

Abstract
Objective:
Chromatin remodeling genes (CRGs) encode components of epigenetic regulatory mechanisms and alterations in these genes have been identified in several tumor types, including gynecologic cancers. In this study, we sought to investigate the prevalence and clinicopathological associations of CRG alterations in endometrial carcinoma (EC).
Methods:
We performed a retrospective analysis of 660 ECs sequenced using a clinical massively parallel sequencing assay targeting up to 468 genes, including 25 CRGs, and defined the presence of somatic CRG alterations. Clinicopathologic features were obtained for all cases. Immunohistochemical interrogation of ARID1A and PTEN proteins was performed in a subset of samples.
Results:
Of the 660 ECs sequenced, 438 (66.4%) harbored CRG alterations covered by our panel. The most commonly altered CRG was ARID1A (46%), followed by CTCF (21%), KMT2D (18%), KMT2B (17%), BCOR (16%), ARID1B (12%) and SMARCA4 (11%). We found that ARID1A genetic alterations were preferentially bi-allelic and often corresponded to altered ARID1A protein expression in ECs. We further observed that ARID1A alterations were often subclonal when compared to PTEN alterations, which were primarily clonal in ECs harboring both mutations. Finally, CRG alterations were associated with an increased likelihood of myometrial and lymphovascular invasion in endometrioid ECs.
Conclusion:
CRG alterations are common in EC and are associated with clinicopathologic features and likely play a crucial role in EC.
Keywords: Chromatin remodeling, DNA, Endometrial carcinoma, Genetics, Mutations, Massively parallel sequencing, RNA, SWI/SNF, Transcriptomics
INTRODUCTION
Chromatin is the complex of chromosomal DNA that is organized by coiling around octamers of histone proteins and forming complexes called nucleosomes. The nucleosomes are in turn packaged together to form the chromatids of chromosomes. The organization and packaging of the nucleosomes along with the epigenetic modifications of the DNA determine the availability of DNA for transcription and replication (1, 2). Proteins involved in regulation of chromatin structure and accessibility are referred to as chromatin remodelers (3), and the genes encoding them are chromatin remodeling genes (CRGs).
Histones have long and flexible N-terminal tails rich in lysine and arginine residues that can undergo extensive modifications (4). One such modification is acetylation which neutralizes the positive charge of the histones and weakens the interaction of histones with DNA leading to transcriptional activation. Acetylation and subsequent gene activation is mediated by histone acetyltransferases (HAT); conversely, the effects of histone deacetyl-transferases (HDAC) lead to gene repression (5). Methylation is another common histone modification, and varying levels and sites of methylation affect the availability of DNA for transcription. For example, H3K4me3 is abundant in active gene promoter sites while H3K9me3 is associated with transcriptional repression (6). Other modifications of histones including phosphorylation, ubiquitination and biotinylation also affect DNA availability. In cancer cells, the balance of histone modification is often altered through mutations of histone modifying enzymes (7, 8).
Another group of chromatin regulators is the SWI/SNF complex, which is involved in altering the position of nucleosomes along the DNA, which in turn affects the organization of DNA and changes the distribution of chromosomal DNA into euchromatin and heterochromatin (9). These alterations lead to spatiotemporal changes in chromatin structure (10). Modifications of the chromatin structure can also occur at the macromolecular level affecting the overall organization of chromatin within the nucleus, including formation of chromosomes and centromeres (11).
In tumors, chromatin remodeling alterations transform the epigenetic and transcriptional programs modulating DNA replication and repair, and cell differentiation and proliferation (12). Massively parallel sequencing analyses revealed that chromatin remodeling genes are recurrently altered in several cancer types (13–15), including endometrial carcinoma (EC). In this study, we sought to define the landscape of CRG alterations and their clinico-pathologic associations in a large cohort of ECs subjected to massively parallel sequencing analysis.
METHODS
Case selection and data extraction
This study was approved by the Institutional Review Board at Memorial Sloan Kettering Cancer Center (MSK). Untreated primary ECs of all histologic types that underwent clinical FDA-authorized tumor-normal targeted massively parallel sequencing of 410–468 cancer-related genes (MSK-Integrated Mutation Profiling of Actionable Cancer Targets; MSK-IMPACT) (16, 17) during the years 2014 to 2019 were evaluated. Classification of ECs into the Cancer Genome Atlas molecular subtypes (18) was performed by employing a surrogate of the ProMisE model, as described previously (19, 20): tumors with hotspot POLE exonuclease domain mutations (n=50) were assigned to the ‘POLE ultra-mutated’ category; tumors with DNA mismatch repair (MMR)-deficiency were defined either by immunohistochemistry and/or genomic determination (high MSIsensor score; n=140) (21); tumors with aberrant p53 immunohistochemical expression and/or TP53 genetic alterations (n=237) were assigned to the p53 altered, non-hypermutated category (as a surrogate for copy number-high tumors); and the remainder of the samples (n=233) were assigned to the ECs of no specific molecular subtype (NSMP, a surrogate for copy number-low tumors).
Genomic data extraction
Genomic data were extracted from the MSK-IMPACT assay, including somatic mutations, copy number (CN) alterations, structural variants, fraction of genome altered and tumor mutational burden, as previously described (20). The mutational data included data on chromosomal location, base-pair change, protein change, predicted functional impact of the mutation and the associated variant allele frequency (VAF). The mutations were further annotated using the OncoKB database (22). Data on allele specific CN alterations and ploidy were extracted using the ‘facets’ R package (version 0.5.14) (23). Cancer cell fractions (CCFs, i.e. the bioinformatically inferred percentage of tumor cells harboring a given mutation in a sample, indicating clonality) of all mutations were inferred using ABSOLUTE (v1.0.6), as described previously (24). To identify genes that were mutated more often than expected by chance given background mutation processes we used the MutSig (MutSigCV) Matlab package (25). Evaluation for mutual exclusivity/co-occurrence was performed using the ‘DISCOVER’ R package (v0.9) (26).
The MSK-IMPACT assay includes 25 CRGs, which are listed in Supplementary Table 1; the designation of these CRGs to various categories was determined based on previously described criteria (3, 27, 28).
Pathology review
Histopathologic and morphologic data were extracted from the synoptic pathology reports. Scanned pathology slides of the sequenced tumors were reviewed by one author (AMB) to confirm the findings in the pathology report. To mitigate the effect of suboptimal interobserver concordance in histologic typing (12, 13), we confined this study to a single institution in which all tumors had been reviewed by a group of experienced gynecologic pathologists. Biweekly diagnostic consensus conferences encouraged a uniform diagnostic approach within the group, as did frequent review of cases for Tumor Board meetings and quality assurance, as previously described (20).
As a proof of concept, 18 cases were randomly chosen for testing by immunohistochemistry (IHC); IHC analyses for PTEN (clone 6h2.1, Ventana) and ARID1A (clone CL3595, AMAb91192, Atlas) proteins were performed. Loss of expression was considered as abnormal and extent and distribution of loss of expression was documented in the tumor and corresponding normal tissue. ARID1A and PTEN IHC analysis was performed on the same tissue blocks from which DNA for the sequencing assay was obtained.
Statistical analysis and clustering
Comparisons of quantitative data between the groups were performed using ANOVA with post-hoc Tukey tests and comparison of qualitative data including associations between clinicopathologic features and molecular data were performed using chi-squared tests with Fisher’s exact p-value calculation. P-values of <0.05 were considered statistically significant. Disease-free survival (DFS) and overall survival (OS) were analyzed using the log-rank test to compare subgroups, with the start date set as the date of initial diagnostic biopsy. Univariate and multivariate Cox proportional hazards analyses were performed to determine hazard ratios (HRs). RNA expression data (in the FPKM format) from The Cancer Genome Atlas EC cohort (18) were obtained from the Genomic Data Commons Data Portal (https://portal.gdc.cancer.gov) and the iDEP R package was used for analysis of differentially expressed genes (DEG) and unsupervised clustering and dimensionality reduction (29). For DEG; genes were selected using a paired t test with P < 0.05 and |log2(fold change)| > 1.
RESULTS
CRG alterations in ECs
The 660 primary untreated ECs included the following histologic subtypes: endometrioid carcinoma (n=418); serous carcinoma (n=131); clear cell carcinoma (n=22); mesonephric-like carcinoma (n=9); dedifferentiated carcinoma (n=3); undifferentiated carcinoma (n=3); corded and hyalinized EC (n=2); 72 ECs were diagnosed as high-grade endometrial carcinoma (not otherwise specified) based on ambiguous morphologic features and absence of type-specific immunophenotypes, which precluded confident histologic subclassification.
