

Abstract
Background:
Heart failure (HF) is a debilitating disease affecting more than 64 million people worldwide. In addition to impaired cardiovascular performance and associated systemic complications, most patients with HF suffer from depression and/or substantial cognitive decline. Although neuroinflammation and brain hypoperfusion occur in humans and rodents with heart failure (HF), the underlying neuronal substrates, mechanisms, and their relative contribution to cognitive deficits in HF remains unknown.
Methods:
To address this critical gap in our knowledge, we used a well-established HF rat model that mimics clinical outcomes observed in the human population, along with a multidisciplinary approach combining behavioral, electrophysiological, neuroanatomical, molecular and systemic physiological approaches.
Results:
Our studies support neuroinflammation, hypoperfusion/hypoxia and neuronal deficits in the hippocampus of HF rats, which correlated with the progression and severity of the disease. An increased expression of angiotensin II (AngII) AT1a receptors (AT1aRs) in hippocampal microglia preceded the onset of neuroinflammation. Importantly, blockade of AT1Rs with a clinically used therapeutic drug (Losartan), and delivered in a clinically-relevant manner, efficiently reversed neuroinflammatory endpoints (but not hypoxia ones), resulting in turn in improved cognitive performance in HF rats. Finally, we show than circulating AngII can leak and access the hippocampal parenchyma in HF rats, constituting a possible source of AngII initiating the neuroinflammatory signaling cascade in HF.
Conclusions:
In this study, we identified a neuronal substrate (hippocampus), a mechanism (angiotensin II-driven neuroinflammation) and a potential neuroprotective therapeutic target (angiotensin II 1a receptors, AT1aRs) for the treatment of cognitive deficits in HF.
Keywords: Angiotensin II, neuroinflammation, microglia, heart failure, hippocampus, cognition
Graphical Abstract
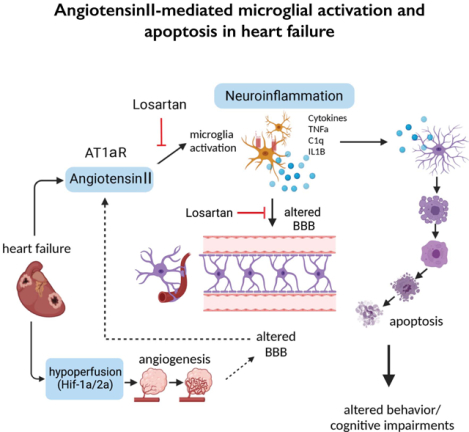
Introduction
Heart failure (HF) is a debilitating disease affecting more than 64 million people worldwide1. In addition to cardiovascular complications, most patients with HF suffer from depression2 and/or cognitive decline3. Studies in HF patients and animal models support overactivation of the renin-angiotensin system (RAS) as a critical mechanism underlying sympathohumoral activation and associated cardiovascular compromise in HF4–6. Notably, pharmacological blockade of the RAS is an effective treatment for the improvement of cardiovascular compromise in HF7–9.
Conversely, the specific neural substrates and mechanisms contributing to emotional and cognitive decline in HF remain to be determined. The hippocampus plays a critical role in cognitive performance10, including spatial and emotional memory11. Moreover, hippocampal abnormalities as a result of aging12 or in Alzheimer’s Disease13 have been shown to contribute to cognitive deficits, standing thus as a candidate substrate contributing to cognitive deficits in HF.
Two major pathological factors have been proposed to contribute to neurological dysfunction in HF, including end-organ hypoperfusion/hypoxia14–18 a state associated also to cognitive impairment and neurodegeneration such as Alzheimer’s19. Additionally, recent studies support neuroinflammation as a common finding in numerous brain regions during HF20–23. Still, whether these two pathological processes are causally interrelated, and what their relative contribution to cognitive deficits in HF are, remains largely unknown.
Neuroinflammation is a process regulated by microglial and astrocytic interactions24. Microglia, the resident macrophages of the brain parenchyma, monitor and protect neurons under normal conditions25. Conversely, sustained microglial activation during pathological conditions induces a neurotoxic astrocyte phenotype24, which eventually results in neuronal apoptosis and death. The circulating peptide angiotensin II (AngII), acting on its AT1a receptors (AT1aR) is a potent proinflammatory molecule26 shown to contribute to hypothalamic neuroinflammation and sympathohumoral activation in hypertension27,28 and HF21,23,29–33. Given this, and the fact that AT1aRs are also expressed in hippocampal microglia34, we used an established ischemic HF rat model and a highly multidisciplinary approach to test the hypothesis that the AngII/AT1aR-signaling pathway contributes to microglia activation, hippocampal neuroinflammation, neuronal apoptosis and cognitive deficits in HF.
Materials and methods
The data that support the findings of this study are available from the corresponding author upon reasonable request. All experiments were approved and carried out in agreement to the Georgia State University Institutional Animal Care and Use Committee (IACUC) guidelines. A detailed description of experimental procedures can be found in the supplemental materials.
Animals
We used male Wistar rats (5–7 weeks old at HF surgery, 180–200g, Envigo, Indianapolis, IN, USA) for all experiments (n=151). Rats were housed under constant temperature (22 ± 2°C) and humidity (55 ± 5%) on a 12-h light cycle (lights on: 08:00–20:00) ad libitum access to food and water.
Heart failure surgery and Echocardiography
The ischemic HF surgical model and echocardiography was performed as previously described35. A summary of all assessed parameters is shown in Table S1. HF rats were categorized into an early (6–10 weeks) and late (10–16 weeks) progressive stages of the disease.
Immunohistochemistry
Following pentobarbital-induced anesthesia rats were intracardially perfused and fixed with 4% paraformaldehyde20. Conventional fluorescence immunohistochemistry was performed in fixed brain sections as described in the supplementary Methods section.
RNAScope in situ hybridization
RNAScope reagents were purchased from acdbio (PN320881). Nuclease-free water and PBS were purchased from Fisher Scientific. Brains were processed as described under Immunohistochemistry using nuclease-free PBS, water, PBS and sucrose, following manufacturer’s protocol. For analysis, microglia were considered mRNA-positive if they displayed three or more fluorescently-labeled voxels within the respective soma.
Confocal microscopy and 3D IMARIS analysis
Confocal images were obtained using a Zeiss LSM 780 confocal microscope (1024×1024 pixel, 16-bit depth, pixel size 0.63-micron, zoom 0.7). Three-dimensional reconstruction of microglia, astrocytes or axons was performed as previously described20. For the quantification of mRNA transcripts and subsequent correlation with microglial morphology, spheres precisely engulfing the microglial soma were manually placed on individual microglia as described previously36. The fluorescent intensity within the respective spheres was measured and then correlated with microglial complexity assessed via Sholl analysis.
Analysis of cellular density, signal intensity and density
Cellular density (microglia or astrocytes), signal intensity and density were assessed blindly using the cell counter plugin and a thresholding approach in Fiji as previously described27. To differentiate neurons from glia (astrocytes and microglia) for the cCasp3 study (Figure 3 and S3), we ran a separate set of experiments in which we immunohistochemically labeled for astrocytes (glutamine synthetase antibody), microglia cells (IBA1 antibody) and neurons (NeuN antibody) along with DAPI nuclear staining. Using ImageJ algorithms, the nuclear size (DAPI area) of individually identified cell types was measured, and a frequency distribution histogram was built.
Reverse transcription polymerase chain reaction (RT-PCR) and quantitative real time PCR (qPCR)
RNA extraction and isolation, cDNA synthesis and qPCR were performed using the miRNAeasy Mini kit (Qiagen, Cat. No. 217004), QIAzol Lysis Reagent (Qiagen, Mat. No. 1023537) and iScript™ gDNA Clear cDNA Synthesis Kit (BIO RAD, cat. no. 1725035) and the SimpliAmp Thermal Cycler (applied biosystems, Thermo Fisher Scientific) according to manufacturer protocol. All results are expressed as fold changes in HF relative to control (sham) rats unless indicated otherwise and as previously reported20.
Measurement of tissue partial pressure of oxygen (pO2) and cerebral blood flow (CBF)
Partial pressure of tissue O2 (pO2) and CBF in the hippocampus were recorded in urethane anesthetized rats positioned in a stereotaxic apparatus using an optical fluorescence probe (250 μm tip diameter; OxyLite (pO2) and OxyFlo (CBF) systems; Oxford Optronix) that allows real-time recording of absolute values of these parameters.
Assessment microglial AngIIfluo uptake
Rats were anesthetized with Ketamine/Xylazine (60/8 mg/mL, respectively) and a non-occluding catheter filled was inserted into the left internal carotid artery27. AngIIfluo (3μmol/L, Anaspec, CA) was infused at 2.86μl/g/rat and allowed to circulate for 30 mins. Rats were then decapitated, and brains were fixed and sectioned using a Cryostat at 40μm-thick for confocal imaging. Imaris was used to detect and quantify the amount of microglial AngIIfluo.
Losartan treatment
HF rats were randomly allocated two either HF or HF + Losartan groups. Losartan (20mg/kg/day in the drinking water started 1 week after the HF surgery until rats were sacrificed for analysis 13-weeks post-surgery.
Acute slice patch clamp electrophysiology
Conventional whole-cell patch-clamp recordings from dorsal hippocampus CA1 neurons were obtained from acutely obtained slices from Sham and HF rats as previously described37. CA1 neurons were identified by their anatomical location in the pyramidal laminar layer.
Behavioral studies
Spontaneous Alternation and Inhibitory Avoidance
Spontaneous alternation testing and inhibitory avoidance testing were performed as previously described38.
Statistical analyses
All statistical analyses were performed using GraphPad Prism 9 (GraphPad Software, California, USA). Student’s t-test, Chi square test or one- or two-way analysis of variance (ANOVA) were used to compare the groups followed by Tukey post-hoc tests. Chi-square tests were used to compare incidence of effects. Results are expressed as mean ± standard error of the mean (SEM). Results were considered statistically significant if p<0.05 and are presented as * for p<0.05, ** for p<0.01 and *** for p<0.0001 in the respective Figures.
Results
Heart failure induces a pro-inflammatory microglial phenotype in the hippocampus
All experimental procedures and anatomical analyses were performed both in the dorsal ventral hippocampus unless indicated otherwise. Given that we obtained very similar results between both hippocampal subregions, we are only reporting findings in the dorsal hippocampus (DH), given its known direct involvement39 in the cognitive deficits we reported here and previousl38.
An Imaris-assisted microglia morphometry analysis20 revealed significant changes in microglia morphometry in the DH of HF rats that are consistent with a pro-inflammatory state40. These included larger microglia somatic volumes along with significant changes in microglia surface area, cell volume, and filament length compared to sham rats (n=8, Figure 1A–C and Figure S1B-D). No differences in the number of microglia cells were observed between the two groups, indicative of lack of microglia cell proliferation during HF (Figure S1E). These morphological changes were found to be dependent on the progression of the disease (Figure 1C), as animals at later stages of HF displayed more profound changes in microglial morphology. We next quantified the percentage of activated microglia using a morphological threshold established in our previous studies20 and found a roughly 2.5-fold increase in activated microglia in HF rats (p<0.0001, Chi-square test, Figure 1D). Changes in hippocampal microglia morphometry in HF rats were further confirmed by Sholl analysis and heatmaps that displayed the maximum filament length and Sholl values for each microglia (n= 256 randomly selected microglia, 32 per animal) (Figure 1E), showing a reduced reach (i.e. maximum filament extension) and diminished maximum complexity (peak Sholl value) in microglia of HF rats.
Figure 1-. Microglial activation and increased cytokine levels in the hippocampus of HF rats.
