

Abstract
Cancer is a systemic disease that involves malignant cell-intrinsic and extrinsic metabolic adaptations. Most studies have tended to focus on elucidating the metabolic vulnerabilities in the primary tumor microenvironment, leaving the metastatic microenvironment less explored. In this Opinion, we discuss the current understanding of the metabolic crosstalk between the cancer cells and the tumor microenvironment, both at local and systemic levels. We explore the possible influence of the primary tumor secretome to metabolically and epigenetically-rewire the non-malignant distant organs during prometastatic niche formation and successful metastatic colonization by the cancer cells. In an attempt to understand the process of prometastatic niche formation, we have speculated how cancer may hijack the inherent regenerative propensity of tissue parenchyma during metastatic colonization.
Keywords: prometastatic niche, wound response, stroma, metabolism, tissue regeneration, metastasis
Cancer-cell intrinsic and extrinsic “rewiring” during metastasis
Metastasis, the major cause of death in cancer patients, is a multi-step process involving metabolic and epigenetic rewiring in cancer cells (intrinsic) and the host tissue parenchyma (extrinsic), both at the primary and secondary sites of cancer spread [1, 2]. From the days of the “seed and soil” hypothesis of cancer metastasis, some studies have demonstrated early pathological changes in distant organs that co-evolve with the primary tumor and contribute towards “organotropism” and colonization of the disseminated cancer cells [3]. Broadly, such a conducive tissue microenvironment that supports metastatic outgrowth is termed as the (pro/pre)-metastatic niche. Most of the previous work in this area was based on the intrinsic molecular fitness of the cancer cells and the cancer-immune cell crosstalk at the metastatic niche. While several studies have demonstrated that secreted metabolites from malignant cells can influence the differentiation state, phenotype and function of non-malignant cells in the tumor microenvironment [4], it is not known whether such metabolic alterations or secreted factors from the primary tumor can systemically prime the tissue parenchyma of distant organs. The complexity of metabolic alterations in the host at an organismal level in response to malignancy, and the dynamic nature of the metastatic process have limited our ability to make progress in this area. In this Opinion article, we highlight the current advancements in the field with speculative insights on the less-explored avenues of the cancer metastatic cascade: prometastatic niche (PMN) preparation, as well as metabolic adaptations in the malignant cells and tissue-resident cells at the metastatic sites during cancer colonization, dormancy and reactivation. This article also underscores the current knowledge gaps and suggests possible approaches to study the spatiotemporal dynamics of metastasis. Moreover, considering the molecular analogies between physiologic wound-healing and malignant stromal responses, we have set forth the opinion that integrating the interdisciplinary understanding of organ-specific epigenetic and metabolic changes during tissue regeneration will provide a framework to identify and interrogate the molecular determinants that the primary tumor may employ to prime the epigenome of the distal organs, during or prior to metastasis.
Metastasis involves dynamic metabolic switches between cell states
Metastatic cancer cells exist in a spectrum of genetic, epigenetic, and metabolic states that phenotypically manifest as local matrix invasion in the primary tumor, hematological transit, seeding and colonization at secondary sites. During each of these steps in the metastatic trajectory, cancer cells undergo cell-autonomous metabolic reprogramming and are also involved in a complex molecular interplay with the non-malignant tissue parenchyma depending on the spatial distribution of nutrients and oxygen, and the mechanical factors that they encounter [5, 6].
The current understanding in the field indicates a dichotomy in the cancer cell metabolism during proliferation and migration, known as the “grow or go” hypothesis. Recent reports suggest that enhanced mitochondrial biogenesis, elevated OXPHOS [7] and mitochondrial enrichment at the invasive front of metastatic cells [8, 9] are significantly correlated with distant metastasis. Interestingly, it has been shown that during the collective migration of breast cancer cells, the leader cells at the invasive front exhibit higher glycolytic flux than the follower cells, whereas in lung cancer, the leader cells have been shown to exhibit higher OXPHOS than the follower counterparts [10, 11], suggesting a tumor type-specific bioenergetic switch of metastatic cancer cells. During extravasation and intravasation, pancreatic cancer cells face compressive mechanical forces from the stiff tumor microenvironment (TME), hence intracellular ATP:ADP ratio, glycolytic flux and phosphocreatine production remain high in the invading cells to surmount the high energy barrier of the surrounding ECM [12]. Similarly, the surrounding collagen matrix architecture was observed to impact the ATP:ADP ratio and hence the migratory potential of breast cancer cells [13].
While most studies so far have focused on the metabolic heterogeneity of cancer cells that endow metastatic fitness to cells in order to breach the basement membrane in the primary tumor and colonize secondary sites, metabolic determinants of circulating tumor cells are recently starting to gain attention. For example, matrix-detached cells were shown to have altered mitochondrial morphology and a switch of metabolic preference from OXPHOS to glycolysis, with enhanced synthesis of pyruvate and lactate as compared to TCA cycle intermediates. This preferential glycolytic switch is essential to circumvent ROS-mediated cell-death or anoikis of cancer cells in circulation [14–16]. Consistent with this, higher pyruvate and lactate concentrations were observed in the serum of highly metastatic breast cancer [17] and colorectal cancer [18] patients, respectively. However, in Lkb1 mutant lung cancer models, enhanced tumor cell glutaminolysis correlated with anoikis resistance and metastatic burden [19], which may suggest that the matrix-detached cancer cells originating from different organs or with distinct mutational profiles may have unique metabolic demands that are maintained in circulation. Hence it is tempting to speculate that cancer cells retain their metabolic preferences as “metabolic memory” along their metastatic course. Interestingly, the route of dissemination also directs the metabolic adaptations required for metastasis. For example, cancer cells that spread via the lymphatic system utilize exogeneous lipid (lymph contains oleic acid laden vesicles) to form saturated cell membranes with less sensitivity to lipid peroxidation, resulting in higher resistance to ferroptosis than those which spread through blood [20]. Studies to decipher the metabolic plasticity of cancer cells during the processes of intravasation, survival in circulation and extravasation at the secondary sites, are comprehensively reviewed elsewhere [5].
