

Abstract
Recording transcriptional histories of a cell would enable deeper understanding of cellular developmental trajectories and responses to external perturbations. Here, we describe an engineered protein fiber that incorporates diverse fluorescent marks during its growth to store a ticker tape-like history. An embedded HaloTag reporter incorporates user-supplied dyes, leading to colored stripes that map the growth of each individual fiber to wall-clock time. A co-expressed eGFP tag driven by a promoter of interest records a history of transcriptional activation. High-resolution multispectral imaging on fixed samples reads the cellular histories, and interpolation of eGFP marks relative to HaloTag timestamps provides accurate absolute timing. We demonstrate recordings of doxycycline-induced transcription in HEK cells and cFos activation in cultured neurons, with a single-cell absolute accuracy of 30-40 min over a 12-hour recording. The protein-based ticker tape design we present here could be generalized to achieve massively parallel single-cell recordings of diverse physiological modalities.
A longstanding goal in biology is to map the dynamics of gene expression throughout a tissue or organism. Such maps could reveal mechanisms of spatial and temporal patterning, e.g. in brain activity, embryonic development, or disease progression. Fluorescent protein-based markers are a powerful tool for mapping gene expression. However, optical imaging of fluorescent reporters faces a tradeoff between temporal and spatial information. Imaging in fixed tissue can report organ-wide patterns of gene expression, but typically at only one1,2 or, at most, two3 timepoints. Time-lapse in vivo microscopy can report longitudinal gene expression dynamics,4 but only in a small optically accessible region, limited by light-scatter and optical instrumentation, and in vivo imaging typically requires surgery and/or immobilization of the animal. Theoretical analyses have explored the possibility of encoding organ-wide dynamics in DNA or RNA sequences,5,6 but despite progress7 such ideas have not yet been realized. Tools to record the dynamics of large numbers of cells, without constraints from in vivo imaging, could transform our ability to study ensemble dynamics in the nervous system and in other tissues.
Natural histories are written in the patterns of tooth enamel, the structures of pearls, and the thickness of tree rings. The annual bands in tree rings mark the passage of time in a manner that is independent of variations in growth rate between trees or over years. These bands permit one to identify the timing of other events (e.g. a forest fire) with absolute chronological accuracy. Inspired by these natural phenomena, we sought to encode cellular histories in protein microcrystals within individual cells. Protein assemblies can last for months or years and offer a wide array of functionalities which could serve as the basis for recording schemes.
Here we develop a protein-based recording scheme that consists of three elements (Fig. 1a,b). First, a protein scaffold which grows over time and which can incorporate fluorescent marks. Second, a means to impart fiducial timestamps to relate scaffold growth to the timing of events in the outside world. By marking known timepoints on the fiber, one can correct for inevitable variations in fiber growth rate over time and between cells. Third, a fluorescent reporter of transcriptional activity which can be stably incorporated into the scaffold. By measuring the position of the activity-driven marks relative to the fiducial timestamps, one can infer the timing of the transcriptional events with a precision substantially better than the timing between the timestamps and insensitive to cell-to-cell variations in growth rate. This procedure could enable recording of transcriptional activity with high absolute timing accuracy on a cell-by-cell basis.
Figure 1. iPAK4 forms intracellular protein fibers.
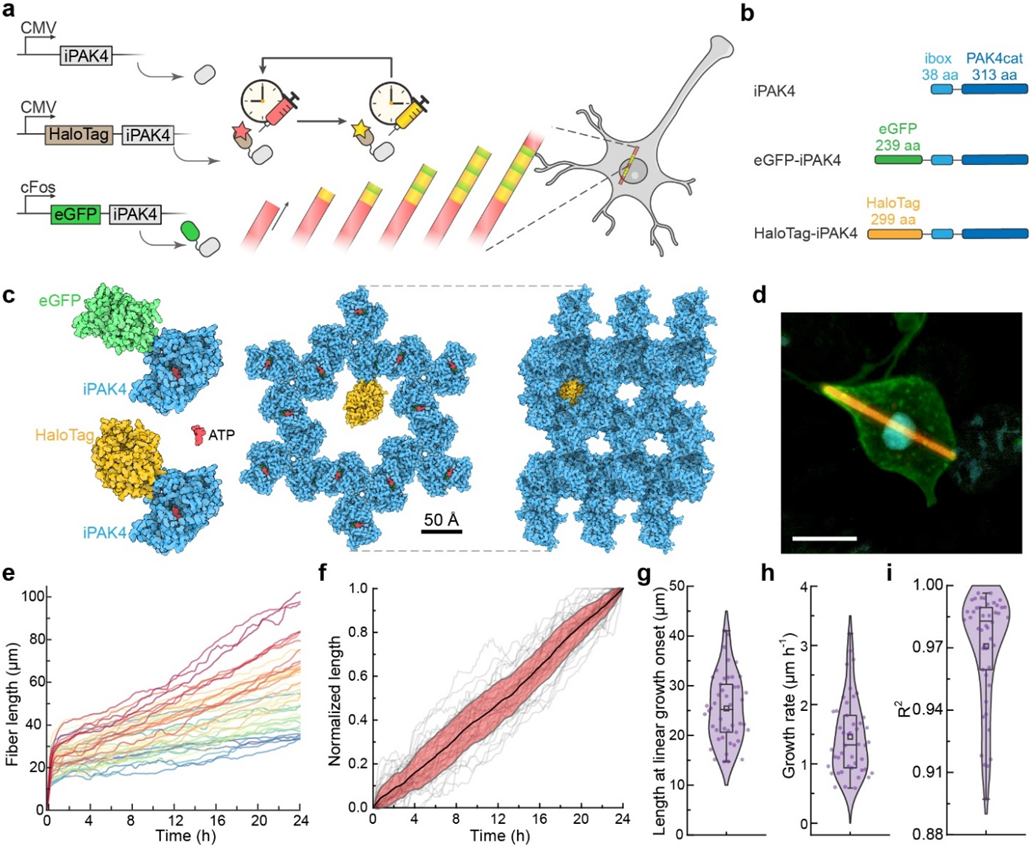
a, Scheme for intracellular recording of cFos activity with fiducial timestamps. iPAK4 forms the fiber scaffold. HaloTag-iPAK4 incorporates HT dyes, permitting labeling of the fiber with fiducial timestamps. Neural activation drives expression of cFos::eGFP-iPAK4, introducing green bands into the fiber. b, Composition of the protein constructs used to label intracellular protein fibers. c, The structures of tagged iPAK4 monomers and the crystal structure with hexagonal pores (from PDB:4XBR) were modeled using Protein Imager software.39 d, Image of a HEK cell expressing CMV::iPAK4 (95%) and CMV::HT-iPAK4 (5%). The fiber was stained with JFX608, the membrane was labeled by expressing GPI-eGFP, and the nucleus was labeled with DAPI. Scale bar 10 μm. e. Growth profiles of iPAK4 fibers in HEK cells (n = 46 fibers). f, Population average (black) and standard deviation (red) of the linear-phase fiber growth (grey lines). Here each fiber’s linear-phase growth profile was mapped to the interval [0, 1]. g-i, Statistics of fiber growth, showing g, fiber length at the transition from initial nucleation to linear growth, h, linear-phase growth rate, and i, R2 of the fit of the linear-phase growth to a straight line. In e-i, fibers were randomly selected from Movie S1. Box bounds: 25th and 75th percentile; whiskers: minimum and maximum; squares: mean; center lines: median. All data points are displayed.
For the protein scaffold, our selection criteria were that it should express in mammalian cells, assemble into a growing structure from a single polypeptide, have a known crystal structure, be unlikely to interfere with cellular physiology, and accommodate decoration with a fluorescent tag without disrupting the structure of the assembly. We considered many possibilities, including endogenous microtubules, bacterial R-bodies,8 plant forisomes,9 amyloid fibrils,10 prions,11 filamentous viruses,12 crystals of the fluorescent protein XpA,13 engineered fiber-forming peptides,14 and other proteins that spontaneously crystallize in cells.15
A fusion of the catalytic domain of the Pak4 kinase and the 38 amino acid iBox domain of its inhibitor Inka1 (hereafter called iPAK4) largely satisfied the selection criteria. This construct has been shown to stably assemble in cells into rod-shaped crystals.16 Remarkably, the crystal structure has a hexagonal array of internal pores large enough to accommodate eGFP or a HaloTag, suggesting the possibility of linear encoding of information via patterned fluorescence (Fig. 1c).
We sought to use a fusion of the HaloTag (HT) to iPAK4 to provide fiducial timestamps. We reasoned that washes with different colored HT-ligand dyes could create color boundaries whose positions would correspond to known times. Even though HT dye washout in vivo occurs over hours, HT dye injection and labeling have fast onset (< 10 minutes in vivo),17 permitting precise demarcation of timestamps by the location of a color transition. A broad palette of bright, photostable, and brain-penetrant Janelia Fluor HT-ligand dyes are available, permitting diverse spectral encodings of fiducial timestamps.18-20
The activity reporter should store a stable mark of cellular activity or physiology upon incorporation into the growing fiber. Here we focus on markers of transcriptional activation, though other modalities are conceivable. First, we mapped the kinetics of activation of a synthetic drug-inducible promoter, the Tet-ON system. Then we mapped the dynamics of an activity-responsive immediate early gene (IEG) in cultured neurons. IEG reporters are a powerful tool for identifying the brain regions and neuronal subtypes activated in a particular context.21 For example, the IEG cFos has been used to map neurons activated during feeding,22 sleep,23 parenting,24 aggression,25 and memory encoding.26 IEG activation is a good target for a ticker-tape recording because the relevant dynamics are often broadly distributed throughout the brain.26 Motivated by potential applications toward brain-wide activity mapping, we used the cFos promoter to drive expression of eGFP-iPAK4 and thereby record time-series of cFos activity.
