

Abstract
Insulin-like growth factor 2 (IGF2) emerged as a critical mechanism of synaptic plasticity and learning and memory. Deficits in IGF2 in the brain, serum, or cerebrospinal fluid are associated with brain diseases including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and amyotrophic lateral sclerosis. Increasing IGF2 levels enhances memories in healthy animals and reverses numerous symptoms in laboratory models of aging, neurodevelopmental disorders, and neurodegenerative diseases. These effects occur via the IGF2 receptor (IGF2R) – a receptor that is highly expressed in neurons and regulates protein trafficking, synthesis, and degradation. Here, I summarize the current knowledge regarding IGF2 expression and functions in the brain, particularly in memory, and propose a novel conceptual model for IGF2/IGF2R mechanisms of action in brain health and diseases.
Keywords: IGF2 receptor, cation-independent mannose 6 phosphate receptor, memory, neurodegenerative disease, neurodevelopmental disorder, protein metabolism
IGF2, an understudied factor with unique tissue distribution, important for developmental growth and tumor suppression
Insulin-like growth factor 2 (IGF2), like the structurally similar IGF1, was discovered as a serum factor with insulin-like activity [1] and, due to its structural similarity to insulin, was named an insulin-like-growth-factor. Insulin, IGFs, their respective high affinity receptors, and IGF-binding proteins that regulate IGF tissue distribution, constitute the IGF/insulin system (Figure 1) (see [2] and [3] for excellent reviews of this system).
Figure 1. Schema depicting the Insulin/IGF system.
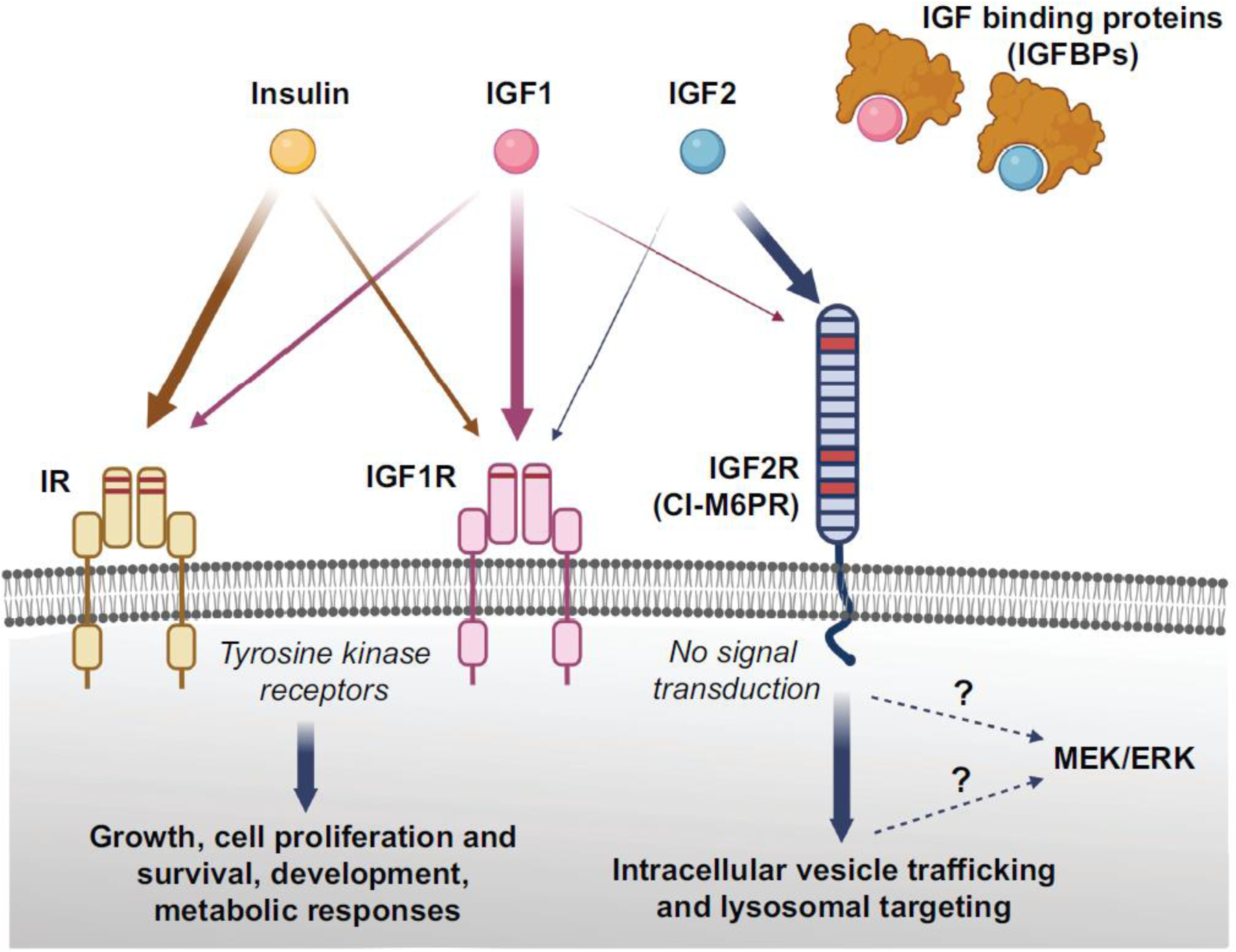
Insulin, IGF1, IGF2, and the relative high affinity receptors insulin receptor (IR), IGF1 receptor (IGF1R), and IGF2 receptor (IGF2R), along with IGF binding proteins are shown. IGF2R is also known as cation-independent mannose 6 phosphate receptor (CI-M6PR). The relative affinity of each ligand for the receptors is represented by the thickness of arrows. Insulin and IGFs can cross-bind their respective high affinity receptors, but with lower affinity, but insulin does not seem to bind to IGF2R. The IR and IGF1R are tyrosine kinase receptors; they consist of an extracellular ligand-binding domain and a cytosolic tyrosine kinase domain that autophosphorylates upon ligand binding and transphosphorylates several substrates that initiate downstream signalling supporting growth, cell proliferation and survival, development, and metabolic responses. IGF2R has a large N-terminal extracellular region of 15 homologous domains, a single membrane-spanning region, and a small cytoplasmic tail, and it lacks intrinsic signalling capability (no signal transduction). However, in neuronal cultures, Igf2/Igf2R-mediated MEK/ERK activation has been reported as critical for synapse formation and spine maturation. Whether IGF2R or downstream intermediate mechanism activate this signalling remains to be determined (indicated by the dotted lines). The binding sites for IGF2 or M6P are located on different segments of the protein. The main known functions of IGF2R are to bind IGF2, activate intracellular vesicle trafficking, and target it to lysosomal degradation, and traffic M6P-conjugated proteins, such as lysosomal enzymes. Figure created using BioRender.com
The two IGFs were initially investigated for their roles in tissue and tumor growth. Gene knockout studies in mice have shown that both IGFs play a key role in the development and growth of a variety of tissues. IGF2 was considered redundant with IGF1 because both are expressed during development and can bind, although with different affinity, to the same receptors —namely the high-affinity receptor for IGF1, or IGF1R, and the high-affinity receptor for IGF2, or IGF2R. Further investigations have, however, shown that the two IGFs play opposite roles in growth regulation via the distinct IGFRs, with IGF1 and IGF2 promoting growth via IGF1R and IGF2 suppressing it via IGF2R [4–7]. Notably, IGF1R and IGF2R are structurally and functionally very distinct (Figure 1): IGF1R, like structurally similar insulin receptors (IRs), is a tyrosine kinase receptor, whereas IGF2R consists of a single transmembrane protein with 15 homologous domains and a small cytoplasmic tail. Moreover, IGF2R lacks intrinsic tyrosine kinase or any enzymatic activity [8]. IGF2R binds, in addition to IGF2, mannose-6-phosphate residues, which intracellularly are conjugated to glycoproteins; thus, IGF2R is also known as cation-independent-mannose-6-phosphate receptor (CIM6PR) [7]. The receptor has three M6P recognition sites and its ability to bind M6P-containing proteins is an ancestral property of the receptor, which has been found starting from molluscs; only in mammals the CIM6PR acquired a binding site for IGF2 [9]. Thus far, IGF2R has been mostly known for its roles in targeting IGF2 to lysosomal degradation and transporting lysosomal enzymes to lysosomes [10] (Figure 2).
Figure 2. Schema depicting IGF2 and IGF2R endocytosis and intracellular trafficking.