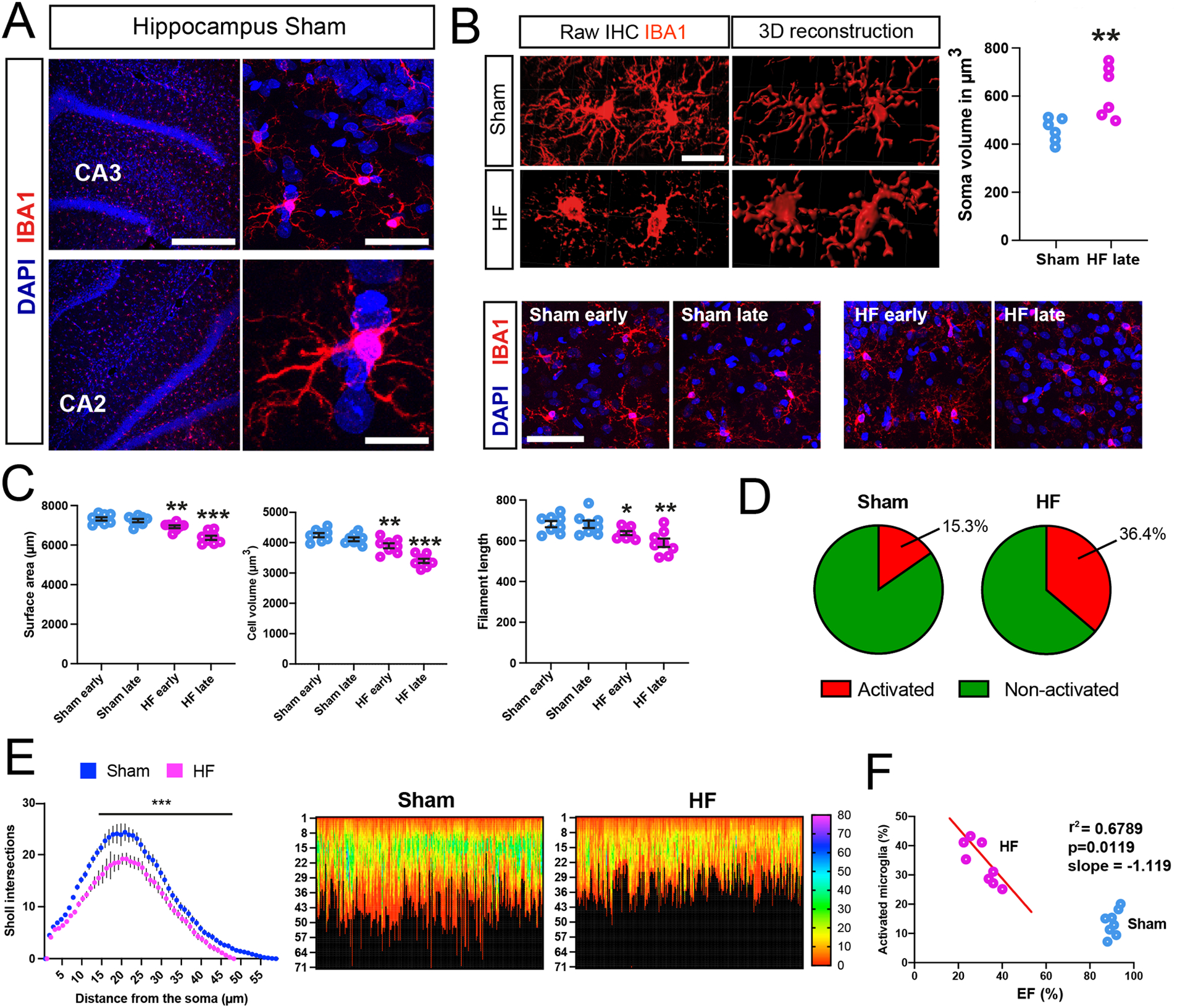
A IBA1-positive microglia in the DH of ham rats. CA2/3= cornu ammonis 2/3. B Images of microglia in sham and HF rats, and their assessment of somatic volume (n=6 per group). C Quantification of microglial morphology at early and late HF stages, showing a de-ramified microglial phenotype with reduced cell volume, surface area and filament length in HF (n=8 per group). D Quantification of activated microglia based on their morphological appearance in sham and HF rats. E Sholl analysis for microglia in sham and HF rats. Heat map analysis (256 microglia, 32 per animal, 8 rats per group) reveals reduced microglial complexity in HF rats. Left numbers indicate microglial reach (in μm), color coding indicates peak Sholl values of individual microglia. F The percentage of activated microglia correlates with the severity of disease (EF%) in HF rats. p<0.05*, p<0.01** and p<0.001*** (n=8/group). Scale bars 300μm (a, top left), 25μm (a, top right), 10μm (a, bottom right), 10μm (b) and 50μm (c).
A significant negative correlation between the degree of microglia cell activation and the degree of HF (i.e., echocardiography EF value, Figure 1F) support that hippocampal microglial status is dependent on the severity of HF.
None of changes reported above were observed in the prelimbic cortex (PLC, Figure S1F-G) nor in the somatosensory cortex (SSC) (S1BF region)20, suggesting that HF-induced microglia changes were region-dependent.
In addition to native brain microglia, IBA1 also labels infiltrating macrophages during systemic inflammation41. Thus, to determine whether the IBA1 staining in the hippocampus reflected resident microglia, we combined IBA1 and TMEM119 staining, a microglia-specific marker41. We found the majority of microglia to colocalize both IBA1 and TMEM119 signals (Sham: 90.2% TMEM119+; HF: 92.8% TMEM119+, Figure S1I,J), supporting that IBA1-stained cells reflect for the most part resident microglia rather than infiltrating macrophages. Together, these findings indicate that hippocampal microglia during HF undergo a morphological transition towards a pro-inflammatory phenotype which is progressive in time, and dependent on the severity of the disease.
Microglial morphological changes correlate with cytokine mRNA expression levels in HF rats
qPCR assessment of mRNA transcripts revealed a significant increase in neuroinflammation-associated genes within the DH of HF rats (Figure 2A), but not in the PLC (Figure S1G). Moreover, measurement of cytokine mRNAs expression at the single microglia level (IBA1 immunohistochemistry+RNAScope hybridization, Figure 2B–D, Figure S2A-D), showed a significant negative correlation between IL1β, TNFα and C1q mRNA levels with microglial morphological complexity. While the cytokine mRNA signal was rather diffuse, likely reflecting the fact that other cell types express cytokines42, our analysis was restricted to IBA1 positive microglial cells (see Figure S2A).
Figure 2-. Cytokine mRNA expression correlates with microglial morphology.
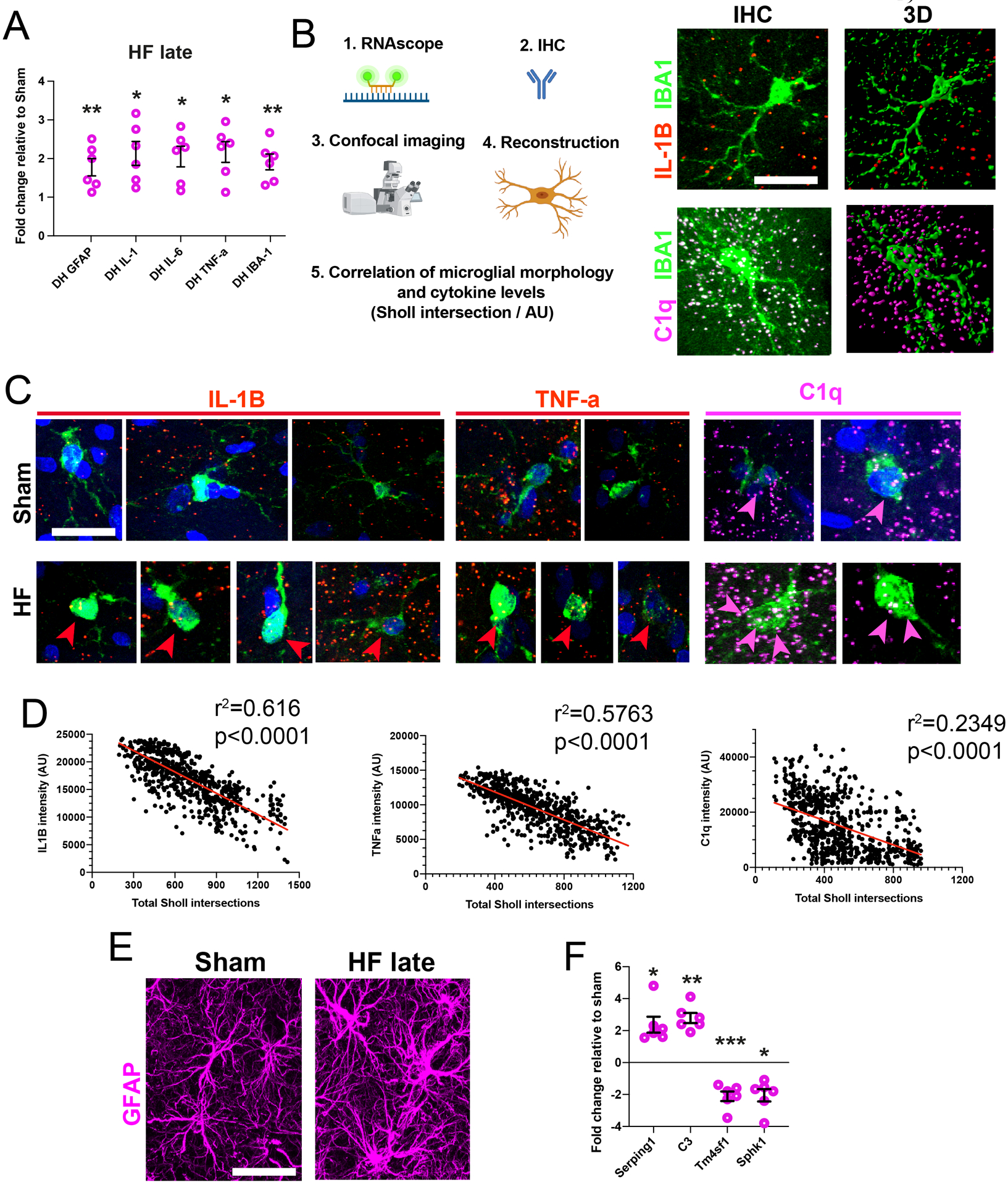
A Quantification of cytokine mRNA levels in HF relative to sham rats (n=6 per group). B Schematic workflow used for correlative assessment of microglial complexity with cytokine mRNA levels. Images show samples of microglia negative for IL-1β (b) and positive for C1q (c). C Images of microglial IL1β, TNF-α or C1q mRNA in Sham and HF rats. Note that less complex microglia in HF rats (bottom rows) have more cytokine mRNA. Arrowheads: co-localization of IBA1 and cytokine mRNA probe. D Cytokine mRNA levels negatively correlate with microglial complexity (each dot represents a single microglia, plot is a pool of microglia obtained from n=4 rats/group). E Images of normal and hypertrophic astrocytes in sham and HF rats respectively. F Quantification of A1/A2 astrocyte mRNA markers in HF rats relative to Shams (n=6 per group). p<0.05*, p<0.01** and p<0.001***. GFAP = glial fibrillary acidic protein; GS = glutamine synthetase. Scale bars 10μm (b, c) and 20μm (e).
Evidence for reactive A1 astrocytes in the hippocampus of HF rats
We found that astrocytes in the DH of HF rats had increased soma volume and swelling of GFAP-labeled processes (Figure 2E, Figure S2E, F), indicative of a hypertrophic phenotype43 and reactive astrocytes44. The number of astrocytes however did not change (Figure S2G). Similarly, we found lower levels of neuroprotective A2- and higher levels of neurotoxic A1-related mRNA transcripts in the DH of HF rats (Figure 2F). Neither astrocytic numbers nor their soma volume were altered in the PLC (Figure S2F,G). Together, these findings support an astrocytic shift from a neuroprotective to a neurotoxic state the hippocampus of HF rats.