Results
iPAK4 fibers grow linearly in HEK cells.
In HEK cells co-transfected with CMV::iPAK4 (95%) and CMV::HT-iPAK4 (5%), fibers began to grow 14-20 h after transfection. Incubation with a HT-ligand dye made the fibers brightly fluorescent (Fig. 1d, S1, Methods). When the fibers grew longer than the cell diameter, the membrane deformed, forming a sheath around the fiber (Fig. S1).
To assess the suitability of iPAK4 fibers as a recording medium, we performed a detailed characterization of their nucleation and growth. We co-expressed CMV::iPAK4 (90%), CMV::eGFP-iPAK4 (5%) and CMV::HT-iPAK4 (5%) in HEK cells using lentiviral transduction and added JF669 to the medium to label the HT. We recorded time-lapse video microscopy over 43 h, starting 20 h after transfection (Methods, Movie S1), and tracked the growth of individual fibers (Methods, Movie S2). Initially, green (eGFP) and red (HT-JF669) fluorescence accumulated in the cytoplasm. Upon nucleation, the fiber growth exhibited two clearly distinct phases: (1) initially, fibers grew quickly (~0.5 μm/min), while the cytoplasmic fluorescence dropped. This phase typically lasted ~1 h. (2) The fibers then transitioned to slower linear growth (Fig. 1e). While there was substantial cell-to-cell variability in the rate of linear growth, the population-average growth rate did not change over 24 h after nucleation (Fig. 1f). A simple mass-action kinetic model predicted the two-phase growth profiles and quantitatively reproduced the observed growth profiles (Supplementary Calculation and Fig. S2).
We quantified the distribution of fiber growth parameters across the population. The length of fibers at the end of the nucleation phase was 26 ± 7 μm (mean ± s.d., n = 46 fibers, Fig. 1g). During the linear growth phase, the growth rate was 1.46 ± 0.64 μm/h (mean ± s.d., Fig. 1h). Within each cell, the fiber growth rate remained remarkably constant during the linear growth phase. During 24 h, the fits of the growth profiles to straight lines yielded a population average R2 = 0.97 ± 0.02 (mean ± s.d., Fig. 1i). Of the cells that had any fibers, most contained only one (of 333 fiber-containing cells, 288 (86%) contained only one fiber). We ascribe the rarity of multiple fibers per cell to the fact that a single growing fiber maintained the soluble iPAK4 concentration below the nucleation threshold. Anecdotally, multi-fiber cells seemed to arise from cells where a fiber broke, leading to two nuclei.
Within the population, we observed cell-to-cell variations in the intensities of the eGFP and HT fluorescence signals, presumably due to variations in gene dosage. We did not observe any correlation between the growth rates and these fluorescence levels, indicating that at the low mole-fractions of fluorescent tags used here (approximately 5% for each fluorescent tag), the fluorescent tags did not affect the growth rate (Fig. S3).
We then asked if the iPAK4 fibers affected cell survival, as the fibers often grew longer than the cell diameters (Fig. S1). In a live-dead assay, we did not observe a difference in survival between HEK cells expressing iPAK4 fibers and control untransfected HEK cells (iPAK4(+): 0.267 ± 0.139% dead cells, n = 24 fields of view comprising 89,341 cells; iPAK4(−): 0.262 ± 0.107% dead cells, n = 24 fields of view comprising 94,202 cells. P = 0.90, student’s t-test. Fig. S4). Though the stiff fibers deforming the cell membranes looked unusual, simple geometrical estimates showed that the fibers perturbed HEK cell surface area by < 15% and volume by < 2%; and the predicted fractional perturbations for neurons were even smaller (Supplementary Calculation 2).
We then tested whether cells containing iPAK4 fibers could divide. In taking time-lapse movies of HEK cells labeled with Hoechst 33342 nuclear stain, we observed occasional cell divisions. The formation of mitotic chromosomes and the process of cell division appeared qualitatively similar in cells with iPAK4 fibers, in cells expressing iPAK4 but without fibers, and in non-expressing cells (Fig. S5), though the number of mitotic events captured (n = 8 divisions in cells containing fibers) was too small for statistical analysis. Possible perturbations to cell division should be assessed separately in any cell type where iPAK4 fibers are used. We note that for applications in neurons cell division is not a concern. Together, these results established that iPAK4 fiber growth during the linear growth phase formed a suitable substrate for cellular recordings, provided one could correct for cell-to-cell variations in growth rate.
HaloTag time-stamps enable accurate timing in iPAK4 fibers.
To test whether fibers could encode HT dye timestamps, we successively washed HEK cells growing HT-iPAK4 fibers with different colors of HT-ligand dyes at Δt = 2 h intervals in the sequence JF503, JF669, JFX608, JF503, JF669 (Fig. 2a; see Fig. S6 for dye structures, photophysical properties, and synthesis of JFX608). We then fixed the cells and imaged them with spectrally resolved confocal microscopy (Methods). We observed clear progression of colored bands matching the sequence of dye additions. The bands followed mirror-image patterns on opposite sides of the fibers, indicating that the fibers grew from both ends (Fig. 2b, S7).
Figure 2. iPAK4 fibers can be sequentially labeled with different dyes.
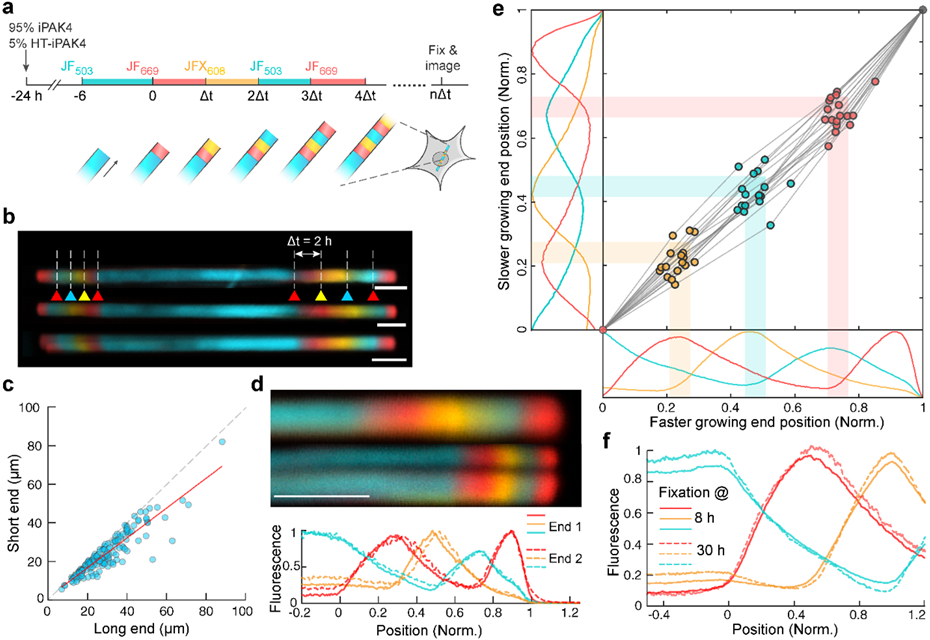
a, Scheme for multi-color labeling of intracellular iPAK4 fibers. b, Images of iPAK4 fibers labeled with four dye transitions on each end. Triangles indicate times of dye addition. Blue: JF503, Red: JF669, Yellow: JFX608. Scale bars 5 μm. c, Comparison of the growth rate on the two ends after one dye switch. The long end and short end correspond to the faster and slower growing end, respectively (N = 153 fibers). The growth rate on the slower-growing end was 0.77 ± 0.05-fold lower than on the faster-growing end (95% C.I.). d, Comparison of two ends of a single fiber. Top: images of the two ends. In this case, the fiber split at one of its ends, an occurrence in approximately 10% of fibers. Bottom: quantification of the fluorescence traces in the two ends, with position normalized to the locations of the first JF669 addition and the end of the fiber. Scale bar 5 μm. e, Comparison of dye transition points on the faster and slower-growing fiber ends. The scatter plot shows the positions of the JFX608, JF503 and JF669 transitions normalized relative to the first JF669 addition (at position 0) and the end of the fiber (at position 1). The plots show the mean fluorescence profiles of N = 17 fibers. Here the number of fibers was set by the requirement for in-focus, unobstructed views of both ends of the fibers. f, Mean fluorescence profiles of fibers exposed to the same sequence of dyes and then grown for different amounts of time (N = 22 fixed at 8 h, 12 fixed at 30 h). The overlap of these profiles indicates negligible monomer exchange over 22 h.
To test the limits of how fast we could encode dye transitions, we made fibers with seven dye switches at Δt = 1 h. Although peaks were still visible at this short interval, their amplitude was suppressed relative to the parts of the fiber with no dye switches, indicating incomplete transitions in dye labeling (Fig. S7e). Analysis of the dye profiles for more widely spaced dye transitions indicated a half-life of soluble HT-iPAK4 of 4.5 h (Fig. S8a), explaining the loss of signal at faster dye switches. These results indicated that fiducial timestamps should be separated by at least Δt = 2 h.