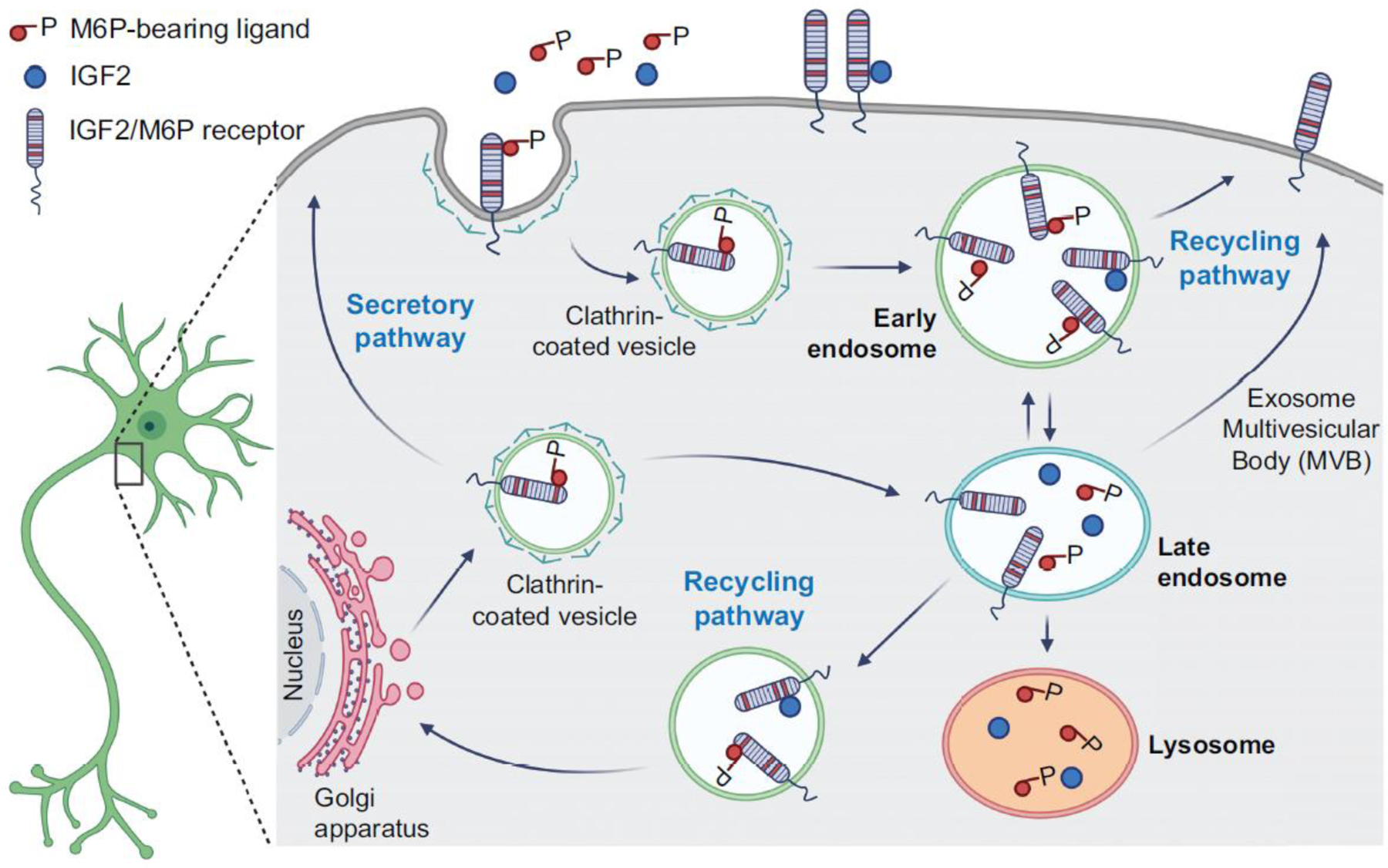
IGF2R is internalized from the plasma membrane and traffics proteins via endosomes to either recycle them to the plasma membrane, or to progress them to late endosomes from which they can travel to the trans Golgi network. IGF2R also traffics proteins from the trans Golgi network to either late endosomes and then lysosomes or back to the plasma membrane. For example, IGF2R binds extracellular IGF2 and transports it to lysosomes for degradation and sorts lysosomal hydrolases to lysosomes via M6P binding. Specifically, lysosomal hydrolase precursors are modified in the cis-Golgi by the addition of M6P. At the trans-Golgi, lysosomal hydrolases bind M6P receptors, including IGF2R, which in turn interact with adaptor proteins to form clathrin-coated vesicles (CCVs). CCVs bud off from the trans-Golgi and fuse with late endosomes. The lysosomal hydrolases are released while the receptors are recycled back to the Golgi for reuse. It is unclear whether these changes in neurons may occur in the cell body as well as in processes, therefore the window’s position is not indicative of a selective cellular compartment. Figure created using BioRender.com
The patterns of expression of IGF1 and IGF2 are also distinct [11,12]. The two IGFs are expressed in different tissues during defined developmental stages, suggesting that they have tissue-specific functions: in mammals, IGF1 is primarily expressed after birth when it is produced mostly by the liver, while IGF2 is typically expressed during early embryonic and fetal development in a variety of somatic tissues [11, 12]. The expression of both IGFs dramatically decays postnatally in most mammalian tissues [13], but the level of IGF2 remains relatively higher throughout life in a few tissues, and particularly in the brain [13]. The main sources of IGF2 in adult mammals are liver and epithelial cells lining the surface of the brain, i.e., the meninges and choroid plexus. Although controlled comparisons require further validation, some studies reported species-related differences in IGF2 expression. For instance, in mice, IGF2 levels decrease in most tissues within a few weeks of birth [11, 14, 15], whereas in humans, IGF2 serum levels remain elevated throughout life [16, 17]. The reasons for these differences are unclear; however, in both humans and rodents the expression regulation of IGF2 in the prenatal and postnatal brain, including choroid plexus are similar [13].
IGF2 expression is subject to genomic imprinting through mechanisms that are still not fully understood. In most tissues, but not in the brain, the Igf2/IGF2 gene is maternally imprinted; hence, it is expressed by the paternal allele [18, 19]. Loss of imprinting of the IGF2 gene in humans is associated with growth disorders, such as that found in Beckwith-Wiedemann syndrome, which combines overgrowth with a variety of malignant tumors. As the mitogenic activity of IGF2 is mediated through IGF1R, it is possible that the growth effects found in Beckwith-Wiedemann syndrome are mediated via this receptor. Conversely, both epimutation and copy number variants of Igf2/IGF2 cause limited growth, which is characteristic of Silver-Russell syndrome [20]. Paternally transmitted IGF2 loss-of-function variants also result in poor growth associated with macrocephaly and short stature at preschool age, feeding problems, neuropsychomotor developmental delays, cardiac and male genital anomalies, and finger deformities, underscoring the critical role of IGF2 in developmental growth regulation. IGF2 variants are not only linked to poor growth but may also decrease the risk of diabetes mellitus type 2 [2], suggesting that IGF2 also plays a role in metabolism regulation, although the underlying mechanisms are unclear.
Compared to its role during fetal development, IGF2 contributions during postnatal growth and metabolism responses are much less understood. IGF2 seems to regulate the development and maintenance of adipocyte [21], pancreatic β islet cells [22], skeletal muscle mass [23] and placenta [24]. IGF2 also plays a key role in various diseases including cancer [25], as well as endocrine, metabolic [6], and cardiovascular diseases [26], although these effects likely implicate different receptors of the IGF system.
Much remains to be understood about IGF2 and its effects via IGF2R, especially in the brain, where it recently emerged as a critical player in cognitive functions. In this review, I will focus on IGF2 and its expression and functions via IGF2R in the central nervous system (CNS). I will summarize and discuss studies in (healthy) animals indicating a role of IGF2 as a cognitive enhancer, and IGF2’s effects in CNS diseases, particularly in neurodevelopmental disorders, and neurodegenerative diseases.
IGF2 expression in the brain and its role in memory and cognition
As mentioned, compared to other tissues, the levels of IGF2 in the brain remain relatively elevated throughout life [13]. The reason for these relatively high levels of IGF2 in the fully developed brain remained unclear until about a decade ago, when we and other research groups identified IGF2’s critical contribution to memory processes [27, 28]. Since then, several studies provided new information regarding the unique role of IGF2 in healthy brain functions as well as its dysregulations in neuropsychiatric diseases.
In the adult brain, like in the fetal brain, IGF2 is primarily produced by the choroid plexus, leptomeninges, and parenchymal vasculature, where it is biallelically expressed [13, 29]. This biallelic expression contrasts with the monoallelic expression of most peripheral tissues, including the liver—the major source of peripheral IGF2-where the gene is maternally imprinted. IGF2 in the brain is also produced by stromal cells, whose allelic expression appears inverted compared to that of peripheral tissues. For example, in many regions of the rat brain, including the hippocampus, anterior cingulate cortex, medial prefrontal cortex, and amygdala, IGF2 mainly derives (>90%) from the maternal allele [30]. The mechanisms and functional meaning of this unique regulation as well as its human counterparts remain to be understood. It is also unknown whether the unique brain allelic expression varies over the lifespan, how it is regulated in different brain cell types in baseline conditions, and whether it changes in response to activity-dependent processes such as learning.
The cell type-specific distribution of the IGF2R in the brain is also poorly characterized. Global genetic knockout of IGF2R leads to death at the time of birth, demonstrating its key role in development [31], but the roles of IGF2R in adult tissues remain to be understood. RNA hybridization and immunohistochemistry studies have shown that IGF2R is enriched in many brain regions such as the olfactory bulb, striatum, pallidum, hypothalamus, thalamus, hippocampus, midbrain, cortex, and cerebellum [8]. At the cellular level, IGF2R is significantly more abundant in neurons compared to astrocytes, microglia, or other brain cells, where its expression seems negligible [8, 32]. This differential neuronal enrichment of IGF2Rs implies that IGF2 in the brain preferentially targets neuronal functions.