Evidence for hippocampal apoptotic signaling and neuronal dysfunction in HF rats
We found a significant increase in hippocampal caspase-3 (cCasp3) immunoreactivity both at early and late stages in HF compared to sham rats (Figure 3A,B), and observed a significant thinning of pyramidal cell layers in the DH of HF rats (Figure S3A-C). The majority of hippocampal cCasp3-staining in HF rats was located within pyramidal cell layers (90.2%) (Figure S3D).
Figure 3-. Apoptotic signaling and altered pyramidal neuronal function in HF rats.
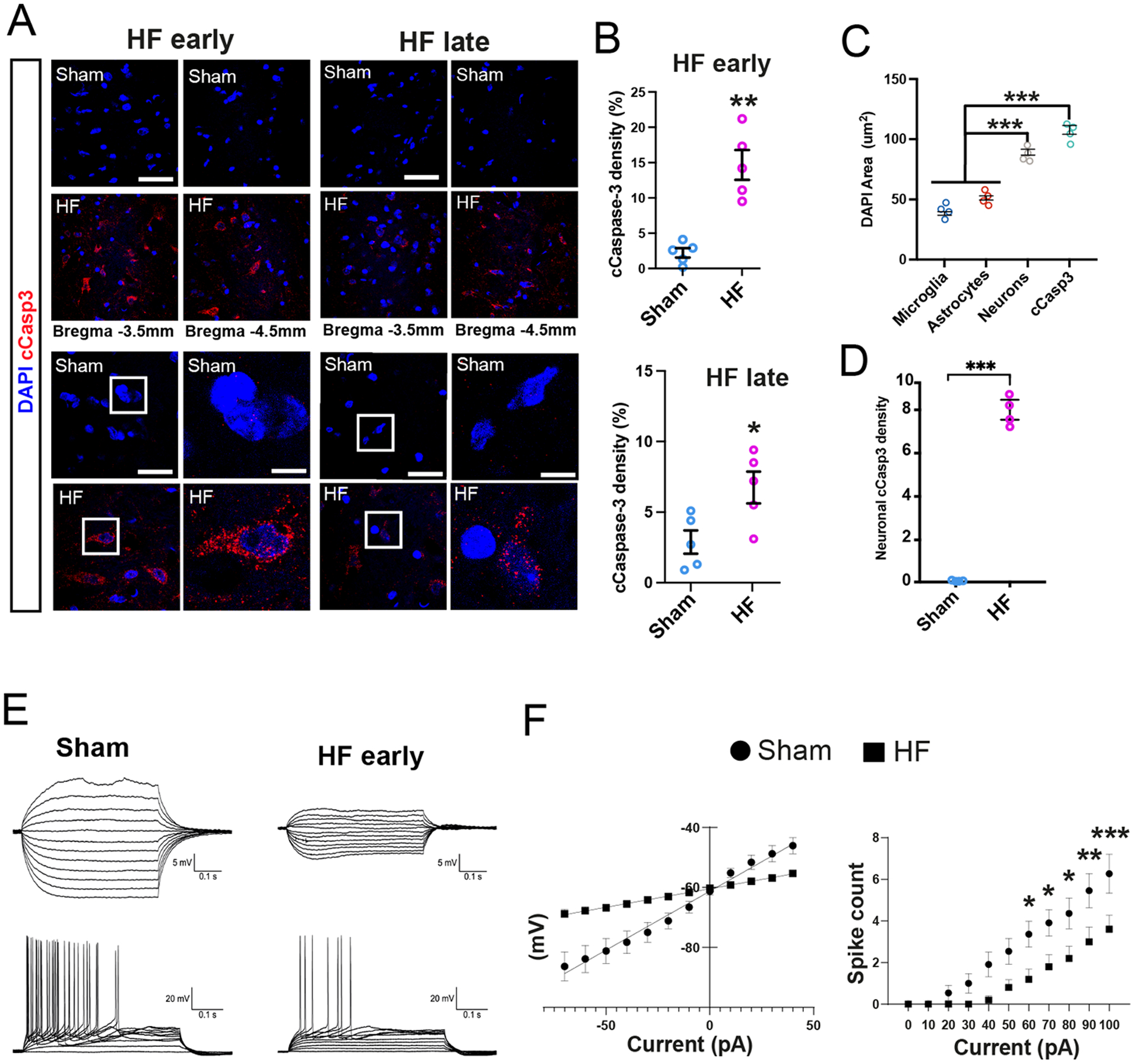
A cCasp3-positive cells at early and late HF stages. Note the absence of cCasp3 staining in sham rats. High magnification images show nuclear deterioration and overlap of DAPI and cCasp3 signal in HF rats. B Quantification of cCasp3-positive signal at early and late HF stages (n=5/group). C Quantification of cell type-specific DAPI nuclear size. D Quantification of mean single cell neuronal cCasp3 signal in sham and HF rats (n=4/group). E Representative whole-cell voltage traces in response to depolarizing/hyperpolarizing current injection in CA1 pyramidal neurons from Sham (left) and HF (right) rats. F Mean I-V curves in sham (circles, n=11 cells/2 rats) and HF (squares, HF n=9 cells animals/ 3 rats. Note the decreased slope (input resistance) in sham rats. Input/output function plots from the same CA1 neurons in D, showing a diminished firing discharge in HF rats p<0.05*, p<0.01** and p<0.001***. Scale bars 100μm (a, overviews), 25μm (a, enlarged images) and 5μm (enlarged insets).
To more conclusively determine if apoptosis was primarily neuronal, we traced individual cell profiles showing positive cCasp3 signal, and using an approach that allows us to differentiate neurons from astrocytes and microglia cells based on their nuclear DAPI size (see Methods and Figure S3E), we found that the mean cCasp3-positive nuclear size was similar to that in neurons, but significantly larger from those of astrocytes and microglia cells (Figure 3C). Importantly, we found a significantly higher cCasp3 signal at the individual neuronal level in HF when compared to sham rats (Figure 3D).
We further assessed neuronal apoptosis via TUNEL staining, and found frequent apoptotic clusters in the DH in early and late stages of HF (Figure S3F-I), which we analyzed with two complementary approaches (Figure S3H-I). Moreover, and in line with the pyramidal layer thinning, we found a reduction in DH neuronal counts in HF rats (Figure S3K).
To further assess for possible hippocampal neuronal functional damage during HF, we performed patch-clamp recordings ex vivo from pyramidal neurons in dorsal CA1 (Sham: 9 neurons from 3 rats; HF early stage, n= 11 neurons from 3 rats). We found a decreased input resistance (i.e., decreased slope in the current/voltage plots, Figure 3E,F and Figure S3F), along with a diminished input/output function (i.e. decreased number of evoked action potentials per stimulation, Figure 3F) (p= 0.0135, 2-way ANOVA) in CA1 neurons in HF rats. Moreover, CA1 neuronal resting membrane potential was significantly more depolarized in HF rats (Figure S3K). No differences were observed in general action potential properties between the two groups (Figure S3K). Together, these findings support HF-induced hippocampal neuronal apoptotic signaling and functional compromise, that manifested as an overall decreased neuronal ability to fire in response to an incoming stimulus.
Evidence for a hippocampal hypoperfusion/hypoxic microenvironment in HF rats
Next, to probe for a potential hippocampal hypoxic state in HF, we assessed mRNA expression of Hif1α and Hif2α, two widely used hypoxia markers, and found significant increases in the DH of HF rats relative to sham controls (Figure 4A,B). Interestingly, the increase in Hif1α and Hif2α diminished and increased over time, respectively, following the induction of HF (Figure 4A,B). in vivo recordings of hippocampal pO2 levels in anesthetized sham and HF rats (n= 4 and 6 respectively) showed significantly lower basal pO2 levels in the latter (Figure 4C). In addition, in separate sets of rats we observed a decreased basal cerebral blood flow (CBF) in the hippocampus of HF, when compared to sham rats (n= 4 and 5 respectively, (Figure 4C, right panel), further supporting a hypoxic/hypo-perfused hippocampal state.
Figure 4-. Evidence for a hypoperfusion/hypoxic state in the hippocampus of HF rats.
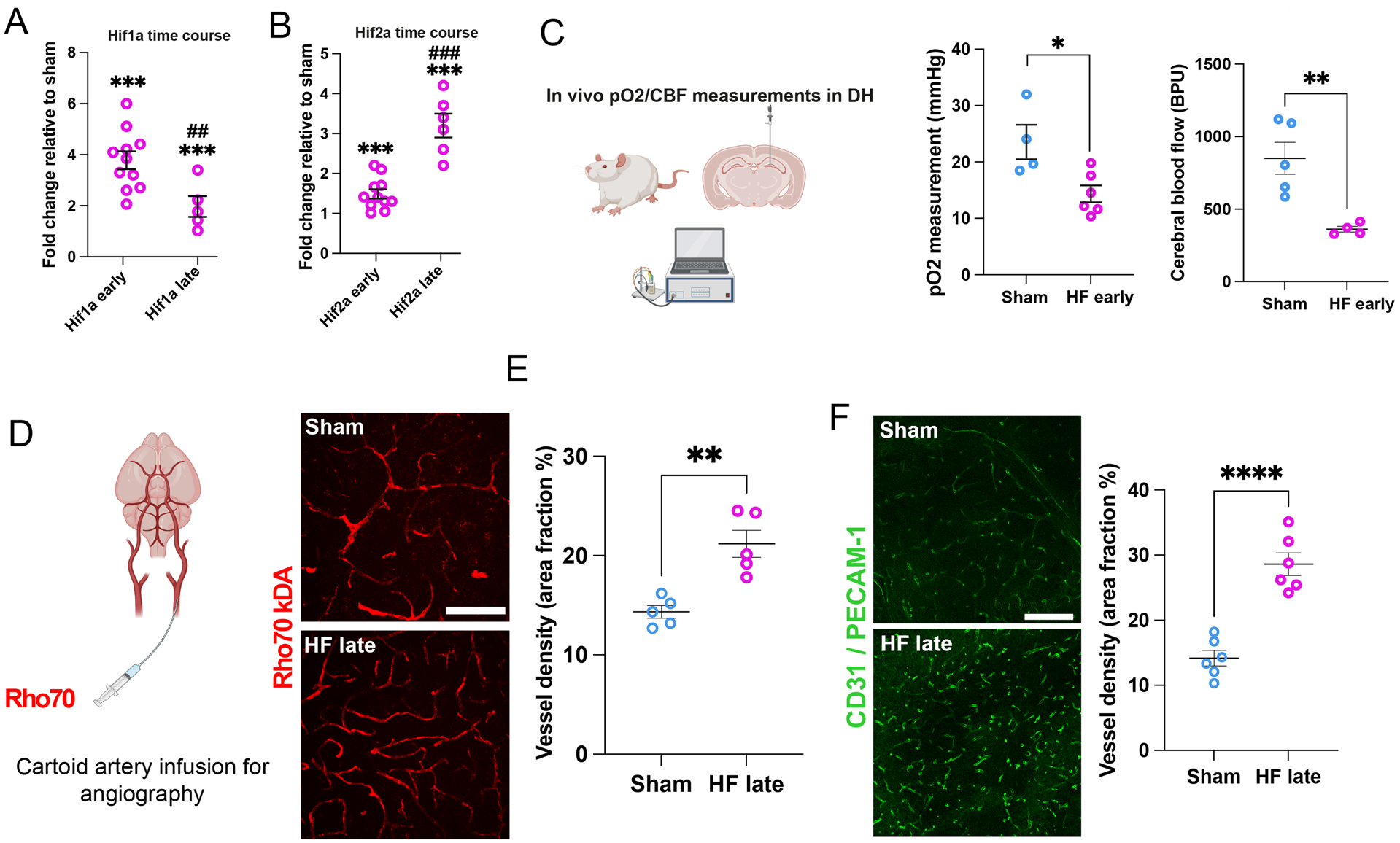
A Changes in hypoxia markers Hif-1α and (B) Hif-2α mRNA in the DH at early and late HF stages relative to Sham (n=5 /group). C Schematic depiction of in vivo measurement of pO2 and cerebral blood flow (CBF) in the DH. HF rats display lower basal hippocampal pO2 levels (n=4 sham, n=6 HF, left panel) as well as decreased basal CBF (n=5 sham, n=4 HF, right panel). D Schematic depiction of angiography via intra-carotid infusion of Rho70 dye. Rho70-labeled blood vessels within the DH of sham and HF rats. E HF rats display significantly increased vessel density. F Confocal images show vascular CD31/PECAM-1 staining in the DH of sham and HF. Note the significantly increased vascular density in HF compared to sham rats. Scale bar 100μm (e) and 200μm (f). p<0.05*, and p<0.001*** vs sham; p<0.01## and p<0.0001### vs HF early.