The presence of disseminated cells in the systemic circulation or distant organs is not always clinically apparent in patients with advanced primary tumors. With advances in lineage tracing and single cell genomic techniques, it is now known that cancer cells can survive as indolent microcolonies in distal organs as early as the premalignant stages at the primary site, as observed in autochthonous models of breast cancer-bone metastasis and pancreatic cancer-liver metastasis models [21–23]. It is known that cancer cells undergo a state of proliferation arrest, metabolic quiescence and immune-masking at the secondary organs, broadly known as metastatic dormancy. A recent effort to identify the metabolic signature associated with dormancy suggested a decline in expression of genes associated with proliferation, protein synthesis, and bioenergetic pathways (TCA cycle, OXPHOS, and the PPP) in the dormant disseminated cancer cells in a pancreatic cancer mouse model and patients with localized pancreatic cancer [24]. Based on single cell transcriptomics and chromatin accessibility maps, the authors observed that the tumor cell transcriptome from primary tumors and “reactivated” cells clustered together and were distinct from quiescent tumor cells. Additionally, they showed that dormancy is associated with large-scale chromatin alterations.
While it is critical for the dormant/newly-seeded cancer cells at the secondary sites to acquire general survival mechanisms, such as mitigating oxidative stress by decelerating bioenergetics and biosynthesis processes, for successful colonization they also need to adopt molecular strategies tailored according to the metabolic nature of the target tissue. For example, breast cancer cells colonizing the lung, a rich source of pyruvate, adapt to utilize higher pyruvate by enhancing mitochondrial alanine aminotransferase 2 (ALT2) activity [25]. The low levels of serine in brain tissues confer a higher expression of PHGDH, the first rate-limiting enzyme of the serine synthesis pathway in breast cancer cells metastasizing to the brain [26]. Similarly, the low availability of lipids in the brain require metastatic breast cancer cells to upregulate fatty acid synthetase (FASN) for successful brain colonization [27]. In contrast, the lipid rich metabolic milieu of the peritoneum requires ovarian cancer cells to upregulate the fatty acid uptake receptor CD36 for metastatic seeding [28]. Lastly, it was shown that primary brain tumors and brain metastasis upregulate acetate utilization to fuel the TCA cycle [29]. Consistent with observations of enhanced OXPHOS in breast cancer cells micro-metastasizing to the lung [30, 31], pancreatic cancer cells forming liver micro-metastases demonstrate elevated succinate dehydrogenase (SDHB) expression as compared to macrometastases [32], suggesting a shift towards mitochondrial metabolism during dormancy at the metastatic site. On the other hand, colorectal cancer cells utilize fructose metabolism by upregulation of aldolase B (ALDOB) to promote metastatic outgrowth in the liver [33]. Another crucial and less-explored aspect that may help in the understanding of the “dormancy-reactivation switch” of cancer cells at the secondary organ, is demystifying the contribution of the biophysical characteristics of the surrounding tissue matrix towards the structure/function of the subcellular organelles and chromatin organization of the malignant cells. Along the same lines, recent work elucidated the role of stiff tumor matrix in regulating mitochondrial structural and functional reprogramming, and hence the ability of breast cancer cells to mitigate redox stress [34]. As previously reviewed, rewiring cellular stress-response metabolism is a crucial determinant of successful colonization and re-activation of disseminated cancer cells at secondary sites [35]. While the biophysical nature of the surrounding matrix as a prime regulator of the cellular actin-cytoskeleton and nuclear topology is gaining attention [36],[37],[38],[39], it will be important to investigate its influence on the (epi)-genetic and metabolic plasticity of the cancer cells undergoing “dormancy-reactivation” switch. Although, the metabolic dynamics during tumor dormancy and reactivation are still elusive, accumulating evidence suggests that the organ-specific nutrient environment plays a crucial role in selection and expansion of the “fit” clones of disseminated cancer cells [40]. Hence, understanding the “support mechanisms” from the surrounding niche has the potential to prolong cancer cell dormancy without reactivation, or increase the sensitivity of indolent cells to therapy after inducing their reawakening from metabolic quiescence.
With the rapid expansion of metastasis research over the last few years, several rewired metabolic pathways as well as metabolites have been identified that provide metastatic fitness to the cancer cells [41, 42]. However, as metastasis is a non-linear process where each stage has a transient window, it is challenging to assess active metabolic fluxes in metastasizing malignant cells. Overcoming these technical roadblocks and defining the tumor stage at which each of these pathways or metabolites bolster the cancer cells will be essential for sculpting diagnostic and therapeutic regimens to intervene appropriately.
Prometastatic niche preparation: local and systemic rewiring of the metabolic milieu of distant sites
A unique pathological signature of solid malignancies is their complex microenvironment, comprised of a heterogenous pool of non-malignant cells such as neurons, immune cells, cancer-associated fibroblasts (CAFs) etc., which participate in a multidirectional nutrient network with the cancer cells to support tumor progression. Several groups have elucidated a variety of “metabolic-symbiosis” mechanisms existing among different cell types in the primary tumor microenvironment, and some of those adaptations have been associated with various mutational profiles of the tumor [43–45]. We had previously demonstrated that CAFs undergo autophagy to provide alanine and hence, support the growth of pancreatic cancer cells [46]. Along the same lines, neurons innervating the pancreatic tumors were shown to secrete serine that supported the cancer cell survival [47]. However, whether such inter-cellular metabolic cross-talks hold true at the metastatic sites is still unknown. Comparing and contrasting the metabolic crosstalk of cancer cells with the resident or recruited cells at the primary and metastatic TME can help in elucidating approaches for co-targeting the tumor and its microenvironment. For example, hypoxic cancer cells and CAFs in the primary tumors are highly glycolytic and provide lactate to the spatially oxygenated tumor area, which supports the reverse Warburg effect in cancer cells [48, 49]. Lactate accumulation promotes an upregulation of Arginase 1 (Arg-1) in tumor-associated macrophages (TAMs) which polarize towards a tumor-promoting and immunosuppressive phenotype [50]. On the other hand, stromal cells at the metastatic sites (lung fibroblasts and brain astrocytes) demonstrate a decrease in pyruvate kinase isozyme M2 (PKM2), resulting in a decrease in glycolytic activity and increase in glucose availability for the metastatic breast cancer cells [51]. Furthermore, cancer-associated adipocytes (CAAs) induce upregulation of fatty acid transporters (CD36 and FATP1) in melanoma and ovarian cancer cells [28, 52], while CAAs undergo lipolysis and promote lipid utilization during omental (omentum being a rich source of adipocytes) metastasis of breast, melanoma and ovarian cancers [53, 54]. In view of metastasis as one of the major barriers to successful cancer management strategies, the field warrants further in-depth investigations on the pro-metastatic TME, taking into account the organ-specific anatomical, cellular and metabolic features.