In total, 438/660 (66.4%) ECs harbored alterations in CRGs covered by our gene panel. Of the 2,229 somatic mutations identified in the 438 samples, 1,021 (45.8%) were loss of function alterations, including nonsense, frame-shift and splice site variants. Among the remainder of the 1,208 missense and inframe alterations, only 82 (6.8%) were classified as oncogenic, likely oncogenic or predicted oncogenic (22). The most commonly altered CRG was ARID1A (n=305, 46%), followed by CTCF (n=141, 21%), KMT2D (n=118, 18%), KMT2B (n=111, 17%), BCOR (n=106, 16%), ARID1B (n=76, 12%) and SMARCA4 (n=74, 11%) (Figure 1A). On average, 27% of the alterations in each gene consisted of multiple hits (2 or more mutations) and on average, a further 29% of the mutations in the CRG genes were accompanied by gene deletion and loss of heterozygosity in the corresponding wild-type allele. ARID1A (65%), and KMT2A (43%) were the genes most often associated with multiple hits or combinations of mutations and loss of heterozygosity/deletion; in contrast, CARM1 and SMARCD1 were least likely to harbor more than one hit (8% and 0% respectively).
Figure 1.
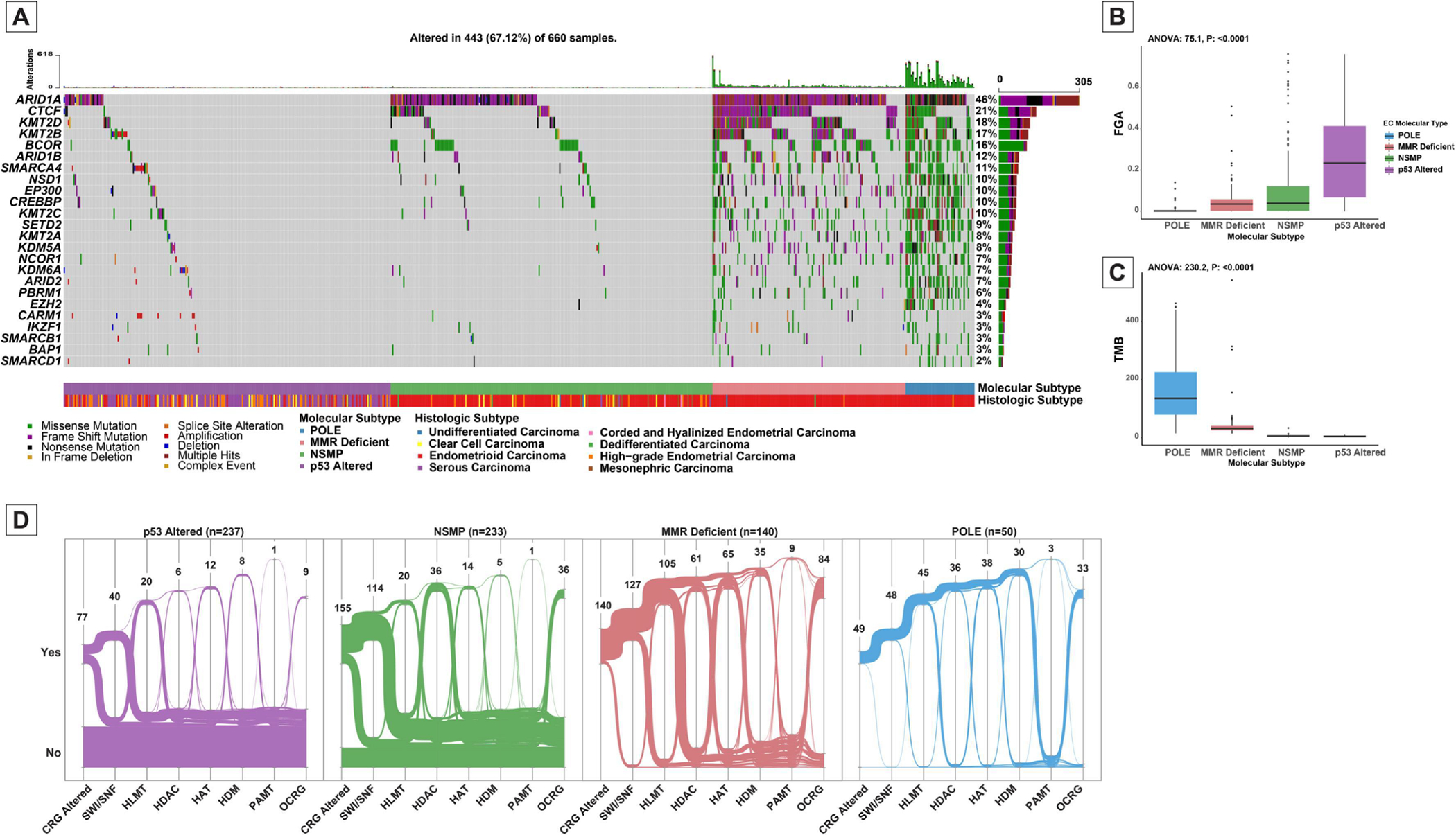
Landscape of somatic chromatin remodeling gene (CRG) alterations in endometrial carcinomas. (A) Oncoprint depicting the most recurrent CRG alterations in endometrial carcinomas. Each column represents a tumor with the bar graphs at the top depicting the number/distribution of alterations per sample and the fraction of genome alterations per sample. The Oncoprint rows show alterations in each gene. The lower part of the graph shows the summary of molecular subtype of endometrial carcinoma and histologic type for each case. The bar graph on the right of the panel shows the number and distribution of alterations for each gene. Mutation types and clinicopathologic features are color-coded according to the legend. (B) Box-plot showing the distribution of fraction of genome altered (FGA) among molecular subtypes of endometrial carcinoma. (C) Box-plot showing the distribution of tumor mutation burden (TMB) among molecular subtypes of endometrial carcinoma. (D) Alluvial graphs showing the distribution of alterations in CRG categories among the four major molecular subtypes of endometrial carcinoma (18).
POLE, POLE exonuclease domain mutated carcinoma; MMR-deficient, DNA mismatch repair deficient carcinoma; NSMP, TP53 wild-type non-hypermutant endometrial carcinomas; p53 Altered, TP53 altered non-hypermutant endometrial carcinomas.
SWI/SNF, SWI/SNF complex; HLMT, Histone lysine methyltransferases; HDAC, Histone deacetylases; HAT, Histone acetyltransferases; HDM, Histone demethylases; PAMT, Protein arginine methyltransferases; OCRG, Other chromatin gene alterations.
Overall, the SWI/SNF family of genes, including ARID1A, ARID1B, ARID2, PBRM1, SMARCA4, SMACRB1, and SMARCD1, were the most frequently altered CRGs in ECs (n=329, 50%). Other CRG complexes that were commonly altered included histone lysine methyltransferases (HLMT; n=190, 29%), histone deacetylases (HDAC; n=139, 21%), histone acetyltransferases (HAT; n=129, 20%) and histone demethylases (HDM; n=78, 12%).
CRG alterations in molecular subtypes of EC
The distribution of these alterations differed among molecular subgroups, e.g., HDAC alterations were common in ECs of POLE, MMR-deficient and NSMP molecular subtypes while these alterations were rare in p53-altered ECs (31% versus 3%; X2 P: <0.0001; Figure 1D). Specifically, among the 50 POLE exonuclease domain-mutated ECs, 49 had at least one CRG mutation. All MMR-deficient ECs (n=140) had at least one CRG mutation. Of the 233 NSMP ECs, 155 (67%) had CRG alterations, whereas only 41% (n=98) of p53-altered ECs harbored CRG alterations. In fact, only 32% (n=77) of p53-altered EC had pathogenic CRG alterations (Figures 1-A, B). In concordance with this observation, CRG-altered tumors were found to have lower fraction of the genome altered (median fraction of genome altered 0.037 versus 0.16; Mann-Whitney U P: <0.001) and conversely a higher tumor mutation burden (median tumor mutation burden 9.7 versus 4.4, Mann-Whitney U P: <0.001).
ARID1A alterations were the most common CRG alteration in POLE (88%), MMR-deficient (90%) and NSMP (45.5%) subgroups, however, they were relatively uncommon in p53-altered ECs (12.2%; X2 P: <0.0001). POLE and MMR-deficient tumors showed a higher abundance of CRG alterations compared to other molecular subtypes across all genes evaluated in the cohort.
ARID1A alterations in ECs
In total, 481 gene mutations and 9 deep deletions of ARID1A were observed in 305 ECs. In general, somatic mutations were observed along the length of the ARID1A gene, but there were recurrent frameshift mutations of codon D1850 (n=30, 10%) and nonsense mutations of codon R1989 (n=20, 7%; Figure 2-A). While the majority (86%; 38/50) of POLE exonuclease domain-mutated ECs harbored truncating (frameshift or nonsense) ARID1A mutations, 87% of the mutations in all other genes in POLE-mutated tumors were missense mutations, showing relative enrichment of loss-of-function ARID1A mutations in this subgroup. Also, the 90% (126/140) MMR-deficient ECs harboring ARID1A gene alterations were in the form of truncating mutations.
