3−/− mice (Figures 3J and 3K). Together, these findings suggest that Tlr3 plays a central role in bone development and calcification in both zebrafish and mice.
Figure 3: Tlr3−/− exhibit osteoporotic phenotype.
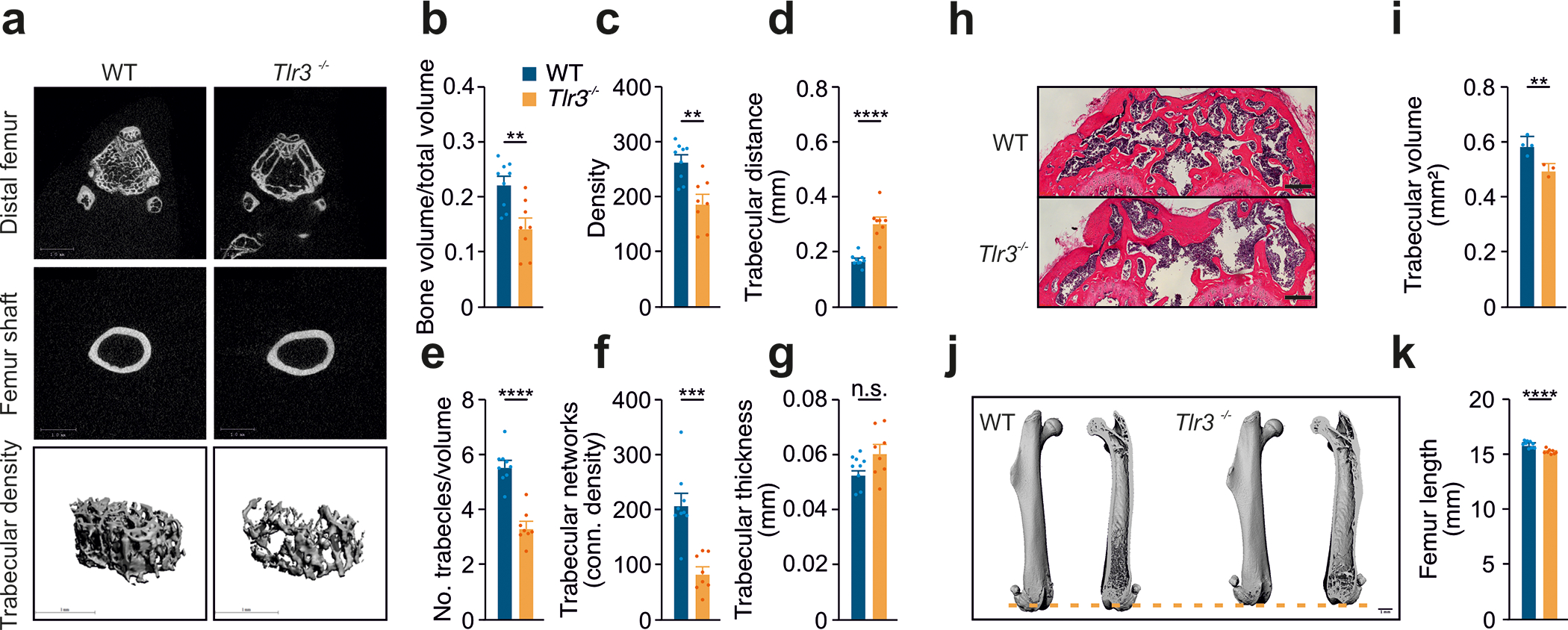
(a) Femurs from 12-week-old wild type and Tlr3−/− (n=8–9 femora) analyzed morphologically via micro-CT for
(b) bone volume,
(c) bone density,
(d) trabecular distance,
(e) number of trabecles/volume,
(f) trabecular networks and
(g) trabecular thickness.
(h) Histological analysis of distal femurs from 12-week-old wild type and Tlr3−/− analyzing
(i) trabecular volume (H.E. staining, n=3–4).
(j,k) Femur length from 12-week-old wild type and Tlr3−/− analyzed via micro-CT (n=8–9 femora).
All data are presented as mean ± SEM. Statistical comparisons: 2-tailed unpaired t-test, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Biglycan is an endogenous TLR3 ligand
To systematically investigate the endogenous TLR3 ligands potentially involved in CAVD and VIC calcification, we first considered modified bioactive lipids including oxidized low-density lipoprotein (oxLDL) and lipoprotein (a) (Lp(a)), which have been associated with CAVD in humans 37,38. In our in vitro experiments, neither oxLDL nor Lp(a) caused direct activation of TLR3 (Fig. S4A+B). Nevertheless, both upregulated TLR3 protein expression in human VICs (Fig. 4A), a finding corroborated in vivo in aortic valves of ApoE−/− mice which are characterized by high plasma levels of oxLDL 39 (Fig. S4C+D). Importantly, ApoE−/− and Tlr3−/− double-knockout mice were protected from hyperlipidemia-induced CAVD (Figure S4E–N).
Figure 4: Biglycan serves as an endogenous ligand of TLR3.
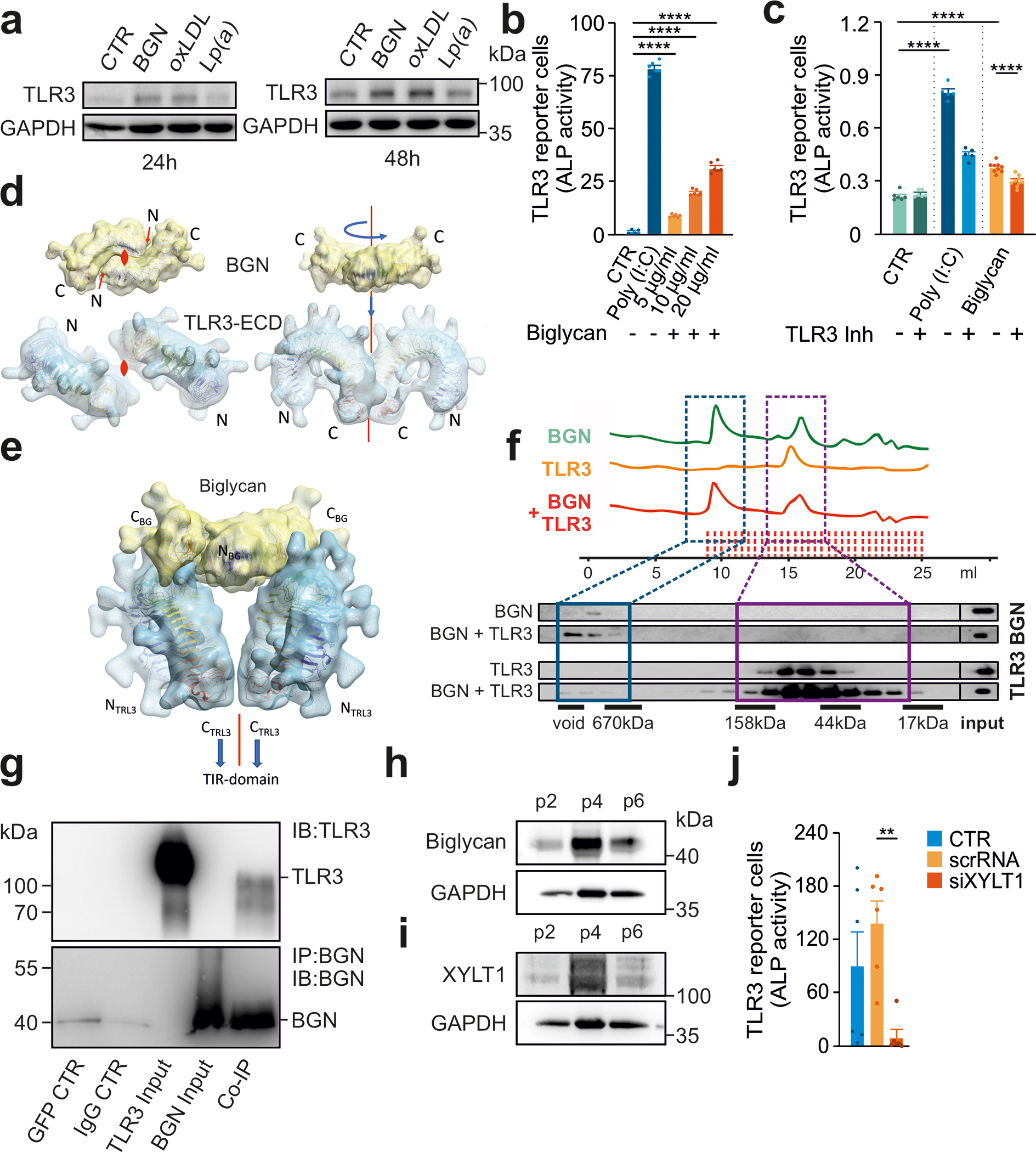
(a) Immunoblot analysis of human VICs treated with biglycan (20 μg/mL), Lp(a) (20 μg/mL) or oxLDL (20 μg/mL) for 24h and 48h for TLR3 protein expression.