Since metastasis is an inefficient process where only a small percentage of extravasated cells can successfully micro-colonize the secondary sites and a large majority of solitary cells were observed to be dormant, it is fair to speculate that metastatic outgrowth is a stochastic biological event in the malignant spectrum [55] [56, 57]. However, several studies over the past decade have indicated a selective pattern of colonization of cancer cells derived from specific primary tumors to select organs, defined as “organotropism”. Moreover, it was observed that secreted factors from the primary tumor can orchestrate modulation of the extracellular matrix, immune milieu, and the tissue-resident cells at distant locales to generate a nutrient-rich, immune-cold and seeding-permissive “pro-metastatic niche” (PMN) before the cancer cells physically arrive [58–60]. Primary tumors release exosomes laden with a plethora of bio-active factors like mi-RNA, integrins and growth factor receptor ligands, which have been shown to educate distant organs such as lung, liver, brain and bone to generate a PMN for successful seeding and colonization of breast, pancreatic, lung and colorectal cancers [61, 62]. Additionally, local crosstalk between diverse cell types at the distant organs can be driven by local enrichment of the tumor secretome, which has been shown to augment the priming of PMN. For example, lysyl oxidase (LOX) secreted by the primary tumor cells modulates collagen remodeling by lung fibroblasts and supports the recruitment of pro-tumorigenic immune cells at the lung PMN [63]. The concerted action of LOX and IL6 from the primary breast tumor modulates the bone matrix via altering the osteoblast and osteoclasts differentiation, generating bone PMNs [64, 65]. Macrophage inhibitory factor (MIF) in pancreatic cancer exosomes has been shown to activate hepatic Kupffer cells to induce TGF-β signaling that promotes fibrosis by the hepatic stellate cells leading to recruitment of tumor-promoting macrophages at the liver PMN [61]. Another recent study showed that IL6 secreted from CAFs in the primary pancreatic tumor can transactivate the signal transducer and activator of transcription 3 (STAT3)-serum amyloid A1, A2 (SAA) axis in the hepatocytes which in turn promotes the formation of liver PMN [66]. While several studies have shown that the primary tumor secretome can influence the immune milieu and induce cellular crosstalk at the systemic level during the formation of PMNs, the possibility of primary tumors to modulate the metabolic microenvironment at distant sites to make it more permissive for colonization by the disseminated cancer cells is relatively unexplored.
The ability of the distant organ to respond to the primary tumor secretome and generate a pro-inflammatory, pro-metastatic microenvironment can be remarkably governed by the age of the tissue [39, 67]. Previous studies suggest that fibroblasts, the functional cellular component responsible for maintaining the structural integrity and homeostasis of tissues, undergo a hyper-secretive phenotype-switch marked by an elevated secretion of pro-inflammatory mediators, growth factors and proteases, which is broadly termed “senescence-associated secretory phenotype” (SASP). Aging fibroblasts are metabolically distinct and tend to have higher ROS levels [68] [69, 70], which along with the inflammatory milieu can provide a conducive environment for tumor aggressiveness. Moreover, recent research suggests an overall difference in the alignment and crosslinking pattern of collagen, elastin and hyaluronan matrix in aged versus a young microenvironment, which is also thought to influence cancer cells’ dissemination [71–73]. While, SASP of aged fibroblasts are thought to reprogram the biophysical, chemical and metabolic nature of the local tissue microenvironment, recent studies have identified secreted factors from aged fibroblasts beyond the SASP that can promote lung metastasis in a melanoma model [74]. Notably, a follow-up study demonstrated the mechanistic interplay between aged dermal and lung fibroblasts’ secretomes in providing a permissive metastatic niche for re-activation of melanoma cells, post dormancy in the lung [75], thereby opening up the possibility of considering epithelial age as another dimension in the quest of understanding PMN formation. Interestingly, a recent study showed that age-induced metabolic alterations, specifically higher levels of methylmalonic acid in the serum of older individuals promoted an EMT-phenotype and metastasis of lung and breast tumors, suggesting a mechanistic link between systemic metabolism, aging and cancer [76].
Metabolic analogy between tissue parenchyma regeneration and pro-metastatic niche preparation
Malignant transformation, tumor development and metastatic outgrowth require support from the tumor-associated, non-malignant stromal compartment that accounts for up to 90% of the bulk tumor volume in various gastric, mammary and pancreatic malignancies [77]. Fibroblasts form the predominant stromal cell population in the TME, which phenotypically mirror those involved in a physiologic wound healing response. The natural course of a wound healing response is conserved across tissue types, and results in parenchyma repair, which is an amalgamation of tissue-specific developmental programs involving chromatin remodeling and metabolic perturbations in the participating fibroblasts and immune cells [78]. Many decades ago, cancer was defined as the “wound that never heals” and since then, various studies have indicated that the TME gene expression pattern closely resembles that of a wound response signature, encompassing a pro-inflammatory and pro-fibrotic phenotype [79]. Accumulating evidence now suggests that the PMN reflects a “reactive stroma”, primed by the primary tumor secretome from a very early stage of cancer progression, followed by recruitment/activation of mesenchymal and myeloid cells [80],[81],[66, 82]. Considering the molecular analogy of benign tissue repair and cancer-associated stromagenesis, it is tempting to speculate that cancer cells impart a constant loop of regenerative phenotype in the PMN as a strategy to sustain the stromal support for their growth and aggressiveness. Figure 1, Key figure depicts some metabolic and epigenetic analogies between physiologic tissue repair and PMN preparation.
Figure 1. Key figure. Molecular analogy in the microenvironment of metastasis and regenerating benign tissue.