A hippocampal angiography (intra-carotid infusion of Rho70) showed an increased vessel volume in HF compared to sham rats (Figure 4D,E). This hypervascularized state was also corroborated using the vessel-specific antibody CD31 (PECAM-1, Figure 4F), as well as with an Aquaporin-4 immunostaining, (labeling astrocytic process enwrapping all these vessels) , showing also a progressive increased degree of vessel bifurcations (Figure S4A-D). Contrasting pro-inflammatory markers, the hypervascularization in HF rats was also observed in the SSC, PVN and the amygdala (CeA) (Figure S4E-G). Together, these findings support a hypoperfusion/hypoxic state in the hippocampus and other brain regions of HF rats.
AT1aR expression in hippocampal microglia is linked to microglial activation during HF
Overactivation of the renin-angiotensin system (RAS) is a hallmark of HF, and exacerbated angiotensin II (AngII) signaling, a pro-inflammatory peptide26, contributes to the pathophysiology of HF21,23,31. To determine whether AT1aRs were upregulated in the DH of HF rats, we first performed non-cell-type specific qPCR. We found a progressive increase of AT1aR expression in the DH of HF rats when compared to age-matched sham controls (Figure 5A). A similar increase was observed in the PVN and CeA (but not in the SSC or PLC) of HF rats (Figure S5A). Moreover, a combination of RNAScope for AT1aR mRNA with IHC staining against IBA1 confirmed a significant increase in the number of DH AT1aR-positive microglia Figure 5B,C) and AT1aR receptor density/microglia (Figure S5-E). We also found that AT1aR positive microglia were morphometrically less complex than their AT1aR-negative counterparts (Figure 5C). These results suggest a potential role for AT1aR in promoting the pro-inflammatory phenotype. This was further supported by the finding that only AT1aR (but not IBA1 and other cytokines) mRNA hippocampal level (qPCR) was already elevated at a very early stage of the disease (10 days post HF surgery) (Figure 5D, left graph). A similar elevation in microglial AT1aR mRNA (RNAscope) was detected at this early stage (Figure 5D, right graph). Conversely, we failed to detect any increases in neuronal AT1aR mRNA in HF (Figure S5D,E). Notably, we did not detect any AT1aRs in hippocampal astrocytes in sham or HF rats (Figure S5F).
Figure 5-. Upregulation of AT1aRs in hippocampal microglia of HF rats.
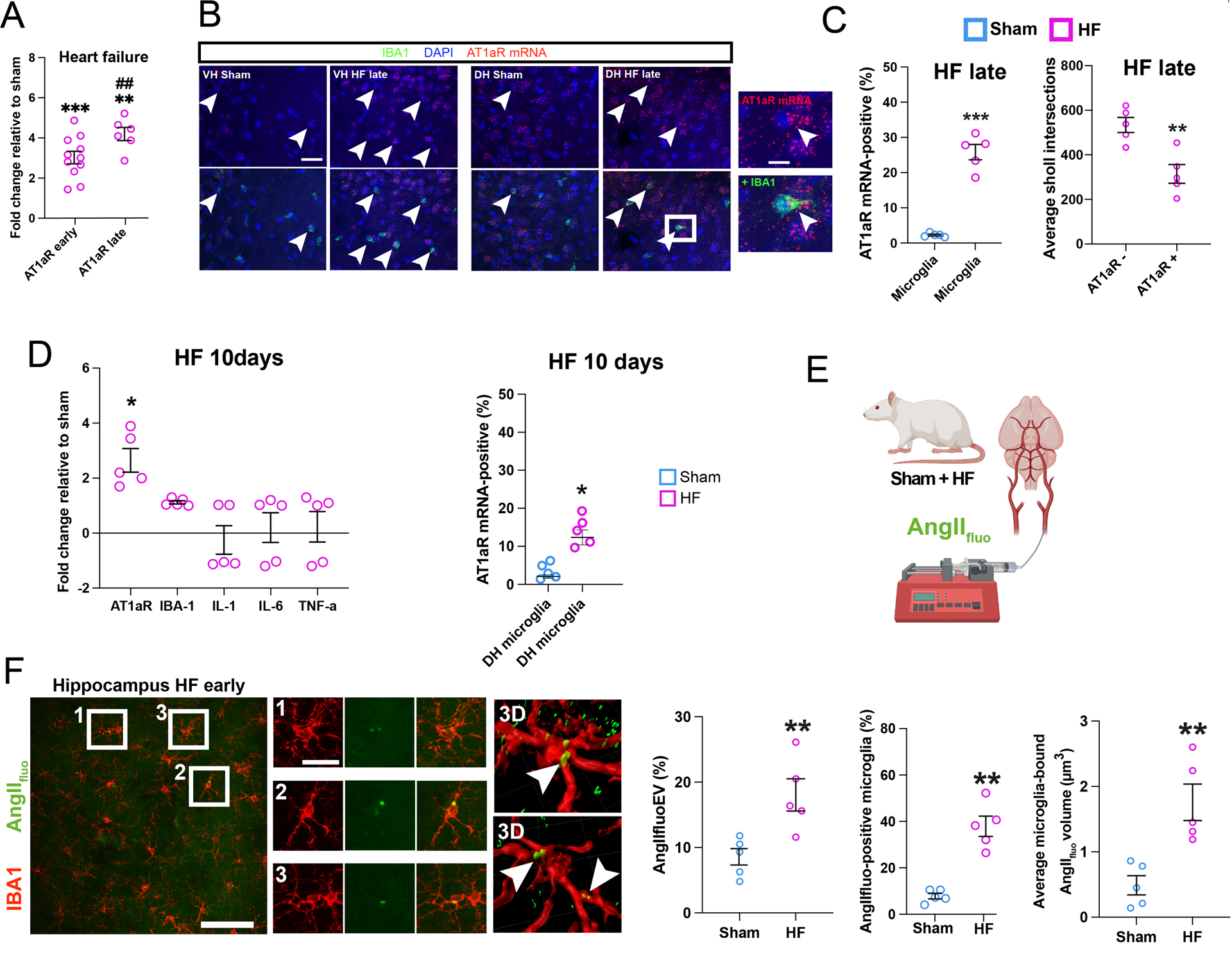
A Changes in AT1a mRNA levels in HF rats relative to Sham at early and late HF stages (n=5/6 rats per group). B Images of AT1aR-positive microglia (white arrowheads) in DH of sham and HF rats. Enlarged inset shows magnification of an exemplary AT1aR-positive microglia. C The number of AT1aR mRNA-positive microglia is increased in HF rats (left). AT1aR-positive microglia are less complex than AT1aR-negative microglia (right) (n=5 rats per group). D Left: Assessment of AT1aR and cytokine mRNA expression via qPCR in HF relative to Sham rats 10 days post HF surgery (n=5/group). Right: Quantification of % microglial showing a positive AT1aR mRNA expression in in sham and HF rats 10 days post-surgery via RNAScope. E Schematic depiction of intra-carotid infusion of AngIIfluo. F Images showing leakage of AngIIflu (green, following i.v. infusion) in the DH of a HF rat. Insets 1–3 (corresponding to squared areas in left panel) show co-localization of AngIIfluo with IBA1-positive microglia. Right panels: Three-dimensional reconstructions showing accumulation of AngIIfluo both in microglia processes and somata (arrowheads). Quantification of hippocampal extravasated (EV) AngIIfluo in sham and HF rats (n=5/group, left panel). Bar graphs showing an overall increase in extravasated hippocampal AngIIfluo (left), increased AngIIfluo positive microglia (middle), and increased AngIIfluo levels within individual microglial cells (right) in the DH of HF compared to sham rats (n=5/group). p<0.05*, p<0.01** and p<0.001***; p<0.01## vs HF early . Scale bars 20μm (b), 5μm (b), 10μm (c), 100μm (e) and 10μm (f).
We finally aimed to investigate whether circulating AngII gained access to the DH in HF rats. Intra-carotid artery infusions of AngIIfluo showed a significant extravasation of AngIIfluo within the DH of HF compared to sham rats (n=5 and 4 respectively, Figure 5E,F). Notably, the leaked AngIIfluo accumulated within IBA1-positive microglia cells, to a much higher degree in HF compared to sham rats (Figure 5F, Figure S5G).
Based on these results, we hypothesized that a leaky BBB could be an underlying cause contributing to the elevated access of circulating AngII to the brain parenchyma in HF rats. To test this hypothesis, and following a procedure we previously established to quantitatively assess altered BBB permeability using intra-carotid infusions of two dyes of different molecular sizes (Rho70kDa and FITC10kDa)27, we observed an increased intraparenchymal leakage of FITC10kDa in the DH, but not the PLC or cortex of HF rats (Figure S5H–J). In line with this finding, we observed a downregulation of mRNA levels of several tight junction proteins associated to normal BBB integrity (e.g., Claudin 1,3,5 and Beta-catenin) in the DH (but not the PLC), of HF rats relative to sham rats, further supporting a compromised BBB integrity in the DH of HF rats, which in turn could contribute to circulating AngII access to this brain region in this condition.
Blockade of AT1Rs improves neuroinflammation and neuronal apoptosis, but not the hypoperfused state in HF rats
To determine whether AT1aRs played a causal role in mediating hippocampal HF-associated pro-inflammatory and hyperperfused states, we assessed the effects of pharmacological blockade of AT1R-mediated signaling on these pathological alterations. We chose the widely clinically-used and orally-delivered AT1R blocker Losartan45–47 (20 mg/kg/day, for a duration of 12 weeks after the myocardial infarction)27. Losartan did not affect cardiac function, body weight gain or total water consumption (Figure S6A-C). Conversely, Losartan reduced mRNA levels for IBA1, GFAP, IL-1, IL-6 and TNF-α, but not AT1aR (Figure 6A). Losartan also ameliorated the hippocampal microglia morphological proinflammatory (Figure 6B,C) and the astrocyte reactive/neurotoxic phenotypes (Figure S6D-G). Importantly, Losartan also diminished cCasp3 hippocampal staining (Figure 6D), as well as the changes in some of the tight junction proteins observed in HF rats (Figure S5K)
Figure 6-. AT1aR blockade improves neuroinflammation and cognitive performance in HF rats.