In one of our early studies, which identified IGF2 as part of the molecular cascade underlying long-term memory, we found that IGF2 expression is upregulated at both mRNA and protein levels following inhibitory avoidance (IA) learning in the adult rat hippocampus. This increase was required for memory consolidation [27] i.e., the process that establishes and stabilizes a long-term memory trace from an initial fragile learned representation. Similar results were found by another group with a different type of learning, contextual fear extinction, and in a different species, the mouse [28]. Furthermore, studies on cell cultures and adult mouse brain showed that IGF2 via IGF2R is a target of the IKK/NF-kB pathway critical for synapse formation and maturation [33]. Recently, IGF2R was reported to have an important role in layer 2/3 of the auditory cortex for the precision of recent memories, the stability of remote memories, and the development of fear generalization [34]. These findings provide new insights into the neural mechanisms that underlie the time-dependent development of fear generalization that may occur over time after a traumatic event. Collectively, these studies indicated that IGF2 has an important role in functions of healthy, fully developed brains, specifically in plasticity, memory, and cognition.
IGF2 exerts important effects beyond its endogenous expression. Administration of recombinant IGF2 at the time of learning or memory recall significantly enhances several types of hippocampal-dependent memories (Table 1, [27, 28, 32, 35–38]) including fear extinction [28, 35], suggesting that IGF2 acts as a potent memory and cognitive enhancer. As the memory-enhancing effects require IGF2R, and not IGF1R, it appears that IGF2R ligands may represent potential therapeutic targets for cognitive impairments. IGF2 crosses the blood brain barrier (BBB), and subcutaneous (s.c.) injections of IGF2, like intracerebral administrations, are highly effective in promoting memory and cognitive enhancement in healthy mice [35, 36, 39–41]. In fact, a s.c. administration of IGF2 in mice significantly increases retention and persistence of several types of hippocampus-dependent memories, including contextual, spatial, and social memories; it also potentiates working memory. Furthermore, potentiated memories following IGF2 treatment remain flexible, as shown by their rate of reversal learning, which is similar to that of normal memories [35]. Notably, in contrast to the significant effects on hippocampus-dependent memories, IGF2 treatment does not appear to change amygdala-dependent long-term memories such as cued conditioning [35–37], suggesting that the effect is selective for certain memory systems.
Table 1.
References, divided by species, for the role of IGF2 in neuronal plasticity, memory formation and enhancement (top) and for effects of IGF2 in aging, neural diseases, and other effects (bottom)
| CONDITION OR DISEASE | REFERENCES | |||
|---|---|---|---|---|
| Species | ||||
| Human | Rat | Mouse | Chick | |
| Memory Formation and Enhancement | [27, 32, 35, 37] | [28, 32, 34, 36] | ||
| Plasticity | [38] | |||
| Aging | [39, 48] | |||
| Neurodevelopmental Disorders | [40, 41, 49] | |||
| Huntington Disease | [51] | [50, 51] | ||
| Alzheimer’s disease | [45, 51, 52, 55, 57–59] | [54, 56, 57, 60] | ||
| Parkinson Disease | [62] | [61, 62] | ||
| Schizophrenia | [63, 64] | |||
| Stroke | [66] | [65] | ||
| Repair from Stress | [67, 68] | |||
| Traumatic Brain Injury or Hemorrhagic Brain Damage | [65, 69, 70] | |||
| Depression | [71, 72] | |||
| Amyotrophic Lateral Sclerosis | [73, 74] | [74] | [73, 74] | |
| Motor Neuron Regeneration | [75, 76] | |||
| Neuroprotection | [67, 77, 78] | |||
| Psychiatric and Neurological Disorders | [5] | [5] | [5] | |
| Lack of effect in Angelman syndrome | [79] | [79] | ||
| Lack of effect on cognitive impairment following TBI | [80] | |||
| Brain metastasis formation in breast cancer | [81] | [81] | ||
| Food intake | [82] | [83] | ||
To enhance memory, IGF2 must be present when the brain system is in an active state, as the effect is seen only within a timeframe close to learning or memory recall. For example, a bilateral injection of IGF2 in the dorsal hippocampus of rats given immediately after IA learning significantly potentiates memory strength and persistence, and the effect is very long lasting. However, if IGF2 is injected 24 hours after learning, it produces no effect. In contrast, if 24 hours after learning, an IGF2 injection is given alongside memory recall, then the memory is significantly enhanced [27]. Thus, IGF2 must recruit mechanisms activated by learning or memory recall in order to potentiate and prolong memory retention.
The understanding of the molecular and cellular mechanisms underlying IGF2’s effect on memory consolidation and enhancement is still very limited. We know that the memory enhancement produced by IGF2 requires IGF2R and not IGF1R. In addition, IGF1 does not have similar memory-enhancing effects, suggesting that the mechanisms underlying IGF2-mediated memory enhancement are distinct from the typical cell ‘growth’ activation mediated by G protein-coupled tyrosine kinase receptors signalling pathways. The unique recruitment of IGF2R also implies that memory-enhancing processes engage this receptor’s downstream cellular mechanisms, i.e., endosomal trafficking and lysosomal targeting. IGF2-mediated memory enhancement also requires hippocampal de novo protein synthesis, the immediate early gene activity-regulated cytoskeleton-associated protein (ARC/ARG3.1), and glycogen synthetase kinase (GSK) [27]. However, it does not engage, at least in the initial phase, the transcription factors cAMP response element binding protein (CREB) and CCAAT enhancer binding protein (C/EBP) —a transcriptional cascade typically required for long-term synaptic plasticity and memory consolidation [27, 42]. In sum, it appears that IGF2 targets, at least in its initial phase of action, synaptic mechanisms of protein synthesis, ARC and receptor metabolism or trafficking, rather than transcription-dependent cascades [27]. On extinction learning it was found that IGF2 promotes the survival of immature neurons apparently via IGF1R [28]; however, these neuronal survival mechanisms were not investigated in the IGF2-mediated extinction enhancement, for which the underlying mechanisms remain to be determined.
At the cellular level, studies conducted in several species showed that IGF2 promotes synaptic plasticity. In the marine mollusk Aplysia californica, it produces an enhancement of both synaptic transmission and neurite outgrowth accompanied by a reduction of excitability [43]. In the rat hippocampus, IGF2 supports the persistence of long-term potentiation (LTP) in an IGF2R-dependent manner [27]. IGF2 also acts as a neuroprotective factor as a target of IKK/NF-kB and via activation of IGF2R and MEK/ERK to promote synapse formation and maturation in adult mice [33]. Despite these important findings, a comprehensive understanding of the cellular and electrophysiological mechanisms underlying IGF2 functions is lacking, and research is urgently needed in this area.
Given the critical role of IGF2R in the memory-enhancing effects evoked by IGF2, some additional mechanistic insights can be inferred from studies on the IGF2R itself in the brain and cognition. Across species, IGF2R in healthy brains is detected mostly in neurons. IGF2R expression is detected in other brain cell types only in some disease states, for instance in microglia nodules of brains infected by viruses such as HIV and in reactive astrocytes associated with senile plaques of Alzheimer disease (AD) brains [44, 45]. Local pharmacological blockade of IGF2R or its genetic knockout in the dorsal hippocampus of rats or mice very rapidly impairs long-term memories without affecting learning or short-term memories, indicating that IGF2R is required for memory consolidation [32].
Two interesting and possibly related observations worth underscoring are IGF2R’s rapid recruitment in memory consolidation [32], and the receptor’s established roles in protein trafficking via endosomes and lysosomal targeting [10]. Based on these considerations, I suggest that learning engages endosomal and lysosomal functions via IGF2R. Also, as hippocampal IGF2R controls the upregulation of de novo protein synthesis induced by learning required for long-term memory formation [32], it is reasonable to hypothesize that this protein synthesis is closely interconnected with IGF2R-mediated endosomal trafficking and lysosomal targeting.
Endosomal trafficking has been at the center of attention of several synaptic plasticity studies, but its role has been mostly investigated in relation to trafficking of neuronal receptors such as the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) glutamate receptors [46]. It is, however, plausible that endosomes play broader roles in brain functions, especially in the highly compartmentalized neuronal cells. For example, the specialized functions of pre- and post-synaptic compartments strongly rely on vesicles, including endosomes and lysosomes, for neurotransmission, receptor recycling and trafficking, and, as discussed further down, possibly also for local protein synthesis. Based on the current knowledge, below I will propose a conceptual model for the mechanisms of action of IGF2 in brain functions and diseases.
While much remains to be explored concerning the underlying mechanisms of IGF2, its behavioural effects as a memory enhancer have led to the hypothesis that IGF2 could be a potential target for developing novel treatments for cognitive impairments [5, 8, 13, 47]. Studies in rats that tested the effect of IGF2 in aging-related cognitive impairment provided support to this idea, and demonstrated a significant recovery from aging-related memory decay after a single injection in the hippocampus [39]. As discussed in the next sections, other experiments investigated the effects of administering or overexpressing IGF2 in a variety of CNS disease laboratory models, suggesting that IGF2 may be beneficial for several disorders.