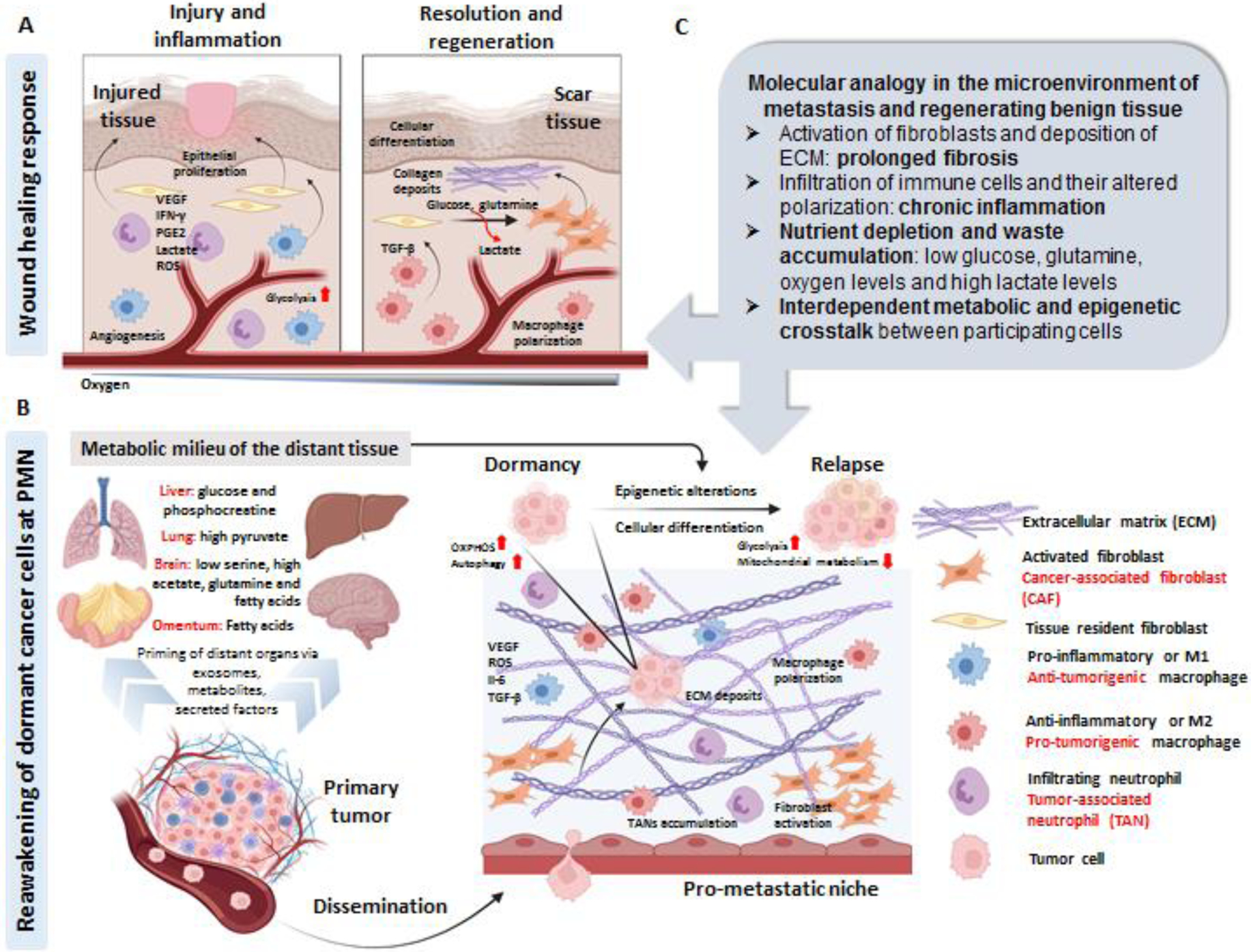
A. During the initial phase of the physiologic wound healing response, there is a cascade of pro-inflammatory events including neutrophil and macrophage infiltration/activation, angiogenesis (or neo-vascularization: generation of new blood vessels) and epithelial proliferation. The accumulated lactate in the microenvironment (due to the high glycolysis rates of the pro-inflammatory cells and poor oxygenation in the wound) polarizes the macrophages to an anti-inflammatory phenotype, which in turn leads to activation of tissue-resident fibroblasts. Highly proliferative, activated (myo)-fibroblasts utilize glucose and glutamine for their energy consumption, leading to more lactate release (that further promotes angiogenesis), with a concurrently ECM deposition to form scar tissue. B. Circulating exosomes, metabolites and secreted factors from the primary tumor primes different organs of the body from a very early stage of cancer progression leading to the formation of prometastatic niche (PMN), which is comprised of activated fibroblasts, ECM deposits, and an inflamed parenchyma (caused by an intricate interplay between the pro and anti-tumorigenic neutrophils, macrophages and T-cells). As the cancer progresses, disseminated tumor cells are passively trapped or actively seeded in the inflamed tissue where they can survive for years as indolent non-proliferative cells, manifested by high autophagy and elevated mitochondrial metabolism (termed as dormancy). Owing to several factors (cancer-cell autonomous: acquired genetic or epigenetic modifications or microenvironment dependent: local metabolic milieu or crosstalk between both), the quiescent cancer cells can “reawake” and undergo cellular differentiation (with altered genetic and epigenetic traits) that phenotypically manifests as a metastatic relapse. Among the known metabolic “support mechanisms” that cancer cells utilize to come out of dormancy in different organs are mentioned in the figure. C. Considering the parallels between reactive-stromal response and metabolic rewiring at the PMN and regenerating tissue parenchyma, it can be speculated that tumor cells can induce a paracrine loop of irreversible (and chronic) tissue inflammation and repair at the distant sites (PMN generation) to prepare for their seeding and colonization. The molecular signaling and metabolic rewiring induced at the distant sites can in turn support the indolent cancer cells to gain epigenetic traits or altered cellular states (differentiation/de-differentiation) to promote their “reawakening” from dormancy. (Index for cell types on the right; black: implicated during physiologic conditions; red: implicated during malignancy). Created with BioRender.com. ECM: Extracellular matrix, PMN: Pro-metastatic niche, VEGF: Vascular endothelial growth factor, IFN-γ: Interferon gamma, PGE2: Prostaglandin E2, ROS: Reactive oxygen species, OXPHOS: Oxidative phosphorylation, TGF-β: Transforming growth factor beta, IL-6: Interleukin-6, TANs: Tumor-associated neutrophils.