(b) HEK293 hTLR3 reporter cells treated with Poly(I:C) (20 μg/ml) and various concentrations of biglycan (5 μg/ml, 10 μg/ml, 20 μg/ml), (n=6).
(c) HEK293 hTLR3 reporter cells treated with Poly(I:C) or biglycan (both at 5 μg/ml) for 6 h in the presence of (R)-2-(3-chloro-6-fluorobenzo[b]thiophene-2-carboxamido)-3-phenylpropanoic acid, a dsRNA/TLR3 complex inhibitor (n=6).
(d) Illustration of the dimer binding mode. The top view (left panel) shows the location of the 2-fold axes perpendicular to the projection plane with dyad symbols, and the right panel shows the assembly of the model maintaining the 2-fold symmetry (red line). The docked, refined, and optimized model (e) is presented as ribbons surrounded by a macro shape which includes the forests core residues of all glycan decorations, none of which cause any steric clashes.
(f) Binding experiments with the recombinant human TLR3 ectodomain and biglycan (size-exclusion chromatography and subsequent immunoblot analysis).
(g) Following the co-immunoprecipitation of purified TLR3 ectodomain and purified BGN, both proteins were detected on immunoblots (n=2 independent experiments, representative blots shown).
(h,i) Increasing levels of BGN and XYLT1 in VICs with increasing passage number analyzed by immunoblotting (n=2 independent western blotting experiments, representative western blot shown)
(j) HEK293 hTLR3 reporter cells treated with supernatants from VICs after siRNA knockdown of XYLT1 or after treatment with a control RNA (scRNA) (n=6).
Statistical comparisons: (b-c,k) One-way ANOVA with Tukey post hoc test: **P<0.01, ****P<0.0001.
We screened other putative ligands previously found to accumulate in human calcific aortic valves (Figure S5) and found that the proteoglycan biglycan (BGN), a structural extracellular matrix (ECM) protein, activated TLR3 signaling in reporter cells in a dose-dependent manner (Figure 4B). Signaling was dependent on TLR3, as it was abolished in reporter cells treated with a pharmacological TLR3 inhibitor (Figure 4C).
The human TLR3 ectodomain (huTLR3-ECD) forms a horseshoe structure that is connected to the transmembrane helix via its C-terminal domain. Binding of a dsRNA ligand leads to huTLR3 homodimer formation and initiates the TLR3 signaling cascade 40,41. The BGN core protein forms obligate dimers 42, suggesting that TLR3 dimerization may be induced upon binding to a BGN dimer (Figure 4D). We identified the most plausible interaction between huTLR3 and huBGN, by modeling the complex whilst maintaining the collinearity of the dimer axes as a restraint in subsequent protein-protein docking and local refinement 43,44 (Figure 4E). Binding-site analysis and the absence of steric interference with glycan decorations supported the proposed interaction model (Figure S6A).
We tested the physical interaction between BGN and TLR3 by incubating purified huTLR3-ECD with BGN and performing size-exclusion chromatography followed by immunoblotting, which demonstrated the co-elution of these two proteins (Figure 4F). The co-immunoprecipitation of BGN and huTLR3-ECD confirmed this interaction (Figure 4G). To substantiate the interaction of the two proteins we have performed double staining of both TLR3 and BGN in murine aortic valves. Co-staining showed co-localization of both proteins, confirming our results previously performed in prespecified buffers, further indicating physical interaction (Figure S6B). These data provide additional evidence in support of a direct interaction between BGN and TLR3.
Maturation of BGN via XYLT1 is crucial for TLR3 activation
The direct interaction of BGN with other proteins is mediated by gylcosaminoglycan chains 45. Indeed, BGN lacking chondroitin sulfate chains (rhBGN) (Figure S6C) did not activate TLR3 in reporter cells (Figure S6D).
Xylosyltransferases (XYLT) add a xylose linker to BGN, which is the crucial step for further BGN maturation. The protein levels of both BGN and XYLT1 increased with higher passage number of cultured VICs (Figure 4H+I). Moreover, XYLT1-deficient VICs showed reduced transcription of Xylt1, Tlr3 and IFN-ß (Figures S7A–C) and supernatant from XYLT1-deficient VICs did not activate TLR3-signaling reporter cells (Figure 4J), independently of applied mechanical strain (Figure S7D). These experiments not only uncovered that maturation of BGN is crucial for TLR3 activation, but also indicated that mechanical strain in heart valves may induce the release of BGN from VICs, resulting in a transcellular activation of this repair and defense mechanism. Overall, our data suggest that the XYLT1-mediated addition of xylose sulfate chains during BGN maturation is essential for TLR3 activation. Intriguingly, XYLT1 mutations appear to impair calcification and chondrocyte maturation 46, supporting a role for the XYLT1-BGN axis in this process.
The BGN-TLR3-IFNAR1 axis induces calcification both in vitro and in vivo
To assess the generality of these effects, we examined human dermal fibroblasts, which have a slightly different expression profile in response to TLR3 stimulation compared to VICs (Figures S8A–E). Importantly, both BGN and Poly(I:C) treatments induced the transcription of TLR3 and IFN-ß in wild-type and empty vector-transformed fibroblasts, but not in TLR3−/− fibroblasts (Figures 5A and B). Both Poly(I:C) and BGN treatments led to IRF3 phosphorylation, IFNAR1 upregulation, phosphorylation of JAK1/STAT3, and also increased levels of RUNX2, in wild-type and empty vector-transformed fibroblasts but not in TLR3−/− fibroblasts (Figure 5C).
Figure 5: The BGN-TLR3-IFNAR1 axis induces aortic valve calcification.
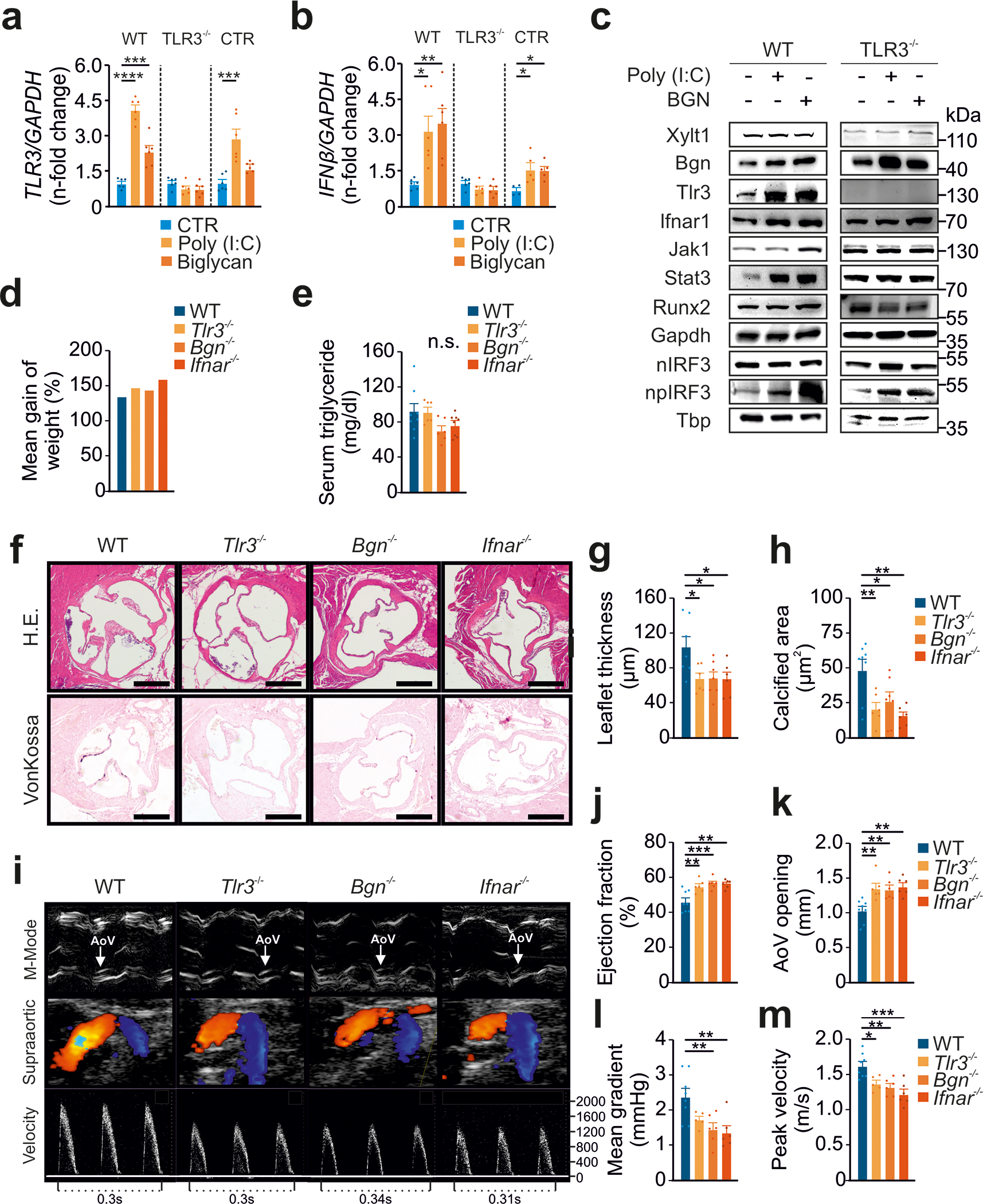
(a) RT-PCR analysis of human wild-type dermal fibroblasts, TLR3−/− CRISPR fibroblasts or empty vector-transformed control fibroblasts (CTR) treated with Poly(I:C) or BGN (both at 5 μg/ml) for 6h and then subjected to an analysis of TLR3 expression (n=6).