The role of IGF2 in CNS diseases, particularly in neurodevelopmental disorders and neurodegenerative diseases
Which CNS disorders are linked to alterations of IGF2? Are there diseases, perhaps even ones without detectable alterations of the IGF2 pathway, that may benefit from IGF2 or IGF2-mimicking treatments? What are the mechanisms underlying the effects of IGF2 in these contexts?
Numerous studies have provided evidence for positive effects of IGF2 in a variety of laboratory models of CNS diseases, including AD, Parkinson disease (PD), Huntington disease (HD), amyotrophic lateral sclerosis (ALS), schizophrenia, neurodevelopmental disorders such autism and Angelman syndrome, depression, stress-related disorders, traumatic brain injury (TBI), and motor neuron degeneration. A list of references reporting these effects, organized by disease and species being investigated, is provided in Table 1 [5, 39–41, 45, 48–83].
Notably, only some of these diseases have been found to have alterations in IGF2 level in serum, cerebrospinal fluid (CSF), and/or brain areas [53, 55, 57, 58, 60, 62–64, 84, 86–88] (see Box 1). Studies in laboratory animal models show that several pathologies may benefit from treatment with IGF2 or other ligands that activate IGF2R even in conditions that do not have any apparent link to IGF2 or IGF2R deficits (see Table 1). Also, the benefits of IGF2 treatment in CNS disease models are not limited to recovering memory or cognitive impairments, but rather extend to improving a variety of other symptoms. Despite the mechanisms underlying these positive effects of IGF2 on a wide spectrum of diseases are still unclear, the behavioural results identify IGF2R as an important potential therapeutic target for a variety of CNS disorders. I will discuss briefly current knowledge of IGF2 effects on neurodevelopmental disorders and neurodegenerative diseases and propose a conceptual model for the underlying mechanisms.
BOX 1. IGF2 levels in diseases.
In schizophrenia, a study on serum IGF2 levels reported decreased IGF2 levels in schizophrenia patients compared to controls (32 and 30 individuals, respectively) [64]. In a subsequent study, correlation analysis in 31 patients relative to 30 controls revealed that the changes of serum IGF-2 levels in patients were significantly correlated with the improvements of negative symptoms following two months of atypical antipsychotic treatment. In fact, after this treatment, a significant improvement was observed in patients, and their serum IGF2 levels significantly increased compared with those at baseline [63]. Conversely, another study reported that a comparison in 53 schizophrenic male patients relative to 52 control males revealed an increased IGF2 serum level [84].
In autism spectrum disorder (ASD), a study on 25 autistic children reported no changes in CSF IGF2 levels compared to the control group of 16 children [85]. The small sample size of this study is a major limitation, and larger-scale studies are needed for more definitive conclusions.
Alzheimer’s disease patients have significant alterations of IGF2 levels. In multiple studies, postmortem tissue or genomic analysis indicated decreased IGF2 in some brain regions, including the hippocampus, frontal cortex, and hypothalamus [53, 57, 60, 86; studies with 9–33 patients/group], although one study reported an increase in Igf2 mRNA levels in the prefrontal cortex of 13 AD patients—without a change at the protein level [87]. In addition, two studies found increased IGF2 in the cerebrospinal fluid (CSF) of AD patients (92 and 32 individuals in each of the studies, respectively) compared to controls (72 and 20 individuals, respectively) [55, 58]. This outcome was also confirmed in a smaller study of 10 AD patients compared to 10 controls [52]. Another study suggests that the IGF2 increase in CSF correlates with amyloid-β (Aβ42) levels (16 AD patients and 15 controls) [88].
Parkinson’s disease patients have altered IGF2 levels as well. Specifically, a study on 43 Parkinson’s disease patients and 41 controls showed lower IGF2 plasma levels in the patients, and a dramatic decrease in IGF2 mRNA and protein levels in their peripheral blood mononuclear cells (PBMCs), along with a downregulation of key components of the initial stages of the autophagy process [62]. Although IGF2 levels were not found to be directly correlated with PD severity, PD PBMC samples showed a correlation between IGF2 levels and autophagy gene profile expression in a sex-dependent fashion [62], suggesting a possible link between IGF2 and autophagy.
A study on Huntington’s disease patients (13 patients and 13 controls) found reduced IGF2 protein levels in their caudate-putamen, and HD-derived PBMCs were found to have approximately a 70% decrease in IGF2 protein levels compared to controls, in addition to a small, but significant, decrease of circulating IGF2 in their plasma [50].
In autism-related laboratory animal models, including BTBR mice (BTBR T+ Itpr3tf/J, a strain of mice that shows phenotypic similarities to traits described in individuals with autism spectrum disorder) and Fmr1 KO mice, a model of Fragile X syndrome, IGF2 treatment ameliorated cognitive impairments. In the BTBR mice IGF2 administration also improved social deficits and repetitive behaviours [40]. In the BTBR mice, the contribution of IGF receptors on the effect of IGF2 was assessed, and it was found that the effects required IGF2R and not IGF1R [40]. In another neurodevelopmental disorder model, a widely employed mouse model of Angelman Syndrome based on the maternal deletion of the E3 ubiquitin ligase (ube3a) gene [89], a systemic injection of IGF2 or M6P reversed most core deficits, including impairments in memory, working memory and flexibility, deficits in motor functions, and repetitive behaviours [41]. IGF2 also significantly protected Angelman Syndrome mice against seizures [41]. Some of these conclusions (the IGF2 effects) were recently disputed in a study that used a new rat model of Angelman Syndrome generated using CRISPR technology [79]. Although the different results may be due to differences of the models and species being used (which future studies should be able to assess), I would like to note some methodological and interpretative caveats of the study in rats [79]. First, some of the required controls, such as a dose-response curve when testing the behavioural effect of s.c. administration of IGF2 in a different species, and validation of the biological activity of the IGF2 preparation being used appear to be lacking. Second, the use of different protocols and behavioural schedules compared to those used in prior studies in mice [41] could contribute to the divergent in outcomes. Lastly, the rat model needs confirmation and validation from other laboratories. A point of direct disagreement between the studies concerns some results reported in [79] on the mouse model of Angelman syndrome, and specifically the novel object recognition (nOR) impairment of Angelman Syndrome mice, a phenotype which study [79] failed to reproduce but that has been reported in several studies from different laboratories [41, 90–92].
In studies of various other disease models by numerous laboratories, IGF2 overexpression or administration has shown positive effects, as discussed next and as summarized in Table 1.
Mouse models of AD. IGF2 virus-mediated expression in the hippocampus ameliorated cognitive impairments in the widely used mouse model of AD Tg2576 [57]. This mouse overexpresses a mutant form of amyloid precursor protein (APP, isoform 695) with the Swedish mutation; hence, it produces high levels of amyloid β(Aβ) peptide, which result in amyloid plaques. IGF2 overexpression enhanced memory in aged wild-type mice, while in Tg2576 mice it promoted dendritic spine formation, rescued behavioural deficits, restored normal hippocampal excitatory synaptic transmission, and significantly reduced the levels of Aβ40 and Aβ42. This amyloid reduction appeared to take place via IGF2R [57]. Relatedly, in AD patients, RNA sequencing data (RNA-seq) revealed reduced IGF2 expression in the brain [60]. In addition, in the hippocampus of the Tg2576 mice, acute IGF2 treatment (although it is unclear from the reporting at which timepoint it was administered) increased the expression of the PI3K and AKT genes and their downstream gene CREB. The IGF2 treatment also mitigated water maze memory impairment and reduced amyloid plaques as well as levels of Aβ40 and Aβ42 [60]. In another AD model, APPswe.PS1dE9 mice, which bear mutant transgenes of APP and presenilin-1 and show progressive age-related development of Aβ accumulation and cognitive deficits, intracerebroventricular IGF2 administrations reversed and prevented several pathophysiologic processes associated with AD, by mechanisms such as reducing the number of hippocampal Aβ40- and Aβ42-positive amyloid plaques and increasing cholinergic markers [56].
traumatic brain injury (TBI). In rats, intracerebroventricular injections of IGF2 or IGF1 reduced the volume of ischemic damage after a permanent occlusion of the middle cerebral artery [69], although in a mouse model of chronic stage of TBI, an acute injection of IGF2 did not ameliorate memory deficits in either nOR or spatial memory tasks [80]. The effects of prolonged or chronic treatments of IGF2R ligands in TBI remain to be tested.
Huntington’s Disease. In mice carrying a genetic disruption of the transcription factor XBP1, the progression of diseases such as HD, ALS, PD, and AD, as well as their associated protein aggregation in their brain, is significantly reduced [51], indicating that elimination of XBP1 may lead to the discovery of potential beneficial mechanisms. Using a mouse model of HD reproducing behavioural symptoms, as well as aggregation and deposition of Huntingtin in the brain, an unbiased screening revealed that IGF2 is one of the most significantly upregulated genes in the brain of HD mice with the XBP1 disruption. IGF2 treatment protected against neurodegeneration as it reduced protein aggregation in cell culture and in vivo in an HD mouse model. The effect did not appear to involve autophagy or proteasome-mediated degradation, but rather the secretion of soluble polyglutamine peptide (polyQ) tract species through exosomes and microvesicles [50].