Interestingly, the harsh metabolic microenvironment faced by the stromal cells during wound-repair profoundly matches with the metabolic niche in a tumor. For example, the extracellular abundance of glucose and glutamine sharply goes down in a wound with a concomitant increase in lactate accumulation and hypoxia prior to angiogenesis, which mirrors the hallmarks of cancer metabolism [83]. The metabolic adaptations involving different cell types in a wound healing response is illustrated in [78]. It is now known that the local nutrient availability or accumulation of metabolic intermediates can influence the chromatin organization at a gene-specific and global scale, defined as the epigenetic state/code of the cells. For example, dietary methionine restriction can be sensed and directed towards the dynamics of histone methylation patterns (particularly H3K4) in mouse liver [84]. Similar observations were reported by the same group in human colorectal cancer cells [85]. The fundamentals and current understanding of metabolism-epigenetic axis have been comprehensively reviewed in [86]. So far, microenvironment-mediated epigenetic reprogramming has been reported in the context of “trained immunity” where innate immune cells in a secondary wound undergo faster functional differentiation owing to long-term histone/DNA modifications upon exposure to prolonged inflammation or altered metabolic milieu in a primary wound [87]. This model of “inflammatory memory” was extended to dermal wound healing in a recent study, which showed that inflammation-trained skin is capable of faster re-epithelialization due to preserved inflammation-induced chromatin states followed by rapid transcription of tissue-repair genes [88]. In this vein, the liver serves as one of the most regenerative organs in the body where chromatin remodeling in the hepatocytes play crucial roles in determining the switch between their proliferative, differentiation and senescence phases post injury [89–91]. Several studies in the past have illustrated the temporal metabolic alterations associated with these phases of priming, proliferation and termination of hepatic regeneration [92, 93]. Such metabolic alterations are also observed during liver steatosis and benign metabolic disorders like non-alcoholic fatty liver diseases, which present the classical hallmarks of chronic wound and fibrosis in the liver parenchyma [94, 95]. Considering the fact that liver serves as the master regulator of organismal metabolic homeostasis, liver resident cells (especially the hepatocytes) can be imagined to be highly sensitive to fine changes to the nutrient availability or metabolic waste accumulation [92], which may suggest that the local metabolic milieu in the liver may “train” the resident cells to respond faster to metabolic alterations by influencing their epigenome (as observed in the case of “immunological memory”). Indeed, the potential bidirectional metabolic-epigenetic axis in the liver is now starting to be studied [91, 96].
Integrating the findings of how metabolic intermediates and inflammatory milieu can influence the epigenome of not only immune cells, but epithelial cells of skin and liver in the context of wound response and tissue regeneration, it is tempting to believe that the altered metabolic or pro-inflammatory milieu can have imprints in the epigenome of the tissue parenchyma of distant organs in a tumor-bearing host. This may be crucial for the PMN formation and determining “metastatic permissiveness” of one organ over the other and may explain why the liver serves as the most prevalent “pro-metastatic” sites for multiple solid malignancies [97]. It was shown before that secreted factors from primary pancreatic tumors can modulate the activation states of liver resident cells and also cause infiltration of pro-inflammatory immune population [54, 60]. Such cellular crosstalk may create a metabolic competition among various cell types (as shown before in case of primary tumors) [28, 98, 99], leading to an altered nutrient microenvironment in the liver during the formation of PMN. This may suggest that the liver resident cells (hepatocytes, stromal cells and macrophages) in the PMN, exposed to prolonged systemic inflammation and metabolic imbalance in a tumor-bearing host, may acquire epigenetic marks that may manifest as tumor-permissive transcriptional reprogramming that is hijacked by the malignant cells for successful colonization. Hence, interrogating the role of the local altered metabolites at the metastatic lesions (after cancer cells’ seeding and forming microcolonies) and the primary tumor secretome (during or prior to seeding of cancer cells at secondary sites) as epigenetic “rheostats” (or regulators to fine-tune the epigenetic code and subsequent transcriptional rewiring) of the host tissue parenchyma may be helpful to identify drug targets to mitigate the molecular symbiosis of tumor cells and its microenvironment at the metastatic sites. We propose that integrating the lessons learned from the studies on metabolic orchestration and the associated epigenetic rewiring that occurs in the participating cells in a wound during the inflammation, resolution and regeneration phases, may serve as a missing piece of the puzzle of understanding PMN formation. This will also pave the way towards identifying the molecular determinants that the primary tumor may employ to “train” the epigenome of the distal organs prior to metastasis.
Concluding remarks
Metastasis has been a challenging area of research owing to the heterogeneity of the disease at multiple levels, starting from the intra-tumoral genetics to organ-dependent physiological/morphological challenges faced by the invading cancer cells at secondary sites. Recently, host-specific metabolic heterogeneity has been identified as an additional layer of complexity in the field of cancer research [60, 100, 101]. In population-based studies, tumor-driven influence on the host organs like adipose tissue, muscles or liver via systemic hormones/growth factors/inflammatory mediators like leptin, insulin, insulin growth factor and cytokines have been shown to associate metabolic conditions like cachexia, glucose insensitivity and deregulated lipid metabolism with cancer [101, 102]. This is particularly important to study in the context of metastasis where the primary tumor secretome is proposed to prime distal organs prior to colonization by the cancer cells. Another critical aspect of metastasis is the long latency period and the strong correlation between chances of tumor relapse and patients’ age in certain malignancies like melanoma. Broadly, inflammaging (persistent low-grade inflammation in the aging epithelia, leading to chronic tissue degeneration) and immunosenesence (age-induced systemic increase in immunosuppressive immune populations) are now thought to promote PMN generation and support metastatic colonization [67, 103]. [104]. On the contrary, breast cancer is more aggressive and likely to recur more in younger patients [105]. This context-specificity may be attributed to the fact that age-derived alterations in the ECM organization have organ-dependent influence on metastatic progression (elaborately reviewed in [67]). Hence, delineating the tissue-specific immunometabolic atlas and the epigenetic plasticity of the target epithelial parenchyma (young versus aged) will be crucial to understand how cancer can potentially corrupt the host’s molecular machinery to repair and regenerate, and thereby design effective co-targeting strategies. Currently, it is difficult to dynamically assess the rapidly evolving genetic, epigenetic and metabolic perturbations that accumulate and evolve as the tumor cells prepare distant “soils” and successfully metastasize. With the advances of systems biology and spatial-omics techniques, it is likely that many of these unanswered questions will be addressed in coming years (see Outstanding questions). Lastly, mechanistic integration of the findings from interdisciplinary research in wound healing and benign metabolic diseases like fatty liver disease, obesity and diabetes, may help to identify common molecular nodes of systemic metabolic vulnerabilities associated with metastatic cancer, and will potentially pave the way for improved diagnostic and therapeutic strategies in patients.
Outstanding questions.