Dye additions appeared as upward-going kinks in the fluorescence profiles (Fig. S8a), which we located as peaks in the second derivative of fluorescence vs. position (Methods). By dividing the transition zone widths (full-width at half-maximum of the peak in the second derivative) by the fiber-specific growth rates, we calculated a mean transition zone duration of 7.5 min (Fig. S8a).
To determine the influence of HT dye labeling kinetics on the widths of these transitions, we measured the labeling of intracellular HT receptors with different HT dyes (Methods). These measurements probed the combined effects of membrane permeation and the HT reaction. The effective time constants at 1 μM dye were: 43 min (JF503), 3.5 min (JF525), 2.0 min (JF552), 12 min (JFX608), and 17 min (JF669) (Fig. S8). The precision of localizing a dye transition in a fiber is not equal to the labeling time constant, but rather the precision with which one can identify the onset of the labeling reaction. For all the JF dyes, this time was much less than 10 min. Together, these results established that HT dye transitions provided a means to timestamp iPAK4 fiber growth with a local precision (i.e. around the time of a time stamp) of ~10 min or better.
Due to the P63 symmetry of the iPAK4 crystal structure,16 the two ends are not chemically equivalent and need not have the same growth rate. However, the asymmetry in growth rate was modest: The growth rate on the slower-growing end was 0.77 ± 0.05 (95% CI) of the growth rate on the faster-growing end (Fig. 2c). We then compared the fluorescence profiles on the two ends. To account for the difference in growth rate between the fiber ends, we mapped the first dye addition (JF669 at t = 0) to x = 0, and the end of the fiber (fixation at t = 8 h) to x = 1. After normalizing the spatial scales, the fast and slow-growing fiber ends showed very similar profiles (Fig. 2d).
To test whether both fiber ends could be useful for recording fiducial timestamps, we analyzed the locations of the dye transitions on both ends of N = 17 fibers. For both the faster and slower-growing ends, the normalized locations of dye transitions at t = 2, 4, and 6 h mapped linearly onto position between the first dye addition (t = 0) and the end of the fiber (t = 8 h, Fig. 2e). The standard deviations in the inferred timing (averaging over the three transitions) were 18.3 min on the fast end and 24.8 min on the slow end. Together, these results established that both ends of the fibers could record fiducial timestamps with an absolute accuracy of better than 25 min over an 8 h baseline.
If the residual errors in timing were driven by a factor shared by the two fiber ends (e.g. variations in iPAK4 expression level), then these errors would lie primarily along the diagonal line Position 1 = Position 2 in Fig. 2e. We calculated the cross-correlation in the timing errors between the two fiber ends, and averaged over all transitions (t = 2, 4, and 6 h) and all fibers. This cross-correlation was only 0.32, implying that dynamic fluctuations in growth rate were primarily driven by factors local to each end. In principle, timing precision could be improved by averaging measurements on the two fiber ends, but we found that often the image quality was better on one end than the other because of differences in the focal plane or the presence of out-of-focus fibers. Consequently, we typically analyzed only one fiber end per cell, and we did not attempt to distinguish between the faster- and slower-growing ends.
A protein ticker tape should stably store its information for extended times. Since the iPAK4 fibers were held together by non-covalent interactions, we tested whether monomer exchange blurred the HT dye boundaries over time. Two dishes were exposed to the same sequence of dye switches at intervals of Δt = 2 h. One dish was fixed at t = 8 h, and the other was returned to the incubator and fixed a day later at t = 30 h. We then compared the fluorescence profiles for the section of the fibers that grew concurrently (from t = 0 to 8 h). The mean profiles were nearly indistinguishable between the early dish (N = 22 fibers) and the late dish (N = 12 fibers, Fig. 2f). Thus, monomer exchange was negligible over one day.
We next studied the precision with which the timing of cellular events could be identified. If each fiber end grew at a constant rate, the timing of cellular events could be linearly interpolated between timestamps with a precision far greater than the interval between the timestamps. However, fluctuations in the growth rate might degrade the precision. To determine the precision empirically, we incubated fibers in JF525, and then switched to JF669 at t = 0. In different dishes we doped in the dye JFX608 at t = 2, 4, 6, 7, 8, or 9 h to simulate the onset of a cellular event. Finally, we switched back to JF525 at t = 10 h, grew the fibers for another 10 h, and fixed the dishes at t = 20 h (Fig. 3a). Low-magnification images showed clear stripes in most fibers, corresponding to the time stamps and the addition of JFX608 (Fig. 3b).
Figure 3. iPAK4 fibers report timing of intracellular events.
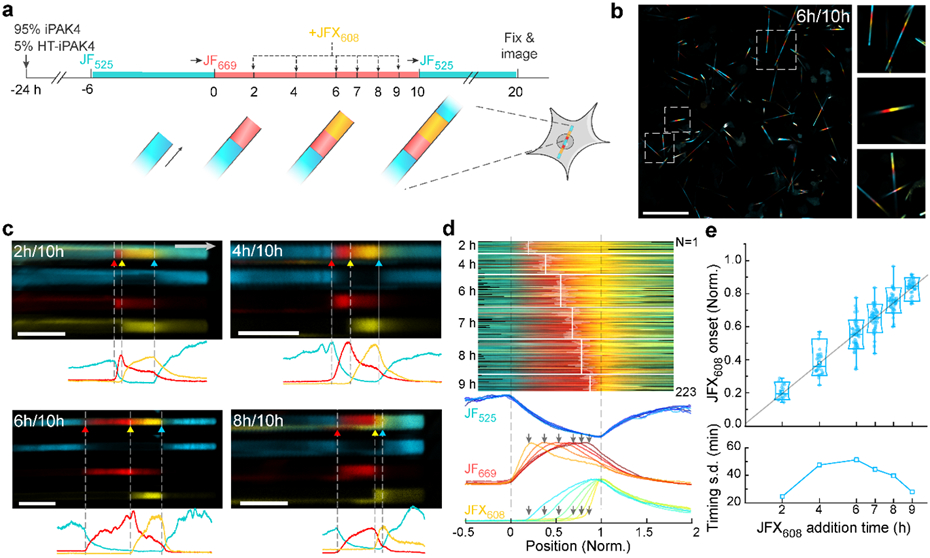
a, Experimental design for testing the precision with which addition of JFX608 could be determined relative to timestamps from addition of JF669 (at t = 0) and JF525 (at t = 10 h). b, Low-magnification (left) and magnified (right) images of iPAK4 fibers in HEK cells. Scale bar 100 μm. c, Top: Images of fibers with JFX608 addition at t = 2, 4, 6, and 8 h. Bottom: fluorescence line profiles. Scale bars 10 μm. d, Top: fluorescence profiles of N = 223 fiber ends with JFX608 addition at different timepoints. The profile lengths have been normalized to line up the timestamps at t = 0 and 10 h. Bottom: Mean fluorescence traces in each of the three dye color channels, for all times of JFX608 addition. e, Top: positions of the JFX608 onset as a function of dye addition time. Bottom: standard deviation in the inferred timing of JFX608 addition for each population of fibers. Lower and upper bounds of the box plot: 10th and 90th percentile; lower and upper whiskers: minimum and maximum; squares: mean; center lines: median. All data points are displayed.
Visual inspection of the fibers with JFX608 addition at different timepoints showed clear onset of yellow staining at the corresponding positions along the red band (Fig. 3c). We quantified the three-color fluorescence profiles of N = 223 fibers (20 to 51 fibers per timepoint of JFX608 addition) and identified the locations of the three dye additions (JF669, JFX608, JF525). As above, we mapped each fluorescence trace so that the first and third dye additions occurred at x = 0 and 1, respectively (Fig. 3d).
The mean traces clearly showed the time-dependent onset of JFX608 fluorescence, which was also evident in low-magnification images of the fiber population (Fig. S9). We then quantified the distributions of JFX608 onset. The distribution linearly mapped dye addition time to normalized position between 0 and 1 (Fig. 3e). Mapping the standard deviation of JFX608 labeling onset onto the 10 h time axis yielded single-fiber precisions between 25 and 51 min (Fig. 3e). The precision was greatest near the fiducial points at t = 0 and 10 h, and lowest in the middle of the trajectory. This observation established that the uncertainty in JFX608 timing was dominated by intracellular fluctuations in the fiber growth rate, as opposed to errors in locating the JFX608 transition points, since localization errors would be statistically similar anywhere along the fiber. Thus, to achieve the greatest temporal precision for detecting an event, one should deposit a fiducial timestamp near the candidate event.
Time-tagged iPAK4 fibers record dynamics of gene expression
As the first test of a physiological recording, we used iPAK4 fibers to report on a transcriptional activator, the tetracycline mediated Tet-ON transcription system.28 We co-infected HEK cells with lentivirus containing CMV::iPAK4 (60%), CMV::rTTA3 (30%), CMV::HT-iPAK4 (5%), and CMV::eGFP-iPAK4 (5%) to establish tetracycline-dependent eGFP-iPAK4 expression (Fig. 4a). After fibers had nucleated (24 h after lentiviral infection), we added JF552 to label the initial nuclei, and then switched to JF669 at t = 0. In different dishes we added doxycycline (DOX, 2 μg/mL) at t = 2, 4, or 6 h to activate expression of eGFP-iPAK4. Then we switched back to JF552 at t = 8 h and grew the fibers for another 8-14 h before fixation and imaging (Fig. 4b).