In summary, IGF2 appears to have beneficial effects in a variety of models of neurodevelopmental disorders and neurodegenerative diseases, suggesting that identifying the underlying mechanisms of action may uncover important information for designing potential therapeutic approaches. How can these mechanisms be best identified? One approach could be to pinpoint and compare the pathways targeted by IGF2R ligands across models and systems, i.e., in healthy animals with memory enhancement following treatment with IGF2R agonists and in animal models of various diseases in which IGF2R ligand treatment attenuated their deficits. Notably, all these diseases carry the commonality of aberrant protein accumulation in the brain. Diseases that accumulate brain proteins in aggregates are defined as proteinopathies or diseases of misfolded proteins [93].
Conceptual model of IGF2-IGF2R mechanisms of action in memory formation and enhancement, and their potential impairments in CNS diseases
In this section, I outline a proposed conceptual model of IGF2 mechanisms of action via IGF2R in the CNS, which may help explain the role of this pathway in plasticity and memory, and its possible dysregulation in neural diseases. I suggest that IGF2R controls neuronal endosomal trafficking networks, thereby regulating protein sorting, recycling, synthesis, and degradation (Figure 3A). The learning-induced activation of IGF2R by IGF2 or other ligands would stimulate vesicular network functions, including endosomal genesis and trafficking and their communication with autophagy pathways, lysosomes, and the Golgi apparatus, providing critical intra-subcellular compartment communications essential for neuronal plasticity. As learning-induced de novo protein synthesis in the hippocampus is coupled to an induction of autophagic flux [94], I also propose that vesicular network regulated via IGF2R balances protein synthesis and degradation. IGF2R may promote genesis or mobilization of endosomes, which could serve as platforms of the de novo mRNA translation. IGF2R-mediated vesicle trafficking may support the translation-autophagy coupling and enhance the autophagic flux, hence autophagy. Vesicle-based mRNA translation coupled to vesicle trafficking including autophagy would be advantageous for protein synthesis taking place at specialized compartments such as synapses as it could organize mRNA trafficking, their translation, and efficient local quality control of the translated proteins. Different ligands of the IGF2R could also regulate the trafficking of distinct vesicular networks, and vesicles activated via IGF2R may also contribute to intracellular signalling.
Figure 3. Schematic model of (A) IGF2R-dependent mechanisms of action in neurons following IGF2 binding and (B) illustrating points of dysregulations that may lead to protein accumulation, and which may benefit from IGF2R ligand treatments.
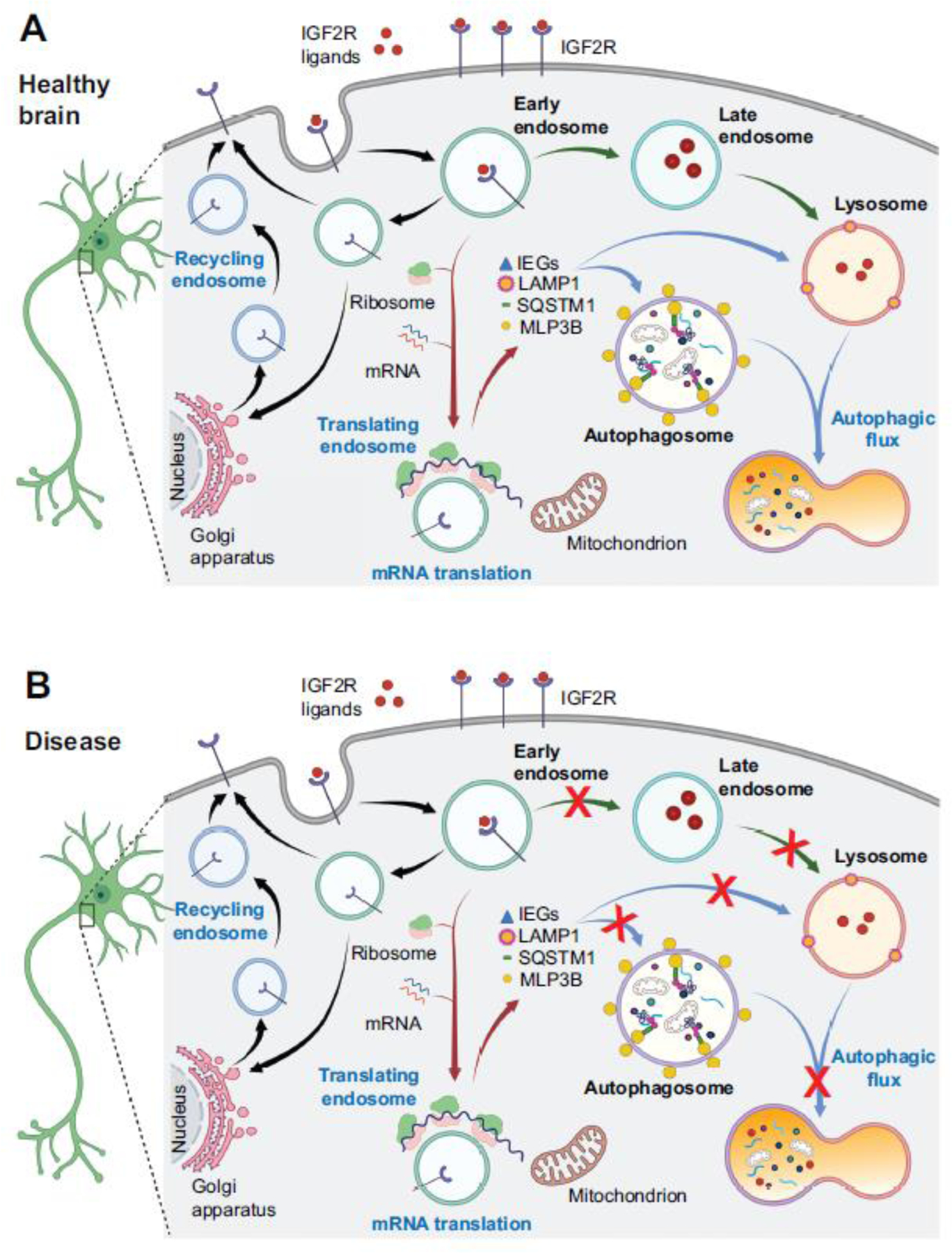
The schematic summarizes the proposed model for IGF2/IGF2R mechanisms of action in brain health and disease. (A) Binding of IGF2 to the IGF2R on the plasma membrane initiates IGF2R-bearing endosomal trafficking, beginning with the generation of early endosomes (EE). These EE can proceed to late endosomes and then lysosomes, or traffic to the Golgi apparatus, from which recycling endosomes can return to the plasma membrane. As blocking IGF2 blocks learning-induced mRNA translation [32], I hypothesize that a pool of endosomes is recruited to serve as platform for this de novo mRNA translation, including the translation of immediate early genes (IEGs) and proteins involved in autophagy such as MLP3B, SQSTM1, and LAMP1. Since learning-induced mRNA translation is coupled to autophagy [94], I suggest that the endosomes supporting translation are in direct communication with autophagosome formation and activation of autophagy. Protein degradation underlying memory and neuronal plasticity may also occur via the IGF2-bearing endosome-lysosomal classical path. Many steps and mechanisms of this model are still unclear and future studies should be able to experimentally test them. (B) A summary of possible major cell biological steps (indicated by a red X) that could be impaired in disease conditions of altered de novo protein synthesis, autophagy, and endosomal trafficking leading to protein accumulation. These diseases include neurodevelopmental disorders and neurodegenerative diseases [93, 94, 98]. Studies have shown that in brain plasticity and memory, de novo protein synthesis is coupled to autophagy [94] and that in laboratory animal models IGF2 has beneficial effects in neurodegenerative and neurodevelopmental disorders (Table 1). These results suggest that the common cell biological deficits of those diseases may include impaired autophagic flux and lysosomal targeting. The potential therapeutic-like effect produced by the activation of IGF2Rs could be in fact an increase in autophagic flux that, by enhancing protein degradation, may prevent and/or reverse protein accumulation. Figure created using BioRender.com
The idea of endosomes serving as a platform of neuronal translation is in line with recent evidence from Xenopus retinal ganglion cells showing that axonally transported late endosomes act as mRNA translation platforms [95]. It also agrees with findings showing that local mRNA translation required for synaptic plasticity is accompanied by trafficking of membranous organelles [46]. Given its vesicular localization, the IGF2R could also be involved in the genesis and trafficking of exosomes, multivesicular bodies (MVBs), and extracellular vesicles (EVs) - an area that is awaiting to be explored. One may speculate that administration of IGF2 during learning or memory recall could amplify or accelerate the endosomal trafficking associated with learning-induced synapse potentiation that has engaged the endogenous IGF2R; this potentiation of vesicular functions could strengthen memory consolidation or reconsolidation, thereby producing memory enhancement.