(b) RT-PCR analysis of human wild-type dermal fibroblasts, TLR3−/− CRISPR fibroblasts or empty vector-transformed control fibroblasts (CTR) were treated with Poly(I:C) or BGN (both at 5 μg/ml) for 6 h and then analyzed for IFN-ß expression (n=6).
(c) Immunoblot analysis of human wild-type dermal fibroblasts and TLR3−/− CRISPR fibroblasts treated with Poly(I:C) or BGN (both at 5 μg/ml) for 6h.
(d) Mean weight gain after four months on a high-fat diet, for wild-type, Tlr3−/−, Bgn−/− and Ifnar1−/− animals (n=6–8).
(e) Serum triglyceride concentration after 4 months on a high-fat diet, for wild-type, Tlr3−/−, Bgn−/− and Ifnar1−/− animals (n=6–8 animals per group).
(f) Histological evaluation of aortic valves from wild-type, Tlr3−/−, Bgn−/− or Ifnar1−/− animals fed a high-fat diet for four weeks by H.E. and vonKossa staining.
(g) Analysis of aortic valve leaflet thickness in wild-type, Tlr3−/−, Bgn−/− and Ifnar1−/− animals after 4 months on a high-fat diet (n=6–8).
(h) Quantification of aortic valve calcification in wild-type, Tlr3−/−, Bgn−/− and Ifnar1−/− animals after 4 months on a high-fat diet (n=6–8).
(i) Assessment, by transthoracic echocardiography, of aortic valve function after 4 months on a high-fat diet, for wild-type, Tlr3−/−, Bgn−/− and Ifnar1−/− animals (n=6–8).
(j) Analysis of ejection fraction in wild-type, Tlr3−/−, Bgn−/− and Ifnar1−/− animals after 4 months on a high-fat diet (n=6–8).
(k) Aortic valve opening in wild-type, Tlr3−/−, Bgn−/− and Ifnar1−/− animals after 4 months on a high-fat diet (n=6–8).
(l) Mean transvalvular pressure gradient in wild-type, Tlr3−/−, Bgn−/− and Ifnar1−/− animals after 4 months on a high-fat diet (n=5–8).
(m) Transvalvular peak velocities in wild-type, Tlr3−/−, Bgn−/− and Ifnar1−/− animals after 4 months on a high-fat diet (n=5–8).
All data are presented as the mean ± SEM. Statistical comparisons: (a), (b), (e), (g), (h), (j), (k), (l), (m) One-way ANOVA with Dunnett post hoc tests: *P<0.05, **P<0.01, ***P<0.05, ****P<0.000
To probe the physiological role of the BGN-TLR3-IFNAR1 axis in vivo, we analyzed gene expression of the pathway in human tissue. IFN expression and RUNX2 expression were both increased in tissue from CAVD patients (Figure S8F). To further explore the role of the axis in vivo, we used hypercholesterinemia to induce CAVD in wild-type mice, as well as in Tlr3−/−, Bgn−/−, and Ifnar1−/− mice. We fed animals a high-fat diet for four months, and then analyzed their valvular phenotype 47. Mean weight gain and serum triglyceride levels were similar between groups (Figure 5D and 5E), but Tlr3−/−, Bgn−/−, and Ifnar1−/− mice displayed significantly reduced aortic valve leaflet thickness compared to wild-type mice (Figure 5F+G). In addition, hypercholesterinemia induced aortic valve calcification in wild-type but not Tlr3−/−, Bgn−/− and Ifnar1−/− mice (Figure 5H). A functional analysis of the aortic valves by transthoracic echocardiography confirmed that deficiencies of Tlr3, Bgn or Ifnar1 protected the mice from the valvular thickening and hemodynamic consequences of CAVD, as these animals did not display the hypercholesterinemia-induced changes in leaflet thickness, aortic valve opening, mean transvalvular gradient or peak velocity observed in wild-type animals (Figure 5I–M). These results provide strong support for the involvement of the BGN-TLR3-IFNAR1 axis in CAVD development.
Finally, we investigated the effect of the BGN-TLR3-IFNAR1 axis on mouse bone structure by performing micro-computed tomography on femurs from Bgn−/−, and Ifnar1−/− mice. Knockout animals had a clear osteoporotic phenotype, with a lower bone volume and bone density compared to WT animals (Figure S9).
Genetic association of TLR3 signaling with CAVD
To determine whether genetic variation at genes implicated by the uncovered TLR3 signaling pathway are associated with aortic stenosis in humans, we examined two large-scale cohorts (Genetic Epidemiology on Adult Health and Aging (GERA), n = 55,192 with 3,469 aortic stenosis cases; UK Biobank, n = 257,231 with 2,213 aortic stenosis cases; genotyping methods are available in the supplement). We discovered 307 variants that were nominally significant (p ≤ 0.05) for aortic stenosis in a meta-analysis of the UK Biobank and GERA, including 16 variants in the JAK1, TLR3, IFNB1, IFNA1, XYLT1, and IFNAR1 loci, representing 13 independent signals, that demonstrated strong associations (p ≤ 1 × 10−3) and/or two-fold or greater (up to 5.86-fold) odds of aortic stenosis (Figure 6A and Table 1). Of the 16 variants, all were rare (minor allele frequency < 0.01) except for three variants at the XYLT1 locus. Variants throughout the XYLT1 locus demonstrated significant associations with aortic stenosis in the meta-analysis (Figure 6B). Mendelian randomization analysis identified that XYLT1 expression in the aorta was associated with greater odds of aortic stenosis, but this was only significant with the Penalized Weighted Median method (Supplemental Tables 1 and 2). Mendelian randomization consistently indicated that XYLT1 expression in the aorta was associated with greater odds of aortic valve replacement (indicating progression to severe aortic stenosis) among aortic stenosis cases in a meta-analysis of the GERA and UK Biobank cohorts (odds ratio per unit of normalized expression, 2.42; 95% CI 1.53 to 3.83; p = 1.7 × 10−4) (Table 2, Table 3, Fig. 6C).
Figure 6: Association of TLR3 pathway variants in aortic stenosis patients.