Figure 4. Protein ticker tape recordings of doxycycline (DOX) activation of Tet-ON system in HEK cells.
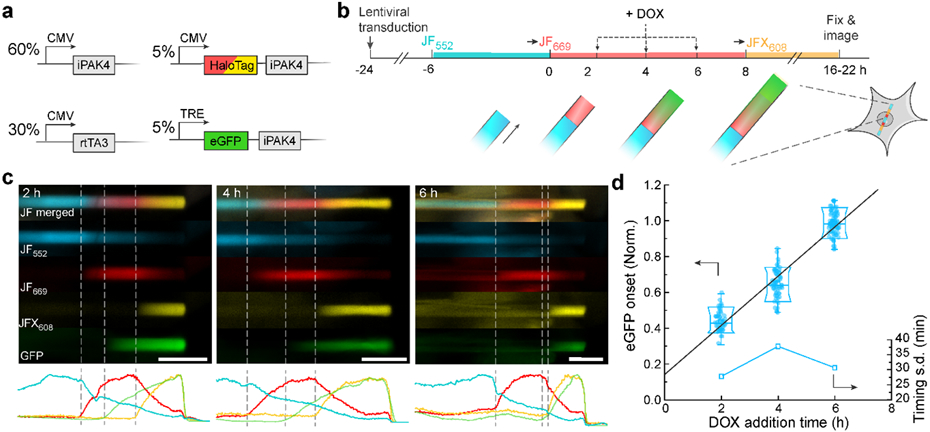
a, Genetic constructs for recording time-tagged transcription activation. b, Experimental protocol for recording time-tagged transcription activation. Transitions to JF669 at t = 0 and to JFX608 at t = 8 h provided fiducial timestamps. Transcription was activated via addition of DOX at t = 2, 4, or 6 h. c, Top: representative images of fibers with DOX addition at t = 2, 4, or 6 h. Bottom: fluorescence line profiles. Scale bars: 10 μm. d, Normalized positions of eGFP onset relative to fiducial timestamps at t = 0 and 8 h. The linear fit has a y-intercept of 1.2 h, indicative of the delay between DOX addition and protein synthesis. Lower and upper bounds of the box plot: 10th and 90th percentile; lower and upper whiskers: minimum and maximum; squares: mean; center lines: median. All data points are displayed.
DOX addition led to formation of green bands in the fibers, with onset at positions linearly related to DOX addition time (N = 191 fibers, Fig. 4c, d). Based on the fiber-to-fiber variations in the positions of the green onsets, we inferred an absolute timing accuracy of 30-40 min over an 8 h baseline. A linear fit to the band positions yielded an apparent offset of 1.2 h between DOX addition and the start of the green stripe. We ascribe this delay to the time required for transcription, translation, and protein folding. Similar experiments where cells were exposed to a 30-minute pulse of DOX caused eGFP-iPAK4 expression to rise for approximately 8 h and then fall, leading to a ~14 h window of protein expression (Fig. S10). These experiments demonstrate that the iPAK4 ticker tape system can quantify the dynamics of promoter activity in single cells.
The Tet-ON system also provides a means to control the onset of iPAK4 nucleation, allowing a separation between gene delivery and the start of a recording. We co-expressed CMV::rtTA3 (30%), TRE::iPAK4 (65%) and TRE::eGFP-iPAK4 (5%) in HEK cells using lentiviral transduction, and added DOX after 7 days (Fig. S11). In the first 7 days, we observed negligible eGFP signal, indicating minimal background expression of the Tet-ON system. After DOX addition, iPAK4 fibers were observed starting on day 8, with clear elongation of the fibers over the following day. Inducible ticker tape expression could be a powerful tool for defining recording windows to coincide with specific experimental perturbations.
Time-tagged iPAK4 fibers record neural activation
Finally, we tested the ability of iPAK4 ticker tapes to report activation of the IEG cFos in cultured rat hippocampal neurons. To establish iPAK4 fiber recordings in neurons, we first verified that: (1) the fibers grew linearly in neurons; (2) a dye switch introduced fiducial time stamps in neurons; and (3) the fibers did not introduce cytotoxicity or changes to neuronal electrophysiology.
To characterize fiber growth in neurons, we performed time-lapse imaging over 33 h in a primary neuron culture lentivirally infected with CMV::iPAK4 (90%) and CMV::HT-iPAK4 (10%), (Movie S4). Fibers of iPAK4 nucleated and grew in neurons following a similar trajectory to in HEK cells, comprising a fast-growing nucleation phase followed by slower linear growth (Fig. S12, Movie S5). We occasionally observed fibers stall for a few hours, or sometimes stop growing altogether. Stalled fibers were flagged by requiring a mean growth rate of > 0.8 μm/hr. This excluded 11 of 33 tracked fibers (33%). A key point is that the growth rate threshold could be applied based on a single terminal measurement of fiber length, and thus did not require time-lapse imaging.
Of the fibers that passed the growth rate criterion, the fiber length at onset of linear growth was 31 ± 12 μm (mean ± s.d., N = 22 fibers). The growth rate in the linear phase was 2.5 ± 1.5 μm/h. During the linear growth phase, the fit to a straight line growth profile had a mean R2 = 0.95 ± 0.04, over a 24 h interval. Of the neurons that had fibers, most (73%) had only one fiber, although many neurons did not have fibers nucleated at the time of the recording (Fig. S13). We also performed dye switch experiments in neurons and confirmed the ability of switches in HT dye labeling to impart fiducial time stamps on HT-iPAK4 fibers in neurons (Figs. S14, S15).
To test for effects of iPAK4 fibers on neuronal survival, we lentivirally expressed iPAK4 (95%) and HT-iPAK4 (5%) in cultured neurons, allowed the fibers to grow for 6 days, and then performed a live-dead assay (Fig. S16). There was no significant difference in the fraction of dead cells between dishes expressing iPAK4 fibers and dishes not infected with any lentivirus. The iPAK4-expressing neurons had typical soma and neurite structures, similar to those observed in healthy neurons.
We then tested whether iPAK4 fibers affected neuronal electrophysiology. Neurons infected with lentivirus encoding CMV::iPAK4 (90%) and CMV::eGFP-iPAK4 (10%) grew single straight fibers (Fig. 5a). We used patch clamp recordings to compare the electrophysiology of neurons with and without fibers. Although in many cases the fiber length was several times longer than the soma diameter, neurons with fibers spiked normally (Fig. 5b), and had membrane resistance, membrane capacitance, resting potential, and rheobase which were statistically indistinguishable from neurons without fibers (N = 11 neurons with fibers, N = 12 without, two-sided Student’s t-test, Fig. 5c).
Figure 5. Protein ticker tape recordings of cFos activation in neurons.
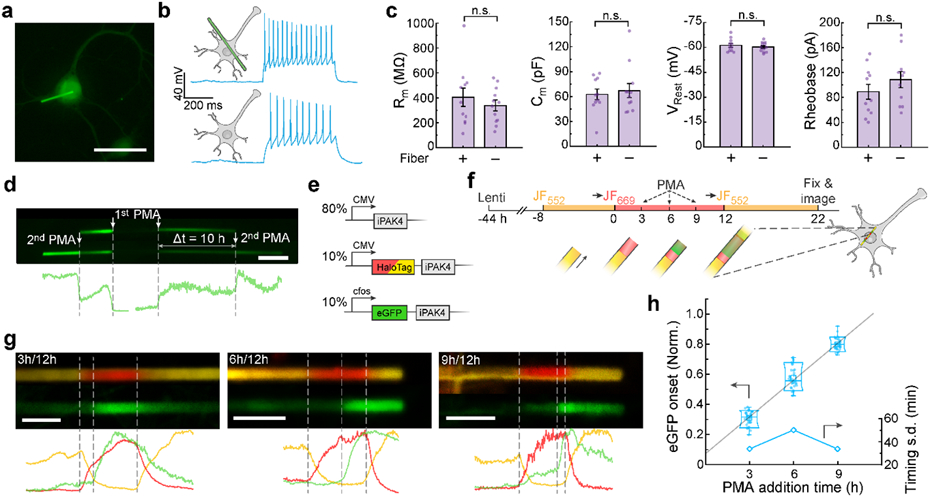
a, Image of a cultured neuron expressing lentiviral CMV::iPAK4 (90%) and CMV::eGFP-iPAK4 (10%). Scale bar: 50 μm. b, Representative patch clamp recordings in neurons with or without an iPAK4 fiber. Spikes were evoked by a current injection of 100 pA. c, There were no significant differences between neurons with or without fibers in membrane resistance (404 ± 73 MΩ vs 339 ± 43 MΩ, p = 0.43), membrane capacitance (63 ± 6 pF vs 67 ± 9, p = 0.67), resting potential (−61.2 ± 1.2 mV vs −60.2 ± 0.8 mV, p = 0.48), or rheobase (89 ± 12 pA vs 109 ± 13 pA, p = 0.27, N = 11 neurons with fibers, 12 neurons without). Error bars show mean ± s.e.m.. Two-sided Student’s t-test was employed for data comparison. d, Fiber in a neuron expressing lentiviral CMV::iPAK4 (90%) and cFos::eGFP-iPAK4 (10%). The top image shows the fiber after a first PMA addition, the bottom image shows the same fiber after a second PMA addition. Scale bar 5 μm. e, Genetic constructs for recording time-tagged cFos activation in neurons. f, Experimental protocol for recording time-tagged cFos activation in neurons. Transitions to JF669 at t = 0 and to JF552 at t = 12 h provided fiducial timestamps. cFos was activated via addition of PMA at t = 3, 6, or 9 h. g, Top: representative images of fibers with PMA addition at t = 3, 6, or 9 h. Bottom: fluorescence line profiles. Scale bar 5 μm. h, Normalized positions of eGFP onset relative to fiducial timestamps at t = 0 and 12 h. Lower and upper bounds of the box plot: 10th and 90th percentile, respectively; lower and upper whiskers: minimum and maximum, respectively; squares: mean; center lines: median. The linear fit has a y-intercept of ca. 1.0 h. All data points are displayed.