Figure 2.
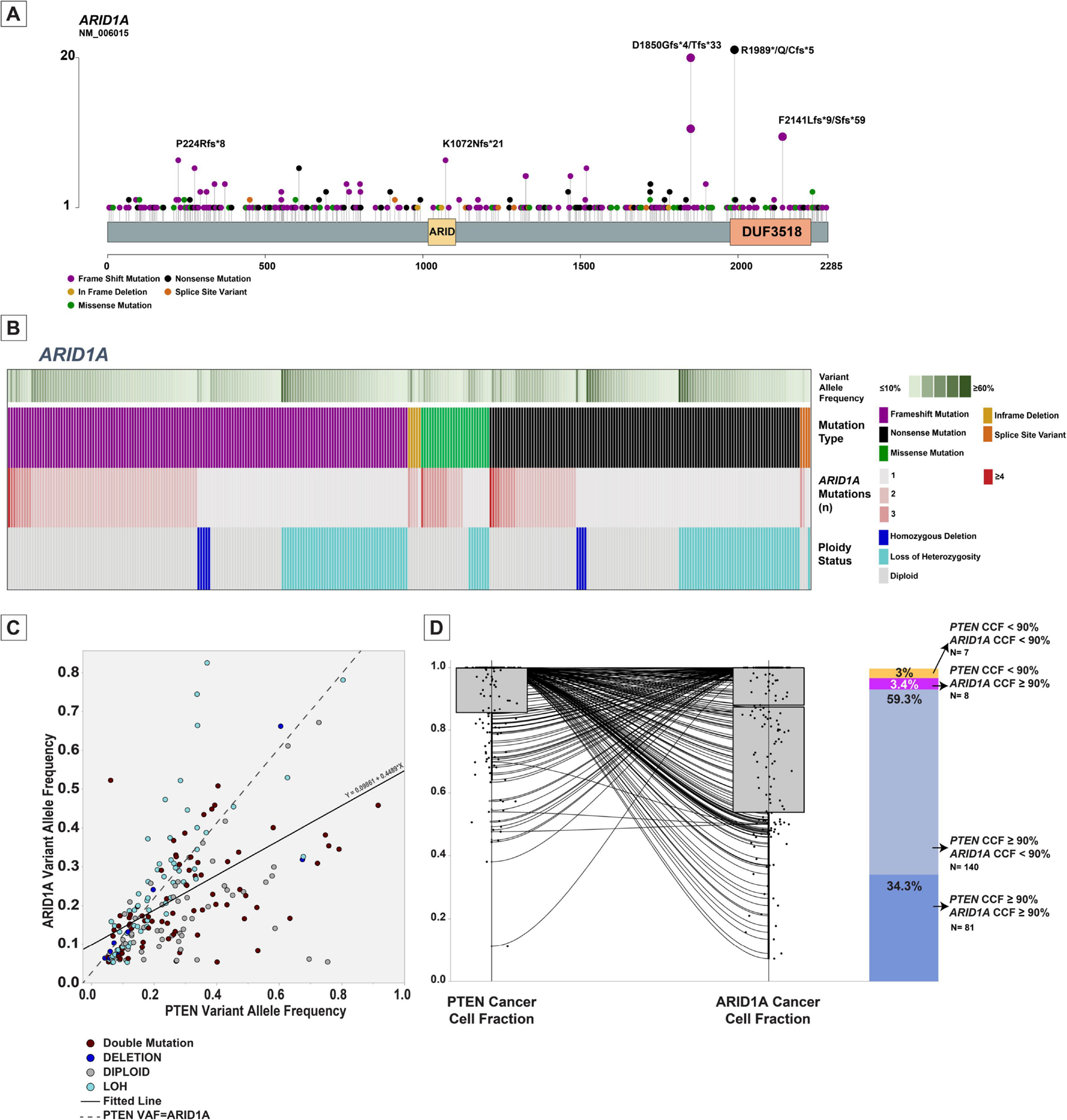
Overview of ARID1A alterations in endometrial carcinomas. (A) Lollipop plot showing the distribution of mutations involving ARID1A in endometrial carcinomas. Each vertical line shows the location of the mutation, and the height of the lines corresponds to number of samples with the corresponding mutation. Mutation types are color-coded according to the legend. (B) Oncoprint showing the distribution of ARID1A mutation types, variant allele frequency (VAF) of the ARID1A mutation in the sample, number of ARID1A alterations per sample and corresponding ARID1A ploidy status in each sample. Color codes are provided in the legend. (C) Scattergram comparing VAF of ARID1A alterations to VAF of PTEN alterations in samples harboring both alterations. Each circle corresponds to a sample. The circles are color-coded based on ARID1A alteration status. The dashed line represents the hypothetical perfect correlation fit. The solid line represents the fitted line for the comparison of VAF between the alterations. (D) Box and whisker plots comparing the cancer cell fraction of PTEN to ARID1A in samples harboring both alterations. The dots represent individual samples. The lines connect the VAF of the two alterations in each sample. The bar-plot on the right depicts the proportion of samples with various cancer cell fractions of PTEN and ARID1A alterations in endometrial carcinomas.
VAF, Variant allele frequency; LOH, Loss of heterozygosity; CCF, Cancer cell fraction; N, Number.
ARID1A is thought to have a tumor-suppressive role and we therefore assessed whether both ARID1A alleles were affected in the ECs studied. Of the 305 ARID1A-mutant ECs, 104 (34.1%) showed either loss of heterozygosity (LOH) or copy neutral LOH of the wild-type allele, and 9 ECs (2.9%) harbored ARID1A homozygous deletions in addition to mutations. In addition, 128 (42%) ECs harbored at least two mutations in ARID1A, either pairs of loss-of-function mutations or missense mutations involving the -cis and -trans alleles. In total, 241 ECs (79%) harbored biallelic ARID1A genomic alterations. The proportion of endometrioid ECs harboring more than one genomic alteration of ARID1A was 73% (Figure 2-B), in contrast to only 9% in non-endometrioid ECs (X2 P: <0.0001). When assessing the mutation status by molecular subtype, we observed that among the POLE exonuclease domain-mutated ECs with ARID1A alterations (n=44), 27 (61%) harbored bi-allelic ARID1A alterations with 16 (36%) having more than one truncating mutation and 11 (25%) displaying LOH or copy neutral LOH. The prevalence of biallelic ARID1A mutations in MMR-deficient ECs was also high (75/126, 59.5%), with 57 having more than one truncating mutation (45%) and 18 (14%) having LOH or copy neutral LOH of the wild-type allele. Of note, while the median tumor mutation burden for POLE exonuclease domain-mutated ECs and MMR-deficient ECs were 135.1 and 31.6 mutations/Mb respectively, the median mutation burden for the ARID1A gene in these molecular subtypes was 285.7 and 266.3, respectively (P: <0.001 for both). Furthermore, evaluation by MutSig showed that among CRG genes, the mutation rate in ARID1A and CTCF is significantly higher than the background mutational process (P <0.0001 and q <0.0001 for both).
Alterations in PTEN were the most common genomic alterations in ECs, and were identified in all but the p53-altered molecular subtype. Of the 660 ECs included in our study, 54% harbored alterations involving the PTEN gene. The majority of ARID1A-mutant ECs had a co-occurring PTEN alteration (245/305, 80.3%, DISCOVER P: <0.00001). PTEN alterations are believed to be the initiating event in pathogenesis of many ECs, especially endometrioid carcinomas (30). Consequently, we decided to compare the clonality of the PTEN and ARID1A mutations in endometrioid ECs harboring alterations in both genes (n=236). We observed that the ratio of the variant allele frequency (VAF) of ARID1A versus PTEN was 0.45 in endometrioid ECs harboring both mutations (Figure 2-C). In addition, the mean cancer cell fractions (CCFs) of PTEN were significantly higher than those of ARID1A mutations (0.91 versus 0.78; Wilcoxon signed-rank test P: <0.0001), providing evidence to suggest that PTEN mutations are more frequently clonal (n=221, 93.6%) as compared to ARID1A mutations, which are commonly subclonal (n=147, 62.3%) (Figure 2-D).