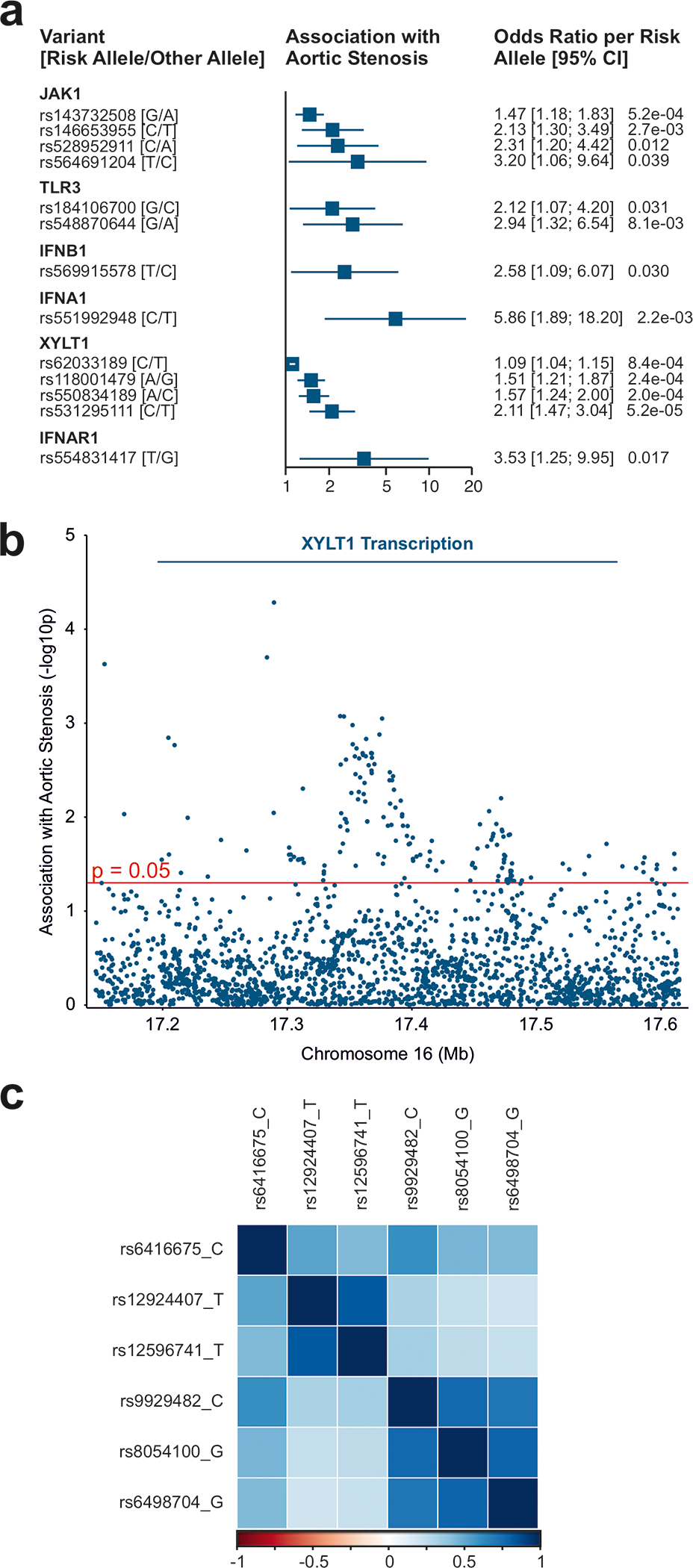
(a) Association with aortic stenosis (meta-analysis of UK Biobank and GERA) of variants within 50 kb of 6 autosomal genes. Variants shown are independent (r2 < 0.01) and have an association of 1) p ≤ 1 × 10−3 or 2) p ≤ 0.05 and odds ratio ≥ 2. (See Table 1 for a complete list of significant variants.)
(b) Association with aortic stenosis (meta-analysis of UK Biobank and GERA) of variants within 50 kb of the XYLT1 transcript (Manhattan plot).
(c) Correlation of variants (r2) in the UK Biobank participants of variants associated with XYLT1 expression.
Table 1:
Associations* with aortic stenosis in a meta-analysis of UK Biobank and GERA cohorts of variants within 50 kb of 6 autosomal genes.
| Gene | Position (GRCh37) | Variant | Meta-Analysis Risk Allele (Freq.) | GERA Odds Ratio per Risk Allele (95% CI) | GERA p | UKB Odds Ratio per Risk Allele (95% CI) | UKB p | Meta-Analysis Odds Ratio per Risk Allele (95% CI) | Meta-Analysis p |
|---|---|---|---|---|---|---|---|---|---|
| JAK1 | 1:65335640 | rs143732508 | G (0.0087) | 1.23 (0.89, 1.68) | 0.21 | 1.75 (1.29, 2.37) | 3.1E-04 | 1.47 (1.18, 1.83) | 5.2E-04 |
| JAK1 | 1:65342993 | rs564691204 | T (0.9984) | 2.11 (0.57, 7.82) | 0.26 | 8.80 (1.14, 67.77) | 0.037 | 3.20 (1.06, 9.64) | 0.039 |
| JAK1 | 1:65347527 | rs528952911 | C (0.0012) | 3.06 (1.53, 6.09) | 1.5E-03 | 0.23 (0.03, 1.67) | 0.15 | 2.31 (1.20, 4.42) | 0.012 |
| JAK1 | 1:65380580 | rs146653955 | C (0.0013) | 1.20 (0.47, 3.04) | 0.70 | 2.67 (1.49, 4.78) | 9.8E-04 | 2.13 (1.30, 3.49) | 2.7E-03 |
| TLR3 | 4:186953463 | rs548870644 | G (0.9981) | 2.38 (0.96, 5.90) | 0.062 | 6.09 (1.14, 32.50) | 0.035 | 2.94 (1.32, 6.54) | 8.1E-03 |
| TLR3 | 4:187028029 | rs184106700 | G (0.9983) | 2.84 (1.08, 7.47) | 0.035 | 1.59 (0.60, 4.17) | 0.35 | 2.12 (1.07, 4.20) | 0.031 |
| IFNB1 | 9:21119979 | rs569915578 | T (0.9986) | 2.45 (0.79, 7.59) | 0.12 | 2.76 (0.74, 10.28) | 0.13 | 2.58 (1.09, 6.07) | 0.030 |
| IFNB1 | 9:21120058 | rs7555350581 | T (0.9986) | 2.48 (0.79, 7.76) | 0.12 | 2.74 (0.73, 10.24) | 0.13 | 2.59 (1.09, 6.13) | 0.031 |
| IFNA1 | 9:21457591 | rs551992948 | C (0.9980) | 13.34 (1.49, 119.69) | 0.021 | 4.35 (1.16, 16.32) | 0.029 | 5.86 (1.89, 18.20) | 2.2E-03 |
| XYLT1 | 16:17153381 | rs118001479 | A (0.0064) | 1.56 (1.16, 2.10) | 3.0E-03 | 1.44 (1.04, 1.99) | 0.027 | 1.51 (1.21, 1.87) | 2.4E-04 |
| XYLT1 | 16:17283730 | rs550834189 | A (0.0054) | 1.48 (1.05, 2.09) | 0.026 | 1.66 (1.19, 2.31) | 2.6E-03 | 1.57 (1.24, 2.00) | 2.0E-04 |
| XYLT1 | 16:17289368 | rs531295111 | C (0.0018) | 1.71 (0.98, 2.97) | 0.059 | 2.48 (1.54, 4.01) | 2.0E-04 | 2.11 (1.47, 3.04) | 5.2E-05 |
| XYLT1 | 16:17342509 | rs62033189 | C (0.1530) | 1.10 (1.02, 1.17) | 0.010 | 1.09 (1.01, 1.18) | 0.032 | 1.09 (1.04, 1.15) | 8.4E-04 |
| XYLT1 | 16:17345488 | rs345883332 | A (0.2161) | 1.07 (1.01, 1.14) | 0.028 | 1.10 (1.02, 1.18) | 0.011 | 1.08 (1.03, 1.14) | 8.5E-04 |
| XYLT1 | 16:17376126 | rs9363462 | C (0.5907) | 1.05 (1.00, 1.11) | 0.059 | 1.10 (1.03, 1.16) | 3.5E-03 | 1.07 (1.03, 1.11) | 8.9E-04 |
| IFNAR1 | 21:34683984 | rs554831417 | T (0.9986) | 4.19 (0.96, 18.22) | 0.056 | 2.99 (0.69, 12.84) | 0.14 | 3.53 (1.25, 9.95) | 0.017 |
Abbreviations: GERA, Genetic Epidemiology Research on Adult Health and Aging; UKB, UK Biobank.
In linkage disequilibrium with rs569915578 (r2 = 1).
In linkage disequilibrium with rs62033189 (r2 ≥ 0.098).