In neurons co-expressing CMV::iPAK4 (90%) and cFos::eGFP-iPAK4 (10%), fibers initially grew with little green fluorescence. Addition of phorbol 12-myristate 13-acetate (PMA, 1 μM), an activator of cFos expression,29 led to bands of bright green fluorescence (Fig. S8). Sequential additions of PMA to a single dish led to distinct bands of eGFP fluorescence, clearly resolved by sharp eGFP boundaries (Fig. 5d).
To measure timing of cFos activation relative to fiducial timestamps, we co-expressed CMV::iPAK4 (80%), CMV::HT-iPAK4 (10%), and cFos::eGFP-iPAK4 (10%; Fig. 5e). We stained the neurons with JF552, and then switched to JF669 at t = 0. We then added PMA (1 μM) at t = 3, 6, or 9 h, and introduced a second fiducial timestamp by switching back to JF552 at t = 12 h. Fibers were grown until t = 22 h and then fixed and imaged (Fig. 5f).
We observed clear green bands indicating cFos-driven expression of eGFP-iPAK4. To infer an onset time for each band, we mapped the onset of eGFP fluorescence relative to the two dye switches. The slope of the plot of inferred eGFP onset time vs nominal PMA addition time was 0.98± 0.10 (mean ± 95% CI, N = 34 fibers at 3 h, 32 fibers at 6 h, 40 fibers at 9 h). Extrapolating this fit to t = 0 implied that eGFP onset was delayed by 53 minutes relative to the timing of PMA addition (Fig. 5h). We interpret this delay as the time for PMA to activate the cFos promoter plus delays of transcription, translation, and protein folding of eGFP-iPAK4. The standard deviations in the inferred eGFP onset times were, 34 min (3 h), 50 min (6 h), and 34 min (9 h), implying an average absolute timing accuracy of 39 minutes over a measurement with 12 h between fiducial time stamps.
Discussion
Slowly growing protein assemblies are a promising substrate for massively parallel cellular recordings. Our recording strategy relies on three key elements: a protein scaffold, a means to impart fiducial timestamps, and a fluorescent reporter of cellular activity which is irreversibly incorporated into the scaffold during growth. Here we validated the system with externally applied perturbations (drug-induced gene expression) which led to synchronous population-wide responses; but in principle the system is also capable of recording spontaneous or asynchronous activation of reporter genes.
The fiducial timestamps are a particularly important conceptual and practical aspect of our approach. Timestamps permit detection, and in some cases correction, of many sources of variability that could otherwise confound the recordings. Here, we used the timestamps to correct for cell-to-cell variations in the linear-phase growth rate, via a simple linear interpolation scheme. The timestamps could also flag individual cells in which there were substantial fluctuations in growth rate during a recording. These would manifest as distinct stripes having widths out of proportion to the corresponding dye incubation times. The timestamps could also flag fibers that nucleated after the first dye switch: such fibers would have their central portion tagged with a mixture of dyes instead of solely with the first dye. Fibers which fractured or aggregated would have timestamps that deviated from the anticipated color order. Thus, the timestamps provide a robust means of calibration and quality control within each cell.
There are opportunities to broaden and improve upon our approach regarding each of the three key elements: the scaffold, the timestamps, and the physiological signal. For applications in vivo, the most critical need is to modify the scaffold so that it does not deform the cells. One approach is to engineer thinner fibers, either by modifying the iPAK4 scaffold to enhance the ratio of axial to radial growth, or by using a different fiber-forming protein scaffold.14 Thinner fibers would undergo Euler buckling at the cell membrane rather than deforming the cell, though if the fiber becomes so thin that the thermal persistence length is smaller than a few microns, optical tracking of the fiber backbone could be difficult. Alternatively, one could explore alternate ticker-tape geometries, such as helical aggregates (thereby achieving a ratio of contour length to axial length that exceeds 1), or spherical onion-type aggregates.
A second improvement to the scaffolds would be to decrease the ratio between the soluble iPAK4 concentration at fiber nucleation, Cnuc, and the concentration during steady state growth, CSS. Fits to the simple kinetic model of fiber growth (Fig. S2) suggest that this ratio is currently ~100. Consequences of this high ratio are that (a) there is a highly variable delay of approximately 12 h between onset of iPAK4 transcription and fiber nucleation, and (b) the fibers undergo a period of fast and nonlinear growth during the first ~hour after nucleation (Fig. 1e). On the other hand, to ensure that most cells only contain a single fiber, one wants Cnuc/CSS > 1. Thus, a protein engineering goal is to make this ratio slightly larger than 1.
Cultured neurons could sustain iPAK4 fiber growth for at least 6 days (Fig. S13), suggesting the possibility of multi-day recordings. To tune the dynamic range of ticker tape recordings, one would like small-molecule or light-induced control over the onset and rate of ticker tape scaffold growth. Putting the iPAK4 scaffold under control of the Tet-ON system gave control over the onset of fiber growth (Fig. S11). However, the slow transcriptional response to changes in DOX concentration (Fig. S10), together with the high ratio of Cnuc/CSS, meant that subsequent tuning of the growth rate by Dox concentration was not feasible by this approach. Achieving control over the rate of fiber growth would require either lowering Cnuc/CSS or developing alternate means of regulating protein assembly into the fiber, e.g. by pharmacological or optical regulation of protein stability31, or of protein-protein interactions. Finally, one may wish to express both the iPAK4 and the HT-iPAK4 from a single vector, either by making use of endogenous RNA splicing machinery or by applying a partially effective self-cleaving peptide.
At present, the spacing of the fiducial timestamps is constrained to ~2 h by the ~4.5 h half-life of soluble HT-iPAK4. If one could shorten this half-life, one could place time stamps closer together, and thereby achieve greater absolute timing accuracy. One might shorten the half-life of the soluble HT-iPAK4 by attaching ubiquitination domains or other tags to facilitate proteolytic turnover of the soluble subunits.32 Alternatively, one might consider a variety of optogenetic caging strategies to reversibly control the availability of time-stamp monomers.33 Optogenetic modulation of protein availability can occur over seconds, suggesting the possibility to introduce extremely precise fiducial time stamps.
Finally, the modular design of iPAK4 fibers could be adapted to accommodate diverse recording modalities. A simple generalization of the present results would be to record the simultaneous dynamics of multiple promoters by using each to drive expression of iPAK4 fused to a spectrally distinct fluorescent protein. There also exist diverse fluorescent reporters of covalent enzymatic modifications, e.g. of kinase, phosphatase, or protease activity.34,35 Such reporters are likely protected from enzymatic modification within the fiber, and thereby might be a means to record the state of the cell at the moment of incorporation into the fiber.
A related protein-based recording technique was introduced concurrent with our work, termed expression recording islands (XRIs).27 The primary conceptual difference between the XRIs and our time-tagged ticker tapes is that our HaloTag-based time-stamps give our ticker tapes an absolute time-base in each single cell.
Despite the broad scope for improvements on the existing ticker tape system, we believe that these tools are already suitable for applications in vitro. For instance, they could be used to map transcriptional dynamics during stem cell differentiation, in in vitro tumor models, or in diverse organoid models. In each new cellular preparation, it will be critical to test whether expression of iPAK4 fibers perturbs the physiological dynamics of interest. The introduction of protein-based ticker tapes in our work and the Expression Recording Islands in the companion paper 27 should spur diverse efforts on improving and applying these tools.
Methods
Cloning and molecular biology
All iPAK4 constructs were cloned into a second-generation lentiviral backbone (Addgene: 136636) with either a CMV or a cFos promoter (Addgene: 47907) using standard Gibson Assembly.36 Briefly, the vector was linearized by double digestion (BamHl and EcoRl for CMV driven constructs, Pacl and EcoRl for cFos driven constructs) and purified by the GeneJET gel extraction kit (ThermoFisher). Gene fragments and cFos promoter were generated by PCR amplification and then combined with the linearized backbones by Gibson ligation. EGFP-iPAK4 was a gift from B. Cui (Stanford), and HaloTag was cloned from pCAG-Voltron (Addgene: 119033). The eGFP and iPAK4 were connected with a SGGS linker, while the HaloTag and iPAK4 were connected with a SGS linker. All plasmids were verified by full sequencing around the cloned regions.