Most alterations in the ARID1A gene are predicted to be truncating events; to verify whether ARID1A mutations indeed lead to loss of protein expression, we determined the levels of ARID1A protein expression using IHC in 18 randomly selected endometrioid ECs harboring both PTEN and ARID1A alterations (Figure 3). We also stained the tumors for PTEN to serve as a comparator. All 18 ECs showed some loss of PTEN expression in tumor cells: 11 tumors displayed complete loss of PTEN protein expression in the presence of an internal control; two tumors showed subclonal loss of expression; and 5 tumors showed variable loss of PTEN protein expression (Figure 3 and 4). ARID1A IHC analysis revealed complete loss of expression in 6 tumors, subclonal loss in 3 tumors, variable loss in 6 tumors and retained expression in 3 tumors. As expected, given their lower CCFs, loss of ARID1A expression was observed in a lower fraction of tumor cells compared to PTEN in 10/18 cases. However, 3 endometrioid ECs showed complete loss of ARID1A and PTEN expression and 3 further cases showed complete loss of ARID1A expression with variable loss of PTEN expression (Figure 4-A, Supplementary Table 2). 2 cases showed variable/subclonal staining for both ARID1A and PTEN.
Figure 3.
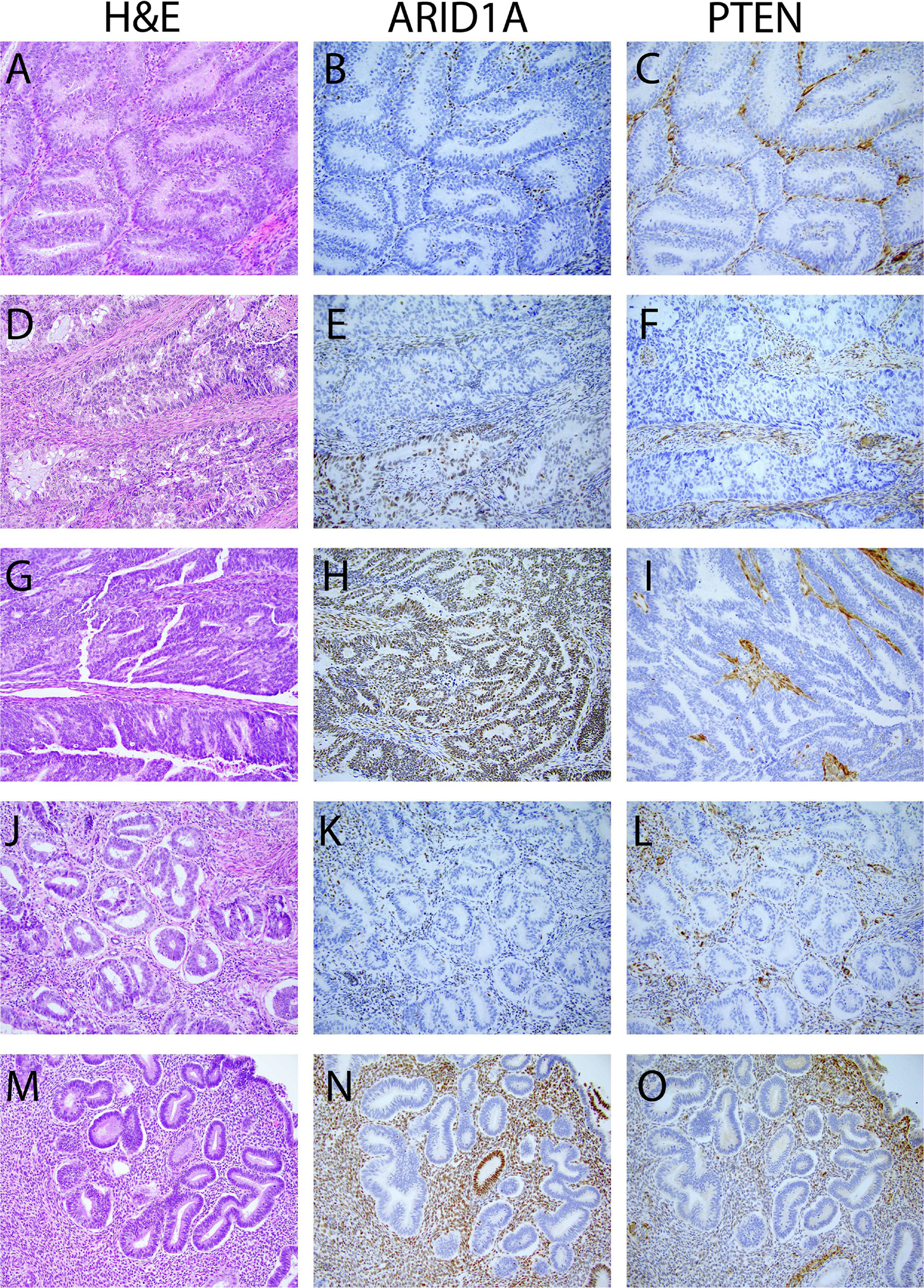
Examples of immunohistochemical evaluation of ARID1A and PTEN protein expression in endometrial samples. The figures in the left panels represent images of hematoxylin-eosin-stained tissue samples. The middle panel and the right panel show the corresponding images of the ARID1A and PTEN immunohistochemical stained slides. (A-C) Uterine endometrioid carcinoma with complete loss of ARID1A and PTEN expression. (D-F) Uterine endometrioid carcinoma with complete loss of PTEN and subclonal loss of ARID1A expression. (G-H) Uterine endometrioid carcinoma with complete loss of PTEN and retained ARID1A expression. (J-K) Atypical endometrial hyperplasia with complete loss of PTEN and ARID1A expression. (M-O) Loss of PTEN and ARID1A expression in morphologically normal endometrium.
Figure 4.
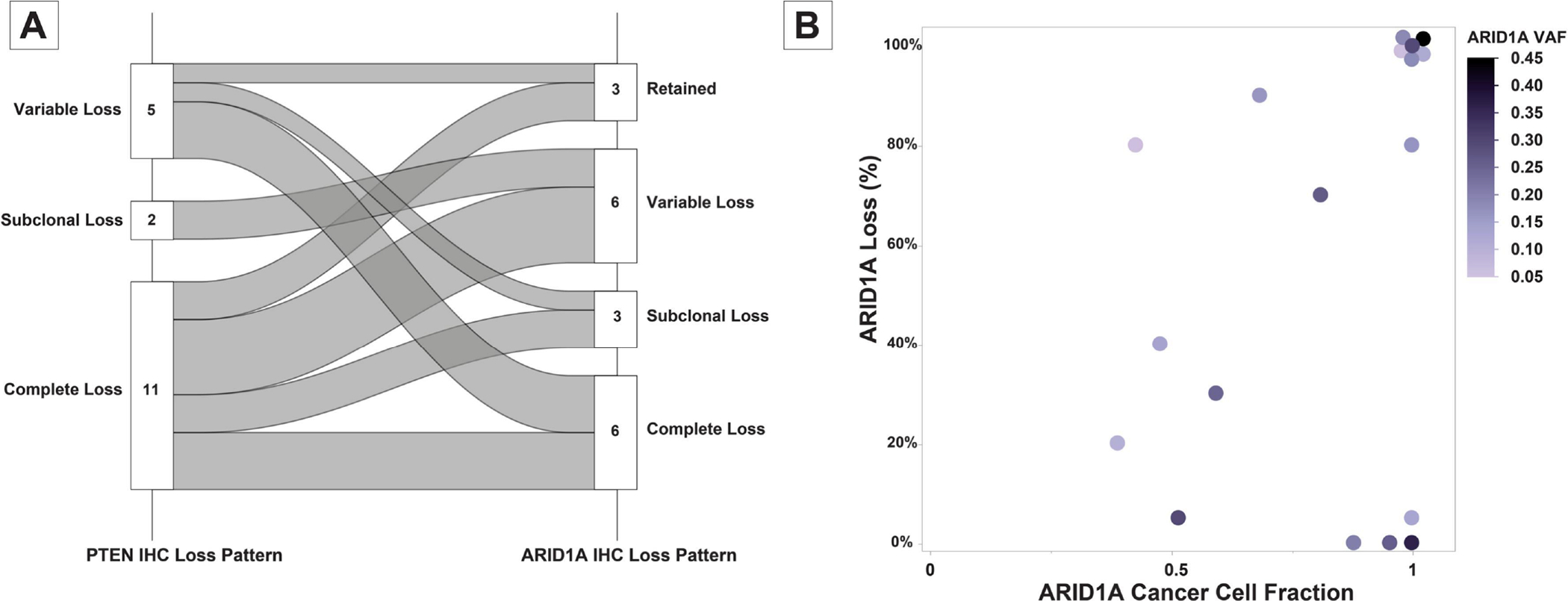
Summary of immunohistochemical evaluation of ARID1A and PTEN protein expression in endometrial carcinoma samples. (A) Alluvial graph showing the relationship of immunohistochemical expression patterns of PTEN and ARID1A in the interrogated samples. (B) Scattergram comparing the cancer cell fractions of ARID1A alterations in a sample with corresponding immunohistochemical expression status of ARID1A in the corresponding samples. Each circle represents a sample; the colors represent the ARID1A variant allele frequency (VAF).