Associations with either 1) p ≤ 1 × 10−3 or 2) p ≤ 0.05 and odds ratio ≥ 2 are provided
Table 2:
Mendelian randomization for aortic valve replacement of XYLT1 expression in the aorta.
| Method | Odds Ratio per Unit of Normalized Expression (95% CI) | p |
|---|---|---|
| Inverse-variance weighted | 2.42 (1.53, 3.83) | 1.7 × 10−4 |
| Penalized weighted median | 2.55 (1.51, 4.32) | 5.0 × 10−4 |
Table 3.
Associations With XYLT1 Expression in the Aorta and With Aortic Valve Replacement (for Variants in the Mendelian Randomization Instrument for Aortic Valve Replacement)
| Chromosomal Position (GRCh37) | Variant | Expression-Increasing Allele | Other Allele | Expression in Normalized Effect Size (95% CI) | Expression p | Aortic Valve Replacement Odds Ratio (95% CI) | Aortic Valve Replacement p |
|---|---|---|---|---|---|---|---|
| 16:17415463 | rs6416675 | C | T | 0.19 (0.09, 0.28) | 1.3E-04 | 1.21 (1.10, 1.34) | 1.3E-04 |
| 16:17436344 | rs12924407 | T | C | 0.19 (0.09, 0.28) | 8.4E-05 | 1.16 (1.06, 1.28) | 1.7E-03 |
| 16:17499765 | rs8054100 | G | A | 0.18 (0.09, 0.28) | 2.1E-04 | 1.19 (1.07, 1.32) | 1.1E-03 |
Variants are correlated (0.23 ≤ r2 ≤ 0.84; see Fig. S7). Estimates for aortic valve replacement were from the meta-analysis of the GERA and UK Biobank cohorts. Abbreviations: AVR, aortic valve replacement; GERA, Genetic Epidemiology Research on Adult Health and Aging
Discussion
This study identifies TLR3 as a key element in a conserved pathway of aortic valve calcification. Here, we show that a central component of the ECM, BGN, induces the TLR3 signaling cascade, resulting in a phenotypic switch of VICs to bone-forming osteoblast-like cells. BGN is a proteoglycan consisting of a core protein with two glycosaminoglycan side chains. It stabilizes the ECM and is released from cells subjected to stress 48, leading to the recruitment of macrophages and dendritic cells via TLR2/TLR4, potentially contributing to inflammaging and CAVD 49.
We for the first time provide evidence for a direct interaction between BGN and TLR3 in the cell type primarily responsible for aortic valve calcification. Our results suggest that BGN may govern the TLR3-dependent levels of basal type I IFN in human fibroblasts. They also demonstrate that only a mature form of BGN can activate TLR3, substituted with glycosaminoglycan side chains playing a key role in the biological activity of this proteoglycan. Enzymes responsible for the modification of BGN side chains, such as XYLT1, may therefore play a crucial role in determining the fate of BGN and the initiation and progression of CAVD. From an evolutionary perspective, with the aim of understanding the involvement of the newly identified XYLT1-BGN-TLR3-IFNAR1 pathway in the context of calcification per se, it is important to note that the loss of XYLT1 results in impaired chondrocyte maturation and skeletal defects 46. Consistent with this observation, we found that Tlr3−/− mice and zebrafish treated with a Tlr3 inhibitor displayed severe impairments of skeletal calcification. Moreover, we found that variants of XYLT1 significantly associate with CAVD in humans.
A correlation between cardiovascular calcification and osteoporosis has been suggested in some side-by-side clinical studies. Bioactive modified lipoproteins promote inflammation and the production of inflammatory cytokines, facilitating aortic valve calcification and bone demineralization at the same time50. A recent study showed that the antiresorptive treatment with denosumab and alendronate successfully inhibited bone resorption, but had no effect on the progression of aortic valve calcification51. The potential effect of a medical therapy targeting the uncovered BGN-TLR3-IFN pathway on bone metabolism appears promising and has to be determined.
By connecting the fate of an ECM-derived protein to Type I IFN signaling, the uncovered calcification pathway is thus at the interface of innate immunity and bone formation. Moreover, our results suggest that type I IFN may be involved in the physiological and pathological calcification of connective tissues in general, including musculoskeletal tissue and the cardiovascular system. This finding is consistent with recent reports linking interferonopathies with elevated basal interferon levels, to pathological calcifications in the brain and heart 17.
We suggest that pharmacological inhibition of the discovered XYLT1-BGN-TLR3-IFNAR1 axis might be a potentially promising approach to the treatment of CAVD. TLR3 inhibition has been successfully achieved in experimental settings, with small-molecule inhibitors or blocking antibodies 52. Besides inhibiting TLR3 directly, inhibition of its ligand BGN e.g. by the use of monoclonal antibodies directed against its Toll-binding site might represent a therapeutic strategy.
An alternative approach would involve the inhibition of type I IFN. Both TLR3 inhibition and inhibition of interferon signaling have been used in clinical studies for patients with asthma, inflammatory bowel disease or systemic lupus without reporting serious adverse events in their setting53,54. However, the targeting of TLR3 or IFNAR1 signaling might entail increased risk for viral infections leading to Herpes simplex encephalitis, influenza or COVID-19, which has yet to be assessed 55. It certainly remains a major challenge to identify which part of the pathway can be tackled to effectively inhibit aortic stenosis with the least adverse events.
In conclusion, we reveal a novel mechanism driving CAVD, in which mature BGN, modified by XYLT1, constitutes a selective and potent endogenous TLR3 ligand, inducing valvular calcification via a type I IFN-dependent switch of phenotype in VICs, from a fibroblast to an osteoblast-like phenotype. Our data identify the XYLT1-BGN-TLR3-IFNAR1 axis as an evolutionarily conserved pathway of morphogenesis and calcification, paving the way for novel therapeutic strategies for detecting and treating CAVD in humans.
Supplementary Material
Clinical perspective.
What is new?
Toll-like receptor 3 (Tlr3), biglycan (Bgn) and Interferon-alpha/beta receptor alpha chain (Ifnar1) mutant mice are protected from aortic valve calcification.
Maturation of the extracellular matrix protein biglycan (BGN) via xylosyltransferase 1 (XYLT1) is crucial for TLR3 activation.
Human genetic association analysis reveals that aortic valve calcification associates with variants in the XYLT1-BGN-TLR3 pathway.
What are clinical implications?
Our data identify the XYLT1-BGN-TLR3-IFNAR1 axis as an evolutionarily conserved pathway of morphogenesis and calcification, paving the way for novel therapeutic strategies for detecting and treating calcific aortic valve disease in humans.
Acknowledgments
Funding
This work was supported by grants from the “Gesellschaft zur Förderung der Herz-Kreislaufforschung in Tirol“ (to CGT), Bayer Grants4Targets (No.2014-08-1162), the Austrian Science Fund (FWF) to CGT, JH and IT (P 32821), “Medizinischer Forschungsfond Tirol” (no.257) to CGT and JH. It was also supported in part by the Austrian Science Fund (FWF) under project P28395-B26 to BR, project I-3321 grants to GW and by the City of Vienna Competence Team Signal Tissue (MA23#18-;08) and City of Vienna Competence Team Aging Tissue (MA23#29-07). This study is supported by VASCage – Research Centre on Vascular Ageing and Stroke. VASCage is a COMET Centre within the Competence Centers for Excellent Technologies (COMET) programme and funded by the Federal Ministry for Climate Action, Environment, Energy, Mobility, Innovation and Technology, the Federal Ministry of Labour and Economy, and the federal states of Tyrol, Salzburg and Vienna. This work was partially supported by funding to GT from the Canadian Institutes of Health Research, National Institutes of Health/National Heart Lung and Blood Institute (HL128550), the Heart and Stroke Foundation of Canada and the “Fonds de Recherche Québec – Santé”.
Non-standard Abbreviations and Acronyms
- ALP
alkaline phosphatase
- BGN
Biglycan
- CAVD
Calcific aortic valve disease
- ECM
extracellular matrix
- GERA
Genetic Epidemiology Research on Adult Health and Aging
- GOF
gain-of-function
- GSEA
gene set enrichment analysis
- HEK
human embryonic kidney
- HF/HC
high-fat/high-carbohydrate
- HTX
heart transplantation
- huTLR3-ECD
human TLR3 ectodomain
- IFN
Interferon
- IFNAR
Interferen alpha/beta receptor alpha chain
- LP(a)
Lipoprotein (a)
- LPS
Lipoplysaccharide
- MEF
mouse embryonic fibroblasts
- oxLDL
oxidizied low-density lipoprotein
- Poly(I:C)
Polyinosinic:polycytidylic acid
- PSLAX
parasternal long axis
- TLR
Toll-like receptor
- VIC
Valvular interstitial cell
- VitD3
Vitamin D3
- XYLT1
Xylosyltransferase 1
Footnotes
Conflict of Interest Disclosures
None.