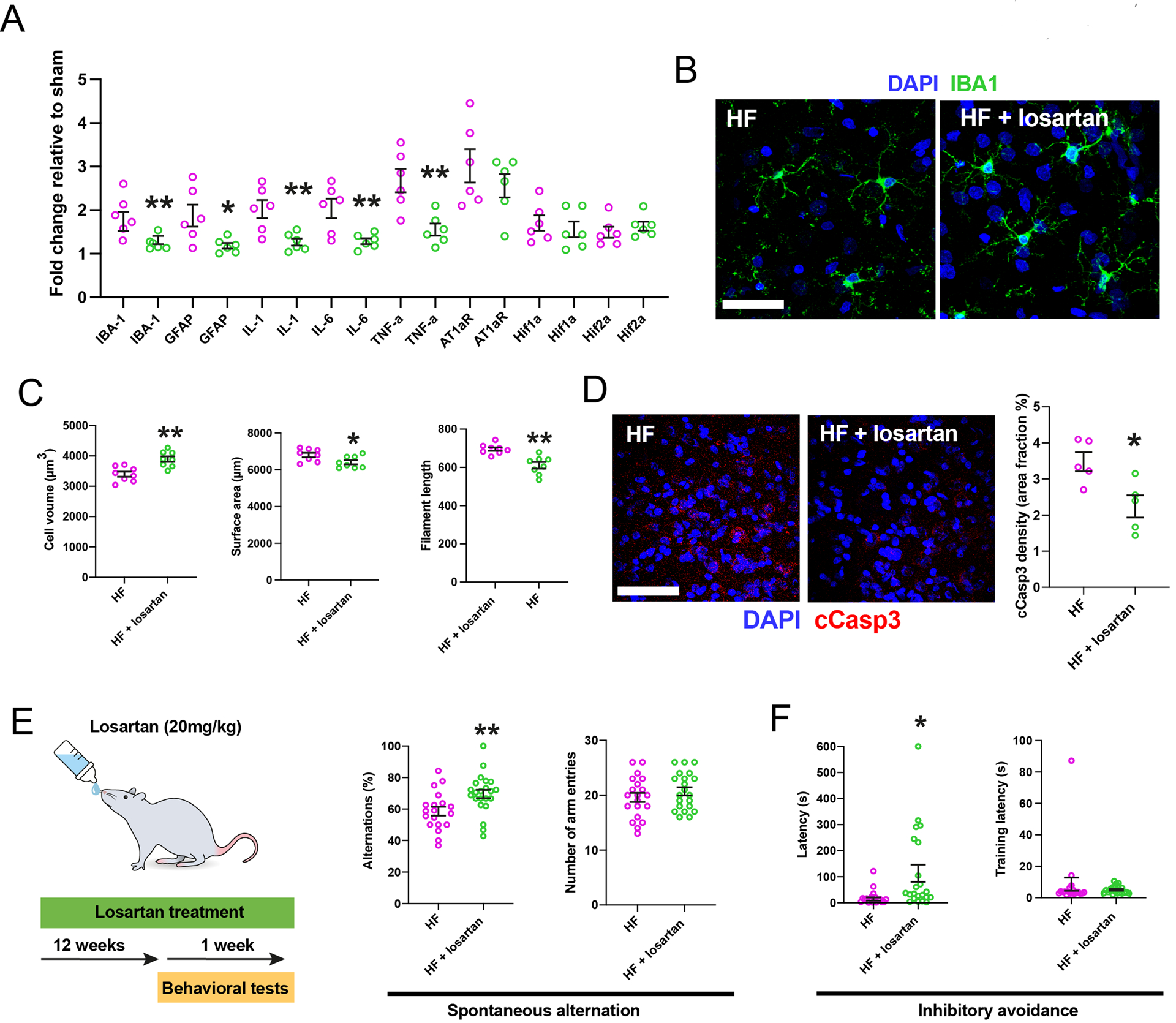
A AT1aR blockade (Losartan) reduced mRNA levels of IBA1, GFAP and various cytokines, but not AT1aR, Hif-1α or Hif-2α in late stage HF rats (n=6 per group). B Images of microglia in HF rats with and without Losartan. C Assessment of microglial morphometry in HF rats with and without Losartan (n=6/group, HF late). D Losartan significantly reduced cCasp3 immunoreactivity in HF rats (n=5/group). E, HF rats subjected to Losartan displayed significantly more spontaneous alternations without changes in the total number of arm entries. F, Losartan-treated HF rats showed higher retention latency without difference in training latencies during inhibitory avoidance testing (n=19 HF, n=22 HF + Losartan). p<0.05*, p<0.01** and p<0.001*** Scale bars 25 μm (b) and 150 μm (d).
Interestingly, Losartan treatment did not change any the increased hypoxia markers (Hif1/2a mRNA) (Figure 6A) or the hypervascularized state (Figure S6H-K) observed in HF rats. Together, these results support a critical contribution of AT1Rs to HF-induced hippocampal neuroinflammation, but not to the hypoxic state.
Blockade of AT1Rs improves cognitive impairment in HF rats
We recently reported that HF rats displayed signs of spatial and emotional memory impairments38 both of which are associated with altered DH function48,49. To determine whether AT1aR activation also contributed to these effects, we repeated the spontaneous alternation (SA) and inhibitory avoidance (IA) tests (for spatial and emotional memory, respectively) in HF and Losartan-treated HF rats (Figure 6E). Losartan-treated rats displayed significantly more percent alternations, compared to non-treated HF rats (Figure 6E), suggestive of improved spatial working memory. Although Losartan affected the sequence of arm entries (i.e., percent alternation), it did not affect the number of arms the rats entered in the maze (Figure 6E), ruling out that changes were due to altered activity levels. Losartan-treated HF rats had significantly higher retention latencies during the IA memory test, suggesting that it also improved emotional memory (Figure 6F). Of note, there were no differences in the training latencies (Figure 6F), further supporting that Losartan did not affect overall activity level. Finally, to rule out a general effect of Losartan per se, we performed a similar set of behavioral experiments in healthy control rats, in which we observed no effects of Losartan on spontaneous alternations or inhibitory avoidance latency (Figure S6L, M),
Together, these findings support that blockade of AT1Rs using a clinically-relevant drug via a clinically- relevant delivery route ameliorated key neuroinflammatory and apoptotic processes during HF, which we propose to contribute in turn to the improvements in cognitive impairments observed in this condition.
Discussion
A growing body of clinical studies supports a high degree of comorbidity between cardiovascular diseases and cognitive decline50–52. In fact, 20–40% of all HF patients develop major depression and elevated anxiety2,50,51,53, along with memory-associated symptoms, which appear later than the cardiovascular and autonomic-related symptoms3. These cognitive and mood disorders have also been observed in experimental animal models of HF, including the rat and mouse left coronary ligation model54–56, a widely accepted model that replicates the cardiac, systemic and neurohumoral pathology observed in patients57. Moreover, we38 and others58 recently showed that HF rats displayed cognitive and emotional deficits, supporting that this model is well-suited to study underlying mechanisms and substrates involved in these deficits. It is important to highlight that the majority of existing studies, both HF patients and animal models, have focused thus far on pathophysiological mechanisms that contribute to cardiac/cardiovascular59,60 and sympathohumoral activation61, whereas those involved in cognitive/mood deficits still remain largely unexplored.
HF induces hippocampal pro-inflammatory microglia, reactive neurotoxic A1 astrocytes and neuronal apoptosis
Using our recently validated morphometric profiler to assess microglial morphology changes in disease states20, we found significant microglial process retraction, somatic swelling and reduction of surface area, all of which support a pro-inflammatory microglial phenotype20,62,63. Importantly, these changes were progressive in time, and dependent on the severity of cardiac compromise in HF. Moreover, the fact that the pro-inflammatory state was brain region-specific supports topographical differences in the susceptibility and resilience of the brain to HF-induced neuroinflammation.
Cytokines such as C1q, TNFα and IL1β play an important role in microglia-to-microglia communication and are pivotal to microglia-mediated maintenance of a pro-inflammatory state24. In addition to finding increased expression of these cytokines in the hippocampus of HF rats, we report a robust and significant negative correlation at the single-cell level between cytokine level expression (e.g. IL1β and TNFα) and microglial morphometric complexity, further supporting the microglia pro-inflammatory status in the hippocampus of HF rats. It is important to note that we observed substantial “diffuse” cytokine mRNA signal that was not confined to microglia, which could reflect cytokine production by other neighboring cells. The facts that we observed changes in mRNA expression of selective cytokines, and that other mRNA signals (e.g., AT1aR) detected using the same approach provided a much more restricted staining, argue against a non-specific mRNA staining provided by the RNAScope approach.
Another core feature of neuroinflammation is the intricate interaction between microglia and astrocytes24. Microglia release C1q, TNFα and IL-1α to activate astrocytes, which in turn become neurotoxic (A1 phenotype) leading to apoptosis in neighboring neurons24. We report here swelling of hippocampal astrocyte processes, as well as an astrocytic shift from a neuroprotective to a neurotoxic state. In line with this, and as previously reported64, we show evidence for HF-induced hippocampal neuronal apoptosis, along with shrinkage of pyramidal cell layers. Moreover, ex vivo electrophysiological recordings showed dampened membrane excitability and blunted input/output function in CA1 pyramidal neurons in HF rats. While the precise mechanisms leading to these changes remain to be determined, our studies support an overall blunted ability of CA1 pyramidal neurons during HF to process incoming inputs to mount a proper action potential firing output. Altogether, these findings support neuronal dysfunction and neurotoxicity in the hippocampus of HF rats.
AngII-AT1R signaling contributes to hippocampal neuroinflammation, apoptosis and cognitive deficits in HF rats
We27,28,31,65 and others6,21,23,29,30 have shown that the pro-inflammatory peptide AngII is linked to neuroinflammation and autonomic changes in HF and hypertension. In addition, a recent study showed that systemic administration of AngII induced neuroinflammation in the mouse hippocampus66. Still, whether endogenously elevated levels of AngII contribute to hippocampal neuroinflammation and a state of hypoperfusion/hypoxia in HF remained unknown. We recently reported that AngII type 1a receptors (AT1aR) are present in hypothalamic microglia27,33,67, while others have shown the same in hippocampal microglia34.
Several pieces of evidence from our study support indeed a critical role for AngII-AT1aRs in HF-induced neuroinflammation. These include i) a gradual and progressive increase of AT1aR expression in the hippocampus of HF rats prior to elevated cytokine mRNA levels; ii) a 6-fold increase in AT1aR-positive microglia in HF rats and iii) virtually all de-ramified microglia in HF rats were AT1aR-positive.
Most importantly, a causal link between AngII-AT1aR and neuroinflammation is more compellingly supported by the fact that treating HF rats with the AT1R antagonist Losartan substantially reversed most neuroinflammatory endpoints including microglial morphometric changes and the elevated cytokine levels. These results are in line with a previous study showing that candesartan (another AT1R blocker) ameliorated brain inflammation following LPS injection68. Notably, AT1R blockade was able to diminish hippocampal apoptosis, supporting a neuroprotective effect of Losartan. This agrees with recent studies showing that targeting the RAS slowed down the progression of clinical Alzheimer’s Disease69–71. We chose and prioritized to use a drug and a route of administration (i.e. orally-administered Losartan) that mimic those used in human patients, and are thus clinically relevant. There are several caveats however that need to be considered in this approach. Firstly, it is controversial whether Losartan can cross the BBB, with studies arguing in favor72 or against it73. Nonetheless, our results showing a compromised BBB integrity in the hippocampus of HF rats (see more below) would further support the ability of Losartan to access the hippocampal parenchyma. Still, we acknowledge that changes observed in the DH following Losartan treatment could be secondary to peripheral actions of this drug. Secondly, our approach is not cell-type or region specific. Thus, despite that our studies support a key contribution of AT1Rs in hippocampal microglial cells, future studies will be needed to more conclusively determine the specific cell-type and topographical location of the receptors mediating Losartan’s neuroprotective effects. Similarly, we cannot completely rule out the contribution of neuronal AT1aRs in HF-induced neuroinflammation. The facts that we observed no changes in neuronal AT1aRs, that we failed to detect AT1aRs in astrocytes, and that AngII-mediated neuronal dysfunction has not yet been reported in the absence of glial cell activation74, would argue against a neuronal/astrocyte critical contribution. On the other hand, it is worth acknowledging that AngII can suppress LTP in hippocampal neurons75 and that other mechanisms, including leaky ryanodine receptors76, could also contribute to cognitive impairment in this HF rat model.