Is there a link between metabolic heterogeneity among cancer cells in the primary tumor and dormancy-phenotype at metastatic lesions?
Does the formation of a prometastatic niche at distal organs depend on the age of the host?
Is the “permissive code” of metastatic colonization engrained in the host tissue’s epigenome? Can decoding that help in designing epigenetic preventive measures against cancer metastasis or tumor relapse?
Does heterogeneity in host metabolism at a systemic level account for relapse after therapy in some patients versus others?
Highlights.
Metabolic contributions from the tissue-resident cells at metastatic sites (such as hepatocytes, stellate cells, alveolar epithelial cells, omental adipocytes, osteoclasts etc.) are now acknowledged as important drivers for the colonization process of disseminated cancer cells. However, the stroma-derived extrinsic signals that may drive the “re-activation” of the dormant cells are still elusive.
The ability of the primary tumor secretome to influence the immune milieu at distant organs and hence prepare the permissive-soil for colonization of circulating tumor cells is starting to be identified. Further study is needed to decipher the possible mechanisms by which the primary tumor may systemically “re-educate” the metabolic and epigenetic states of the resident cells in the host organs.
We propose a framework whereby tumors hijack the inherent regenerative capacity of the tissue parenchyma to form the prometastatic niche.
Acknowledgments
This work was supported by NCI Grants P01CA117969, R35CA232124, P30CA016087, 1R01CA251726-01A1; the Lustgarten Foundation, and SU2C to A.C.K.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
A.C.K. has financial interests in Vescor Therapeutics and is an inventor on patents pertaining to KRAS-regulated metabolic pathways, redox control pathways in pancreatic cancer, targeting GOT1 as a therapeutic approach, targeting alanine transporters, and the autophagic control of iron metabolism. A.C.K. is on the scientific advisory board of Rafael/Cornerstone Pharmaceuticals, OncoRev and has been a consultant for Deciphera and Abbvie. The other author declares no competing interests.
References
- 1.Bergers G and Fendt S-M, The metabolism of cancer cells during metastasis. Nature Reviews Cancer, 2021. 21(3): p. 162–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suhail Y, et al. , Systems biology of cancer metastasis. Cell systems, 2019. 9(2): p. 109–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoshino A, et al. , Tumour exosome integrins determine organotropic metastasis. Nature, 2015. 527(7578): p. 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elia I and Haigis MC, Metabolites and the tumour microenvironment: from cellular mechanisms to systemic metabolism. Nature metabolism, 2021. 3(1): p. 21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elia I, Doglioni G, and Fendt S-M, Metabolic hallmarks of metastasis formation. Trends in cell biology, 2018. 28(8): p. 673–684. [DOI] [PubMed] [Google Scholar]
- 6.Zanotelli MR, Zhang J, and Reinhart-King CA, Mechanoresponsive metabolism in cancer cell migration and metastasis. Cell metabolism, 2021. 33(7): p. 1307–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.LeBleu VS, et al. , PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nature cell biology, 2014. 16(10): p. 992–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cunniff B, et al. , AMPK activity regulates trafficking of mitochondria to the leading edge during cell migration and matrix invasion. Molecular biology of the cell, 2016. 27(17): p. 2662–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Porporato PE, et al. , A mitochondrial switch promotes tumor metastasis. Cell reports, 2014. 8(3): p. 754–766. [DOI] [PubMed] [Google Scholar]
- 10.Zhang J, et al. , Energetic regulation of coordinated leader–follower dynamics during collective invasion of breast cancer cells. Proceedings of the National Academy of Sciences, 2019. 116(16): p. 7867–7872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Commander R, et al. , Subpopulation targeting of pyruvate dehydrogenase and GLUT1 decouples metabolic heterogeneity during collective cancer cell invasion. Nature communications, 2020. 11(1): p. 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papalazarou V, et al. , The creatine–phosphagen system is mechanoresponsive in pancreatic adenocarcinoma and fuels invasion and metastasis. Nature metabolism, 2020. 2(1): p. 62–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zanotelli MR, et al. , Regulation of ATP utilization during metastatic cell migration by collagen architecture. Molecular biology of the cell, 2018. 29(1): p. 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piskounova E, et al. , Oxidative stress inhibits distant metastasis by human melanoma cells. Nature, 2015. 527(7577): p. 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guadamillas MC, Cerezo A, and Del Pozo MA, Overcoming anoikis–pathways to anchorage-independent growth in cancer. Journal of cell science, 2011. 124(19): p. 3189–3197. [DOI] [PubMed] [Google Scholar]
- 16.Wiel C, et al. , BACH1 stabilization by antioxidants stimulates lung cancer metastasis. Cell, 2019. 178(2): p. 330–345. e22. [DOI] [PubMed] [Google Scholar]
- 17.Jobard E, et al. , A serum nuclear magnetic resonance-based metabolomic signature of advanced metastatic human breast cancer. Cancer letters, 2014. 343(1): p. 33–41. [DOI] [PubMed] [Google Scholar]
- 18.Wei Y, et al. , Prognostic significance of serum lactic acid, lactate dehydrogenase, and albumin levels in patients with metastatic colorectal cancer. BioMed Research International, 2018. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin L, et al. , The PLAG1-GDH1 axis promotes anoikis resistance and tumor metastasis through CamKK2-AMPK signaling in LKB1-deficient lung cancer. Molecular cell, 2018. 69(1): p. 87–99. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ubellacker JM, et al. , Lymph protects metastasizing melanoma cells from ferroptosis. Nature, 2020. 585(7823): p. 113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harper KL, et al. , Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature, 2016. 540(7634): p. 588–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rhim AD, et al. , EMT and dissemination precede pancreatic tumor formation. Cell, 2012. 148(1–2): p. 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muzumdar MD, et al. , Clonal dynamics following p53 loss of heterozygosity in Kras-driven cancers. Nature communications, 2016. 7(1): p. 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dudgeon C, et al. , A novel model of pancreatic cancer dormancy reveals mechanistic insights and a dormancy gene signature with human relevance. BioRxiv, 2020. [Google Scholar]