Supplemental Materials
References
- 1.Danielsen R, Aspelund T, Harris TB, Gudnason V. The prevalence of aortic stenosis in the elderly in Iceland and predictions for the coming decades: the AGES-Reykjavik study. Int J Cardiol. 2014;176:916–922. doi: 10.1016/j.ijcard.2014.08.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lindman BR, Clavel MA, Mathieu P, Iung B, Lancellotti P, Otto CM, Pibarot P. Calcific aortic stenosis. Nat Rev Dis Primers. 2016;2:16006. doi: 10.1038/nrdp.2016.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rajamannan NM, Evans FJ, Aikawa E, Grande-Allen KJ, Demer LL, Heistad DD, Simmons CA, Masters KS, Mathieu P, O’Brien KD, et al. Calcific aortic valve disease: not simply a degenerative process: A review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: Calcific aortic valve disease-2011 update. Circulation. 2011;124:1783–1791. doi: 10.1161/CIRCULATIONAHA.110.006767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu AC, Joag VR, Gotlieb AI. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am J Pathol. 2007;171:1407–1418. doi: 10.2353/ajpath.2007.070251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Driscoll K, Cruz AD, Butcher JT. Inflammatory and Biomechanical Drivers of Endothelial-Interstitial Interactions in Calcific Aortic Valve Disease. Circ Res. 2021;128:1344–1370. doi: 10.1161/CIRCRESAHA.121.318011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karikó K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem. 2004;279:12542–12550. doi: 10.1074/jbc.M310175200 [DOI] [PubMed] [Google Scholar]
- 7.Bsibsi M, Bajramovic JJ, Vogt MH, van Duijvenvoorden E, Baghat A, Persoon-Deen C, Tielen F, Verbeek R, Huitinga I, Ryffel B, et al. The microtubule regulator stathmin is an endogenous protein agonist for TLR3. J Immunol. 2010;184:6929–6937. doi: 10.4049/jimmunol.0902419 [DOI] [PubMed] [Google Scholar]
- 8.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863 [DOI] [PubMed] [Google Scholar]
- 9.Zhan Q, Song R, Zeng Q, Yao Q, Ao L, Xu D, Fullerton DA, Meng X. Activation of TLR3 induces osteogenic responses in human aortic valve interstitial cells through the NF-kappaB and ERK1/2 pathways. Int J Biol Sci. 2015;11:482–493. doi: 10.7150/ijbs.10905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang X, Fullerton DA, Su X, Ao L, Cleveland JC Jr., Meng X. Pro-osteogenic phenotype of human aortic valve interstitial cells is associated with higher levels of Toll-like receptors 2 and 4 and enhanced expression of bone morphogenetic protein 2. J Am Coll Cardiol. 2009;53:491–500. doi: 10.1016/j.jacc.2008.09.052 [DOI] [PubMed] [Google Scholar]
- 11.Zeng Q, Song R, Fullerton DA, Ao L, Zhai Y, Li S, Ballak DB, Cleveland JC, Jr., Reece TB, McKinsey TA, et al. Interleukin-37 suppresses the osteogenic responses of human aortic valve interstitial cells in vitro and alleviates valve lesions in mice. Proc Natl Acad Sci U S A. 2017;114:1631–1636. doi: 10.1073/pnas.1619667114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lopez J, Fernandez-Pisonero I, Duenas AI, Maeso P, Roman JA, Crespo MS, Garcia-Rodriguez C. Viral and bacterial patterns induce TLR-mediated sustained inflammation and calcification in aortic valve interstitial cells. Int J Cardiol. 2012;158:18–25. doi: 10.1016/j.ijcard.2010.12.089 [DOI] [PubMed] [Google Scholar]
- 13.Niessner A, Sato K, Chaikof EL, Colmegna I, Goronzy JJ, Weyand CM. Pathogen-sensing plasmacytoid dendritic cells stimulate cytotoxic T-cell function in the atherosclerotic plaque through interferon-alpha. Circulation. 2006;114:2482–2489. doi: 10.1161/circulationaha.106.642801 [DOI] [PubMed] [Google Scholar]
- 14.Gao D, Ciancanelli MJ, Zhang P, Harschnitz O, Bondet V, Hasek M, Chen J, Mu X, Itan Y, Cobat A, et al. TLR3 controls constitutive IFN-β antiviral immunity in human fibroblasts and cortical neurons. The Journal of Clinical Investigation. 2021;131. doi: 10.1172/JCI134529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takayanagi H, Kim S, Matsuo K, Suzuki H, Suzuki T, Sato K, Yokochi T, Oda H, Nakamura K, Ida N, et al. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-beta. Nature. 2002;416:744–749. doi: 10.1038/416744a [DOI] [PubMed] [Google Scholar]
- 16.Li J. JAK-STAT and bone metabolism. JAKSTAT. 2013;2:e23930. doi: 10.4161/jkst.23930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crow YJ, Manel N. Aicardi–Goutières syndrome and the type I interferonopathies. Nature Reviews Immunology. 2015;15:429–440. doi: 10.1038/nri3850 [DOI] [PubMed] [Google Scholar]
- 18.Rutsch F, MacDougall M, Lu C, Buers I, Mamaeva O, Nitschke Y, Rice Gillian I, Erlandsen H, Kehl Hans G, Thiele H, et al. A Specific IFIH1 Gain-of-Function Mutation Causes Singleton-Merten Syndrome. The American Journal of Human Genetics. 2015;96:275–282. doi: 10.1016/j.ajhg.2014.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aicardi J, Goutières F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol. 1984;15:49–54. doi: 10.1002/ana.410150109 [DOI] [PubMed] [Google Scholar]
- 20.Crow Y, Keshavan N, Barbet JP, Bercu G, Bondet V, Boussard C, Dedieu N, Duffy D, Hully M, Giardini A, et al. Cardiac valve involvement in ADAR-related type I interferonopathy. J Med Genet. 2020;57:475–478. doi: 10.1136/jmedgenet-2019-106457 [DOI] [PubMed] [Google Scholar]
- 21.Meng X, Ao L, Song Y, Babu A, Yang X, Wang M, Weyant MJ, Dinarello CA, Cleveland JC Jr., Fullerton DA. Expression of functional Toll-like receptors 2 and 4 in human aortic valve interstitial cells: potential roles in aortic valve inflammation and stenosis. Am J Physiol Cell Physiol. 2008;294:C29–35. doi: 10.1152/ajpcell.00137.2007 [DOI] [PubMed] [Google Scholar]
- 22.Gollmann-Tepeköylü C, Lobenwein D, Theurl M, Primessnig U, Lener D, Kirchmair E, Mathes W, Graber M, Pölzl L, An A, et al. Shock Wave Therapy Improves Cardiac Function in a Model of Chronic Ischemic Heart Failure: Evidence for a Mechanism Involving VEGF Signaling and the Extracellular Matrix. Journal of the American Heart Association. 2018;7. doi: 10.1161/jaha.118.010025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weiss RM, Ohashi M, Miller JD, Young SG, Heistad DD. Calcific aortic valve stenosis in old hypercholesterolemic mice. Circulation. 2006;114:2065–2069. doi: 10.1161/CIRCULATIONAHA.106.634139 [DOI] [PubMed] [Google Scholar]
- 24.Chen HY, Dufresne L, Burr H, Ambikkumar A, Yasui N, Luk K, Ranatunga DK, Whitmer RA, Lathrop M, Engert JC, et al. Association of LPA Variants With Aortic Stenosis: A Large-Scale Study Using Diagnostic and Procedural Codes From Electronic Health Records. JAMA Cardiol. 2018;3:18–23. doi: 10.1001/jamacardio.2017.4266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12:e1001779. doi: 10.1371/journal.pmed.1001779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pryshchep O, Ma-Krupa W, Younge BR, Goronzy JJ, Weyand CM. Vessel-specific Toll-like receptor profiles in human medium and large arteries. Circulation. 2008;118:1276–1284. doi: 10.1161/CIRCULATIONAHA.108.789172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tadros TM, Klein MD, Shapira OM. Ascending aortic dilatation associated with bicuspid aortic valve: pathophysiology, molecular biology, and clinical implications. Circulation. 2009;119:880–890. doi: 10.1161/CIRCULATIONAHA.108.795401 [DOI] [PubMed] [Google Scholar]
- 28.Eveborn GW, Schirmer H, Heggelund G, Lunde P, Rasmussen K. The evolving epidemiology of valvular aortic stenosis. the Tromso study. Heart. 2013;99:396–400. doi: 10.1136/heartjnl-2012-302265 [DOI] [PubMed] [Google Scholar]