It is worth highlighting that neuroinflammatory markers were evident only in brain regions where an expression/upregulation of AT1aR was observed (e.g., hippocampus/PVN, but not PLC or SSC). Thus, the brain region specificity of the neuroinflammatory response during HF could be dependent, at least in part, on a concomitant upregulation of AT1aRs in those regions.
We recently reported that HF rats were impaired in the spontaneous alternation (SA) and inhibitory avoidance (IA) memory tasks used in the present experiment38. Importantly, we extend those previous results by showing that Losartan-treated HF rats showed significant improvements in these memory tests compared to their non-treated counterparts, as shown by increased percent alternation scores and IA retention latencies that were not associated with changes in activity levels. These findings are consistent with the observation that intracerebroventricular (ICV) infusions of Losartan reverse SA and IA memory deficits produced by ICV infusions of Ang II77. Given that successful performance in both the SA and IA tasks requires an intact hippocampus39,78–82, our findings suggest that HF impairs hippocampal function, and that Losartan improves memory in HF rats via an effect on this brain region. This interpretation is supported by findings showing that systemic administration of Losartan reverses deficits in hippocampal synaptic plasticity75,83 and that systemic doses of Losartan that have no effect in control animals prevented LPS-induced hippocampal-dependent memory impairments, including IA retention deficits84. Together, these findings support exacerbated AngII-AT1aR signaling in the hippocampus as a pivotal mechanism driving neuroinflammation and cognitive impairment after myocardial infarction.
Hippocampal hypoxia is not linked to AT1R-mediated neuroinflammation
In addition to neuroinflammation21, myocardial infarction with reduced ejection fraction has been associated with brain hypoperfusion, a process that could lead to changes in brain vascularization, metabolism and ultimately brain dysfunction14,17,85. Still, to what extent hypoperfusion is mechanistically linked to neuroinflammation, and whether it contributes to cognitive deficits in HF is unknown. Our findings showing increased expression of Hif1α/Hif2α, lower hippocampal tissue pO2 levels, lower basal hippocampal CBF, along with hypervascularization (a compensatory response to tissue hypoxia86) in all brain regions explored, support a rather global hypoxic state in the brain of HF rats. Intriguingly however, while AT1R blockade reversed most neuroinflammatory endpoints, the hypoxia-associated markers remained unaffected. This suggests that AT1aR-mediated neuroinflammation and brain hypoxia are two relatively independent pathophysiological processes during HF, and that the former is a key contributor to cognitive deficits in this condition, at least at the time points of the disease assessed in the present study.
Circulating AngII as a possible source activating hippocampal AT1Rs in HF
While our studies support a contribution of AngII-AT1Rs signaling to hippocampal neuroinflammation and cognitive deficits in HF, the source of AngII driving these effects within the hippocampus is unknown. Because AngII is highly hydrophilic, the general consensus is that circulating AngII does not cross the blood brain barrier (BBB), acting only on circumventricular organs (CVOs), which have an incomplete BBB87. However, we recently showed that chronically elevated levels of AngII during hypertension leads to BBB disruption and its own access to the brain27,28,88. Here, we provide evidence for a leaky hippocampal BBB in HF rats, along with altered expression of mRNA for tight junction proteins that are critical for BBB integrity. Moreover, we show that systemically-infused fluorescently-labeled AngII leaked into the hippocampal parenchyma of rats with HF. This indicates that the chronically elevated levels circulating AngII in HF6,21,59 could constitute a source leading to the AT1R-mediated neuroinflammatory cascade. Notably, we found the leaked AngII in hypertensive27,28,88 and HF rats were mostly bound to microglial cells. While these results indicate that exogenously applied AngII can leak into the DH of HF rats, they do not necessarily prove that the same is true for endogenous circulating AngII. Moreover, alternative sources of AngII, such as local production by astrocytes89,90 could also contribute to the neuroinflammatory cascade.
Together, our results provide additional evidence to support the hippocampal AngII-ATa1R signaling cascade as a critical pathophysiological mechanism contributing to neuroinflammation and cognitive deficits in HF, standing in turn as a novel neuroprotective therapeutic target to combat brain-related deficits in this highly prevalent cardiovascular disease.
Perspective
HF is a prevalent disease that poses debilitating burdens on affected individuals and particularly aging societies in the western world. Although both neuroinflammation and hypoxia have been described as potential mechanisms contributing to memory loss and mood changes in HF patients, their respective role in the pathophysiology of the disease is poorly understood. Previous studies showed that the pro-inflammatory peptide Angiotensin II might be a pivotal contributor to microglial activation, especially in HF. Here we expand on these findings by showing microglial and astrocytic activation, as well as apoptotic events and neuronal deterioration in the hippocampus of HF rats. In addition, we found that the clinically-approved AT1aR antagonist Losartan, ameliorates microglial and astrocytic activation and neuroinflammation-associated changes. Importantly, Losartan treatment rescued HF-induced memory deficits and cognitive impairments. We believe these results are important in the context of the ongoing Prospective Evaluation of Cognitive Function in Heart Failure: Efficacy and Safety of Entresto (LCZ696, Sacubitril/Valsartan) compared to Valsartan on Cognitive Function in Patients with Chronic Heart Failure and Preserved Ejection Fraction (PERSPECTIVE, NCT02884206) which aims at addressing the potential beneficial cognitive effects of this new angiotensin receptor blocker. Thus, we believe our study provides important mechanistic insights regarding the potential outcome of this study.
Supplementary Material
Pathophysiological Novelty and Relevance.
- What is new?
- AngII-driven neuroinflammation in the hippocampus of rats with heart failure leads to microglia activation, neuronal apoptosis and cognitive impairment.
- What is relevant?
- Elucidating the precise cellular and signaling mechanisms contributing to cognitive impairment in heart failure is critical for the development of novel and more efficient therapeutic approaches.
- Clinical/Pathophysiological Implications?
- Blockade of AngII-AT1Rs in rats with heart failure ameliorated hippocampal neuroinflammation and neuronal apoptosis, improving in turn cognitive outcomes.
Acknowledgements
The authors cordially thank Marina Eliava (Central Institute of Mental Health, Mannheim) for help with the scientific illustrations. The authors also thank Atit Patel and Daniel Cox (Neuroscience Institute, Georgia State University) for assistance with the qPCR analysis. We thank Eric G. Krause and Justin Smith (Department of Pharmacodynamics, College of Pharmacy, University of Florida) for helping us to troubleshoot the RNAScope assay.
Sources of Funding
This work was supported by DFG Postdoc Fellowship AL 2466/1–1 to FA; National Heart, Lung, and Blood Institute Grant NIH HL090948 to JES, National Institute of Neurological Disorders and Stroke Grant NIH NS094640 to JES, American Heart Association. Grant ID: 916907 to RKR and funding provided by the Center for Neuroinflammation and Cardiometabolic Diseases (CNCD) at Georgia State University.
List of abbreviations
- AngII
Angiotensin II
- AT1aR
Angiotensin II receptor type 1a
- BBB
Blood brain barrier
- CeA
Central amygdala
- CVOs
Circumventricular organs
- DH
Dorsal hippocampus
- HF
Heart failure
- IA
Inhibitory avoidance
- ICV
Intracerebroventricular
- PLC
Prelimbic cortex
- PVN
Paraventricular nucleus
- RAS
Renin angiotensin system
- SA
Spotaneous alternation
- SSC
Somatosensory cortex
References
- 1.Groenewegen A, Rutten FH, Mosterd A, Hoes AW. Epidemiology of heart failure. Eur J Heart Fail. 2020;22:1342–1356. doi: 10.1002/ejhf.1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Konstam V, Moser DK, De Jong MJ. Depression and anxiety in heart failure. J Card Fail. 2005;11:455–463. doi: 10.1016/j.cardfail.2005.03.006 [DOI] [PubMed] [Google Scholar]
- 3.Hammond CA, Blades NJ, Chaudhry SI, Dodson JA, Longstreth WT Jr., Heckbert SR, Psaty BM, Arnold AM, Dublin S, Sitlani CM, et al. Long-Term Cognitive Decline After Newly Diagnosed Heart Failure: Longitudinal Analysis in the CHS (Cardiovascular Health Study). Circ Heart Fail. 2018;11:e004476. doi: 10.1161/CIRCHEARTFAILURE.117.004476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zucker IH, Schultz HD, Li YF, Wang Y, Wang W, Patel KP. The origin of sympathetic outflow in heart failure: the roles of angiotensin II and nitric oxide. Prog Biophys Mol Biol. 2004;84:217–232. doi: 10.1016/j.pbiomolbio.2003.11.010 [DOI] [PubMed] [Google Scholar]
- 5.Zucker IH, Xiao L, Haack KK. The central renin-angiotensin system and sympathetic nerve activity in chronic heart failure. Clin Sci (Lond). 2014;126:695–706. doi: 10.1042/CS20130294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang BS, Leenen FH. The brain renin-angiotensin-aldosterone system: a major mechanism for sympathetic hyperactivity and left ventricular remodeling and dysfunction after myocardial infarction. Curr Heart Fail Rep. 2009;6:81–88. doi: 10.1007/s11897-009-0013-9 [DOI] [PubMed] [Google Scholar]
- 7.Konstam MA, Neaton JD, Dickstein K, Drexler H, Komajda M, Martinez FA, Riegger GA, Malbecq W, Smith RD, Guptha S, et al. Effects of high-dose versus low-dose Losartan on clinical outcomes in patients with heart failure (HEAAL study): a randomised, double-blind trial. Lancet. 2009;374:1840–1848. doi: 10.1016/S0140-6736(09)61913-9 [DOI] [PubMed] [Google Scholar]
- 8.Shearer F, Lang CC, Struthers AD. Renin-angiotensin-aldosterone system inhibitors in heart failure. Clin Pharmacol Ther. 2013;94:459–467. doi: 10.1038/clpt.2013.135 [DOI] [PubMed] [Google Scholar]
- 9.Crozier I, Ikram H, Awan N, Cleland J, Stephen N, Dickstein K, Frey M, Young J, Klinger G, Makris L, et al. Losartan in heart failure. Hemodynamic effects and tolerability. Losartan Hemodynamic Study Group. Circulation. 1995;91:691–697. doi: 10.1161/01.cir.91.3.691 [DOI] [PubMed] [Google Scholar]
- 10.Bird CM, Burgess N. The hippocampus and memory: insights from spatial processing. Nat Rev Neurosci. 2008;9:182–194. doi: 10.1038/nrn2335 [DOI] [PubMed] [Google Scholar]
- 11.Strange BA, Witter MP, Lein ES, Moser EI. Functional organization of the hippocampal longitudinal axis. Nat Rev Neurosci. 2014;15:655–669. doi: 10.1038/nrn3785 [DOI] [PubMed] [Google Scholar]