Comparison of loss of ARID1A expression with the predicted CCFs of ARID1A mutation showed good concordance in 13 cases. In the 5 outliers, little or no loss of ARID1A protein expression was observed despite a high predicted CCF of ARID1A mutation in the tumor (Figure 4-B). When assessing the mutation types, we found that the cases with discordant ARID1A CCFs and ARID1A IHC levels of expression all either harbored mono-allelic and non-pathogenic missense mutations (p.H2090R and p.R1658W) or mono-allelic frameshift mutations. Of note, we found that 8 ECs also had normal endometrium present in the tissue block; one of these cases showed subclonal loss of ARID1A and PTEN expression in the normal endometrium and two other samples showed subclonal loss of PTEN expression in the normal endometrium (the results of PTEN and ARID1A IHC are summarized in Supplementary Table 2).
BCOR alterations
BCOR is another commonly altered CRG in ECs (n=106, 16%). BCOR alterations were found to be more common in endometrioid than non-endometrioid ECs (24% vs 3%, X2 P: <0.0001). Of the BCOR-altered tumors, 62 (58%) harbored p.N1459S hotspot mutations including 33 NSMP, 27 MMR-deficient, 1 POLE and 1 p53-altered ECs (Figure 5-A). While the functional consequences of this hotspot alteration are not known, in the TCGA EC (TCGA-UCEC) cohort, 27 of 517 samples (5%) harbored the p.N1459S BCOR mutation, including 11/147 (7%) NSMP [CN-L] EC and 16/148 (10.8%) MMR-deficient [MSI] carcinomas (18). To identify whether this alteration leads to any changes in the transcriptomic landscape of tumors, we performed hierarchical clustering and principal component analysis of the normalized RNA expression in the NSMP subgroup of the TCGA cohort which revealed that the 7 (7/11) of the BCOR-mutated ECs clustered together, and in a branch separate from most BCOR wild-type ECs, suggesting transcriptomic differences between BCOR-mutated and the other tumors in the cohort (Figure 5-B, C). Furthermore, differential expression analysis showed 41 differentially expressed genes (31 upregulated genes and 10 downregulated genes) in the 11 EC harboring pathogenic BCOR mutations as compared to those lacking these alterations (n=136; Figure 5-D, Supplementary Table 3).
Figure 5.
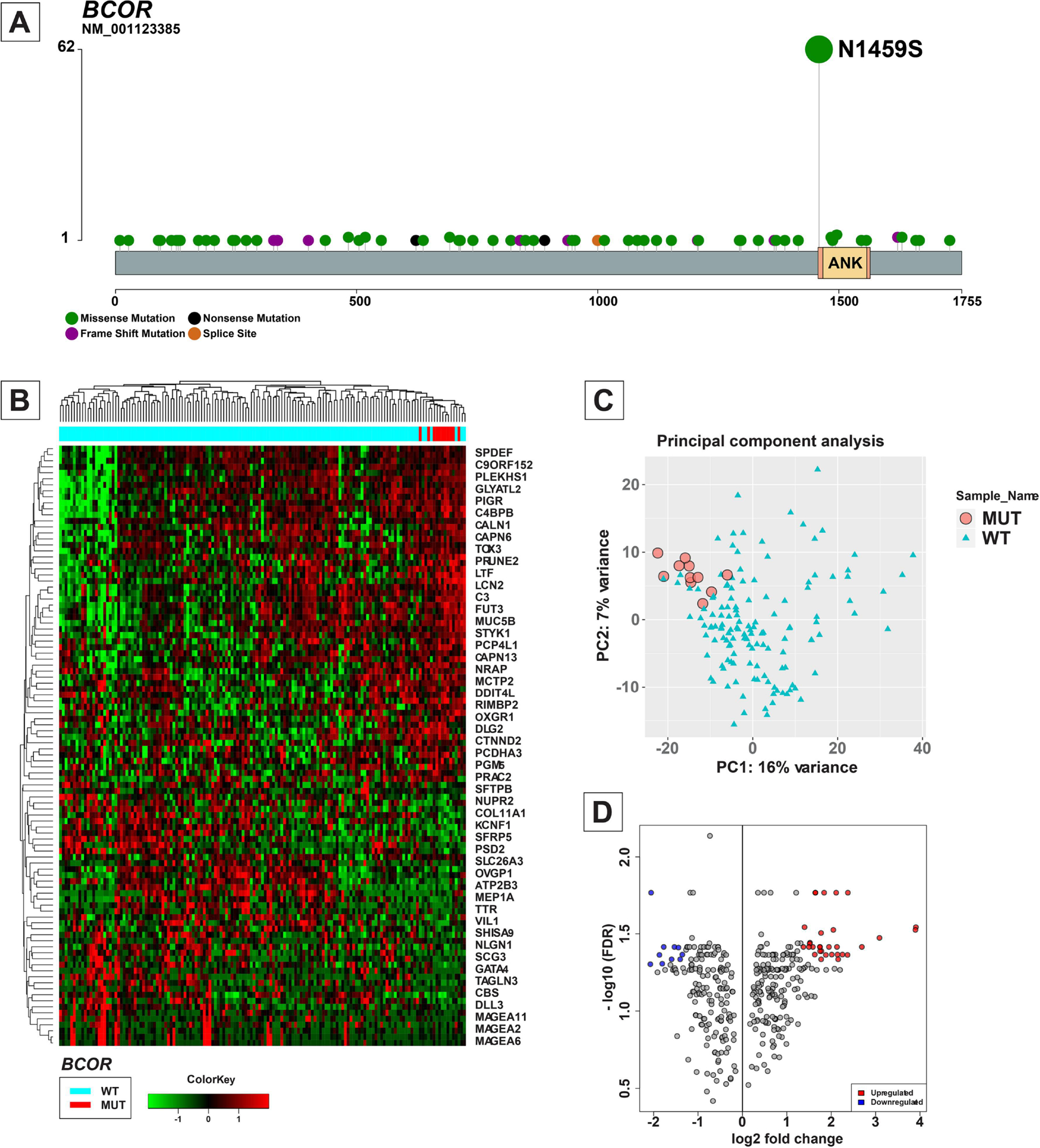
Overview of BCOR alterations in endometrial carcinomas. (A) Lollipop plot showing the distribution of mutations involving BCOR in endometrial carcinomas. Each vertical line shows the location of the mutation, and the height of the lines corresponds to number of samples with the corresponding mutation. Mutation types are color-coded according to the legend. (B) Heatmap and hierarchical clustering based on mRNA expression data of the 100 most variably expressed genes in the copy number low molecular subtype of endometrial carcinomas in The Cancer Genome Atlas (TCGA) cohort (18). The columns mark individual samples, and the rows represent individual genes. The expression data is normalized with the color scale shown at the bottom. The bar at the top indicates BCOR wild-type and BCOR-altered samples. (C) Scattergram showing the principal component analysis based on the mRNA expression data of the 100 most variably expressed genes in the copy number-low molecular subtype of endometrial carcinomas in TCGA cohort. The pink circles represent BCOR-mutated samples and green triangles represent BCOR-wild-type samples. (D) Volcano-plot comparing the mRNA expression of various genes among samples with and without BCOR alterations in TCGA cohort.
WT, Wild-type; MUT, Mutant; PC, Principal component.
Clinicopathologic associations of CRG alterations in ECs
We explored whether the presence of CRG alterations was associated with specific clinicopathologic features. While CRG alterations are seen in all histologic subtypes of endometrial carcinoma, the frequency of CRG alterations was found to be higher in endometrioid ECs (80%) compared to non-endometrioid ECs (27%, X2 P: 0.001). CRG-wild-type ECs occurred in older patients compared with CRG-mutant tumors (median age: 67 versus 63 years, Mann-Whitney U P: <0.001).
Information on myometrial invasion was available in 397/418 endometrioid ECs. In these tumors, the presence of CRG alterations was associated with increased likelihood of myometrial invasion (75%, 242/324 CRG-altered versus 51%, 38/75 no CRG alterations; X2 P: <0.001; Figure 6-A). The presence of CRG alterations was associated also with greater depth of myometrial invasion in endometrioid ECs (median depth of myometrial invasion as proportion of myometrial thickness: 25% versus 6%; Mann-Whitney U P: 0.02). Of the 280 endometrioid ECs with myometrial invasion, CRG-altered tumors were more likely to exhibit a microcystic, elongated and fragmented (MELF) pattern of invasion (49, 20.2% versus 2, 5.3%, X2 P: 0.026). The association of myometrial invasion and MELF pattern of invasion in endometrioid ECs was also seen when focusing on tumors with ARID1A alterations only (74% versus 55%, X2 P: 0.001 and 16% versus 5%, X2 P:0.012, respectively). Endometrioid ECs with CRG alterations were more likely to have lymphovascular invasion (45%, 146/324 versus 29%, 22/75; X2 P: 0.018). In endometrioid ECs with known pelvic lymph node status (233/418), the presence of CRG alterations was associated with increased likelihood of pelvic lymph node metastasis (12%, 18/155 versus 3%, 2/78, X2 P: 0.038). CRG alteration status did not show any correlation with non-lymphoid extrauterine disease spread at presentation or at recurrence (X2 P > 0.05 for both). None of the CRG alterations showed statistically significant associations with clinical stage, recurrence rate, recurrence/metastatic site, disease free survival or overall survival (P >0.05 for all comparisons).