- 25.Elia I, et al. , Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature, 2019. 568(7750): p. 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ngo B, et al. , Limited Environmental Serine and Glycine Confer Brain Metastasis Sensitivity to PHGDH InhibitionPHGDH Inhibition Suppresses Brain Metastasis. Cancer discovery, 2020. 10(9): p. 1352–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferraro GB, et al. , Fatty acid synthesis is required for breast cancer brain metastasis. Nature cancer, 2021. 2(4): p. 414–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ladanyi A, et al. , Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene, 2018. 37(17): p. 2285–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mashimo T, et al. , Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell, 2014. 159(7): p. 1603–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis RT, et al. , Transcriptional diversity and bioenergetic shift in human breast cancer metastasis revealed by single-cell RNA sequencing. Nature cell biology, 2020. 22(3): p. 310–320. [DOI] [PubMed] [Google Scholar]
- 31.Andrzejewski S, et al. , PGC-1α promotes breast cancer metastasis and confers bioenergetic flexibility against metabolic drugs. Cell metabolism, 2017. 26(5): p. 778–787. e5. [DOI] [PubMed] [Google Scholar]
- 32.Fabian A, et al. , Metastasis of pancreatic cancer: An uninflamed liver micromilieu controls cell growth and cancer stem cell properties by oxidative phosphorylation in pancreatic ductal epithelial cells. Cancer Letters, 2019. 453: p. 95–106. [DOI] [PubMed] [Google Scholar]
- 33.Bu P, et al. , Aldolase B-mediated fructose metabolism drives metabolic reprogramming of colon cancer liver metastasis. Cell metabolism, 2018. 27(6): p. 1249–1262. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tharp KM, et al. , Adhesion-mediated mechanosignaling forces mitohormesis. Cell metabolism, 2021. 33(7): p. 1322–1341. e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Senft D and Ze’ev AR, Adaptive stress responses during tumor metastasis and dormancy. Trends in cancer, 2016. 2(8): p. 429–442. [DOI] [PubMed] [Google Scholar]
- 36.Tharp KM, et al. , Actomyosin-mediated tension orchestrates uncoupled respiration in adipose tissues. Cell metabolism, 2018. 27(3): p. 602–615. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Swift J, et al. , Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science, 2013. 341(6149): p. 1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uhler C and Shivashankar G, Nuclear mechanopathology and cancer diagnosis. Trends in cancer, 2018. 4(4): p. 320–331. [DOI] [PubMed] [Google Scholar]
- 39.Fane M and Weeraratna AT, How the ageing microenvironment influences tumour progression. Nature Reviews Cancer, 2020. 20(2): p. 89–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghajar CM, Metastasis prevention by targeting the dormant niche. Nature Reviews Cancer, 2015. 15(4): p. 238–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DeBerardinis RJ and Chandel NS, Fundamentals of cancer metabolism. Science advances, 2016. 2(5): p. e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chaffer CL and Weinberg RA, A perspective on cancer cell metastasis. science, 2011. 331(6024): p. 1559–1564. [DOI] [PubMed] [Google Scholar]
- 43.Lyssiotis CA and Kimmelman AC, Metabolic interactions in the tumor microenvironment. Trends in cell biology, 2017. 27(11): p. 863–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li F and Simon MC, Cancer cells don’t live alone: Metabolic communication within tumor microenvironments. Developmental cell, 2020. 54(2): p. 183–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Encarnación-Rosado J and Kimmelman AC, Harnessing metabolic dependencies in pancreatic cancers. Nature Reviews Gastroenterology & Hepatology, 2021. 18(7): p. 482–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sousa CM, et al. , Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature, 2016. 536(7617): p. 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Banh RS, et al. , Neurons release serine to support mRNA translation in pancreatic cancer. Cell, 2020. 183(5): p. 1202–1218. e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pavlides S, et al. , The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell cycle, 2009. 8(23): p. 3984–4001. [DOI] [PubMed] [Google Scholar]
- 49.Sonveaux P, et al. , Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. The Journal of clinical investigation, 2008. 118(12): p. 3930–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Colegio OR, et al. , Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature, 2014. 513(7519): p. 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fong MY, et al. , Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nature cell biology, 2015. 17(2): p. 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang M, et al. , Adipocyte-Derived Lipids Mediate Melanoma Progression via FATP ProteinsAdipocyte-Derived Lipids Drive Melanoma Progression via FATP. Cancer discovery, 2018. 8(8): p. 1006–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nieman KM, et al. , Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nature medicine, 2011. 17(11): p. 1498–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Doglioni G, Parik S, and Fendt S-M, Interactions in the (pre) metastatic niche support metastasis formation. Frontiers in oncology, 2019. 9: p. 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luzzi KJ, et al. , Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. The American journal of pathology, 1998. 153(3): p. 865–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheung KJ and Ewald AJ, A collective route to metastasis: Seeding by tumor cell clusters. Science, 2016. 352(6282): p. 167–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cameron MD, et al. , Temporal progression of metastasis in lung: cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer research, 2000. 60(9): p. 2541–2546. [PubMed] [Google Scholar]
- 58.Oudin MJ, et al. , Tumor Cell–Driven Extracellular Matrix Remodeling Drives Haptotaxis during Metastatic ProgressionHaptotaxis and Metastasis. Cancer discovery, 2016. 6(5): p. 516–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sullivan WJ, et al. , Extracellular matrix remodeling regulates glucose metabolism through TXNIP destabilization. Cell, 2018. 175(1): p. 117–132. e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McAllister SS and Weinberg RA, The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nature cell biology, 2014. 16(8): p. 717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Costa-Silva B, et al. , Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nature cell biology, 2015. 17(6): p. 816–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gopal SK, et al. , Extracellular vesicles: their role in cancer biology and epithelial–mesenchymal transition. Biochemical Journal, 2017. 474(1): p. 21–45. [DOI] [PubMed] [Google Scholar]
- 63.Wu S, et al. , Matrix stiffness-upregulated LOXL2 promotes fibronectin production, MMP9 and CXCL12 expression and BMDCs recruitment to assist pre-metastatic niche formation. Journal of Experimental & Clinical Cancer Research, 2018. 37(1): p. 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reynaud C, et al. , Lysyl Oxidase Is a Strong Determinant of Tumor Cell Colonization in BoneLOX Triggers Tumor Cell Colonization in Bone. Cancer Research, 2017. 77(2): p. 268–278. [DOI] [PubMed] [Google Scholar]