All plasmids are available on Addgene:
| Plasmid ID | Description | Addgene ID |
|---|---|---|
| DL015: CMV::iPAK4 | ibox-PAK4cat (iPAK4) fusion with CMV promoter, cloned into a lentivirus backbone | 177880 |
| DL016: CMV::eGFP-iPAK4 | eGFP-IPAK4 fusion with CMV promoter, cloned into a lentivirus backbone | 177881 |
| DL017: CMV::HaloTag-iPAK4 | HT-iPAK4 fusion with CMV promoter, cloned into a lentivirus backbone | 177882 |
| DL033: cFos::eGFP-iPAK4 | eGFP-iPAK4 fusion with cFos promoter, cloned into a lentivirus backbone | 177883 |
| DL034: cFos::HaloTag-iPAK4 | HaloTag-iPAK4 fusion with cFos promoter, cloned into a lentivirus backbone | 177884 |
| DL145: TRE::iPAK4 | Tet-On inducible expression of iPAK4 in a lentiviral backbone | 187445 |
| DL146: TRE::eGFP-iPAK4 | Tet-On inducible expression of eGFP-iPAK4 in a lentiviral backbone | 187446 |
| DL147: TRE::HaloTag-iPAK4 | Tet-On inducible expression of HaloTag-iPAK4 in a lentiviral backbone | 187447 |
Synthesis of JFX608-HaloTag
General synthetic methods.
Commercial reagents were obtained from reputable suppliers and used as received. All solvents were purchased in septum-sealed bottles stored under an inert atmosphere. All reactions were sealed with septa through which a nitrogen atmosphere was introduced unless otherwise noted. Reactions were conducted in round-bottomed flasks or septum-capped crimp-top vials containing Teflon-coated magnetic stir bars. Heating of reactions was accomplished with a silicon oil bath or an aluminum reaction block on top of a stirring hotplate equipped with an electronic contact thermometer to maintain the indicated temperatures. Reactions were monitored by thin layer chromatography (TLC) on precoated TLC glass plates (silica gel 60 F254, 250 μm thickness) or by LC/MS (Phenomenex Kinetex 2.1 mm × 30 mm 2.6 μm C18 column; 5 μL injection; 5–98% MeCN/H2O, linear gradient, with constant 0.1% v/v HCO2H additive; 6 min run; 0.5 mL/min flow; ESI; positive ion mode). TLC chromatograms were visualized by UV illumination or developed with p-anisaldehyde, ceric ammonium molybdate, or KMnO4 stain. Reaction products were purified by flash chromatography on an automated purification system using pre-packed silica gel columns or by preparative HPLC (Phenomenex Gemini–NX 30 × 150 mm 5 μm C18 column). Analytical HPLC analysis was performed with an Agilent Eclipse XDB 4.6 × 150 mm 5 μm C18 column under the indicated conditions. High-resolution mass spectrometry was performed by the High Resolution Mass Spectrometry Facility at the University of Iowa. NMR spectra were recorded on a 400 MHz spectrometer. 1H and 13C chemical shifts were referenced to TMS or residual solvent peaks. Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublets, m = multiplet), coupling constant (Hz), integration. Data for 13C NMR spectra are reported by chemical shift (δ ppm) with hydrogen multiplicity (C, CH, CH2, CH3) information obtained from DEPT spectra.
6-tert-Butoxycarbonyl-JFX608 (2).
A vial was charged with 6-tert-butoxycarbonyl-carbofluorescein ditriflate18 (1; 250 mg, 0.346 mmol), Pd2dba3 (31.7 mg, 34.6 μmol, 0.1 eq), XPhos (49.5 mg, 0.104 mmol, 0.3 eq), and Cs2CO3 (316 mg, 0.969 mmol, 2.8 eq). The vial was sealed and evacuated/backfilled with nitrogen (3×). Dioxane (2 mL) was added, and the reaction was flushed again with nitrogen (3×). Following the addition of azetidine-2,2,3,3,4,4-d6 (52.4 mg, 0.830 mmol, 2.4 eq), the reaction was stirred at 100 °C for 4 h. It was subsequently cooled to room temperature, filtered through Celite with CH2Cl2, and concentrated to dryness. Purification by silica gel chromatography (20–100% EtOAc/hexanes, linear gradient) afforded 6-tert-butoxycarbonyl-JFX608 (2) as a blue-green solid (181 mg, 95%). 1H NMR (CDCl3, 400 MHz) δ 8.14 (dd, J = 8.0, 1.3 Hz, 1H), 8.00 (dd, J = 8.0, 0.8 Hz, 1H), 7.62 (dd, J = 1.2, 0.7 Hz, 1H), 6.58 (d, J = 2.4 Hz, 2H), 6.54 (d, J = 8.6 Hz, 2H), 6.21 (dd, J = 8.6, 2.3 Hz, 2H), 1.83 (s, 3H), 1.73 (s, 3H), 1.53 (s, 9H); 13C NMR (CDCl3, 101 MHz) δ 170.2 (C), 164.6 (C), 155.6 (C), 152.5 (C), 146.8 (C), 137.8 (C), 130.3 (C), 130.1 (CH), 128.9 (CH), 125.1 (CH), 124.8 (CH), 119.8 (C), 110.5 (CH), 108.0 (CH), 88.8 (C), 82.3 (C), 38.5 (C), 35.5 (CH3), 32.8 (CH3), 28.2 (CH3); Analytical HPLC: tR = 13.3 min, >99% purity (10–95% MeCN/H2O, linear gradient, with constant 0.1% v/v TFA additive; 20 min run; 1 mL/min flow; ESI; positive ion mode; detection at 600 nm); HRMS (ESI) calcd for C34H25D12N2O4 [M+H]+ 549.3501, found 549.3503.
6-Carboxy-JFX608 (3).
6-tert-Butoxycarbonyl-JFX608 (2; 310 mg, 0.565 mmol) was taken up in CH2Cl2 (10 mL), and trifluoroacetic acid (2 mL) was added. The reaction was stirred at room temperature for 6 h. Toluene (10 mL) was added; the reaction mixture was concentrated to dryness and then azeotroped with MeOH three times to provide 6-carboxy-JFX608 (3) as a dark blue solid (323 mg, 94%, TFA salt). Analytical HPLC and NMR indicated that the material was >95% pure and did not require further purification prior to amide coupling. 1H NMR (CD3OD, 400 MHz) δ 8.36 – 8.27 (m, 2H), 7.86 – 7.78 (m, 1H), 6.91 (d, J = 9.1 Hz, 2H), 6.81 (d, J = 2.3 Hz, 2H), 6.38 (dd, J = 9.1, 2.3 Hz, 2H), 1.82 (s, 3H), 1.70 (s, 3H); 13C NMR (CD3OD, 101 MHz) δ 168.0 (C), 167.5 (C), 158.0 (C), 157.0 (C), 139.5 (C), 137.6 (CH), 136.2 (C), 135.5 (C), 132.4 (CH), 132.3 (CH), 131.4 (CH), 121.8 (C), 111.9 (CH), 109.7 (CH), 42.8 (C), 35.6 (CH3), 32.0 (CH3); Analytical HPLC: tR = 10.5 min, >99% purity (10–95% MeCN/H2O, linear gradient, with constant 0.1% v/v TFA additive; 20 min run; 1 mL/min flow; ESI; positive ion mode; detection at 600 nm); HRMS (ESI) calcd for C30H17D12N2O4 [M+H]+ 493.2875, found 493.2873.
JFX608-NHS (4).
6-Carboxy-JFX608 (3, TFA salt; 125 mg, 0.206 mmol) was combined with DSC (116 mg, 0.453 mmol, 2.2 eq) in DMF (5 mL). After adding Et3N (172 μL, 1.24 mmol, 6 eq) and DMAP (2.5 mg, 20.6 μmol, 0.1 eq), the reaction was stirred at room temperature for 30 min. It was subsequently diluted with 10% w/v citric acid and extracted with EtOAc (2×). The combined organic extracts were washed with water and brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. Flash chromatography (25–100% EtOAc/CH2Cl2, linear gradient) yielded 116 mg (95%) of JFX608-NHS (4) as a dark blue-green solid. 1H NMR (CDCl3, 400 MHz) δ 8.30 (dd, J = 8.0, 1.4 Hz, 1H), 8.11 (dd, J = 8.0, 0.8 Hz, 1H), 7.78 (dd, J = 1.4, 0.7 Hz, 1H), 6.57 (d, J = 2.4 Hz, 2H), 6.50 (d, J = 8.6 Hz, 2H), 6.23 (dd, J = 8.5, 2.4 Hz, 2H), 2.87 (s, 4H), 1.83 (s, 3H), 1.71 (s, 3H); Analytical HPLC: tR = 11.1 min, >99% purity (10–95% MeCN/H2O, linear gradient, with constant 0.1% v/v TFA additive; 20 min run; 1 mL/min flow; ESI; positive ion mode; detection at 600 nm); HRMS (ESI) calcd for C34H20D12N3O6 [M+H]+ 590.3039, found 590.3043.
JFX608–HaloTag ligand (6).