Figure 6.
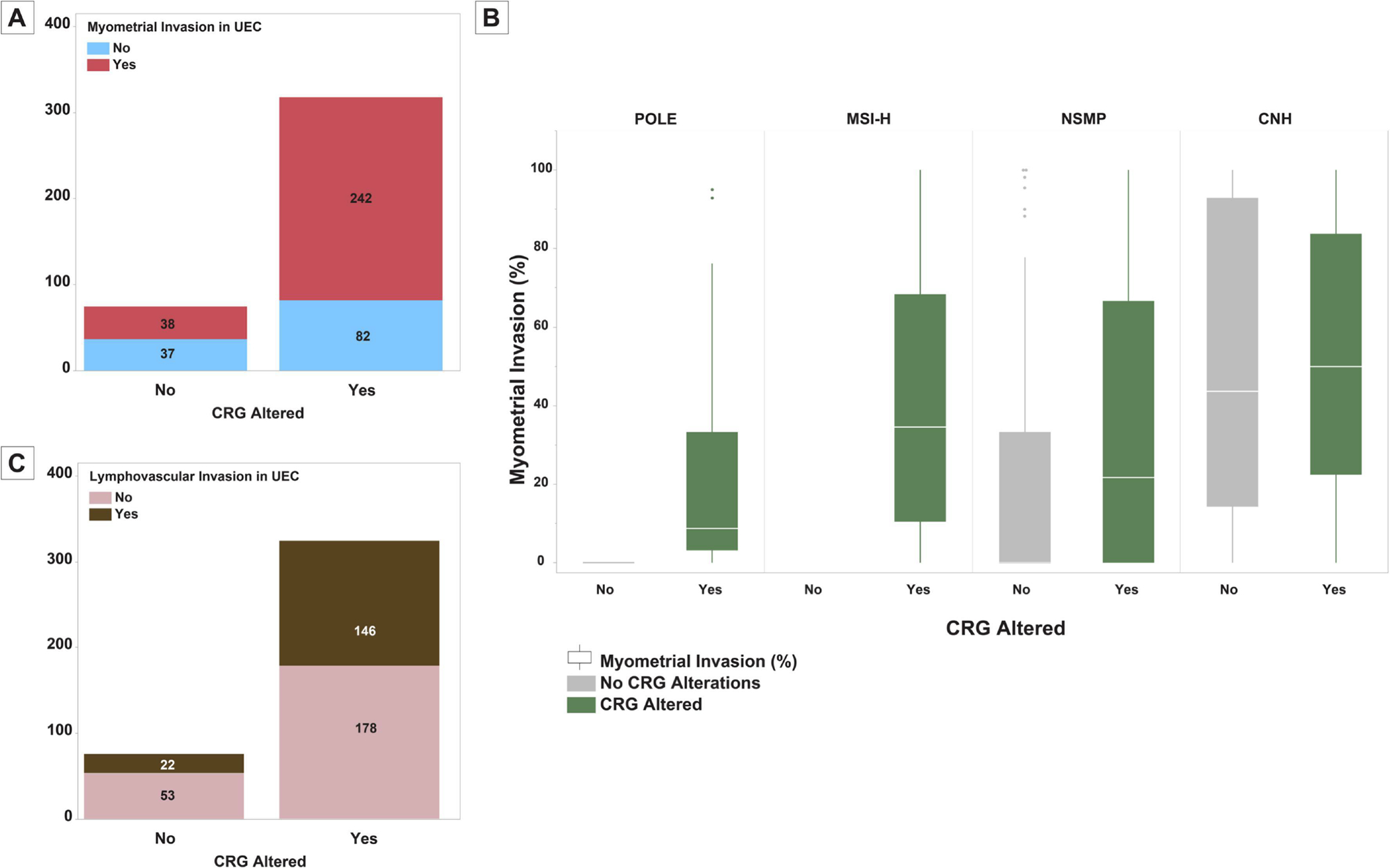
Correlation of chromatin remodeling gene (CRG) alterations in endometrial carcinomas with histopathologic features. (A) Bar-plot showing the distribution of myometrial invasion among CRG-altered and CRG-wild-type uterine endometrioid carcinomas. (B) Bar-plot showing the distribution of lymphovascular invasion among CRG-altered and CRG-wild-type uterine endometrioid carcinomas. (C) Box and whisker plots comparing the depth of myometrial invasion between CRG-altered and CRG-wild-type endometrial carcinomas among the four molecular subtypes of endometrial carcinoma.
UEC, Uterine endometrioid carcinoma; POLE, POLE exonuclease domain mutated carcinoma; MSI-H, DNA mismatch repair deficient carcinoma; CN-L, TP53 wild-type non-hypermutant endometrial carcinomas; CN-H, TP53 altered non-hypermutant endometrial carcinomas.
DISCUSSION
Our analyses show that alterations of CRGs are common events (present in 67%) in ECs, most commonly in endometrioid ECs, but are less prevalent in p53-altered non-hypermutated ECs.
The most commonly altered CRGs in ECs encode proteins involved in the SWI/SNF complex. This complex is composed of many subunits including ARID1A, ARID1B, ARID2, SMARCA2, SMARCA4, SMARCB1, SMARCE1, and DPF2 among others (9, 31). Among our samples, the most frequently altered gene from this group was ARID1A (altered in 46% of tumors) which is in line with prior studies showing frequent occurrence of somatic ARID1A mutations and loss of ARID1A expression in endometrioid ECs (32–35). We have also provided further evidence that ARID1A gene alterations are commonly truncating/inactivating mutations (85%), as previously suggested (14, 18, 36). The rate of ARID1A alterations and CRG alterations in serous ECs was significantly lower than the endometrioid ECs, suggesting that genomic landscape alterations caused by TP53 mutation-associated chromosomal instability in the former group might (to varying degrees) supersede epigenetic landscape modulations and epigenetic instability due to CRG alterations (37). Interestingly, in ECs with either POLE exonuclease domain mutations or microsatellite instability, there is an enrichment of ARID1A mutations as shown by higher mutation burden of the gene compared to other the other cancer genes assessed. Furthermore, even in POLE exonuclease domain mutated carcinomas which mainly harbor missense mutations of various genes, the ARID1A mutations are almost entirely truncating, suggesting an evolutionary selective pressure for loss of ARID1A function.
The AT-rich interactive domain 1A (ARID1A) gene encodes for BAF250, a nuclear protein that participates in SWI/SNF complexes, regulates DNA organization and consequently modulates transcription, DNA repair and replication (9, 38, 39). ARID1A mutations contribute to tumor progression and acquisition of invasive properties (33, 34, 36). Since ARID1A mutations have been shown to result in loss of ARID1A/BAF250 protein expression, immunohistochemical evaluation for ARID1A can serve as a surrogate marker for the gene alterations (40, 41). In our cohort, there was evidence of bi-allelic inactivation of the gene in 65% of ECs and IHC results showed loss of ARID1A expression in some atypical endometrial hyperplasia and even in rare instances of morphologically normal endometrium adjacent to carcinoma in some cases; immunohistochemical interrogation of the ARID1A protein showed generally concordant results between the molecular profile and the tumor immunophenotype. In contrast to EC, ARID1A mutations and loss of expression are less common in EC precursors such as atypical endometrial hyperplasia (33, 34, 36). Nevertheless, loss of ARID1A immunoreactivity in endometrial biopsies showing atypical endometrial hyperplasia has been shown to be highly predictive of subsequent endometrial carcinoma in the hysterectomy specimen (42), suggesting that ARID1A alteration plays a role in the progression of endometrial neoplasia.
While some studies have associated ARID1A mutations with adverse prognostic effects (43, 44), others have not shown any significant clinical differences between ECs harboring ARID1A alterations and ARID1A-wild-type tumors (36). There is also evidence that ARID1A is a tumor suppressor which acts with p53 to regulate CDKN1A and SMAD3, and mutations in this gene lead to endometrial tumor growth (40). ARID1A protein also binds to estrogen receptor enhancer targets and regulates estrogen-dependent transcription; as such ARID1A alterations may affect the response of neoplastic cells to estrogen regulation (45). Furthermore, murine models have shown that ARID1A alterations remove the suppressive effects of TGFβ on tumor cells and consequently promote cell migration and tumor invasion in PTEN-deficient ECs (46). Another study has suggested that ARID1A alterations in presence of PI3K pathway activation leads to enhanced invasion through epithelial mesenchymal transition (38). Our results also show that CRG alterations, especially ARID1A alterations, are associated with increased depth of myometrial invasion and increased frequency of a MELF pattern of invasion, which supports the evidence for increased cell mobility and invasive properties of CRG altered tumors.