- 29.Gomez-Stallons MV, Wirrig-Schwendeman EE, Hassel KR, Conway SJ, Yutzey KE. Bone Morphogenetic Protein Signaling Is Required for Aortic Valve Calcification. Arterioscler Thromb Vasc Biol. 2016;36:1398–1405. doi: 10.1161/atvbaha.116.307526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: A Transcriptional Activator of Osteoblast Differentiation. Cell. 1997;89:747–754. doi: 10.1016/S0092-8674(00)80257-3 [DOI] [PubMed] [Google Scholar]
- 31.Zhao W, Qi J, Wang L, Zhang M, Wang P, Gao C. LY294002 inhibits TLR3/4-mediated IFN-beta production via inhibition of IRF3 activation with a PI3K-independent mechanism. FEBS Lett. 2012;586:705–710. doi: 10.1016/j.febslet.2012.01.016 [DOI] [PubMed] [Google Scholar]
- 32.Hessle L, Johnson KA, Anderson HC, Narisawa S, Sali A, Goding JW, Terkeltaub R, Millan JL. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci U S A. 2002;99:9445–9449. doi: 10.1073/pnas.142063399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ducy P, Schinke T, Karsenty G. The osteoblast: a sophisticated fibroblast under central surveillance. Science. 2000;289:1501–1504. doi: 10.1126/science.289.5484.1501 [DOI] [PubMed] [Google Scholar]
- 34.Paré AC, Vichas A, Fincher CT, Mirman Z, Farrell DL, Mainieri A, Zallen JA. A positional Toll receptor code directs convergent extension in Drosophila. Nature. 2014;515:523–527. doi: 10.1038/nature13953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson KV, Bokla L, Nusslein-Volhard C. Establishment of dorsal-ventral polarity in the Drosophila embryo: the induction of polarity by the Toll gene product. Cell. 1985;42:791–798. [DOI] [PubMed] [Google Scholar]
- 36.Aceto J, Nourizadeh-Lillabadi R, Maree R, Dardenne N, Jeanray N, Wehenkel L, Alestrom P, van Loon JJ, Muller M. Zebrafish Bone and General Physiology Are Differently Affected by Hormones or Changes in Gravity. PLoS One. 2015;10:e0126928. doi: 10.1371/journal.pone.0126928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thanassoulis G, Campbell CY, Owens DS, Smith JG, Smith AV, Peloso GM, Kerr KF, Pechlivanis S, Budoff MJ, Harris TB, et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368:503–512. doi: 10.1056/NEJMoa1109034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng KH, Tsimikas S, Pawade T, Kroon J, Jenkins WSA, Doris MK, White AC, Timmers N, Hjortnaes J, Rogers MA, et al. Lipoprotein(a) and Oxidized Phospholipids Promote Valve Calcification in Patients With Aortic Stenosis. J Am Coll Cardiol. 2019;73:2150–2162. doi: 10.1016/j.jacc.2019.01.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kato R, Mori C, Kitazato K, Arata S, Obama T, Mori M, Takahashi K, Aiuchi T, Takano T, Itabe H. Transient increase in plasma oxidized LDL during the progression of atherosclerosis in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2009;29:33–39. doi: 10.1161/ATVBAHA.108.164723 [DOI] [PubMed] [Google Scholar]
- 40.Leonard JN, Ghirlando R, Askins J, Bell JK, Margulies DH, Davies DR, Segal DM. The TLR3 signaling complex forms by cooperative receptor dimerization. Proc Natl Acad Sci U S A. 2008;105:258–263. doi: 10.1073/pnas.0710779105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y, Liu L, Davies DR, Segal DM. Dimerization of Toll-like receptor 3 (TLR3) is required for ligand binding. J Biol Chem. 2010;285:36836–36841. doi: 10.1074/jbc.M110.167973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scott PG, Dodd CM, Bergmann EM, Sheehan JK, Bishop PN. Crystal structure of the biglycan dimer and evidence that dimerization is essential for folding and stability of class I small leucine-rich repeat proteoglycans. J Biol Chem. 2006;281:13324–13332. doi: 10.1074/jbc.M513470200 [DOI] [PubMed] [Google Scholar]
- 43.Lyskov S, Gray JJ. The RosettaDock server for local protein–protein docking. Nucleic Acids Research. 2008;36:W233–W238. doi: 10.1093/nar/gkn216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abagyan R, Totrov M, Kuznetsov D. ICM: a new method for protein modeling and design. Applications to docking and structure prediction from the distorted native conformation. J Comp Chem. 1994;15:488–506. [Google Scholar]
- 45.Tufvesson E, Malmstrom J, Marko-Varga G, Westergren-Thorsson G. Biglycan isoforms with differences in polysaccharide substitution and core protein in human lung fibroblasts. Eur J Biochem. 2002;269:3688–3696. doi: 10.1046/j.1432-1033.2002.03058.x [DOI] [PubMed] [Google Scholar]
- 46.Mis EK, Liem KF, Kong Y, Schwartz NB, Domowicz M, Weatherbee SD. Forward genetics defines Xylt1 as a key, conserved regulator of early chondrocyte maturation and skeletal length. Dev Biol. 2014;385:67–82. doi: 10.1016/j.ydbio.2013.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Drolet MC, Roussel E, Deshaies Y, Couet J, Arsenault M. A high fat/high carbohydrate diet induces aortic valve disease in C57BL/6J mice. J Am Coll Cardiol. 2006;47:850–855. doi: 10.1016/j.jacc.2005.09.049 [DOI] [PubMed] [Google Scholar]
- 48.Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–837. doi: 10.1038/nri2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moreth K, Brodbeck R, Babelova A, Gretz N, Spieker T, Zeng-Brouwers J, Pfeilschifter J, Young MF, Schaefer RM, Schaefer L. The proteoglycan biglycan regulates expression of the B cell chemoattractant CXCL13 and aggravates murine lupus nephritis. J Clin Invest. 2010;120:4251–4272. doi: 10.1172/JCI42213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Demer LL, Tintut Y. Hearts of Stone: Calcific Aortic Stenosis and Antiresorptive Agents for Osteoporosis. Circulation. 2021;143:2428–2430. doi: 10.1161/CIRCULATIONAHA.121.054823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pawade TA, Doris MK, Bing R, White AC, Forsyth L, Evans E, Graham C, Williams MC, Beek EJRv, Fletcher A, et al. Effect of Denosumab or Alendronic Acid on the Progression of Aortic Stenosis: A Double-Blind Randomized Controlled Trial. Circulation. 2021;143:2418–2427. doi: 10.1161/CIRCULATIONAHA.121.053708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cheng K, Wang X, Yin H. Small-molecule inhibitors of the TLR3/dsRNA complex. J Am Chem Soc. 2011;133:3764–3767. doi: 10.1021/ja111312h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Silkoff PE, Flavin S, Gordon R, Loza MJ, Sterk PJ, Lutter R, Diamant Z, Turner RB, Lipworth BJ, Proud D, et al. Toll-like receptor 3 blockade in rhinovirus-induced experimental asthma exacerbations: A randomized controlled study. J Allergy Clin Immunol. 2018;141:1220–1230. doi: 10.1016/j.jaci.2017.06.027 [DOI] [PubMed] [Google Scholar]
- 54.Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, Bae SC, Brohawn PZ, Pineda L, Berglind A, et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N Engl J Med. 2020;382:211–221. doi: 10.1056/NEJMoa1912196 [DOI] [PubMed] [Google Scholar]
- 55.Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317:1522–1527. doi: 10.1126/science.1139522 [DOI] [PubMed] [Google Scholar]
- 56.Takemura N, Kawasaki T, Kunisawa J, Sato S, Lamichhane A, Kobiyama K, Aoshi T, Ito J, Mizuguchi K, Karuppuchamy T, et al. Blockade of TLR3 protects mice from lethal radiation-induced gastrointestinal syndrome. Nature communications. 2014;5:3492. doi: 10.1038/ncomms4492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Holfeld J, Tepekoylu C, Reissig C, Lobenwein D, Scheller B, Kirchmair E, Kozaryn R, Albrecht-Schgoer K, Krapf C, Zins K, et al. Toll-like receptor 3 signalling mediates angiogenic response upon shock wave treatment of ischaemic muscle. Cardiovasc Res. 2016;109:331–343. doi: 10.1093/cvr/cvv272 [DOI] [PubMed] [Google Scholar]