JFX608-NHS (4; 50 mg, 84.8 μmol) and 2-(2-((6-chlorohexyl)oxy)ethoxy)ethanamine (5, “HaloTag(O2)amine,” TFA salt; 43.0 mg, 0.127 mmol, 1.5 eq) were combined in DMF (3 mL), and DIEA (44.3 μL, 0.254 mmol, 3 eq) was added. After stirring the reaction at room temperature for 1 h, it was diluted with saturated NaHCO3 and extracted with EtOAc (2×). The combined organic extracts were washed with water and brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. Purification of the crude product by silica gel chromatography (50–100% EtOAc/toluene, linear gradient) provided JFX608–HaloTag ligand (6) as a blue foam (47 mg, 79%). 1H NMR (CDCl3, 400 MHz) δ 8.02 (dd, J = 8.0, 0.7 Hz, 1H), 7.94 (dd, J = 7.9, 1.4 Hz, 1H), 7.44 – 7.39 (m, 1H), 6.73 (t, J = 4.8 Hz, 1H), 6.57 (d, J = 2.4 Hz, 2H), 6.52 (d, J = 8.6 Hz, 2H), 6.20 (dd, J = 8.6, 2.4 Hz, 2H), 3.64 – 3.56 (m, 6H), 3.54 – 3.48 (m, 4H), 3.38 (t, J = 6.6 Hz, 2H), 1.83 (s, 3H), 1.78 – 1.73 (m, 2H), 1.72 (s, 3H), 1.55 – 1.48 (m, 2H), 1.45 – 1.38 (m, 2H), 1.34 – 1.27 (m, 2H); Analytical HPLC: tR = 12.6 min, >99% purity (10–95% MeCN/H2O, linear gradient, with constant 0.1% v/v TFA additive; 20 min run; 1 mL/min flow; ESI; positive ion mode; detection at 600 nm); HRMS (ESI) calcd for C40H37D12ClN3O5 [M+H]+ 698.4108, found 698.4118.
Fiber expression in HEK cells
HEK293T cells (ATCC; CRL-11268) were grown and split following standard protocols as described previously.37 HEK cells at low-passage-number (< 10 passages) were plated at a confluence of 30% onto 10-cm dishes coated with gelatin (Stemcell Technologies; 07903) or 14 mm glass bottom dishes (CellVis, D35-14-1.5-N) coated with 40 μg/ml poly-L-lysine-coated (P8920, Sigma-Aldrich). Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS and penicillin/streptomycin was used as the culture medium. Cells were grown at 37 °C and 5% CO2.
When cells reached 50-70% confluence, genes were delivered by either lentivirus or TransIT-293 (Mirus; MIR2700) transfection kit. In lentiviral transduction, the high-titer lentiviral vectors were first pre-mixed at the designated ratio and diluted to 10% of the initial concentration by DMEM medium. The cells’ culture medium was then replaced by the fresh lentivirus-containing DMEM medium, and the cells were further incubated at 37 °C and 5% CO2 for ~24 h. In the TransIT-293 transfection, 1 μg plasmids at the designated ratio were first diluted by 100 μl Opti-MEM medium, followed by the addition of 3 μl TransIT-293 reagent. The cocktail was incubated at room temperature for 15 min and diluted 10-fold into DMEM on the culture dishes. The cells were then further incubated at 37 °C and 5% CO2 for ~24 h.
Fiber labeling
The in-cellulo fibers were labeled with HT-ligand Janelia Fluor (JF) dyes. In this work the cell-permeable dyes were: JF503, JF525, JF552, JFX608, and JF669. The JF dyes were first diluted into 1 mM stock solution as described previously,17 which was aliquoted and stored at −20 °C. The 1 mM solution was further diluted to 1 μM in 37 °C DMEM medium for HEK cells or BPNM/SM1 medium for neurons. The dyed medium was used to replace the original medium in the culture dishes at timed staining. In the dye switching processes, the medium containing the original dye was fully removed, followed by thorough wash of the culture dishes 5 times with 37 °C culture medium. Then, the medium with 1 μM new dye was added to the cells, and the cells were returned to the incubator at 37 °C and 5% CO2.
Measurements of HT dye labeling kinetics in HEK cells
HEK cells were transfected with an inducible nuclear-localized HT protein (HT-NLS, Addgene #82518). Doxycycline (DOX, 2 μg/ml) was added 12 h after transfection to induce the expression of HT-NLS. 24 h after transfection, cells were incubated with 0.1 μM of the indicated dye and nuclear fluorescence was monitored via wide-field epifluorescence microscopy as a function of time.
Primary neuron culture
All procedures involving animals were in accordance with the US National Institutes of Health Guide for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee at Harvard University.
Before the plating of primary hippocampal neurons, 14 mm glass-bottom dishes were first incubated with 40 μg/ml poly-D-lysine (PDL) in PBS at room temperature for 1 h and subsequentially with 20 μg/ml laminin (Fisher Scientific; 23-017-015) at 4 °C overnight, followed by thorough wash with PBS. Hippocampi (BrainBits; SKU: SDEHP) from embryonic day 18 (E18) rats were dissected and resuspended in Brainphys™ medium (BPNM, Stemcell Technologies; 05790) supplemented with 2% SM1 (Stemcell Technologies; 05792), 5 mM L-Glutamine (Stemcell Technologies; 07100), and 35 μg/ml L-Glutamic Acid (Sigma Aldrich; 49449), to a final concentration of 3.0 ×106 cells/ml. The neurons were then plated at a density of 30,000 cells/cm2 on the pretreated glass-bottom dishes, with subsequent addition of 2 ml BPNM with 2% SM1 (BPNM/SM1). Neuronal health was monitored daily from DIV1 to DIV 7. Every 3-4 days, 1 ml of the medium in each dish was replaced with 37 °C fresh BPNM/SM1 medium.
Patch clamp electrophysiology
Whole-cell recordings were performed in extracellular buffer containing (in mM): 125 NaCl, 2.5 KCl, 15 HEPES, 25 D-glucose, 1 MgCl2, and 2 CaCl2 (pH = 7.2-7.3 with NaOH). Fiber-forming and non-forming (control) neurons were visualized with a home-built inverted epifluorescence microscope. Experiments were made at 23 °C under ambient atmosphere. The whole-cell internal solution comprised (in mM): 8 NaCl, 130 KMeSO3, 10 HEPES, 5 KCl, 0.5 EGTA, 4 Mg-ATP, and 0.3 Na3-GTP. The pH was adjusted to 7.2-7.3 with KOH and osmolarity was set to 290-295 mOsm/L. Borosilicate glass pipettes were used with a resistance of 3-5 MΩ (1.5 mm OD). Signals were acquired and filtered at 4 kHz with the internal Bessel filter using a Multiclamp 700B (Molecular Devices) and digitized with PCIe-6323 (National Instruments) at 10 kHz. Following the whole-cell configuration, membrane capacitance (Cm), and membrane resistance (Rm) were estimated under voltage-clamp mode. Measurements of resting membrane potential (Vrest), rheobase, and spike rates were made under current-clamp mode. Rheobase was defined as the minimum current step (in 500 ms duration) required for any spike onset. Whole-cell recordings were monitored and analyzed using a custom code written in LabView and Matlab.
Lentivirus production
Lentivirus production in HEK cells.
Plasmids of CMV::iPAK4, CMV::HaloTag-iPAK4, and cFos::eGFP-iPAK4 were used to produce lentivirus according to published methods.38 Briefly, low passage-number HEK293T cells (ATCC; CRL-11268) were plated onto gelatin-coated (Stemcell Technologies; 07903) 10-cm dishes. When HEK cells reached 80% confluence, the medium was exchanged to a serum-free DMEM. After 0.5–1 h, cells were transfected using polyethylenimine (PEI; Sigma; 408727). 7 μg of the vector plasmid, 4 μg of the second-generation packaging plasmid psPAX2 (Addgene; 12260), and 2 μg of viral entry protein VSV-G plasmid pMD2.G (Addgene; 12259) were mixed into 600 μl of serum-free DMEM, and 20 μl of 1 mg/ml PEI was then added. The mixture was incubated at room temperature for 15 min and added dropwise to the plate. After 3-4 h, the medium was exchanged back to 10 ml of DMEM10. The supernatant was harvested at 36 h post-transfection, and another 10 ml of DMEM10 was added to the cells and incubated for another 24 h. At 60 h post-transfection, the supernatant was harvested again and combined with the first batch of supernatant, centrifuged for 5 min at 500 g, and filtered through a 0.45-μm filter (EMD Millipore; SE1M003M00).
Lentivirus concentration.
1 part of Lenti-X™ concentrator (TaKaRa; 631232) was first mixed with 3 parts of supernatant and incubated at 4 °C overnight for lentivirus precipitation. The mixture was then centrifuged at 1,500 g for 45 min at 4°C. The supernatant was gently removed, and the off-white pellet was resuspended in 200 μl neurobasal-based medium. The concentrated virus was titrated in neurons, aliquoted, and stored at −80 °C for neuronal transduction.
Fiber expression in neurons
Genes were delivered to neurons by lentiviral transduction at DIV 7-10. The lentiviral vectors of CMV::iPAK4, CMV::HaloTag-iPAK4, and cFos::eGFP-iPAK4 were first mixed at the designated ratio, which was further diluted to 10% of the original concentration by fresh BPNM/SM1 medium. The dilution was then used to replace the original medium in neuronal culture. The neurons were incubated in the lentivirus-containing medium at 37 °C and 5% CO2 for 12 h, followed by medium replacement with lentivirus-free medium.
Chemical activation of neuronal activity
The cFos promoter was activated by phorbol 12-myristate 13-acetate (PMA; Sigma Aldrich P8139). Briefly, the PMA was first diluted with DMSO to form a 1 mM stock solution. At the designated time, 2 μl PMA stock was directly added to each 14 mm glass-bottom culture dish, which contained 2 ml BPNM/SM1 medium. The dishes were then stirred gently to mix the PMA and medium. After PMA addition, the dishes were returned to an incubator at 37 °C with 5% CO2.