ARID1A alterations commonly co-occur with alterations that activate the PI3K pathway (18), with evidence at proteomic level for activation of PI3K pathway in ARID1A mutant endometrial cell lines (47). The synergistic effects of PTEN and ARID1A loss lead to increased proliferation rate in neoplastic endometrial cells (36). Unlike PI3K pathway alterations which are most commonly truncal and clonal mutations, ARID1A alterations commonly occur in phylogenic branches and are subclonal events, with frequent heterogeneity of the mutations in this gene among spatially sampled endometrial carcinomas (30). Other SWI/SNF complex alterations also show phylogenetic heterogeneity in various cancers (48, 49). This finding suggests that chromatin remodeling alterations occur later in tumor evolution, the resultant epigenetic transformation furthering tumor growth and progression (30). Despite subclonal and heterogenous mutations in ARID1A, null pattern ARID1A immunophenotype in advanced lesions suggests phenotypic convergence of tumors towards loss of functional ARID1A protein (30, 35, 50). The results of our study also suggest that ARID1A alterations commonly occur later in endometrioid tumor evolution than PI3K pathway alterations, with CCFs of ARID1A alterations often being lower than those of PTEN alterations. However, we also found that in 37.7% of tumors, ARID1A alterations have a >90% CCF, which may indicate clones which expand to dominate the tumor as a result of evolutionary benefit conferred by these alterations (a phenomenon termed ‘clonal sweep’) (51).
Other components of the SWI/SNF complex are also frequently altered in ECs - 50% of ECs showed at least one alteration in the SWI/SNF complex proteins. Loss of expression of other members of the SWI/SNF family, especially SMARCA4 and SMARCA2 have been associated with undifferentiated and dedifferentiated carcinomas of the endometrium (52). Alterations of the members of SWI/SNF complex other than ARID1A are preferentially seen in POLE exonuclease domain-mutated tumors and DNA mismatch repair-deficient tumors (53), findings which were also corroborated in our cohort.
Another frequent CRG alteration in ECs were CTCF mutations. Alterations in this gene were observed almost exclusively in endometrioid carcinomas. CTCF encodes 11-ZF DNA binding protein, which has extensive and diverse roles in organization of chromatin structure and regulates inter- and intra-chromosomal interactions, nucleosome positioning, alternative splicing among other functions (54). This protein is called the ‘master weaver of the genome’ because of its multifaceted role (54, 55). Loss of CTCF has been associated with silencing of tumor suppressor genes and activation of oncogenes (56). CTCF knockdown in cell models has been shown to produce sustained proliferative signaling, anti-apoptotic properties, and epigenetic dysregulation as well as deregulation of endometrial cancer spheroid polarity (54).
BCOR was another commonly altered gene in our cohort, almost exclusively in endometrioid ECs. BCOR encodes for a transcriptional co-repressor (57). Translocations of the gene are seen in some lymphomas (58) and translocation and internal tandem duplications of the gene are seen in soft tissue sarcomas (59) and endometrial stromal sarcomas (60). Somatic mutations of the gene have been previously reported in endometrial carcinomas with almost all reported mutations involving the N1459S hotspot (15), a finding corroborated by our results. Evaluation of the TCGA RNA expression data shows that this hotspot mutation is associated with transcriptional changes in endometrioid ECs, but the functional significance of this mutation is yet to be determined.
Other commonly altered CRGs included the KMT2 family of genes including KMT2D and KMT2B (61); these genes encode for histone lysine methyltransferase proteins that regulate gene transcription by altering the methylation status of the lysine 4 residue of histone 3 tail (H3K4) leading to transcriptional activation (62). Mutations in these genes commonly occur in endometrial carcinomas (63). Loss of function of these genes has been to be associated with weaker capacity of myometrial invasion by tumors (15) However, this effect was not observed in our cohort.
One limitation of our study is the limited number of CRGs included in our targeted sequencing panel. Given this, cases designated as non-CRG-altered in our cohort may harbor alterations in other CRGs not covered by our panel. Further studies using whole exome or whole genome sequencing are needed to comprehensively evaluate the role of CRG alterations in endometrial cancers.
In summary, we have shown that CRG alterations are common in ECs; these alterations are especially common in endometrioid carcinomas and appear to confer myometrial and angiolymphatic invasive properties. Functional and mechanistic studies are needed to further elucidate the roles played by these alterations in the evolution and tumorigenesis of ECs.
Supplementary Material
Research Highlights:
ARID1A is the most commonly altered chromatin remodeling gene in endometrial carcinomas (n=305, 46%).
ARID1A alterations are often biallelic (n=241, 79%) likely causing loss of function.
CRG alterations are less frequent in p53-altered endometrial carcinoma (n=77, 32%).
BCOR p.N1459S mutation is a hotspot mutation exclusively occurring in endometrioid carcinomas.
In endometrioid carcinomas, CRG alterations are associated with an increased likelihood of myometrial invasion and lymphovascular invasion.
ACKNOWLEDGEMENTS
B. Weigelt is funded in part by NIH/NCI P50 CA247749 01, Cycle for Survival and Breast Cancer Research Foundation grants. LHE is funded in part by a Cycle for Survival grant.
Research reported in this publication was supported in part by a Cancer Center Support Grant of the NIH/NCI (Grant No. P30CA008748).
Footnotes
CONFLICTS OF INTEREST
B.W. reports ad hoc membership of the scientific advisory board of REPARE Therapeutics, outside the current work. R.A.S. reports medical legal consultant (Shook Hardy Bacon); royalties from Modern Pathology/USCAP, AFIP/ARP, Cambridge Univ Press and Springer publishing. C.A. reports membership of advisory boards/ personal fees from Tesaro, Eisai/Merck, Mersana Therapeutics, Roche/Genentech, Abbvie, AstraZeneca/Merck, Repare Therapeutics, and grants from Clovis, Genentech, AbbVie, Astra Zeneca, all outside the submitted work. N.R. Abu-Rustum reports Stryker/ Novadaq and GRAIL grants paid to the institution, outside the current study. The remaining authors have no relevant conflicts.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Hübner MR, Eckersley-Maslin MA, Spector DL. Chromatin organization and transcriptional regulation. Current opinion in genetics & development. 2013;23(2):89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Graaf CA, van Steensel B. Chromatin organization: form to function. Current opinion in genetics & development. 2013;23(2):185–90. [DOI] [PubMed] [Google Scholar]
- 3.Zhang P, Torres K, Liu X, Liu C-g, E Pollock R. An overview of chromatin-regulating proteins in cells. Current Protein and Peptide Science. 2016;17(5):401–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGinty RK, Tan S. Nucleosome structure and function. Chemical reviews. 2015;115(6):2255–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu N, Li S, Wu N, Cho K-S. Acetylation and deacetylation in cancer stem-like cells. Oncotarget. 2017;8(51):89315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Du J, Johnson LM, Jacobsen SE, Patel DJ. DNA methylation pathways and their crosstalk with histone methylation. Nature reviews Molecular cell biology. 2015;16(9):519–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sawan C, Herceg Z. Histone modifications and cancer. Advances in genetics. 2010;70:57–85. [DOI] [PubMed] [Google Scholar]
- 8.Lorch Y, Maier-Davis B, Kornberg RD. Mechanism of chromatin remodeling. Proceedings of the National Academy of Sciences. 2010;107(8):3458–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mittal P, Roberts CW. The SWI/SNF complex in cancer—biology, biomarkers and therapy. Nature Reviews Clinical Oncology. 2020;17(7):435–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prakash K, Fournier D. Histone code and higher-order chromatin folding: A hypothesis. Genomics and computational biology. 2017;3(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Braun S, Madhani HD. Shaping the landscape: mechanistic consequences of ubiquitin modification of chromatin. EMBO reports. 2012;13(7):619–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Valencia AM, Kadoch C. Chromatin regulatory mechanisms and therapeutic opportunities in cancer. Nature cell biology. 2019;21(2):152–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nair SS, Kumar R. Chromatin remodeling in cancer: a gateway to regulate gene transcription. Molecular oncology. 2012;6(6):611–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones S, Li M, Parsons DW, Zhang X, Wesseling J, Kristel P, et al. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Human mutation. 2012;33(1):100–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.García-Sanz P, Triviño JC, Mota A, Perez Lopez M, Colás E, Rojo-Sebastián A, et al. Chromatin remodelling and DNA repair genes are frequently mutated in endometrioid endometrial carcinoma. International journal of cancer. 2017;140(7):1551–63. [DOI] [PubMed] [Google Scholar]