- 58.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 61.Kamburov A, Pentchev K, Galicka H, Wierling C, Lehrach H, Herwig R. ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res. 2011;39:D712–717. doi: 10.1093/nar/gkq1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bosse Y, Miqdad A, Fournier D, Pepin A, Pibarot P, Mathieu P. Refining molecular pathways leading to calcific aortic valve stenosis by studying gene expression profile of normal and calcified stenotic human aortic valves. Circ Cardiovasc Genet. 2009;2:489–498. doi: 10.1161/CIRCGENETICS.108.820795 [DOI] [PubMed] [Google Scholar]
- 64.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sturn A, Quackenbush J, Trajanoski Z. Genesis: cluster analysis of microarray data. Bioinformatics. 2002;18:207–208. [DOI] [PubMed] [Google Scholar]
- 66.Lobenwein D, Tepekoylu C, Kozaryn R, Pechriggl EJ, Bitsche M, Graber M, Fritsch H, Semsroth S, Stefanova N, Paulus P, et al. Shock Wave Treatment Protects From Neuronal Degeneration via a Toll-Like Receptor 3 Dependent Mechanism: Implications of a First-Ever Causal Treatment for Ischemic Spinal Cord Injury. Journal of the American Heart Association. 2015;4:e002440. doi: 10.1161/JAHA.115.002440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tepekoylu C, Lobenwein D, Urbschat A, Graber M, Pechriggl EJ, Fritsch H, Paulus P, Grimm M, Holfeld J. Shock wave treatment after hindlimb ischaemia results in increased perfusion and M2 macrophage presence. J Tissue Eng Regen Med. 2018;12:e486–e494. doi: 10.1002/term.2317 [DOI] [PubMed] [Google Scholar]
- 68.Hinton RB Jr., Alfieri CM, Witt SA, Glascock BJ, Khoury PR, Benson DW, Yutzey KE. Mouse heart valve structure and function: echocardiographic and morphometric analyses from the fetus through the aged adult. Am J Physiol Heart Circ Physiol. 2008;294:H2480–2488. doi: 10.1152/ajpheart.91431.2007 [DOI] [PubMed] [Google Scholar]
- 69.Casaclang-Verzosa G, Enriquez-Sarano M, Villaraga HR, Miller JD. Echocardiographic Approaches and Protocols for Comprehensive Phenotypic Characterization of Valvular Heart Disease in Mice. J Vis Exp. 2017. doi: 10.3791/54110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dharmarajan S, Speer MY, Pierce K, Lally J, Leaf EM, Lin ME, Scatena M, Giachelli CM. Role of Runx2 in Calcific Aortic Valve Disease in Mouse Models. Front Cardiovasc Med. 2021;8:687210. doi: 10.3389/fcvm.2021.687210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hildebrand T, Rüegsegger P. A new method for the model-independent assessment of thickness in three-dimensional images. J Microsc. 1997;185:67–75. doi: 10.1046/j.1365-2818.1997.1340694.x [DOI] [Google Scholar]
- 72.Walker MB, Kimmel CB. A two-color acid-free cartilage and bone stain for zebrafish larvae. Biotech Histochem. 2007;82:23–28. doi: 10.1080/10520290701333558 [DOI] [PubMed] [Google Scholar]
- 73.Fleming A, Sato M, Goldsmith P. High-throughput in vivo screening for bone anabolic compounds with zebrafish. Journal of biomolecular screening. 2005;10:823–831. doi: 10.1177/1087057105279952 [DOI] [PubMed] [Google Scholar]
- 74.Pierce BG, Wiehe K, Hwang H, Kim B-H, Vreven T, Weng Z. ZDOCK server: interactive docking prediction of protein–protein complexes and symmetric multimers. Bioinformatics. 2014;30:1771–1773. doi: 10.1093/bioinformatics/btu097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Choe J, Kelker MS, Wilson IA. Crystal structure of human toll-like receptor 3 (TLR3) ectodomain. Science. 2005;309:581–585. doi: 10.1126/science.1115253 [DOI] [PubMed] [Google Scholar]
- 76.Liu L, Botos I, Wang Y, Leonard JN, Shiloach J, Segal DM, Davies DR. Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science. 2008;320:379–381. doi: 10.1126/science.1155406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bell JK, Botos I, Hall PR, Askins J, Shiloach J, Segal DM, Davies DR. The molecular structure of the Toll-like receptor 3 ligand-binding domain. Proc Natl Acad Sci U S A. 2005;102:10976–10980. doi: 10.1073/pnas.0505077102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim JH, Song DH, Youn SJ, Kim JW, Cho G, Kim SC, Lee H, Jin MS, Lee JO. Crystal structures of mono- and bi-specific diabodies and reduction of their structural flexibility by introduction of disulfide bridges at the Fv interface. Sci Rep. 2016;6:34515. doi: 10.1038/srep34515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. 2010;D66:486–501. doi: 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Naschberger A, Fürnrohr B, Lenac Rovis T, Malic S, Scheffzek K, Dieplinger H, Rupp B. The N14 anti-afamin antibody Fab: a rare VL1 CDR glycosylation, crystallographic re-sequencing, molecular plasticity, and conservative versus enthusiastic modelling. Acta Crystallogr. 2016;D72:1267–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fernandez-Recio J, Totrov M, Skorodumov C, Abagyan R. Optimal docking area: A new method for predicting protein–protein interaction sites. Proteins: Structure, Function, and Bioinformatics. 2005;58:134–143. doi: 10.1002/prot.20285 [DOI] [PubMed] [Google Scholar]
- 82.Krissinel E, Henrick K. Inference of Macromolecular Assemblies from Crystalline State. J Mol Biol. 2007;372:774–797. [DOI] [PubMed] [Google Scholar]
- 83.Krissinel E. Crystal contacts as nature’s docking solutions. Journal of Computational Chemistry. 2010;31:133–143. doi: 10.1002/jcc.21303 [DOI] [PubMed] [Google Scholar]
- 84.Garcia-Cattaneo A, Gobert FX, Muller M, Toscano F, Flores M, Lescure A, Del Nery E, Benaroch P. Cleavage of Toll-like receptor 3 by cathepsins B and H is essential for signaling. Proc Natl Acad Sci U S A. 2012;109:9053–9058. doi: 10.1073/pnas.1115091109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hoffmann TJ, Zhan Y, Kvale MN, Hesselson SE, Gollub J, Iribarren C, Lu Y, Mei G, Purdy MM, Quesenberry C, et al. Design and coverage of high throughput genotyping arrays optimized for individuals of East Asian, African American, and Latino race/ethnicity using imputation and a novel hybrid SNP selection algorithm. Genomics. 2011;98:422–430. doi: 10.1016/j.ygeno.2011.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kvale MN, Hesselson S, Hoffmann TJ, Cao Y, Chan D, Connell S, Croen LA, Dispensa BP, Eshragh J, Finn A, et al. Genotyping Informatics and Quality Control for 100,000 Subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) Cohort. Genetics. 2015;200:1051–1060. doi: 10.1534/genetics.115.178905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi: 10.1038/s41586-018-0579-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.O’Connell J, Sharp K, Shrine N, Wain L, Hall I, Tobin M, Zagury JF, Delaneau O, Marchini J. Haplotype estimation for biobank-scale data sets. Nat Genet. 2016;48:817–820. doi: 10.1038/ng.3583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, Donnelly P, Eichler EE, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, Kang HM, Fuchsberger C, Danecek P, Sharp K, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48:1279–1283. doi: 10.1038/ng.3643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huang J, Howie B, McCarthy S, Memari Y, Walter K, Min JL, Danecek P, Malerba G, Trabetti E, Zheng H-F, et al. Improved imputation of low-frequency and rare variants using the UK10K haplotype reference panel. Nature communications. 2015;6:8111. doi: 10.1038/ncomms9111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Consortium GT. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yavorska OO, Burgess S. MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int J Epidemiol. 2017;46:1734–1739. doi: 10.1093/ije/dyx034 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All high-throughput data are available via GEO. Accession numbers are GSE138360 and GSE223543. The datasets generated and/or analyzed during this study are available from the corresponding author on reasonable request except for GERA and UK Biobank data, which are available directly from those cohorts upon application.