Multispectral imaging
Multispectral images were acquired using ZEISS LSM 980 confocal microscope with Airyscan 2. Lambda scan mode was used to image fibers with multi-color labeling. The excitation laser wavelengths were 488 nm (eGFP, JF503, JF525), 561 nm (JF552, JFX608), and 639 nm (JF669). In each Lambda scan, 32 channels in the range of 414-688 nm were simultaneously acquired to obtain a hyperspectral stack of images. The images were then unmixed with the built-in linear unmixing algorithm in Zen Blue software. Reference images of individual fluorescent labels were taken in the same instrumental configuration to train the linear unmixing algorithm. The spectral unmixing typically produced negligible residual signals.
Time-lapse microscopy
HEK cells expressing the target constructs were grown on 14 mm glass bottom culture dishes (CellVis, D35-14-1.5-N) and were monitored under a Zeiss Elyra microscope with 488 nm laser and a 10x air objective in an environmental chamber at 37 °C and 5% CO2. Images were acquired at 1% laser power, 300 ms exposures, 10 min intervals over 10-23 hours post-transfection.
Image processing and data analysis
Images of individual fibers were rotated to align the long axis to the x-axis. Fluorescence profiles were then calculated as the median fluorescence in each spectral channel across the width of the fiber. Fibers which were not in focus or where two or more fibers crossed each other near a dye transition were excluded from analysis. Dye transitions were identified as local maxima in the second derivative of the dye fluorescence as a function of position. To avoid spurious peaks due to noise, the second derivative signal was smoothed with a kernel of typically 10 min, though this smoothing was omitted when calculating the width of the dye transition (Fig. S3a).
For tracking fibers during time-lapse recordings, a region of interest (ROI) was manually defined on a maximum intensity projection of the image stack, to select individual fibers and to encompass the entire fiber in all frames of the movie. A Radon transform was then calculated on the selected ROI for each video frame. The peak of the Radon transform was associated with the fiber. The corresponding line in the real-space movie was used to calculate the fiber intensity profile. Nearby parallel lines on either side of the fiber were averaged and used for background subtraction. The fiber ends were then found by applying a simple threshold to the plot of fluorescence vs. position. The fluorescence of the cytoplasm was determined by summing the intensity from pixels that were on-cell but off-fiber. Fiber length trajectories were median filtered (kernel = 1 hr) to suppress small, high-frequency fluctuations caused by segmentation noise.
Supplementary Material
Acknowledgments:
We thank D. Kim, Y. Baskaran and E. Manser for helpful discussions. We thank B. Cui for the PAK4 plasmid and C. Hellriegel at the Harvard Center for BioImaging for assistance with microscopy. We thank S. Begum, A. Preecha, and J. Koob for technical assistance. This work was supported by Schmidt Futures (AEC, XL, EM, PP, BT), a Vannevar Bush Faculty Fellowship (AEC), the Howard Hughes Medical Institute (AEC, JBG, NF, LDL, HS, DB), National Institutes of Health grant 1-R21-1EY033669 (AEC, DL, XL), a John S. LaDue Fellowship (BJ), and the Harvard Brain Initiative (DL).
Footnotes
Competing interests: AEC, DL, and XL have filed a U.S. patent application on protein-based tickertapes for intracellular recordings.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
- 1.Renier N et al. Mapping of Brain Activity by Automated Volume Analysis of Immediate Early Genes. Cell 165, 1789–1802 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vetere G et al. Chemogenetic Interrogation of a Brain-wide Fear Memory Network in Mice. Neuron 94, 363–374.e4 (2017). [DOI] [PubMed] [Google Scholar]
- 3.DeNardo LA et al. Temporal evolution of cortical ensembles promoting remote memory retrieval. Nat. Neurosci. 22, 460–469 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mahringer D et al. Expression of c-Fos and Arc in hippocampal region CA1 marks neurons that exhibit learning-related activity changes. bioRxiv 644526 (2019) doi: 10.1101/644526. [DOI] [Google Scholar]
- 5.Zamft BM et al. Measuring Cation Dependent DNA Polymerase Fidelity Landscapes by Deep Sequencing. PloS One 7, e43876 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glaser JI et al. Statistical Analysis of Molecular Signal Recording. PLOS Comput. Biol 9, e1003145 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodriques SG et al. RNA timestamps identify the age of single molecules in RNA sequencing. Nat. Biotechnol 39, 320–325 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pond FR, Gibson I, Lalucat J & Quackenbush RL R-body-producing bacteria. Microbiol. Mol. Biol. Rev 53, 25–67 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pélissier HC, Peters WS, Collier R, van Bel AJE & Knoblauch M GFP Tagging of Sieve Element Occlusion (SEO) Proteins Results in Green Fluorescent Forisomes. Plant Cell Physiol. 49, 1699–1710 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.LeVine H [18] Quantification of β-sheet amyloid fibril structures with thioflavin T. in Methods in Enzymology vol. 309 274–284 (Academic Press, 1999). [DOI] [PubMed] [Google Scholar]
- 11.Barmada SJ & Harris DA Visualization of Prion Infection in Transgenic Mice Expressing Green Fluorescent Protein-Tagged Prion Protein. J. Neurosci 25, 5824–5832 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shukla S et al. Molecular farming of fluorescent virus-based nanoparticles for optical imaging in plants, human cells and mouse models. Biomater. Sci 2, 784–797 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Tsutsui H et al. A Diffraction-Quality Protein Crystal Processed as an Autophagic Cargo. Mol. Cell 58, 186–193 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Shen H et al. De novo design of self-assembling helical protein filaments. Science 362, 705–709 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nguyen TK et al. In-Cell Engineering of Protein Crystals with Nanoporous Structures for Promoting Cascade Reactions. ACS Appl. Nano Mater 4, 1672–1681 (2021). [Google Scholar]
- 16.Baskaran Y et al. An in cellulo -derived structure of PAK4 in complex with its inhibitor Inka1. Nat. Commun 6, 8681 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abdelfattah AS et al. Bright and photostable chemigenetic indicators for extended in vivo voltage imaging. Science 365, 699 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Grimm JB et al. A general method to fine-tune fluorophores for live-cell and in vivo imaging. Nat. Methods 14, 987–994 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grimm JB et al. A general method to optimize and functionalize red-shifted rhodamine dyes. Nat. Methods 17, 815–821 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grimm JB et al. A General Method to Improve Fluorophores Using Deuterated Auxochromes. JACS Au 1, 690–696 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeNardo L & Luo L Genetic strategies to access activated neurons. Curr. Opin. Neurobiol 45, 121–129 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu Q et al. The Temporal Pattern of cfos Activation in Hypothalamic, Cortical, and Brainstem Nuclei in Response to Fasting and Refeeding in Male Mice. Endocrinology 155, 840–853 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Sherin JE, Shiromani PJ, McCarley RW & Saper CB Activation of Ventrolateral Preoptic Neurons During Sleep. Science 271, 216–219 (1996). [DOI] [PubMed] [Google Scholar]
- 24.Gammie SC & Nelson RJ cFOS and pCREB activation and maternal aggression in mice. Brain Res. 898, 232–241 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Haller J, Tóth M, Halasz J & De Boer SF Patterns of violent aggression-induced brain c-fos expression in male mice selected for aggressiveness. Physiol. Behav 88, 173–182 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Josselyn SA & Tonegawa S Memory engrams: Recalling the past and imagining the future. Science 367, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Linghu C et al. Recording of cellular physiological histories along optically readable self-assembling protein chains. 2021.10.13.464006 (2021) doi: 10.1101/2021.10.13.464006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das AT, Tenenbaum L & Berkhout B Tet-On Systems For Doxycycline-inducible Gene Expression. Curr. Gene Ther 16, 156–167 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amemiya T, Kambe T, Fukumori R & Kubo T Role of protein kinase Cβ in phorbol ester-induced c-fos gene expression in neurons of normotensive and spontaneously hypertensive rat brains. Brain Res. 1040, 129–136 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Furth PA et al. Temporal control of gene expression in transgenic mice by a tetracycline-responsive promoter. Proc. Natl. Acad. Sci. 91, 9302–9306 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iwamoto M, Björklund T, Lundberg C, Kirik D & Wandless TJ A general chemical method to regulate protein stability in the mammalian central nervous system. Chem. Biol 17, 981–988 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miyamae Y, Chen L-C, Utsugi Y, Farrants H & Wandless TJ A Method for Conditional Regulation of Protein Stability in Native or Near-Native Form. Cell Chem. Biol 27, 1573–1581.e3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Q & Tucker CL Engineering genetically-encoded tools for optogenetic control of protein activity. Curr. Opin. Chem. Biol 40, 17–23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hertel F & Zhang J Monitoring of post-translational modification dynamics with genetically encoded fluorescent reporters. Biopolymers 101, 180–187 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Specht EA, Braselmann E & Palmer AE A Critical and Comparative Review of Fluorescent Tools for Live-Cell Imaging. Annu. Rev. Physiol 79, 93–117 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Gibson DG et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Thomas P & Smart TG HEK293 cell line: A vehicle for the expression of recombinant proteins. J. Pharmacol. Toxicol. Methods 51, 187–200 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Geraerts M, Michiels M, Baekelandt V, Debyser Z & Gijsbers R Upscaling of lentiviral vector production by tangential flow filtration. J. Gene Med 7, 1299–1310 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Tomasello G, Armenia I & Molla G The Protein Imager: a full-featured online molecular viewer interface with server-side HQ-rendering capabilities. Bioinformatics 36, 2909–2911 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.