- 12.Mota C, Taipa R, das Neves SP, Monteiro-Martins S, Monteiro S, Palha JA, Sousa N, Sousa JC, Cerqueira JJ. Structural and molecular correlates of cognitive aging in the rat. Sci Rep. 2019;9:2005. doi: 10.1038/s41598-019-39645-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allen G, Barnard H, McColl R, Hester AL, Fields JA, Weiner MF, Ringe WK, Lipton AM, Brooker M, McDonald E, et al. Reduced hippocampal functional connectivity in Alzheimer disease. Arch Neurol. 2007;64:1482–1487. doi: 10.1001/archneur.64.10.1482 [DOI] [PubMed] [Google Scholar]
- 14.Tublin JM, Adelstein JM, Del Monte F, Combs CK, Wold LE. Getting to the Heart of Alzheimer Disease. Circ Res. 2019;124:142–149. doi: 10.1161/CIRCRESAHA.118.313563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.David H, Ughetto A, Gaudard P, Plawecki M, Paiyabhroma N, Zub E, Colson P, Richard S, Marchi N, Sicard P. Experimental Myocardial Infarction Elicits Time-Dependent Patterns of Vascular Hypoxia in Peripheral Organs and in the Brain. Front Cardiovasc Med. 2020;7:615507. doi: 10.3389/fcvm.2020.615507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vallabhajosyula S, Dunlay SM, Prasad A, Kashani K, Sakhuja A, Gersh BJ, Jaffe AS, Holmes DR Jr., Barsness GW. Acute Noncardiac Organ Failure in Acute Myocardial Infarction With Cardiogenic Shock. J Am Coll Cardiol. 2019;73:1781–1791. doi: 10.1016/j.jacc.2019.01.053 [DOI] [PubMed] [Google Scholar]
- 17.Kaplan A, Yabluchanskiy A, Ghali R, Altara R, Booz GW, Zouein FA. Cerebral blood flow alteration following acute myocardial infarction in mice. Biosci Rep. 2018;38. doi: 10.1042/BSR20180382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mamalyga ML, Mamalyga LM. Effect of Progressive Heart Failure on Cerebral Hemodynamics and Monoamine Metabolism in CNS. Bull Exp Biol Med. 2017;163:307–312. doi: 10.1007/s10517-017-3791-1 [DOI] [PubMed] [Google Scholar]
- 19.Nishimura T, Hashikawa K, Fukuyama H, Kubota T, Kitamura S, Matsuda H, Hanyu H, Nabatame H, Oku N, Tanabe H, et al. Decreased cerebral blood flow and prognosis of Alzheimer’s disease: a multicenter HMPAO-SPECT study. Ann Nucl Med. 2007;21:15–23. doi: 10.1007/BF03033995 [DOI] [PubMed] [Google Scholar]
- 20.Althammer F, Ferreira-Neto HC, Rubaharan M, Roy RK, Patel AA, Murphy A, Cox DN, Stern JE. Three-dimensional morphometric analysis reveals time-dependent structural changes in microglia and astrocytes in the central amygdala and hypothalamic paraventricular nucleus of heart failure rats. J Neuroinflammation. 2020;17:221. doi: 10.1186/s12974-020-01892-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diaz HS, Toledo C, Andrade DC, Marcus NJ, Rio RD. Neuroinflammation in heart failure: NEW insights for an old disease. J Physiol. 2019. doi: 10.1113/JP278864 [DOI] [PubMed] [Google Scholar]
- 22.Najjar F, Ahmad M, Lagace D, Leenen FHH. Role of Myocardial Infarction-Induced Neuroinflammation for Depression-Like Behavior and Heart Failure in Ovariectomized Female Rats. Neuroscience. 2019;415:201–214. doi: 10.1016/j.neuroscience.2019.07.017 [DOI] [PubMed] [Google Scholar]
- 23.Yu Y, Wei SG, Weiss RM, Felder RB. Angiotensin II Type 1a Receptors in the Subfornical Organ Modulate Neuroinflammation in the Hypothalamic Paraventricular Nucleus in Heart Failure Rats. Neuroscience. 2018;381:46–58. doi: 10.1016/j.neuroscience.2018.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. doi: 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cserep C, Posfai B, Lenart N, Fekete R, Laszlo ZI, Lele Z, Orsolits B, Molnar G, Heindl S, Schwarcz AD, et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science. 2020;367:528–537. doi: 10.1126/science.aax6752 [DOI] [PubMed] [Google Scholar]
- 26.Phillips MI, Kagiyama S. Angiotensin II as a pro-inflammatory mediator. Curr Opin Investig Drugs. 2002;3:569–577. [PubMed] [Google Scholar]
- 27.Biancardi VC, Son SJ, Ahmadi S, Filosa JA, Stern JE. Circulating angiotensin II gains access to the hypothalamus and brain stem during hypertension via breakdown of the blood-brain barrier. Hypertension. 2014;63:572–579. doi: 10.1161/HYPERTENSIONAHA.113.01743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Biancardi VC, Stern JE. Compromised blood-brain barrier permeability: novel mechanism by which circulating angiotensin II signals to sympathoexcitatory centres during hypertension. J Physiol. 2016;594:1591–1600. doi: 10.1113/JP271584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang YM, Ma Y, Elks C, Zheng JP, Yang ZM, Francis J. Cross-talk between cytokines and renin-angiotensin in hypothalamic paraventricular nucleus in heart failure: role of nuclear factor-kappaB. Cardiovasc Res. 2008;79:671–678. doi: 10.1093/cvr/cvn119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pyner S The paraventricular nucleus and heart failure. Exp Physiol. 2014;99:332–339. doi: 10.1113/expphysiol.2013.072678 [DOI] [PubMed] [Google Scholar]
- 31.Stern JE, Son S, Biancardi VC, Zheng H, Sharma N, Patel KP. Astrocytes Contribute to Angiotensin II Stimulation of Hypothalamic Neuronal Activity and Sympathetic Outflow. Hypertension. 2016;68:1483–1493. doi: 10.1161/HYPERTENSIONAHA.116.07747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng H, Li YF, Wang W, Patel KP. Enhanced angiotensin-mediated excitation of renal sympathetic nerve activity within the paraventricular nucleus of anesthetized rats with heart failure. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1364–1374. doi: 10.1152/ajpregu.00149.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Biancardi VC, Stranahan AM, Krause EG, de Kloet AD, Stern JE. Cross talk between AT1 receptors and Toll-like receptor 4 in microglia contributes to angiotensin II-derived ROS production in the hypothalamic paraventricular nucleus. Am J Physiol Heart Circ Physiol. 2016;310:H404–415. doi: 10.1152/ajpheart.00247.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun H, Wu H, Yu X, Zhang G, Zhang R, Zhan S, Wang H, Bu N, Ma X, Li Y. Angiotensin II and its receptor in activated microglia enhanced neuronal loss and cognitive impairment following pilocarpine-induced status epilepticus. Mol Cell Neurosci. 2015;65:58–67. doi: 10.1016/j.mcn.2015.02.014 [DOI] [PubMed] [Google Scholar]
- 35.Biancardi VC, Son SJ, Sonner PM, Zheng H, Patel KP, Stern JE. Contribution of central nervous system endothelial nitric oxide synthase to neurohumoral activation in heart failure rats. Hypertension. 2011;58:454–463. doi: 10.1161/HYPERTENSIONAHA.111.175810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang Y, Benusiglio D, Lefevre A, Hilfiger L, Althammer F, Bludau A, Hagiwara D, Baudon A, Darbon P, Schimmer J, et al. Social touch promotes interfemale communication via activation of parvocellular oxytocin neurons. Nat Neurosci. 2020;23:1125–1137. doi: 10.1038/s41593-020-0674-y [DOI] [PubMed] [Google Scholar]
- 37.Bischofberger J, Engel D, Li L, Geiger JR, Jonas P. Patch-clamp recording from mossy fiber terminals in hippocampal slices. Nat Protoc. 2006;1:2075–2081. doi: 10.1038/nprot.2006.312 [DOI] [PubMed] [Google Scholar]
- 38.Parent MB, Ferreira-Neto HC, Kruemmel AR, Althammer F, Patel AA, Keo S, Whitley KE, Cox DN, Stern JE. Heart Failure Impairs Mood and Memory in Male Rats and down-Regulates the Expression of Numerous Genes Important for Synaptic Plasticity in Related Brain Regions. Behav Brain Res. 2021:113452. doi: 10.1016/j.bbr.2021.113452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lalonde R The neurobiological basis of spontaneous alternation. Neurosci Biobehav Rev. 2002;26:91–104. doi: 10.1016/s0149-7634(01)00041-0 [DOI] [PubMed] [Google Scholar]
- 40.Prinz M, Jung S, Priller J. Microglia Biology: One Century of Evolving Concepts. Cell. 2019;179:292–311. doi: 10.1016/j.cell.2019.08.053 [DOI] [PubMed] [Google Scholar]
- 41.Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, Mulinyawe SB, Bohlen CJ, Adil A, Tucker A, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A. 2016;113:E1738–1746. doi: 10.1073/pnas.1525528113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turner MD, Nedjai B, Hurst T, Pennington DJ. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta. 2014;1843:2563–2582. doi: 10.1016/j.bbamcr.2014.05.014 [DOI] [PubMed] [Google Scholar]
- 43.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, Steinhauser C, Volterra A, Carmignoto G, Agarwal A, et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. 2021;24:312–325. doi: 10.1038/s41593-020-00783-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S, et al. Effects of Losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861–869. doi: 10.1056/NEJMoa011161 [DOI] [PubMed] [Google Scholar]
- 46.Goa KL, Wagstaff AJ. Losartan potassium: a review of its pharmacology, clinical efficacy and tolerability in the management of hypertension. Drugs. 1996;51:820–845. doi: 10.2165/00003495-199651050-00008 [DOI] [PubMed] [Google Scholar]
- 47.Sica DA, Gehr TW, Ghosh S. Clinical pharmacokinetics of Losartan. Clin Pharmacokinet. 2005;44:797–814. doi: 10.2165/00003088-200544080-00003 [DOI] [PubMed] [Google Scholar]
- 48.Fanselow MS, Dong HW. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron. 2010;65:7–19. doi: 10.1016/j.neuron.2009.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Broadbent NJ, Squire LR, Clark RE. Spatial memory, recognition memory, and the hippocampus. Proc Natl Acad Sci U S A. 2004;101:14515–14520. doi: 10.1073/pnas.0406344101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mbakwem A, Aina F, Amadi C. Expert Opinion-Depression in Patients with Heart Failure: Is Enough Being Done? Card Fail Rev. 2016;2:110–112. doi: 10.15420/cfr.2016:21:1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parissis JT, Fountoulaki K, Paraskevaidis I, Kremastinos D. Depression in chronic heart failure: novel pathophysiological mechanisms and therapeutic approaches. Expert Opin Investig Drugs. 2005;14:567–577. doi: 10.1517/13543784.14.5.567 [DOI] [PubMed] [Google Scholar]
- 52.Rustad JK, Stern TA, Hebert KA, Musselman DL. Diagnosis and treatment of depression in patients with congestive heart failure: a review of the literature. Prim Care Companion CNS Disord. 2013;15. doi: 10.4088/PCC.13r01511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rutledge T, Reis VA, Linke SE, Greenberg BH, Mills PJ. Depression in heart failure a meta-analytic review of prevalence, intervention effects, and associations with clinical outcomes. J Am Coll Cardiol. 2006;48:1527–1537. doi: 10.1016/j.jacc.2006.06.055 [DOI] [PubMed] [Google Scholar]
- 54.Frey A, Popp S, Post A, Langer S, Lehmann M, Hofmann U, Siren AL, Hommers L, Schmitt A, Strekalova T, et al. Experimental heart failure causes depression-like behavior together with differential regulation of inflammatory and structural genes in the brain. Front Behav Neurosci. 2014;8:376. doi: 10.3389/fnbeh.2014.00376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Prickaerts J, Raaijmakers W, Blokland A. Effects of myocardial infarction and captopril therapy on anxiety-related behaviors in the rat. Physiol Behav. 1996;60:43–50. [DOI] [PubMed] [Google Scholar]
- 56.Schoemaker RG, Smits JF. Behavioral changes following chronic myocardial infarction in rats. Physiol Behav. 1994;56:585–589. [DOI] [PubMed] [Google Scholar]