The mechanisms connecting endosomes and other intracellular or extracellular membranous network compartments are still largely unknown, particularly in neurons. There is lack of a clear understanding of the genesis, interaction, and specialization of the various vesicle populations that connect compartments such as the endoplasmic reticulum (ER), Golgi apparatus, plasma membrane, and EVs as well as of their local regulation in neuronal subcellular compartments such as soma vs. synapses. Yet, vesicular structures are highly enriched in neurons, and vesicle-mediated mechanisms have been implicated in CNS diseases, especially in neurodevelopmental disorders [96] and neurodegenerative diseases [97–100]. Neurodegeneration is accompanied by alterations of autophagy, lysosomal delivery or autolysosomal acidification, particularly as the brain ages [97–100]. Notably, many aspects of these diseases recapitulated in laboratory animal models appear to benefit from IGF2 treatment.
Based on the proposed model (Figure 3A), I suggest that the therapeutic effects of activating IGF2Rs as seen in preclinical models stems from the receptor’s ability to regulate vesicle trafficking, particularly the vesicular flux linked to protein degradation. Activation of IGF2R may be therefore beneficial for treating CNS proteinopathies because, by accelerating endolysosomal and autophagic flux, it would prevent and/or reverse the accumulation of proteins (Figure 3B). The flux acceleration may also generate other pools of vesicles such as EV or MVBs. One important question to be addressed by future research is whether the potential therapeutic effects of activating IGF2R can reverse compromised endosomal/autophagy flux in unhealthy neurons, or functionally compensate by potentiating pools of vesicles in healthy neurons or compartments. Finally, one should mention in this context the mitochondrion – another organelle that is linked to mRNA translation, endosomes, and autophagy, and whose function is impaired in neurodegenerative and neurodevelopmental diseases. Mitochondria and endosomes are both intimately linked to mRNA transport and translation [101], and mitochondria provide the energy demand of the de novo local protein synthesis [102]. Recent work on models of Parkinson disease, as well as studies in tissues outside the CNS like skeletal muscle and liver indicate that IGF2 may protect healthy mitochondrial functions and prevent oxidative and cellular (including neuronal) damage [103–107].
The proposed model agrees with the correlations reported between defective IGF2 levels and reduced levels of autophagy markers in PD blood samples [62] as well as with the positive effects of IGF2 in HD models, although these effects did not appear to involve protein degradation but rather trafficking of vesicles such as exosomes and microvesicles [50].
Concluding remarks and future perspectives
In rodents, IGF2 is required for memory formation, and its administration promotes memory enhancement. In humans, altered levels of brain or circulating IGF2 accompany neurodegenerative diseases, suggesting an important role for IGF2 in maintaining CNS health and efficient brain functions. IGF2 administration ameliorates numerous symptoms in animal models of neurodevelopmental and neurodegenerative diseases, most of which seem to have protein accumulation in the brain as a common pathophysiological mechanism. The effects of IGF2 occur via the IGF2R - a receptor highly enriched in neurons and involved in endosomal trafficking, protein synthesis, and lysosomal targeting. Unfortunately, the functions of IGF2 and IGF2R in the brain across species are still poorly characterized. Future studies should elucidate their expression distribution over the lifespan and their mechanisms of action both in healthy brains and in brains of neuropsychiatric diseases (see Outstanding Questions). Based on the current research on IGF2R and IGF2, I proposed new hypotheses in a conceptual model for the IGF2/IGF2R mechanisms of action. Further studies are critically needed to test elements of the model proposed, and to ultimately unravel the role of IGF2 and IGF2R in the brain, the understanding of which may inspire novel strategies to treat a variety of brain disorders.
Outstanding Questions.
Limited knowledge is available on the tissue and cell-type sources of insulin like growth factor 2 (IGF2). Most studies have thus far investigated expression and sources of IGF2 only in early development. Which tissues and cell types produce IGF2, and how does IGF2 expression change over the lifespan?
Which tissues and cell types express IGF2 receptor (IGF2R)? How does IGF2R expression change over the lifespan?
Which mechanisms underlie the effects of IGF2 in memory enhancement, as observed in animal models?
Are other receptors of the IGF-Insulin system involved in the roles of IGF2 in brain functions?
Which mechanisms underlie the effect of IGF2 in diseases of the central nervous system, particularly in neurodevelopmental disorders and neurodegenerative diseases?
Highlights.
Insulin like growth factor 2 (IGF2) plays a critical role in memory and cognition in adult rodents, while its alterations contribute to CNS diseases. Yet, the mechanisms by which IGF2 is involved in these processes are unclear.
In rodents, IGF2 administration promotes memory enhancement and persistence. IGF2 administration restores impaired memory functions in aged rats and reverses cognitive impairments and several other deficits in rodent models of neurodevelopmental and neurodegenerative disorders.
The beneficial effects of IGF2 occur via its high affinity receptor, the IGF2R, which may represent a target for potential therapeutic development.
Current knowledge of IGF2R-mediated mechanisms in both heathy brains and diseases remains limited. Here, I propose a conceptual model for these mechanisms, in which IGF2 or other IGF2R ligands promote vesicle network communications and protein metabolism quality control. This framework may explain how treatments based on IGF2R ligands could counteract diseases characterized by protein accumulation in the brain.
Acknowledgments
I thank Anna Lisa Scardovi for her valuable help in putting together the figures and manuscript formatting, and Jessica Gaunt and Camille Casino for proofreading the manuscript. I thank all the members of my lab who contributed to the discovery and understanding of IGF2 and IGF2R in the brain, memory processes, and brain diseases. The work from my lab reported in this review has been supported by the grant MH065635, FAST-US, and FAST-Italia.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests:
CMA is the Founder and current President of Ritrova Therapeutics, Inc., a start-up that stemmed from IGF2 and IGF2R studies and aims at developing treatment for neurodevelopmental disorders and neurodegenerative diseases.
References
- 1.Rinderknecht E and Humbel RE (1978) Primary structure of human insulin-like growth factor II. FEBS Lett. 89(2), 283–286 [DOI] [PubMed] [Google Scholar]
- 2.LeRoith D et al. (2021) Insulin-like growth factors: Ligands, binding proteins, and receptors. Mol. Metab 52, 101245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lewitt MS and Boyd GW (2019) The Role of Insulin-Like Growth Factors and Insulin-Like Growth Factor-Binding Proteins in the Nervous System. Biochem. Insights 12, 1178626419842176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chao W and D’Amore PA (2008) IGF2: epigenetic regulation and role in development and disease. Cytokine Growth Factor Rev. 19(2), 111–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pardo M et al. (2019) Insulin growth factor 2 (IGF2) as an emergent target in psychiatric and neurological disorders. Review. Neurosci. Res 149, 1–13 [DOI] [PubMed] [Google Scholar]
- 6.Livingstone C and Borai A (2014) Insulin-like growth factor-II: its role in metabolic and endocrine disease. Clin. Endocrinol 80(6), 773–781 [DOI] [PubMed] [Google Scholar]
- 7.El-Shewy HM and Luttrell LM (2009) Insulin-like growth factor-2/mannose-6 phosphate receptors. Vitam. Horm 80, 667–697 [DOI] [PubMed] [Google Scholar]
- 8.Wang Y et al. (2017) Insulin-Like Growth Factor-II/Cation-Independent Mannose 6-Phosphate Receptor in Neurodegenerative Diseases. Mol. Neurobiol 54(4), 2636–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nadimpalli SK and Amancha PK (2010) Evolution of mannose 6-phosphate receptors (MPR300 and 46): lysosomal enzyme sorting proteins. Curr Protein Pept Sci. 1:68–90. [DOI] [PubMed] [Google Scholar]
- 10.Ghosh P et al. (2003) Mannose 6-phosphate receptors: New twists in the tale. Nat. Rev. Mol. Cell Biol 4, 202–212 [DOI] [PubMed] [Google Scholar]
- 11.DeChiara TM. et al. (1990) A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature 345, 78–80 [DOI] [PubMed] [Google Scholar]
- 12.Louvi A et al. (1997) Growth-promoting interaction of IGF-II with the insulin receptor during mouse embryonic development. Dev. Biol 189(1), 33–48 [DOI] [PubMed] [Google Scholar]
- 13.Beletskiy A et al. (2021). Insulin-Like Growth Factor 2 As a Possible Neuroprotective Agent and Memory Enhancer-Its Comparative Expression, Processing and Signaling in Mammalian CNS. Int. J. Mol. Sci 22(4), 1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lui JC and Baron J (2013) Evidence that Igf2 down-regulation in postnatal tissues and up-regulation in malignancies is driven by transcription factor E2f3. Proc Natl Acad Sci U S A, 110(15):6181–6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee JE. et al. (1990) Pattern of the insulin-like growth factor II gene expression during early mouse embryogenesis. Development 110: 151–159 [DOI] [PubMed] [Google Scholar]