- 65.Cox TR, et al. , The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature, 2015. 522(7554): p. 106–110. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 66.Lee JW, et al. , Hepatocytes direct the formation of a pro-metastatic niche in the liver. Nature, 2019. 567(7747): p. 249–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fane M and Weeraratna AT, Normal aging and its role in cancer metastasis. Cold Spring Harbor Perspectives in Medicine, 2020. 10(9): p. a037341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Balliet RM, et al. , Mitochondrial oxidative stress in cancer-associated fibroblasts drives lactate production, promoting breast cancer tumor growth: understanding the aging and cancer connection. Cell cycle, 2011. 10(23): p. 4065–4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martinez-Outschoorn UE, et al. , Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment. Cell cycle, 2010. 9(17): p. 3515–3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Coppé J-P, et al. , The senescence-associated secretory phenotype: the dark side of tumor suppression. Annual review of pathology: mechanisms of disease, 2010. 5: p. 99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mavrogonatou E, et al. , Extracellular matrix alterations in senescent cells and their significance in tissue homeostasis. Matrix Biology, 2019. 75: p. 27–42. [DOI] [PubMed] [Google Scholar]
- 72.Kaur A, et al. , Remodeling of the collagen matrix in aging skin promotes melanoma metastasis and affects immune cell motility. Cancer discovery, 2019. 9(1): p. 64–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ecker BL, et al. , Age-Related Changes in HAPLN1 Increase Lymphatic Permeability and Affect Routes of Melanoma MetastasisAge-Induced Lymphatic Permeability Increases Metastasis. Cancer discovery, 2019. 9(1): p. 82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kaur A, et al. , sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature, 2016. 532(7598): p. 250–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fane ME, et al. , Stromal changes in the aged lung induce an emergence from melanoma dormancy. Nature, 2022. 606(7913): p. 396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gomes AP, et al. , Age-induced accumulation of methylmalonic acid promotes tumour progression. Nature, 2020. 585(7824): p. 283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Casazza A, et al. , Tumor stroma: a complexity dictated by the hypoxic tumor microenvironment. Oncogene, 2014. 33(14): p. 1743–1754. [DOI] [PubMed] [Google Scholar]
- 78.Eming SA, Murray PJ, and Pearce EJ, Metabolic orchestration of the wound healing response. Cell metabolism, 2021. 33(9): p. 1726–1743. [DOI] [PubMed] [Google Scholar]
- 79.Dvorak HF, Tumors: wounds that do not heal—redux. Cancer immunology research, 2015. 3(1): p. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aiello NM, et al. , Metastatic progression is associated with dynamic changes in the local microenvironment. Nature communications, 2016. 7(1): p. 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sai B, et al. , Cancer-educated mesenchymal stem cells promote the survival of cancer cells at primary and distant metastatic sites via the expansion of bone marrow-derived-PMN-MDSCs. Cell Death & Disease, 2019. 10(12): p. 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Deguchi A and Maru Y, Inflammation-associated premetastatic niche formation. Inflammation and Regeneration, 2022. 42(1): p. 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schwörer S, Vardhana SA, and Thompson CB, Cancer metabolism drives a stromal regenerative response. Cell metabolism, 2019. 29(3): p. 576–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mentch SJ, et al. , Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell metabolism, 2015. 22(5): p. 861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dai Z, et al. , Methionine metabolism influences genomic architecture and gene expression through H3K4me3 peak width. Nature communications, 2018. 9(1): p. 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dai Z, Ramesh V, and Locasale JW, The evolving metabolic landscape of chromatin biology and epigenetics. Nature Reviews Genetics, 2020. 21(12): p. 737–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Netea MG, et al. , Trained immunity: a program of innate immune memory in health and disease. Science, 2016. 352(6284): p. aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Naik S, et al. , Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature, 2017. 550(7677): p. 475–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang S, et al. , Epigenetic compensation promotes liver regeneration. Developmental cell, 2019. 50(1): p. 43–56. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li W, et al. , A homeostatic Arid1a-dependent permissive chromatin state licenses hepatocyte responsiveness to liver-injury-associated YAP signaling. Cell Stem Cell, 2019. 25(1): p. 54–68. e5. [DOI] [PubMed] [Google Scholar]
- 91.Aloia L, Epigenetic regulation of cell-fate changes that determine adult liver regeneration after injury. Frontiers in Cell and Developmental Biology, 2021. 9: p. 643055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Solhi R, et al. , Metabolic hallmarks of liver regeneration. Trends in Endocrinology & Metabolism, 2021. 32(9): p. 731–745. [DOI] [PubMed] [Google Scholar]
- 93.Caldez MJ, et al. , Metabolic remodeling during liver regeneration. Developmental cell, 2018. 47(4): p. 425–438. e5. [DOI] [PubMed] [Google Scholar]
- 94.Khomich O, Ivanov AV, and Bartosch B, Metabolic hallmarks of hepatic stellate cells in liver fibrosis. Cells, 2019. 9(1): p. 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Eslam M, Valenti L, and Romeo S, Genetics and epigenetics of NAFLD and NASH: clinical impact. Journal of hepatology, 2018. 68(2): p. 268–279. [DOI] [PubMed] [Google Scholar]
- 96.Feng D, et al. , A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science, 2011. 331(6022): p. 1315–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gao Y, et al. , Metastasis organotropism: redefining the congenial soil. Developmental cell, 2019. 49(3): p. 375–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chang C-H, et al. , Metabolic competition in the tumor microenvironment is a driver of cancer progression. cell, 2015. 162(6): p. 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ho P-C, et al. , Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell, 2015. 162(6): p. 1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.McAllister SS and Weinberg RA, Tumor-host interactions: a far-reaching relationship. Journal of clinical oncology, 2010. 28(26): p. 4022–4028. [DOI] [PubMed] [Google Scholar]
- 101.Taylor SR, et al. , Developing dietary interventions as therapy for cancer. Nature Reviews Cancer, 2022: p. 1–15. [DOI] [PubMed] [Google Scholar]
- 102.Pavlova NN, Zhu J, and Thompson CB, The hallmarks of cancer metabolism: Still emerging. Cell Metabolism, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fulop T, et al. , Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Frontiers in immunology, 2018. 8: p. 1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fane ME, et al. , Stromal changes in the aged lung induce an emergence from melanoma dormancy. Nature, 2022: p. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Beadle BM, et al. , Ten-year recurrence rates in young women with breast cancer by locoregional treatment approach. International Journal of Radiation Oncology* Biology* Physics, 2009. 73(3): p. 734–744. [DOI] [PMC free article] [PubMed] [Google Scholar]