- 57.Francis J, Weiss RM, Wei SG, Johnson AK, Felder RB. Progression of heart failure after myocardial infarction in the rat. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1734–1745. doi: 10.1152/ajpregu.2001.281.5.R1734 [DOI] [PubMed] [Google Scholar]
- 58.Toledo C, Lucero C, Andrade DC, Diaz HS, Schwarz KG, Pereyra KV, Arce-Alvarez A, Lopez NA, Martinez M, Inestrosa NC, et al. Cognitive impairment in heart failure is associated with altered Wnt signaling in the hippocampus. Aging (Albany NY). 2019;11:5924–5942. doi: 10.18632/aging.102150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Metra M, Teerlink JR. Heart failure. Lancet. 2017;390:1981–1995. doi: 10.1016/S0140-6736(17)31071-1 [DOI] [PubMed] [Google Scholar]
- 60.Teerlink JR, Pfeffer JM, Pfeffer MA. Progressive ventricular remodeling in response to diffuse isoproterenol-induced myocardial necrosis in rats. Circ Res. 1994;75:105–113. doi: 10.1161/01.res.75.1.105 [DOI] [PubMed] [Google Scholar]
- 61.Potapenko ES, Biancardi VC, Zhou Y, Stern JE. Astrocytes modulate a postsynaptic NMDA-GABAA-receptor crosstalk in hypothalamic neurosecretory neurons. J Neurosci. 2013;33:631–640. doi: 10.1523/JNEUROSCI.3936-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Torres-Platas SG, Comeau S, Rachalski A, Bo GD, Cruceanu C, Turecki G, Giros B, Mechawar N. Morphometric characterization of microglial phenotypes in human cerebral cortex. J Neuroinflammation. 2014;11:12. doi: 10.1186/1742-2094-11-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Davis BM, Salinas-Navarro M, Cordeiro MF, Moons L, De Groef L. Characterizing microglia activation: a spatial statistics approach to maximize information extraction. Sci Rep. 2017;7:1576. doi: 10.1038/s41598-017-01747-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Suzuki H, Sumiyoshi A, Matsumoto Y, Duffy BA, Yoshikawa T, Lythgoe MF, Yanai K, Taki Y, Kawashima R, Shimokawa H. Structural abnormality of the hippocampus associated with depressive symptoms in heart failure rats. Neuroimage. 2015;105:84–92. doi: 10.1016/j.neuroimage.2014.10.040 [DOI] [PubMed] [Google Scholar]
- 65.de Kloet AD, Liu M, Rodriguez V, Krause EG, Sumners C. Role of neurons and glia in the CNS actions of the renin-angiotensin system in cardiovascular control. Am J Physiol Regul Integr Comp Physiol. 2015;309:R444–458. doi: 10.1152/ajpregu.00078.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Iulita MF, Vallerand D, Beauvillier M, Haupert N, C AU, Gagne A, Vernoux N, Duchemin S, Boily M, Tremblay ME, et al. Differential effect of angiotensin II and blood pressure on hippocampal inflammation in mice. J Neuroinflammation. 2018;15:62. doi: 10.1186/s12974-018-1090-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Biancardi VC, Stern J. Angiotensin II contributes to microglial cell activation in the PVN of hypertensive rats. The FASEB Journal. 2013;27:1 supplement, 699.618–699.618 [Google Scholar]
- 68.Benicky J, Sanchez-Lemus E, Honda M, Pang T, Orecna M, Wang J, Leng Y, Chuang DM, Saavedra JM. Angiotensin II AT1 receptor blockade ameliorates brain inflammation. Neuropsychopharmacology. 2011;36:857–870. doi: 10.1038/npp.2010.225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wharton W, Goldstein FC, Tansey MG, Brown AL, Tharwani SD, Verble DD, Cintron A, Kehoe PG. Rationale and Design of the Mechanistic Potential of Antihypertensives in Preclinical Alzheimer’s (HEART) Trial. J Alzheimers Dis. 2018;61:815–824. doi: 10.3233/JAD-161198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Davies NM, Kehoe PG, Ben-Shlomo Y, Martin RM. Associations of anti-hypertensive treatments with Alzheimer’s disease, vascular dementia, and other dementias. J Alzheimers Dis. 2011;26:699–708. doi: 10.3233/JAD-2011-110347 [DOI] [PubMed] [Google Scholar]
- 71.Wharton W, Stein JH, Korcarz C, Sachs J, Olson SR, Zetterberg H, Dowling M, Ye S, Gleason CE, Underbakke G, et al. The effects of ramipril in individuals at risk for Alzheimer’s disease: results of a pilot clinical trial. J Alzheimers Dis. 2012;32:147–156. doi: 10.3233/JAD-2012-120763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li Z, Bains JS, Ferguson AV. Functional evidence that the angiotensin antagonist Losartan crosses the blood-brain barrier in the rat. Brain Res Bull. 1993;30:33–39. doi: 10.1016/0361-9230(93)90036-b [DOI] [PubMed] [Google Scholar]
- 73.Bui JD, Kimura B, Phillips MI. Losartan potassium, a nonpeptide antagonist of angiotensin II, chronically administered p.o. does not readily cross the blood-brain barrier. Eur J Pharmacol. 1992;219:147–151. doi: 10.1016/0014-2999(92)90593-s [DOI] [PubMed] [Google Scholar]
- 74.Min LJ, Mogi M, Iwanami J, Sakata A, Jing F, Tsukuda K, Ohshima K, Horiuchi M. Angiotensin II and aldosterone-induced neuronal damage in neurons through an astrocyte-dependent mechanism. Hypertens Res. 2011;34:773–778. doi: 10.1038/hr.2011.38 [DOI] [PubMed] [Google Scholar]
- 75.Wayner MJ, Armstrong DL, Polan-Curtain JL, Denny JB. Role of angiotensin II and AT1 receptors in hippocampal LTP. Pharmacol Biochem Behav. 1993;45:455–464. doi: 10.1016/0091-3057(93)90265-u [DOI] [PubMed] [Google Scholar]
- 76.Marks AR. Targeting ryanodine receptors to treat human diseases. J Clin Invest. 2023;133. doi: 10.1172/JCI162891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bonini JS, Bevilaqua LR, Zinn CG, Kerr DS, Medina JH, Izquierdo I, Cammarota M. Angiotensin II disrupts inhibitory avoidance memory retrieval. Horm Behav. 2006;50:308–313. doi: 10.1016/j.yhbeh.2006.03.016 [DOI] [PubMed] [Google Scholar]
- 78.Kraeuter AK, Guest PC, Sarnyai Z. The Y-Maze for Assessment of Spatial Working and Reference Memory in Mice. Methods Mol Biol. 2019;1916:105–111. doi: 10.1007/978-1-4939-8994-2_10 [DOI] [PubMed] [Google Scholar]
- 79.Phillmore LS, Klein RM. The puzzle of spontaneous alternation and inhibition of return: How they might fit together. Hippocampus. 2019;29:762–770. doi: 10.1002/hipo.23102 [DOI] [PubMed] [Google Scholar]
- 80.Izquierdo I, Furini CR, Myskiw JC. Fear Memory. Physiol Rev. 2016;96:695–750. doi: 10.1152/physrev.00018.2015 [DOI] [PubMed] [Google Scholar]
- 81.Giovannini MG, Lana D, Pepeu G. The integrated role of ACh, ERK and mTOR in the mechanisms of hippocampal inhibitory avoidance memory. Neurobiol Learn Mem. 2015;119:18–33. doi: 10.1016/j.nlm.2014.12.014 [DOI] [PubMed] [Google Scholar]
- 82.Izquierdo I, Medina JH. Role of the amygdala, hippocampus and entorhinal cortex in memory consolidation and expression. Braz J Med Biol Res. 1993;26:573–589. [PubMed] [Google Scholar]
- 83.Hosseini M, Salmani H, Baghcheghi Y. Losartan improved hippocampal long-term potentiation impairment induced by repeated LPS injection in rats. Physiol Rep. 2021;9:e14874. doi: 10.14814/phy2.14874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Salmani H, Hosseini M, Baghcheghi Y, Moradi-Marjaneh R, Mokhtari-Zaer A. Losartan modulates brain inflammation and improves mood disorders and memory impairment induced by innate immune activation: The role of PPAR-gamma activation. Cytokine. 2020;125:154860. doi: 10.1016/j.cyto.2019.154860 [DOI] [PubMed] [Google Scholar]
- 85.Hauck EF, Apostel S, Hoffmann JF, Heimann A, Kempski O. Capillary flow and diameter changes during reperfusion after global cerebral ischemia studied by intravital video microscopy. J Cereb Blood Flow Metab. 2004;24:383–391. doi: 10.1097/00004647-200404000-00003 [DOI] [PubMed] [Google Scholar]
- 86.Yang Y, Torbey MT. Angiogenesis and Blood-Brain Barrier Permeability in Vascular Remodeling after Stroke. Curr Neuropharmacol. 2020;18:1250–1265. doi: 10.2174/1570159X18666200720173316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Daneman R The blood-brain barrier in health and disease. Ann Neurol. 2012;72:648–672. doi: 10.1002/ana.23648 [DOI] [PubMed] [Google Scholar]
- 88.Mowry FE, Peaden SC, Stern JE, Biancardi VC. TLR4 and AT1R mediate blood-brain barrier disruption, neuroinflammation, and autonomic dysfunction in spontaneously hypertensive rats. Pharmacol Res. 2021:105877. doi: 10.1016/j.phrs.2021.105877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Thomas WG, Sernia C. Immunocytochemical localization of angiotensinogen in the rat brain. Neuroscience. 1988;25:319–341. doi: 10.1016/0306-4522(88)90029-2 [DOI] [PubMed] [Google Scholar]
- 90.Morimoto S, Cassell MD, Beltz TG, Johnson AK, Davisson RL, Sigmund CD. Elevated blood pressure in transgenic mice with brain-specific expression of human angiotensinogen driven by the glial fibrillary acidic protein promoter. Circ Res. 2001;89:365–372. doi: 10.1161/hh1601.094988 [DOI] [PubMed] [Google Scholar]
- 91.Haruwaka K, Ikegami A, Tachibana Y, Ohno N, Konishi H, Hashimoto A, Matsumoto M, Kato D, Ono R, Kiyama H, et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat Commun. 2019;10:5816. doi: 10.1038/s41467-019-13812-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Biancardi VC, Son SJ, Sonner PM, Zheng H, Patel KP, Stern JE. Contribution of central nervous system endothelial nitric oxide synthase to neurohumoral activation in heart failure rats. Hypertension. 2011;58:454–463. doi: 10.1161/HYPERTENSIONAHA.111.175810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ferreira-Neto HC, Stern JE. Functional coupling between NMDA receptors and SK channels in rat hypothalamic magnocellular neurons: altered mechanisms during heart failure. J Physiol. 2019. doi: 10.1113/JP278910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ferreira-Neto HC, Biancardi VC, Stern JE. A reduction in SK channels contributes to increased activity of hypothalamic magnocellular neurons during heart failure. J Physiol. 2017;595:6429–6442. doi: 10.1113/JP274730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stern JE, Potapenko ES. Enhanced NMDA receptor-mediated intracellular calcium signaling in magnocellular neurosecretory neurons in heart failure rats. Am J Physiol Regul Integr Comp Physiol. 2013;305:R414–422. doi: 10.1152/ajpregu.00160.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Garrott K, Dyavanapalli J, Cauley E, Dwyer MK, Kuzmiak-Glancy S, Wang X, Mendelowitz D, Kay MW. Chronic activation of hypothalamic oxytocin neurons improves cardiac function during left ventricular hypertrophy-induced heart failure. Cardiovasc Res. 2017;113:1318–1328. doi: 10.1093/cvr/cvx084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Potapenko ES, Biancardi VC, Florschutz RM, Ryu PD, Stern JE. Inhibitory-excitatory synaptic balance is shifted toward increased excitation in magnocellular neurosecretory cells of heart failure rats. J Neurophysiol. 2011;106:1545–1557. doi: 10.1152/jn.00218.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Roy RK, Althammer F, Seymour AJ, Du W, Biancardi VC, Hamm JP, Filosa JA, Brown CH, Stern JE. Inverse neurovascular coupling contributes to positive feedback excitation of vasopressin neurons during a systemic homeostatic challenge. Cell Rep. 2021;37:109925. doi: 10.1016/j.celrep.2021.109925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.McGaugh JL. The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu Rev Neurosci. 2004;27:1–28. doi: 10.1146/annurev.neuro.27.070203.144157 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.