- 16.Sussenbach JS, et al. (1993) Transcriptional and post-transcriptional regulation of the human IGF-II gene expression. Adv. Exp. Med. Biol 343, 63–71 [DOI] [PubMed] [Google Scholar]
- 17.Daughaday WH. and Rotwein P (1989) Insulin-like growth factors I and II. Peptide, messenger ribonucleic acid and gene structures, serum, and tissue concentrations. Endocr. Rev 10, 68–91 [DOI] [PubMed] [Google Scholar]
- 18.DeChiara TM. et al. (1991) Parental imprinting of the mouse insulin-like growth factor II gene. Cell 64, 849–859 [DOI] [PubMed] [Google Scholar]
- 19.Giannoukakis N et al. (1993) Parental genomic imprinting of the human IGF2 gene. Nat. Genet 4, 98–101 [DOI] [PubMed] [Google Scholar]
- 20.Begemann M et al. (2012) Clinical significance of copy number variations in the 11p15.5 imprinting control regions: new cases and review of the literature. J. Med. Genet 49(9), 547–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alfares MN et al. (2018) Insulin-like growth factor-II in adipocyte regulation: depot-specific actions suggest a potential role limiting excess visceral adiposity. Am. J. Physiol. Endocrinol. Metab 315(6), E1098–E1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sandovici I et al. (2021) Autocrine IGF2 programmes β-cell plasticity under conditions of increased metabolic demand. Sci Rep. 11(1):7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zierath JR et al. (1992) Wallberg-Henriksson H. Insulin-like growth factor II stimulates glucose transport in human skeletal muscle. FEBS Lett. 307(3), 379–382 [DOI] [PubMed] [Google Scholar]
- 24.Sandovici I et al. (2022) The imprinted Igf2-Igf2r axis is critical for matching placental microvasculature expansion to fetal growth. Dev Cell. 57(1), 63–79.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scalia P et al. (2020) The IGF-II-Insulin Receptor Isoform-A Autocrine Signal in Cancer: Actionable Perspectives. Cancers (Basel) 12(2), 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faienza MF et al. (2010) IGF2 gene variants and risk of hypertension in obese children and adolescents. Pediatr. Res 67(4), 340–4 [DOI] [PubMed] [Google Scholar]
- 27.Chen DY et al. (2011) A critical role for IGF-II in memory consolidation and enhancement. Nature 469(7331), 491–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agis-Balboa RC et al. (2011) A hippocampal insulin-growth factor 2 pathway regulates the extinction of fear memories. EMBO J. 30(19), 4071–4083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sélénou C et al. (2022) IGF2: Development, Genetic and Epigenetic Abnormalities. Cells 11(12), 1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ye X et al. (2015). Insulin Like Growth Factor 2 Expression in the Rat Brain Both in Basal Condition and following Learning Predominantly Derives from the Maternal Allele. PLoS One 10(10), e0141078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lau MM et al. (1994) Loss of the imprinted IGF2/cation-independent mannose 6-phosphate receptor results in fetal overgrowth and perinatal lethality. Genes Dev. 8(24), 2953–2963 [DOI] [PubMed] [Google Scholar]
- 32.Yu XW et al. (2020) A role for CIM6P/IGF2 receptor in memory consolidation and enhancement. eLife 9, e54781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmeisser MJ. et al. (2012) IκB kinase/nuclear factor κB-dependent insulin-like growth factor 2 (Igf2) expression regulates synapse formation and spine maturation via Igf2 receptor signaling. J. Neurosci 32(16), 5688–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Concina G et al. (2021) Expression of IGF-2 Receptor in the Auditory Cortex Improves the Precision of Recent Fear Memories and Maintains Detailed Remote Fear Memories Over Time. Cereb. Cortex 31(12), 5381–5395 [DOI] [PubMed] [Google Scholar]
- 35.Stern SA et al. (2014) Enhancement of memories by systemic administration of insulin-like growth factor II. Neuropsychopharmacology 39(9), 2179–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stern SA et al. (2014) The effect of insulin and insulin-like growth factors on hippocampus- and amygdala-dependent long-term memory formation. Learn. Mem 21(10), 556–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duan Q et al. (2020) BET proteins inhibitor JQ-1 impaired the extinction of remote auditory fear memory: An effect mediated by insulin like growth factor 2. Neuropharmacology 177, 108255. [DOI] [PubMed] [Google Scholar]
- 38.Luo Y et al. (2020) The developmental and experience-dependent expression of IGF-2 in mice visual cortex. Neurosci. Lett 721, 134828. [DOI] [PubMed] [Google Scholar]
- 39.Steinmetz AB et al. (2016) Insulin-like growth factor 2 rescues aging-related memory loss in rats. Neurobiol. Aging 44, 9–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steinmetz AB et al. (2018) Insulin-Like Growth Factor II Targets the mTOR Pathway to Reverse Autism-Like Phenotypes in Mice. J. Neurosci 38(4), 1015–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cruz E et al. (2021) CIM6P/IGF-2 Receptor Ligands Reverse Deficits in Angelman Syndrome Model Mice. Autism Res. 14(1), 29–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alberini CM and Kandel ER (2014) The regulation of transcription in memory consolidation. Cold Spring Harb. Perspect. Biol 7(1), a021741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kukushkin NV. et al. (2019) Neurotropic and modulatory effects of insulin-like growth factor II in Aplysia. Sci. Rep 9(1), 14379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suh HS, Cosenza-Nashat M, Choi N, Zhao ML, Li JF, Pollard JW, Jirtle RL, Goldstein H, Lee SC. Insulin-like growth factor 2 receptor is an IFNgamma-inducible microglial protein that facilitates intracellular HIV replication: implications for HIV-induced neurocognitive disorders. Am J Pathol. 2010. Nov;177(5):2446–58. doi: 10.2353/ajpath.2010.100399. Epub 2010 Oct 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kar S et al. (2006) Cellular distribution of insulin-like growth factor-II/mannose-6-phosphate receptor in normal human brain and its alteration in Alzheimer’s disease pathology. Neurobiol. Aging, 27(2), 199–210 [DOI] [PubMed] [Google Scholar]
- 46.Rajgor D, et al. (2021) The Coordination of Local Translation, Membranous Organelle Trafficking, and Synaptic Plasticity in Neurons. Front. Cell Dev. Biol 9, 711446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alberini CM and Chen DY (2012) Memory enhancement: consolidation, reconsolidation and insulin-like growth factor 2. Trends Neurosci. 35(5), 274–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lim PH et al. (2018) Premature hippocampus-dependent memory decline in middle-aged females of a genetic rat model of depression. Behav. Brain. Res 353, 242–249 [DOI] [PubMed] [Google Scholar]
- 49.Pardo M et al. (2017) Intranasal siRNA administration reveals IGF2 deficiency contributes to impaired cognition in Fragile X syndrome mice. JCI insight 2(6), e91782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.García-Huerta P et al. (2020) Insulin-like growth factor 2 (IGF2) protects against Huntington’s disease through the extracellular disposal of protein aggregates. Acta Neuropathol. 140(5), 737–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Troncoso-Escudero P and Vidal RL (2021) Insulin-like Growth Factor 2: Beyond its Role in Hippocampal-dependent Memory. J. Cell. Immunol 3(1), 46–52 [Google Scholar]
- 52.Tham A et al. (1993) Insulin-like growth factors and insulin-like growth factor binding proteins in cerebrospinal fluid and serum of patients with dementia of the Alzheimer type. J. Neural. Transm. Park. Dis. Dement. Sect 5(3), 165–176 [DOI] [PubMed] [Google Scholar]
- 53.Rivera EJ et al. (2005) Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: link to brain reductions in acetylcholine. J. Alzheimers Dis 8(3), 247–268 [DOI] [PubMed] [Google Scholar]
- 54.Amritraj A et al. (2009) Altered levels and distribution of IGF-II/M6P receptor and lysosomal enzymes in mutant APP and APP + PS1 transgenic mouse brains. Neurobiol. Aging 30(1), 54–70 [DOI] [PubMed] [Google Scholar]
- 55.Hertze J et al. (2014) Changes in cerebrospinal fluid and blood plasma levels of IGF-II and its binding proteins in Alzheimer’s disease: an observational study. BMC Neurol. 14, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mellott TJ et al. (2014) IGF2 ameliorates amyloidosis, increases cholinergic marker expression and raises BMP9 and neurotrophin levels in the hippocampus of the APPswePS1dE9 Alzheimer’s disease model mice. PloS one 9(4), e94287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pascual-Lucas M et al. (2014) Insulin-like growth factor 2 reverses memory and synaptic deficits in APP transgenic mice. EMBO Mol. Med 6(10), 1246–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Åberg D et al. (2015) Increased Cerebrospinal Fluid Level of Insulin-like Growth Factor-II in Male Patients with Alzheimer’s Disease. J. Alzheimers Dis 48(3), 637–646 [DOI] [PubMed] [Google Scholar]
- 59.Liou CJ et al. (2019). Altered Brain Expression of Insulin and Insulin-Like Growth Factors in Frontotemporal Lobar Degeneration: Another Degenerative Disease Linked to Dysregulation of Insulin Metabolic Pathways. ASN Neuro. 11, 1759091419839515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xia L et al. (2019) Genome-wide RNA sequencing analysis reveals that IGF-2 attenuates memory decline, oxidative stress and amyloid plaques in an Alzheimer’s disease mouse model (AD) by activating the PI3K/AKT/CREB signaling pathway. Int. Psychogeriatr 31(7), 947–959 [DOI] [PubMed] [Google Scholar]