- 16.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nature medicine. 2017;23(6):703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. The Journal of molecular diagnostics. 2015;17(3):251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levine DA. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Talhouk A, McConechy M, Leung S, Li-Chang H, Kwon J, Melnyk N, et al. A clinically applicable molecular-based classification for endometrial cancers. British journal of cancer. 2015;113(2):299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Momeni-Boroujeni A, Dahoud W, Vanderbilt CM, Chiang S, Murali R, Rios-Doria EV, et al. Clinicopathologic and genomic analysis of tp53-mutated endometrial carcinomas. Clinical Cancer Research. 2021;27(9):2613–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Middha S, Zhang L, Nafa K, Jayakumaran G, Wong D, Kim HR, et al. Reliable pan-cancer microsatellite instability assessment by using targeted next-generation sequencing data. JCO precision oncology. 2017;1:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol. 2017;2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic acids research. 2016;44(16):e131–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roth A, Khattra J, Yap D, Wan A, Laks E, Biele J, et al. PyClone: statistical inference of clonal population structure in cancer. Nature methods. 2014;11(4):396–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Canisius S, Martens JW, Wessels LF. A novel independence test for somatic alterations in cancer shows that biology drives mutual exclusivity but chance explains most co-occurrence. Genome biology. 2016;17(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell research. 2011;21(3):381–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao Z, Shilatifard A. Epigenetic modifications of histones in cancer. Genome biology. 2019;20(1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ge SX, Son EW, Yao R. iDEP: an integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC bioinformatics. 2018;19(1):1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gibson WJ, Hoivik EA, Halle MK, Taylor-Weiner A, Cherniack AD, Berg A, et al. The genomic landscape and evolution of endometrial carcinoma progression and abdominopelvic metastasis. Nature genetics. 2016;48(8):848–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mohrmann L, Verrijzer CP. Composition and functional specificity of SWI2/SNF2 class chromatin remodeling complexes. Biochimica et Biophysica Acta (BBA)-Gene Structure and Expression. 2005;1681(2–3):59–73. [DOI] [PubMed] [Google Scholar]
- 32.Jones S, Wang T-L, Shih I-M, Mao T-L, Nakayama K, Roden R, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330(6001):228–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao X, Tate P, Hu P, Tjian R, Skarnes WC, Wang Z. ES cell pluripotency and germ-layer formation require the SWI/SNF chromatin remodeling component BAF250a. Proceedings of the National Academy of Sciences. 2008;105(18):6656–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guan B, Mao T-L, Panuganti PK, Kuhn E, Kurman RJ, Maeda D, et al. Mutation and loss of expression of ARID1A in uterine low-grade endometrioid carcinoma. The American journal of surgical pathology. 2011;35(5):625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wiegand KC, Lee AF, Al-Agha OM, Chow C, Kalloger SE, Scott DW, et al. Loss of BAF250a (ARID1A) is frequent in high-grade endometrial carcinomas. The Journal of pathology. 2011;224(3):328–33. [DOI] [PubMed] [Google Scholar]
- 36.Rahman M, Nakayama K, Rahman MT, Katagiri H, Katagiri A, Ishibashi T, et al. Clinicopathologic analysis of loss of AT-rich interactive domain 1A expression in endometrial cancer. Human pathology. 2013;44(1):103–9. [DOI] [PubMed] [Google Scholar]
- 37.Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008;135(4):1079–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson MR, Reske JJ, Holladay J, Wilber GE, Rhodes M, Koeman J, et al. ARID1A and PI3-kinase pathway mutations in the endometrium drive epithelial transdifferentiation and collective invasion. Nature communications. 2019;10(1):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X, NAGL JR, Wilsker D, Van Scoy M, Pacchione S, Yaciuk P, et al. Two related ARID family proteins are alternative subunits of human SWI/SNF complexes. Biochemical Journal. 2004;383(2):319–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guan B, Wang T-L, Shih I-M. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer research. 2011;71(21):6718–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Samartzis EP, Samartzis N, Noske A, Fedier A, Caduff R, Dedes KJ, et al. Loss of ARID1A/BAF250a-expression in endometriosis: a biomarker for risk of carcinogenic transformation? Modern Pathology. 2012;25(6):885–92. [DOI] [PubMed] [Google Scholar]
- 42.Yen T-T, Miyamoto T, Asaka S, Chui MH, Wang Y, Lin S-F, et al. Loss of ARID1A expression in endometrial samplings is associated with the risk of endometrial carcinoma. Gynecologic oncology. 2018;150(3):426–31. [DOI] [PubMed] [Google Scholar]
- 43.Toumpeki C, Liberis A, Tsirkas I, Tsirka T, Kalagasidou S, Inagamova L, et al. The role of ARID1A in endometrial cancer and the molecular pathways associated with pathogenesis and cancer progression. in vivo. 2019;33(3):659–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu G, Xu P, Fu Z, Hua X, Liu X, Li W, et al. Prognostic and Clinicopathological Significance of ARID1A in Endometrium-Related Gynecological Cancers: A Meta-Analysis. Journal of cellular biochemistry. 2017;118(12):4517–25. [DOI] [PubMed] [Google Scholar]
- 45.Hu H, Chen Z, Ji L, Wang Y, Yang M, Lai R, et al. ARID1A-dependent permissive chromatin accessibility licenses estrogen-receptor signaling to regulate circadian rhythms genes in endometrial cancer. Cancer Letters. 2020;492:162–73. [DOI] [PubMed] [Google Scholar]
- 46.Rahmanto YS, Shen W, Shi X, Chen X, Yu Y, Yu Z-C, et al. Inactivation of Arid1a in the endometrium is associated with endometrioid tumorigenesis through transcriptional reprogramming. Nature communications. 2020;11(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liang H, Cheung LW, Li J, Ju Z, Yu S, Stemke-Hale K, et al. Whole-exome sequencing combined with functional genomics reveals novel candidate driver cancer genes in endometrial cancer. Genome research. 2012;22(11):2120–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nature genetics. 2014;46(3):225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abedalthagafi MS, Bi WL, Merrill PH, Gibson WJ, Rose MF, Du Z, et al. ARID1A and TERT promoter mutations in dedifferentiated meningioma. Cancer genetics. 2015;208(6):345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Werner HM, Berg A, Wik E, Birkeland E, Krakstad C, Kusonmano K, et al. ARID1A loss is prevalent in endometrial hyperplasia with atypia and low-grade endometrioid carcinomas. Modern Pathology. 2013;26(3):428–34. [DOI] [PubMed] [Google Scholar]
- 51.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481(7381):306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Köbel M, Hoang LN, Tessier-Cloutier B, Meng B, Soslow RA, Stewart CJ, et al. Undifferentiated endometrial carcinomas show frequent loss of core switch/sucrose nonfermentable complex proteins. The American journal of surgical pathology. 2018;42(1):76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karnezis AN, Hoang LN, Coatham M, Ravn S, Almadani N, Tessier-Cloutier B, et al. Loss of switch/sucrose non-fermenting complex protein expression is associated with dedifferentiation in endometrial carcinomas. Modern Pathology. 2016;29(3):302–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marshall AD, Bailey CG, Rasko JE. CTCF and BORIS in genome regulation and cancer. Current opinion in genetics & development. 2014;24:8–15. [DOI] [PubMed] [Google Scholar]
- 55.Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137(7):1194–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Witcher M, Emerson BM. Epigenetic silencing of the p16INK4a tumor suppressor is associated with loss of CTCF binding and a chromatin boundary. Molecular cell. 2009;34(3):271–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huynh KD, Fischle W, Verdin E, Bardwell VJ. BCoR, a novel corepressor involved in BCL-6 repression. Genes & development. 2000;14(14):1810–23. [PMC free article] [PubMed] [Google Scholar]
- 58.Wagner SD, Ahearne M, Ferrigno PK. The role of BCL6 in lymphomas and routes to therapy. British journal of haematology. 2011;152(1):3–12. [DOI] [PubMed] [Google Scholar]
- 59.Kao Y-C, Owosho AA, Sung Y-S, Zhang L, Fujisawa Y, Lee J-C, et al. BCOR-CCNB3-fusion positive sarcomas: a clinicopathologic and molecular analysis of 36 cases with comparison to morphologic spectrum and clinical behavior of other round cell sarcomas. The American journal of surgical pathology. 2018;42(5):604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Momeni-Boroujeni A, Chiang S. Uterine mesenchymal tumours: recent advances. Histopathology. 2020;76(1):64–75. [DOI] [PubMed] [Google Scholar]
- 61.Rao RC, Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nature Reviews Cancer. 2015;15(6):334–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kouzarides T Chromatin modifications and their function. Cell. 2007;128(4):693–705. [DOI] [PubMed] [Google Scholar]
- 63.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.