- 61.Claros S et al. (2021) Insulin-like Growth Factor II Prevents MPP+ and Glucocorticoid Mitochondrial-Oxidative and Neuronal Damage in Dopaminergic Neurons. Antioxidants (Basel) 11(1), 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sepúlveda D et al. (2022) Insulin-like growth factor 2 and autophagy gene expression alteration arise as potential biomarkers in Parkinson’s disease. Sci. Rep 12(1), 2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chao XL et al. (2020) Changes of Serum Insulin-like Growth Factor-2 Response to Negative Symptom Improvements in Schizophrenia Patients Treated with Atypical Antipsychotics. Curr. Med. Sci 40(3), 563–569 [DOI] [PubMed] [Google Scholar]
- 64.Yang YJ et al. (2020) Altered insulin-like growth factor-2 signaling is associated with psychopathology and cognitive deficits in patients with schizophrenia. PloS one 15(3), e0226688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vafaee F et al. (2020) Insulin-Like Growth Factor 2 (IGF-2) Regulates Neuronal Density and IGF-2 Distribution Following Hippocampal Intracerebral Hemorrhage. J. Stroke. Cerebrovasc. Dis 29(10), 105128. [DOI] [PubMed] [Google Scholar]
- 66.Åberg D et al. (2021) Insulin-Like Growth Factor-II and Ischemic Stroke-A Prospective Observational Study. Life (Basel) 11(6), 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Martín-Montañez E et al. (2017) IGF-II promotes neuroprotection and neuroplasticity recovery in a long-lasting model of oxidative damage induced by glucocorticoids. Redox Biol. 13, 69–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guan SZ et al. (2021) The mechanism of enriched environment repairing the learning and memory impairment in offspring of prenatal stress by regulating the expression of activity-regulated cytoskeletal-associated and insulin-like growth factor-2 in hippocampus. Environ. Health Prev. Med 26(1), 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mackay KB et al. (2003) Neuroprotective effects of insulin-like growth factor-binding protein ligand inhibitors in vitro and in vivo. J. Cereb. Blood Flow Metab 23, 1160–1167 [DOI] [PubMed] [Google Scholar]
- 70.Guan J et al. (1996) The effects of insulin-like growth factor (IGF)-1, IGF-2, and des-IGF-1 on neuronal loss after hypoxic-ischemic brain injury in adult rats: Evidence for a role for IGF binding proteins. Endocrinology 137, 893–898 [DOI] [PubMed] [Google Scholar]
- 71.Li Y et al. (2017) The behavioral deficits and cognitive impairment are correlated with decreased IGF-II and ERK in depressed mice induced by chronic unpredictable stress. Int. J. Neurosci 127(12), 1096–1103 [DOI] [PubMed] [Google Scholar]
- 72.Poggini S et al. (2019) Combined Fluoxetine and Metformin Treatment Potentiates Antidepressant Efficacy Increasing IGF2 Expression in the Dorsal Hippocampus. Neural Plast. 2019, 4651031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Allodi I et al. (2016) Differential neuronal vulnerability identifies IGF-2 as a protective factor in ALS. Sci. Rep 6, 25960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Osborn TM et al. (2018) Increased motor neuron resilience by small molecule compounds that regulate IGF-II expression. Neurobiol. Dis 110, 218–230 [DOI] [PubMed] [Google Scholar]
- 75.Near SL et al. (1992) Insulin-like growth factor II stimulates motor nerve regeneration. Proc. Natl. Acad. Sci. USA 89(24), 11716–11720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Glazner GW et al. (1993) Insulin-like growth factor II increases the rate of sciatic nerve regeneration in rats. Neuroscience 54(3), 791–797 [DOI] [PubMed] [Google Scholar]
- 77.Hedlund E et al. (2010) Global gene expression profiling of somatic motor neuron populations with different vulnerability identify molecules and pathways of degeneration and protection. Brain 133(Pt 8), 2313–2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Martin-Montañez E et al. (2014) Involvement of IGF-II receptors in the antioxidant and neuroprotective effects of IGF-II on adult cortical neuronal cultures. Biochim. Biophys. Acta 1842(7), 1041–1051 [DOI] [PubMed] [Google Scholar]
- 79.Berg EL et al. (2021) Insulin-like growth factor-2 does not improve behavioral deficits in mouse and rat models of Angelman Syndrome. Mol. Autism 12(1), 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Redell JB et al. (2021) Insulin-Like Growth Factor-2 (IGF-2) Does Not Improve Memory in the Chronic Stage of Traumatic Brain Injury in Rodents. Neurotrauma Rep. 2(1), 453–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Molnár K et al. (2020) Pericyte-secreted IGF2 promotes breast cancer brain metastasis formation. Mol. Oncol 14(9), 2040–2057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lauterio TJ. et al. (1987) Marson L, Daughaday WH, Baile CA. Evidence for the role of insulin-like growth factor II (IGF-II) in the control of food intake. Physiol Behav. 40(6):755–8 [DOI] [PubMed] [Google Scholar]
- 83.Honda K et al. (2021) Central administration of insulin-like growth factor-2 suppresses food intake in chicks. Neurosci. Lett 751, 135797. [DOI] [PubMed] [Google Scholar]
- 84.Akanji AO et al. (2007) Associations of blood levels of insulin-like growth factor (IGF)-I, IGF-II and IGF binding protein (IGFBP)-3 in schizophrenic Arab subjects. Clin. Chem. Lab. Med 45(9), 1229–1231 [DOI] [PubMed] [Google Scholar]
- 85.Riikonen R et al. (2006) Cerebrospinal fluid insulin-like growth factors IGF-1 and IGF-2 in infantile autism. Dev. Med. Child. Neurol 48(9), 751–755 [DOI] [PubMed] [Google Scholar]
- 86.Steen E et al. (2005) Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes? J. Alzheimers Dis 7(1), 63–80 [DOI] [PubMed] [Google Scholar]
- 87.Agbemenyah HY et al. (2014) Insulin growth factor binding protein 7 is a novel target to treat dementia. Neurobiol. Dis 62, 135–143 [DOI] [PubMed] [Google Scholar]
- 88.Heywood WE et al. (2015) Identification of novel CSF biomarkers for neurodegeneration and their validation by a high-throughput multiplexed targeted proteomic assay. Mol. Neurodegener 10, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jiang YH et al. (1998) Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 21:799. [DOI] [PubMed] [Google Scholar]
- 90.Born HA. et al. (2017) Strain-dependence of the Angelman Syndrome phenotypes in Ube3a maternal deficiency mice. Sci. Rep 7(1):8451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stoppel DC. and Anderson MP. (2017) Hypersociability in the Angelman syndrome mouse model. Exp. Neurol 293:137–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kumar V et al. (2019) Simvastatin Restores HDAC1/2 Activity and Improves Behavioral Deficits in Angelman Syndrome Model Mouse. Front. Mol. Neurosci 12, 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Soto C (2003) Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci 4(1), 49–60. [DOI] [PubMed] [Google Scholar]
- 94.Pandey K et al. (2021) Autophagy coupled to translation is required for long-term memory. Autophagy 17(7), 1614–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cioni JM. et al. (2019) Late Endosomes Act as mRNA Translation Platforms and Sustain Mitochondria in Axons. Cell 176(1–2), 56–72.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Saitoh S (2022) Endosomal Recycling Defects and Neurodevelopmental Disorders. Cells 11(1), 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nixon RA (2013) The role of autophagy in neurodegenerative disease. Nature Med. 19(8), 983–997 [DOI] [PubMed] [Google Scholar]
- 98.Nixon RA (2017) Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer’s disease: inseparable partners in a multifactorial disease. FASEB J. 31(7), 2729–2743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wong E and Cuervo AM (2010) Autophagy gone awry in neurodegenerative diseases. Nature Neurosci. 13(7), 805–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lee JH et al. (2022) Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques. Nature Neurosci. 25(6), 688–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Müntjes K et al. (2021) Linking transport and translation of mRNAs with endosomes and mitochondria. EMBO Rep. 5;22(10):e52445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rangaraju V et al. (2019) Spatially stable mitochondrial compartments fuel local translation during plasticity. Cell. 176(1–2), 73–84 [DOI] [PubMed] [Google Scholar]
- 103.Zhu Y et al. (2021) IGF2 deficiency causes mitochondrial defects in skeletal muscle. Clinical Science. 135(7), 979–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gui W et al. (2021) Knockdown of the insulin-like growth factor 2 gene disrupts mitochondrial functions in the liver. Journal of Molecular Cell Biology. 13(8), 543–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Martin-Montanez E et al. (2021) Insulin-like growth factor II prevents oxidative and neuronal damage in cellular and mice models of Parkinson’s disease. Redox Biol. 46:102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Garcia-Fernandez M et al. (2011) Liver mitochondrial dysfunction is reverted by insulin-like growth factor II (IGF-II) in aging rats. J Transl Med. 9, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Castilla-Cortazar I et al. (2011) Hepatoprotection and neuroprotection induced by low doses of IGF-II in aging rats. J. Trans Med 9, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]